

Evaluation of the Applicability of Voltammetric Modes in Scanning Electrochemical Microscopy for In Situ Corrosion Characterisation of Copper-Based Materials
 ,
,  and
and
Abstract
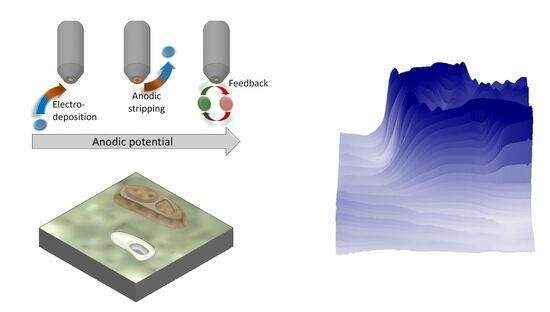
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results and Discussion
3.1. Electrochemical Behaviour of Gold Electrodes for Copper Collection and Redissolution
3.2. Imaging of Copper Corrosion Using SECM Coupled with a Voltammetric Mode
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pistorius, P.C.; Burstein, G.T. Metastable pitting corrosion of stainless steel and the transition to stability. Philos. Trans. R. Soc. London. Ser. A Phys. Eng. Sci. 1992, 341, 531–559. [Google Scholar]
- Montemor, M.F. Corrosion issues in joining lightweight materials: A review of the latest achievements. Phys. Sci. Rev. 2016, 1, 20150011. [Google Scholar] [CrossRef]
- Liu, W.; Yang, H.; Li, X.; Zhang, Z.; Lin, Y.; Deng, K. Effect of chloride and iodide on the corrosion behavior of 13Cr stainless steel. Metals 2022, 12, 1833. [Google Scholar] [CrossRef]
- Zhou, P.; Deng, L.; Guo, P.; Rao, W.; Wang, X.; Zhang, M. Influence of microstructure heterogeneity on the corrosion resistance and microhardness of 5052 Al-Mg alloy. J. Mater. 2020, 72, 4305–4314. [Google Scholar]
- Li, X.-R.; Meng, X.-Z.; Zhang, Q.-H.; Wu, L.-K.; Sun, Q.-Q.; Deng, H.-Q.; Sun, S.-J.; Cao, F.-H. Insight into oxygen reduction activity and pathway on pure titanium using scanning electrochemical microscopy and theoretical calculations. J. Colloid Interface Sci. 2023, 643, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Raj Xavier, J.; Ramesh, B. A study on the effect of multifunctional tantalum carbide nanofillers incorporated graphene oxide structure in the epoxy resin for the applications in the shipbuilding industry. Mater. Sci. Eng. B 2023, 289, 116234. [Google Scholar] [CrossRef]
- Payne, N.A.; Stephens, L.I.; Mauzeroll, J. The application of scanning electrochemical microscopy to corrosion research. Corrosion 2017, 73, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Traxler, I.; Singewald, T.D.; Schimo-Aichhorn, G.; Hild, S.; Valtiner, M. Scanning electrochemical microscopy methods (SECM) and ion-selective microelectrodes for corrosion studies. Corros. Rev. 2022, 40, 515–542. [Google Scholar] [CrossRef]
- Kwak, J.; Bard, A.J. Scanning electrochemical microscopy. Theory of the feedback mode. Anal. Chem. 1989, 61, 1221–1227. [Google Scholar] [CrossRef]
- Bard, A.J.; Denuault, G.; Lee, C.; Mandler, D.; Wipf, D.O. Scanning electrochemical microscopy—A new technique for the characterization and modification of surfaces. Acc. Chem. Res. 1990, 23, 357–363. [Google Scholar] [CrossRef]
- Bard, A.J. Introduction and principles. In Scanning Electrochemical Microscroscopy, 3rd ed.; Bard, A.J., Mirkin, M.V., Eds.; CRC Press: Boca Raton, FL, USA, 2022; pp. 1–10. [Google Scholar]
- Mirkin, M.V.; Nogala, W.; Velmurugan, J.; Wang, Y. Scanning electrochemical microscopy in the 21st century. Update 1: Five years after. Phys. Chem. Chem. Phys. 2011, 13, 21196–21212. [Google Scholar] [CrossRef] [PubMed]
- Noël, J.-M.; Kanoufi, F. Probing the reactive intermediate species generated during electrocatalysis by scanning electrochemical microscopy. Curr. Opin. Electrochem. 2022, 35, 101071. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, T.; Lang, J.; Fu, W.; Li, F. Scanning electrochemical microscopy of living cells. Curr. Opin. Electrochem. 2020, 22, 178–185. [Google Scholar] [CrossRef]
- Hill, C.M.; Pan, S. SECM techniques for locally interrogating the photocatalytic activity of semiconducting materials for solar-driven transformations. In Scanning Electrochemical Microscroscopy, 3rd ed.; Bard, A.J., Mirkin, M.V., Eds.; CRC Press: Boca Raton, FL, USA, 2022; pp. 361–378. [Google Scholar]
- Kranz, C.; Demaille, C. Hybrid scanning electrochemical techniques: Methods and applications. In Scanning Electrochemical Microscroscopy, 3rd ed.; Bard, A.J., Mirkin, M.V., Eds.; CRC Press: Boca Raton, FL, USA, 2022; pp. 513–580. [Google Scholar]
- Heurtault, S.; Robin, R.; Rouillard, F.; Vivier, V. On the propagation of open and covered pit in 316L stainless steel. Electrochim. Acta 2016, 203, 316–325. [Google Scholar] [CrossRef]
- Hampel, M.; Schenderlein, M.; Schary, C.; Dimper, M.; Ozcan, O. Efficient detection of localized corrosion processes on stainless steel by means of scanning electrochemical microscopy (SECM) using a multi-electrode approach. Electrochem. Commun. 2019, 101, 52–55. [Google Scholar] [CrossRef]
- Blanc, C.; Pébère, N.; Tribollet, B.; Vivier, V. Galvanic coupling between copper and aluminium in a thin-layer cell. Corros. Sci. 2010, 52, 991–995. [Google Scholar] [CrossRef]
- Yan, Z.-Z.; Zhang, Q.-H.; Cai, H.-R.; Li, X.-R.; Wu, L.-K.; Luo, Z.-Z.; Cao, F.-H. Study on the galvanic corrosion of titanium and stainless steel couple with the synergistic effect of proton and fluoride ion. Corros. Sci. 2022, 206, 110541. [Google Scholar] [CrossRef]
- Pal, A.; Krishna, N.G.; Shantar, A.R.; Philip, J. High contrast corrosion mapping of dissimilar metal weld joints using alternating current scanning electrochemical microscopy: A case study with Zr-4–Ti–304L SS weld. Corros. Sci. 2023, 221, 111345. [Google Scholar] [CrossRef]
- Martinez-Lombardia, E.; Gonzalez-Garcia, Y.; Lapeire, L.; De Graeve, I.; Verbeken, K.; Kestens, L.; Mol, J.M.C.; Terryn, H. Scanning electrochemical microscopy to study the effect of crystallographic orientation on the electrochemical activity of pure copper. Electrochim. Acta 2014, 116, 89–96. [Google Scholar] [CrossRef]
- Lin, E.; Li, X.; Kure-Chu, S.-Z.; Li, X.; Xiao, X. Effect of electrical parameters on the microstructure and corrosion resistance of anodized film of Mg-1Zn-1Gd alloy based on orthogonal experiment method. J. Mater. Eng. Perform. 2023, in press. [Google Scholar] [CrossRef]
- Nickchi, T.; Rostron, P.; Barsoum, I.; Alfantazi, A. Measurement of local galvanic surface corrosion using scanning electrochemical microscopy on ductile cast iron. J. Mater. Sci. 2019, 54, 9213–9221. [Google Scholar] [CrossRef]
- Sidane, D.; Devos, O.; Puiggali, M.; Touzet, M.; Tribollet, B.; Vivier, V. Electrochemical characterization of a mechanically stressed passive layer. Electrochem. Commun. 2011, 13, 1361–1364. [Google Scholar] [CrossRef]
- Grandy, L.; Mauzeroll, J. Localising the electrochemistry of corrosion fatigue. Curr. Opin. Colloid Interface 2022, 61, 101628. [Google Scholar] [CrossRef]
- Pust, S.E.; Scharnweber, D.; Kirchner, C.N.; Wittstock, G. Electron transfer kinetics at oxide films on metallic biomaterials. Adv. Mater. 2007, 19, 878–882. [Google Scholar] [CrossRef]
- Meiszterics, Z.; Kiss, A.; Nagy, L.; Zsebe, T.; Vasvári, G.F.; Csonka, D.C.; Nagy, G. Investigation of the regeneration of the passive surface oxide layer of TiAl6V4 alloy printed by WAAM technology using different electrochemical test methods. Electroanalysis 2023, 35, e202200506. [Google Scholar] [CrossRef]
- Xia, D.H.; Wang, J.; Wu, Z.; Qin, Z.; Xu, L.; Hu, W.; Behnamian, Y.; Luo, J.L. Sensing corrosion within an artificial defect in organic coating using SECM. Sens. Actuat. B Chem. 2019, 280, 235–242. [Google Scholar] [CrossRef]
- Santana, J.J.; Izquierdo, J.; Souto, R.M. Uses of scanning electrochemical microscopy (SECM) for the characterization with spatial and chemical resolution of thin surface layers and coating systems applied on metals: A review. Coatings 2022, 12, 637. [Google Scholar] [CrossRef]
- Liang, J.; Liu, S.; Peng, Z.; Li, R.; Wang, B. Galvanic corrosion behavior of AZ31 Mg alloy coupled with mild steel: Effect of coatings. J. Mater. Res. Technol. 2023, 24, 7745–7755. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Jamal, S.S.; Hinton, B.R.W.; Moulton, S.E.; Wallace, G.G.; Forsyth, M. The effect of treatment time on the ionic liquid surface film formation: Promising surface coating for Mg alloy AZ31. Surf. Coat. Technol. 2016, 296, 192–202. [Google Scholar] [CrossRef]
- Xavier, J.R. Superior surface protection, mechanical and hydrophobic properties of silanized tungsten carbide nanoparticles encapsulated epoxy nanocomposite coated steel structures in marine environment. Silicon 2022, 14, 11147–11161. [Google Scholar] [CrossRef]
- Niaz, A.; Al Fuhaid, A.F.; Faraz, M.I. Understanding corrosion degradation processes of a multi-component CoNiCrAlY-coating system. Coatings 2022, 12, 1396. [Google Scholar] [CrossRef]
- Al-Jeda, M.; Mena-Morcillo, E.; Chen, A. Micro-sized pH sensors based on scanning electrochemical probe microscopy. Micromachines 2022, 13, 2143. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Liang, R.; Wang, F.; Ding, J.; Qin, W. An all-solid-state potentiometric microelectrode for detection of copper in coastal sediment pore water. Sens. Actuat. B-Chem. 2019, 279, 369–373. [Google Scholar] [CrossRef]
- Varvara, S.; Caniglia, G.; Izquierdo, J.; Bostan, R.; Găină, L.; Bobis, O.; Souto, R.M. Multiscale electrochemical analysis of the corrosion control of bronze in simulated acid rain by horse-chestnut (Aesculus hippocastanum L.) extract as green inhibitor. Corros. Sci. 2020, 165, 108381. [Google Scholar] [CrossRef]
- Izquierdo, J.; Fernández-Pérez, B.M.; Eifert, A.; Souto, R.M.; Kranz, C. Simultaneous atomic force-scanning electrochemical microscopy (AFM-SECM) imaging of copper dissolution. Electrochim. Acta 2016, 201, 320–332. [Google Scholar] [CrossRef]
- Izquierdo, J.; Eifert, A.; Kranz, C.; Souto, R.M. In situ investigation of copper corrosion in acidic chloride solution using atomic force-scanning electrochemical microscopy. Electrochim. Acta 2017, 247, 588–599. [Google Scholar] [CrossRef]
- Schrock, D.S.; Baur, J.E. Chemical imaging with combined fast-scan cyclic voltammetry-scanning electrochemical microscopy. Anal. Chem. 2007, 79, 7053–7061. [Google Scholar] [CrossRef]
- Barton, Z.J.; Rodríguez-López, J. Cyclic voltammetry probe approach curves with alkali amalgams at mercury sphere-cap scanning electrochemical microscopy probes. Anal. Chem. 2017, 89, 2708–2715. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R.; White, H.S. Electrochemical Methods: Fundamentals and Applications, 3rd ed.; John Wiley & Sons: New York, NY, USA, 2022. [Google Scholar]
- Harris, K.R.; Woolf, L.A. Pressure and temperature dependence of the self diffusion coefficient of water and oxygen-18 water. J. Chem. Soc. Faraday Trans. 1980, 76, 377–385. [Google Scholar] [CrossRef]
- Yu, Y.; Gao, Y.; Hu, K.; Blanchard, P.Y.; Noël, J.M.; Nareshkumar, T.; Phani, K.L.; Friedman, G.; Gogotsi, Y.; Mirkin, M.V. Electrochemistry and electrocatalysis at single gold nanoparticles attached to carbon nanoelectrodes. ChemElectroChem 2015, 2, 58–63. [Google Scholar] [CrossRef]
- Papaderakis, A.; Anastopoulos, A.G.; Sotiropoulos, S. Electrochemical studies of processes occurring at the polycrystalline Cu electrode/methanol interface. J. Electroanal. Chem. 2016, 783, 217–225. [Google Scholar] [CrossRef]
- Krznarić, D.; Goričnik, T. Reactions of copper on the Au(111) surface in the underpotential deposition region from chloride solutions. Langmuir 2001, 17, 4347–4351. [Google Scholar] [CrossRef]
- Cornut, R.; Lefrou, C. New analytical approximation of feedback approach curves with a microdisk SECM tip and irreversible kinetic reaction at the substrate. J. Electroanal. Chem. 2008, 621, 178–184. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration/mM | Q1/nC | Q2/nC | Corrected Q1/nC V−1 | Corrected Q2/nC V−1 |
|---|---|---|---|---|
| 0.90 | 3.25 | 0.84 | 9.66 | 7.66 |
| 1.12 | 4.55 | 4.65 | 22.1 | 10.9 |
| 1.77 | 9.32 | 8.03 | 38.5 | 20.7 |
| 2.83 | 13.3 | 18.7 | 70.3 | 29.2 |
| 3.62 | 23.3 | 28.5 | 113 | 50.8 |
| 3.19 | 23.1 | 28.4 | 104 | 46.9 |
| 4.12 | 24.1 | 38.6 | 120 | 46.2 |
| Concentration/mM | Q1/nC | Q2/nC | Corrected Q1/nC V−1 | Corrected Q2/nC V−1 |
|---|---|---|---|---|
| 1 | 13.9 | 2.4 | 19.9 | 3.42 |
| 10 | 219 | 124 | 260 | 148 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Concepción, B.; Méndez-Guerra, A.; Souto, R.M.; Izquierdo, J. Evaluation of the Applicability of Voltammetric Modes in Scanning Electrochemical Microscopy for In Situ Corrosion Characterisation of Copper-Based Materials. Metals 2023, 13, 1965. https://doi.org/10.3390/met13121965
Hernández-Concepción B, Méndez-Guerra A, Souto RM, Izquierdo J. Evaluation of the Applicability of Voltammetric Modes in Scanning Electrochemical Microscopy for In Situ Corrosion Characterisation of Copper-Based Materials. Metals. 2023; 13(12):1965. https://doi.org/10.3390/met13121965
Chicago/Turabian StyleHernández-Concepción, Brenda, Adrián Méndez-Guerra, Ricardo M. Souto, and Javier Izquierdo. 2023. "Evaluation of the Applicability of Voltammetric Modes in Scanning Electrochemical Microscopy for In Situ Corrosion Characterisation of Copper-Based Materials" Metals 13, no. 12: 1965. https://doi.org/10.3390/met13121965