
Discovery of a 2′-Fluoro,2′-Bromouridine Phosphoramidate Prodrug Exhibiting Anti-Yellow Fever Virus Activity in Culture and in Mice
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus, and Media
2.2. Synthesis of Compounds
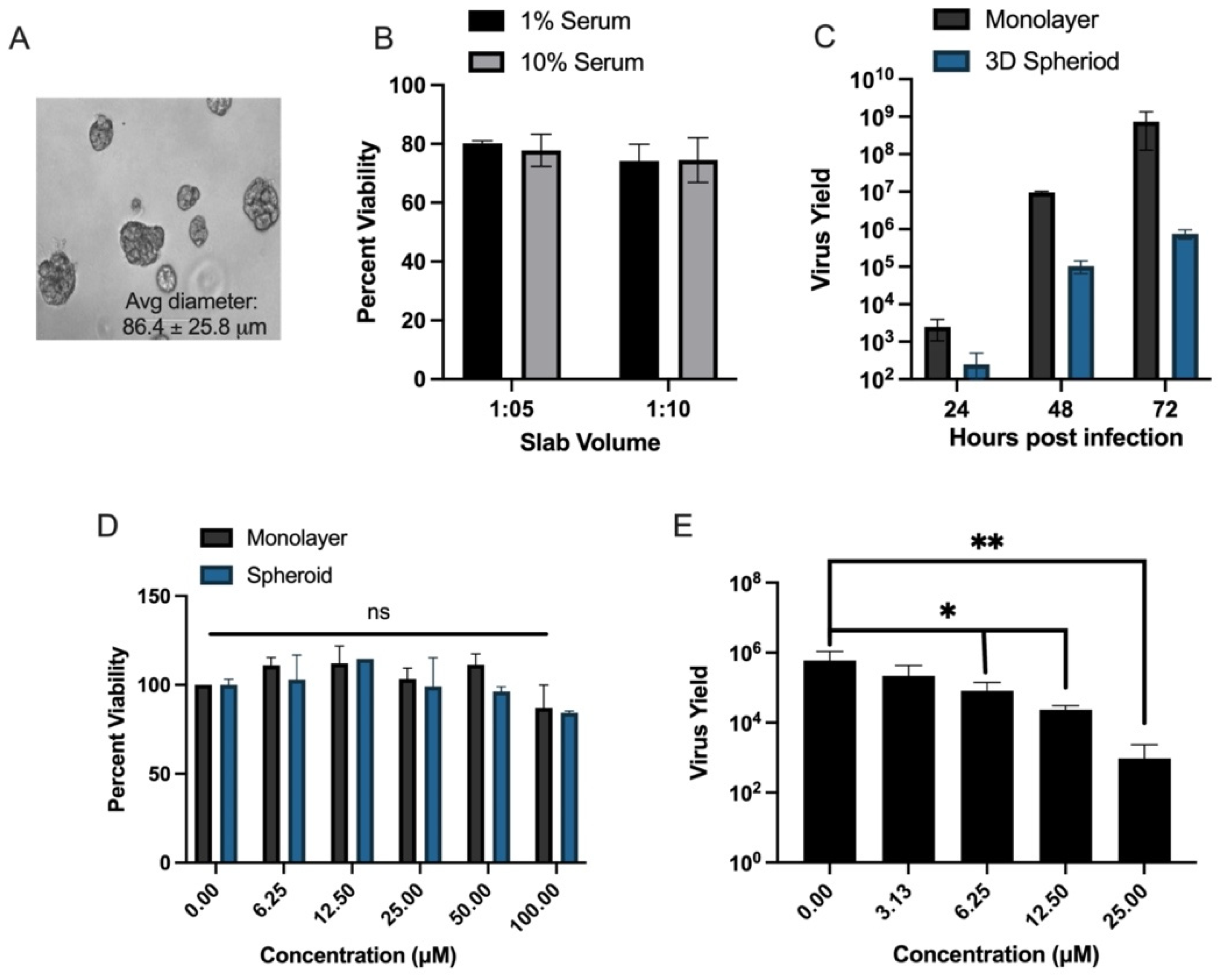
2.3. 3D Hepatoma Spheroid Generation
2.4. MTS Cytotoxicity Assay
2.5. Antiviral Evaluation Assay
2.6. qRT-PCR
2.7. Cellular Pharmacology Assay
2.8. Molecular Modeling
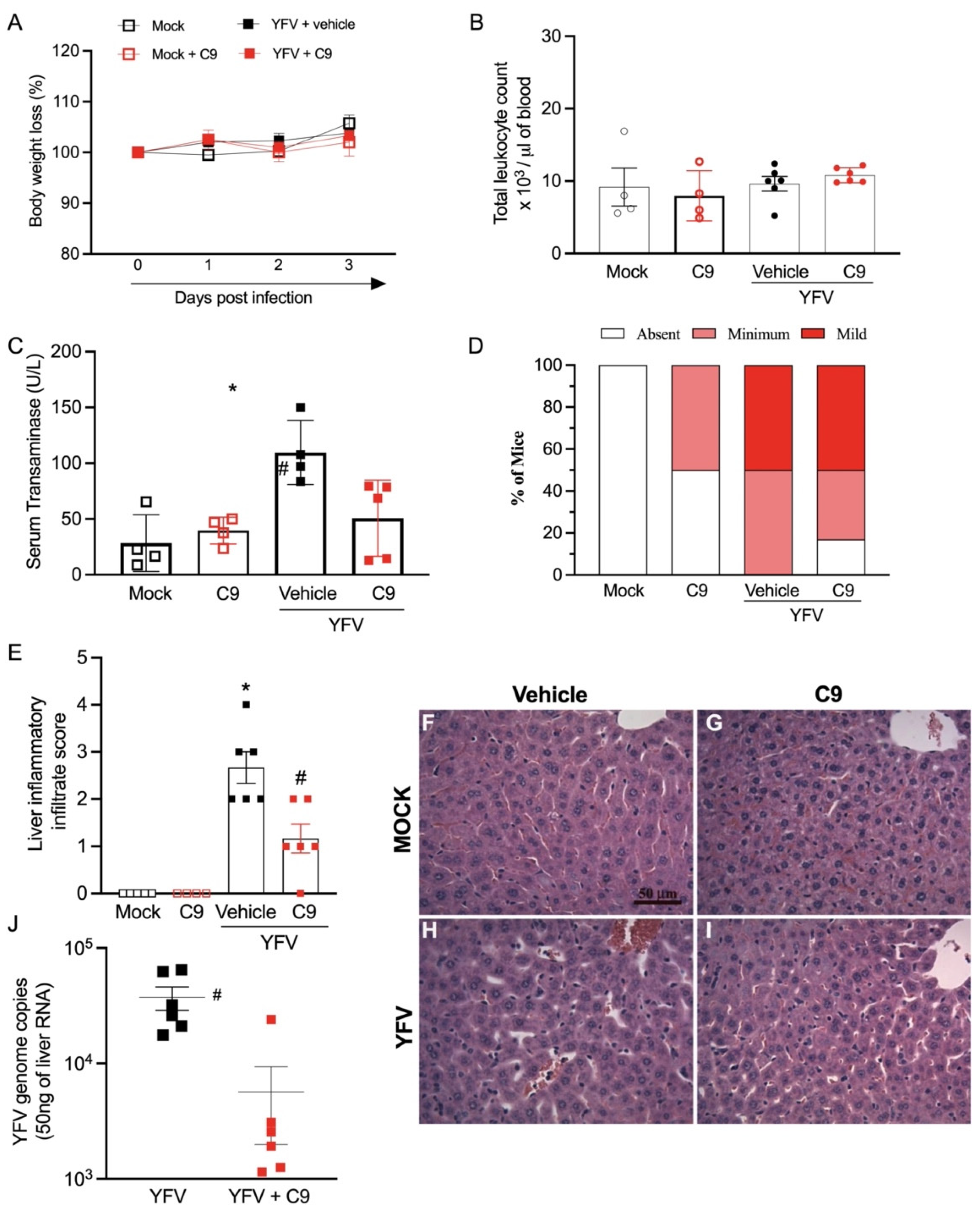
2.9. In Vivo Infection of A129 Mice with YFV 17D
2.9.1. Ethics Statement
2.9.2. Mice and YFV Mouse Infection
2.9.3. Hematological Analysis
2.9.4. Alanine Aminotransferase Measurement
2.9.5. Tissue-Specific Real-Time RT-PCR
2.9.6. Histopathological Liver Analysis
2.9.7. Statistical Analysis
3. Results
3.1. Antiviral and Cytotoxicity of Compounds 1–11
3.2. In Vitro Cellular Metabolism of Lead Compounds
3.3. Prediction of Binding Mode of C1(Sofosbuvir) and C8/9 Compounds
3.4. Evaluation of In Vitro Efficacy of Lead Compound C9 for Anti-YFV Activity in 3D Hepatoma Spheroid Model and Primary Human Macrophages
3.5. In Vivo Anti-YFV Activity of C9 in A129 Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Pan American Health Organization. Yellow Fever. Available online: https://www.paho.org/en/topics/yellow-fever (accessed on 10 November 2021).
- Center for Disease Control and Prevention. Yellow Fever. Available online: https://www.cdc.gov/yellowfever/index.html (accessed on 10 November 2021).
- World Health Organization. World Health Organization Initiative: Eliminate Yellow Fever (EYE) Epidemics 2017–2026. Available online: https://www.who.int/initiatives/eye-strategy (accessed on 10 November 2021).
- Servadio, J.L.; Munoz-Zanzi, C.; Convertino, M. Estimating case fatality risk of severe yellow fever cases: Systematic literature review and meta-analysis. BMC Infect. Dis. 2021, 21, 819. [Google Scholar] [CrossRef] [PubMed]
- Gubler, D.J.; Kuno, G.; Markoff, L. Flaviviruses. In Fields Virology, 5th ed.; Knipe, D.M.a.H., Peter, M., Eds.; Lippincott Williams & Williams: Philadelphia, PA, USA, 2007; Volume 1. [Google Scholar]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.K.; Seley-Radtke, K.L. The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antivir. Res. 2019, 162, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Stuyver, L.J.; McBrayer, T.R.; Tharnish, P.M.; Clark, J.; Hollecker, L.; Lostia, S.; Nachman, T.; Grier, J.; Bennett, M.A.; Xie, M.Y.; et al. Inhibition of hepatitis C replicon RNA synthesis by beta-D-2′-deoxy-2′-fluoro-2′-C-methylcytidine: A specific inhibitor of hepatitis C virus replication. Antivir. Chem. Chemother. 2006, 17, 79–87. [Google Scholar] [CrossRef]
- Mendes, E.A.; Pilger, D.R.B.; Santos Nastri, A.C.S.; Malta, F.M.; Pascoalino, B.D.S.; Carneiro D’Albuquerque, L.A.; Balan, A.; Freitas, L.H.G., Jr.; Durigon, E.L.; Carrilho, F.J.; et al. Sofosbuvir inhibits yellow fever virus in vitro and in patients with acute liver failure. Ann. Hepatol. 2019, 18, 816–824. [Google Scholar] [CrossRef]
- de Freitas, C.S.; Higa, L.M.; Sacramento, C.Q.; Ferreira, A.C.; Reis, P.A.; Delvecchio, R.; Monteiro, F.L.; Barbosa-Lima, G.; James Westgarth, H.; Vieira, Y.R.; et al. Yellow fever virus is susceptible to sofosbuvir both in vitro and in vivo. PLoS Negl. Trop. Dis. 2019, 13, e0007072. [Google Scholar] [CrossRef] [Green Version]
- Zandi, K.; Amblard, F.; Amichai, S.; Bassit, L.; Tao, S.; Jiang, Y.; Zhou, L.; Ollinger Russell, O.; Mengshetti, S.; Schinazi, R.F. Nucleoside analogs with antiviral activity against yellow fever virus. Antimicrob. Agents Chemother. 2019, 63, e00889-19. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Mahmoud, S.; Liu, P.; Zhou, L.; Ehteshami, M.; Bassit, L.; Tao, S.; Domaoal, R.A.; Sari, O.; Schutter, C.; et al. 2′-chloro,2′-fluoro ribonucleotide prodrugs with potent pan-genotypic activity against hepatitis C virus replication in culture. J. Med. Chem. 2017, 60, 5424–5437. [Google Scholar] [CrossRef]
- Mengshetti, S.; Zhou, L.; Sari, O.; De Schutter, C.; Zhang, H.; Cho, J.H.; Tao, S.; Bassit, L.C.; Verma, K.; Domaoal, R.A.; et al. Discovery of a series of 2′-alpha-fluoro,2′-beta-bromo-ribonucleosides and their phosphoramidate prodrugs as potent pan-genotypic inhibitors of hepatitis C virus. J. Med. Chem. 2019, 62, 1859–1874. [Google Scholar] [CrossRef]
- Pinho, P.; Kalayanov, G.; Westerlind, H.; Rosenquist, A.; Wahling, H.; Sund, C.; Almeida, M.; Ayesa, S.; Tejbrant, J.; Targett-Adams, P.; et al. Discovery of beta-D-2′-deoxy-2′-dichlorouridine nucleotide prodrugs as potent inhibitors of hepatitis C virus replication. Bioorg. Med. Chem. Lett. 2017, 27, 3468–3471. [Google Scholar] [CrossRef]
- Haile, W.B.; Gavegnano, C.; Tao, S.; Jiang, Y.; Schinazi, R.F.; Tyor, W.R. The Janus kinase inhibitor ruxolitinib reduces HIV replication in human macrophages and ameliorates HIV encephalitis in a murine model. Neurobiol. Dis. 2016, 92, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.C.; Andersen, M.N.; Moller, H.J. Monocyte isolation techniques significantly impact the phenotype of both isolated monocytes and derived macrophages in vitro. Immunology 2020, 159, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Zandi, K.; Amblard, F.; Musall, K.; Downs-Bowen, J.; Kleinbard, R.; Oo, A.; Cao, D.; Liang, B.; Russell, O.O.; McBrayer, T.; et al. Repurposing nucleoside analogs for human coronaviruses. Antimicrob. Agents Chemother. 2020, 65, e01652-20. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; McArthur, M.A.; Cohen, M.; Jahrling, P.B.; Janosko, K.B.; Josleyn, N.; Kang, K.; Zhang, T.; Holbrook, M.R. Characterization of yellow fever virus infection of human and non-human primate antigen presenting cells and their interaction with CD4+ T cells. PLoS Negl. Trop. Dis. 2016, 10, e0004709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovadia, R.; Khalil, A.; Li, H.; De Schutter, C.; Mengshetti, S.; Zhou, S.; Bassit, L.; Coats, S.J.; Amblard, F.; Schinazi, R.F. Synthesis and anti-HCV activity of beta-D-2′-deoxy-2′-alpha-chloro-2′-beta-fluoro and beta-D-2′-deoxy-2′-alpha-bromo-2′-beta-fluoro nucleosides and their phosphoramidate prodrugs. Bioorg. Med. Chem. 2019, 27, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cox, B.D.; Garnier-Amblard, E.C.; McBrayer, T.R.; Coats, S.J.; Schinazi, R.F.; Amblard, F. Synthesis and anti-HCV activity of a series of beta-D-2′-deoxy-2′-dibromo nucleosides and their corresponding phosphoramidate prodrugs. Bioorg. Med. Chem. Lett. 2017, 27, 5296–5299. [Google Scholar] [CrossRef] [PubMed]
- Cory, A.H.; Owen, T.C.; Barltrop, J.A.; Cory, J.G. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991, 3, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Tao, S.; Zandi, K.; Bassit, L.; Ong, Y.T.; Verma, K.; Liu, P.; Downs-Bowen, J.A.; McBrayer, T.; LeCher, J.C.; Kohler, J.J.; et al. Comparison of anti-SARS-CoV-2 activity and intracellular metabolism of remdesivir and its parent nucleoside. Curr. Res. Pharmacol. Drug Discov. 2021, 2, 100045. [Google Scholar] [CrossRef]
- Tao, S.; Zhou, L.; Zhang, H.; Zhou, S.; Amiralaei, S.; Shelton, J.; Ehteshami, M.; Jiang, Y.; Amblard, F.; Coats, S.J.; et al. Intracellular metabolism and potential cardiotoxicity of a beta-D-2′-C-methyl-2,6-diaminopurine ribonucleoside phosphoramidate that inhibits hepatitis C virus replication. Nucleosides Nucleotides Nucleic Acids 2020, 39, 204–224. [Google Scholar] [CrossRef]
- Eldrup, A.B.; Prhavc, M.; Brooks, J.; Bhat, B.; Prakash, T.P.; Song, Q.; Bera, S.; Bhat, N.; Dande, P.; Cook, P.D.; et al. Structure-activity relationship of heterobase-modified 2′-C-methyl ribonucleosides as inhibitors of hepatitis C virus RNA replication. J. Med. Chem. 2004, 47, 5284–5297. [Google Scholar] [CrossRef] [PubMed]
- Olsen, D.B.; Eldrup, A.B.; Bartholomew, L.; Bhat, B.; Bosserman, M.R.; Ceccacci, A.; Colwell, L.F.; Fay, J.F.; Flores, O.A.; Getty, K.L.; et al. A 7-deaza-adenosine analog is a potent and selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties. Antimicrob. Agents Chemother. 2004, 48, 3944–3953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.L.; Hollecker, L.; Mason, J.C.; Stuyver, L.J.; Tharnish, P.M.; Lostia, S.; McBrayer, T.R.; Schinazi, R.F.; Watanabe, K.A.; Otto, M.J.; et al. Design, synthesis, and antiviral activity of 2′-deoxy-2′-fluoro-2′-C-methylcytidine, a potent inhibitor of hepatitis C virus replication. J. Med. Chem. 2005, 48, 5504–5508. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.S.; Tomassini, J.E.; Bosserman, M.; Getty, K.; Stahlhut, M.W.; Eldrup, A.B.; Bhat, B.; Hall, D.; Simcoe, A.L.; LaFemina, R.; et al. Inhibition of hepatitis C virus RNA replication by 2′-modified nucleoside analogs. J. Biol. Chem. 2003, 278, 11979–11984. [Google Scholar] [CrossRef] [Green Version]
- Pierra, C.; Benzaria, S.; Amador, A.; Moussa, A.; Mathieu, S.; Storer, R.; Gosselin, G. NM-283, an efficient prodrug of the potent anti-HCV agent 2′-C-methylcytidine. Nucleosides Nucleotides Nucleic Acids 2005, 24, 767–770. [Google Scholar] [CrossRef]
- Murakami, E.; Bao, H.; Ramesh, M.; McBrayer, T.R.; Whitaker, T.; Micolochick Steuer, H.M.; Schinazi, R.F.; Stuyver, L.J.; Obikhod, A.; Otto, M.J.; et al. Mechanism of activation of beta-D-2′-deoxy-2′-fluoro-2′-C-methylcytidine and inhibition of hepatitis C virus NS5B RNA polymerase. Antimicrob. Agents Chemother. 2007, 51, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Sofia, M.J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P.G.; Ross, B.S.; Wang, P.; Zhang, H.R.; et al. Discovery of a beta-D-2′-deoxy-2′-alpha-fluoro-2′-beta-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 2010, 53, 7202–7218. [Google Scholar] [CrossRef]
- Dubankova, A.; Boura, E. Structure of the yellow fever NS5 protein reveals conserved drug targets shared among flaviviruses. Antivir. Res. 2019, 169, 104536. [Google Scholar] [CrossRef]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.S.; Malindisa, S.T.; Ntwasa, M. Two-dimensional (2D) and three-dimensional (3D) cell culturing in drug discovery. In Cell Culture; Mehanna, R.M., Ed.; IntechOpen: London, UK, 2018; Volume 2, pp. 1–22. [Google Scholar]
- Meier, K.C.; Gardner, C.L.; Khoretonenko, M.V.; Klimstra, W.B.; Ryman, K.D. A mouse model for studying viscerotropic disease caused by yellow fever virus infection. PLoS Pathog. 2009, 5, e1000614. [Google Scholar] [CrossRef]
- Lemos, F.O.; Franca, A.; Lima Filho, A.C.M.; Florentino, R.M.; Santos, M.L.; Missiaggia, D.G.; Rodrigues, G.O.L.; Dias, F.F.; Souza Passos, I.B.; Teixeira, M.M.; et al. Molecular mechanism for protection against liver failure in human yellow fever infection. Hepatol. Commun. 2020, 4, 657–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coats, S.J.; Garnier-Amblard, E.C.; Amblard, F.; Ehteshami, M.; Amiralaei, S.; Zhang, H.; Zhou, L.; Boucle, S.R.; Lu, X.; Bondada, L.; et al. Chutes and ladders in hepatitis C nucleoside drug development. Antivir. Res. 2014, 102, 119–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Zhang, H.; Li, C.; De Schutter, C.; Sari, O.; Mengshetti, S.; Zhou, S.; Kasthuri, M.; Coats, S.J.; Schinazi, R.F.; et al. Diastereoselective synthesis of 2′-dihalopyrimidine ribonucleoside inhibitors of hepatitis C virus replication. ACS Omega 2022, 7, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Del Sarto, J.L.; Rocha, R.P.F.; Bassit, L.; Olmo, I.G.; Valiate, B.; Queiroz-Junior, C.M.; Pedrosa, C.; Ribeiro, F.M.; Guimaraes, M.Z.; Rehen, S.; et al. 7-Deaza-7-fluoro-2′-C-methyladenosine inhibits Zika virus infection and viral-induced neuroinflammation. Antivir. Res. 2020, 180, 104855. [Google Scholar] [CrossRef] [PubMed]
- Zandi, K.; Bassit, L.; Amblard, F.; Cox, B.D.; Hassandarvish, P.; Moghaddam, E.; Yueh, A.; Libanio Rodrigues, G.O.; Passos, I.; Costa, V.V.; et al. Nucleoside analogs with selective antiviral activity against dengue fever and Japanese encephalitis viruses. Antimicrob. Agents Chemother. 2019, 63, e00397-19. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Anti-YFV Activity a (Huh-7) | Cytotoxicity b: CC50 (µM) c,e | ||||
|---|---|---|---|---|---|---|
| EC50 (µM) d | EC90 (µM) d | PBM | CEM | Vero | Huh-7 | |
| Sofosbuvir 1 | 0.6 ± 0.6 | 3.5 ± 1.5 | >100 | >100 | >100 | 88.5 ± 11.6 |
| 2 | >10 | >10 | >100 | >100 | >100 | >100 |
| 3 | 0.4 ± 0.5 | 3.2 ± 2.7 | >100 | >100 | >100 | >100 |
| 4 | >10 | >10 | >100 | >100 | >100 | >100 |
| 5 | 0.4 ± 0.2 | 6.5 ± 2.6 | >100 | >100 | >100 | 46.7 ± 13.3 |
| 6 | >10 | >10 | >100 | >100 | >100 | >100 |
| 7 | >10 | >10 | >100 | >100 | >100 | 2.2 ± 0.9 |
| 8 | >10 | >10 | >100 | >100 | >100 | >100 |
| 9 | 0.9 ± 0.8 | 7.8 ± 3.6 | >100 | >100 | >100 | 68.9 ± 6.3 |
| 10 | >10 | >10 | >100 | >100 | >100 | >100 |
| 11 | >10 | >10 | >100 | >100 | >100 | 8.7 ± 1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
LeCher, J.C.; Zandi, K.; Costa, V.V.; Amblard, F.; Tao, S.; Patel, D.; Lee, S.; da Silva Santos, F.R.; Goncalves, M.R.; Queroz-Junior, C.M.; et al. Discovery of a 2′-Fluoro,2′-Bromouridine Phosphoramidate Prodrug Exhibiting Anti-Yellow Fever Virus Activity in Culture and in Mice. Microorganisms 2022, 10, 2098. https://doi.org/10.3390/microorganisms10112098
LeCher JC, Zandi K, Costa VV, Amblard F, Tao S, Patel D, Lee S, da Silva Santos FR, Goncalves MR, Queroz-Junior CM, et al. Discovery of a 2′-Fluoro,2′-Bromouridine Phosphoramidate Prodrug Exhibiting Anti-Yellow Fever Virus Activity in Culture and in Mice. Microorganisms. 2022; 10(11):2098. https://doi.org/10.3390/microorganisms10112098
Chicago/Turabian StyleLeCher, Julia C., Keivan Zandi, Vivian Vasconcelos Costa, Franck Amblard, Sijia Tao, Dharmeshkumar Patel, Sujin Lee, Felipe Rocha da Silva Santos, Matheus Rodrigues Goncalves, Celso Martins Queroz-Junior, and et al. 2022. "Discovery of a 2′-Fluoro,2′-Bromouridine Phosphoramidate Prodrug Exhibiting Anti-Yellow Fever Virus Activity in Culture and in Mice" Microorganisms 10, no. 11: 2098. https://doi.org/10.3390/microorganisms10112098