Effects of Short-Range Order on the Magnetic and Mechanical Properties of FeCoNi(AlSi)x High Entropy Alloys


1
Shenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, Shenyang 110016, China
2
College of Science, University of Science and Technology Liaoning, Anshan 114051, China
3
University of Chinese Academy of Sciences, Beijing 100049, China
4
College of Science, Northeastern University, Shenyang 110819, China
*
Author to whom correspondence should be addressed.
Metals 2017, 7(11), 482; https://doi.org/10.3390/met7110482
Submission received: 19 September 2017
/
Revised: 17 October 2017
/
Accepted: 2 November 2017
/
Published: 6 November 2017
(This article belongs to the Special Issue Microstructure based Modeling of Metallic Materials)
Abstract
:The properties of a material are sensitive to chemically-ordered structure in multi-element alloys. Understanding the effects of chemical short-range order (SRO) on magnetic and mechanical properties is important. In this work, we use the Monte Carlo method in combination with density functional theory to investigate atomic nearest neighbor distribution, magnetic moment and elastic modulus in FeCoNi (AlSi)x alloys. It is found that the prominent feature of the FeCoNi (AlSi)x alloys is the change of SRO parameters: the SRO parameters are positive between Al-Al, Al-Si, Si-Si pairs and negative between Ni-Al, Co-Si, Fe-Co, Ni-Si and Fe-Si pairs. The Al and Si elements tend to bond with Fe, Co, Ni elements to form an SRO structure. The change of the atomic nearest neighbor environment leads to a reduction in the atomic magnetic moments of magnetic elements. The calculated saturation magnetizations by considering the effect of SRO are in good accord with the experimental values. We further show that SRO leads to an increase of the elastic modulus, by sacrificing ductility and isotropy. In the study of the structure and properties of high entropy alloys, the effect of SRO should not be ignored.
1. Introduction
In recent years, there have been extensive experimental investigations of multi-component alloys after the new alloy design concept, high entropy alloys (HEAs), was proposed [1,2]. HEAs consist of at least five principal elements, each element with a 5–35% concentration. These alloys have many exceptional properties compared with the traditional alloys due to the high mixing entropy, sluggish diffusion and lattice distortion effects, including good thermal stability, high mechanical strength and excellent corrosion resistance [3,4,5]. The properties of HEAs, such as hardness, ductility and magnetic properties can be tuned by changing the type and concentration of components [6,7,8]. These novel properties of HEAs have potential applications in refractory materials, wear-resistant materials and magnetic materials.
In general, HEAs are considered to be random solid solutions (RSS), and all constituent atoms randomly distribute on the available lattice sites. But is this really true? In fact, the diversity in atomic radii and the different attractive interactions between the constituent elements will result in a short-range ordered structure [9]. The SRO structures have been found in many engineering materials such as FeV, FeAl and FeCr alloys [10,11,12]. It will significantly affect the structural stability, magnetic and mechanical properties. The presence of SRO in these binary alloys suggests that some ordered structures may also exist in HEAs. Manzoni et al. reported that there is an Al-Ni rich region in AlCoCrFeNi alloy [13]. Zuo et al. found that there are Fe-Co and Al-Ni-Si rich phases in FeCoNi(AlSi)0.2 alloy [14]. Zhang et al. provided the experimental evidence of SRO in NiCoCr alloy [15]. Their work showed that Cr atoms separate from each other, but bond with Ni and Co atoms to form SRO structures. The SRO will decrease the configurational entropy and change the free energy of high entropy alloys [16]. It is very important to study the formation of SRO structure and investigate the effects of SRO on the properties of HEAs. However, the study of SRO is relatively scarce in high entropy alloys [17], which is due to the difficulty of probing SRO behavior by conventional X-ray scattering or neutron diffraction, and due to the complex analysis of experimental data. Reverse Monte Carlo (RMC) modelling [18] is a useful method to fit atomic scale structure by inverting experimental diffraction data, using which researchers can further understand the relationship between local atomic structure and material properties. Kaban et al. studied Co43Fe20Ta5.5B31.5 metallic glass with RMC by resolving ten partial pair distribution functions (PDF) to determine the SRO [19]. However, with the increase of constituent elements, it becomes more difficult to resolve the partial distribution function and calculate the total structural factor, which limits its application in HEAs.
First-principles calculation based on density functional theory (DFT) is a powerful tool to explore the structure and properties of materials [20,21]. By constructing appropriate atomic structures, researchers can study the structural stability and alloy properties. Recently, some hybrid Monte Carlo (MC) methods in combination with density functional theory calculation were performed to study SRO behavior in high entropy alloys [9,16]. Widom et al. studied refractory metal Mo-Nb-Ta-W with hybrid Monte Carlo/molecular dynamics method. The simulation result showed that a cesium-chloride ordered structure emerge between the mixed (Mo,W) sites and the mixed (Nb,Ta) sites. Tamm et al. investigated the SRO behavior in NiCrCo and NiCrCoFe alloys and found obvious short-range order between Ni-Cr, Cr-Co and Ni-Fe pairs. Compared to the classical Monte Carlo method combined with empirical interaction potentials, the first-principles calculation can obtain more accurate potential energy and the Monte Carlo swaps of different atomic species, allowing researchers to probe the SRO in high entropy alloys. The hybrid method makes the atomic structure evolve toward the global energy minimum in the simulation. These results demonstrated that the hybrid method is suitable for the study of SRO in HEAs.
Recently, some FeCoNi-based high entropy alloys with attractive soft magnetic properties have been obtained [22,23,24]. In the FeCoNi(AlSi)x (x is in the molar ratio of Al and Si elements) alloys, by adding Al and Si elements into FeCoNi alloy, researchers can adjust lattice distortion and increase electrical resistivity, thus forming appealing soft magnetic alloys. However, Al and Si elements have strong attractive interactions with Fe, Co and Ni elements, which can be seen from the more negative binary mixing enthalpies of Ni-Si, Co-Si and Ni-Al pairs with −40, −38 and −22 kJ/mol, respectively. Al and Si atoms prefer to be surrounded by Fe, Co and Ni atoms to reduce the potential energy, which will lead to the formation of an SRO structure. In this work, we investigate the SRO behavior of FeCoNi(AlSi)x alloys by using the MC method combined with density functional theory calculation. The MC method is carried out to search for the lower energy structure, and density functional theory is used for structure relaxation and the calculation of system energy. We calculate the formation enthalpies, SRO parameters, magnetic moments, and elastic moduli of both initial structures and the structures with SRO. It is expected that the present work can deepen the understanding of SRO behavior in high entropy alloys.
2. Methodology
The two competitive structures for FeCoNi(AlSi)x alloys are the face centered cubic (FCC) phase and body centered cubic (BCC) phase. The FCC and BCC supercell structures of FeCoNi(AlSi)x alloys (x = 0, 0.1, 0.2, 0.4, 0.5 and 0.8) were built according to the maximum entropy (MaxEnt) method. Details of the MaxEnt method can be found in References [25,26,27]. The supercell structures consist of 108 atoms for the FCC phase and 128 atoms for the BCC phase. For comparison, the 92-atom FCC and BCC special quasi-random structures (SQS) of FeCoNi(AlSi)0.8 alloy were generated by using the mcsqs code with the Alloy-Theoretic Automated Toolkit (ATAT) package [28].
The structure relaxation and energy calculation were performed using DFT implemented with the Vienna Ab-Initio Simulation Package (VASP, University of Vienna, version: 5.4.1.05, Vienna, Austria) [21]. The exchange-correlation functional was treated within the generalized gradient approximation as described by Perdew–Burke–Ernzerhof (PBE) [29]. Energy cutoffs for the plane wave in all calculations were set to 300 eV. The reciprocal space energy integration was performed by the Monkhorst-Pack technique with 2 × 2 × 2 mesh. The self-consistence convergence criterion for electron iterations was set to 10−5 eV per atom. The valence electrons of Fe, Co, Ni, Al and Si atoms were eight, nine, 10, three and four, respectively. From Wikipedia, we know that the Curie temperature of Fe, Co and Ni are 1043, 1388 and 628 K. Körmann et al. predicted the Curie temperature of FeCoNi is 868 K [30]. The Curie temperature of Co2FeSi and Co2FeAl are all above 1000 K. Dobrzynski et al. reported that the Curie temperature of Fe2.94Al0.38Si0.68 is 794 K [31]. There is not any Curie temperature of FeCoNi (AlSi)x alloys reported. However, Zhang et al. showed that the saturation magnetizations of FeCoNi (AlSi)x alloys range from 1.32 to 0.46 T (x = 0 to 0.8) [22]. We predict that the Curie temperature of FeCoNi (AlSi)x alloy is above 300 K. Thus, the magnetic state must be considered. All the calculations done were spin-polarized, with the magnetic moments of Fe, Co and Ni atoms initialized with 3, 2 -and 1 , respectively. The calculations in this work did not include the orbital magnetic moment.
In order to probe SRO in FeCoNi (AlSi)x alloys, the Monte Carlo method was carried out in the simulation. The generation of a new atomic configuration is a two-step process. Firstly, the potential energy with the current atomic structure was calculated. In the next step, two different kinds of constituent atoms were selected and the potential energy after the swap of their positions was calculated. The position swap is based on the Metropolis–Hastings sampling [32] with the acceptance probability ; is the energy difference between swapped and not-swapped structures, The FeCoNi (FCC), FeCoNi(AlSi)0.2 (FCC), FeCoNi(AlSi)0.4 (BCC) and FeCoNi(AlSi)0.8 (BCC) structures generated by the MaxEnt method, and FeCoNi(AlSi)0.8 (BCC) generated by the mcsqs method, were selected as the initial starting point in the MC simulations. Each MC simulation ran for 3000 MC steps. The structure relaxations and energy calculations for the final structures with SRO and the initial structures were fully conducted with a higher number of k-points () to increase accuracy.
The Warren–Cowley SRO parameter [33] was used to described the degree of short-range order in the FeCoNi(AlSi)x alloys. The nearest neighbor SRO parameter can be calculated by Equation (1), where cj is the molar fraction of the type j element and is the probability of finding the type j element around the type i element in the nearest neighbor shell. The SRO parameter vanishes if , meaning that there is no site preference between the type i and type j element. The negative indicates the increase in the number of i and j pairs, while the positive value corresponds to the opposite.
Formation enthalpy is defined in Equation (2), where is the zero-temperature total energy per atom, ci is the molar fraction of the type i element. is the ground state energy of type i element in its stable phase and is calculated by using the same DFT code and pseudopotential.
3. Results and Discussion
3.1. Single-Phase High Entropy Alloy (HEA) Stability
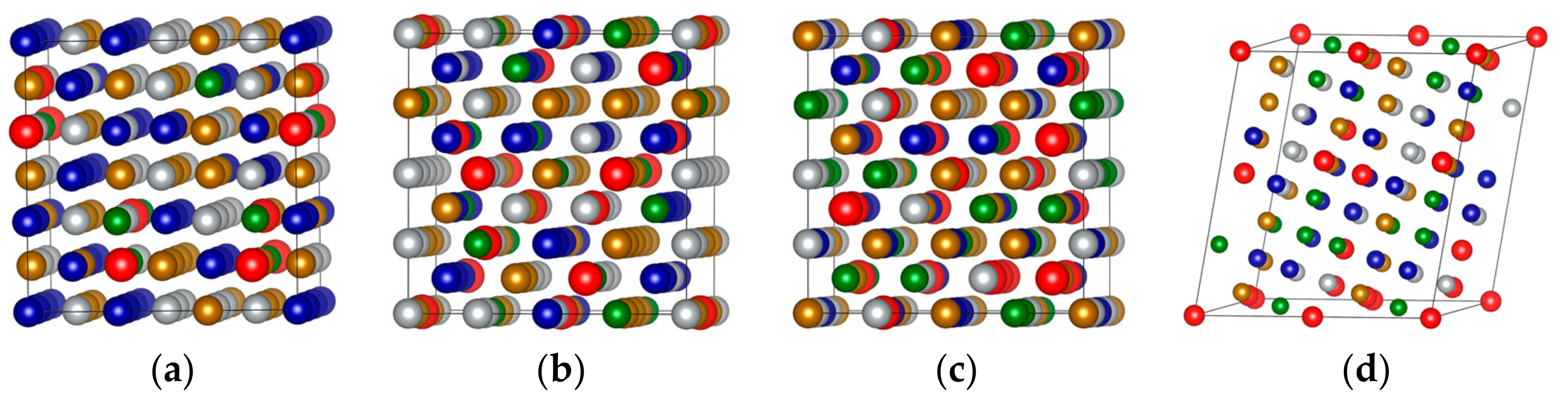
It was reported that non-equimolar FeCoNi(AlSi)x alloys form the FCC phase for and the BCC phase for [22]. In Figure 1, we present the MaxEnt structures of FeCoNi (AlSi)x for x = 0.2 (FCC), x = 0.4 (BCC) and x = 0.8 (BCC) and SQS structure with x = 0.8 (BCC) in their ideal, unrelaxed forms. We performed the structure relaxation and total energy calculation by using the settings described in Section 2. Table 1 shows the lattice parameters, formation enthalpies with BCC and FCC structures and the energy difference between the BCC and FCC phases . The positive indicates that the FCC structure is more stable, and the negative shows that the BCC structure is more stable. As shown in Table 1, when the fraction of Al and Si elements is smaller , the energy difference is positive, indicating that the FCC structure is more stable. When the fraction of Al and Si elements is larger (), the energy difference is negative, demonstrating that the BCC structure is more stable. The experimental lattice parameters are Å for x = 0 [34] and Å for x = 0.2 [14]. The calculated results are Å (x = 0) and Å (x = 0.2). When x = 0.8, there is no experimental lattice parameter available, however, the calculated lattice parameter Å with the MaxEnt structure and Å with the SQS structure were obtained. The consistency of the calculated and experimental values validates the MaxEnt models employed.
3.2. Short-Range Order (SRO) in FeCoNi(AlSi)x Alloys
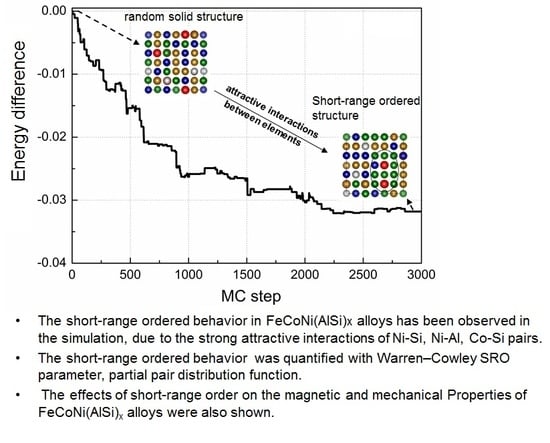
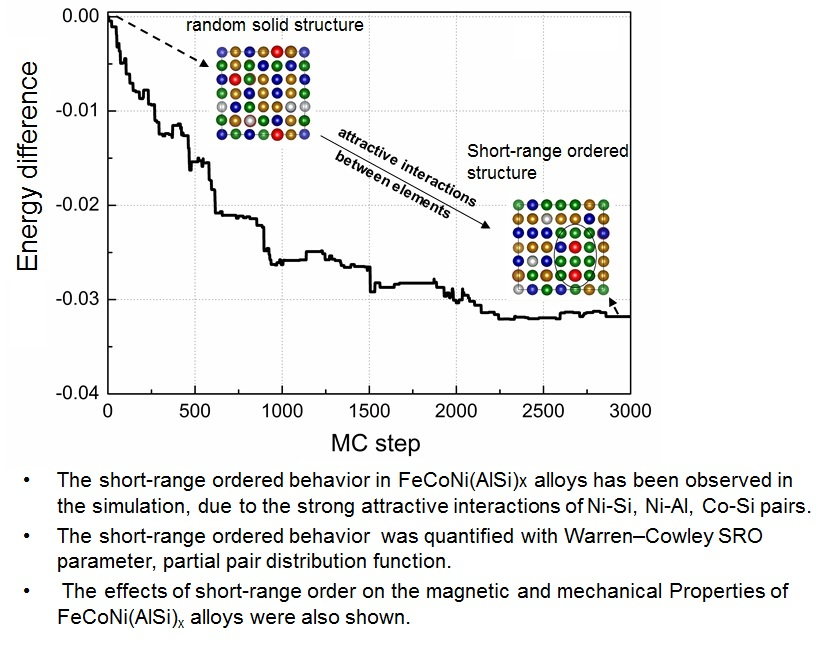
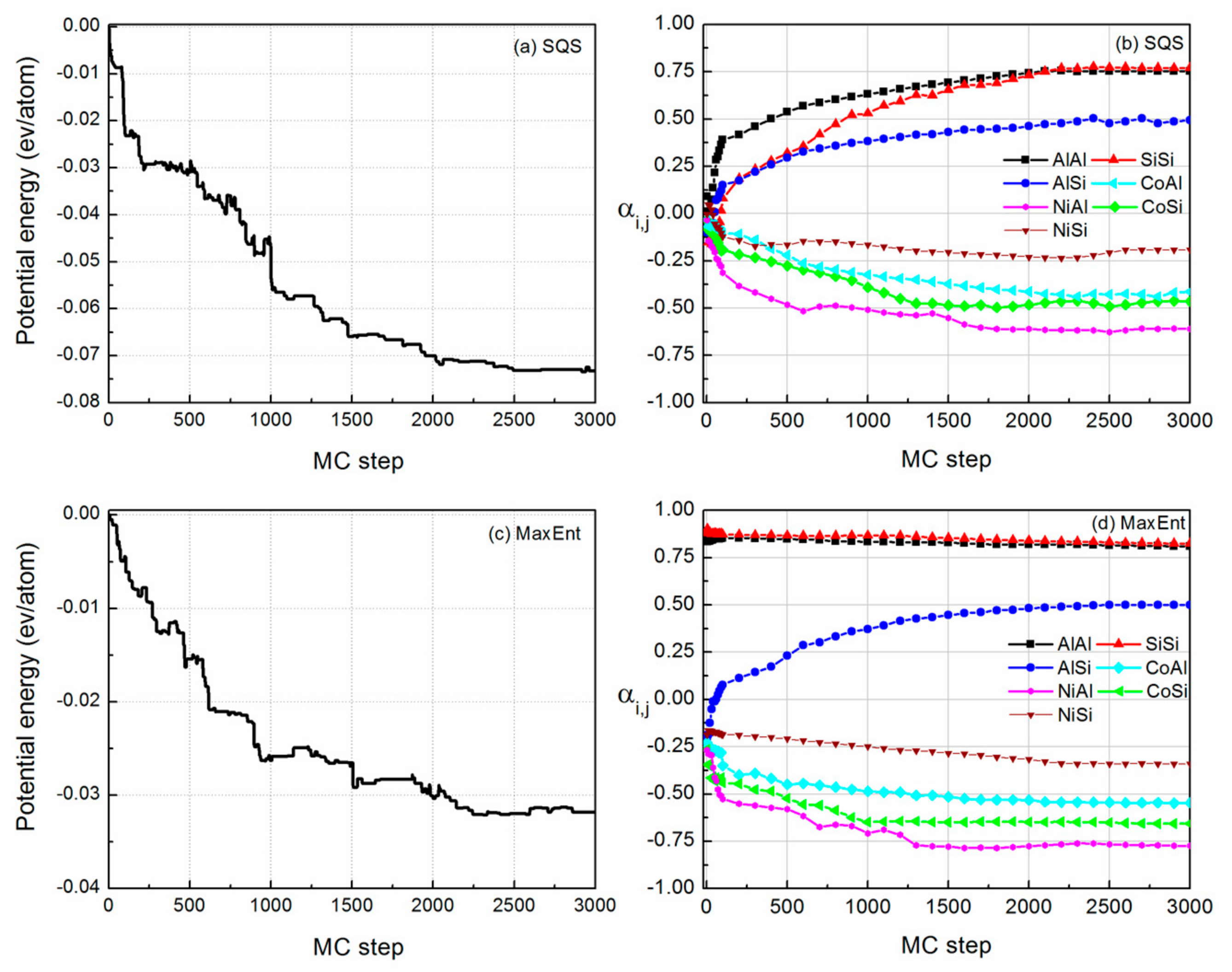
The evolution of the relative potential energy and SRO parameters of FeCoNi (AlSi)0.8 alloy are shown in Figure 2. The relative potential energy is defined as the energy difference between the potential energy of the ith MC step and the initial potential energy. In the case that the 92-atom SQS structure is selected as the initial starting structure, all the constituent atoms are randomly distributed in the lattice positions, and (Figure 2a,b). Figure 2b shows that there are major deviations of SRO parameters between the finial structure and the SQS structure. The average numbers of Al-Al and Si-Si pairs are reduced by ~80%, the nearest neighbor number changes from 2.1 for the random case to about 0.5 for the SRO case, and the average number of Al-Si pairs is reduced by ~50%, indicating Al (or Si) atoms separate from each other. Meanwhile, it can be seen that there is rapid growth of the average number of Ni-Al pairs, followed by Co-Si, Co-Al and Ni-Si pairs. The rapid growth of Ni-Al pairs shows that the Ni-Al pair first formed during the solidification from the liquid phase. The Ni-Al pairs will form a B2-orderd parent crystal structure, which is an ordered structure based on BCC with Pearson symbol of cP2. Si element also have negative mixing enthalpies with Ni, Co and Fe elements, the binary mixing enthalpies of Ni-Si, Co-Si and Fe-Si pairs are −40, −38 and −35 kJ/mol, respectively. Si atoms prefer to separate with Al and Si atoms, and bond with Ni, Co and Fe atoms. Raja et al. studied the structural properties of Fe3-xCoxSi alloys () using X-ray powder diffraction [35]. The results revealed that Si atoms occupy the body-centered positions, Fe and Co atoms occupy the body corners and form B2 and L21 ordered phases. The L21 phase is an ordered structure based on BCC with Pearson symbol of cF16. Just like Al, Si also tends to stabilize BCC structure. In the FeCoNi (AlSi)0.4 alloy, although the fraction of Al element (x = 0.4) is smaller as compared to the case in AlxCoCrFeNi alloy (BCC phase in x), there will be a large amount of Ni-Al and Co-Si pairs first formed and other constituents will dissolve in the parent crystal structure due to the mixing entropy effect. Thus, the system forms a BCC structure.
In the case where the MaxEnt structure is selected as the initial starting structure (Figure 2c,d), since each constituent atoms in the system strives for the maximum free space, and for FeCoNi(AlSi)0.8 alloy. We obtained the lower potential energy at the 2200 MC step. After 800 MC steps searches, the MC method did not find the lower energy structure. The emergence of the SRO reduced the potential energy, which made the potential energy change from −325 to −359 meV per atom. In Figure 2d, the SRO parameters of and decrease slightly. changes from 0.92 to 0.86, and changes from 0.92 to 0.84. We started from two different initial starting points to probe the change of the atomic nearest neighbor environment in FeCoNi (AlSi)0.8 alloy, and obtained a similar trend to the SRO. It can be concluded that the potential energy curve is relatively converged after 3000 MC steps simulation. From the above comparison, it can be seen that in the process of searching for the SRO structure, using the MaxEnt model as the initial starting structure is more efficient. This is based on the following two points: on the one hand, the potential energy of the MaxEnt structure is lower than that of the SQS structure, which can be validated from the difference in formation energy listed in Table 1. On the other hand, the SRO parameters and of the MaxEnt structure are closer to the SRO parameters of the finial SRO structure. Therefore, in the following probing of SRO, and the calculation of the magnetic and mechanical properties, the MaxEnt structure is selected as the initial structure.
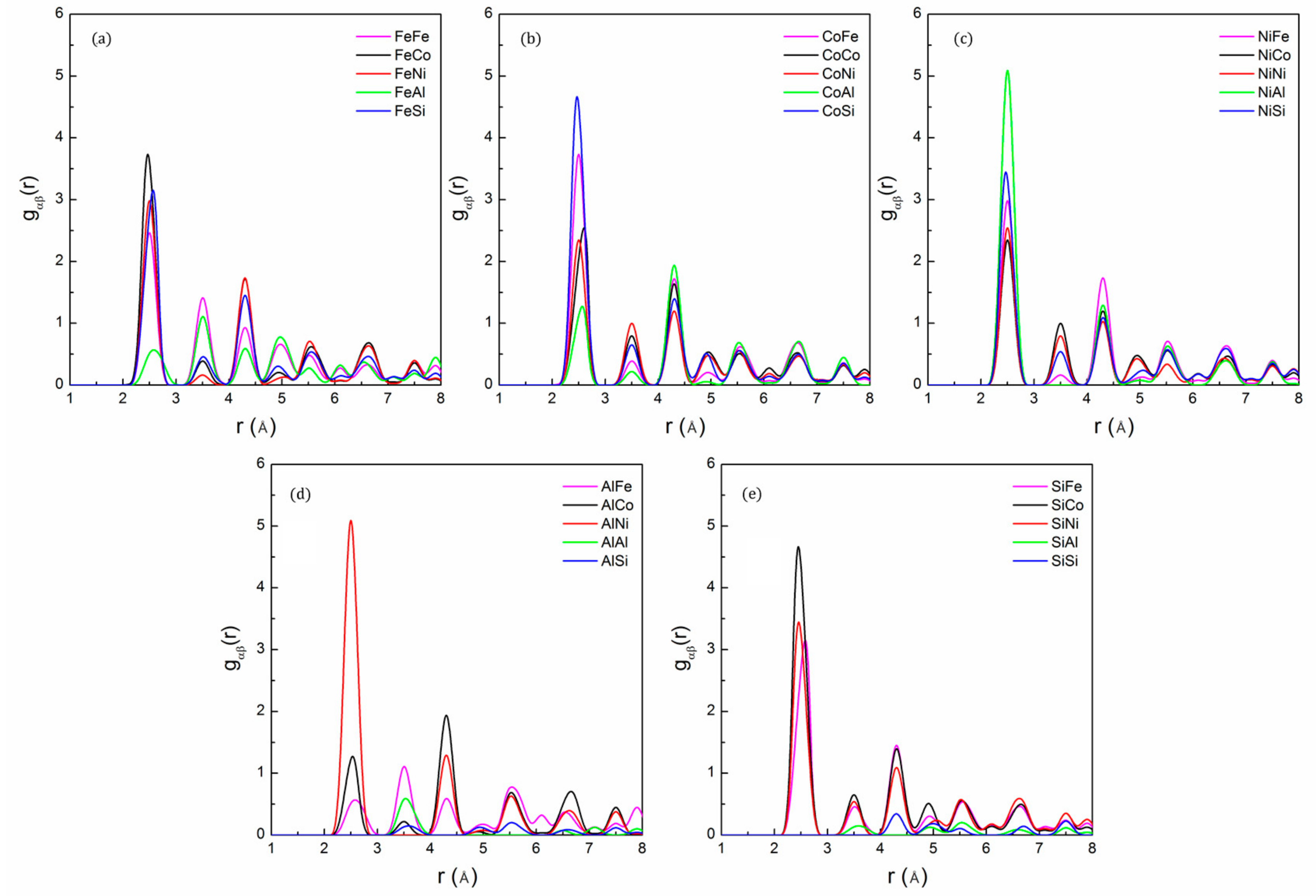
The partial pair distribution function can be used to describe the relative positional preferences of different constituent elements. The shape and position of the peaks provide detailed information about the atomic local environment [36]. We used pair PDF to show the SRO structure of the FeCoNi(AlSi)0.2 alloy, and the corresponding data are plotted in Figure 3. We observe that there is no obvious lattice distortion in the alloy. The average bond lengths of Co-Si and Ni-Si are r ~2.45 Å, which is shorter than the metal–metal bonds because the Si atom has a smaller radius than the metal atoms (Fe, Co, Ni, and Al). The Ni-Al pair has a higher peak intensity, followed by the Co-Si and Fe-Co, Ni-Si pairs. The peaks of the Al-Al and Al-Si pairs have moved to the second nearest neighbor shell, and the peak of the Si-Si pair has moved to the third nearest neighbor shell, indicating that Al and Si prefer to bond with other elements rather than themselves. The results suggest the existence of the preferred Ni-Al, Co-Si, Fe-Co and Ni-Si pairs in the FeCoNi(AlSi)0.2 alloy.
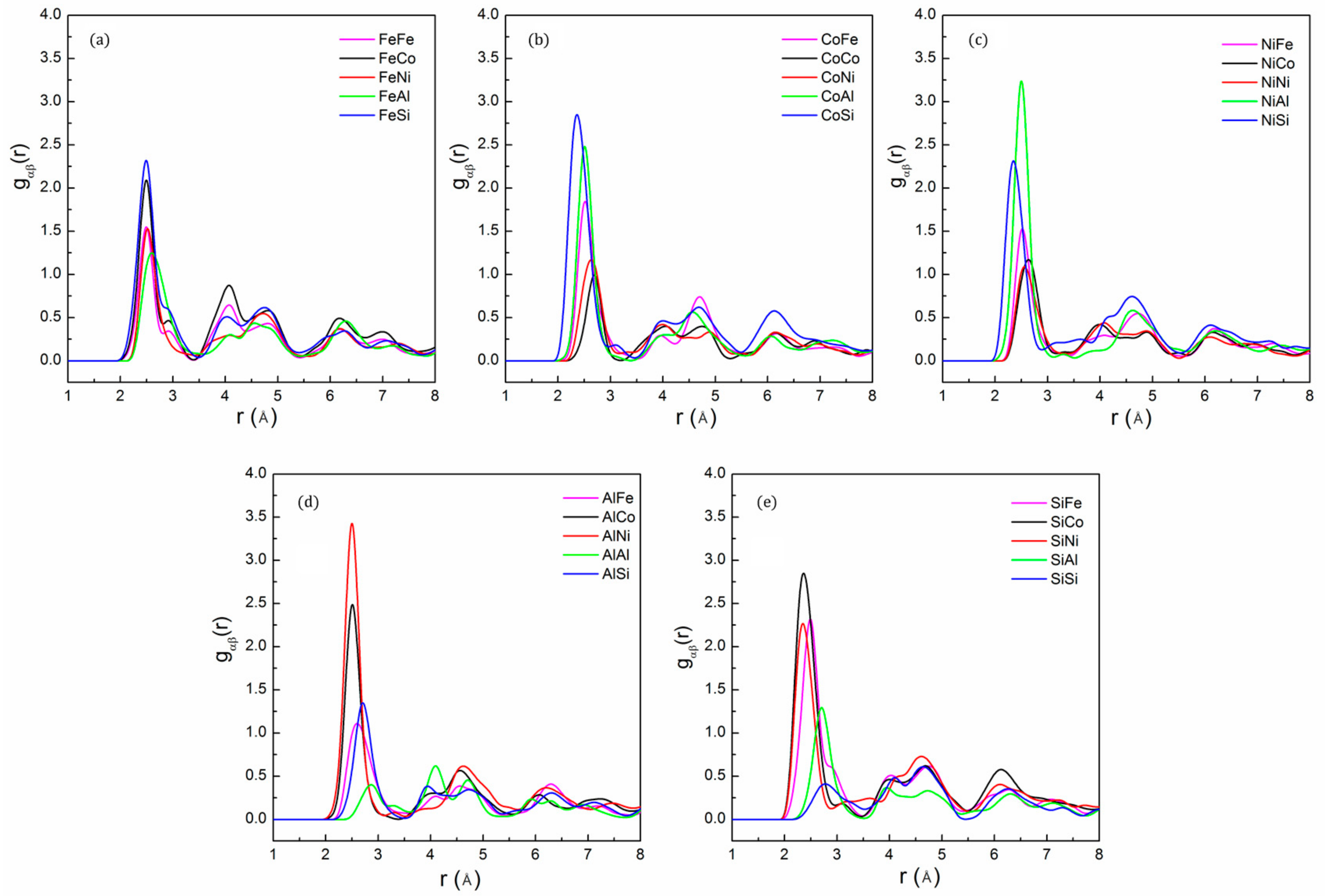
The partial PDF of the FeCoNi(AlSi)0.8 alloy after the 3000 MC steps is shown in Figure 4. It can be seen that the most preferred pair is Ni-Al, followed by the Co-Si, Co-Al, Fe-Si and Ni-Si pairs. The least favored pairs are the Si-Si, Al-Al pairs. Al and Si atoms prefer to bond with other elements to lower the potential energy. The results suggest the existence of a preferred short-range order of Ni-Al, Co-Si, Co-Al, Fe-Si and Ni-Si pairs in the alloy. In addition, there is obvious lattice distortion when the fraction of Al and Si atoms is larger. The lattice distortion makes the second intensity peak smear out and become less distinct, indicating that the atoms in the alloy deviate from the ideal lattice positions. The lattice distortion can significantly scatter free electrons, shortened electrons mean free paths, which will reduce the thermal and electrical conductivity of the alloy.
In Table 2, the SRO parameters of the FeCoNi(AlSi)x alloys as a function of Al and Si fraction x averaged after 3000 MC steps are shown. For FeCoNi alloy, we observed that the SRO parameter of the Fe-Ni pair is negative, and the Fe-Fe, Ni-Ni and Co-Co pairs are positive. Fe atoms prefer to bond with Ni atoms and form an SRO structure. Tamm et al. investigated the SRO behavior of NiCrCoFe alloys and found a negative Ni–Fe pair and positive Fe-Fe, Ni-Ni and Co-Co pairs [16]. When Al and Si atoms are added to the FeCoNi-based alloy, the Fe-Ni pair become positive. Al and Si elements have relative stronger attractive interaction with other constituent elements. They will separate from each other and bond with Fe, Co and Ni elements to form an SRO structure. The strength of the attractive interaction can be seen from the binary mixing enthalpies [37], that is, the more negative the binary mixing enthalpy, the stronger the attractive interaction between the binary pair. This is applicable to other HEAs as well. The SRO depends on the type of constituent elements present in the HEAs. If a constituent element has a stronger attractive interaction with the other constituent elements, the atoms of this element will separate from each other and bond with other constituent elements to form an SRO structure. Also, the concentration of the constituent elements can change the degree of SRO. We observed that the SRO parameter of Co-Al changes from 0.34 (x = 0.2) to −0.56 (x = 0.8) and Co-Co changes from 0.15 (x = 0.2) to 0.84 (x = 0.8). It indicates that the element concentrations also have a certain influence on the degree of SRO. We think that a possibility for the formation of SRO in the HEAs is the enrichment of the preferred element pairs during solidification. At high temperature, the atomistic structure of liquid alloy is generally thought to be totally random due to the high mixing entropy effect. As the temperature decreases, the enthalpy effect becomes more important, and the non-random configuration shows a tendency toward phase separation or chemical short-range order, and some ordered phases may form during the solidification. Santodonato et al. studied the structural evolution of Al1.3CoCrCuFeNi alloy from the high temperature liquid phase to the room temperature phase [38]. The results demonstrated that the alloy is a liquid above 1315 K with Al-Ni, Cr-Fe and Cu-Cu the preferred nearest-neighbor pairs. The results demonstrated that the alloy is a liquid above 1315 K with Al-Ni, Cr-Fe and Cu-Cu preferred nearest-neighbor pairs. During the cooling of the melt, the ordered phases will be preserved.
Due to the preferred Ni-Al, Co-Al and Fe-Si pairs in the BCC phase of FeCoNi (AlSi)0.4 and FeCoNi (AlSi)0.8 alloys, a B2-ordered phase structure may be formed. In the multi-component ordered BCC alloys, the crystal structure can be divided in two interpenetrating sublattices, designed by an sublattice, and a sublattice. and denote the molar fraction of the ith element on and sublattices. The B2-ordered parameter was used to quantify the degree of ordering of the ith element in the sublattices [38]. is described using:
The total order parameter is calculated from with the equation:
where is the overall molar fraction. indicates the random solid solution, and shows the fully B2-ordered structure.
The mixing entropy for the case of a B2 structure can be calculated:
In Table 3, we presented the ordering parameter , and the mixing entropy for the ideal, and taking into account the B2-ordered structure. For the FeCoNi (AlSi)0.4 alloy, , and , indicating that Al prefers to occupy the sublattice and Ni and Si prefer the sublattice to form the B2 phase. The total B2-ordered parameter was 0.294 when x = 0.8, , and , indicating that Al prefers to occupy the sublattice and Ni and Co prefer the sublattice to form the B2 phase. With the increase of Al and Si content, the total B2-ordered parameter reaches 0.427. The emergence of the B2 phase in the alloys reduces the configuration entropy from 1.521 to 1.475 R for the FeCoNi (AlSi)0.4 alloy, forming 1.606 R to 1.509 R for the FeCoNi(AlSi)0.8 alloy.
3.3. SRO on Magnetic Properties
As a reference, a series of calculations were carried out to determine the magnetic moments of Fe with BCC, Ni with FCC and Co with HEX structure, respectively. The calculated magnetic moments per Fe, Co and Ni atom are 2.18 , 1.67 , and 0.62 , which are in good agreement with the experimental values [39]. The deviations between the calculated and the experimental lattice parameters (a) are within 1%. Chandran et al. [40] investigated Fe1−xCox alloys in BCC structures and found that with the increase of Co contents, the magnetic moment per Fe atom increased from 2.22 (x = 0) to 2.76 (x = 0.5). Apiñaniz et al. [41] studied the magnetic properties of ordered FexAl1-x alloys and found that the magnetic moment per Fe atom decreases from 2.22 to 0.64 with the increase in Al contents. The FeM, CoM and NiM binary ordered alloys (M = Fe, Co, Ni, Al and Si) with BCC primitive cells were constructed. Taking the FeM alloy as an example, the Fe atom was assigned to the body-centered position and the M atom to the body corners. Table 4 listed the calculated magnetic moments per Fe, Co and Ni atoms with the M atom in the nearest neighbor shell. It can be seen that Co and Ni atoms in the nearest neighbor shell of Fe atoms greatly increase the magnetic moment of Fe atoms, while Al and Si atoms in the nearest neighbor shell of magnetic atoms will drastically decrease the magnetic moments of magnetic atoms. Therefore, the atomic nearest neighbor environment has considerable influence on the atomic magnetic moments in alloys.
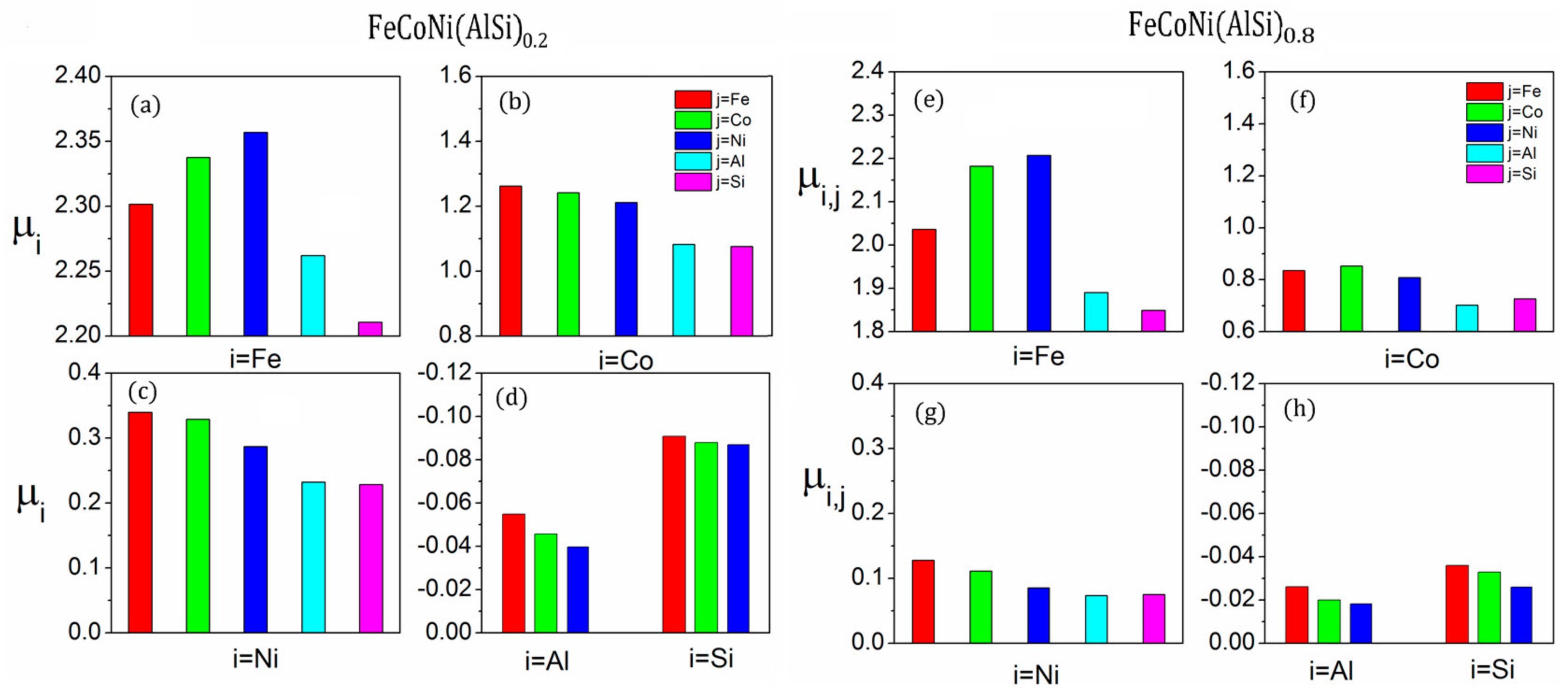
There is obvious SRO behavior in the FeCoNi (AlSi)x alloys. The SRO will significantly change the atomic nearest neighbor environment, which has an impact on the magnetic properties of the alloys. The saturation magnetizations and average atomic magnetic moments of the FeCoNi (AlSi)x alloys with MaxEnt and SRO structures are summarized in Table 5. For the FeCoNi alloy, the average atomic magnetic moments per Fe, Co and Ni atoms are 2.65, 1.64 and 0.62 with the initial structure. The Co and Ni atoms in the nearest neighbor of Fe atoms increase the magnetic moment of the Fe atoms. The effect of SRO on the magnetic moments of the FeCoNi alloy is relatively small. The calculated magnetic moments (= 1.65 T) are consistent with the previous calculated values ( = 1.62 T) [22]. For the FeCoNi(AlSi)0.2 alloy, the SRO parameter of the Ni-Al pair reduces by −100% and the nearest neighbor number of the Ni to Al atom changes from 3.5 to 7 for the SRO structure. Similarly, the SRO parameters of the Co-Si, Ni-Si and Fe-Co pairs are also negative. According to the atomic coordinate analysis, Al atoms occupy the body corners, while Ni atoms occupy the face-centered positions and form an Ni-Al rich region. The negative SRO parameters of Ni-Al, Ni-Si and Fe-Co pairs present a similar trend with the experimental work [14] that the dendritic area is rich in Fe and Co atoms, while the inter-dendritic area is rich in Al, Ni, and Si atoms in FeCoNi (AlSi)0.2 alloy. In Figure 5, the average atomic magnetic moment of the FeCoNi (AlSi)0.2 and FeCoNi (AlSi)0.8 alloys are presented. is the average magnetic moment of the type i element, which is calculated that there is type j element in its nearest neighbor shell. The average magnetic moments , and vary with different nearest neighbor elements. The results are different from the case of random solid solution. If each lattice is randomly occupied and the probability is proportional to the concentration of the element, the magnetic moment will be homogeneous. The SRO behavior decreases the atomic magnetic moments and the saturation magnetization of the system. The average magnetic moment of the Fe atom changes from 2.50 to 2.32 , the Co atom from 1.43 to 1.19 and the Ni atom from 0.51 to 0.32 . Compared to the previously-calculated saturation magnetization value = 1.31 T with the random structure model [22], the saturation magnetization with SRO is = 1.17 T, which is closer to the experimental value = 1.15 T [22].
The FeCoNi(AlSi)0.8 phase contains more Al and Si components. The SRO parameters of Ni-Al, Co-Si, Co-Al, Fe-Si and Ni-Si pairs are more negative. It is obvious that the Fe, Co and Ni atoms prefer to gather around the Si atom, while the Ni and Co atoms prefer to gather around the Al atom. The SRO greatly alters the local environment of the magnetic atoms, which will further reduce the average magnetic moments of the magnetic atoms. The average magnetic moment of the Fe atoms changes from 2.10 to 2.03 , Co atoms from 0.94 to 0.76 . Due to the enrichment of Al and Si atoms in the nearest neighboring shell of Ni atoms, the magnetic moment of Ni atoms vanishes. The saturation magnetization changes from 0.67 T with the random structure to 0.6 T with the SRO structure. The saturation magnetization with SRO is closer to the experimental value = 0.46 T [22].
3.4. SRO on Elastic Properties
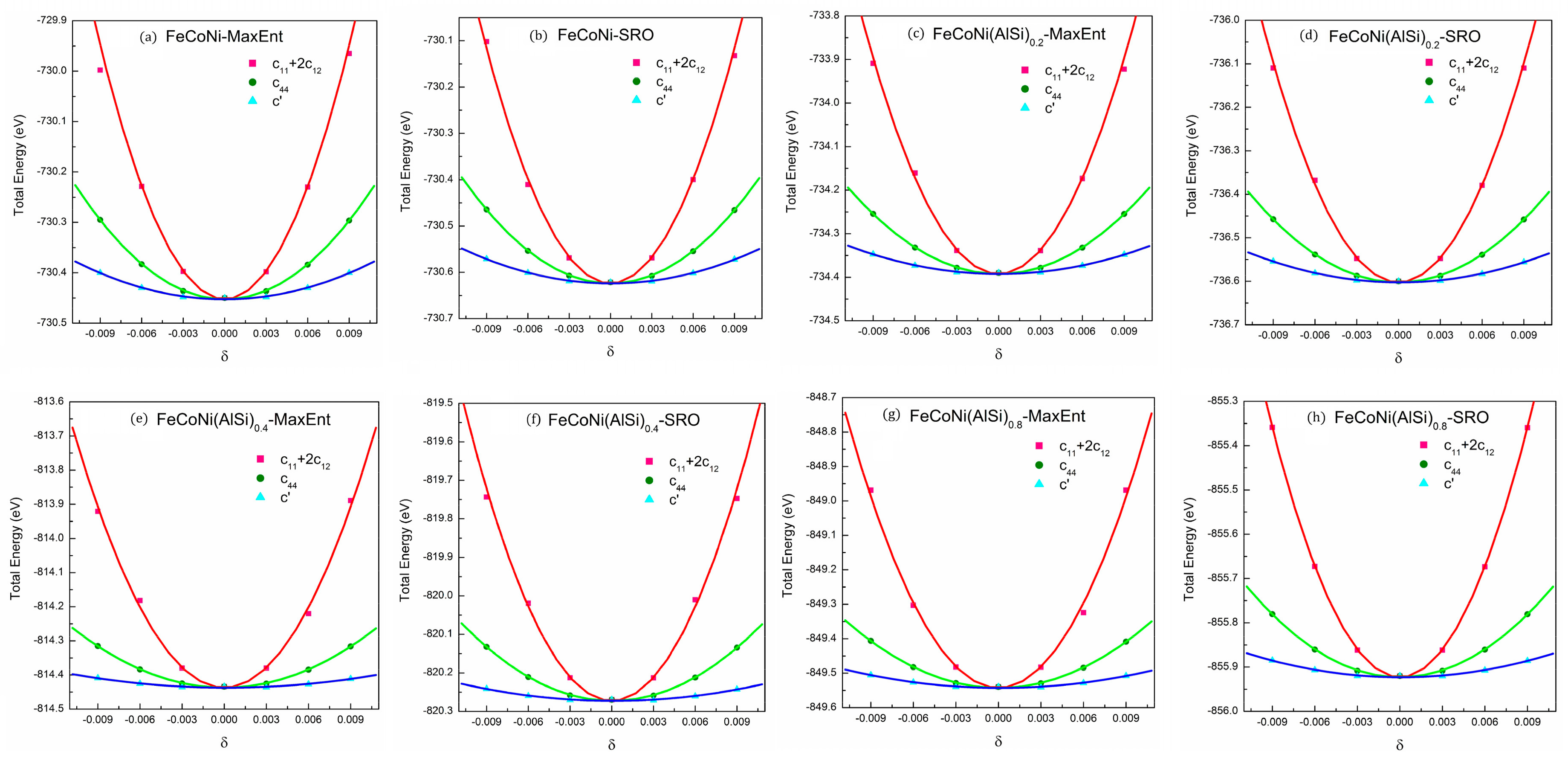
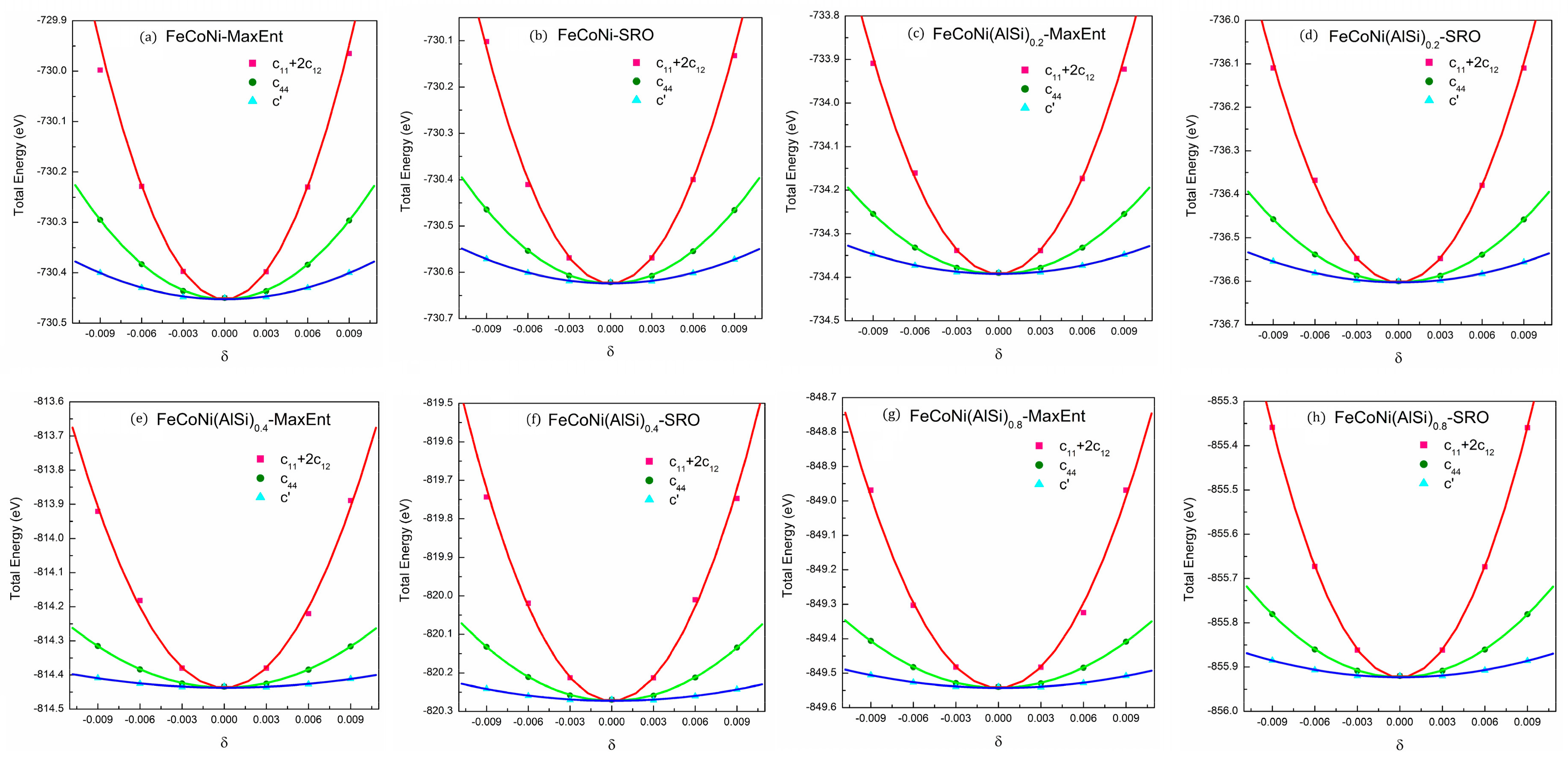
Mechanical properties are a vital aspect in material selection. It is important to understand the relationship of component, structure and mechanical properties for material applications. FeCoNi(AlSi)x alloys form FCC or BCC phase structures with different Al and Si fractions. There are three independent elastic constants , , and for the present cubic lattice. and can be determined from the bulk modulus () and tetragonal shear modulus () with , and . The bulk modulus can be obtained by fitting energy-volume data with the three-order Birch–Murnaghan equation of state [42]. Tetragonal shear modulus can be extracted from by applying orthorhombic strain () to the cubic lattice. The elastic constant can be obtained from fitting the energy-strain equation: by applying the monoclinic strain () to the base lattice [43]. The shear strain, strain matrix and energy-strain equations are shown in Table 6. In order to keep the elastic behavior of crystals, the applied strains should be relatively small, so the strains adopted are = −0.009, −0.006, −0.003, 0, 0.003, 0.006, 0.009. The MaxEnt structure is a cubic lattice structure. We modified the basis vector matrix according to the strain matrix to apply different deformations to the supercell structures. The calculations of the total energy were conducted with k-points to increase the accuracy. The energy curves of different deformations for the FeCoNi(AlSi)x alloys are shown in Figure 6.
The shear modulus is calculated by the Hill average , and the Voigt and Reuss bounds shear modulus can be obtained by using Equations (6) and (7):
Young’s modulus () and Poisson’s ratio () are calculated from the bulk modulus () and the shear modulus ():
The calculated lattice parameters, three elastic moduli , , and derived elastic moduli of the FeCoNi(AlSi)x alloys with the MaxEnt and SRO structures are presented in Table 7. From Table 7, all the calculated elastic constants (, and ) fulfill the mechanical stability criteria:, and , which demonstrates that the FeCoNi (AlSi)x alloys are mechanically stable. The experimental information on the elastic modulus is very limited. The calculated lattice parameter for the FeCoNi alloy is Å, and the experimental value is 3.599 Å [22]. The calculated lattice parameter is closer to the value Å calculated with EMTO [44]. The calculated bulk modulus is , shear modulus , and Young’s modulus . The corresponding results calculated with EMTO are , and
The bulk modulus is a measure of the resistance to compressibility of a material. From the calculations, it can be seen that the fractions of Al and Si have significant influence on bulk modulus of FeCoNi (AlSi)x alloys. It is observed that the elastic constants, and and the three elastic moduli (and) decrease with increase of Si and Al fractions. The bulk modulus changes from 184.7 (x = 0) to 157.1 (x = 0.8). Note that Al and Si have a smaller bulk modulus compared to Fe, Co and Ni components. Also the addition of Al and Si make the alloy structure change from the FCC to BCC phase (the packing factor changes from 0.74 to 0.68), which will result in a decrease of the bulk modulus. Taking into account the effect of SRO, SRO behavior prefers to make the affinity atoms locate together, resulting in an increase of the average bonding strength and a decrease of alloy volume. The FeCoNi(AlSi)x alloys with SRO have a higher bulk modulus as compared to that of random solid structures. We also observed that SRO has similar effects on shear modulus and Young’s modulus.
The Pugh’s ratio () [45] and Poisson’s ratio () [46] can be used to qualify the ductile properties of a material. It was reported that an alloy material is ductile when > 1.75 and, otherwise it is brittle. For FeCoNi(AlSi)x alloys, the Cauchy pressure (), Pugh’s ratio and Poisson’s ratio increase with the increase of Al and Si fractions. The trends of Cauchy pressure, Pugh’s ratio and Poisson’s ratio indicate the addition of Al and Si to FeCoNi alloys improves the ductility of alloys. However, it is observed that the SRO behavior reduces the Pugh’s ratio, Poisson’s ratio and Cauchy pressure, showing that SRO results in a reduction in the ductility of the material. The Zener ratio and the ratio are used to describe the isotropy property of FeCoNi (AlSi)x alloys., and . For an isotropic cubic material, and . The values of indicate the relative degree of the elastic anisotropy. For FeCoNi(AlSi)x alloys, the Zener ratio changes from 2.94 to 4.29 and changes from 0.13 to 0.23 with the increase of Al and Si fractions. The Al and Si enhance the anisotropy of the FeCoNi (AlSi)x alloys. When taking into account the effect of SRO, the changes from 2.99 to 4.36 and the changes from 0.14 to 0.24. The SRO behavior further enhances the anisotropy of the FeCoNi (AlSi)x alloys. From the above results, it can be seen that although SRO has a positive effect on B, G and E, it has a negative effect on the ductility and isotropic properties of the materials.
4. Conclusions
Using Monte Carlo simulation in combination with density functional theory, we investigated the SRO behavior of FeCoNi-based high entropy alloys. We took the SQS and MaxEnt structures as the initial starting point to probe the SRO structure in the system and found that using the MaxEnt structure as the initial starting point was more efficient. The calculated results show that there are obvious SRO structures in the FeCoNi (AlSi)x alloys. The binary mixing enthalpies between constituent elements were found to be the key factor in controlling the formation of SRO in a material. Al and Si elements have more negative binary mixing enthalpies with Fe, Co and Ni elements. The average numbers of Al-Al, Al-Si and Si-Si pairs decrease significantly, while those of Ni-Al, Co-Si, Fe-Si, Ni-Si and Fe-Co pairs correspondingly increase. The changes in the SRO parameters indicate that Al and Si tend to bond with Fe, Co, and Ni atoms to lower the potential energy. The element concentrations also have a certain influence on the degree of SRO. The emergence of SRO will change the properties of high entropy alloys, such as reducing the formation energy, altering the equilibrium volume of the system and decreasing the average atomic magnetic moments. The SRO further reduces the saturation magnetization and damages the ductility and isotropy. The present work shows that SRO behavior has an important influence on the magnetic and mechanical properties of high entropy alloys.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 51471164), the National Key R&D Program of China (No. 2016YFB0701302), and the CAS Frontier Science Research Project (No. QYZDJ-SSW-JSC015). The authors thank the computational support from the Informalization Construction Project of Chinese Academy of Sciences during the 11th Five-Year Plan Period (No. INFO-115-B01). The Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase) are also highly acknowledged. Some of the calculations in this study were done on the Tianhe-II high performance computer system in the National Supercomputer Center in Guangzhou, China.
Author Contributions
Shaoqing Wang conceived the modeling approach. Wenqiang Feng performed the calculations and wrote the paper; Yang Qi read and corrected the paper.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Yeh, J.W.; Lin, S.J.; Chin, T.S.; Gan, J.Y.; Chen, S.K.; Shun, T.T.; Tsau, C.H.; Chou, S.Y. Formation of simple crystal structures in Cu-Co-Ni-Cr-Al-Fe-Ti-V alloys with multiprincipal metallic elements. Metall. Mater. Trans. A 2004, 35, 2533–2536. [Google Scholar] [CrossRef]
- Cantor, B.; Chang, I.T.H.; Knight, P.; Vincent, A.J.B. Microstructural development in equiatomic multicomponent alloys. Mater. Sci. Eng. A 2004, 375, 213–218. [Google Scholar] [CrossRef]
- Tasan, C.C.; Deng, Y.; Pradeep, K.G.; Yao, M.J.; Springer, H.; Raabe, D. Composition Dependence of Phase Stability, Deformation Mechanisms, and Mechanical Properties of the CoCrFeMnNi High-Entropy Alloy System. JOM 2014, 66, 1993–2001. [Google Scholar] [CrossRef]
- Gludovatz, B.; Hohenwarter, A.; Catoor, D.; Chang, E.H.; George, E.P.; Ritchie, R.O. A fracture-resistant high-entropy alloy for cryogenic applications. Science 2014, 345, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, X.; Zhu, R.; Zhang, Y. Corrosion and Serration Behaviors of TiZr0.5NbCr0.5VxMoy High Entropy Alloys in Aqueous Environments. Metals 2014, 4, 597–608. [Google Scholar] [CrossRef]
- Zuo, T.T.; Yang, X.; Liaw, P.K.; Zhang, Y. Influence of Bridgman solidification on microstructures and magnetic behaviors of a non-equiatomic FeCoNiAlSi high-entropy alloy. Intermetallics 2015, 67, 171–176. [Google Scholar] [CrossRef]
- Wang, W.R.; Wang, W.L.; Wang, S.C.; Tsai, Y.C.; Lai, C.H.; Yeh, J.W. Effects of Al addition on the microstructure and mechanical property of AlxCoCrFeNi high-entropy alloys. Intermetallics 2012, 26, 44–51. [Google Scholar] [CrossRef]
- Li, P.P.; Wang, A.; Liu, C.T. A ductile high entropy alloy with attractive magnetic properties. J. Alloy. Compd. 2017, 694, 55–60. [Google Scholar] [CrossRef]
- Widom, M.; Huhn, W.P.; Maiti, S.; Steurer, W. Hybrid Monte Carlo/Molecular Dynamics Simulation of a Refractory Metal High Entropy Alloy. Metall. Mater. Trans. A 2013, 45, 196–200. [Google Scholar] [CrossRef]
- Munoz, J.A.; Lucas, M.S.; Delaire, O.; Winterrose, M.L.; Mauger, L.; Li, C.W.; Sheets, A.O.; Stone, M.B.; Abernathy, D.L.; Xiao, Y.; et al. Positive vibrational entropy of chemical ordering in FeV. Phys. Rev. Lett. 2011, 107, 115501. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T.; Jena, A.P.; Sanyal, B.; Sonomura, H.; Fukuda, T.; Kakeshita, T.; Mukhopadhyay, P.K.; Mookerjee, A. Effect of short range ordering on the magnetism in disordered Fe:Al alloy. J. Alloy. Compd. 2014, 613, 306–311. [Google Scholar] [CrossRef]
- Jena, A.P.; Sanyal, B.; Mookerjee, A. Study of the effect of short ranged ordering on the magnetism in FeCr alloys. J. Magn. Magn. Mater. 2014, 349, 156–158. [Google Scholar] [CrossRef]
- Manzoni, A.; Daoud, H.; Volkl, R.; Glatzel, U.; Wanderka, N. Phase separation in equiatomic AlCoCrFeNi high-entropy alloy. Ultramicroscopy 2013, 132, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.T.; Ren, S.B.; Liaw, P.K.; Zhang, Y. Processing effects on the magnetic and mechanical properties of FeCoNiAl0.2Si0.2 high entropy alloy. Int. J. Min. Met. Mater. 2013, 20, 549–555. [Google Scholar] [CrossRef]
- Zhang, F.X.; Zhao, S.; Jin, K. Local Structure and Short-Range Order in a NiCoCr Solid Solution Alloy. Phys. Rev. Lett. 2017, 118, 205501. [Google Scholar] [CrossRef] [PubMed]
- Tamm, A.; Aabloo, A.; Klintenberg, M.; Stocks, M.; Caro, A. Atomic-scale properties of Ni-based FCC ternary, and quaternary alloys. Acta Mater. 2015, 99, 307–312. [Google Scholar] [CrossRef]
- Owen, L.R.; Playford, H.Y.; Stone, H.J.; Tucker, M.G. A new approach to the analysis of short-range order in alloys using total scattering. Acta Mater. 2016, 115, 155–166. [Google Scholar] [CrossRef]
- McGreevy, R.L. Reverse Monte Carlo modelling. J. Phys. Condens. Matter. 2001, 13, R877–R913. [Google Scholar] [CrossRef]
- Kaban, I.; Jóvári, P.; Stoica, M.; Eckert, J.; Hoyer, W.; Beuneu, B. Topological and chemical ordering in Co43Fe20Ta5.5B31.5 metallic glass. Phys. Rev. B 2009, 79, 1377–1381. [Google Scholar] [CrossRef]
- Hautier, G.; Jain, A.; Ong, S.P. From the computer to the laboratory: materials discovery and design using first-principles calculations. J. Mater. Sci. 2012, 47, 7317–7340. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. J. Phys. Condens. Matter. 1996, 54, 11169–11186. [Google Scholar]
- Zhang, Y.; Zuo, T.T.; Cheng, Y.Q.; Liaw, P.K. High-entropy alloys with high saturation magnetization, electrical resistivity, and malleability. Sci. Rep. 2013, 3, 1455. [Google Scholar] [CrossRef] [PubMed]
- Li, P.P.; Wang, A.; Liu, C.T. Composition dependence of structure, physical and mechanical properties of FeCoNi(MnAl)x high entropy alloys. Intermetallics 2017, 87, 21–26. [Google Scholar] [CrossRef]
- Zhang, Q.; Xu, H.; Tan, X.H.; Hou, X.L.; Wu, S.W.; Tan, G.S.; Yu, L.Y. The effects of phase constitution on magnetic and mechanical properties of FeCoNi(CuAl)x (x = 0–1.2) high-entropy alloys. J. Alloy. Compd. 2017, 693, 1061–1067. [Google Scholar] [CrossRef]
- Feng, W.Q.; Zheng, S.M.; Qi, Y.; Wang, S.Q. Periodic Maximum Entropy Random Structure Models for High-Entropy Alloys. Mater. Sci. Forum 2017, 898, 611–621. [Google Scholar] [CrossRef]
- Wang, S.Q. Atomic Structure Modeling of Multi-Principal-Element Alloys by the Principle of Maximum Entropy. Entropy 2013, 15, 5536–5548. [Google Scholar] [CrossRef]
- Wang, S.Q. Paracrystalline property of high-entropy alloys. AIP Adv. 2013, 3, 102105. [Google Scholar] [CrossRef]
- Van de Walle, A.; Tiwary, P.; de Jong, M.; Olmsted, D.L.; Asta, M.; Dick, A.; Shin, D.; Wang, Y.; Chen, L.Q.; Liu, Z.K. Efficient stochastic generation of special quasirandom structures. Calphad 2013, 42, 13–18. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Körmann, F.; Ma, D.; Belyea, D.D.; Lucas, M.S.; Miller, C.W.; Grabowski, B.; Sluiter, M.H.F. “Treasure maps” for magnetic high-entropy-alloys from theory and experiment. Appl. Phys. Lett. 2015, 107, 142404. [Google Scholar] [CrossRef]
- Dobrzyński, L.; Wiśniewski, A.; Uemura, Y.J.; Shapiro, S.M.; Wicksted, J.P. Inelastic neutron scattering from sendust. Phys. Rev. B 1988, 37, 7175–7181. [Google Scholar] [CrossRef]
- Hastings, W.K. Monte Carlo sampling methods using Markov chains and their applications. Biometrika 1970, 57, 97–109. [Google Scholar] [CrossRef]
- Cowley, J.M. Short-Range Order and Long-Range Order Parameters. Phys. Rev. 1965, 138, A1384–A1389. [Google Scholar] [CrossRef]
- Tsau, C.H.; Lin, S.X.; Fang, C.H. Microstructures and corrosion behaviors of FeCoNi and CrFeCoNi equimolar alloys. Mater. Chem. Phys. 2017, 186, 534–540. [Google Scholar] [CrossRef]
- Raja, M.M.; Kamat, S.V. Structure, Magnetic, and Electrical Properties of Heusler-Type Fe3-xCoxSi Ferromagnetic Alloys. Metall. Mater. Trans. A 2015, 46, 4688–4697. [Google Scholar] [CrossRef]
- Gao, M.; Alman, D. Searching for Next Single-Phase High-Entropy Alloy Compositions. Entropy 2013, 15, 4504–4519. [Google Scholar] [CrossRef]
- Takeuchi, A.; Inoue, A. Classification of Bulk Metallic Glasses by Atomic Size Difference, Heat of Mixing and Period of Constituent Elements and Its Application to Characterization of the Main Alloying Element. Mater. Trans. 2005, 46, 2817–2829. [Google Scholar] [CrossRef]
- Santodonato, L.J.; Zhang, Y.; Feygenson, M.; Parish, C.M.; Gao, M.C.; Weber, R.J.; Neuefeind, J.C.; Tang, Z.; Liaw, P.K. Deviation from high-entropy configurations in the atomic distributions of a multi-principal-element alloy. Nat. Commun. 2015, 6, 5964. [Google Scholar] [CrossRef] [PubMed]
- Billas, I.M.; Chatelain, A.; de Heer, W.A. Magnetism from the atom to the bulk in iron, cobalt, and nickel clusters. Science 1994, 265, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- Chandran, M.; Iorio, L.E.; Subramanian, P.R. Effect of nitrogen on the magnetic moment of α-Fe and FeCo alloys from first-principle calculations. J. Appl. Phys. 2007, 101, 033912–033916. [Google Scholar] [CrossRef]
- Apiñaniz, E.; Plazaola, F.; Garitaonandia, J.S. Influence of disorder on the magnetic properties of FeAl alloys: Theory. J. Non-Cryst. Solids 2001, 287, 302–307. [Google Scholar] [CrossRef]
- Lejaeghere, K.; Speybroeck, V.V.; Oost, G.V.; Cottenier, S. Error Estimates for Solid-State Density Functional Theory Predictions: An Overview by Means of the Ground-State Elemental Crystals. Crit. Rev. Solid State 2014, 39, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.Y.; Varga, L.K.; Chen, N.; Delczeg, L.; Vitos, L. Ab initio investigation of high-entropy alloys of 3d elements. Phys. Rev. B 2013, 87, 075144. [Google Scholar] [CrossRef]
- Tian, F.Y.; Varga, L.K.; Shen, J.; Vitos, L. Calculating elastic constants in high-entropy alloys using the coherent potential approximation: Current issues and errors. Comp. Mater. Sci. 2016, 111, 350–358. [Google Scholar] [CrossRef]
- Pugh, S.F. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos. Mag. 2009, 45, 823–843. [Google Scholar] [CrossRef]
- Gu, X.J.; McDermott, A.G.; Poon, S.J.; Shiflet, G.J. Critical Poisson’s ratio for plasticity in FeMoCBLn bulk amorphous steel. Appl. Phys. Lett. 2006, 88, 211905. [Google Scholar] [CrossRef]
Figure 1.
Crystal structures of the MaxEnt structures and special quasi-random structures (SQS) structure in their ideal, unrelaxed forms: (a) MaxEnt-108 for x = 0.2; (b) MaxEnt-128 for x = 0.4; (c) MaxEnt-128 for x = 0.8; (d) SQS-92 for x = 0.8.
Figure 1.
Crystal structures of the MaxEnt structures and special quasi-random structures (SQS) structure in their ideal, unrelaxed forms: (a) MaxEnt-108 for x = 0.2; (b) MaxEnt-128 for x = 0.4; (c) MaxEnt-128 for x = 0.8; (d) SQS-92 for x = 0.8.
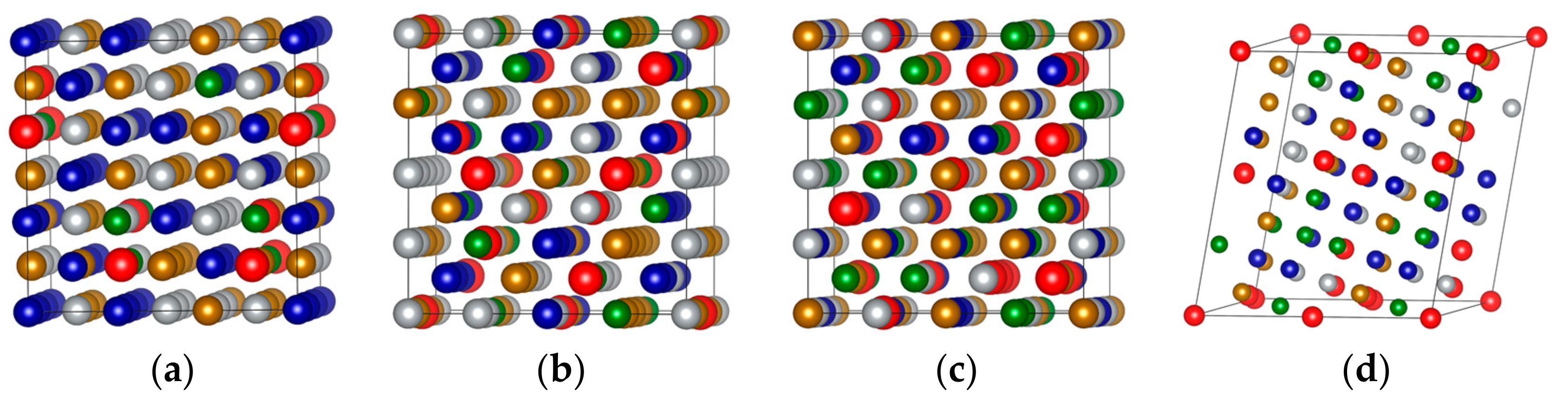
Figure 2.
The evolution of relative potential energy and the corresponding short-range order (SRO) parameters of FeCoNi(AlSi)0.8 alloy: (a,b) with special quasi-random structures (SQS) structure; (c,d) with maximum entropy(MaxEnt) structure as the initial structure.
Figure 2.
The evolution of relative potential energy and the corresponding short-range order (SRO) parameters of FeCoNi(AlSi)0.8 alloy: (a,b) with special quasi-random structures (SQS) structure; (c,d) with maximum entropy(MaxEnt) structure as the initial structure.

Figure 3.
The partial pair distribution function for: (a) Fe; (b) Co; (c) Ni; (d) Al and (e) Si of FeCoNi(AlSi)0.2 alloy after the 3000 MC step, showing short-range order between Ni-Al, Co-Si and Fe-Co pairs.
Figure 3.
The partial pair distribution function for: (a) Fe; (b) Co; (c) Ni; (d) Al and (e) Si of FeCoNi(AlSi)0.2 alloy after the 3000 MC step, showing short-range order between Ni-Al, Co-Si and Fe-Co pairs.
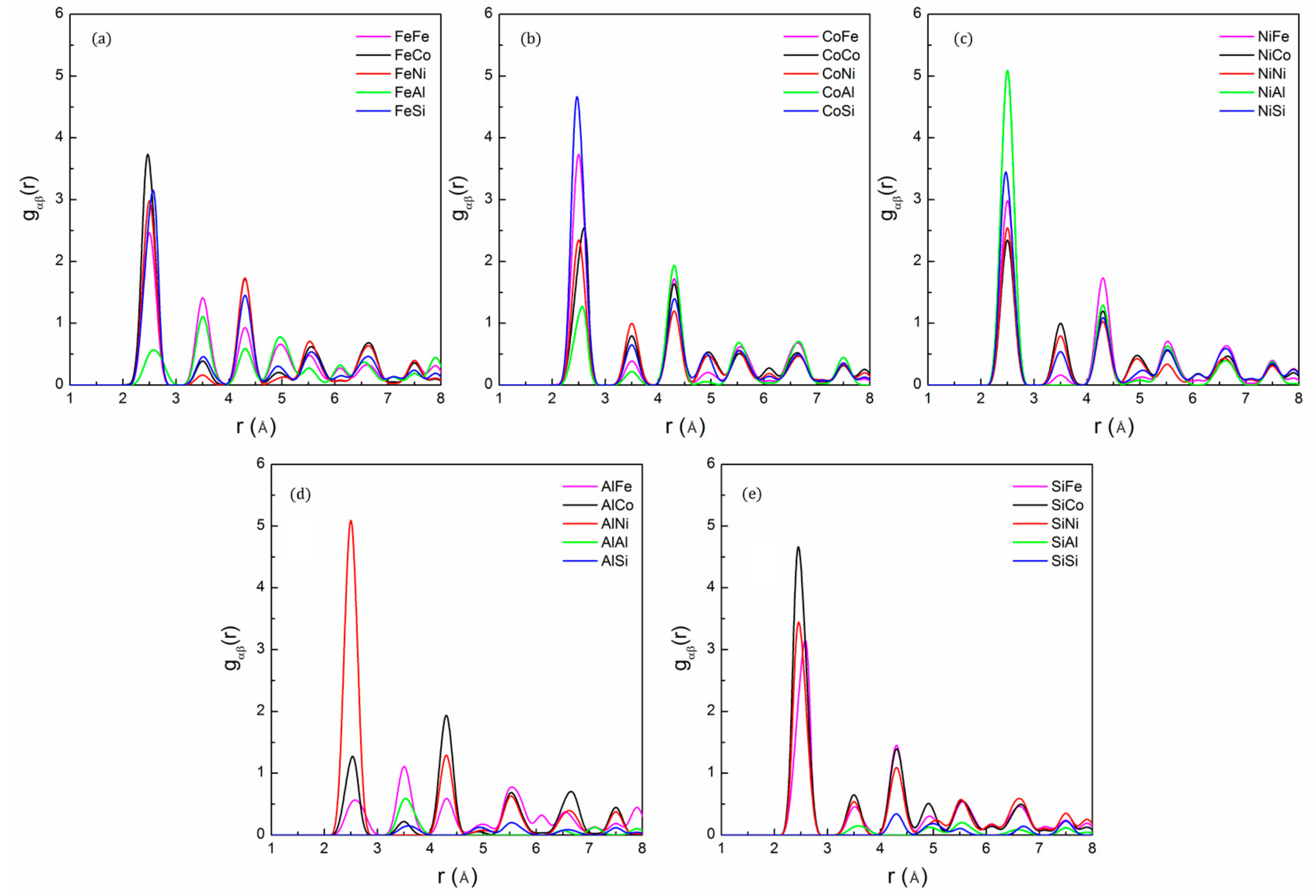
Figure 4.
The partial pair distribution function: (a) Fe; (b) Co; (c) Ni; (d) Al and (e) Si of FeCoNi(AlSi)0.8 alloy after the 3000 MC step, indicating short-range order between Ni-Al, Co-Si, Co-Al and Fe-Si pairs.
Figure 4.
The partial pair distribution function: (a) Fe; (b) Co; (c) Ni; (d) Al and (e) Si of FeCoNi(AlSi)0.8 alloy after the 3000 MC step, indicating short-range order between Ni-Al, Co-Si, Co-Al and Fe-Si pairs.

Figure 5.
The average magnetic moments (unit in ) with different nearest neighbor elements: (a–d) for FeCoNi (AlSi)0.2 alloy; (e–h) for FeCoNi (AlSi)0.8 alloy.
Figure 5.
The average magnetic moments (unit in ) with different nearest neighbor elements: (a–d) for FeCoNi (AlSi)0.2 alloy; (e–h) for FeCoNi (AlSi)0.8 alloy.
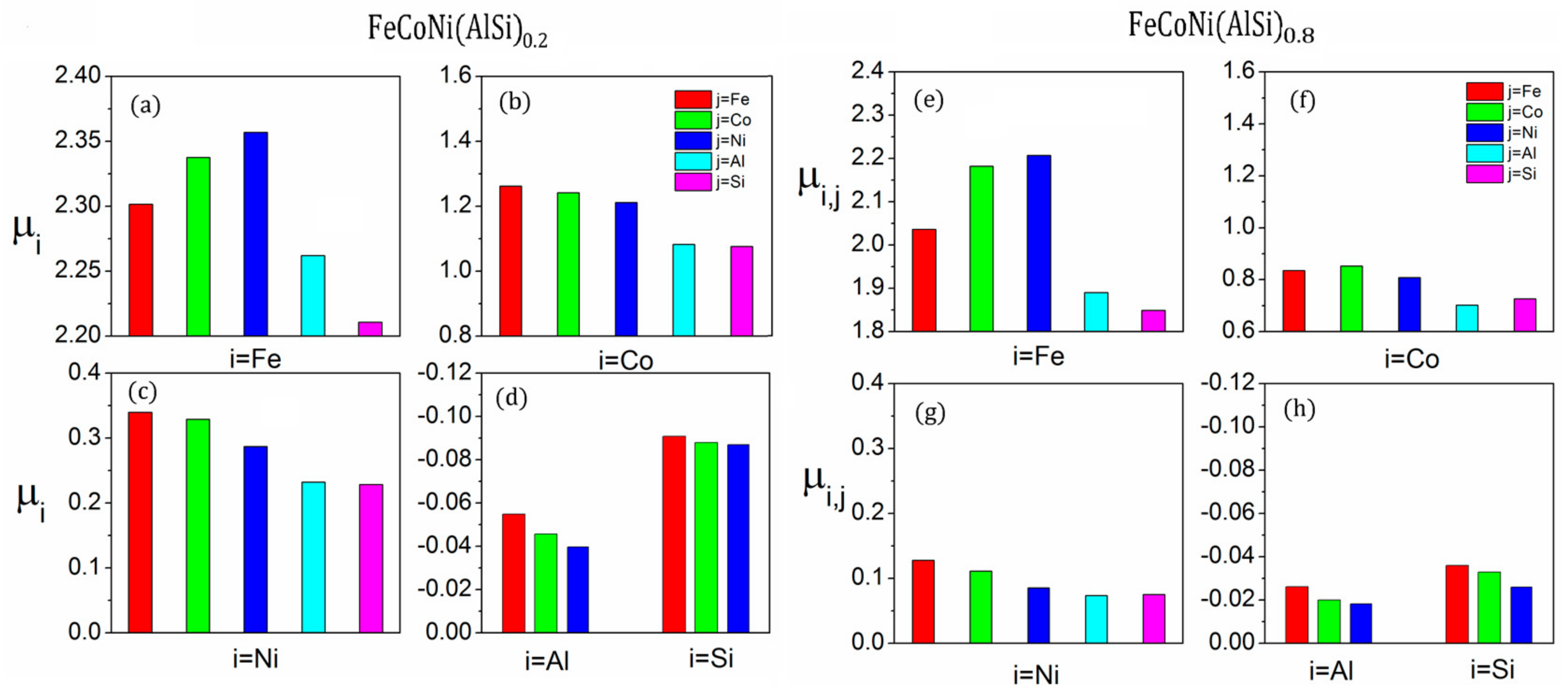
Figure 6.
The total energies curves of different deformations for .

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated lattice parameters (a), formation enthalpies and the energy differences between face-centered cubic (FCC) phase and body-centered cubic (BCC) phases as a function of Al and Si fraction x.
Table 1.
Calculated lattice parameters (a), formation enthalpies and the energy differences between face-centered cubic (FCC) phase and body-centered cubic (BCC) phases as a function of Al and Si fraction x.
| Alloy | Structure | (FCC) (Å) | (BCC) (Å) | (eV/atom) | (eV/atom) | (eV/atom) |
|---|---|---|---|---|---|---|
| FeCoNi | MaxEnt | 3.545 | 2.821 | 0.015 | −0.027 | 0.042 |
| FeCoNi(AlSi)0.1 | MaxEnt | 3.547 | 2.826 | −0.047 | −0.067 | 0.020 |
| FeCoNi(AlSi)0.2 | MaxEnt | 3.554 | 2.832 | −0.132 | −0.145 | 0.012 |
| FeCoNi(AlSi)0.4 | MaxEnt | 3.561 | 2.836 | −0.209 | −0.191 | −0.018 |
| FeCoNi(AlSi)0.5 | MaxEnt | 3.569 | 2.838 | −0.240 | −0.219 | −0.021 |
| FeCoNi(AlSi)0.8 | MaxEnt | 3.581 | 2.853 | −0.325 | −0.296 | −0.029 |
| FeCoNi(AlSi)0.8 | SQS | 3.598 | 2.874 | −0.298 | −0.265 | −0.033 |
Table 2.
Short-range order (SRO) parameters of the FeCoNi (AlSi)x alloys as a function of Al and Si fraction x.
Table 2.
Short-range order (SRO) parameters of the FeCoNi (AlSi)x alloys as a function of Al and Si fraction x.
| Pair | x = 0 | x = 0.2 | x = 0.4 | x = 0.8 | Pair | x = 0 | x = 0.2 | x = 0.4 | x = 0.8 |
|---|---|---|---|---|---|---|---|---|---|
| FeFe | 0.35 | 0.26 | −0.07 | −0.16 | CoSi | −0.37 | −0.54 | −0.72 | |
| FeCo | −0.04 | −0.34 | −0.24 | −0.08 | NiNi | 0.37 | −0.12 | −0.07 | 0.23 |
| FeNi | −0.31 | 0.04 | 0.30 | 0.23 | NiAl | −1.04 | −0.84 | −0.78 | |
| FeAl | 0.32 | 0.30 | 0.36 | NiSi | −0.31 | −0.28 | −0.19 | ||
| FeSi | −0.28 | −0.32 | −0.37 | AlAl | 1.00 | 0.95 | 0.84 | ||
| CoCo | 0.11 | 0.15 | 0.53 | 0.84 | AlSi | 1.00 | 0.72 | 0.54 | |
| CoNi | −0.06 | 0.19 | 0.22 | 0.45 | SiSi | 1.00 | 0.96 | 0.86 | |
| CoAl | 0.34 | −0.38 | −0.56 |
Table 3.
The order parameter , the mixing entropy for the ideal, taking into account the B2-ordered structure for FeCoNi (AlSi)0.4 and FeCoNi (AlSi)0.8 alloys.
Table 3.
The order parameter , the mixing entropy for the ideal, taking into account the B2-ordered structure for FeCoNi (AlSi)0.4 and FeCoNi (AlSi)0.8 alloys.
| Alloy | ||||||||
|---|---|---|---|---|---|---|---|---|
| FeCoNi(AlSi)0.4 | 0.176 | −0.06 | −0.294 | 0.692 | −0.231 | 0.294 | 1.475 | 1.521 |
| FeCoNi(AlSi)0.8 | 0.259 | −0.428 | −0.481 | 0.652 | 0.130 | 0.427 | 1.509 | 1.606 |
Table 4.
The calculated magnetic moments of Fe, Co and Ni in binary FeM-, CoM- and NiM-ordered alloys.
Table 4.
The calculated magnetic moments of Fe, Co and Ni in binary FeM-, CoM- and NiM-ordered alloys.
| Element | Fe () | Co () | Ni () | Al () | Si () |
|---|---|---|---|---|---|
| Fe | 2.18 | 2.75 | 2.79 | 0.67 | 0 |
| Co | 1.69 | 1.62 | 1.60 | 0 | 0.53 |
| Ni | 0.69 | 0.63 | 0.61 | 0 | 0 |
Table 5.
The saturation magnetizations , the errors of calculated saturation magnetizations to experimental values and average magnetic moments for FeCoNi(AlSi)x alloys with the MaxEnt structures and the finial structures with SRO, the calculated and experimental data from previous publications are also shown.
Table 5.
The saturation magnetizations , the errors of calculated saturation magnetizations to experimental values and average magnetic moments for FeCoNi(AlSi)x alloys with the MaxEnt structures and the finial structures with SRO, the calculated and experimental data from previous publications are also shown.
| Alloy | Ordering | Phase | Error | () | () | () | () | () | |
|---|---|---|---|---|---|---|---|---|---|
| FeCoNi | RSM 1 | FCC | 1.62 | 22.73% | 2.65 | 1.66 | 0.62 | − | |
| FeCoNi | MaxEnt | FCC | 1.63 | 23.48% | 2.65 | 1.64 | 0.62 | − | − |
| FeCoNi | SRO | FCC | 1.65 | 25.00% | 2.67 | 1.63 | 0.63 | − | − |
| FeCoNi | Exp 2 | FCC | 1.32 | ||||||
| FeCoNi(AlSi)0.2 | RSM 1 | FCC | 1.31 | 13.91% | 2.48 | 1.50 | 0.52 | −0.05 | −0.06 |
| FeCoNi(AlSi)0.2 | MaxEnt | FCC | 1.32 | 14.78% | 2.50 | 1.43 | 0.51 | −0.05 | −0.06 |
| FeCoNi(AlSi)0.2 | SRO | FCC | 1.17 | 1.74% | 2.32 | 1.19 | 0.32 | −0.04 | −0.09 |
| FeCoNi(AlSi)0.2 | Exp 2 | FCC | 1.15 | ||||||
| FeCoNi(AlSi)0.4 | MaxEnt | BCC | 1.14 | 21.28% | 2.48 | 1.39 | 0.35 | −0.03 | −0.04 |
| FeCoNi(AlSi)0.4 | SRO | BCC | 1.06 | 12.77% | 2.43 | 1.25 | 0.26 | −0.03 | −0.04 |
| FeCoNi(AlSi)0.4 | Exp 2 | BCC | 0.94 | ||||||
| FeCoNi(AlSi)0.8 | RSM 1 | BCC | 0.67 | 45.65% | 2.08 | 0.88 | 0.21 | −0.02 | −0.03 |
| FeCoNi(AlSi)0.8 | MaxEnt | BCC | 0.66 | 43.48% | 2.10 | 0.94 | 0.21 | −0.03 | −0.04 |
| FeCoNi(AlSi)0.8 | SRO | BCC | 0.60 | 30.43% | 2.03 | 0.76 | 0.08 | −0.02 | −0.03 |
| FeCoNi(AlSi)0.8 | Exp 1 | BCC | 0.46 | error |
Table 6.
The shear strain, strain matrix and the corresponding energy-strain equation to calculate , and for the cubic structure.
Table 6.
The shear strain, strain matrix and the corresponding energy-strain equation to calculate , and for the cubic structure.
| Shear Strain | Strain Matrix | Energy-Strain Equation | |
|---|---|---|---|
Table 7.
Calculated lattice parameters (Å), elastic constants ( and ), bulk modulus , shear modulus , Young’s modulus , and derived elastic moduli of the FeCoNi(AlSi)x alloys. The unit for the elastic moduli is
Table 7.
Calculated lattice parameters (Å), elastic constants ( and ), bulk modulus , shear modulus , Young’s modulus , and derived elastic moduli of the FeCoNi(AlSi)x alloys. The unit for the elastic moduli is
| Alloy | Ordering | B/G | |||||||||||
| X = 0 | MaxEnt | 3.545 | 243.6 | 155.3 | 130.2 | 25.1 | 184.7 | 84.5 | 219.8 | 0.302 | 2.19 | 0.13 | 2.94 |
| X = 0 | SRO | 3.543 | 244.8 | 156.6 | 131.9 | 24.7 | 186.0 | 85.1 | 221.5 | 0.302 | 2.19 | 0.14 | 2.99 |
| X = 0.2 | MaxEnt | 3.554 | 230.8 | 154.9 | 113.7 | 41.2 | 180.2 | 73.3 | 193.7 | 0.321 | 2.46 | 0.14 | 2.99 |
| X = 0.2 | SRO | 3.551 | 236.3 | 156.9 | 119.3 | 37.6 | 183.4 | 76.8 | 202.2 | 0.316 | 2.39 | 0.15 | 3.01 |
| X = 0.4 | MaxEnt | 2.836 | 201.7 | 150.2 | 92.9 | 57.3 | 167.4 | 55.8 | 150.5 | 0.350 | 3.00 | 0.17 | 3.61 |
| X = 0.4 | SRO | 2.831 | 207.1 | 153.3 | 98.1 | 52.2 | 171.2 | 58.6 | 157.9 | 0.346 | 2.92 | 0.18 | 3.65 |
| X = 0.8 | MaxEnt | 2.853 | 182.6 | 144.4 | 81.9 | 62.5 | 157.1 | 46.1 | 125.9 | 0.366 | 3.41 | 0.23 | 4.29 |
| X = 0.8 | SRO | 2.847 | 193.3 | 149.9 | 94.6 | 55.3 | 164.3 | 52.9 | 143.3 | 0.355 | 3.10 | 0.24 | 4.36 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Feng, W.; Qi, Y.; Wang, S. Effects of Short-Range Order on the Magnetic and Mechanical Properties of FeCoNi(AlSi)x High Entropy Alloys. Metals 2017, 7, 482. https://doi.org/10.3390/met7110482
AMA Style
Feng W, Qi Y, Wang S. Effects of Short-Range Order on the Magnetic and Mechanical Properties of FeCoNi(AlSi)x High Entropy Alloys. Metals. 2017; 7(11):482. https://doi.org/10.3390/met7110482
Chicago/Turabian StyleFeng, Wenqiang, Yang Qi, and Shaoqing Wang. 2017. "Effects of Short-Range Order on the Magnetic and Mechanical Properties of FeCoNi(AlSi)x High Entropy Alloys" Metals 7, no. 11: 482. https://doi.org/10.3390/met7110482
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.