Cerebrospinal Fluid Biomarkers in Alzheimer’s Disease—From Brain Starch to Bench and Bedside


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Amyloid-β
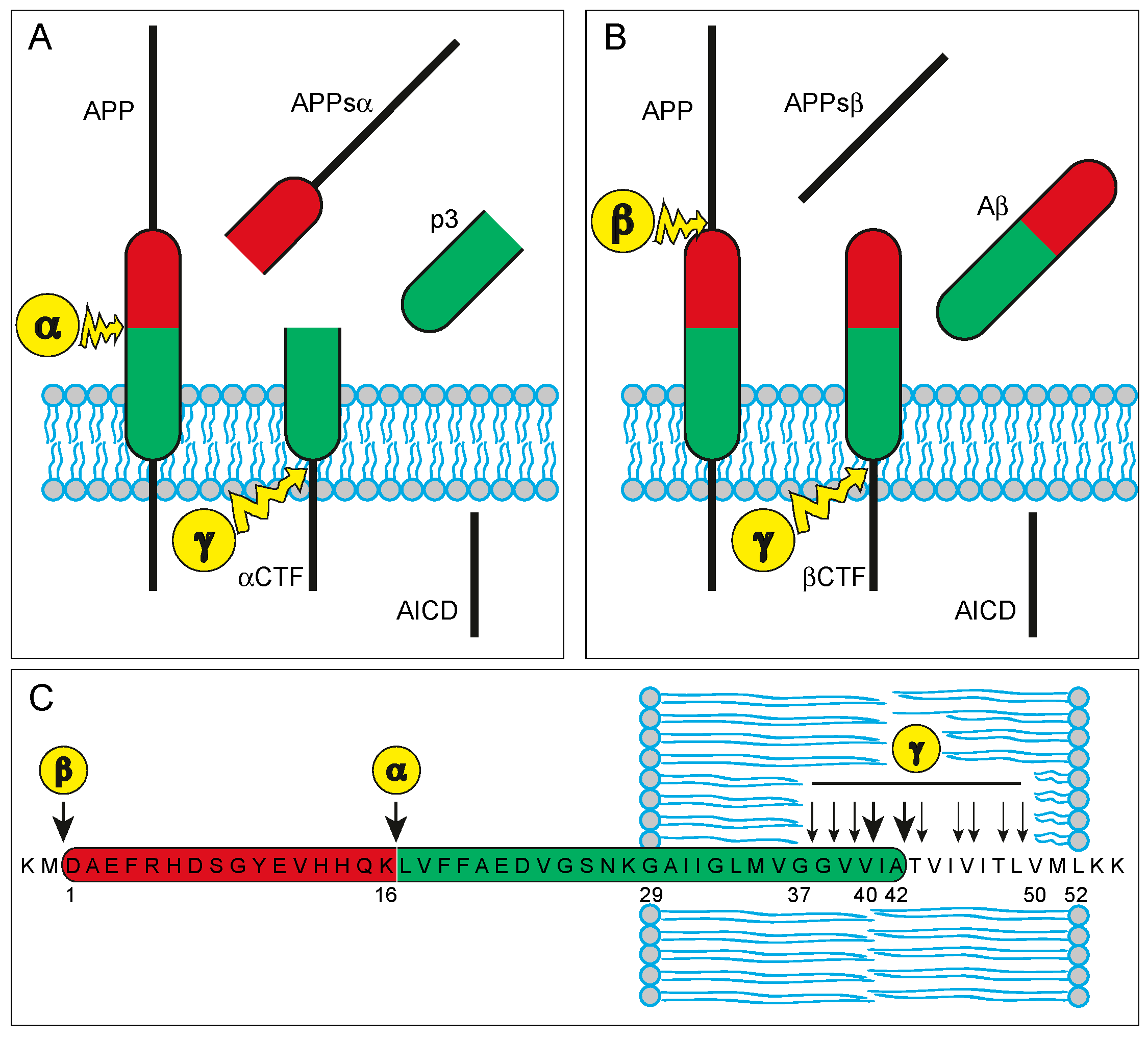
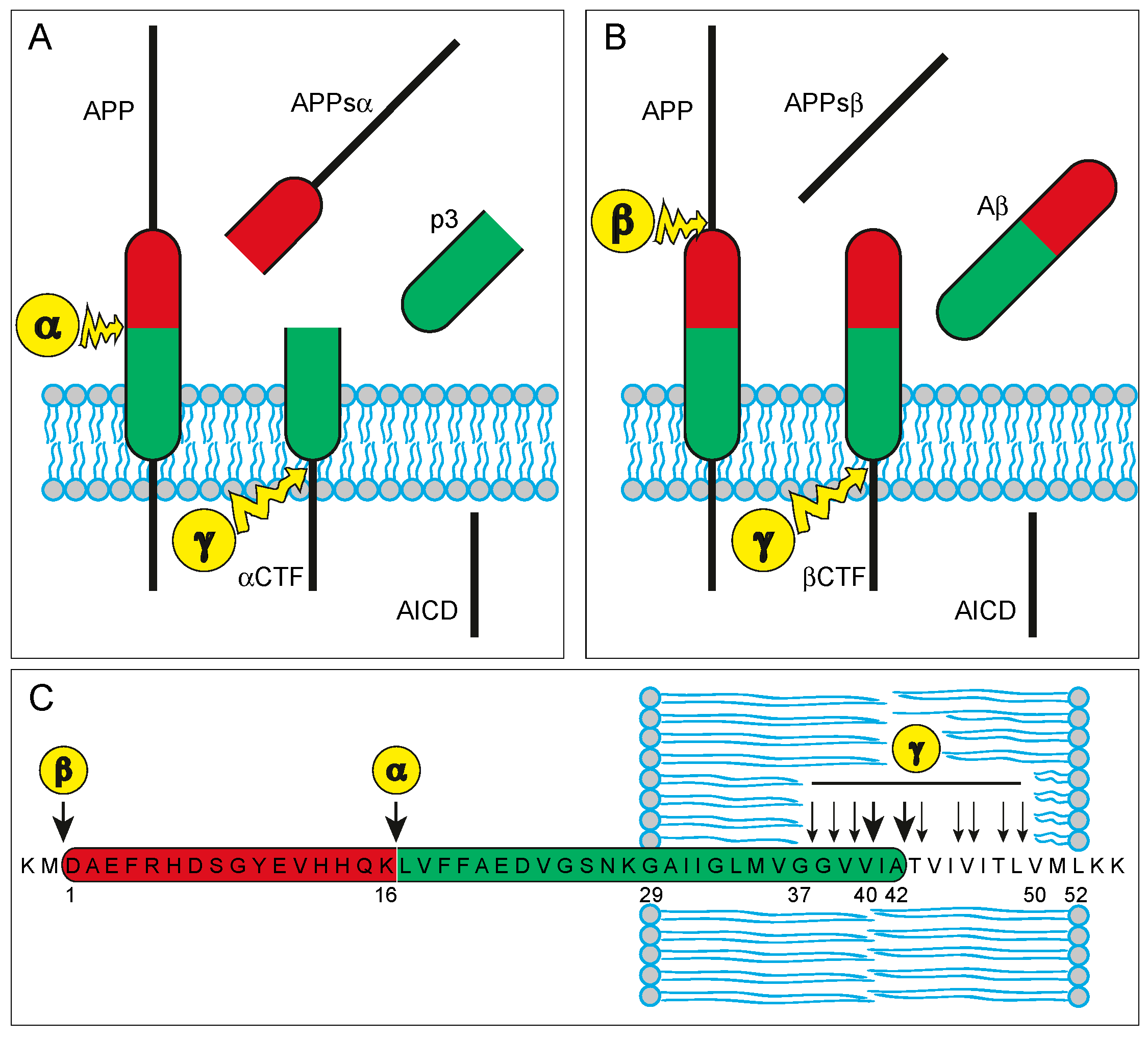
2.1. APP and Aβ—Biochemistry and Physiology
2.2. APP and Aβ—Their Role in the Neuropathology of AD
2.3. From Brain Starch to the Use of Aβ as Biomarker for Alzheimer’s Disease—Historical Context
2.4. Aβ as CSF Biomarker for Alzheimer’s Disease
3. Tau
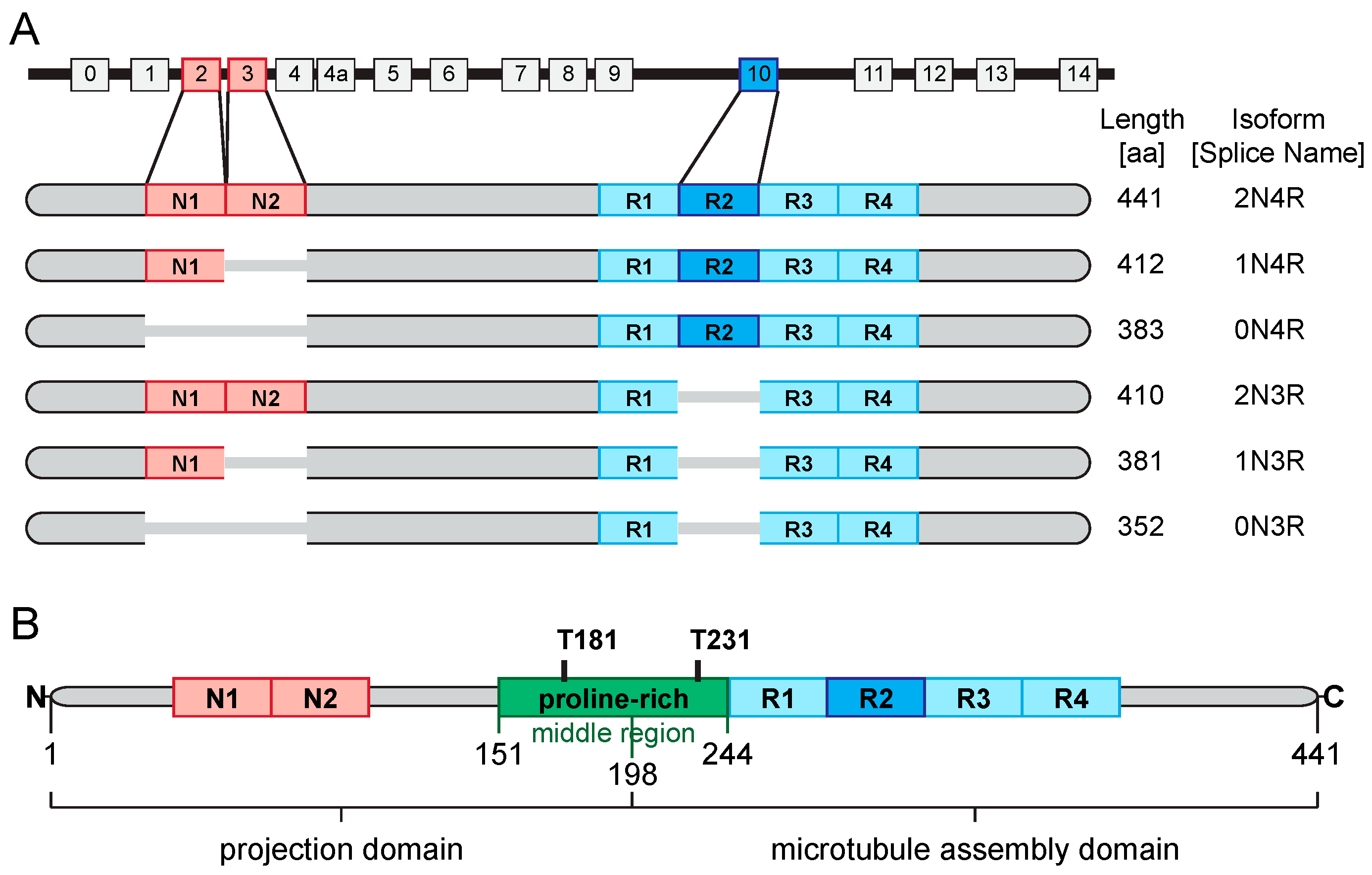
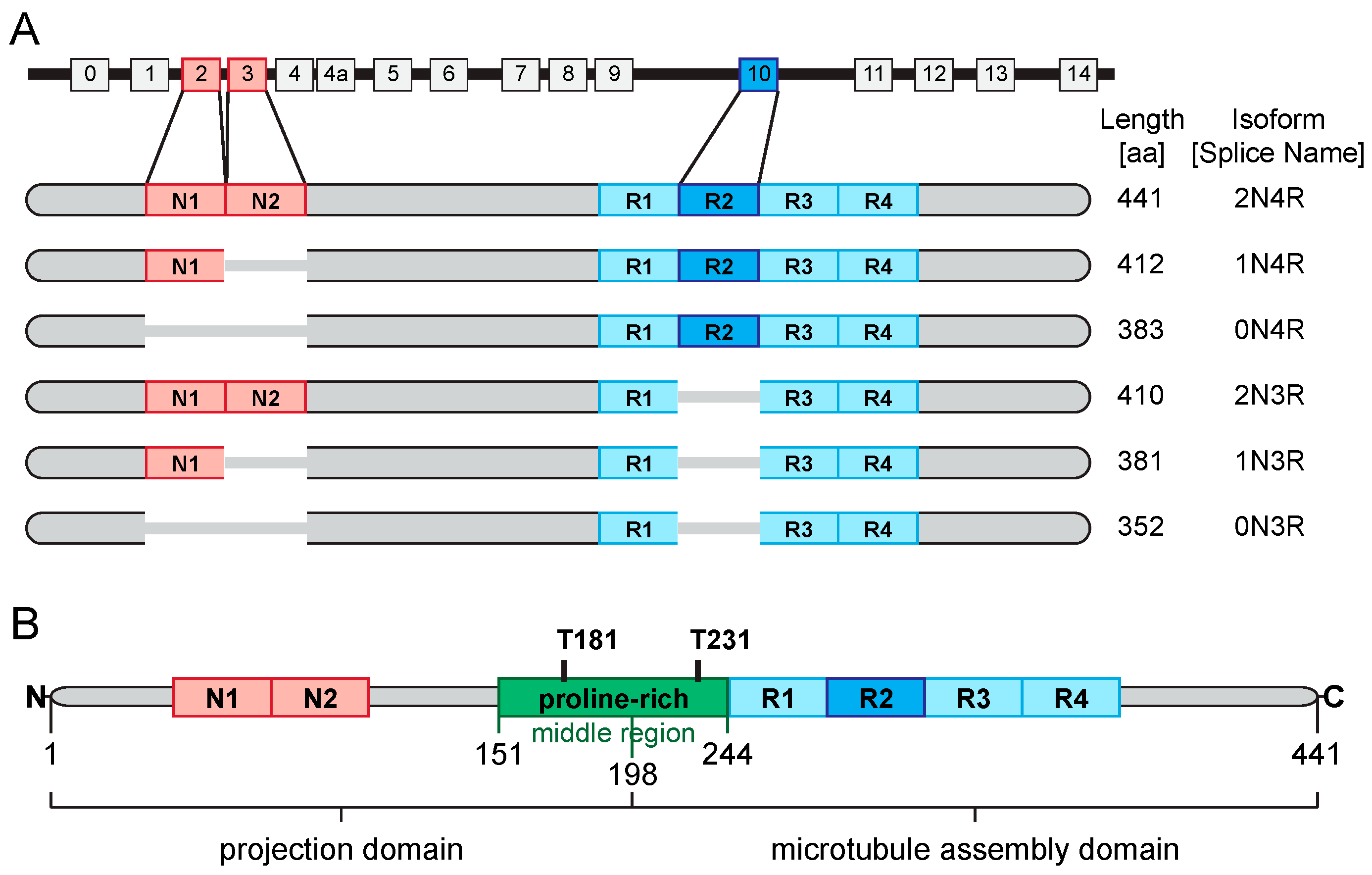
3.1. Tau—Biochemistry and Physiology
3.2. Tau—Its Role in the Neuropathology of AD
3.3. From Paired Helical Filaments to the Use of Tau as Biomarker for Alzheimer’s Disease—Historical Context
3.4. Tau as CSF Biomarker for Alzheimer’s Disease
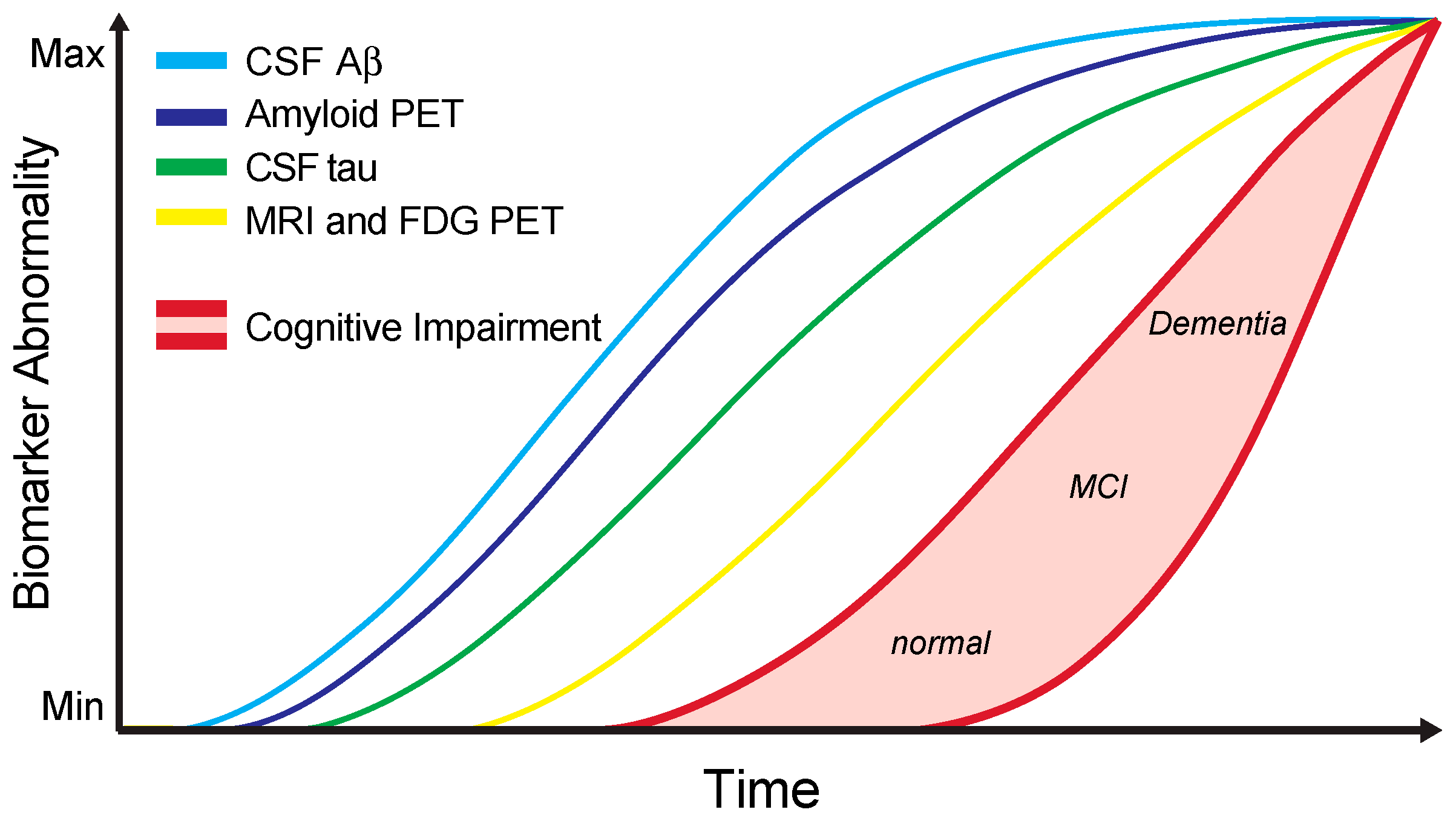
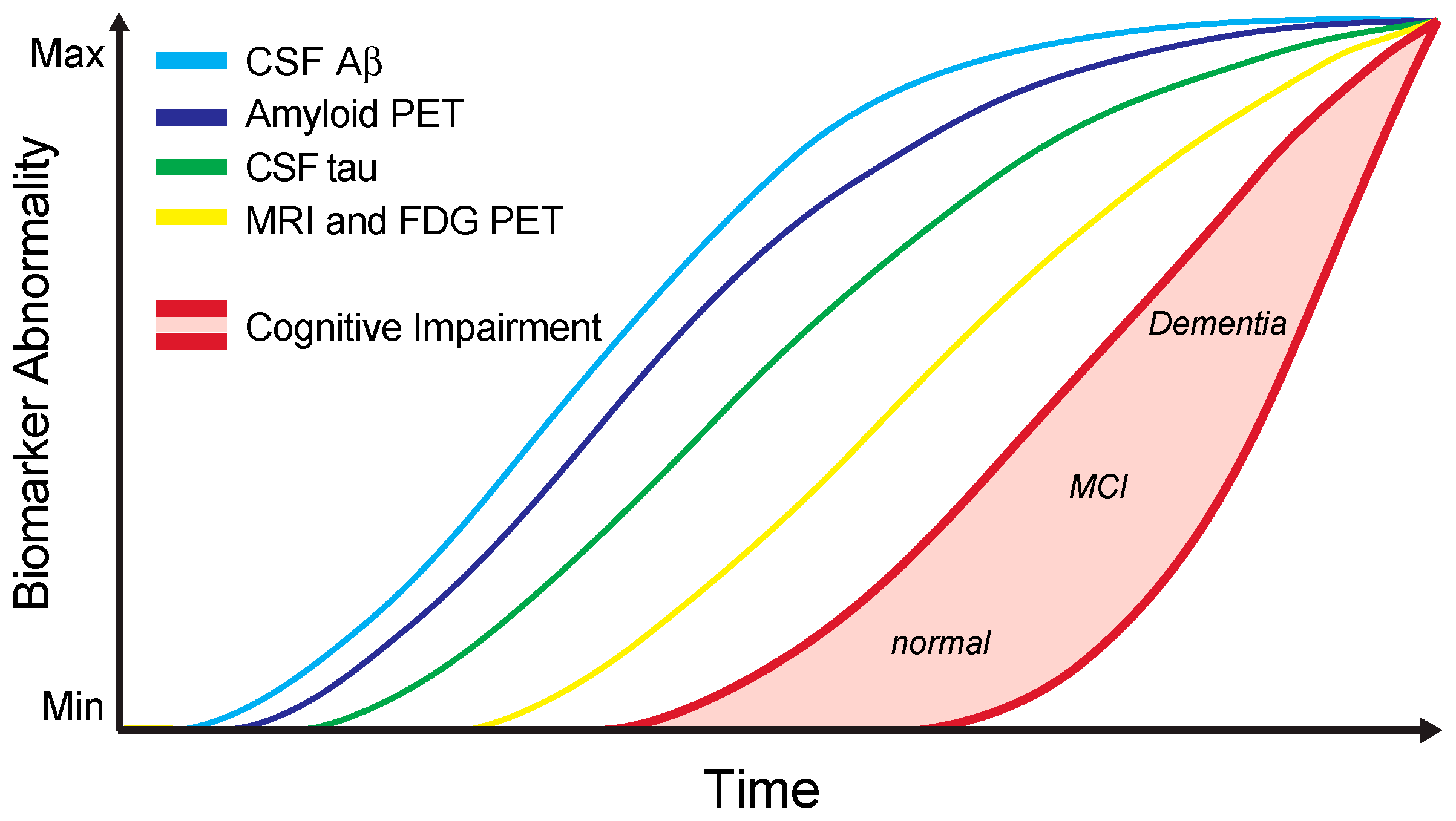
4. Alzheimer’s Disease CSF Core Biomarkers in Clinical Practice
5. Additional Candidate Biomarkers
Conflicts of Interest
References
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509. [Google Scholar]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Weintraub, S.; Wicklund, A.H.; Salmon, D.P. The neuropsychological profile of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006171. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Storandt, M.; McKeel, D.W.; Rubin, E.H.; Price, J.L.; Grant, E.A.; Berg, L. Cerebral amyloid deposition and diffuse plaques in “normal” aging: Evidence for presymptomatic and very mild Alzheimer’s disease. Neurology 1996, 46, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.; Basi, G.S.; Pangalos, M.N. Treatment strategies targeting amyloid β-protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006387. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.-Y.; Brunden, K.R.; Hutton, M.; Trojanowski, J.Q. Developing therapeutic approaches to tau, selected kinases, and related neuronal protein targets. Cold Spring Harb. Perspect. Med. 2011, 1, a006437. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Zetterberg, H.; Fagan, A.M. Fluid biomarkers in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006221. [Google Scholar] [CrossRef] [PubMed]
- Siemers, E.R. How can we recognize “disease modification” effects? J. Nutr. Health Aging 2009, 13, 341–343. [Google Scholar] [CrossRef] [PubMed]
- The Ronald and Nancy Reagan Research Institute of the Alzheimer’s Association; National Institute on Aging Working Group. Consensus report of the Working Group on: “Molecular and Biochemical Markers of Alzheimer’s Disease”. Neurobiol. Aging 1998, 19, 109–116. [Google Scholar]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef] [PubMed]
- Müller, U.C.; Zheng, H. Physiological functions of APP family proteins. Cold Spring Harb. Perspect. Med. 2012, 2, a006288. [Google Scholar] [CrossRef] [PubMed]
- Slunt, H.H.; Thinakaran, G.; Von Koch, C.; Lo, A.C.; Tanzi, R.E.; Sisodia, S.S. Expression of a ubiquitous, cross-reactive homologue of the mouse β-amyloid precursor protein (APP). J. Biol. Chem. 1994, 269, 2637–2644. [Google Scholar] [PubMed]
- Kaether, C.; Skehel, P.; Dotti, C.G. Axonal membrane proteins are transported in distinct carriers: A two-color video microscopy study in cultured hippocampal neurons. Mol. Biol. Cell 2000, 11, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Koo, E.H.; Sisodia, S.S.; Archer, D.R.; Martin, L.J.; Weidemann, A.; Beyreuther, K.; Fischer, P.; Masters, C.L.; Price, D.L. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc. Natl. Acad. Sci. USA 1990, 87, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Selkoe, D.J.; Koo, E.H. Trafficking of cell surface β-amyloid precursor protein: Retrograde and transcytotic transport in cultured neurons. J. Cell Biol. 1995, 129, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Saido, T.; Leissring, M.A. Proteolytic degradation of amyloid β-protein. Cold Spring Harb. Perspect. Med. 2012, 2, a006379. [Google Scholar] [CrossRef] [PubMed]
- Clarris, H.J.; Cappai, R.; Heffernan, D.; Beyreuther, K.; Masters, C.L.; Small, D.H. Identification of heparin-binding domains in the amyloid precursor protein of Alzheimer’s disease by deletion mutagenesis and peptide mapping. J. Neurochem. 1997, 68, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Beher, D.; Hesse, L.; Masters, C.L.; Multhaup, G. Regulation of amyloid protein precursor (APP) binding to collagen and mapping of the binding sites on APP and collagen type I. J. Biol. Chem. 1996, 271, 1613–1620. [Google Scholar] [CrossRef] [PubMed]
- Kibbey, M.C.; Jucker, M.; Weeks, B.S.; Neve, R.L.; van Nostrand, W.E.; Kleinman, H.K. β-Amyloid precursor protein binds to the neurite-promoting IKVAV site of laminin. Proc. Natl. Acad. Sci. USA 1993, 90, 10150–10153. [Google Scholar] [CrossRef] [PubMed]
- Soba, P.; Eggert, S.; Wagner, K.; Zentgraf, H.; Siehl, K.; Kreger, S.; Löwer, A.; Langer, A.; Merdes, G.; Paro, R.; et al. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005, 24, 3624–3634. [Google Scholar] [CrossRef] [PubMed]
- Dalva, M.B.; McClelland, A.C.; Kayser, M.S. Cell adhesion molecules: Signalling functions at the synapse. Nat. Rev. Neurosci. 2007, 8, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Young-Pearse, T.L.; Chen, A.C.; Chang, R.; Marquez, C.; Selkoe, D.J. Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin β1. Neural Dev. 2008, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Ashley, J.; Packard, M.; Ataman, B.; Budnik, V. Fasciclin II signals new synapse formation through amyloid precursor protein and the scaffolding protein dX11/Mint. J. Neurosci. 2005, 25, 5943–5955. [Google Scholar] [CrossRef] [PubMed]
- Allinquant, B.; Hantraye, P.; Mailleux, P.; Moya, K.; Bouillot, C.; Prochiantz, A. Downregulation of amyloid precursor protein inhibits neurite outgrowth in vitro. J. Cell Biol. 1995, 128, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Hérard, A.S.; Besret, L.; Dubois, A.; Dauguet, J.; Delzescaux, T.; Hantraye, P.; Bonvento, G.; Moya, K.L. siRNA targeted against amyloid precursor protein impairs synaptic activity in vivo. Neurobiol. Aging 2006, 27, 1740–1750. [Google Scholar] [CrossRef] [PubMed]
- Hornsten, A.; Lieberthal, J.; Fadia, S.; Malins, R.; Ha, L.; Xu, X.; Daigle, I.; Markowitz, M.; O’Connor, G.; Plasterk, R.; et al. APL-1, a Caenorhabditis elegans protein related to the human β-amyloid precursor protein, is essential for viability. Proc. Natl. Acad. Sci. USA 2007, 104, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.M.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Sastre, M.; Steiner, H.; Fuchs, K.; Capell, A.; Multhaup, G.; Condron, M.M.; Teplow, D.B.; Haass, C. Presenilin-dependent gamma-secretase processing of β-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2001, 2, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Südhof, T.C. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 2001, 293, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid β-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A. Amyloidosis: A convoluted story. Br. J. Haematol. 2001, 114, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Tanskanen, M. “Amyloid”—Historical Aspects. In Amyloidosis; InTechOpen: Rijeka, Croatia, 2013. [Google Scholar]
- Virchow, R. Über eine in Gehirn und Rückenmark des Menschen aufgefundene Substanz mit der chemischen Reaction der Cellulose (in German). Virchows Arch. Pathol. Anat. Klin. Med. 1854, 6, 135–138. [Google Scholar] [CrossRef]
- Virchow, R. Über den Gang der amyloiden Degeneration (in German). Virchows Arch. Pathol. Anat. Klin. Med. 1854, 8, 364–368. [Google Scholar] [CrossRef]
- Alzheimer, A. Über eine eigenartige Erkrankung der Hirnrinde (in German). Allg. Z. Psychiatr. Psychiatr. Med. 1907, 64, 146–148. [Google Scholar]
- Divry, P. Etude histo-chimique des plaques seniles (in French). J. Belg. Neurol. Psychiatr. 1927, 27, 643–657. [Google Scholar]
- Bennhold, H. Eine spezifische Amyloidfärbung mit Kongorot (in German). Münchner Med. Wochenschr. 1922, 69, 1537–1538. [Google Scholar]
- Divry, P.; Florkin, M. Sur les proprietes optiques de l’amyloide (in French). Comptes Rendus Soc. Biol. 1927, 97, 1808–1810. [Google Scholar]
- Cohen, A.S.; Calkins, E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature 1959, 183, 1202–1203. [Google Scholar] [CrossRef] [PubMed]
- Pras, M.; Schubert, M.; Zucker-Franklin, D.; Rimon, A.; Franklin, E.C. The characterization of soluble amyloid prepared in water. J. Clin. Invest. 1968, 47, 924–933. [Google Scholar] [PubMed]
- Cohen, A.S.; Calkins, E. The isolation of amyloid fibrils and a study of the effect of collagenase and hyaluronidase. J. Cell Biol. 1964, 21, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Bladen, H.A. Purification and reconstitution of the periodic fibril and unit structure of human amyloid. Science 1966, 154, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Eanes, E.D.; Glenner, G.G. X-ray diffraction studies on amyloid filaments. J. Histochem. Cytochem. 1968, 16, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Bonar, L.; Cohen, A.S.; Skinner, M.M. Characterization of the amyloid fibril as a cross-β protein. Proc. Soc. Exp. Biol. Med. 1969, 131, 1373–1375. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Terry, W.; Harada, M.; Isersky, C.; Page, D. Amyloid fibril proteins: Proof of homology with immunoglobulin light chains by sequence analyses. Science 1971, 172, 1150–1151. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.D.; Cohen, A.S. Review: History of the amyloid fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Goldgaber, D.; Lerman, M.I.; McBride, O.W.; Saffiotti, U.; Gajdusek, D.C. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science 1987, 235, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- St George-Hyslop, P.H.; Haines, J.L.; Farrer, L.A.; Polinsky, R.; van Broeckhoven, C.; Goate, A.; McLachlan, D.R.; Orr, H.; Bruni, A.C.; Sorbi, S.; et al. FAD Collaborative Study Group Genetic linkage studies suggest that Alzheimer’s disease is not a single homogeneous disorder. Nature 1990, 347, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Amyloid β-peptide is produced by cultured cells during normal metabolism: A reprise. J. Alzheimers Dis. 2006, 9, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Schlossmacher, M.G.; Hung, A.Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B.L.; Lieberburg, I.; Koo, E.H.; Schenk, D.; Teplow, D.B. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 1992, 359, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Seubert, P.; Vigo-Pelfrey, C.; Esch, F.; Lee, M.; Dovey, H.; Davis, D.; Sinha, S.; Schlossmacher, M.; Whaley, J.; Swindlehurst, C. Isolation and quantification of soluble Alzheimer’s β-peptide from biological fluids. Nature 1992, 359, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K. Cerebrospinal fluid protein biomarkers for Alzheimer’s disease. NeuroRx 2004, 1, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.; Ghetti, B.; Benson, M.D.; Farrow, J.S.; van Nostrand, W.E.; Wagner, S.L. Low cerebrospinal-fluid concentrations of soluble amyloid β-protein precursor in hereditary Alzheimer’s disease. Lancet 1992, 340, 453–454. [Google Scholar] [CrossRef]
- Van Nostrand, W.E.; Wagner, S.L.; Shankle, W.R.; Farrow, J.S.; Dick, M.; Rozemuller, J.M.; Kuiper, M.A.; Wolters, E.C.; Zimmerman, J.; Cotman, C.W. Decreased levels of soluble amyloid β-protein precursor in cerebrospinal fluid of live Alzheimer disease patients. Proc. Natl. Acad. Sci. USA 1992, 89, 2551–2555. [Google Scholar] [CrossRef] [PubMed]
- Tabaton, M.; Nunzi, M.G.; Xue, R.; Usiak, M.; Autilio-Gambetti, L.; Gambetti, P. Soluble amyloid β-protein is a marker of Alzheimer amyloid in brain but not in cerebrospinal fluid. Biochem. Biophys. Res. Commun. 1994, 200, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, W.A.; Kuiper, M.A.; Walstra, G.J.; Wolters, E.C.; Bolhuis, P.A. Concentrations of amyloid β protein in cerebrospinal fluid of patients with Alzheimer’s disease. Ann. Neurol. 1995, 37, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Motter, R.; Vigo-Pelfrey, C.; Kholodenko, D.; Barbour, R.; Johnson-Wood, K.; Galasko, D.; Chang, L.; Miller, B.; Clark, C.; Green, R. Reduction of β-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann. Neurol. 1995, 38, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Southwick, P.C.; Yamagata, S.K.; Echols, C.L.; Higson, G.J.; Neynaber, S.A.; Parson, R.E.; Munroe, W.A. Assessment of amyloid β protein in cerebrospinal fluid as an aid in the diagnosis of Alzheimer’s disease. J. Neurochem. 1996, 66, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.; Schröder, J.; Blomberg, M.; Engvall, B.; Pantel, J.; Ida, N.; Basun, H.; Wahlund, L.O.; Werle, E.; Jauss, M.; et al. Cerebrospinal fluid A β42 is increased early in sporadic Alzheimer’s disease and declines with disease progression. Ann. Neurol. 1999, 45, 504–511. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Strozyk, D.; Blennow, K.; White, L.R.; Launer, L.J. CSF Aβ 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology 2003, 60, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Tapiola, T.; Alafuzoff, I.; Herukka, S.; Parkkinen, L.; Hartikainen, P.; Soininen, H.; Pirttilä, T. Cerebrospinal fluid β-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch. Neurol. 2009, 66, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Grimmer, T.; Riemenschneider, M.; Förstl, H.; Henriksen, G.; Klunk, W.E.; Mathis, C.A.; Shiga, T.; Wester, H.-J.; Kurz, A.; Drzezga, A. Beta amyloid in Alzheimer’s disease: Increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biol. Psychiatry 2009, 65, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Fagan, A.M.; Mintun, M.A.; Mach, R.H.; Lee, S.-Y.; Dence, C.S.; Shah, A.R.; LaRossa, G.N.; Spinner, M.L.; Klunk, W.E.; Mathis, C.A.; et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Aβ42 in humans. Ann. Neurol. 2006, 59, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Tolboom, N.; van der Flier, W.M.; Yaqub, M.; Boellaard, R.; Verwey, N.A.; Blankenstein, M.A.; Windhorst, A.D.; Scheltens, P.; Lammertsma, A.A.; van Berckel, B.N.M. Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. J. Nucl. Med. 2009, 50, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, A.; Engler, H.; Almkvist, O.; Blomquist, G.; Hagman, G.; Wall, A.; Ringheim, A.; Långström, B.; Nordberg, A. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol. Aging 2008, 29, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.-M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, K.; Takio, K.; Miura, R.; Titani, K.; Ihara, Y. Fetal-type phosphorylation of the tau in paired helical filaments. J. Neurochem. 1992, 58, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Köpke, E.; Tung, Y.C.; Shaikh, S.; Alonso, A.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [PubMed]
- Matsuo, E.S.; Shin, R.W.; Billingsley, M.L.; Van deVoorde, A.; O’Connor, M.; Trojanowski, J.Q.; Lee, V.M. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron 1994, 13, 989–1002. [Google Scholar] [CrossRef]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Papasozomenos, S.C.; Binder, L.I. Phosphorylation determines two distinct species of Tau in the central nervous system. Cell. Motil. Cytoskelet. 1987, 8, 210–226. [Google Scholar] [CrossRef] [PubMed]
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [Google Scholar] [CrossRef] [PubMed]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L.F. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Frandemiche, M.L.; De Seranno, S.; Rush, T.; Borel, E.; Elie, A.; Arnal, I.; Lanté, F.; Buisson, A. Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-β oligomers. J. Neurosci. 2014, 34, 6084–6097. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.; Bell, M.; Lyons, D.N.; Rodriguez-Rivera, J.; Ingram, A.; Fontaine, S.N.; Mechas, E.; Chen, J.; Wolozin, B.; LeVine, H.; et al. Pathological Tau Promotes Neuronal Damage by Impairing Ribosomal Function and Decreasing Protein Synthesis. J. Neurosci. 2016, 36, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Loomis, P.A.; Howard, T.H.; Castleberry, R.P.; Binder, L.I. Identification of nuclear tau isoforms in human neuroblastoma cells. Proc. Natl. Acad. Sci. USA 1990, 87, 8422–8426. [Google Scholar] [CrossRef] [PubMed]
- Poorkaj, P.; Bird, T.D.; Wijsman, E.; Nemens, E.; Garruto, R.M.; Anderson, L.; Andreadis, A.; Wiederholt, W.C.; Raskind, M.; Schellenberg, G.D. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 1998, 43, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Murrell, J.R.; Goedert, M.; Farlow, M.R.; Klug, A.; Ghetti, B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. USA 1998, 95, 7737–7741. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015, 18, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.-C.; Wang, B.Y.; Serrano-Pozo, A.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 146. [Google Scholar] [CrossRef] [PubMed]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.-L.; et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Decker, J.M.; Krüger, L.; Sydow, A.; Zhao, S.; Frotscher, M.; Mandelkow, E.; Mandelkow, E.-M. Pro-aggregant Tau impairs mossy fiber plasticity due to structural changes and Ca2+ dysregulation. Acta Neuropathol. Commun. 2015, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; Diamond, M.I. Prion-like properties of Tau protein: The importance of extracellular Tau as a therapeutic target. J. Biol. Chem. 2014, 289, 19855–19861. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell. Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Lindwall, G.; Cole, R.D. The purification of tau protein and the occurrence of two phosphorylation states of tau in brain. J. Biol. Chem. 1984, 259, 12241–12245. [Google Scholar] [PubMed]
- Lindwall, G.; Cole, R.D. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [PubMed]
- Lace, G.L.; Wharton, S.B.; Ince, P.G. A brief history of tau: The evolving view of the microtubule-associated protein tau in neurodegenerative diseases. Clin. Neuropathol. 2007, 26, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Delacourte, A.; Defossez, A. Alzheimer’s disease: Tau proteins, the promoting factors of microtubule assembly, are major components of paired helical filaments. J. Neurol. Sci. 1986, 76, 173–186. [Google Scholar] [CrossRef]
- Brion, J.P.; Flament-Durand, J.; Dustin, P. Alzheimer’s disease and tau proteins. Lancet 1986, 2, 1098. [Google Scholar] [CrossRef]
- Ihara, Y.; Nukina, N.; Miura, R.; Ogawara, M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J. Biochem. 1986, 99, 1807–1810. [Google Scholar] [CrossRef] [PubMed]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [PubMed]
- Montejo de Garcini, E.; Serrano, L.; Avila, J. Self assembly of microtubule associated protein tau into filaments resembling those found in Alzheimer disease. Biochem. Biophys. Res. Commun. 1986, 141, 790–796. [Google Scholar] [CrossRef]
- Wood, J.G.; Mirra, S.S.; Pollock, N.J.; Binder, L.I. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc. Natl. Acad. Sci. USA 1986, 83, 4040–4043. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature 1963, 197, 192–193. [Google Scholar] [CrossRef] [PubMed]
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 1986, 387, 271–280. [Google Scholar] [CrossRef]
- Wolozin, B.L.; Pruchnicki, A.; Dickson, D.W.; Davies, P. A neuronal antigen in the brains of Alzheimer patients. Science 1986, 232, 648–650. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Crowther, R.A.; Cohen, P.; Vanmechelen, E.; Vandermeeren, M.; Cras, P. Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer’s disease: Identification of phosphorylation sites in tau protein. Biochem. J. 1994, 301, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Harrington, C.R.; Mukaetova-Ladinska, E.B.; Hills, R.; Edwards, P.C.; Montejo de Garcini, E.; Novak, M.; Wischik, C.M. Measurement of distinct immunochemical presentations of tau protein in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 5842–5846. [Google Scholar] [CrossRef] [PubMed]
- Delacourte, A.; Flament, S.; Dibe, E.M.; Hublau, P.; Sablonnière, B.; Hémon, B.; Shérrer, V.; Défossez, A. Pathological proteins Tau 64 and 69 are specifically expressed in the somatodendritic domain of the degenerating cortical neurons during Alzheimer’s disease. Demonstration with a panel of antibodies against Tau proteins. Acta Neuropathol. 1990, 80, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.D.; Thal, L.; Wisniewski, H.M.; Grundke-Iqbal, I.; Iqbal, K. Paired helical filament antigen in CSF. Lancet 1985, 2, 35. [Google Scholar] [CrossRef]
- Wang, G.P.; Iqbal, K.; Bucht, G.; Winblad, B.; Wisniewski, H.M.; Grundke-Iqbal, I. Alzheimer’s disease: Paired helical filament immunoreactivity in cerebrospinal fluid. Acta Neuropathol. 1991, 82, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Davies, P. Alzheimer-related neuronal protein A68: Specificity and distribution. Ann. Neurol. 1987, 22, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Wallin, A.; Agren, H.; Spenger, C.; Siegfried, J.; Vanmechelen, E. Tau protein in cerebrospinal fluid: A biochemical marker for axonal degeneration in Alzheimer disease? Mol. Chem. Neuropathol. 1995, 26, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Van Hoesen, G.W.; Wolozin, B.L.; Davies, P.; Kromer, L.J.; Damasio, A.R. Alz-50 antibody recognizes Alzheimer-related neuronal changes. Ann. Neurol. 1988, 23, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Bancher, C.; Brunner, C.; Lassmann, H.; Budka, H.; Jellinger, K.A.; Wiche, G.; Seitelberger, F.; Grundke-Iqbal, I.; Iqbal, K.; Wisniewski, H.M. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res. 1989, 477, 90–99. [Google Scholar] [CrossRef]
- Greenberg, S.G.; Davies, P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc. Natl. Acad. Sci. USA 1990, 87, 5827–5831. [Google Scholar] [CrossRef] [PubMed]
- Vandermeeren, M.; Mercken, M.; Vanmechelen, E.; Six, J.; van de Voorde, A.; Martin, J.J.; Cras, P. Detection of tau proteins in normal and Alzheimer’s disease cerebrospinal fluid with a sensitive sandwich enzyme-linked immunosorbent assay. J. Neurochem. 1993, 61, 1828–1834. [Google Scholar] [CrossRef] [PubMed]
- Portelius, E.; Hansson, S.F.; Tran, A.J.; Zetterberg, H.; Grognet, P.; Vanmechelen, E.; Höglund, K.; Brinkmalm, G.; Westman-Brinkmalm, A.; Nordhoff, E.; et al. Characterization of tau in cerebrospinal fluid using mass spectrometry. J. Proteome Res. 2008, 7, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Hesse, C.; Rosengren, L.; Vanmechelen, E.; Vanderstichele, H.; Jensen, C.; Davidsson, P.; Blennow, K. Cerebrospinal fluid markers for Alzheimer’s disease evaluated after acute ischemic stroke. J. Alzheimers Dis. 2000, 2, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Hesse, C.; Rosengren, L.; Andreasen, N.; Davidsson, P.; Vanderstichele, H.; Vanmechelen, E.; Blennow, K. Transient increase in total tau but not phospho-tau in human cerebrospinal fluid after acute stroke. Neurosci. Lett. 2001, 297, 187–190. [Google Scholar] [CrossRef]
- Ost, M.; Nylén, K.; Csajbok, L.; Ohrfelt, A.O.; Tullberg, M.; Wikkelsö, C.; Nellgård, P.; Rosengren, L.; Blennow, K.; Nellgård, B. Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology 2006, 67, 1600–1604. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Hietala, M.A.; Jonsson, M.; Andreasen, N.; Styrud, E.; Karlsson, I.; Edman, A.; Popa, C.; Rasulzada, A.; Wahlund, L.-O.; et al. Neurochemical aftermath of amateur boxing. Arch. Neurol. 2006, 63, 1277–1280. [Google Scholar] [CrossRef] [PubMed]
- Otto, M.; Wiltfang, J.; Tumani, H.; Zerr, I.; Lantsch, M.; Kornhuber, J.; Weber, T.; Kretzschmar, H.A.; Poser, S. Elevated levels of tau-protein in cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Neurosci. Lett. 1997, 225, 210–212. [Google Scholar] [CrossRef]
- Andreasen, N.; Minthon, L.; Clarberg, A.; Davidsson, P.; Gottfries, J.; Vanmechelen, E.; Vanderstichele, H.; Winblad, B.; Blennow, K. Sensitivity, specificity, and stability of CSF-tau in AD in a community-based patient sample. Neurology 1999, 53, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Buerger, K.; Ewers, M.; Pirttilä, T.; Zinkowski, R.; Alafuzoff, I.; Teipel, S.J.; DeBernardis, J.; Kerkman, D.; McCulloch, C.; Soininen, H.; et al. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain 2006, 129, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Blom, E.S.; Giedraitis, V.; Zetterberg, H.; Fukumoto, H.; Blennow, K.; Hyman, B.T.; Irizarry, M.C.; Wahlund, L.-O.; Lannfelt, L.; Ingelsson, M. Rapid progression from mild cognitive impairment to Alzheimer’s disease in subjects with elevated levels of tau in cerebrospinal fluid and the APOE epsilon4/epsilon4 genotype. Dement. Geriatr. Cogn. Disord. 2009, 27, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Snider, B.J.; Fagan, A.M.; Roe, C.; Shah, A.R.; Grant, E.A.; Xiong, C.; Morris, J.C.; Holtzman, D.M. Cerebrospinal fluid biomarkers and rate of cognitive decline in very mild dementia of the Alzheimer type. Arch. Neurol. 2009, 66, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Sämgård, K.; Zetterberg, H.; Blennow, K.; Hansson, O.; Minthon, L.; Londos, E. Cerebrospinal fluid total tau as a marker of Alzheimer’s disease intensity. Int. J. Geriatr. Psychiatry 2010, 25, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Duits, F.H.; Teunissen, C.E.; Bouwman, F.H.; Visser, P.J.; Mattsson, N.; Zetterberg, H.; Blennow, K.; Hansson, O.; Minthon, L.; Andreasen, N.; et al. The cerebrospinal fluid “Alzheimer profile”: Easily said, but what does it mean? Alzheimers Dement. 2014, 10, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.M.; Vanderstichele, H.; Knapik-Czajka, M.; Clark, C.M.; Aisen, P.S.; Petersen, R.C.; Blennow, K.; Soares, H.; Simon, A.; Lewczuk, P.; et al. Cerebrospinal fluid biomarker signature in alzheimer’s disease neuroimaging initiative subjects. Ann. Neurol. 2009, 65, 403–413. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Duits, F.H.; Martinez-Lage, P.; Paquet, C.; Engelborghs, S.; Lleó, A.; Hausner, L.; Molinuevo, J.L.; Stomrud, E.; Farotti, L.; Holmber-Clausen, M.; et al. Performance and complications of lumbar puncture in memory clinics: Results of the multicenter lumbar puncture feasibility study. Alzheimers Dement. 2016, 12, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Vanderstichele, H.; Bibl, M.; Engelborghs, S.; Le Bastard, N.; Lewczuk, P.; Molinuevo, J.L.; Parnetti, L.; Perret-Liaudet, A.; Shaw, L.M.; Teunissen, C.; et al. Standardization of preanalytical aspects of cerebrospinal fluid biomarker testing for Alzheimer’s disease diagnosis: A consensus paper from the Alzheimer’s Biomarkers Standardization Initiative. Alzheimers Dement. 2012, 8, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Andreasson, U.; Persson, S.; Carrillo, M.C.; Collins, S.; Chalbot, S.; Cutler, N.; Dufour-Rainfray, D.; Fagan, A.M.; Heegaard, N.H.H.; et al. CSF biomarker variability in the Alzheimer’s Association quality control program. Alzheimers Dement. 2013, 9, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Molinuevo, J.L.; Blennow, K.; Dubois, B.; Engelborghs, S.; Lewczuk, P.; Perret-Liaudet, A.; Teunissen, C.E.; Parnetti, L. The clinical use of cerebrospinal fluid biomarker testing for Alzheimer’s disease diagnosis: A consensus paper from the Alzheimer’s Biomarkers Standardization Initiative. Alzheimers Dement. 2014, 10, 808–817. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Monsell, S.E.; Phillips, L.E.; Kukull, W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J. Neuropathol. Exp. Neurol. 2012, 71, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Balasa, M.; Gelpi, E.; Antonell, A.; Rey, M.J.; Sánchez-Valle, R.; Molinuevo, J.L.; Lladó, A. Clinical characteristics and APOE genotype of pathologically proven early-onset Alzheimer’s disease. Neurodegener. Dis. 2011, 8, 1720–1725. [Google Scholar]
- Lam, B.; Masellis, M.; Freedman, M.; Stuss, D.T.; Black, S.E. Clinical, imaging, and pathological heterogeneity of the Alzheimer’s disease syndrome. Alzheimers Res. Ther. 2013, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Attems, J. Prevalence of dementia disorders in the oldest-old: An autopsy study. Acta Neuropathol. 2010, 119, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Dekosky, S.T.; Barberger-Gateau, P.; Cummings, J.; Delacourte, A.; Galasko, D.; Gauthier, S.; Jicha, G.; et al. Research criteria for the diagnosis of Alzheimer’s disease: Revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007, 6, 734–746. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef]
- Johnson, K.A.; Fox, N.C.; Sperling, R.A.; Klunk, W.E. Brain imaging in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006213. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, K.; O’Bryant, S.E.; Hampel, H.; Trojanowski, J.Q.; Montine, T.J.; Jeromin, A.; Blennow, K.; Lönneborg, A.; Wyss-Coray, T.; Soares, H.; et al. Blood-Based Biomarker Interest Group The future of blood-based biomarkers for Alzheimer’s disease. Alzheimers Dement. 2014, 10, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhang, Z. Exploring Potential Electrophysiological Biomarkers in Mild Cognitive Impairment: A Systematic Review and Meta-Analysis of Event-Related Potential Studies. J. Alzheimers Dis. 2017, 58, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Yener, G.G.; Başar, E. Biomarkers in Alzheimer’s disease with a special emphasis on event-related oscillatory responses. Suppl. Clin. Neurophysiol. 2013, 62, 237–273. [Google Scholar] [PubMed]
- Scahill, R.I.; Schott, J.M.; Stevens, J.M.; Rossor, M.N.; Fox, N.C. Mapping the evolution of regional atrophy in Alzheimer’s disease: Unbiased analysis of fluid-registered serial MRI. Proc. Natl. Acad. Sci. USA 2002, 99, 4703–4707. [Google Scholar] [CrossRef] [PubMed]
- Foster, N.L.; Chase, T.N.; Fedio, P.; Patronas, N.J.; Brooks, R.A.; Di Chiro, G. Alzheimer’s disease: Focal cortical changes shown by positron emission tomography. Neurology 1983, 33, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Minoshima, S.; Giordani, B.; Berent, S.; Frey, K.A.; Foster, N.L.; Kuhl, D.E. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann. Neurol. 1997, 42, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Furst, A.J.; Rabinovici, G.D.; Rostomian, A.H.; Steed, T.; Alkalay, A.; Racine, C.; Miller, B.L.; Jagust, W.J. Cognition, glucose metabolism and amyloid burden in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Leow, A.D.; Parikshak, N.; Lee, S.; Chiang, M.-C.; Toga, A.W.; Jack, C.R.; Weiner, M.W.; Thompson, P.M. Alzheimer’s Disease Neuroimaging Initiative Tensor-based morphometry as a neuroimaging biomarker for Alzheimer’s disease: An MRI study of 676 AD, MCI, and normal subjects. Neuroimage 2008, 43, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Ridha, B.H.; Anderson, V.M.; Barnes, J.; Boyes, R.G.; Price, S.L.; Rossor, M.N.; Whitwell, J.L.; Jenkins, L.; Black, R.S.; Grundman, M.; et al. Volumetric MRI and cognitive measures in Alzheimer disease: Comparison of markers of progression. J. Neurol. 2008, 255, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Fox, N.C.; Scahill, R.I.; Crum, W.R.; Rossor, M.N. Correlation between rates of brain atrophy and cognitive decline in AD. Neurology 1999, 52, 1687–1689. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, V.A.; Chao, L.L.; Studholme, C.; Yaffe, K.; Miller, B.L.; Madison, C.; Buckley, S.T.; Mungas, D.; Schuff, N.; Weiner, M.W. Brain atrophy associated with baseline and longitudinal measures of cognition. Neurobiol. Aging 2011, 32, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Fagan, A.M.; Perrin, R.J. Upcoming candidate cerebrospinal fluid biomarkers of Alzheimer’s disease. Biomark. Med. 2012, 6, 455–476. [Google Scholar] [CrossRef] [PubMed]
- Piguet, O. Neurodegenerative disease: Frontotemporal dementia—Time to target inflammation? Nat. Rev. Neurol. 2013, 9, 304–305. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawlowski, M.; Meuth, S.G.; Duning, T. Cerebrospinal Fluid Biomarkers in Alzheimer’s Disease—From Brain Starch to Bench and Bedside. Diagnostics 2017, 7, 42. https://doi.org/10.3390/diagnostics7030042
Pawlowski M, Meuth SG, Duning T. Cerebrospinal Fluid Biomarkers in Alzheimer’s Disease—From Brain Starch to Bench and Bedside. Diagnostics. 2017; 7(3):42. https://doi.org/10.3390/diagnostics7030042
Chicago/Turabian StylePawlowski, Matthias, Sven G. Meuth, and Thomas Duning. 2017. "Cerebrospinal Fluid Biomarkers in Alzheimer’s Disease—From Brain Starch to Bench and Bedside" Diagnostics 7, no. 3: 42. https://doi.org/10.3390/diagnostics7030042
APA StylePawlowski, M., Meuth, S. G., & Duning, T. (2017). Cerebrospinal Fluid Biomarkers in Alzheimer’s Disease—From Brain Starch to Bench and Bedside. Diagnostics, 7(3), 42. https://doi.org/10.3390/diagnostics7030042