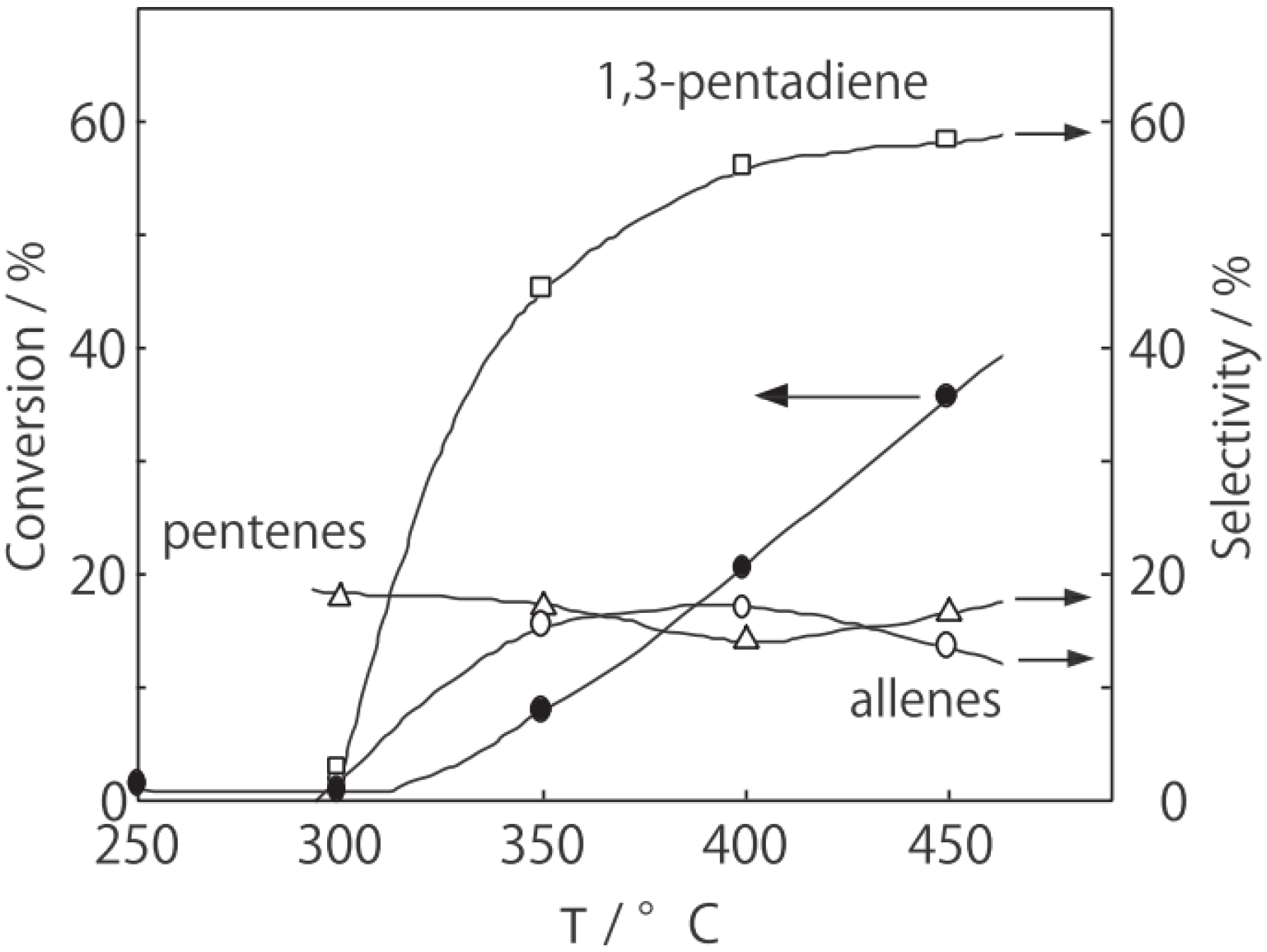
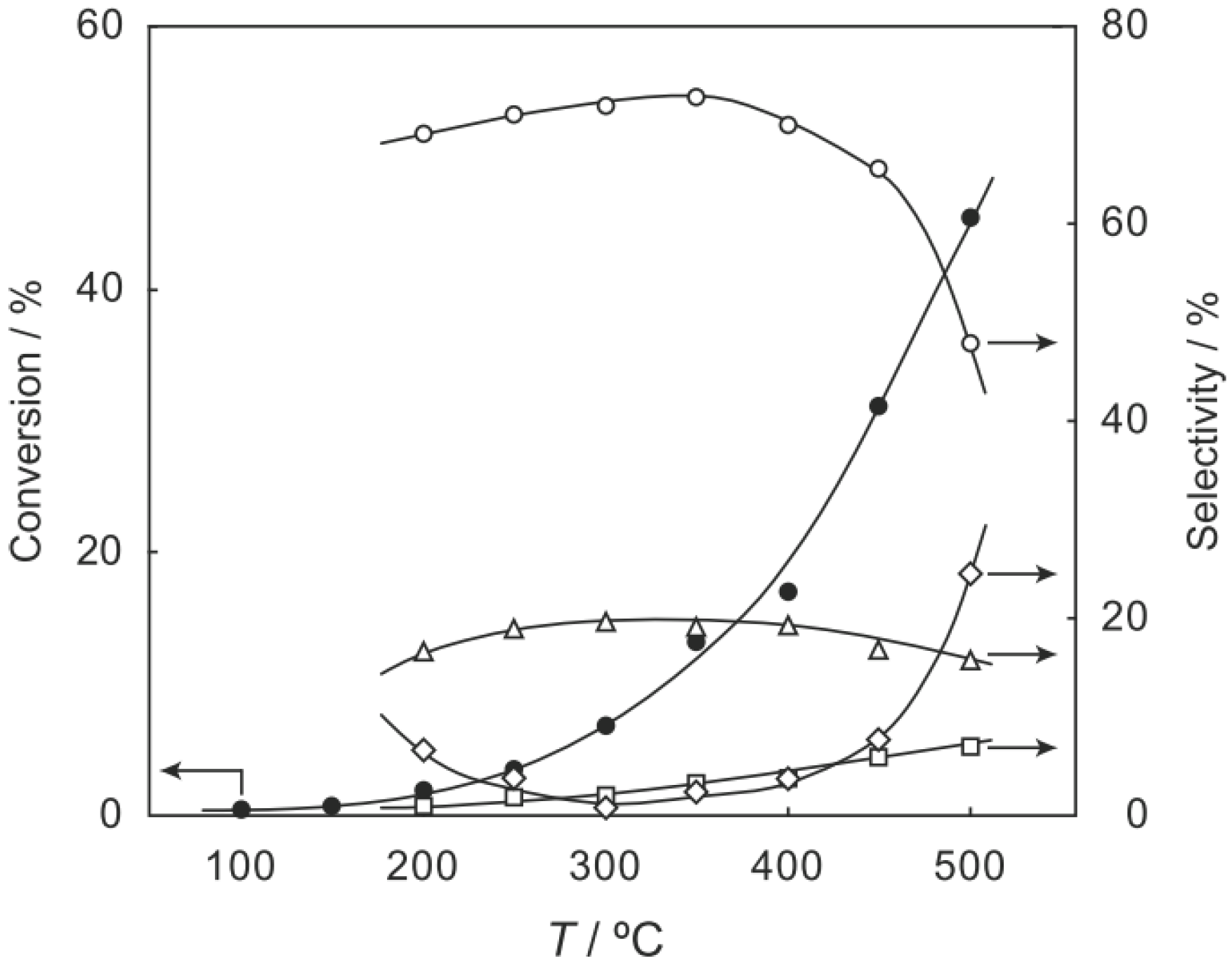
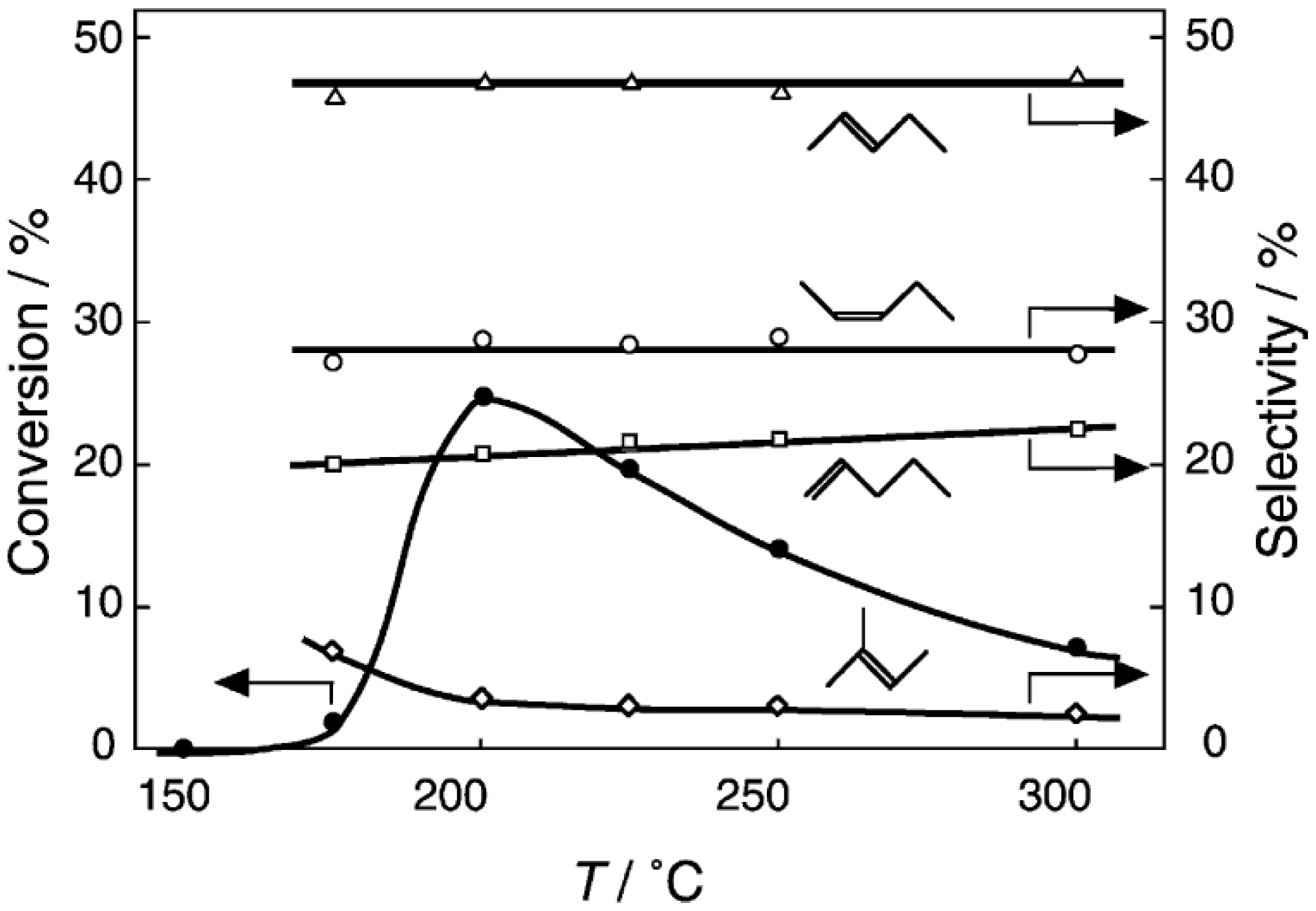
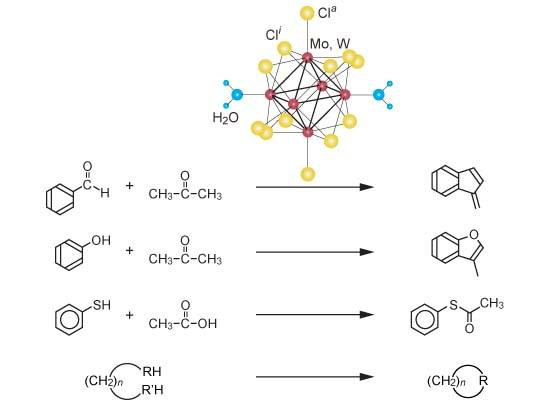
6.1. Isomerization of Diethylbenzene (Equation (15)) [177]
Much literature has appeared on the catalytic ring-attachment isomerization of
o- and
m-xylenes and the catalytic intermolecular transalkylation (disproportionation) of toluene to produce
p-xylene [
90,
178,
179], which is used in its oxidation to form terephthalic acid. Mordenite and ZSM-5 are currently the best catalysts for those catalytic reactions [
180,
181,
182]. On the other hand, very little has been reported on the reactions of diethylbenzenes, even though
p-diethylbenzene is an industrial source of
p-divinylbenzene. Diethylbenzenes disproportionated over solid catalysts, similarly. When Y-zeolite mixed with SiO
2–Al
2O
3 was employed as a catalyst in a neat liquid of
o-,
m-, or
p-diethylbenzene at 170 °C, disproportionation occurred, affording ethylbenzene and 1,2,4-triethylbenzene. Although small yields of
o-,
m-, and
p-diethylbenzenes were formed, they were the secondary disproportionation products from ethylbenzene and 1,2,4-triethylbenzene [
183]. Disproportionation of ethylbenzene to afford benzene and
m- and
p-diethylbenzenes was reported over a ZSM-5 catalyst without yielding
o-diethylbenzene [
184].
Generally, in large-pore zeolites, such as Y-zeolites and mordenite, side chains disproportionate by way of bimolecular transalkylation. Conversely, in medium pore zeolites, such as ZSM-5, disproportionation proceeds by way of successive dealkylation and realkylation [
185]. Thus, ring-attached ethyl groups do not migrate around the aromatic ring by an intramolecular 1,2-shift mechanism over conventional heterogeneous solid acid catalysts.
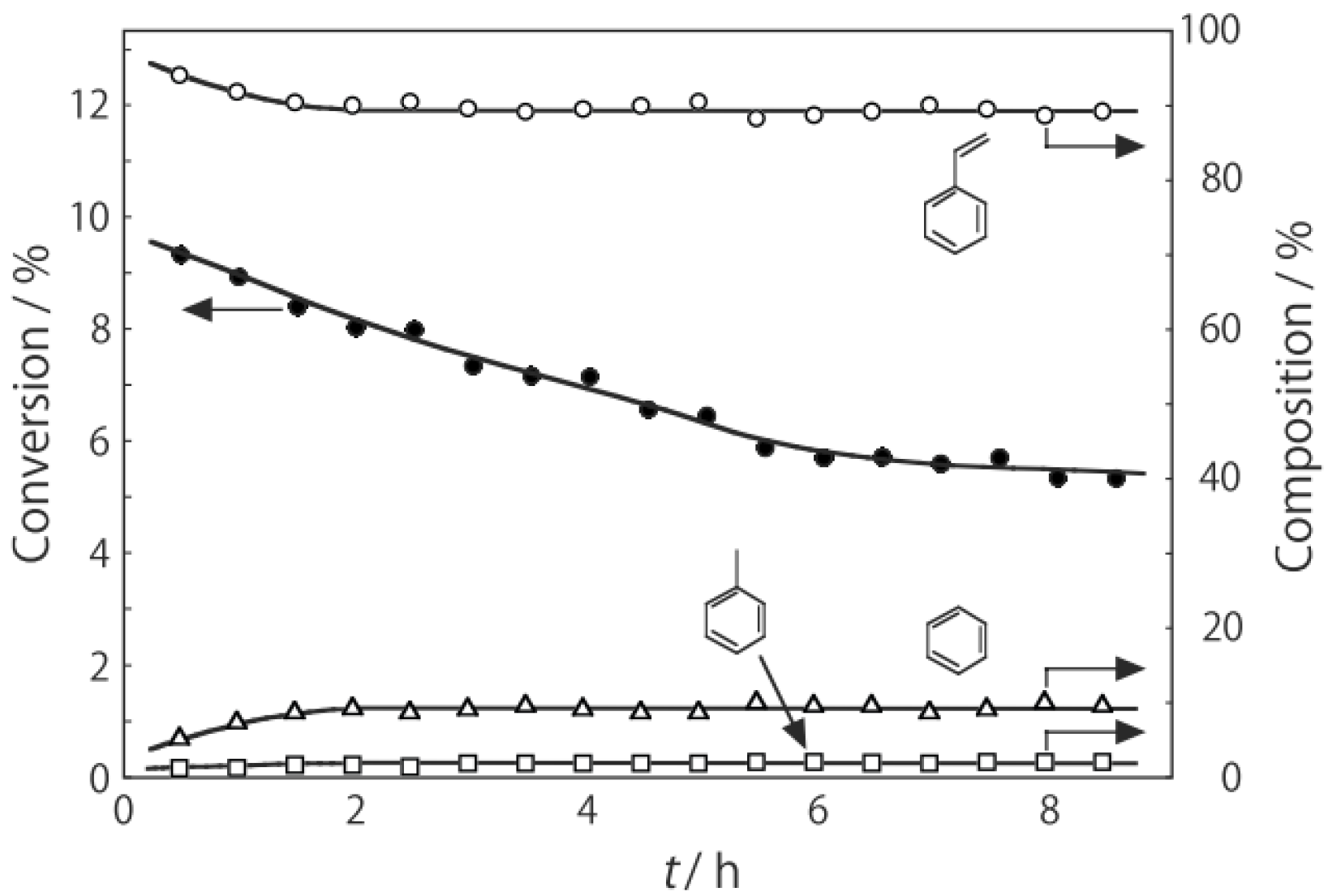
When crushed crystals of (H
3O)
2[(Mo
6Cl
8)Cl
6]·6H
2O (150–200 mesh) were activated in a stream of hydrogen for 1 h, and then allowed to react with
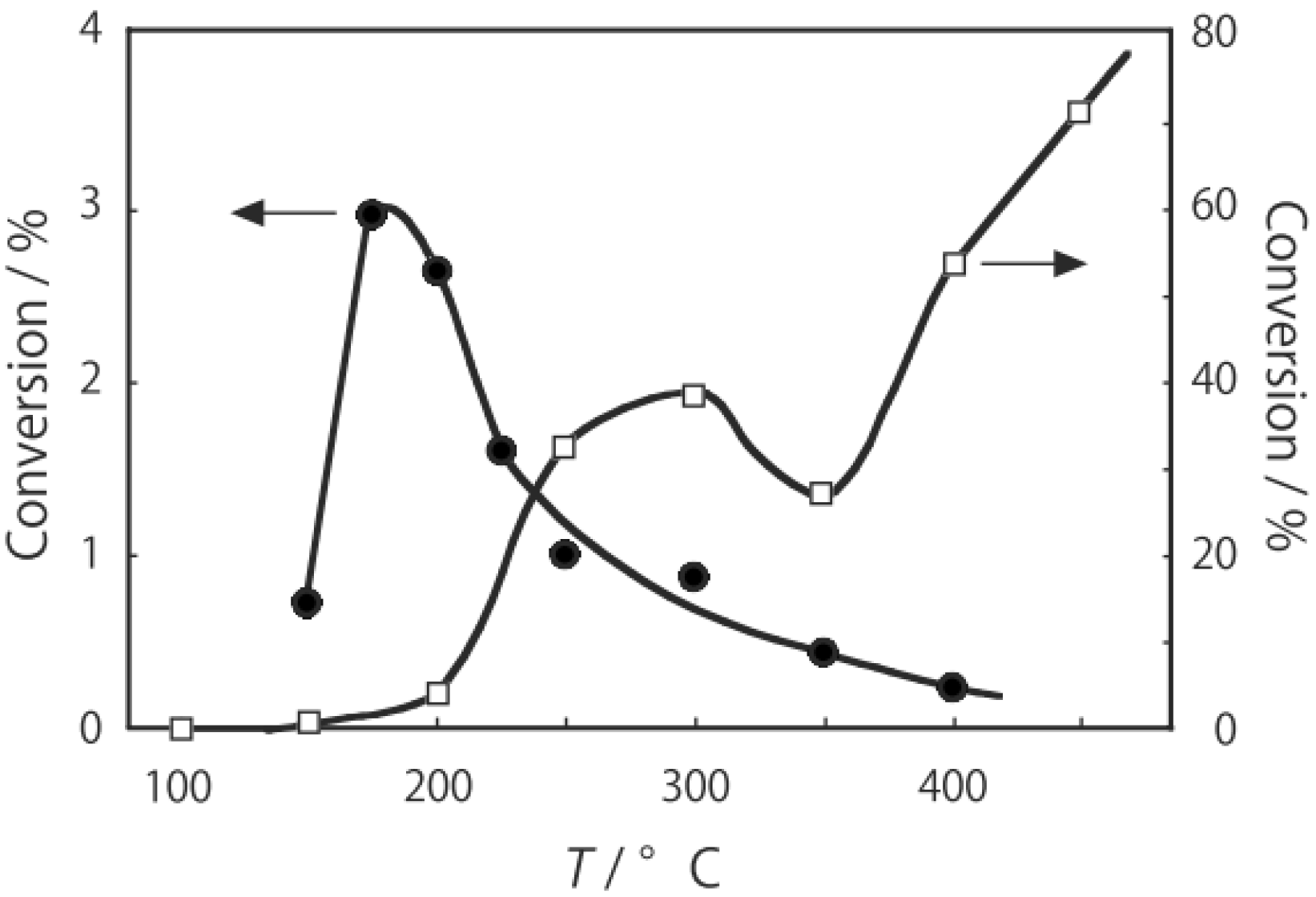
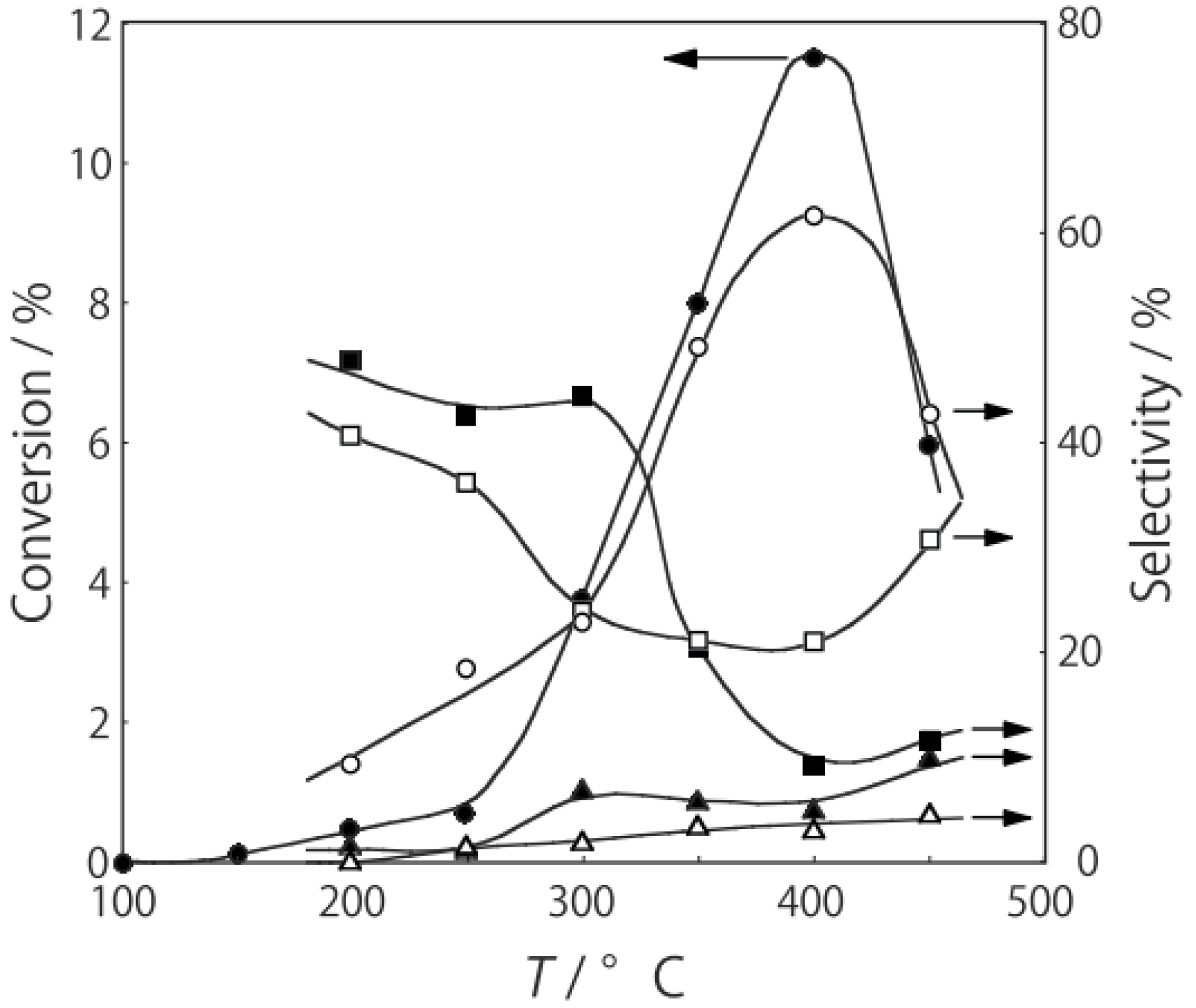
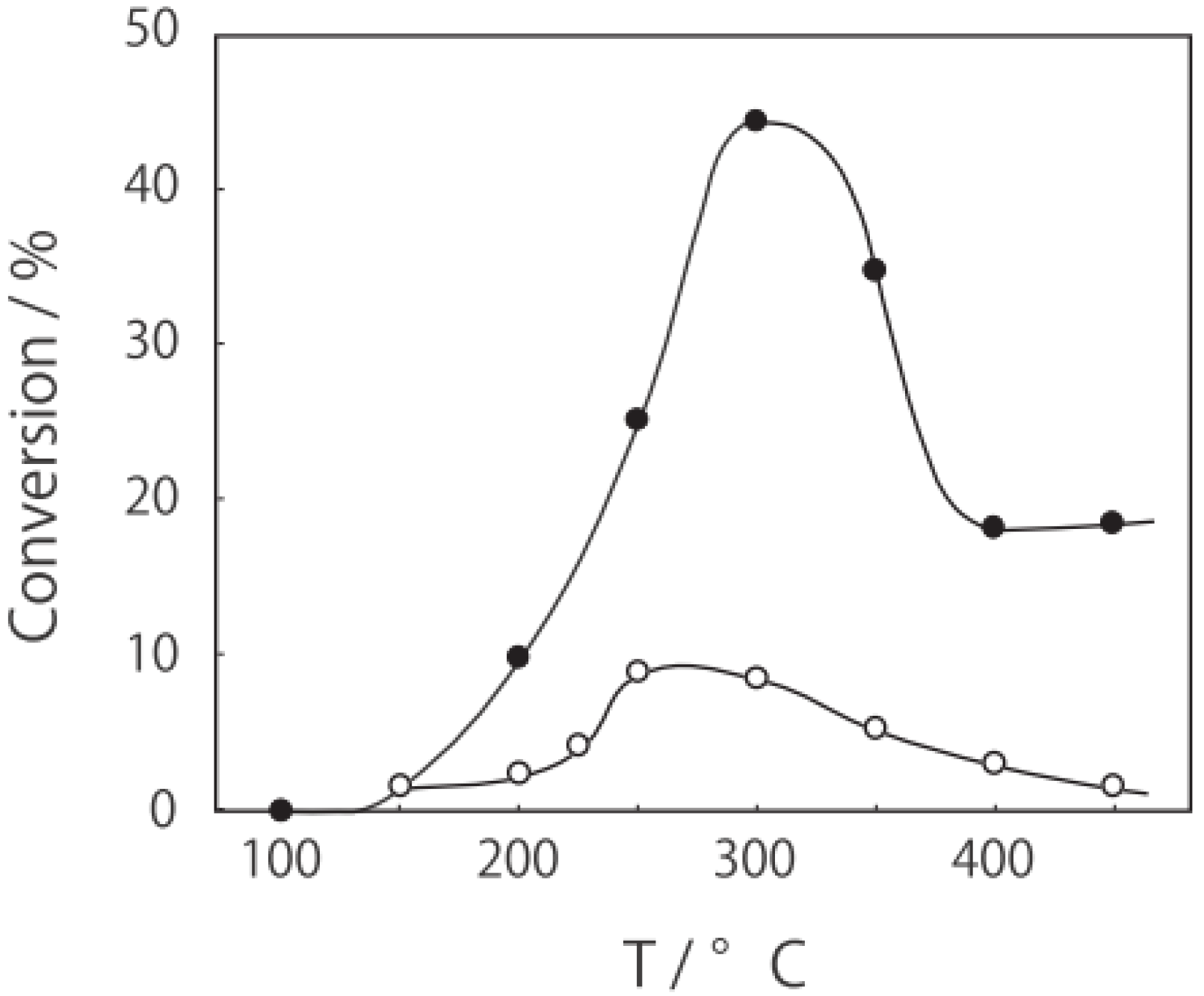
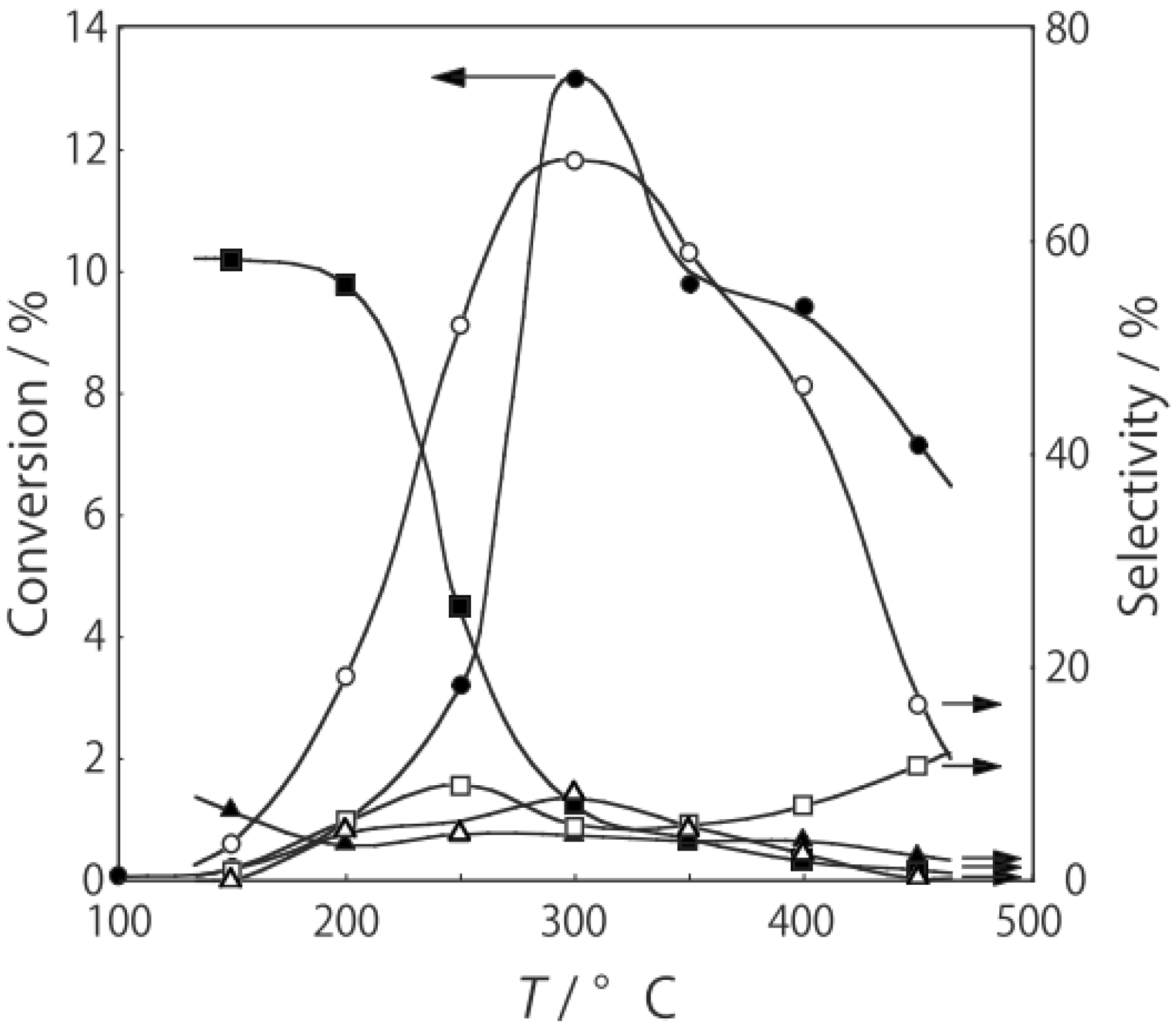
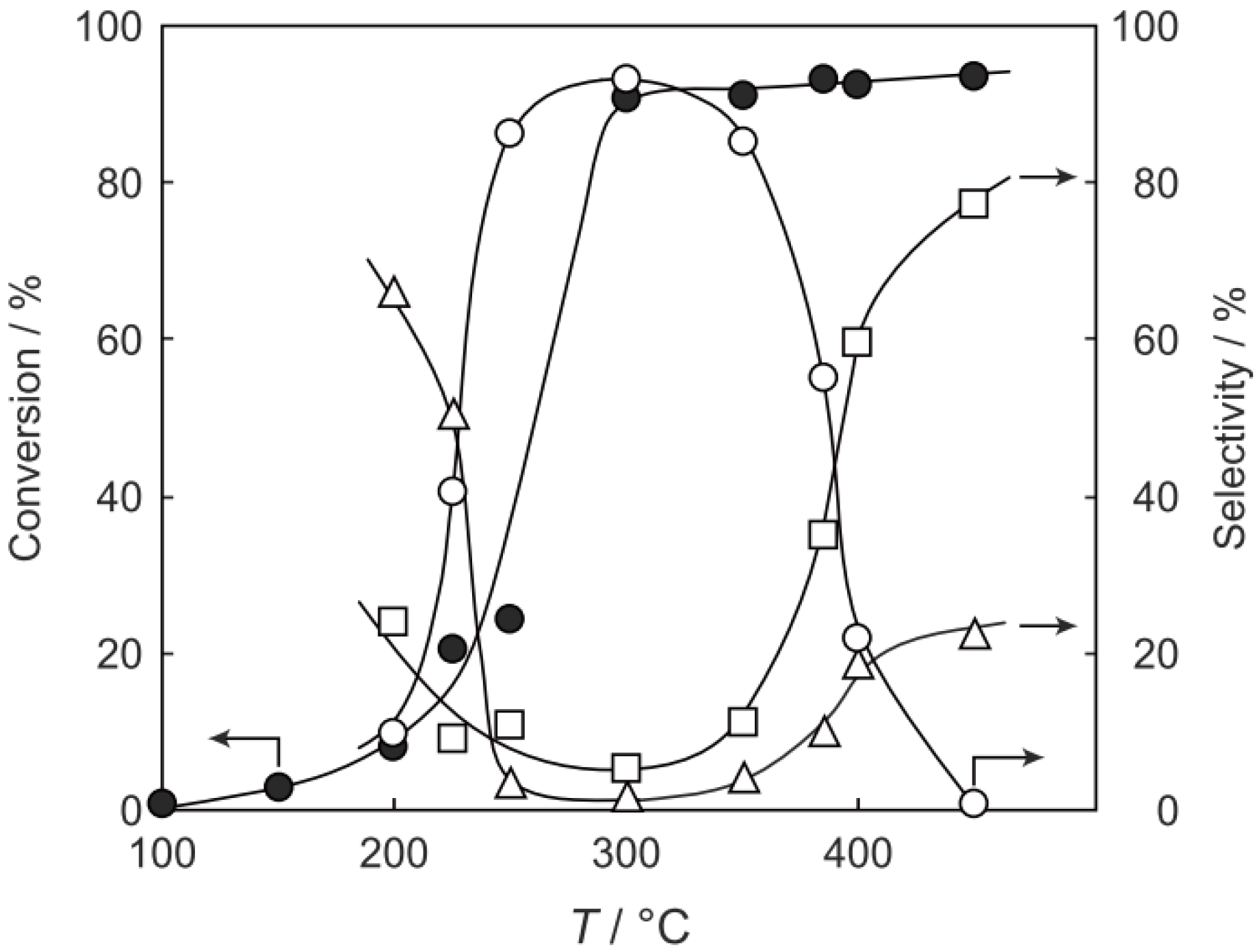
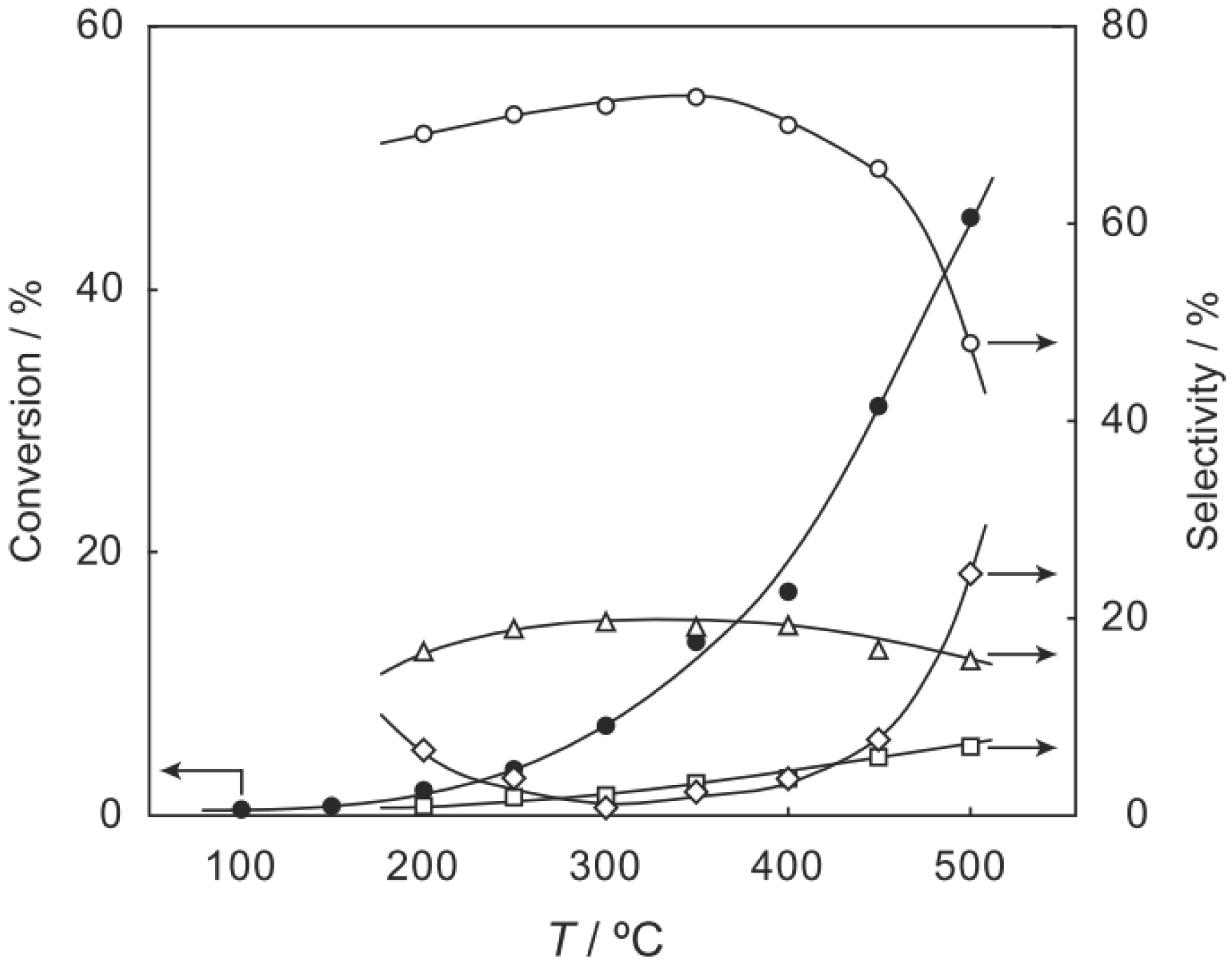
p-diethylbenzene, a catalytic reaction proceeded. The effect of temperature is shown in
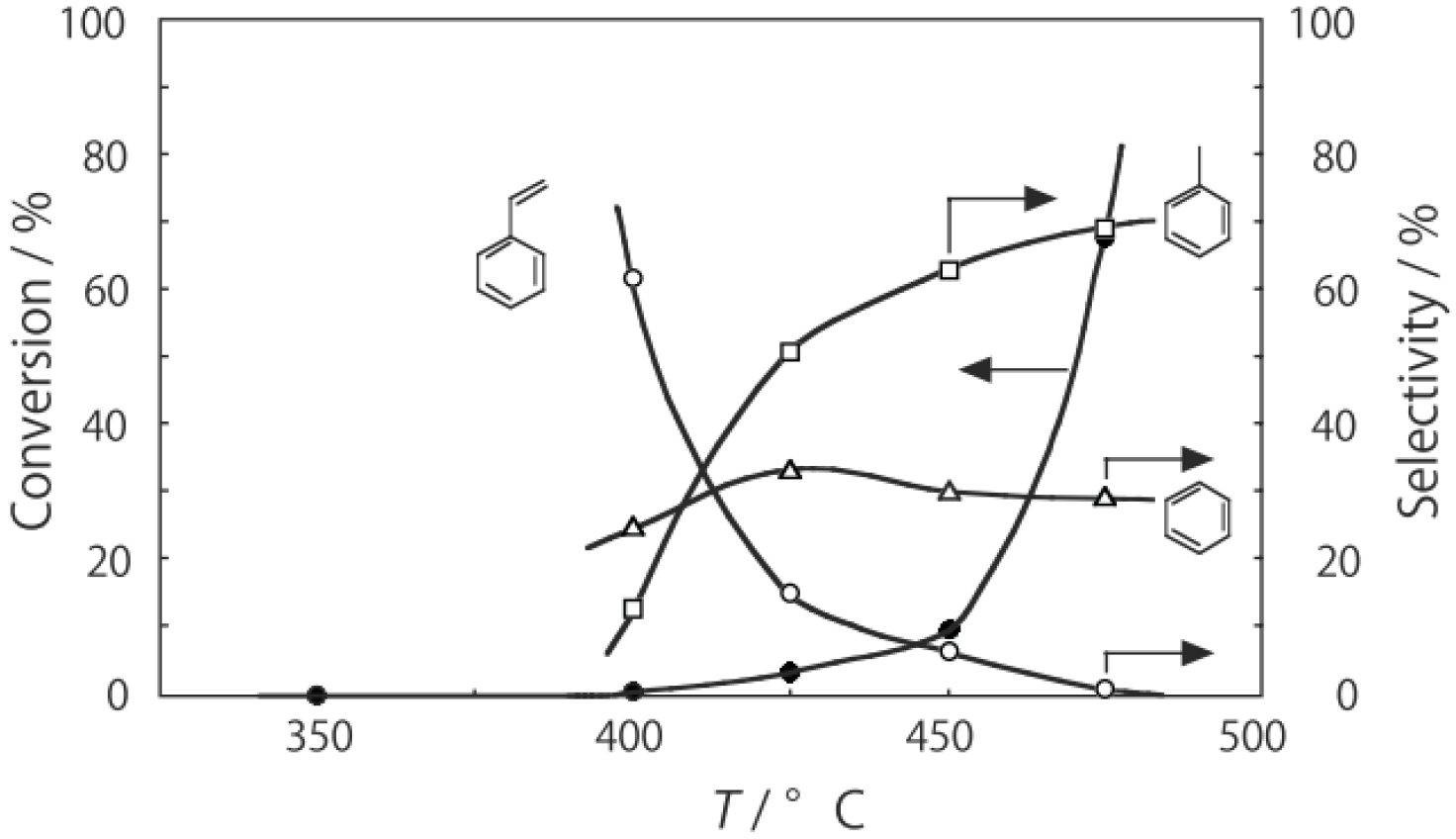
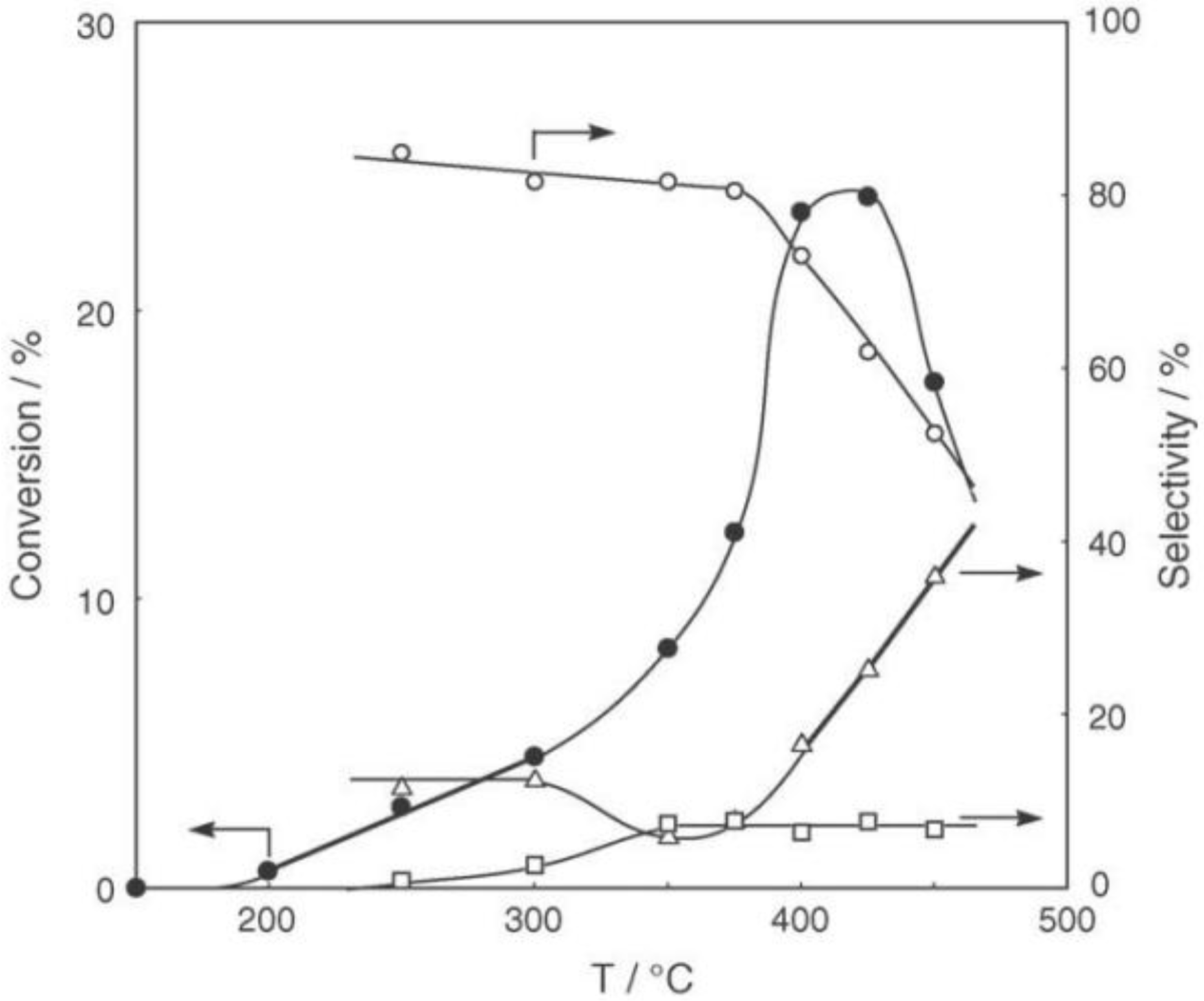
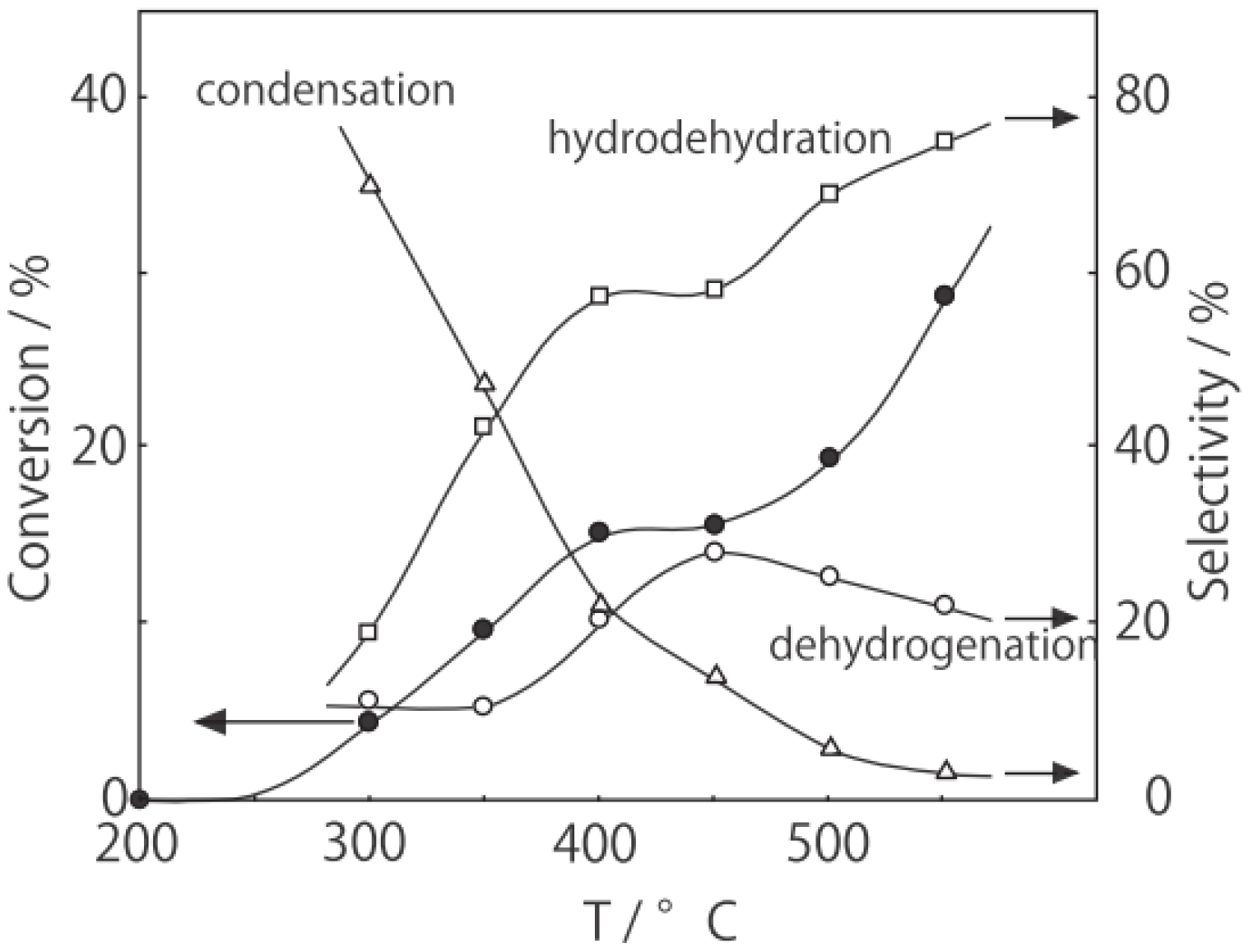
Figure 25. Reactions above 200 °C showed substantial catalytic activity. At 300 °C, the main reaction occurring was dehydrogenation (Equation (31)) to yield
p-ethylstyrene in 90% selectivity. However, at 400–475 °C this changed to ring-attachment isomerization (Equation (32)) to afford
m-diethylbenzene. At 400 °C,
m-diethylbenzene was formed with 74% selectivity, with a smaller yield of
o-diethylbenzene in 8% selectivity. Dealkylation of the side chains (Equation (33)) to yield ethylbenzene and benzene and dehydrogenation (Equation (31)) to yield
p-ethylstyrene proceeded to a small extent. However, no disproportionation (Equation (34)) to yield triethylbenzenes was observed. At higher temperatures, near 500 °C, dealkylation of the side chain (Equation (33)) proceeded more to completion with increasing temperature, yielding ethylbenzene and benzene. Thus, selectivity depended strongly on temperature.
| Dehydrogenation: | (31) |
| Et−C6H4−Et → Et−C6H4−CH=CH2 + H2 |
| Ring-attachment isomerization: | (32) |
| p-Et−C6H4−Et → o-Et−C6H4−Et+ m-Et−C6H4–Et |
| Dealkylation of the side chain (dealkylation of the ring attached alkyl): | (33) |
| Et−C6H4−Et → Et−C6H5 + C2H4 → C6H6 + 2 C2H4 |
| Intermolecular transalkylation (disproportionation): | (34) |
| 2 Et−C6H4−Et → Et−C6H5 + C6H3Et3 |
| Reductive dealkylation (hydrogenolysis ): | (35) |
| Et−C6H4−Et + H2 → Et−C6H4−Me + C6H5−Et + C6H5−Me + C6H6 + CH4 + C2H6 |
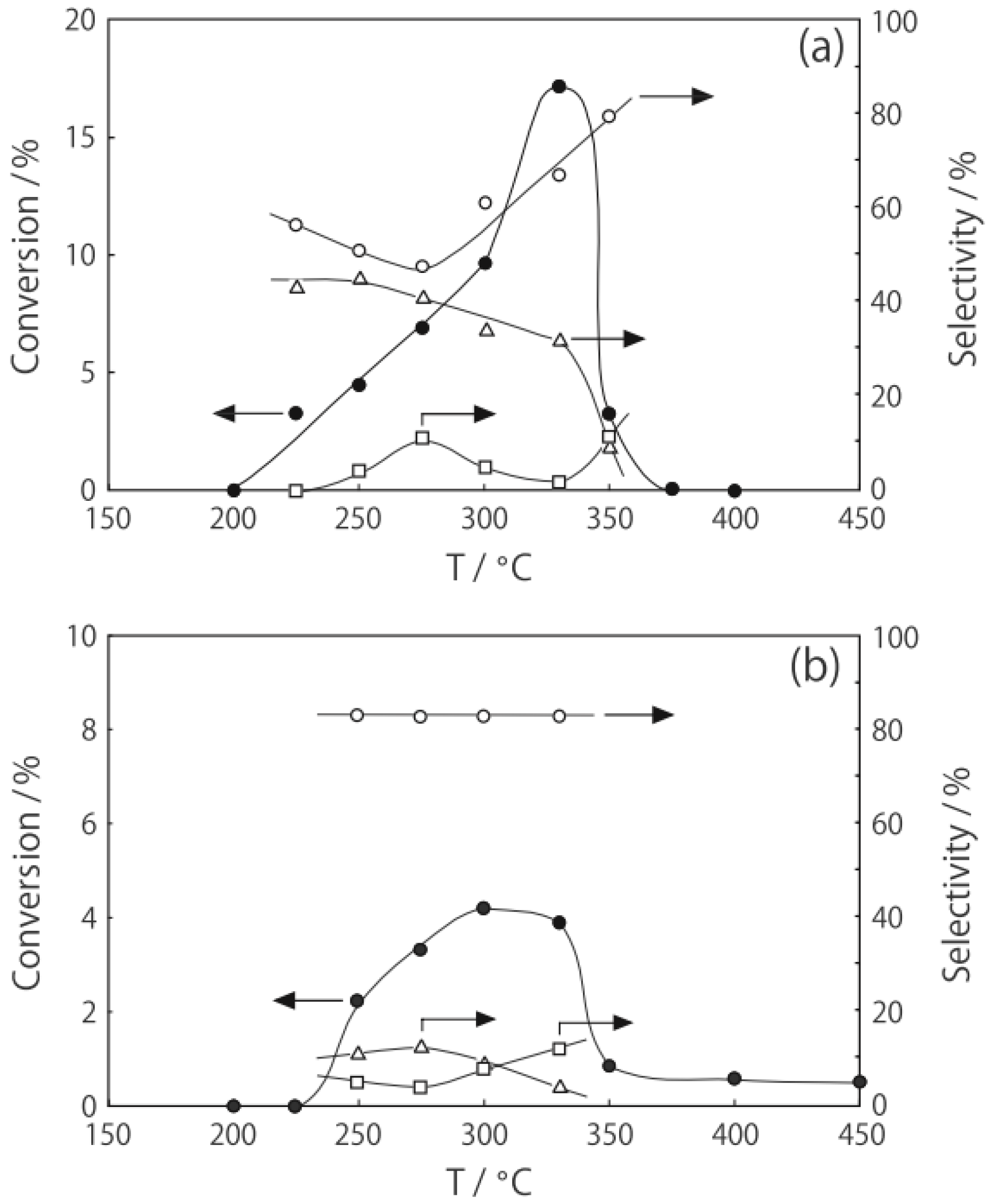
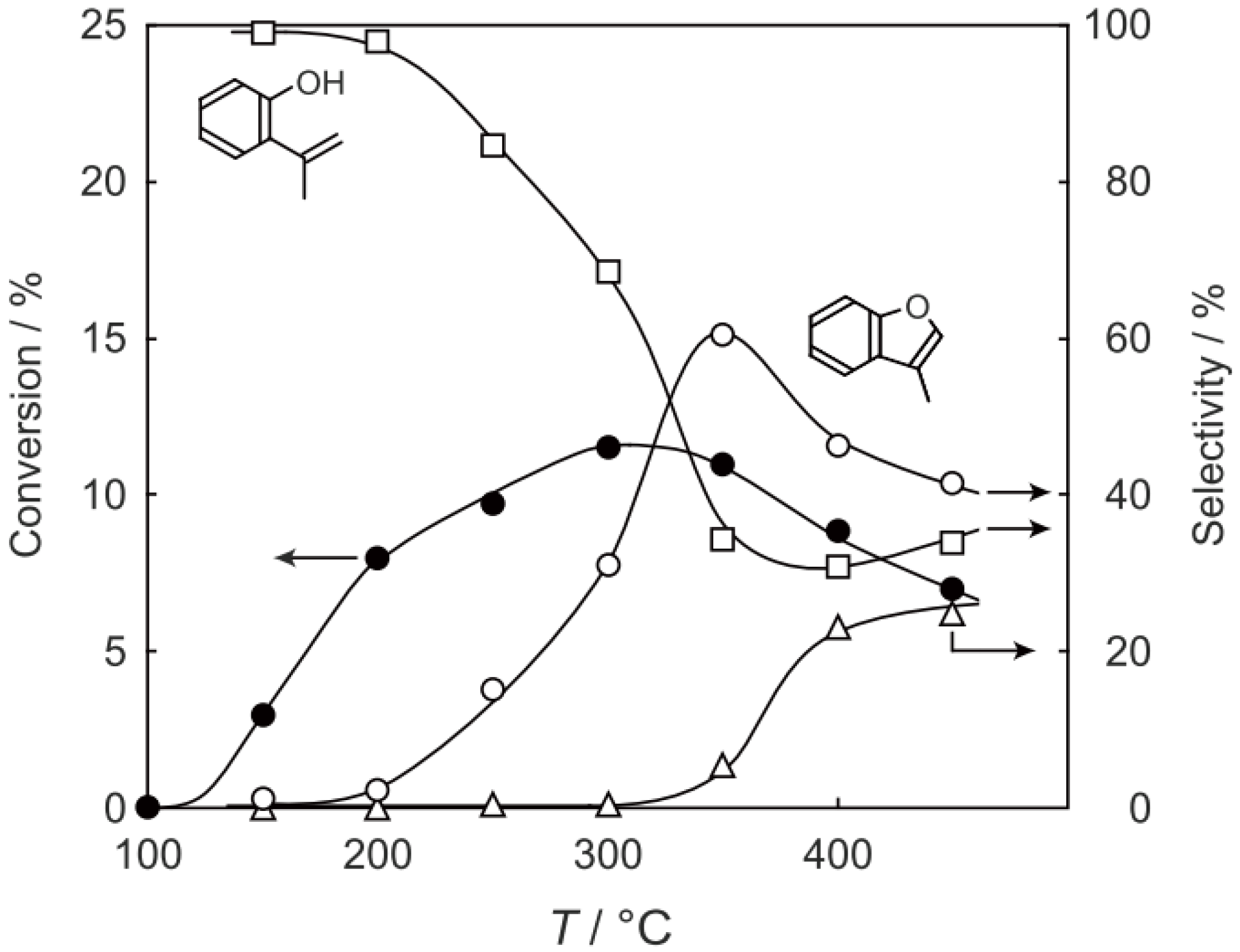
Figure 25.
Effect of temperature on the reactivity of p-diethylbenzene and product distribution over (H3O)2[(Mo6Cl8)Cl6]·6H2O. After activation of the cluster (200 mg, 163 μmol) in a stream of hydrogen (300 mL/h) for 1 h, the reaction was initiated by introducing p-diethylbenzene (0.62 μL/h, 0.40 mmol/h) to the stream without changing the temperature. Conversion of p-diethylbenzene (●); selectivity for m-diethylbenzene (■); selectivity for o-diethylbenzene (▲); selectivity for p-ethylstyrene (○); selectivity for benzene (∆); and selectivity for ethylbenzene (□); at 4 h after the start of the reaction.
The effect of the activation and reaction temperature was examined. When a 400 °C activation of (H
3O)
2[(Mo
6Cl
8)Cl
6]·6H
2O was applied to the reaction at 300 °C, the selectivity was the same as that of (H
3O)
2[(Mo
6Cl
8)Cl
6]·6H
2O treated at 300 °C throughout (
Figure 25). Hence, selectivity does not depend on activation temperature, but on reaction temperature. When the reaction gas was changed from hydrogen to helium under identical conditions, catalytic activity decreased noticeably, and its selectivity changed. Dehydrogenation yielding
p-ethylstyrene (59% selectivity) replaced isomerization to yield
m-diethylbenzene (77% selectivity) as the main reaction, indicating that hydrogen plays an important role in the reaction.
A halide cluster of tungsten, (H3O)2[(W6Cl8)Cl6]·6H2O, exhibited almost the same catalytic activity and selectivity with (H3O)2[(Mo6Cl8)Cl6]·6H2O. On the other hand, a halide cluster of tantalum, [(Ta6Cl12)Cl2(H2O)4]·4H2O, selectively catalyzed the dehydrogenation to yield p-ethylstyrene and a small quantity of p-divinylbenzene. A cluster of niobium, [(Nb6Cl12)Cl2(H2O)4]·4H2O, showed intermediate selectivity with respect to the above clusters, forming both m-dimethylbenzene and p-ethylstyrene in significant quantities.
Some
p-substituted benzenes such as
p-xylene,
p-ethyltoluene, and
p-propyltoluene were reacted over (H
3O)
2[(Mo
6Cl
8)Cl
6]·6H
2O under hydrogen at 400 °C. All substrates tested yielded the corresponding
m-isomer as the main product by ring-attachment isomerization. Three diethylbenzene isomers were reacted over (H
3O)
2[(Mo
6Cl
8)Cl
6]·6H
2O at 400 °C to achieve similar conversions, with the main reaction being ring-attachment isomerization, although the selectivity for the isomerization decreased in the order
p- >
m- >
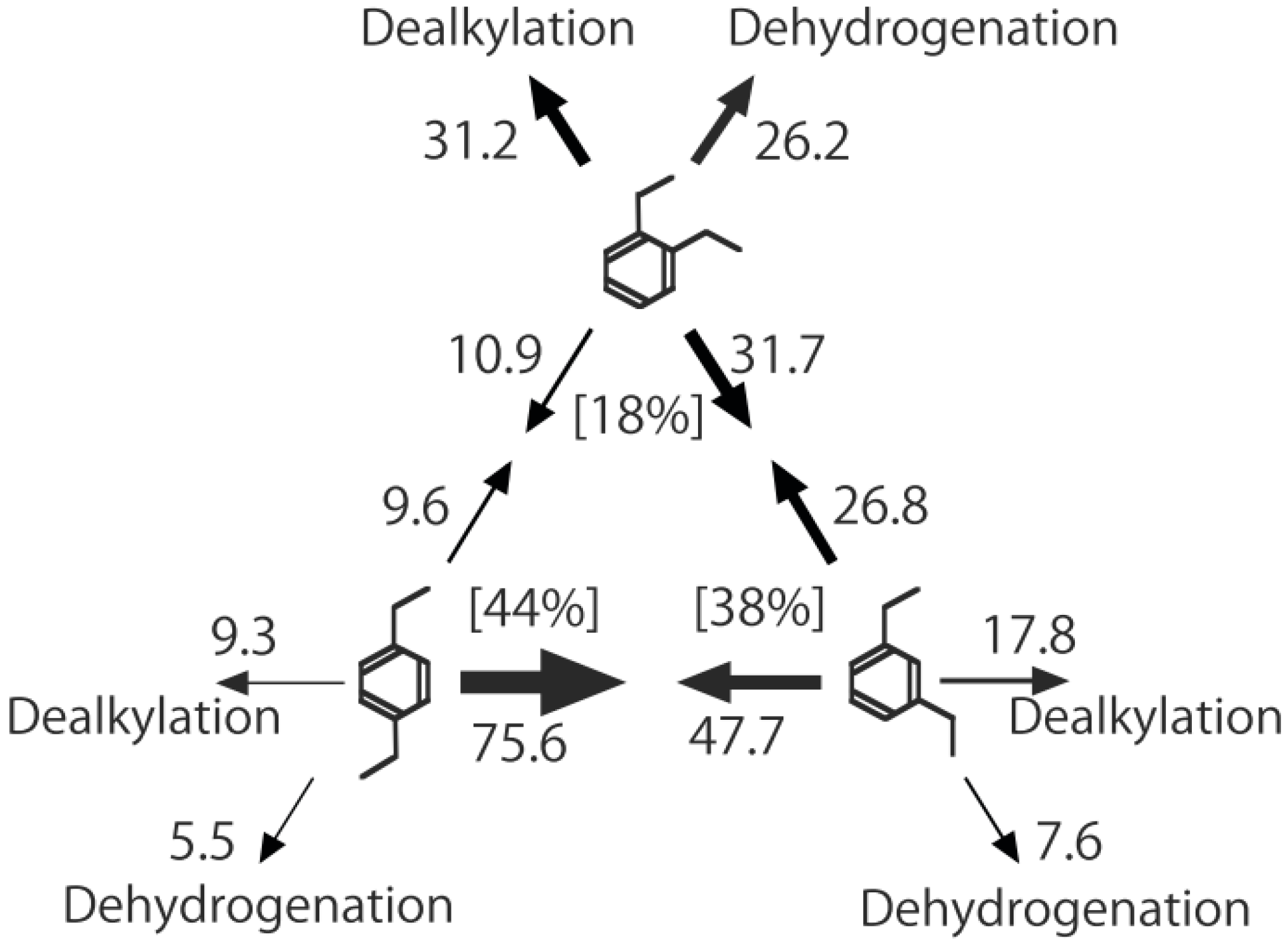
o-isomer. Even traces of triethylbenzenes (Equation (34)) were not detected in all the reactions. The selectivities for mutual interconversion of the three isomers are illustrated in
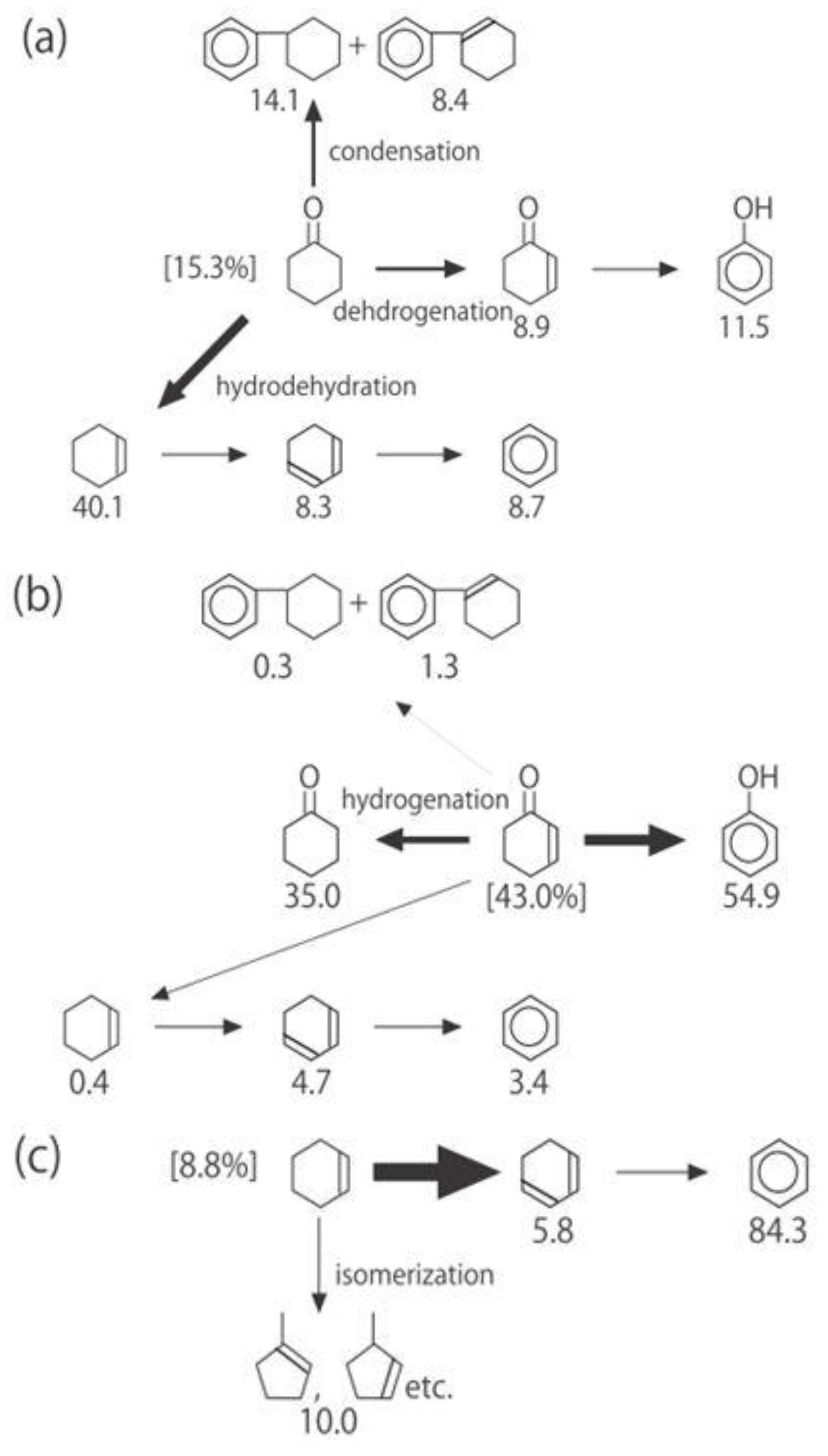
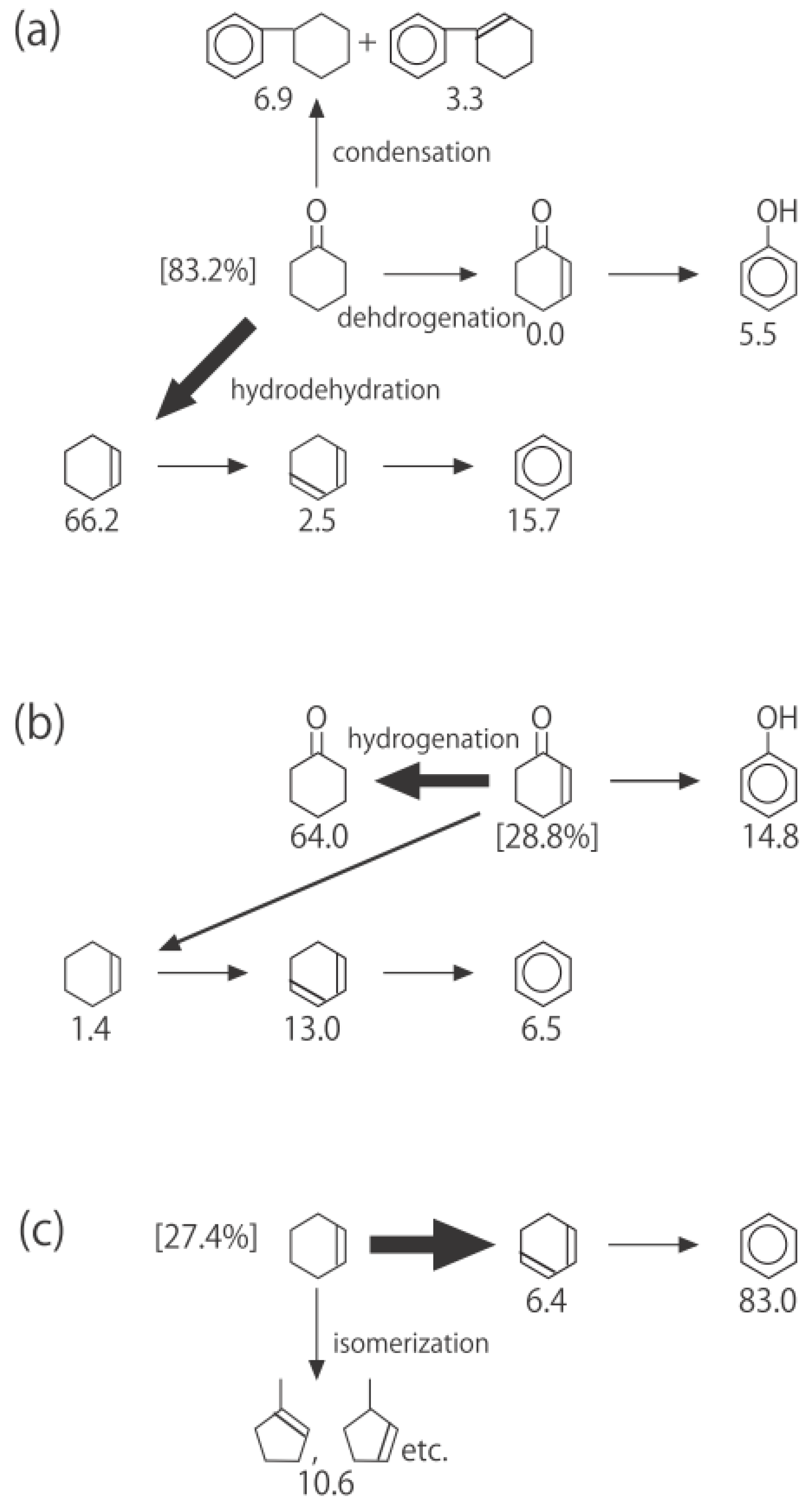
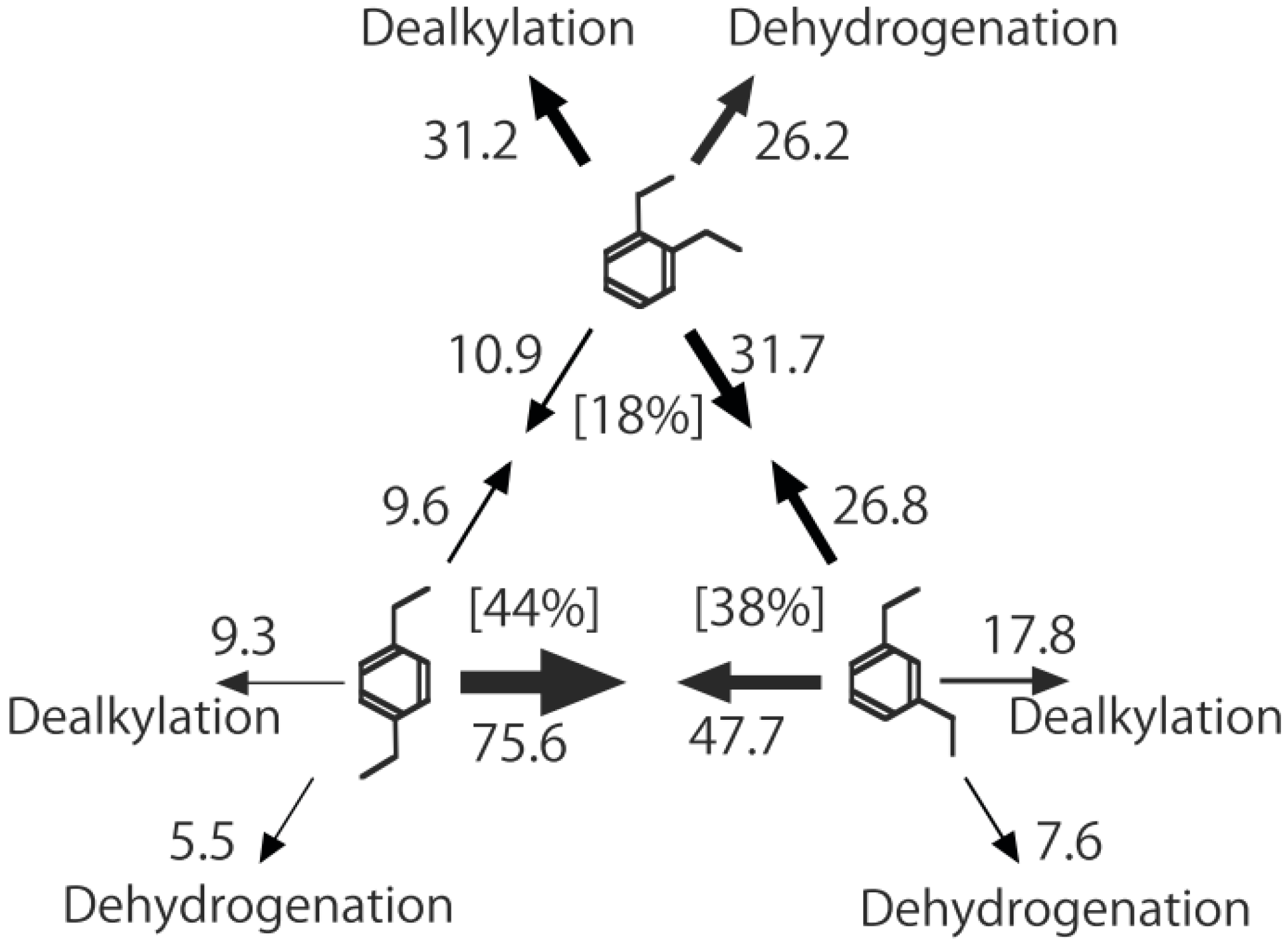
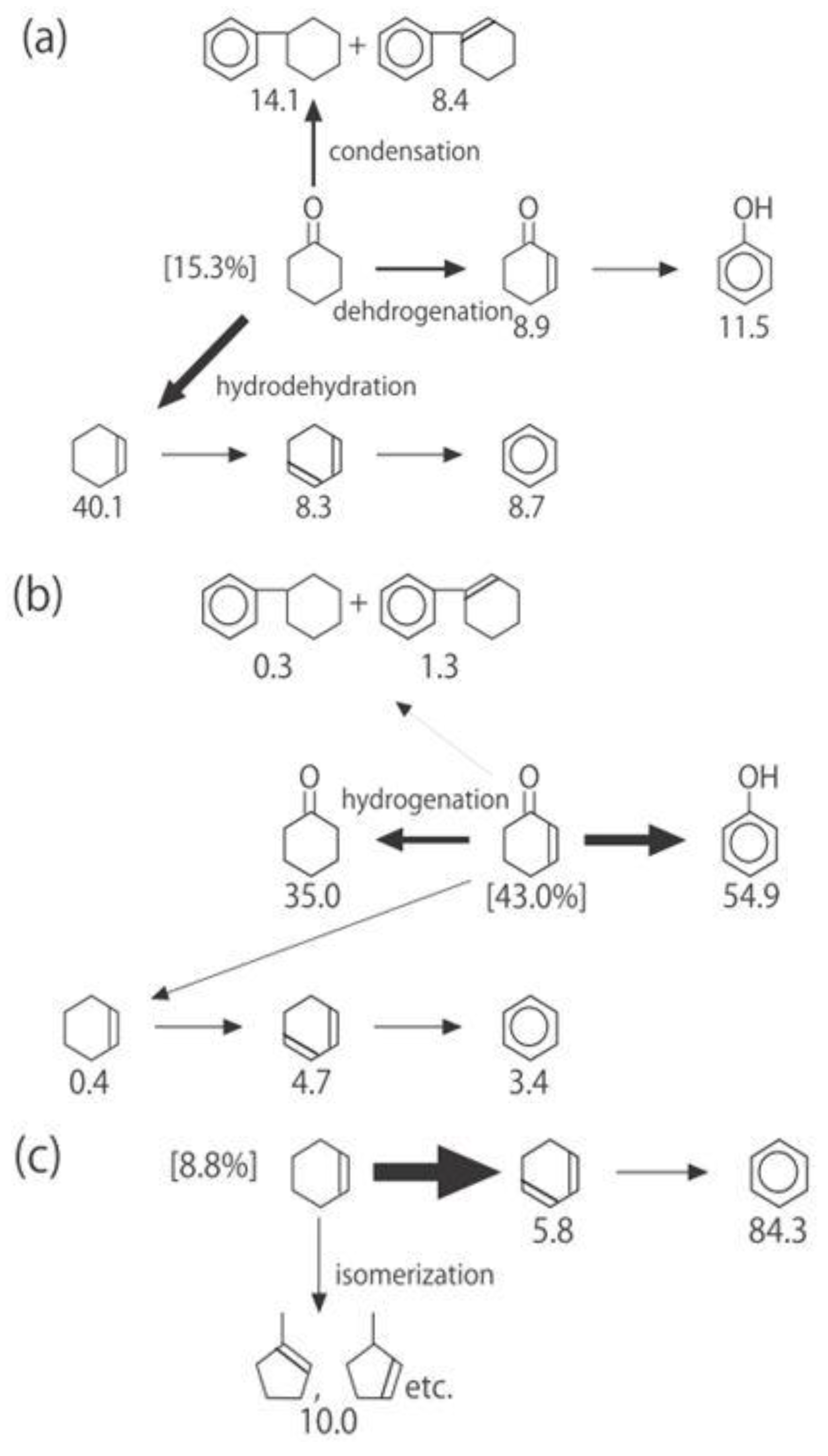
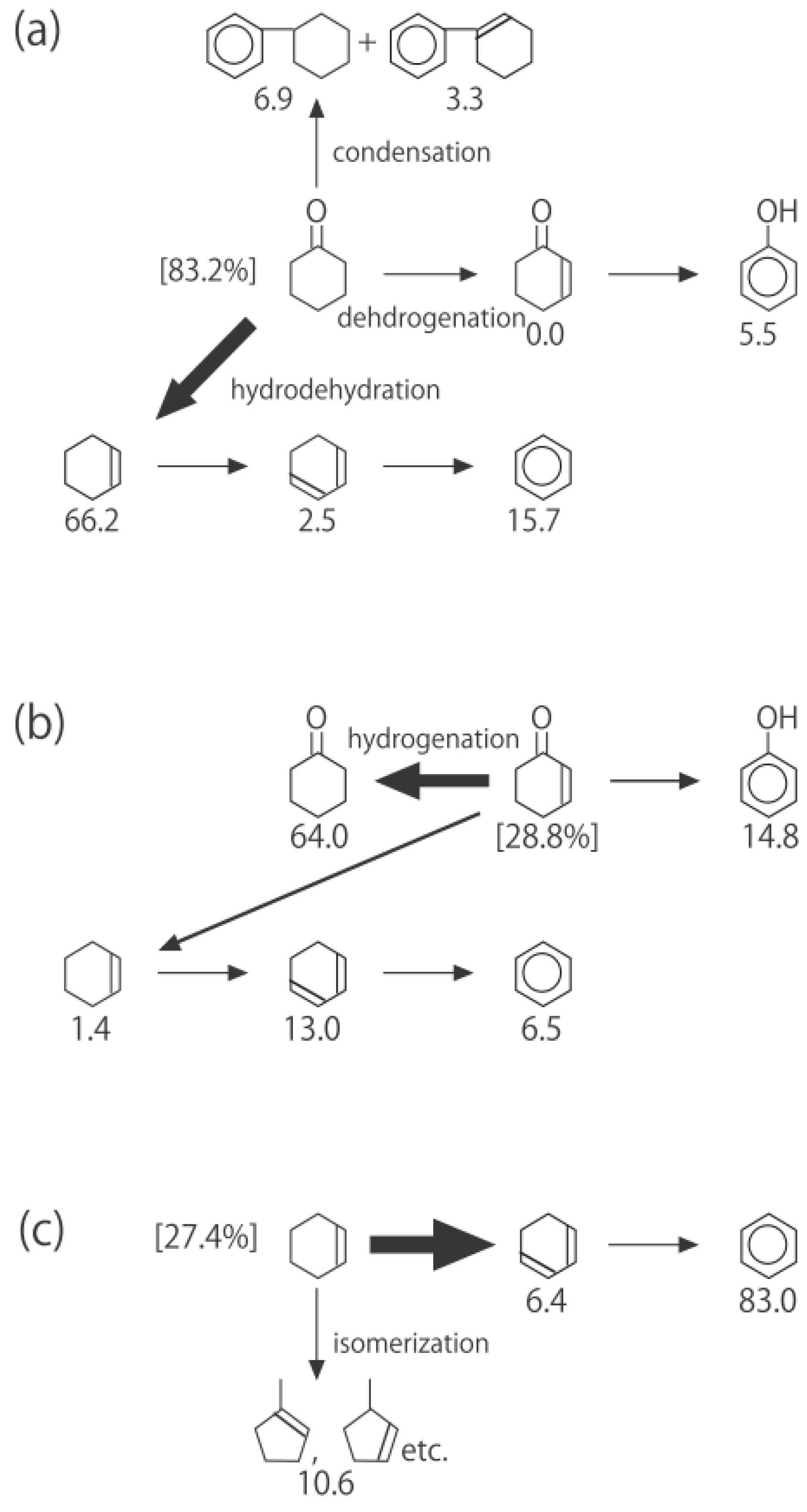
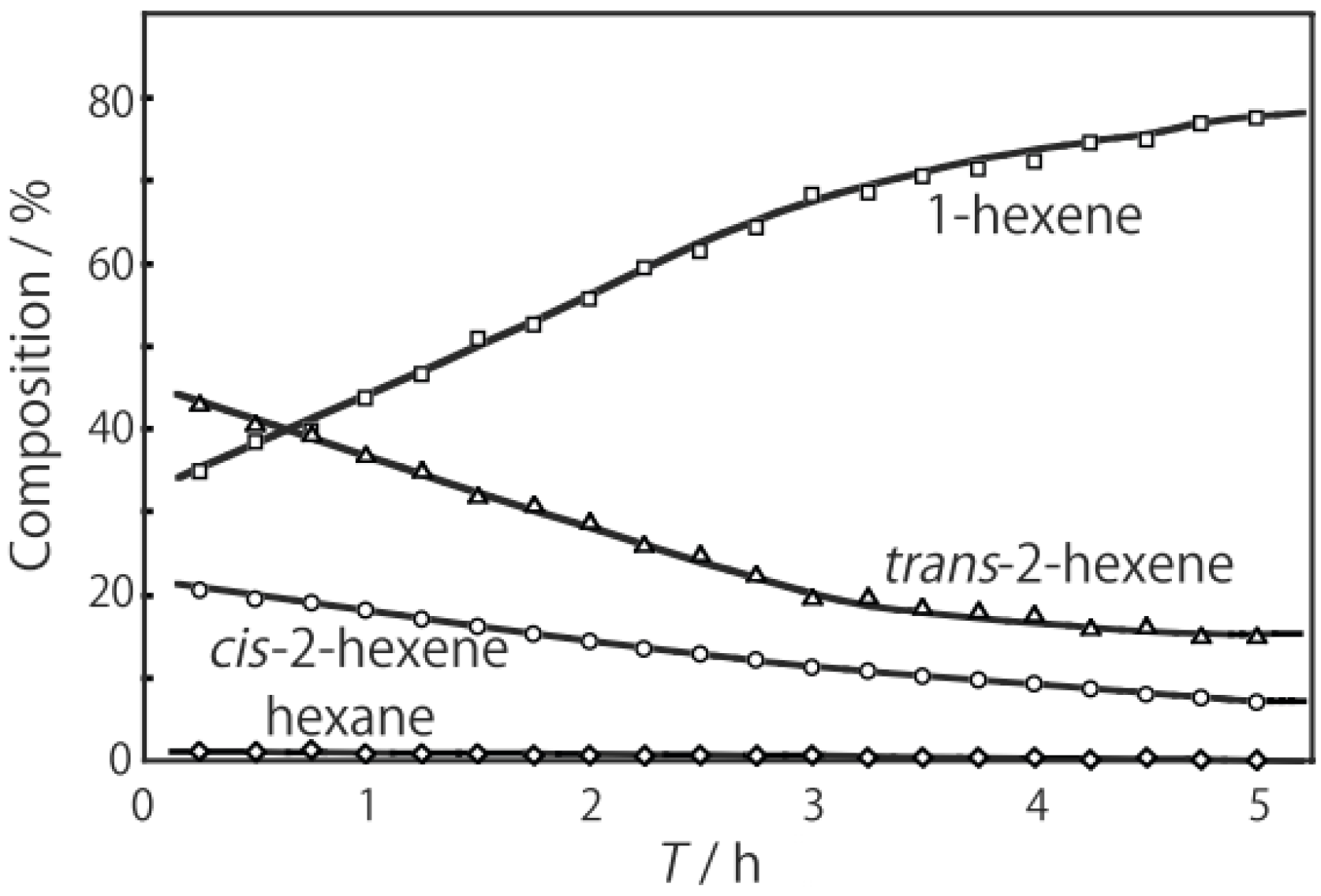
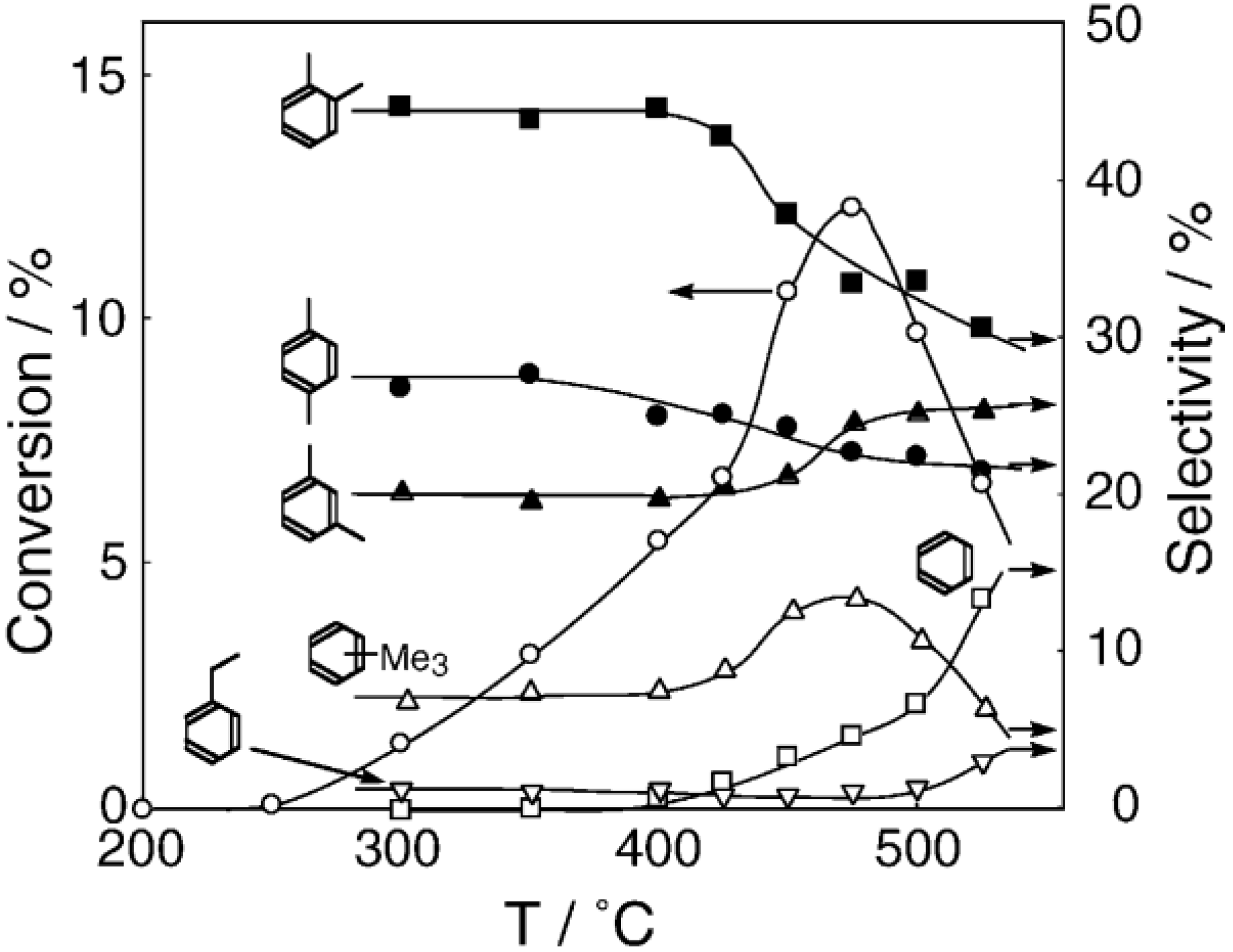
Figure 26. One of the most striking features of the reactions is that
o-diethylbenzene afforded the
m-isomer in a selectivity that was three times that of the
p-isomer, even though from a thermodynamical point of view, the
p-isomer is more stable than the
m-isomer.
p-Diethylbenzene yielded predominantly its
m-isomer, which was only twice as abundant as the equilibrium composition, compared with the
o-isomer. On the other hand, the
m-isomer afforded the sterically hindered
o-isomer more abundantly than expected from the equilibrium composition, when compared with the formation of the
p-isomer. Consequently, the ethyl group preferentially migrated to the neighboring position of the aromatic ring, irrespective of the equilibrium distribution.
Figure 26.
Isomerization of diethylbenzenes over (H
3O)
2[(Mo
6Cl
8)Cl
6]·6H
2O, showing the selectivity values (in %) at 400 °C, 4 h after the reaction was initiated. The other conditions are the same as those described in the caption to
Figure 25. The thickness of each arrow is proportional to the corresponding selectivity. The equilibrium compositions of diethylbenzenes in the vapor phase at 400 °C are shown in brackets.
When a mixture of
p-xylene and
p-diethylbenzene was applied to the reaction, ring-attachment isomerization occurred to yield the corresponding
o- and
m-isomers. However, the formation of
o- and
m-ethyltoluenes, which would be expected from intermolecular alkyl transfer, was not detected. Furthermore, triethylbenzenes were not detected in any of the reactions. These results clearly demonstrate that the isomerization proceeded by way of an intramolecular 1,2-shift of the ethyl group around the aromatic ring. They are distinct from conventional strong acids. The catalytic disproportionation of ethylbenzene in the presence of AlCl
3 yielded benzene,
m- and
p-diethylbenzenes, and 1,2,4-triethylbenzene [
186]. In an AlCl
3–HCl catalytic system, the disproportionation of
m- and
p-diethylbenzenes proceeded to yield ethylbenzene and 1,2,4-triethylbenzene [
187]. A protic acid, CF
3COOH, also catalyzed the disproportionation of
m-diethylbenzene to yield ethylbenzene and 1,2,4-triethylbenzene by an ethylcarbenium rearrangement [
188]. All of these catalytic reactions were disproportionations. The disproportionation of trimethylbenzenes and toluene has been reported to occur on stronger acid sites, while the isomerization of trimethylbenzenes and xylenes have been reported to predominate on weaker acid sites [
189]. In the case of the cluster catalysis, it would be difficult to abstract the ethyl substituent as an ethyl cation intermediate, because the charge density of the counter cluster anion is too low to stabilize the ethyl cation [
1]. The isomerizations over cluster catalysts were always accompanied by an appreciable yield of dealkylation products, particularly over Group 6 metal clusters (
Figure 26). Atmospheric hydrogen, as well as alkyl hydrogen, was activated over these cluster catalysts (
Section 2.1 and
Section 2.2), and used to carry out hydrogenolyses (Equation (35)), which have been reported over platinum group metal catalysts.
Conclusively, the halide clusters developed catalytic activity above 200 °C for p-diethylbenzene. The activity depended on reaction temperature, with dehydrogenation, ring-attachment isomerization, and dealkylation increasing with increasing temperature. At 400–475 °C, halide clusters exhibited a characteristic selective ring-attachment isomerization of diethylbenzenes by the intramolecular 1,2-shift mechanism without yielding disproportionation products. They are distinct from conventional strong acids, isomerization of which is based on a disproportionation mechanism. Another characteristic feature of halide cluster catalysis is that the halide clusters have no micropore structures and, hence, they can be used to catalyze o-diethylbenzene, which is too large to enter the pores of zeolite Y and ZSM-5.
6.2. Reaction of Aliphatic Amine (Equation (16)) [190]
Catalytic dehydrogenation of amines has been investigated extensively, and the dehydrogenation of primary amines usually gives the corresponding nitriles. Sometimes, an
N-substituted Schiff base can be obtained by dehydrogenation followed by condensation with the amine: benzylamine is converted to
N-benzylidenebenzylamine over Mo–V-heteropoly acid in the presence of dioxygen [
191]. On the other hand, catalytic dehydrogenation without oxidizing reagents is rare: molten Zn or Ga metal [
192] and Ga–MFI zeolite [
193] have been used as catalysts in gas-phase reactions. There are several reports on the catalytic and stoichiometric dehydrogenation of activated secondary amines, Ar−CH
2−NH−R (Ar = aryl, R = alkyl or aryl). In this case the dehydrogenation product is unique, and hence, selective reaction can be readily achieved as long as C−N bond cleavage does not occur. In contrast, reports on aliphatic secondary and tertiary amines are few, particularly in the gas phase.
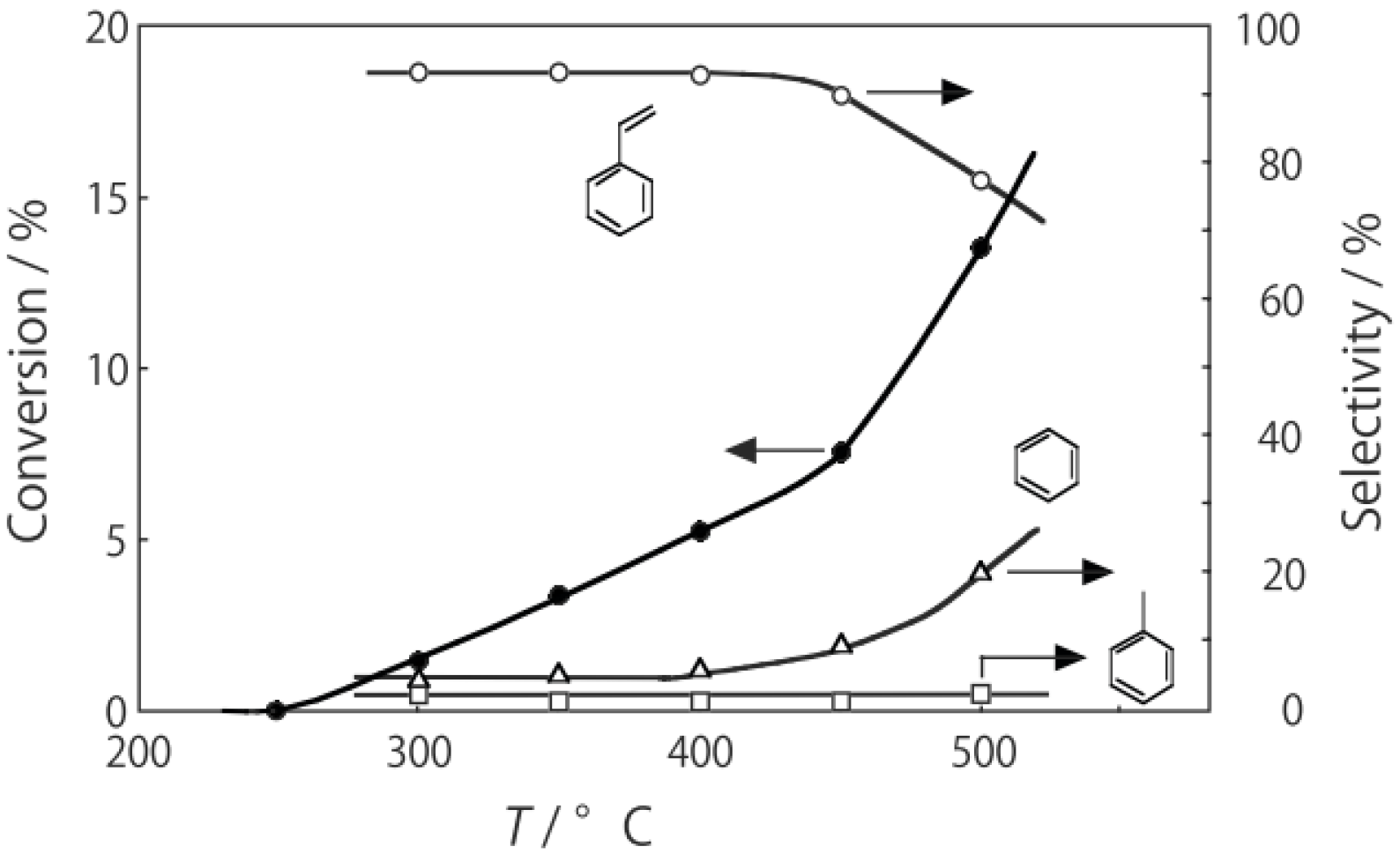
The powdered crystals of (H
3O)
2[(Mo
6Cl
8)Cl
6]·6H
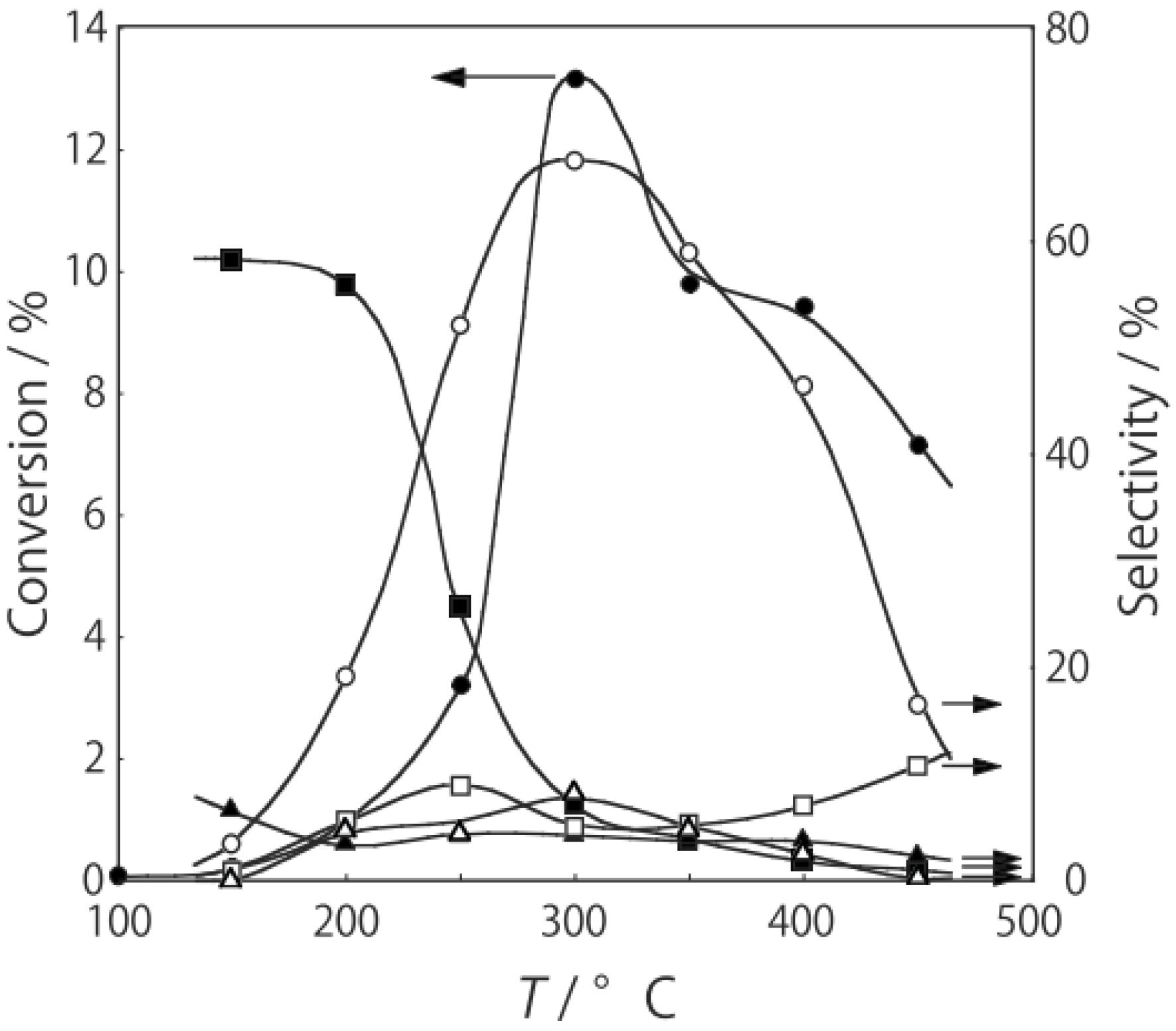
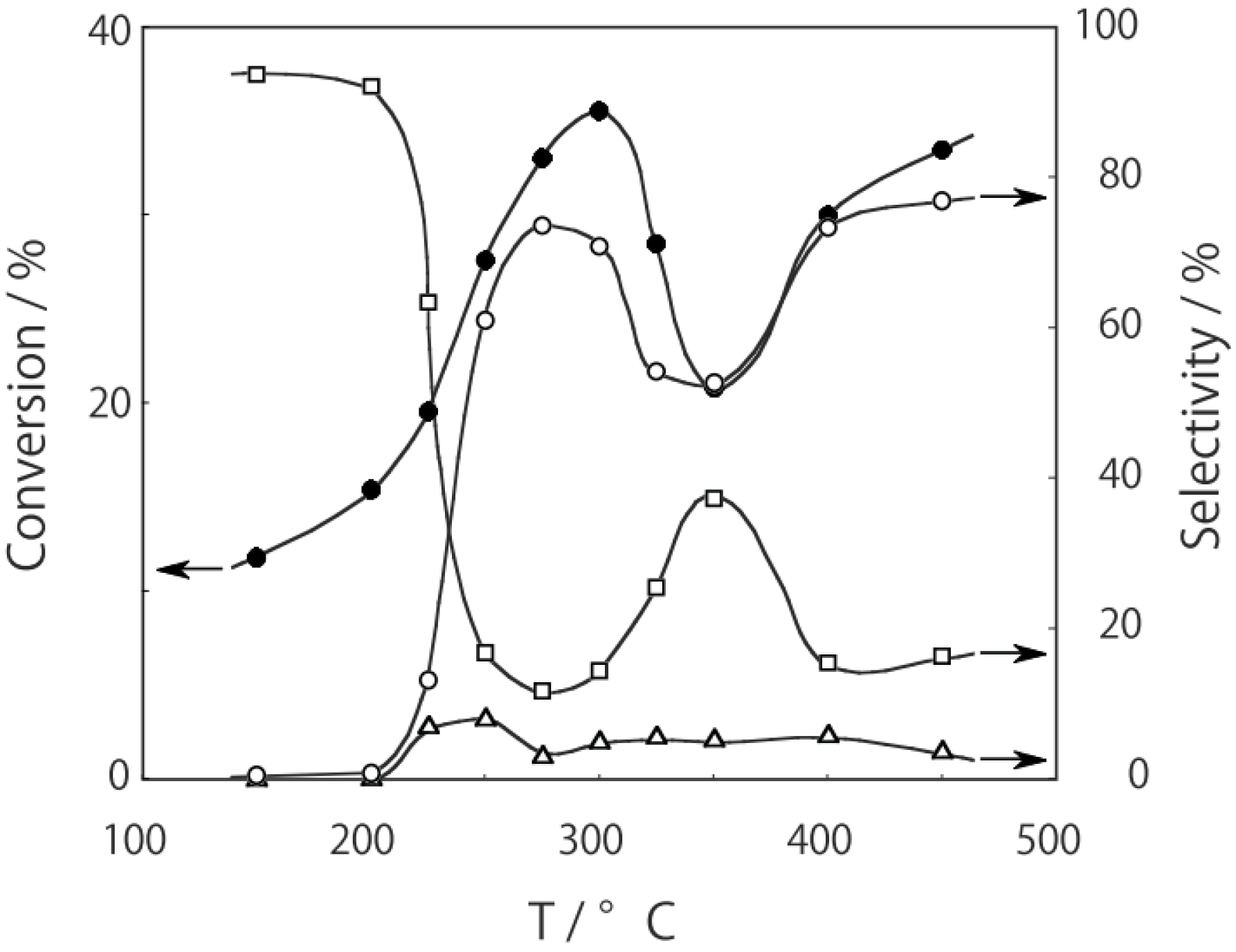
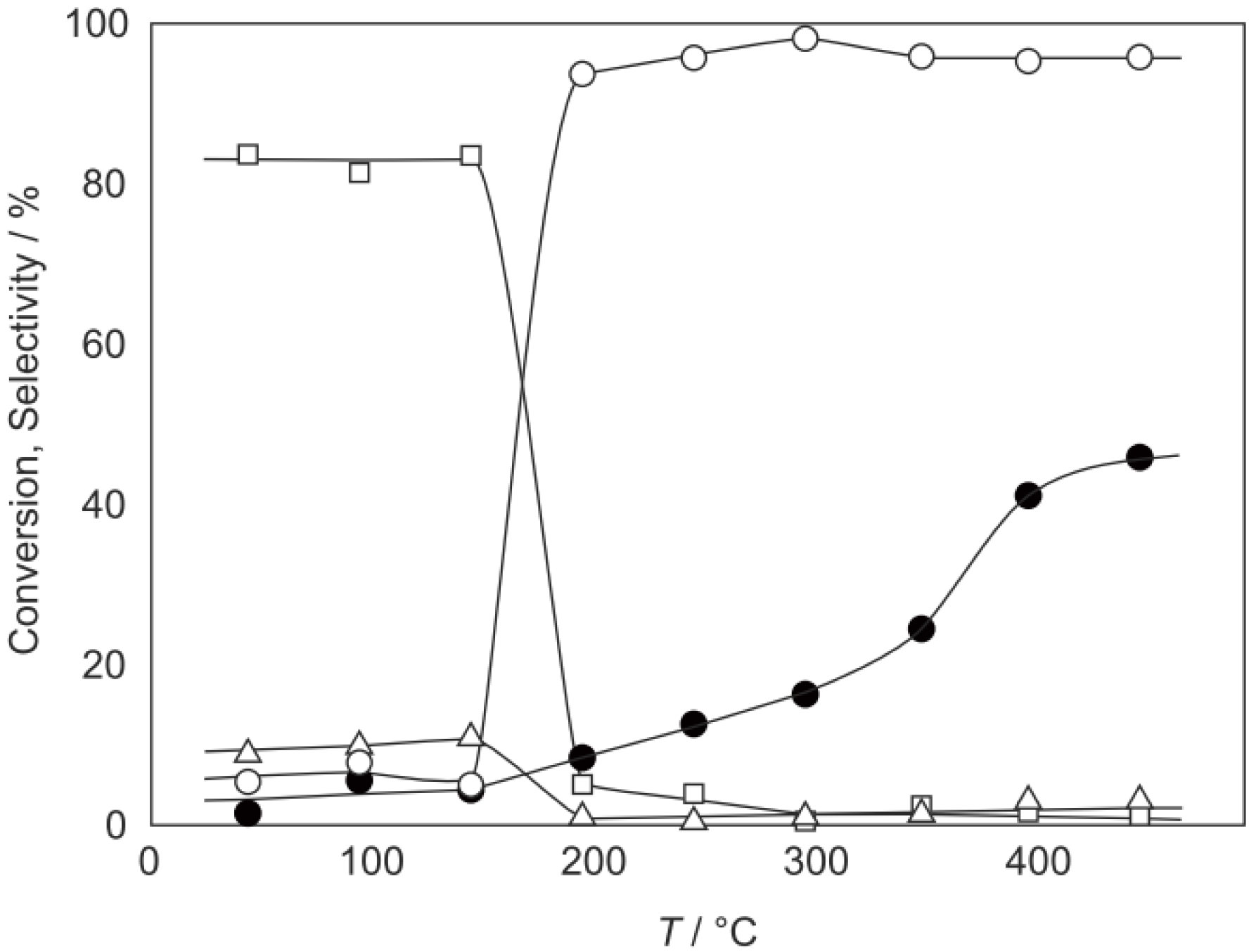
2O were activated in a stream of hydrogen or helium for 1 h. The reaction was started by introducing diethylamine into the stream without changing the temperature. The effect of the reaction temperature is presented in
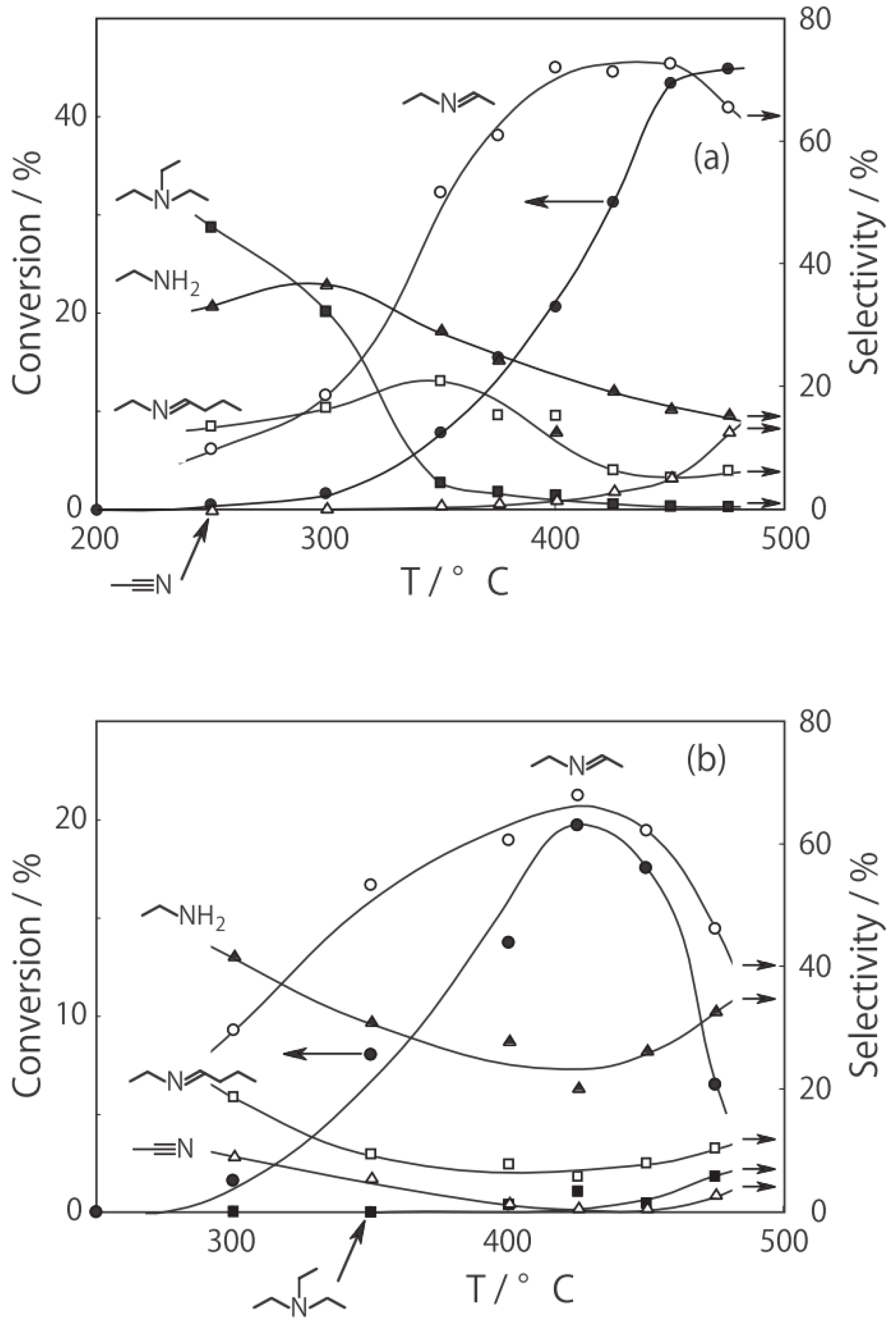
Figure 27. Under hydrogen, the reactivity increased with increasing temperature. At lower temperatures below 300 °C, disproportionation yielding ethylamine and triethylamine was predominant; at higher temperatures at 400–450 °C dehydrogenation providing
N-ethylideneethylamine was the main reaction (72% selectivity at 400 °C). Under helium the catalytic activity was about half that under hydrogen. Hydrogen would prevent dealkylation and the deposition of coke on the active sites [
15,
16]. Under helium, dealkylation yielding ethylamine was noticeable at lower temperatures below 300 °C, and at higher temperatures above 350 °C, dehydrogenation yielding
N-ethylideneethylamine was predominant with a maximum 69% selectivity at 425 °C. Tungsten cluster similarly dehydrogenated to produce
N-ethylideneethylamine with 62% selectivity at 400 °C under hydrogen.
Figure 27.
Effect of temperature on reactivity and product selectivity of diethylamine over (H3O)2[(Mo6Cl8)Cl6]·6H2O in a (a) hydrogen; and (b) helium stream. Following activation of the cluster (30 mg, 25 μmol) in the gas stream (600 mL/h) for 1 h, reaction was started by introduction of diethylamine (0.20 mL/h, 1.9 mmol/h) to the stream without changing the temperature. Conversion and selectivities are based on diethylamine at 3 h after reaction started.
The Group 5 metal clusters preferentially dealkylated diethylamine to yield ethylamine and its dehydrogenation product acetonitrile (44%–59% selectivity under both hydrogen and helium) in conjunction with the transalkylation product triethylamine with more than 12% selectivity. Aliphatic amines usually afford diverse products in catalysis; hence, there are few reports on selective reactions.
The catalytic activity of (H
3O)
2[(Mo
6Cl
8)Cl
6]·6H
2O for some primary amines was examined at 400 °C. Ethylamine was exclusively dehydrogenated to acetonitrile and
N-ethylideneethylamine. The latter, a Schiff base, would be produced by condensation of ethylamine with ethylideneamine, which is a dehydrogenation intermediate to acetonitrile (Equation (36)) [
194].
Butylamine gave the dehydrogenation product butyronitrile with more than 80% selectivity and with a small amount of the condensation product
N-butylidenebutylamine. Thus, the cluster selectively catalyzed dehydrogenation of primary amines to give the corresponding nitriles with their condensation products, Schiff bases. Many catalysts have been applied to aliphatic primary amines. Basic metal oxides, ZrO
2 and MgO, selectively dehydrogenate primary amines to yield the corresponding nitriles [
195]. Solid acids exhibit various activities: Al
2O
3 catalyzes transalkylation, dehydrogenation, condensation, and deamination nonselectively [
196,
197]. Another report states that Al
2O
3 deaminates ethylamine [
198] and various aliphatic primary amines [
194] to give olefins with ammonia. H–MFI and Ga–MFI zeolites catalyze transalkylation of propylamine to provide dipropylamine. In–MFI further catalyzes dehydrogenation to give dipropylamine and
N-propylidenepropylamine. Cu–MFI catalyzes C–C coupling of the alkyl substituents of propylamine and dehydrogenation to yield 2-methylpentanenitrile and propiononitrile as the major products [
193,
199].
The secondary amines, diethylamine and dibutylamine, selectively gave dehydrogenation products (imines) in a hydrogen stream over (H
3O)
2[(Mo
6Cl
8)Cl
6]·6H
2O, although dealkylation proceeded in a helium stream at 400 °C. Several catalytic reactions of aliphatic secondary amines have been reported. Diethylamine is dehydrogenated to
N-ethylideneethylamine at 350 °C over Mo-heteropoly acid [
197]. Solid acid Al
2O
3 [
196] and W-heteropoly acid [
200] nonselectively catalyzes dealkylation, dehydrogenation, and disproportionation. Another report mentions that SiO
2–Al
2O
3 dealkylates and that acid–base bifunctional catalysts, such as SiO
2–Al
2O
3/MgO and ZrO
2, selectively transform diethylamine into acetonitrile [
195]. To the best of our knowledge, the successful application of the platinum metal catalysts to give normal alkylimines has not been reported. Thus, dehydrogenation selectivity of the cluster catalyst for secondary aliphatic amines differs from that of conventional acid or base catalysts but is similar to that of the Mo-heteropoly acid, which is characterized by very weak basicity, great softness, and a large polyhedral anion structure [
201].
Tertiary amines were allowed to react over (H
3O)
2[(Mo
6Cl
8)Cl
6]·6H
2O at 400 °C. In the case of triethylamine, dealkylation and successive dehydrogenation yielded diethylamine and
N-ethylideneethylamine as the major products. The combined selectivity for the monodealkylation and the following dehydrogenation amount to 68% in a hydrogen stream and 55% in a helium stream. Dehydrogenation of trietylamine occurred also in the alkyl chain, yielding
N-vinyldiethylamine with 21%–26% selectivity. Furthermore, tripropylamine and tributylamine gave the corresponding
N-vinylamines as the main products in a hydrogen stream with 52 and 45% selectivity, respectively. There are only a few examples of the catalytic dealkylation or dehydrogenation of tertiary amines. When the acid catalyst SiO
2–Al
2O
3 is applied to triethylamine, dealkylation proceeds nonselectively [
195]. The base catalyst MgO dealkylates and dehydrogenates triethylamine to nonselectively give diethylamine, ethylamine, and acetonitrile, whereas the acid–base bifunctional catalyst SiO
2–Al
2O
3/MgO selectively converts triethylamine to acetonitrile [
195]. Consequently, selective dealkylation of trialkylamine to dialkylamine has not yet been achieved. Furthermore, the formation of even a trace amount of
N-vinyldialkylamine over solid catalysts has not been reported.
One of the active sites on the molybdenum and tungsten clusters is coordinatively unsaturated or, more likely, hydrido ligand coordinated Mo and W atoms, which behave like the platinum group metals [
1]. There are many reports on Group 8–10 transition metals for the exhaustive dehydrogenation of alicyclic amines. However, no reports of success with
n-alkyl amines have been published, presumably because of their high activity, leading to polyunsaturated alkyl chains or their random cleavage. Hence, the characteristic features of the halide clusters to amines cannot be attributed to their similarity to the platinum group metals.
There are fewer cases of dehydrogenation of amines over heteropoly acid, one of which, 12-molybdophosphoric acid, catalyzed the selective dehydrogenation to yield
N-ethylideneethylamine [
200]. The selectivity is similar to that of the halide cluster catalysts. A heteropoly acid has several types of protons, one of which originates from coordinated water [
202,
203]. It has a Brønsted acid site, but no Lewis acid site [
204]. The heteropoly anions are large and hence, have very low charge densities on their surface [
205], as in the case of the large halide cluster anion.
The large anions would not participate in the reaction in either case. Consequently, the reaction proceeded by way of the protonated amines without interaction with the large counter-anions. On the other hand, alumina has been classified as a solid acid. However, it has both acidic and basic sites: incompletely coordinated aluminum ions (strong Lewis acids) and oxide ions (weak Lewis bases) [
49,
50,
51,
52,
53]. Similarly, solid bases such as MgO [
206] and hematite (α-Fe
2O
3) [
207] have both basic sites as oxide ions and acidic surface sites as Mg
2+, Fe
3+, and Fe–OH. All of these sites are small and have high charge densities with participation in the reactions.
In summary, when the halide clusters were treated above 300 °C, the catalytic activity for dehydrogenation of aliphatic amines developed. Primary amines were dehydrogenated to the corresponding nitriles, some of which condensed to the alkylidenealkylamines. Secondary aliphatic amines were dehydrogenated to the corresponding imines. Tertiary amines were dealkylated to dialkylamines, which was followed by dehydrogenation to N-alkylidenealkylamines, or directly dehydrogenated to N-vinyldialkylamines. These selectivities were different from conventional catalysts, such as solid acid–base catalysts or the Group 8–10 transition metals. Spontaneous degradation of the protonated amine at high temperature without interaction with the large cluster counter-anion would be the driving force for these reactions. The initial step of the reactions is simple: from protonated amine, nitrogen-bonded hydrogens are removed completely to yield nitrile or imine; when there is no such hydrogen, either a neighboring hydrogen or the nitrogen-bonded alkyl group is removed to yield vinylamine or alkylideneamine.
6.3. Beckmann Rearrangement (Equation (17)) [208]
The Beckmann rearrangement is used for the industrial synthesis of azepan-2-one (ε
-caprolactam), which is the starting material for nylon 6. The rearrangement is performed in the liquid phase by using an excess amount of fuming sulfuric acid as a promoter, followed by neutralization of the acid with ammonia [
209]. This process has serious disadvantages such as corrosion of equipment by the strong acid and by-production of a large amount of ammonium sulfate. To circumvent these issues, alternative catalytic vapor-phase Beckmann rearrangements have been developed by using solid acids such as silica–alumina [
210], boria [
211], and Ta
2O
5/SiO
2 [
212]. Various zeolites such as HNaY [
213], H-USY [
214], H-beta [
215], H-MCM-41, H-FSM-16 [
216], and MFI [
214,
215] have also been applied to the reaction, and the weak acidity of these catalysts is reported to be favorable for the reaction [
215,
216]. Then, application of halide cluster catalysts to the Beckmann rearrangement was made.
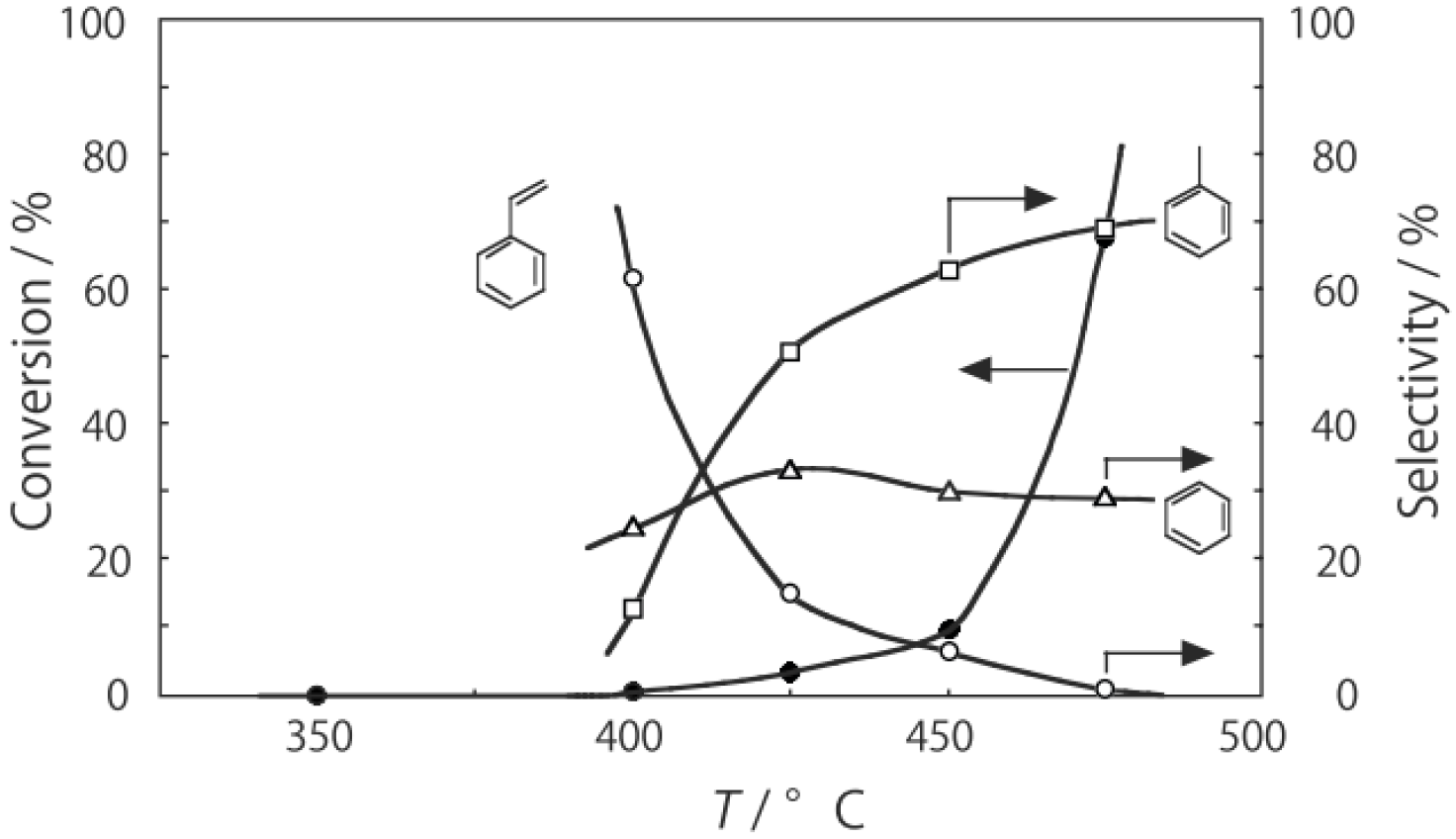
The supported tungsten cluster, (H
3O)
2[(W
6Cl
8)Cl
6]·6H
2O/SiO
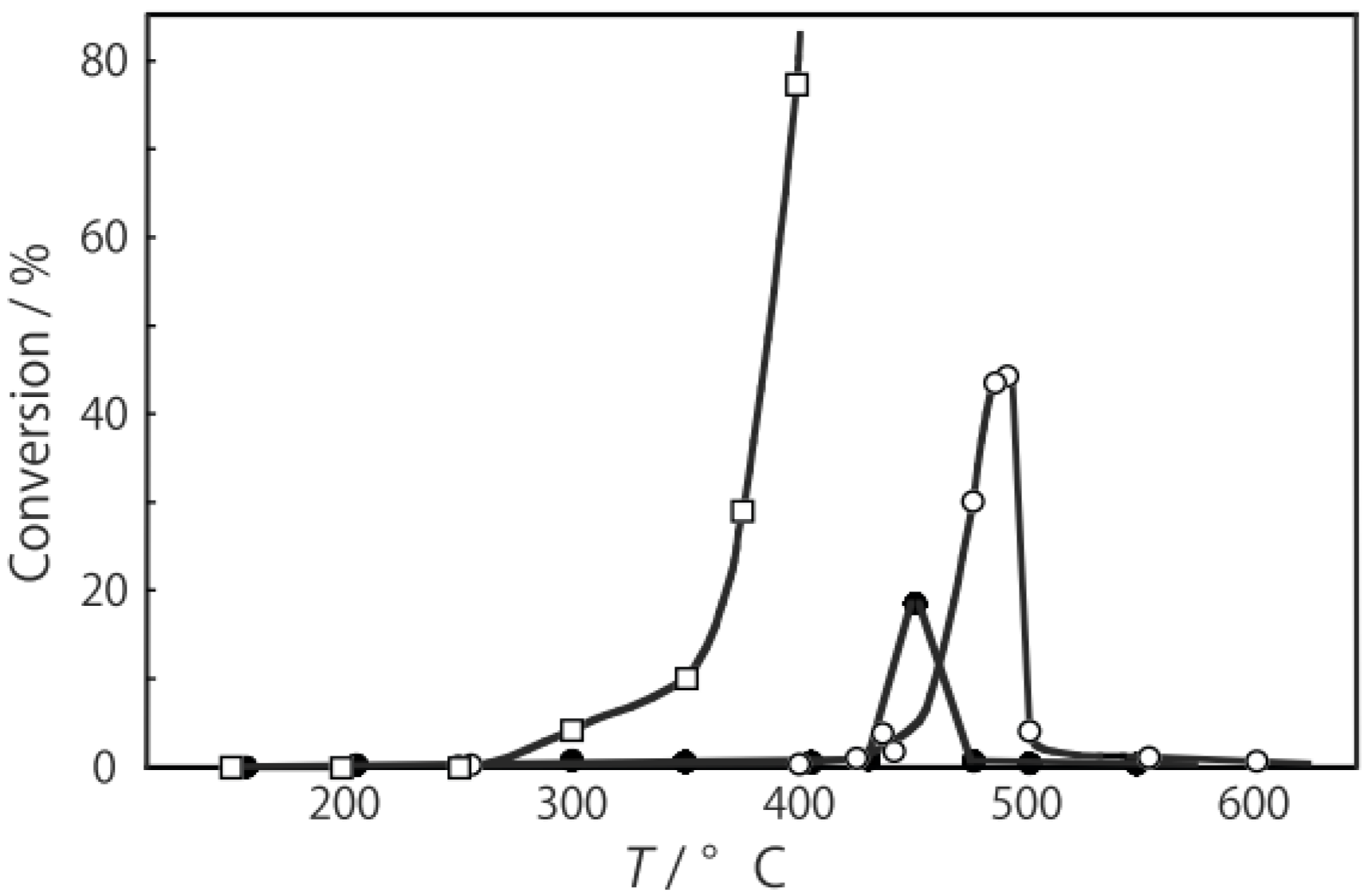
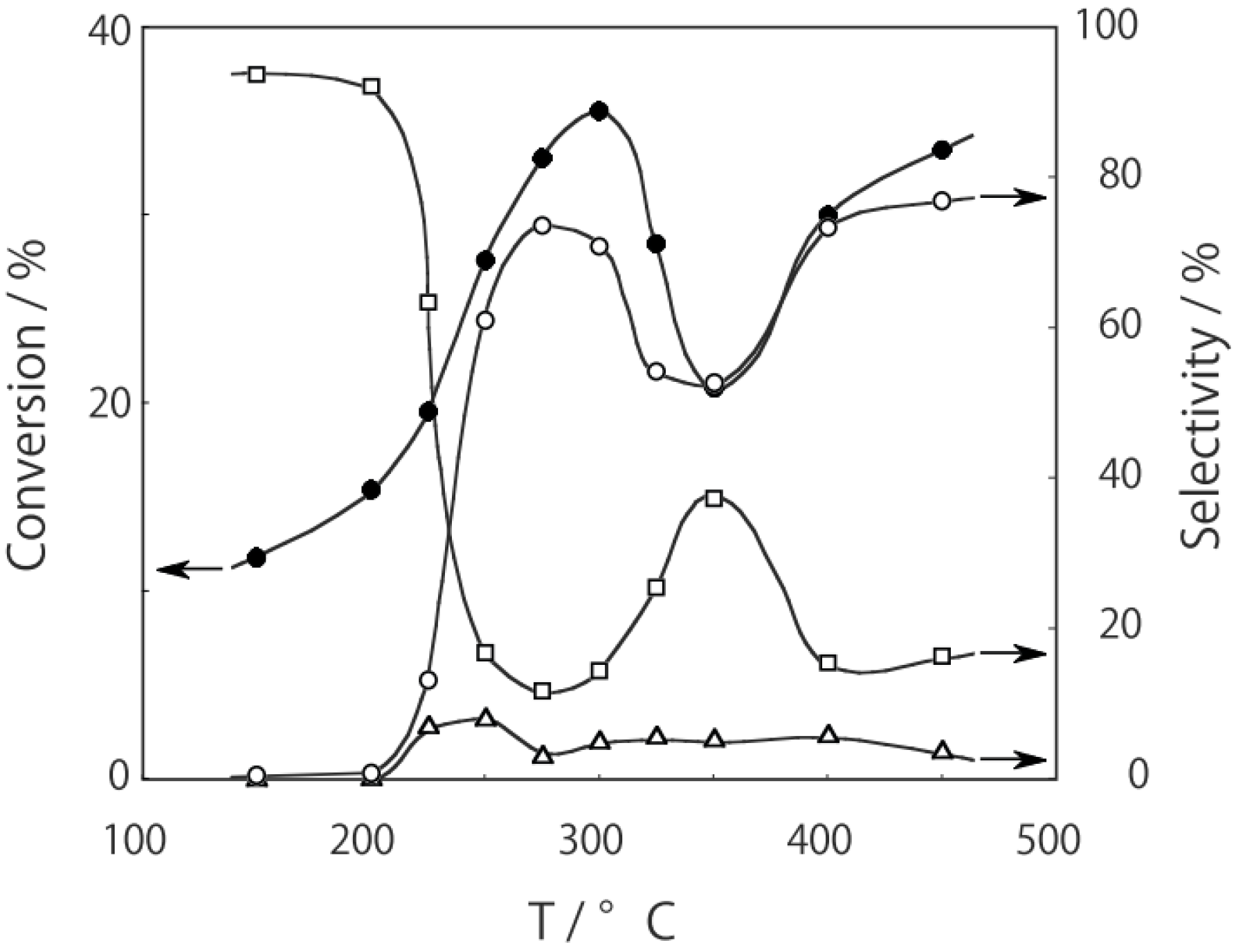
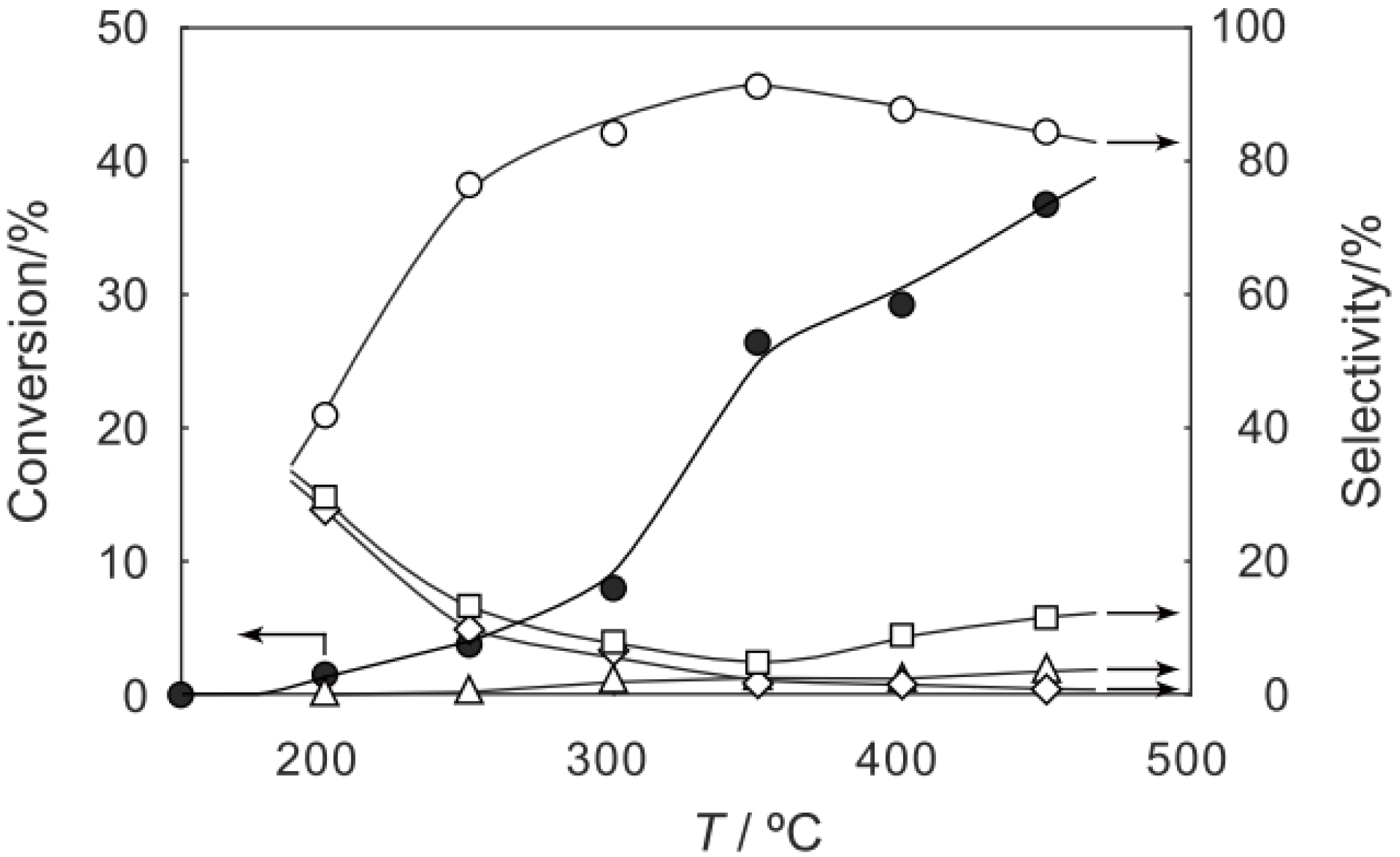
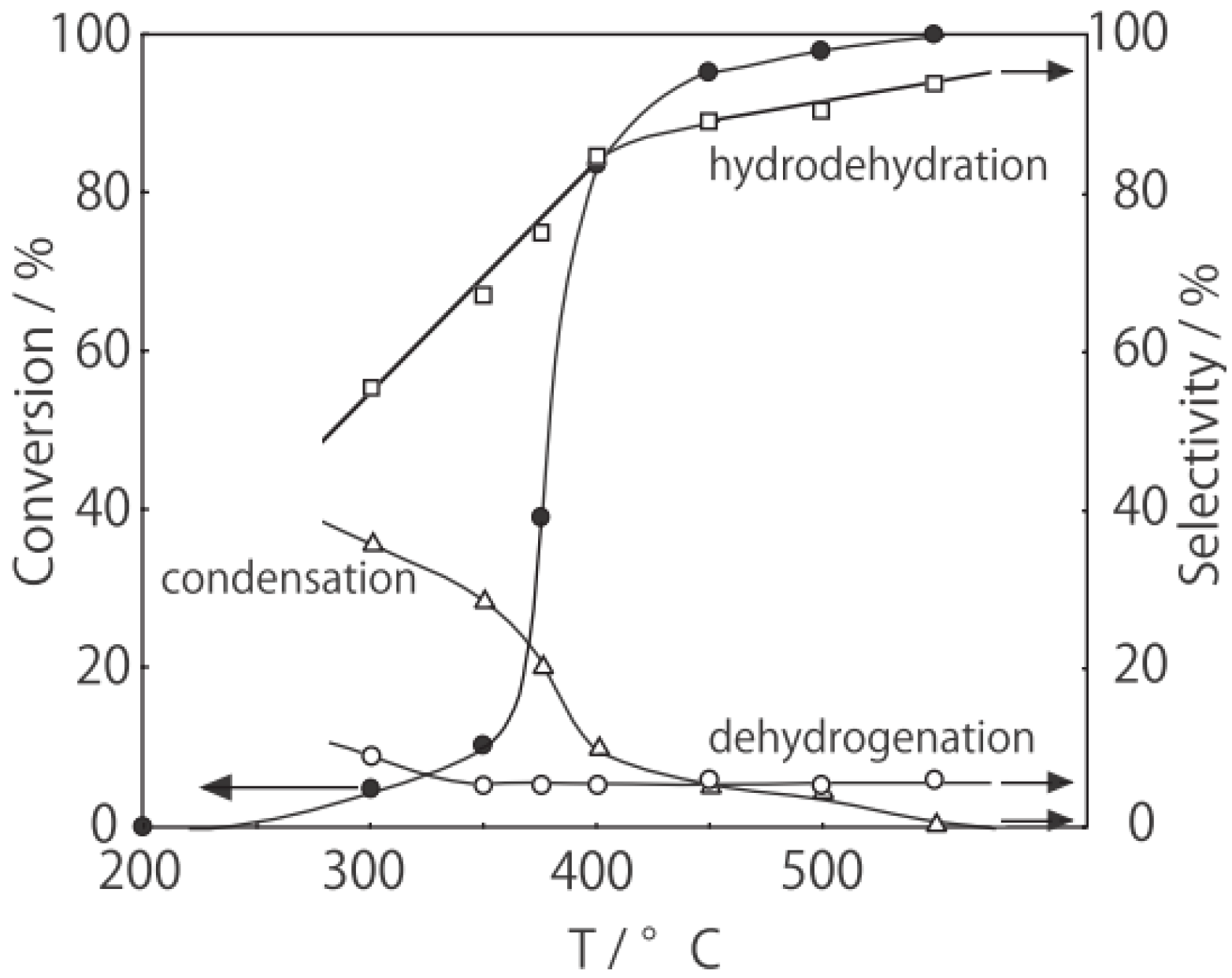
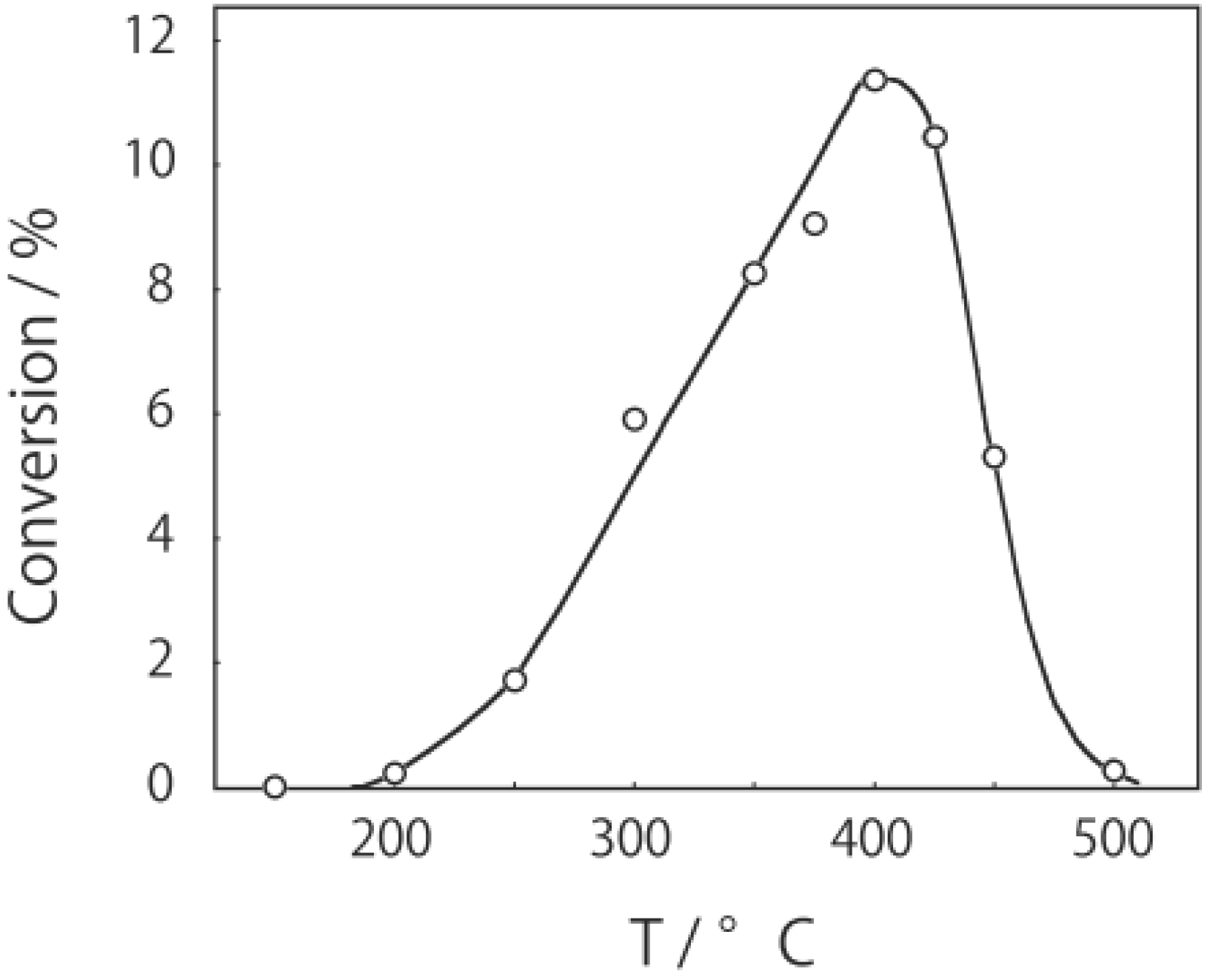
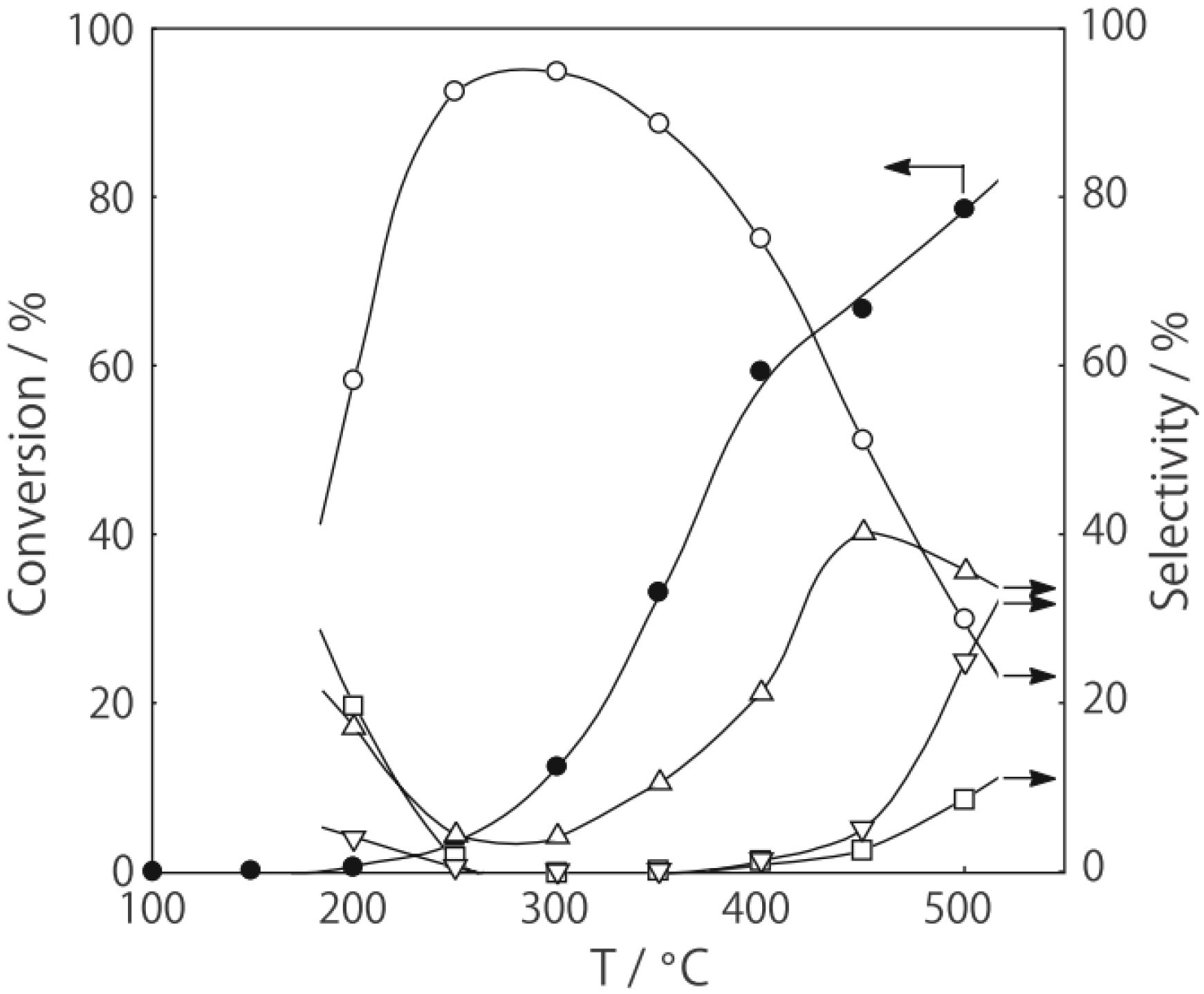
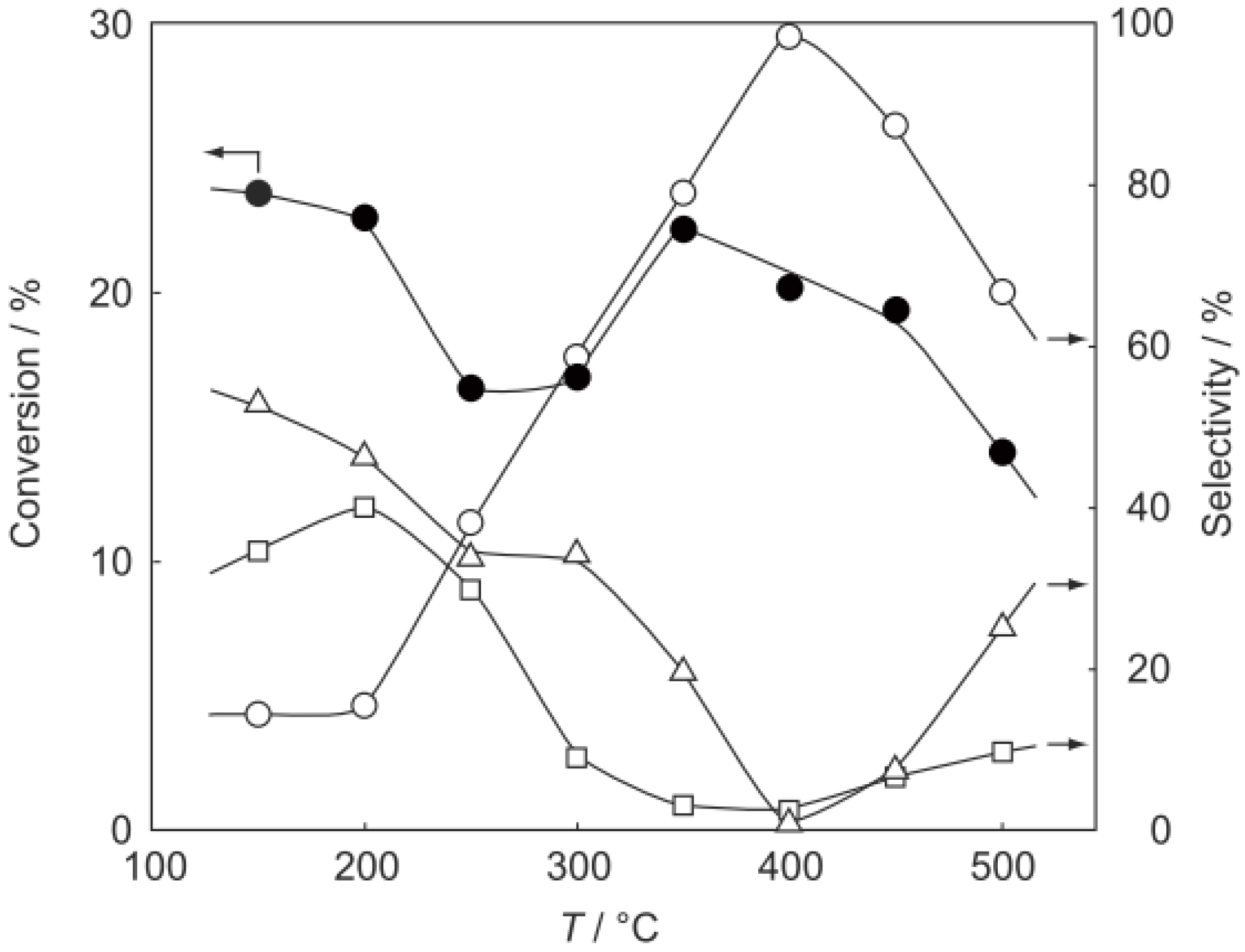
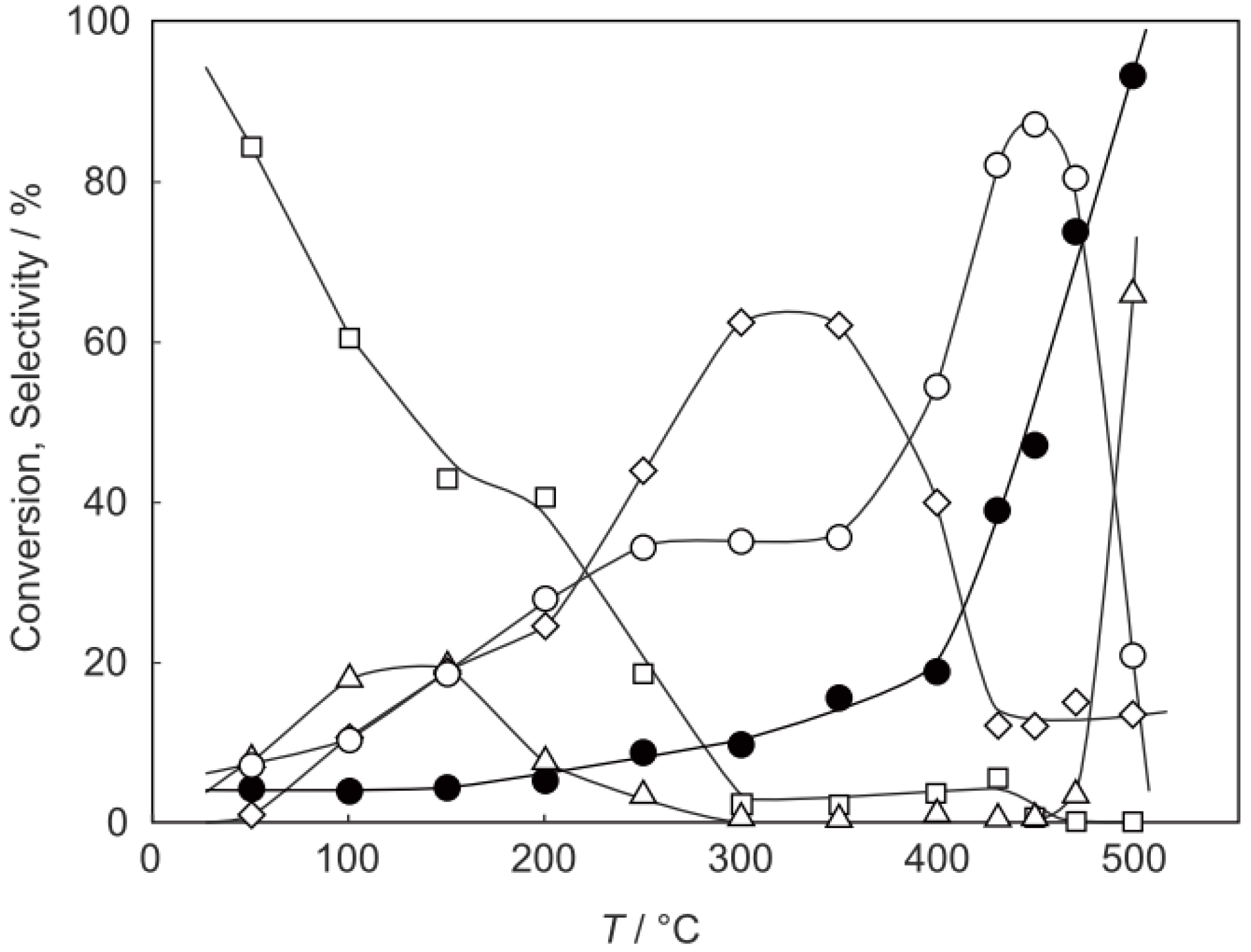
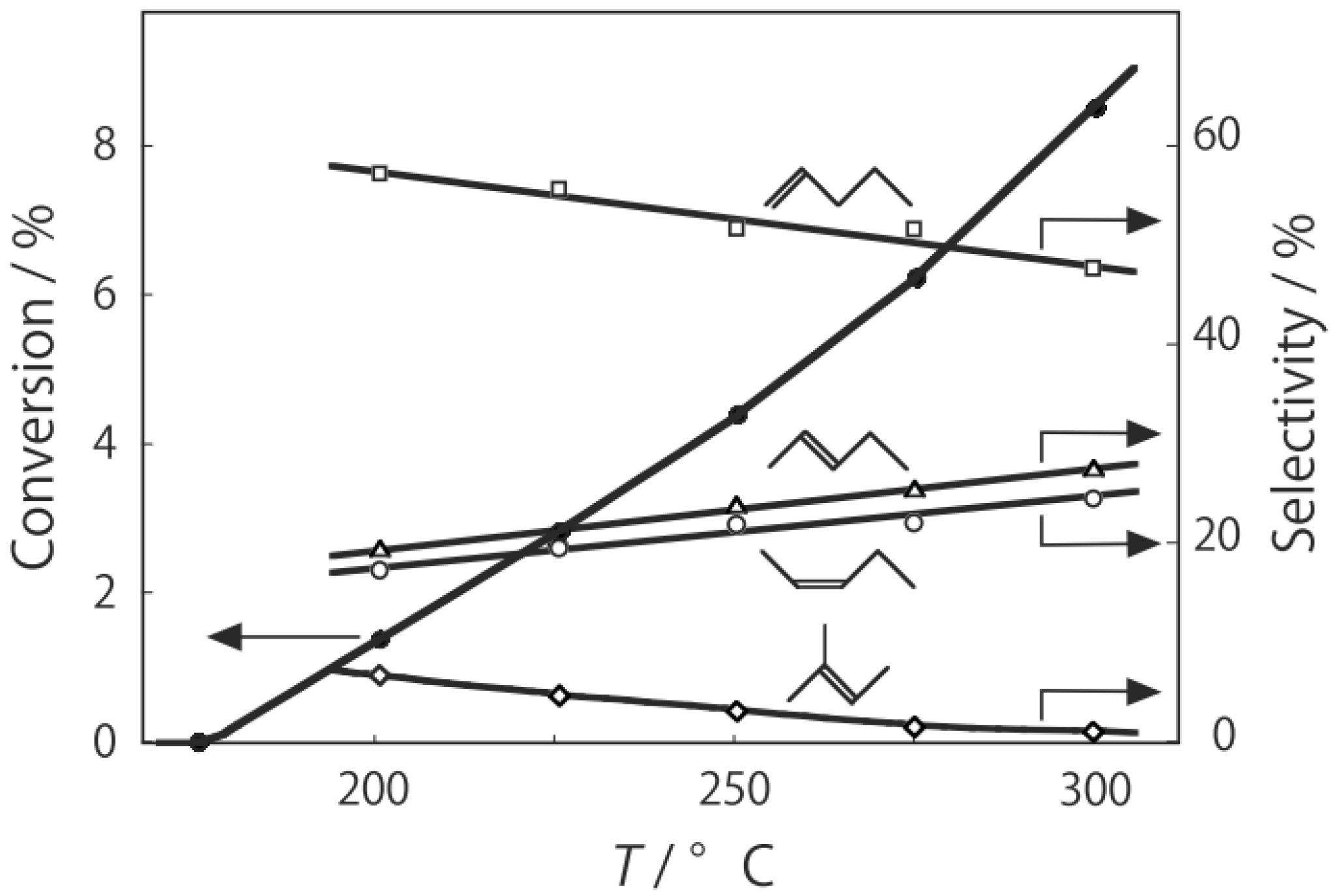
2, was activated at an elevated temperature in a helium stream for 1 h. The reaction was initiated by introduction of a methanol solution of cyclohexanone oxime into the helium stream without changing the temperature. The effect of the reaction temperature on activity and selectivity is shown in
Figure 28. The catalytic activity significantly developed above 250 °C. At 300 °C, the conversion was 91% and the selectivity for ε-caprolactam was as high as 93%. Cyclohexanone, 2-cyclohexen-1-one, hexanenitrile, and hex-5-enenitrile were formed as by-products, and these by-products are also formed in the Beckmann rearrangement over acidic zeolites [
214,
215,
216,
217,
218,
219]. Assuming that all the cluster molecules were active, the turnover frequency per cluster unit at 300 °C during a period of 2–4 h was 0.85 s
−1, which is a very large value for a molecular catalyst. Above 400 °C, selectivity for ε-caprolactam decreased (<22%), and decomposition products such as the nitriles were increased (>60%). A similar increase in the decomposition above 400 °C has been observed in the rearrangement over HY [
220] and B-MFI [
218]. When the supported cluster was activated at 400 °C and the reaction was performed at 300 °C, the selectivity for ε-caprolactam was 93%, indicating the dependence of the reaction temperature.
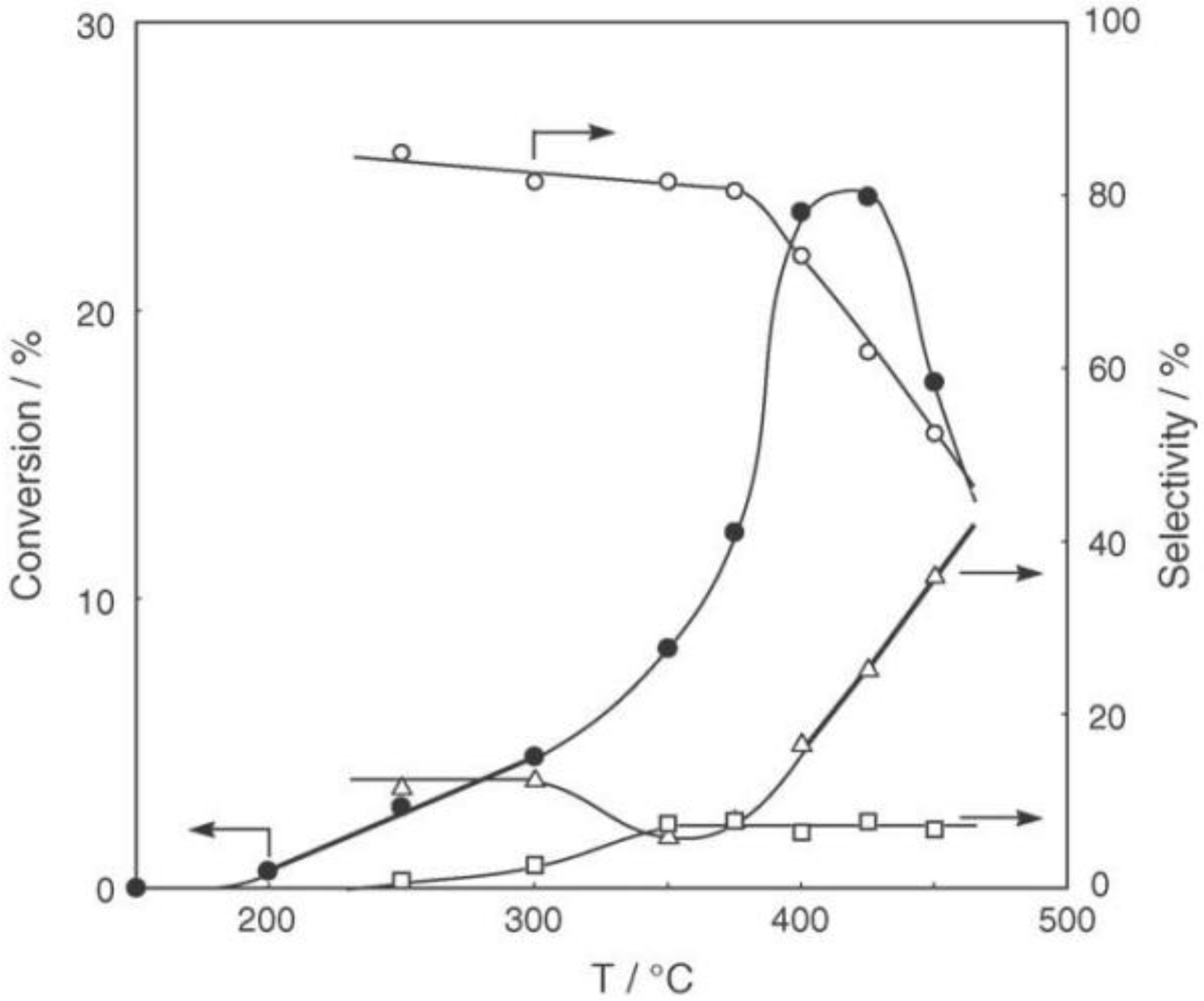
Figure 28.
Effect of temperature on the reaction of cyclohexanone oxime over (H3O)2[(W6Cl8)Cl6]·6H2O/SiO2 in a helium stream. Supported cluster (10 mg, 0.29 μmol) were activated in a helium stream (1.2 L/h) for 1 h, and a methanol solution of cyclohexanone oxime (17 mol%, 200 μL/h, 0.65 mmol/h) was introduced into the helium stream without changing the temperature. Conversion of cyclohexanone oxime (●); selectivity for ε-caprolactam (○); selectivity for cyclohexanone and 2-cyclohexen-1-one (∆); and selectivity for decomposition products including hexanenitrile and hex-5-enenitrile (□); at 3 h after the start of the reaction.
Supported clusters of niobium and tantalum were also active catalysts for the reaction. When acetone oxime was allowed to react over (H3O)2[(W6Cl8)Cl6]·6H2O/SiO2 at 300 °C, N-methylacetamide was obtained with 76% selectivity. Cyclopentanone oxime was also converted to piperidin-2-one (δ-valerolactam) with 81% selectivity. Thus, the halide clusters were effective catalysts for the Beckmann rearrangement of various oximes.
The acid strength of (H
3O)
2[(W
6Cl
8)Cl
6]·6H
2O/SiO
2 that was activated at 300 °C for 1 h was determined using the butylamine titration method with various Hammett indicators. The cluster exhibited an equimolar amount of acid sites at H
0 ≈ 1.3 with an additional equimolar amount of acid sites at H
0 ≈ 4.0−2.0. The weak Brønsted acidity (H
0 ≈ +1.3) of the hydroxo ligand, which is developed on the activated cluster [
1], would be favorable for the rearrangement, since weak acids such as H-beta [
215], H-MCM-41, and H-FSM-16 are reported to be effective catalysts for the Beckmann rearrangement [
216]. On the other hand, HY and H-mordenite, having strong acid sites, exhibit low activity and low selectivity for the reaction [
221]. Weak acid sites on Ta
2O
5/SiO
2 and H-USY effectively catalyze the rearrangement, whereas strong acid sites on the catalysts accelerate formation of by-products [
212,
214]. The rearrangement over HNaY is catalyzed by weak acid sites with a pK
a less than 1.5, whereas the strong acid sites with a pK
a less than −3.0 are not active for the reaction [
213]. Over high-silica MFI, the catalytically active sites are ascribed to nest silanols with very weak acidity, which is undetectable by the temperature-programmed desorption of ammonia (NH
3-TPD) [
217]. Thus, the weak acid sites of the hydroxo ligand on the halide clusters are favorable for the selective vapor-phase Beckmann rearrangement.
6.4. Acylation of Thiol (Equation (18)) [222]
Acylation of alcohols, phenols, amines, and thiols is a frequently used transformation in organic synthesis, and it provides an efficient and inexpensive means of protecting the groups in a multistep synthetic process. Acylation is mostly carried out by use of acid anhydrides or acyl chlorides in the presence of acid catalysts or basic reagents such as 4-(dimethylamino)pyridine, 4-pyrrolidinopyridine, pyridine, or tertiary amines [
223]. Direct acylation with carboxylic acids in the absence of any reagents is the best method for performing the acylation reaction, as the atomic economy is high and only water is produced as a by-product. Another advantage is the relatively cheaper route for industrial purposes when compared with the use of acid anhydrides or acid chlorides. Although many catalytic acylations of alcohols, phenols, and amines with carboxylic acids have been reported, acylation of thiols with carboxylic acids is quite rare. The reactivity of thiol is very much lower than that of an alcohol or amine [
224]. Although, acetyl chloride or acetic anhydride spontaneously reacts with benzenethiol to afford
S-acetylation product at room temperature, acetic acid and methyl acetate did not afford even a trace amount of the thioacetate even in the presence of any catalyst. The single example reported is
S-acetylation of benzenethiol with acetic acid: in a batch reaction system a mixture of benzenethiol and acetic acid at 110 °C for 8 h under neat conditions in the presence of 20 wt% of yttria–zirconia-based Lewis acid catalyst to yield
S-phenyl thioacetate in 94% yield [
225].
S-Acylation with carboxylic ester has not been reported.
As mentioned in
Section 4.5, when alkyl acetates were applied to benzenethiol, selective
S-alkylation or
S-acetylation proceeded depending on the kind of the cluster catalyst (Equation (37)). Further investigation revealed a universally applicable simple and practical method for the acylation of both aliphatic and aromatic thiols with carboxylic acids or their esters over various solid acid catalysts that are stable above 200 °C (Equation (38)).
After activation of (H
3O)
2[(Mo
6Cl
8)Cl
6]·6H
2O/SiO
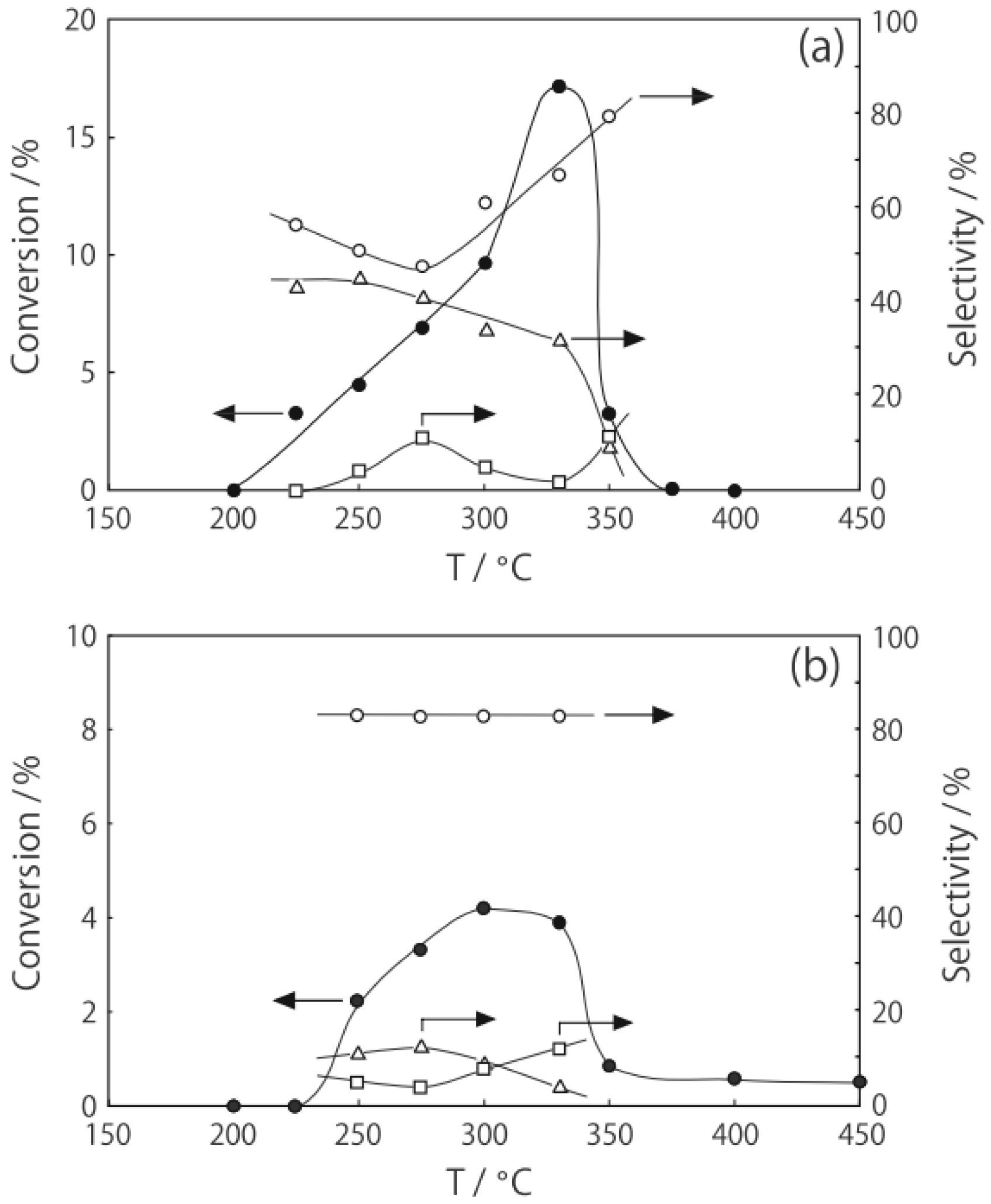
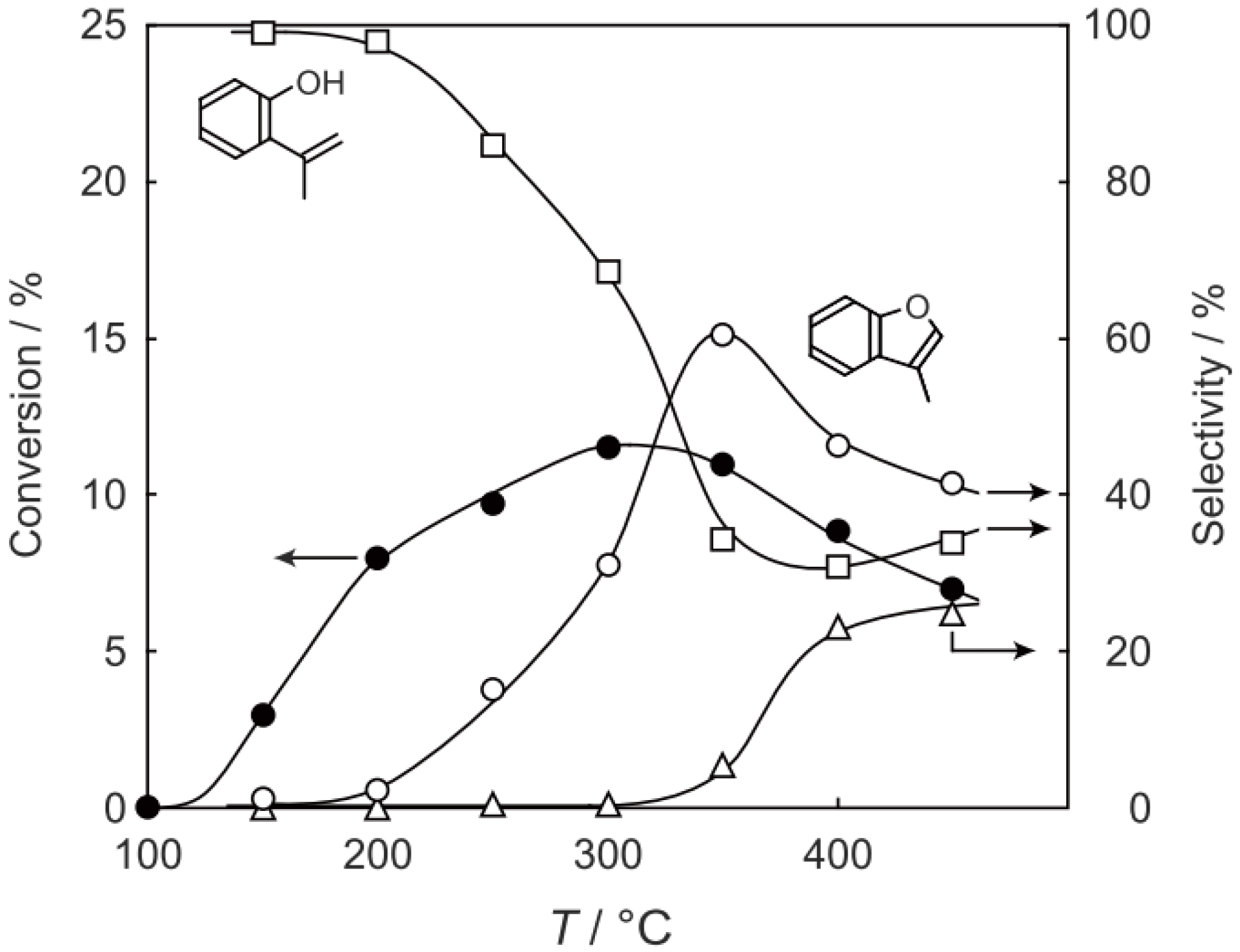
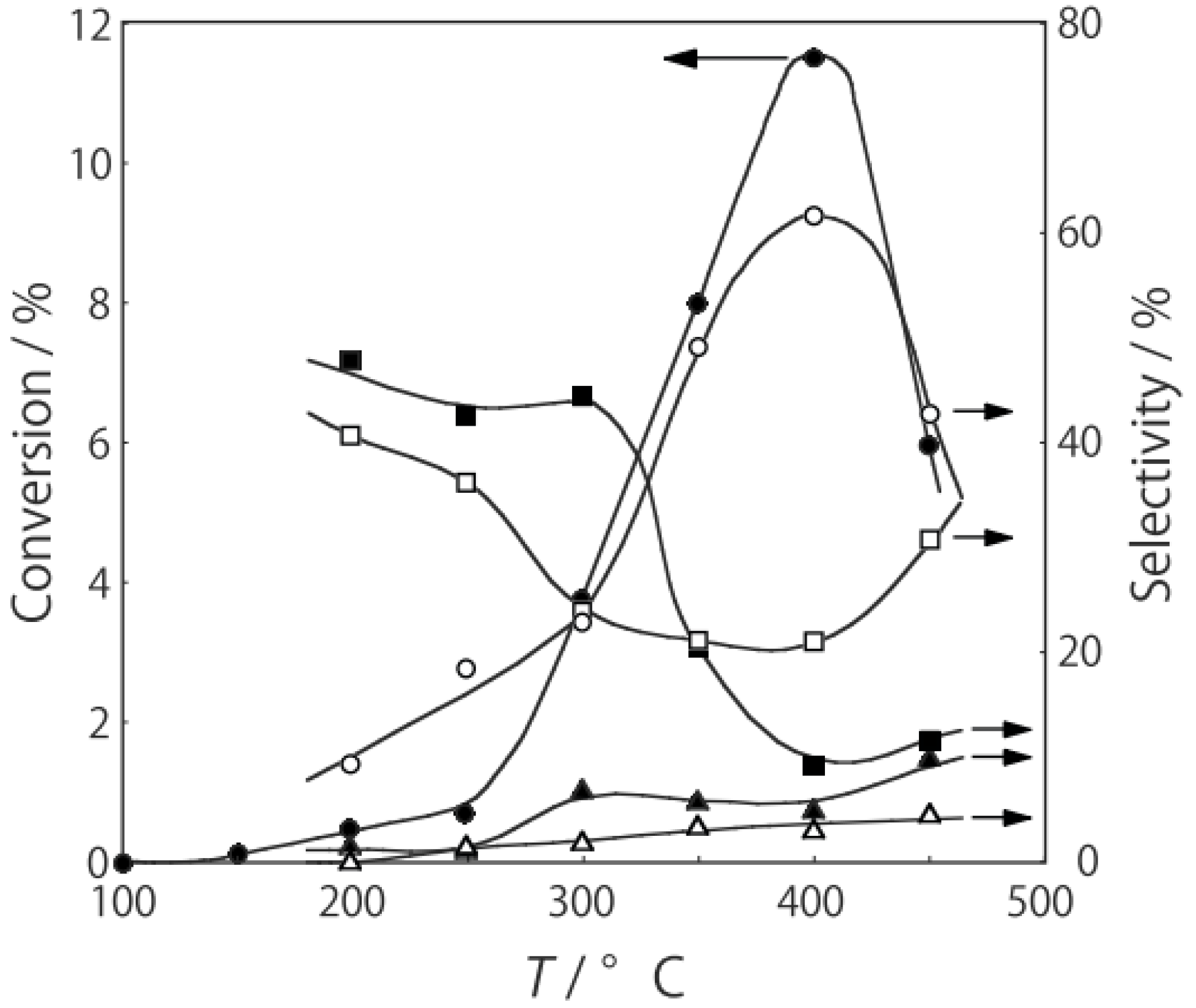
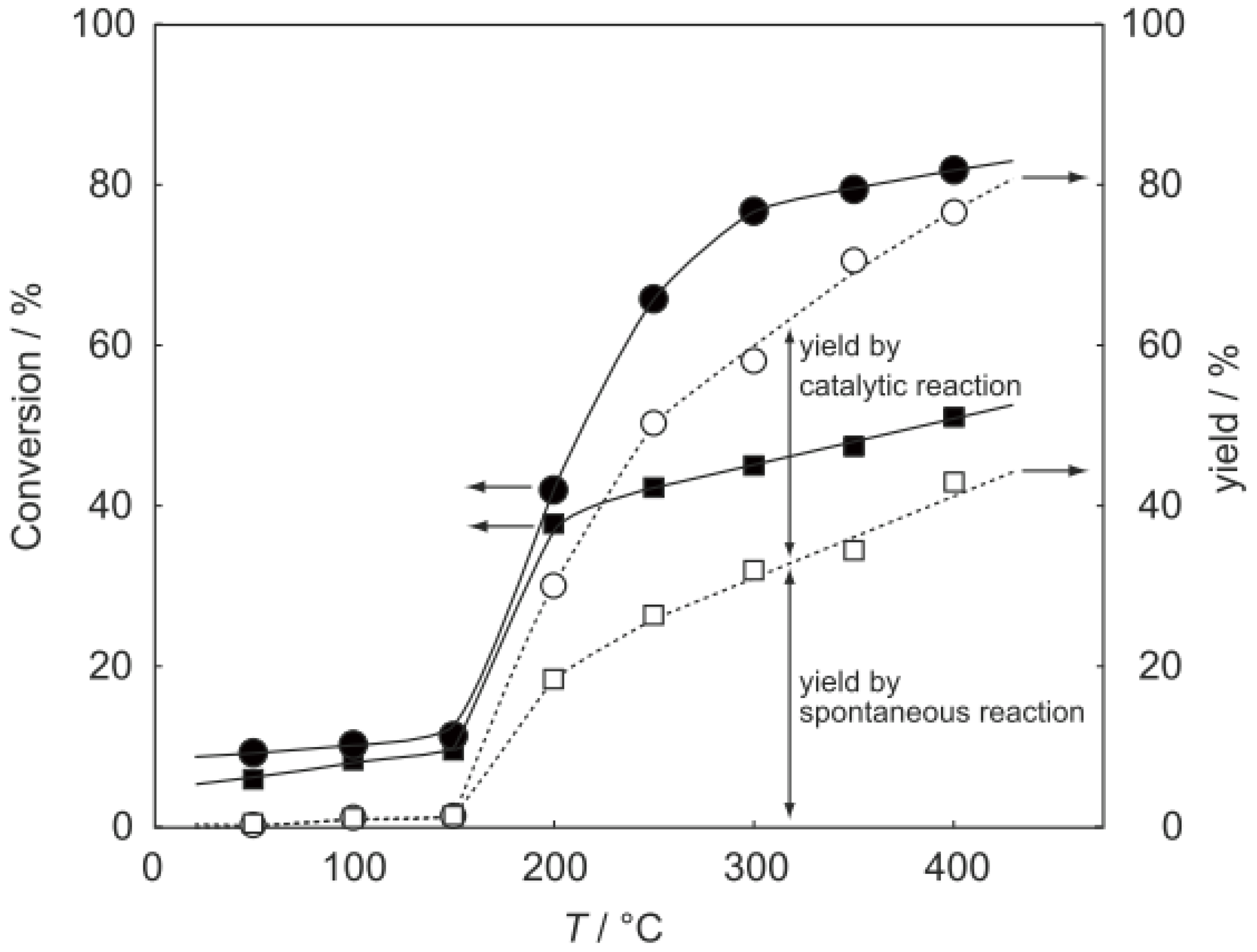
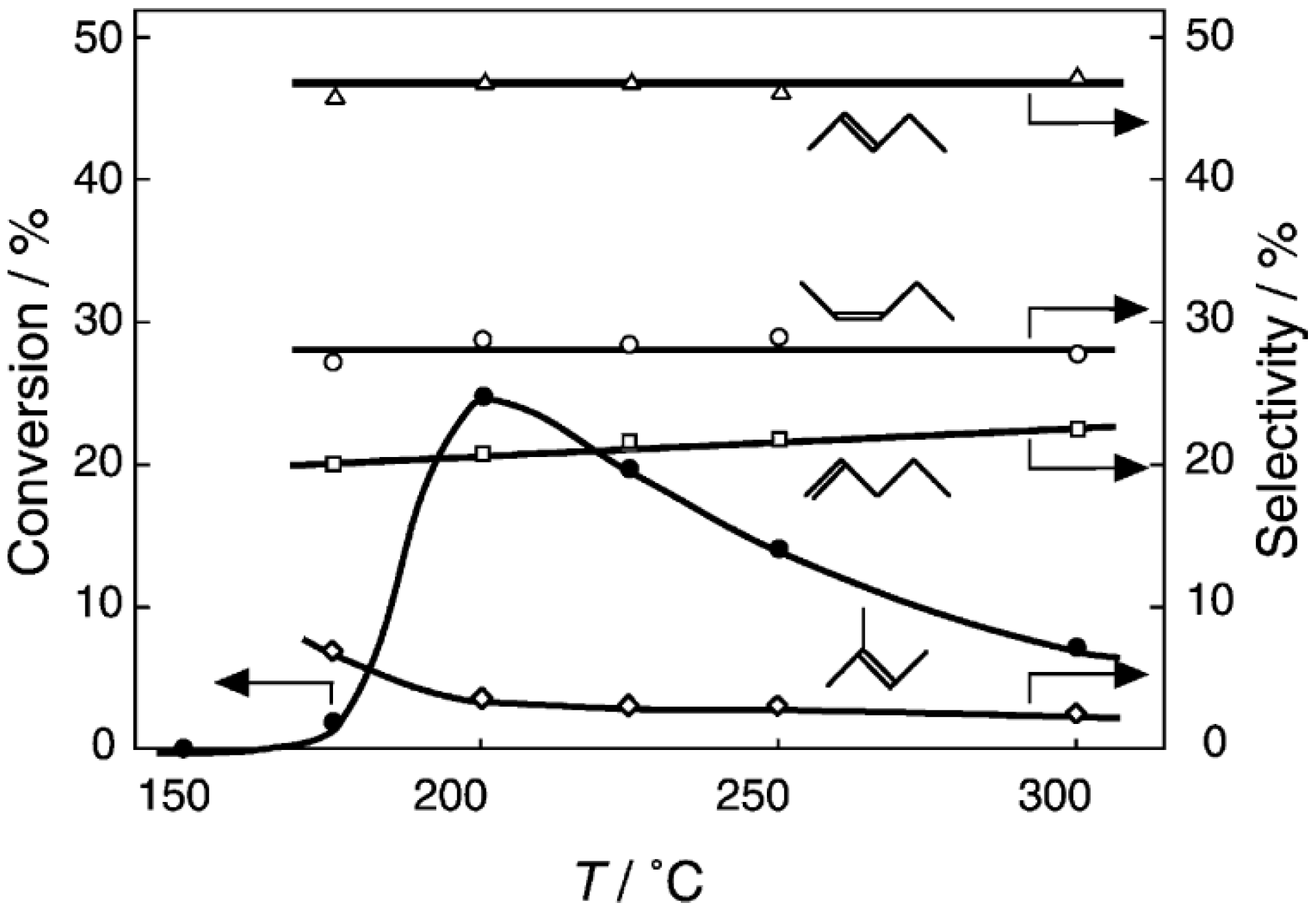
2 at various temperatures in a hydrogen stream for 1 h, reactions were started by introduction of a mixture of benzenethiol with acetic acid without changing the temperature. The effect of the temperature is presented in
Figure 29. Practically no catalytic activity for the
S-acetylation was observed below 150 °C, although there was considerable spontaneous oxidative coupling of benzenethiol to yield diphenyl disulfide. Catalytic activity for the
S-acetylation of benzenethiol to yield
S-phenyl thioacetate developed above 200 °C and increased with increasing temperature. Over the temperature range 200–450 °C, selectivity for
S-phenyl thioacetate was more than 94%. Side reactions were the coupling of benzenethiol yielding diphenyl sulfide and diphenyl disulfide. The turnover frequency per Mo
6 cluster unit during a period of 2–4 h was 150 h
−1, assuming that all of the cluster molecules were active.
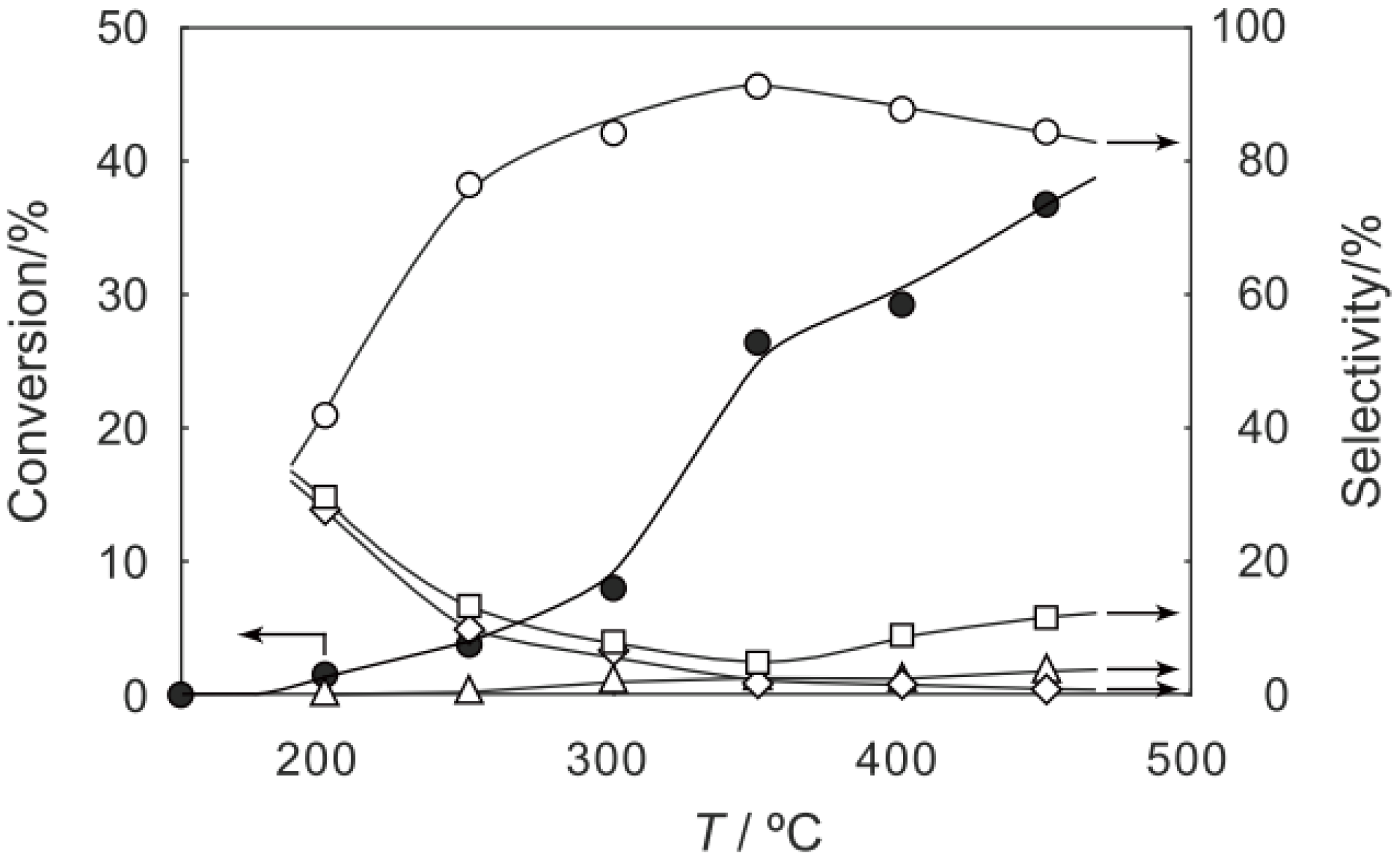
Figure 29.
Effect of temperature on the S-acetylation of benzenethiol with acetic acid over (H3O)2[(Mo6Cl8)Cl6]·6H2O/SiO2. Following the activation of the supported cluster (30 mg, 1.23 μmol) in a hydrogen stream (300 mL/h) for 1 h, reaction was initiated by introduction of a mixture of benzenethiol (102 μL/h, 1.0 mmol/h) and acetic acid (114 μL/h, 2.0 mmol/h) to the hydrogen stream without changing the temperature. Conversion of benzenethiol (●); selectivity for S-phenyl thioacetate (○); selectivity for diphenyl sulfide (∆); and selectivity for diphenyldisulfide (□); at 3 h after the start of the reaction.
Various halide clusters were applied to
S-acetylation of benzenethiol with acetic acid at 300 °C, and the results are summarized in
Table 10. The supported clusters of niobium, tantalum, and tungsten also selectively catalyzed the
S-acetylation. Brønsted acid sites developed on the halide cluster seemed to be active sites, and hence various solid acids were applied to the reaction.
Table 10 also lists the catalytic activities of silica–alumina, zeolites, and heteropoly acids; their acidity is stable above 200 °C. All these acids selectively catalyzed
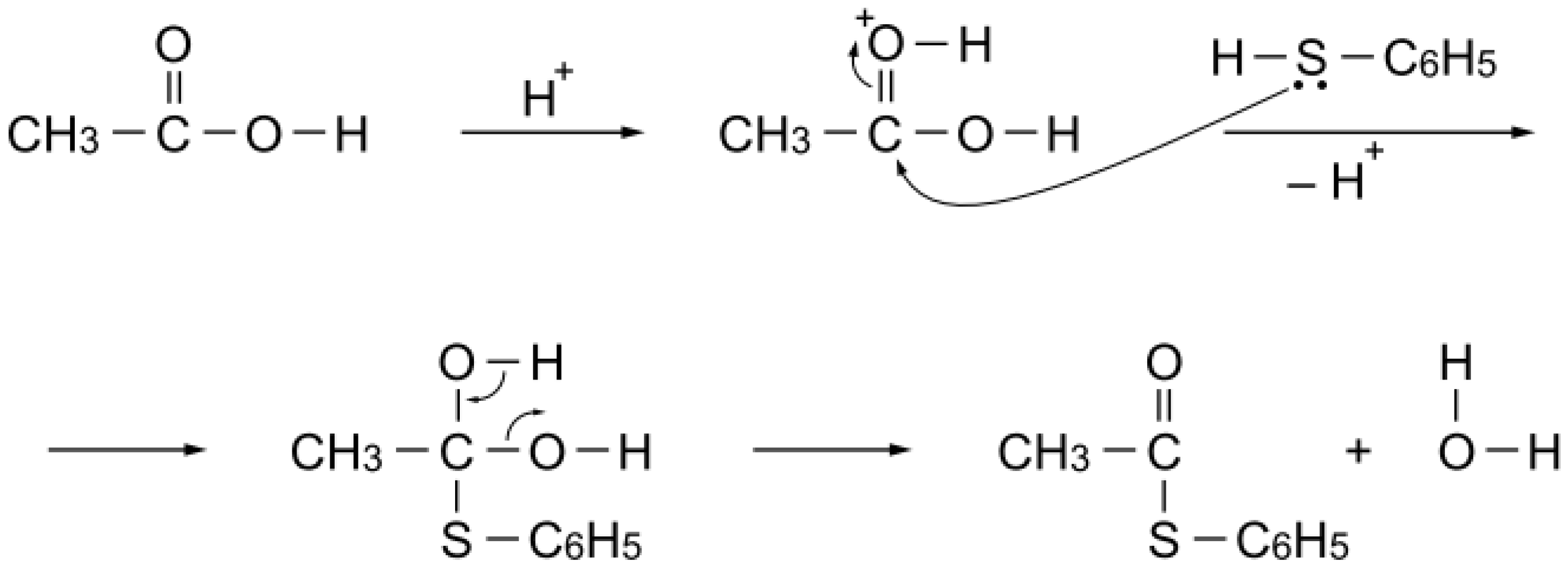
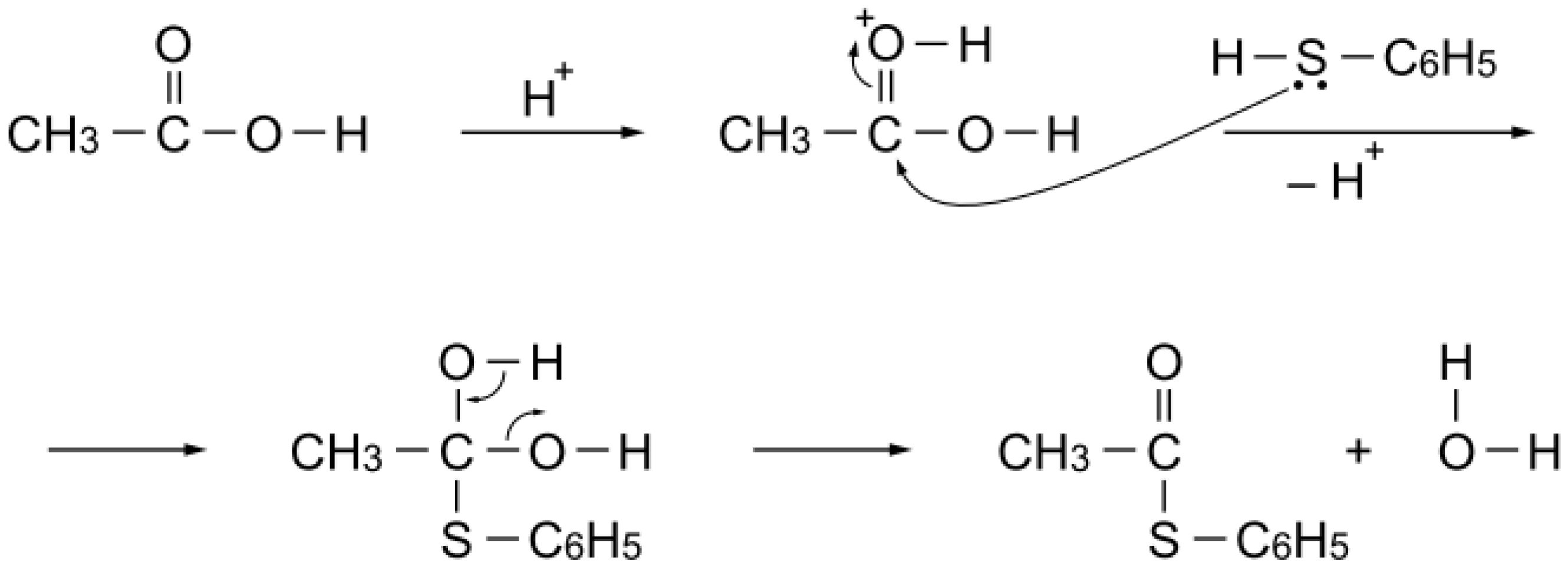
S-acetylation of benzenethiol, as expected. On the other hand, alumina (a Lewis acid) exhibited essentially no catalytic activity for the acetylation. Thus, protonation of the carbonyl group of the acetic acid would facilitate nucleophilic attack of benzenethiol to yield
S-phenyl thioacetate (
Scheme 5). Stability above 200 °C is a requisite of the catalyst for the reaction. Phosphoric acid polymerizes to diphosphoric acid or polyphosphoric acid above 200 °C, and Nafion-H is not stable above 200 °C.
Table 10.
S-Acetylation of benzenethiol with acetic acid over various catalysts a.
| Catalyst | Conversion (%) | Selectivity (%) |
|---|
| S-Phenyl thioacetate | Diphenyl sulfide | Diphenyl disulfide |
|---|
| [(Nb6Cl12)Cl2(H2O)4]·4H2O/SiO2 | 18.5 | 96.9 | 0.4 | 2.7 |
| (H3O)2[(Mo6Cl8)Cl6]·6H2O/SiO2 | 18.7 | 98.2 | 1.2 | 0.6 |
| [(Ta6Cl12)Cl2(H2O)4]·4H2O/SiO2 | 11.9 | 96.1 | 1.7 | 2.3 |
| (H3O)2[(W6Cl8)Cl6]·6H2O/SiO2 | 17.6 | 97.8 | 0.2 | 2.0 |
| SiO2–Al2O3 (high alumina) b | 15.7 | 93.6 | 5.3 | 1.1 |
| H-Y zeolite b | 14.5 | 96.4 | 2.3 | 1.3 |
| H-beta b | 14.2 | 94.3 | 3.4 | 2.3 |
| H-mordenite b | 6.4 | 92.5 | 1.7 | 5.8 |
| H3[PMo12O40]·30H2O c | 2.5 | 88.5 | 3.7 | 7.8 |
| H3[PW12O40]·30H2O c | 23.3 | 92.0 | 5.8 | 2.2 |
| H3[SiW12O40]·24H2O c | 11.4 | 88.1 | 1.8 | 10.1 |
| Al2O3 | 0.9 | 41.6 | 0.0 | 58.4 |
Scheme 5.
S-Acetylation mechanism with acetic acid over a simple Brønsted acid site.
Several aliphatic and aromatic carboxylic acids were applied to the S-acylation of benzenethiol over (H3O)2[(Mo6Cl8)Cl6]·6H2O/SiO2 at 300 °C. S-Acylation with all the saturated aliphatic C2–C5 carboxylic acids proceeded; the longer and bulkier the alkyl chain, the lower the reactivity and selectivity. The selectivities for S-acylation were more than 91% when carboxylic acids bearing α-hydrogen were used. Even carboxylic acids having no α-hydrogen, pivalic acid and benzoic acid, afforded the S-acylation products with selectivities of more than 77%. S-Formylation of benzenethiol was unsuccessful under these reaction conditions, probably because of the thermolability of the product.
Esters, which have higher solubilities and lower melting points compared with the corresponding acids, are occasionally used for acylation of alcohols and amines. Catalytic
S-acylation with esters has not been reported. Methyl acetate was applied to
S-acetylation of benzenethiol in the presence of the [(Nb
6Cl
12)Cl
2(H
2O)
4]·4H
2O at various temperatures, and the results are shown in
Figure 30. Catalytic activity developed above 200 °C and increased with increasing temperature. Over the temperature range 300–400 °C, selective
S-methylation proceeded to yield methyl phenyl sulfide, and at around 450 °C,
S-acetylation was replaced to yield
S-phenyl thioacetate preferentially. Then, methyl acetate was applied to the reaction at 450 °C over various catalysts, and the data are listed in
Table 11. Supported clusters of niobium and tantalum catalyzed
S-acetylation selectively to provide
S-phenyl thioacetate. Silica-alumina, H−Y zeolite, H−beta, H−mordenite, and phosphotungstic acid were also effective catalysts for the reaction. The progress of
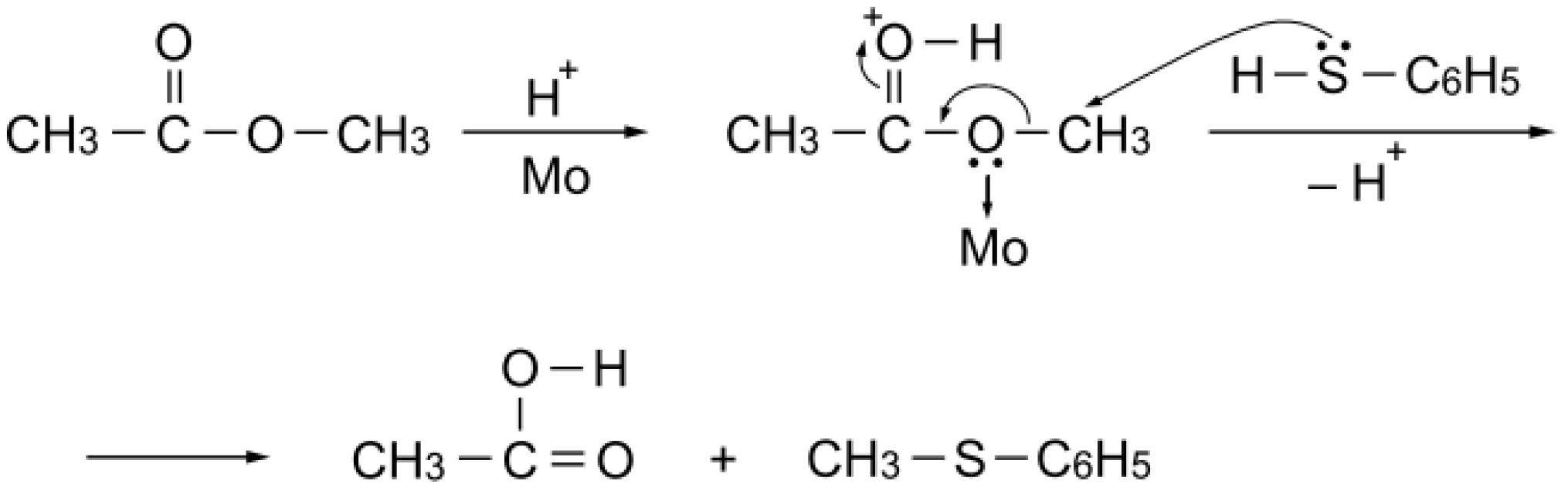
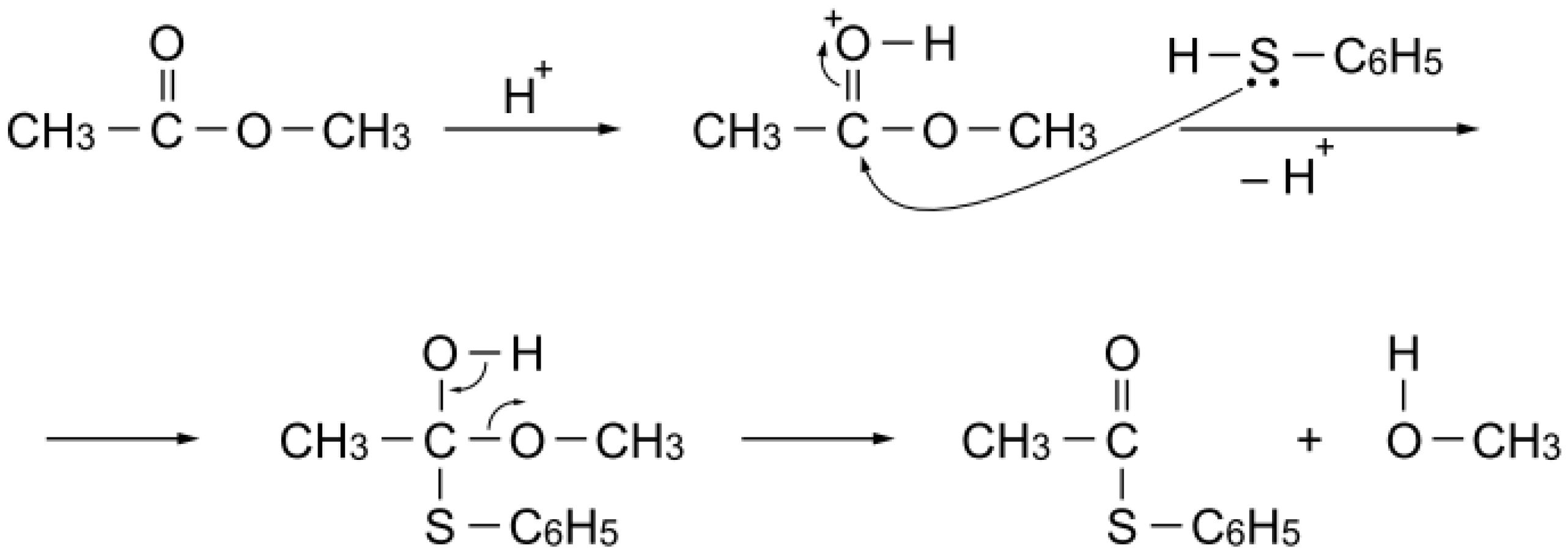
S-acetylation with methyl acetate can be explained as simple Brønsted acid catalysis: the carbonyl group of the ester activated by a proton is attacked by benzenethiol to afford
S-phenyl thioacetate (
Scheme 6).
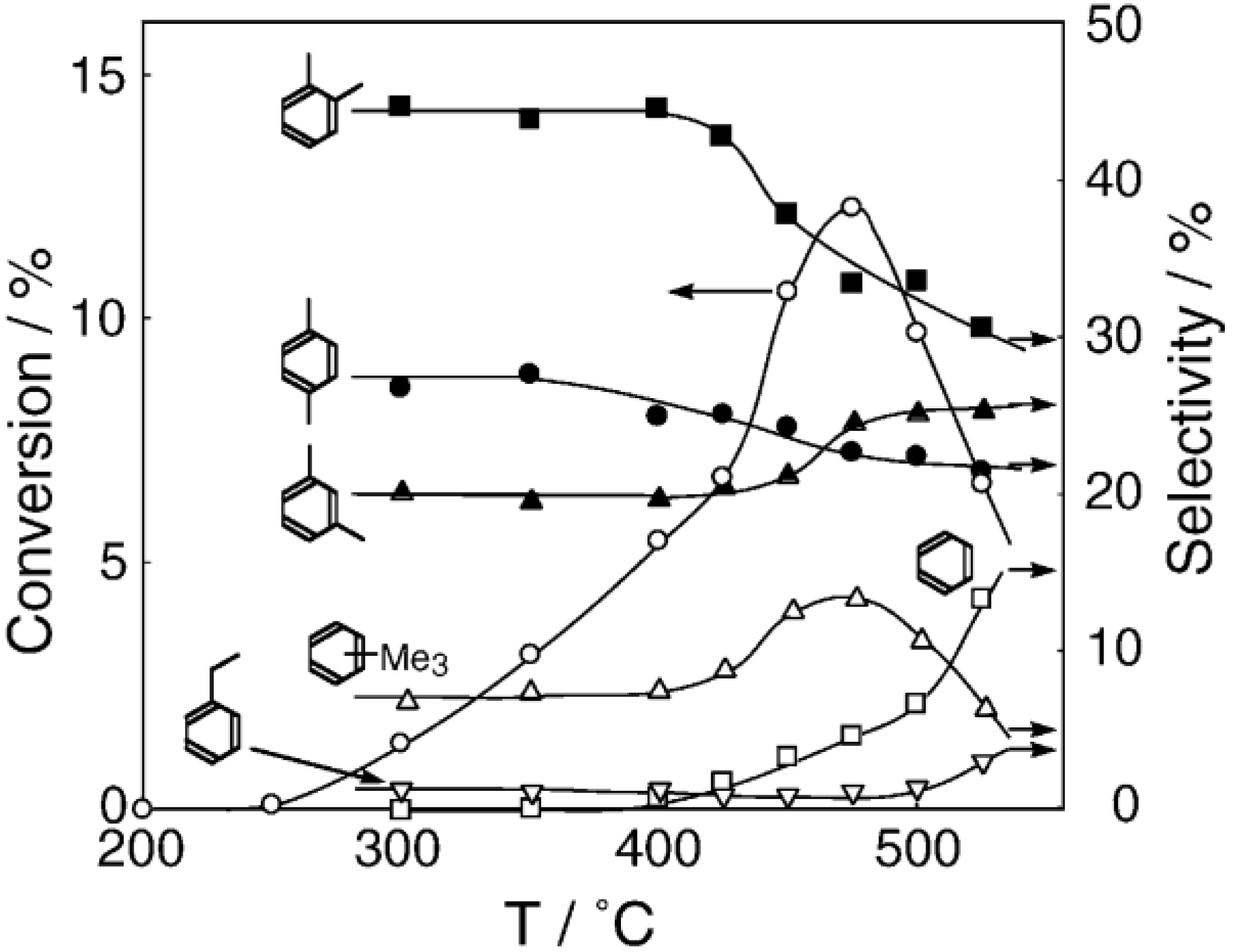
Figure 30.
Effect of temperature on the reaction of benzenethiol with methyl acetate in the presence of [(Nb
6Cl
12)Cl
2(H
2O)
4]·4H
2O/SiO
2. A mixture of benzenethiol (102 μL/h, 1.0 mmol/h) and methyl acetate (795 μL/h, 10.0 mmol/h) was reacted. Other conditions are the same as in
Figure 29. Conversion of benzenethiol (●); selectivity for
S-phenyl thioacetate (○); selectivity for methyl phenyl sulfide (◊); selectivity for diphenyl sulfide (∆); and selectivity for diphenyl disulfide (□); at 3 h after the start of the reaction.
Scheme 6.
S-Acetylation mechanism with methyl acetate over a simple Brønsted acid site.
Table 11.
S-Acetylation and S-methylation of benzenethiol with methyl acetate over various catalysts a.
| Catalyst disulfide | Conversion (%) | Selectivity (%) |
|---|
| S-Phenyl thioacetate | Methyl phenyl sulfide | Diphenyl sulfide | Diphenyl |
|---|
| [(Nb6Cl12)Cl2(H2O)4]·4H2O/SiO2 | 46.8 | 86.9 | 12.0 | 0.6 | 0.5 |
| (H3O)2[(Mo6Cl8)Cl6]·6H2O/SiO2 | 84.7 | 4.1 | 94.7 | 1.0 | 0.2 |
| [(Ta6Cl12)Cl2(H2O)4]·4H2O/SiO2 | 35.8 | 80.6 | 14.8 | 4.1 | 0.6 |
| (H3O)2[(W6Cl8)Cl6]·6H2O/SiO2 | 44.1 | 58.1 | 39.9 | 1.1 | 0.9 |
| SiO2–Al2O3 (high alumina) b | 44.5 | 68.3 | 30.2 | 0.7 | 0.8 |
| H-Y zeolite b | 44.6 | 88.3 | 9.3 | 1.6 | 0.9 |
| H-beta b | 37.7 | 70.8 | 26.9 | 0.7 | 1.6 |
| H-mordenite b | 31.1 | 82.0 | 12.9 | 4.0 | 1.1 |
| H3[PMo12O40]·30H2O c | 28.6 | 19.9 | 74.4 | 4.8 | 0.9 |
| H3[PMo12O40]·30H2O c,d | 5.8 | 14.7 | 9.0 | 13.0 | 63.3 |
| H3[PW12O40]·30H2O c | 13.6 | 57.4 | 14.7 | 18.8 | 9.1 |
| H3[SiW12O40]·24H2O c | 4.5 | 28.0 | 17.1 | 31.4 | 23.5 |
As
Table 11 lists, supported cluster of molybdenum catalyzed
S-methylation to afford methyl phenyl sulfide almost exclusively. Tungsten halide clusters also catalyzed
S-methylation, though less selectively. With heteropoly acids, phosphomolybdic acid catalyzed
S-methylation preferentially. Over molybdenum and tungsten halide clusters, coordinatively unsaturated sites were formed by thermal activation [
1]. With niobium and tantalum clusters, formation of coordinatively unsaturated sites is difficult because of the high bond energy of Nb–O (771.5 kJ mol
−1) and Ta–O (799.1 kJ mol
−1). Under hydrogen, MoO
3 is reported to be reduced to a suboxide of chemical composition between Mo
2O and MoO at 300 °C and to molybdenum metal at 600 °C [
226]. Consequently, coordinatively unsaturated sites should appear in part on phosphomolybdic acid under the reaction conditions. As
Table 11 shows,
S-methylation over phosphomolybdic acid was obviously depressed under a helium stream.
S-Methylation over molybdenum cluster and phosphomolybdic acid under hydrogen can be explained on the basis of a Brønsted acid site in concert with the coordinatively unsaturated site. The carbonyl group of methyl acetate is activated by protonation, and the oxygen atom of the methoxy group coordinates to the molybdenum atom. Then, the electron-deficient methyl group is attacked by benzenethiol to afford the
S-methyl sulfide (
Scheme 7).
Scheme 7.
S-Methylation mechanism with methyl acetate over a combined Brønsted acid site and coordinatively unsaturated site.
Several carboxylic esters were reacted with benzenethiol over [(Nb
6Cl
12)Cl
2(H
2O)
4]·4H
2O/SiO
2 at 450 °C, and the data are listed in
Table 12. Acetyl esters provided
S-acetate almost exclusively. On the other hand, methyl esters of butyric, valeric, and benzoic acids afforded
S-methyl sulfide preferentially. Methyl propionate showed an intermediate selectivity to afford both
S-acyl and
S-alkyl products. Accordingly, a relatively smaller acyl or alkyl moiety of the ester was incorporated into the thiol.
It is known that aliphatic thiols bearing a β-hydrogen atom are susceptible to decomposing to olefins by eliminating hydrogen sulfide at elevated temperatures, particularly in the presence of acid catalysts [
227,
228], as in the case of alcohols decomposing to olefins. When 1-hexanethiol was reacted alone at 300 °C over [(Nb
6Cl
12)Cl
2(H
2O)
4]·4H
2O/SiO
2, the conversion was 36% and decomposition to 1-hexene proceeded with 74% selectivity. However, when 1-hexanethiol was reacted in the presence of acetic acid under identical conditions, the conversion increased to 43% and acetylation to
S-hexyl thioacetate proceeded with 87% selectivity. Halide clusters except molybdenum cluster, acid catalysts such as silica-alumina, H-Y zeolite, and phosphotungstic acid were effective catalysts for the
S-acetylation of 1-hexanethiol. Then, several C
5–C
8 aliphatic thiols were reacted with aromatic and C
2–C
5 aliphatic carboxylic acids over [(Nb
6Cl
12)Cl
2(H
2O)
4]·4H
2O/SiO
2 at 300 °C. All in all, the combination of aliphatic thiol and carboxylic acid afforded the corresponding
S-alkyl carbothioate selectively.
Table 12.
S-Acylation and S-alkylation of benzenethiol with esters over [(Nb6Cl12)Cl2(H2O)4]·4H2O/SiO2 a.
| Reagent | Conversion (%) | Selectivity (%) |
|---|
| S-Phenyl Carbothioate | Alkyl Phenyl Sulfide | Diphenyl Sulfide | Diphenyl Disulfide |
|---|
| Methyl acetate | 46.8 | 86.9 | 12.0 | 0.6 | 0.5 |
| Ethyl acetate | 45.0 | 88.7 | 9.7 | 0.4 | 1.2 |
| n-Propyl acetate | 48.0 | 98.3 | 0.8 | 0.7 | 0.3 |
| Methyl propionate | 36.9 | 69.0 | 30.6 | 0.0 | 0.4 |
| Methyl butyrate | 40.4 | 29.5 | 62.2 | 8.0 | 0.3 |
| Methyl valerate | 44.8 | 27.9 | 62.1 | 8.5 | 1.5 |
| Methyl benzoate | 14.7 | 33.2 | 58.8 | 2.2 | 5.8 |
Thus,
S-acylation of thiols with carboxylic acids or their esters has been difficult even in the presence of catalysts, and there has been only one report of such an acylation by heating the neat mixture at 110 °C for 8 h in the presence of an acid catalyst [
225]. However, halide clusters catalyze
S-acylation of both aliphatic and aromatic thiols with aliphatic and aromatic carboxylic acids or their esters, when the reaction is conducted above 200 °C. Harsh reaction conditions are required for the reaction. The Brønsted acid site developed on the clusters was the active site of the catalyst, and accordingly Brønsted acids such as silica–alumina, zeolites, or heteropoly acids that are stable above 200 °C have also been proved to catalyze the reaction.
6.6. Addition of Water to Alkyne (Equation (20)) [236]
Hydration of alkynes is one of the simplest and convenient ways to obtain carbonyl compounds. This reaction has long been catalyzed by mercury(II) sulfate in aqueous sulfuric acid solution, in which the mercury(II) ion acts as a Lewis acid [
237]. To avoid use of toxicity of mercury ion and strong acidic conditions, various alternative Lewis acids have been applied to the reaction [
238,
239]. Chlorides of iron, ruthenium, rhodium, platinum, and gold are reported to catalyze the reaction. CdO–CaO–P
2O
5 [
240] and zeolites such as CuX, ZnX, and CdX [
241] are used in the vapor phase reaction. Several Brønsted acids have also been applied to the reaction: HY zeolite [
242], heteropoly acids [
243],
p-toluenesulfonic acid [
244], trifluoromethanesulfonic acid, and trifluoromethanesulfonimide [
245]. Halide cluster was applied to this reaction.
The supported tungsten cluster, (H
3O)
2[(W
6Cl
8)Cl
6]·6H
2O/SiO
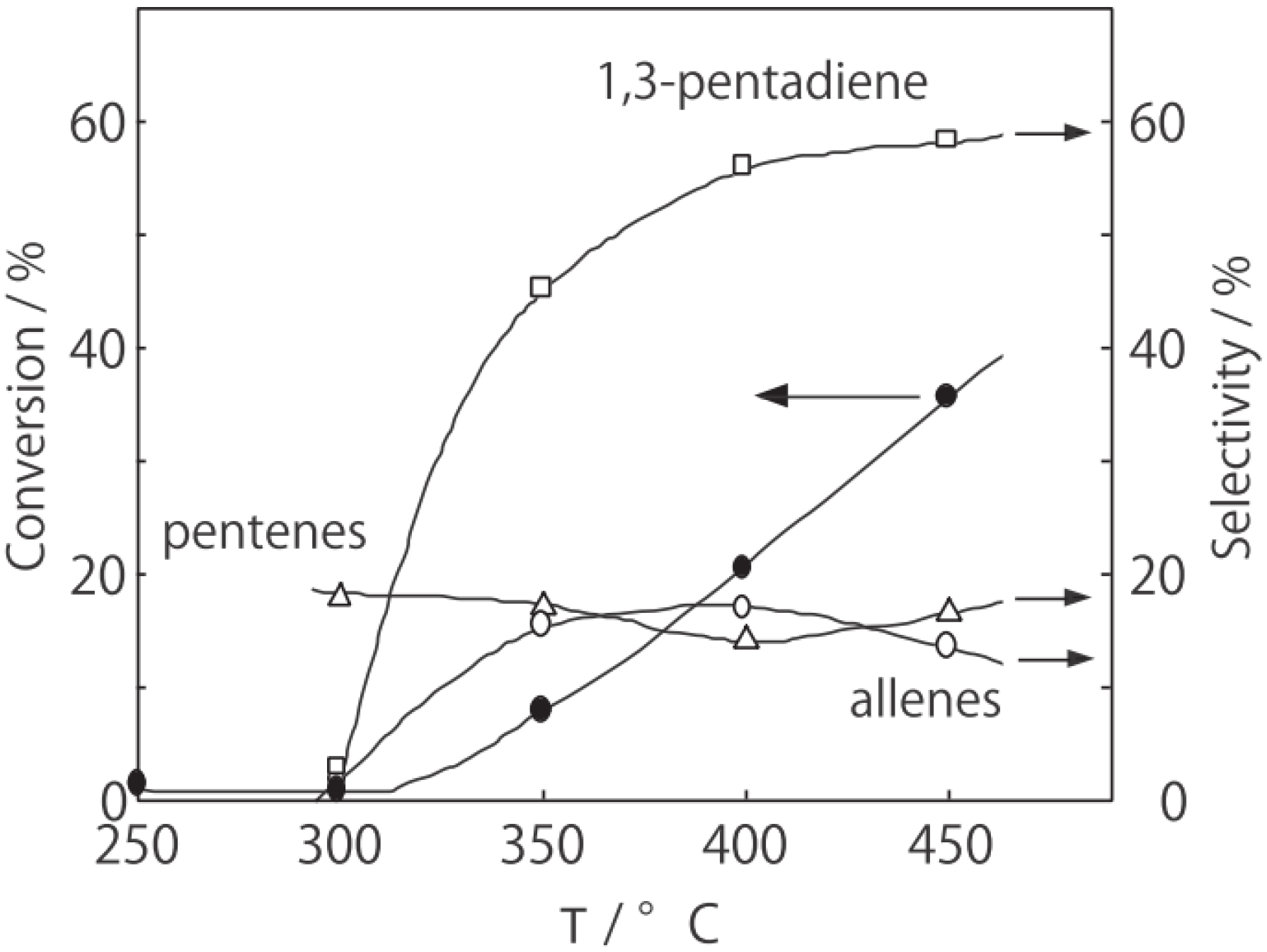
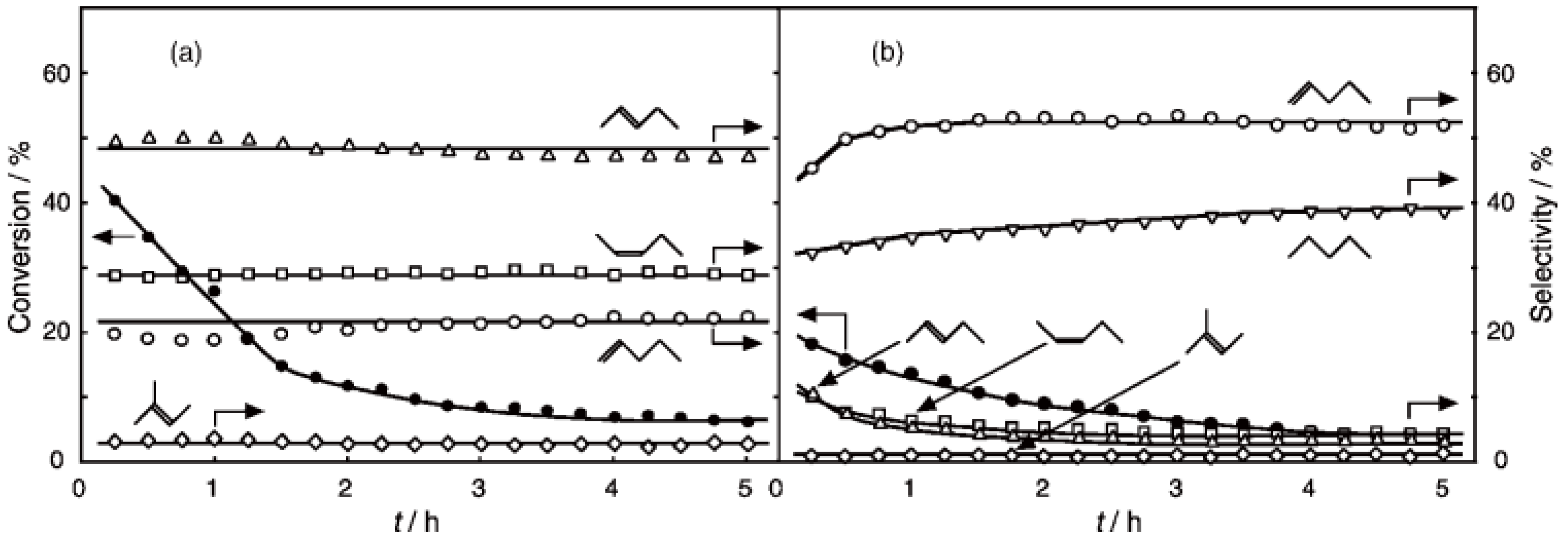
2, was initially activated at an elevated temperature for 1 h in a nitrogen stream, and then reaction was initiated by introduction of an equimolar amount of 1-hexyne and water without changing the temperature. The effect of the reaction temperature on activity and selectivity is presented in
Figure 32. Catalytic activity developed above 200 °C. The selectivity for the hydration yielding 2-hexanone was as high as 70% at temperatures ranging from 200 to 400 °C along with 1,2-hexadiene (allene) with about 20% selectivity. Above 450 °C, decomposition to gaseous products increased. Increase in water ratio to alkyne increased the selectivity for the hydration. The hydration similarly proceeded under hydrogen and helium streams.
The catalytic activity of clusters of Group 5 and 6 metals was examined at 400 °C. The catalytic activity for hydration of the niobium, molybdenum, and tantalum clusters (3%–14% conversion) was lower than that of the tungsten cluster. The selectivity for hydration over those clusters was lower (24%–28% selectivity) than over tungsten cluster. On those clusters, isomerization to 1,2-hexadiene proceeded with moderate selectivities (28%–45%). The isomerization does not involve participation of water molecule. Increase in the electronegativity of a metal atom bound to a hydroxo ligand increases its Brønsted acid strength by reducing electron density of the oxygen, and hence facilitating the release of the hydrogen as a proton [
246]. The higher catalytic activity of the tungsten cluster is attributable to the higher electronegativity of tungsten atom than the other metal atoms.
Figure 32.
Effect of temperature on the reaction of 1-hexyne with water over (H3O)2[(W6Cl8)Cl6]·6H2O/SiO2 in a nitrogen stream. Following activation of the supported cluster (40 mg, 23 μmol) in a nitrogen stream (600 mL/h) for 1 h, reaction was initiated by introduction of 1-hexyne (115 μL/h, 1.0 mmol/h) and water (18 μL/h, 1.0 mmol/h) to the nitrogen stream. Conversion of 1-hexyne (●); selectivity for 2-hexanone (○); selectivity for 1,2-hexadiene (∆); selectivity for 2-hexyne (□); and selectivity for decomposition products such as ethylene, propene, 1-butene, and 1-pentyne (◊); at 3 h after the start of the reaction.
Some alkynes (1-, 2- and 3- hexyne, 1-heptyne, 1-octyne, 1-nonyne, phenylacetylene) were subjected to the reaction over (H3O)2[(W6Cl8)Cl6]·6H2O/SiO2. Both aliphatic and aromatic terminal alkynes were efficiently hydrated to the corresponding 2-ketones (61%–99% selectivity). 2-Hexyne produced 2- and 3-hexanones in equal amounts.
6.7. Dehydrohalogenation of Alkyl Halide (Equation (21)) [247]
Various types of compounds have been reported as catalysts for the dehydrohalogenation of halogenated hydrocarbons. In most cases, a gas-phase reaction system is used for the reaction on solid catalysts. Metal (Ni) [
248,
249], metal oxide (MgO), metal oxychloride, metal sulfate, aluminosilicates [
250], and solid acid and base [
251] have been employed as the catalyst. Metal halides such as MgCl
2, KBr, NaCl, and ZnCl
2 are also the catalysts, and the last one is used as a molten salt. Metal halides used are normal salts that comprise ionic metals and ionic halogens. Halide cluster, in which metal atoms are not ionic but are coordinated with non-ionic halogen ligands, was applied to this reaction.
Powdered crystals of [(Nb
6Cl
12)Cl
2(H
2O)
4]·4H
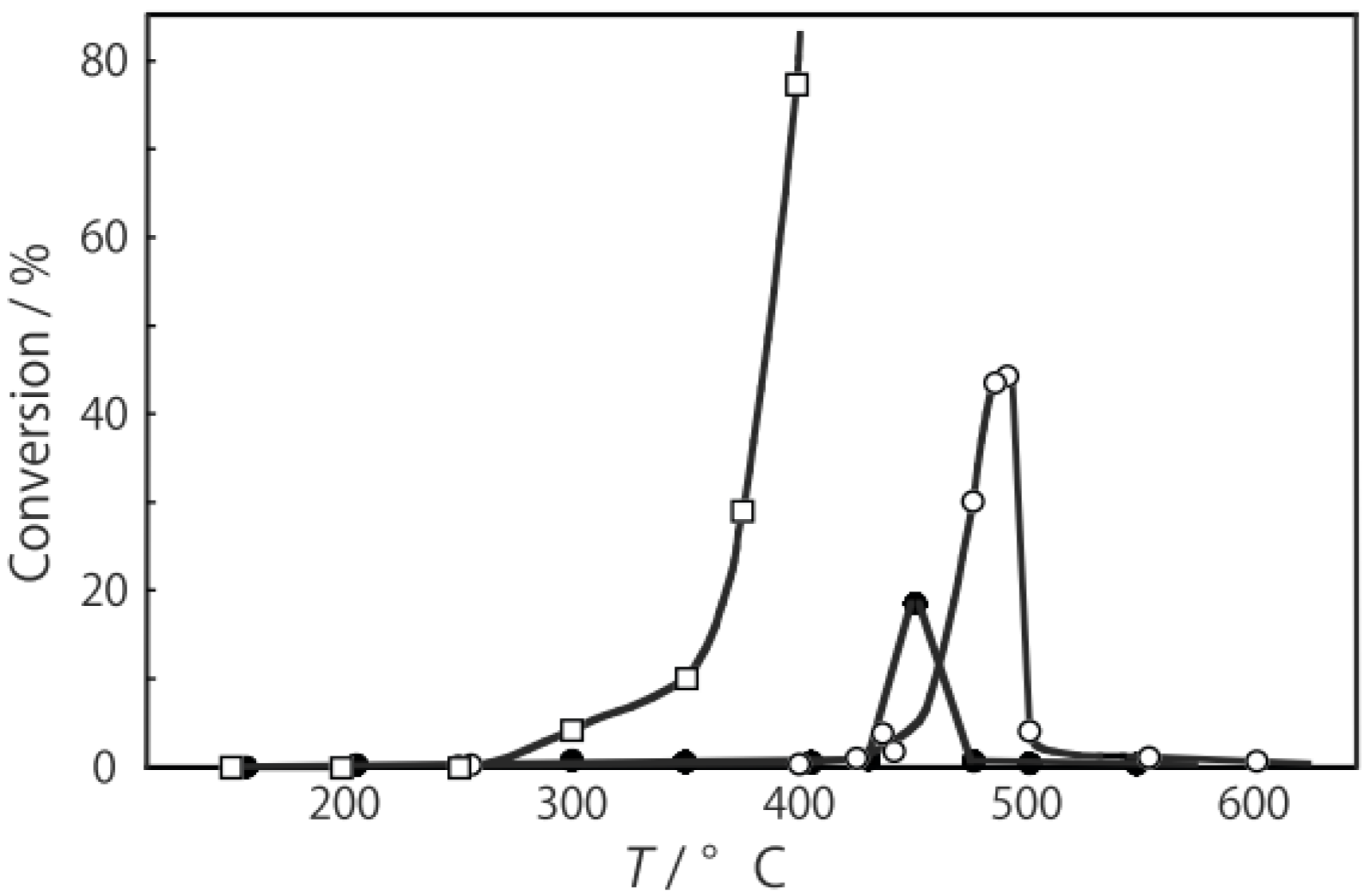
2O were activated in a stream of helium for 1 h, and the reaction was commenced by introduction of 1-chloropentane into the stream without changing the temperature. As
Figure 33 shows, activation above 200 °C brought about the catalytic activity, and the activity increased with increasing temperature up to 300 °C. Blank experiment showed that 1-chloropentane spontaneously decomposed above 325 °C, yielding a variety of products including pentenes as minor products. The selectivity for dehydrohalogenation to yield 1-pentene and
cis- and
trans-2-pentenes was 99% at 300 °C. Hydrodehalogenation (dehalogenation or hydrogenolysis) to yield pentane was not observed at all temperatures tested. As mentioned in
Section 3.2, the halide clusters catalyze isomerization of olefins. Then isomerization in the presence of halogenated hydrocarbon was examined. When 1-hexene in the presence of an equimolar amount of 1-chloropentane was subjected to the reaction over the niobium cluster at 300 °C, no isomerization of 1-hexene to 2-hexenes was observed and only dehydrohalogenation yielding pentenes occurred. Consequently, successive isomerization of the olefin produced by dehydrohalogenation can be ruled out under these reaction conditions.
Figure 33.
Effect of temperature on reactivity and selectivity for dehydrohalogenation of 1-chloropentane over [(Nb6Cl12)Cl2(H2O)4]·4H2O at 5 h after the start of the reaction. After activation of the cluster (30 mg, 17 μmol) under helium (1.2 L/h) for 1 h, reaction was commenced by introduction of 1-chloropentane (0.20 mL/h, 1.66 mmol) without changing the temperature.
Catalytic activities of bromide cluster of niobium, [(Nb6Br12)Br2(H2O)4]·4H2O, and chloride clusters of molybdenum, tantalum, and tungsten were tested for the reaction of 1-chloropentane. All clusters, except tungsten chloride cluster under hydrogen, developed catalytic activity for dehydrohalogenation both under helium and hydrogen streams. Group 5 metal clusters exhibited practically the same catalytic activity and selectivity under helium and hydrogen streams. The selectivities of molybdenum and tungsten of Group 6 metal clusters under helium were similar.
The catalytic activity and selectivity of [(Nb
6Cl
12)Cl
2(H
2O)
4]·4H
2O for 1-fluoro, 1-chloro-, 1-bromo-, and 1-iodopentanes were investigated in helium and hydrogen streams at 300 °C. The niobium cluster dehydrohalogenated all the halogen compounds to 1-pentene,
cis- and
trans-2-pentenes exclusively irrespective of the gas stream. The reactivity order of the halogen substituents was F >> Cl > Br > I, which is the reverse of that observed in catalytic hydrogenolysis over platinum group metals [
27,
56]. Only partial data can be seen: Br-C
2H
5 > I-C
2H
5 over rare-earth-exchanged 13× zeolite to yield ethylene [
250], and generally Cl ≈ Br over solid acids, and Cl < Br over solid bases [
252]. Over [(Nb
6Cl
12)Cl
2(H
2O)
4]·4H
2O, 2-chloropentane was dehydrogenated to yield
cis- and
trans-2-pentenes selectively, and the higher reactivity of 2-chloropentane compared with 1-chloropentane matches the well-known increasing activity order of primary, secondary, and tertiary carbon atoms in dehydrohalogenation [
253].
The reactivity of 1-chloropentane was examined using the rhenium cluster, Re
3Cl
9, in a stream of helium at different temperatures; the results are shown in
Figure 34. The activity appeared by activation at 175 °C, which is a little lower than the corresponding temperature for the niobium cluster. The selectivities remained almost unchanged. The
cis/
trans (0.62) and 1-pentene/2-pentene rations (0.28) are roughly in accord with those of thermal equilibrium (0.61 and 0.13 at 300 °C, respectively), which suggests that the precursors of the products are in equilibrium.
Figure 34.
Effect of temperature on reactivity and selectivity for dehydrohalogenation of 1-chloropentane (0.20 mL/h, 1.66 mmol) over Re
3Cl
9 (30 mg, 34 μmol) in helium at 5 h after the start of the reaction. Other conditions are the same as in
Figure 33.
The effects of the carrier gases, helium and hydrogen, on Re
3Cl
9 at 300 °C are shown in
Figure 35a,b. The activities decreased with time but the selectivities remained substantially constant from the initial stage of the reactions. A remarkable feature of Re
3Cl
9 is that hydrogenolysis also occurred in the hydrogen stream to yield pentane in 38% selectivity, in contrast to the reaction in the stream of helium in which only dehydrohalogenation to yield pentenes occurred. Complex Re
3Cl
9 changed to metallic rhenium by treatment with hydrogen [
1]. In this case, hydrogen participated chemically both as a reagent for the reduction of Re
III3Cl
9 and as a reagent for hydrogenolysis of 1-chloropentane to pentane. Rhenium is located in a neighboring position to the platinum group metals in the periodic table, and is similar to them in some properties. Rhenium metal, as well as its oxide, sulfide, and selenide, is a catalyst for hydrogenation of organic compounds [
90,
254]; however, it has not been reported to catalyze the hydrogenolysis [
255].
Figure 35.
Catalytic activity and selectivity of Re
3Cl
9 (30 mg, 34 μmol) in dehydrohalogenation and hydrogenolysis of 1-chloropentane (0.20 mL/h, 1.66 mmol) in a stream of (
a) helium; and (
b) hydrogen at 300 °C. Other conditions are the same as in
Figure 33.
Various types of halogenated pentanes were allowed to react over Re
3Cl
9 in helium and hydrogen streams at 300 °C, and the results are summarized in
Table 13. Under helium, in which the cluster framework was retained, dehydrohalogenation to yield pentenes proceeded except for iodide, and the reactivity order was F > Cl > Br as in the case of niobium cluster. Under hydrogen, in which the cluster was reduced to metallic rhenium, both dehydrohalogenation to pentenes and hydrogenolysis to pentane for all 1-halogenated pentanes proceeded with almost equal reactivity. Even the C–F bond was hydrogenolyzed by this catalyst. It has been reported in the hydrogenolysis of halogen substituents, RX to RH, that the reactivity order is RI > RBr > RCl and that RF cannot be converted to RH by catalytic means [
27,
56]. However, rhenium metal obtained from reduction of cluster Re
3Cl
9 has caused the hydrogenolysis of the C–F bond.
Table 13.
Dehydrohalogenation and hydrodehalogenation of halogenated pentanes over Re3Cl9 a.
| X-C5H11 | Carrier gas | Conversion/% | Selectivity/% |
|---|
| X = | Pentane | 1-Pentene | cis-2-Pentene | trans-2-Pentene | 2-Methyl-2-butene |
|---|
| F | He | 14.3 | 0.0 | 30.1 | 30.1 | 39.8 | 0.0 |
| 1-Cl | He | 7.1 | 0.0 | 21.6 | 29.0 | 46.8 | 2.6 |
| 2-Cl | He | 29.7 | 0.0 | 17.6 | 35.1 | 47.3 | 0.0 |
| 1-Br | He | 5.7 | 0.0 | 22.9 | 28.2 | 45.0 | 3.9 |
| 1-I | He | 0.0 | | | | | |
| 1-F | H2 | 5.6 | 14.0 | 14.1 | 24.0 | 47.9 | 0.0 |
| 1-Cl | H2 | 5.1 | 38.4 | 52.3 | 4.7 | 3.4 | 1.2 |
| 2-Cl | H2 | 91.6 | 2.2 | 20.2 | 34.3 | 43.3 | 0.0 |
| 1-Br | H2 | 4.7 | 40.9 | 37.2 | 7.6 | 12.7 | 1.6 |
| 1-I | H2 | 4.9 | 37.9 | 43.5 | 9.9 | 8.7 | 0.0 |

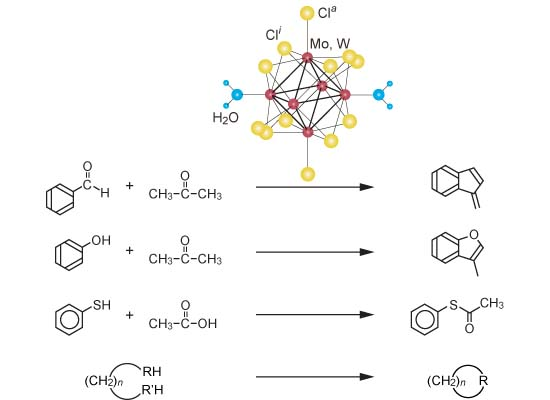






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}