
Nephrotoxicity of Anti-Angiogenic Therapies

 ,
,  ,
,
Abstract
:1. Introduction
2. VEGF, Pro-Angiogenic Factor, Renal Expression
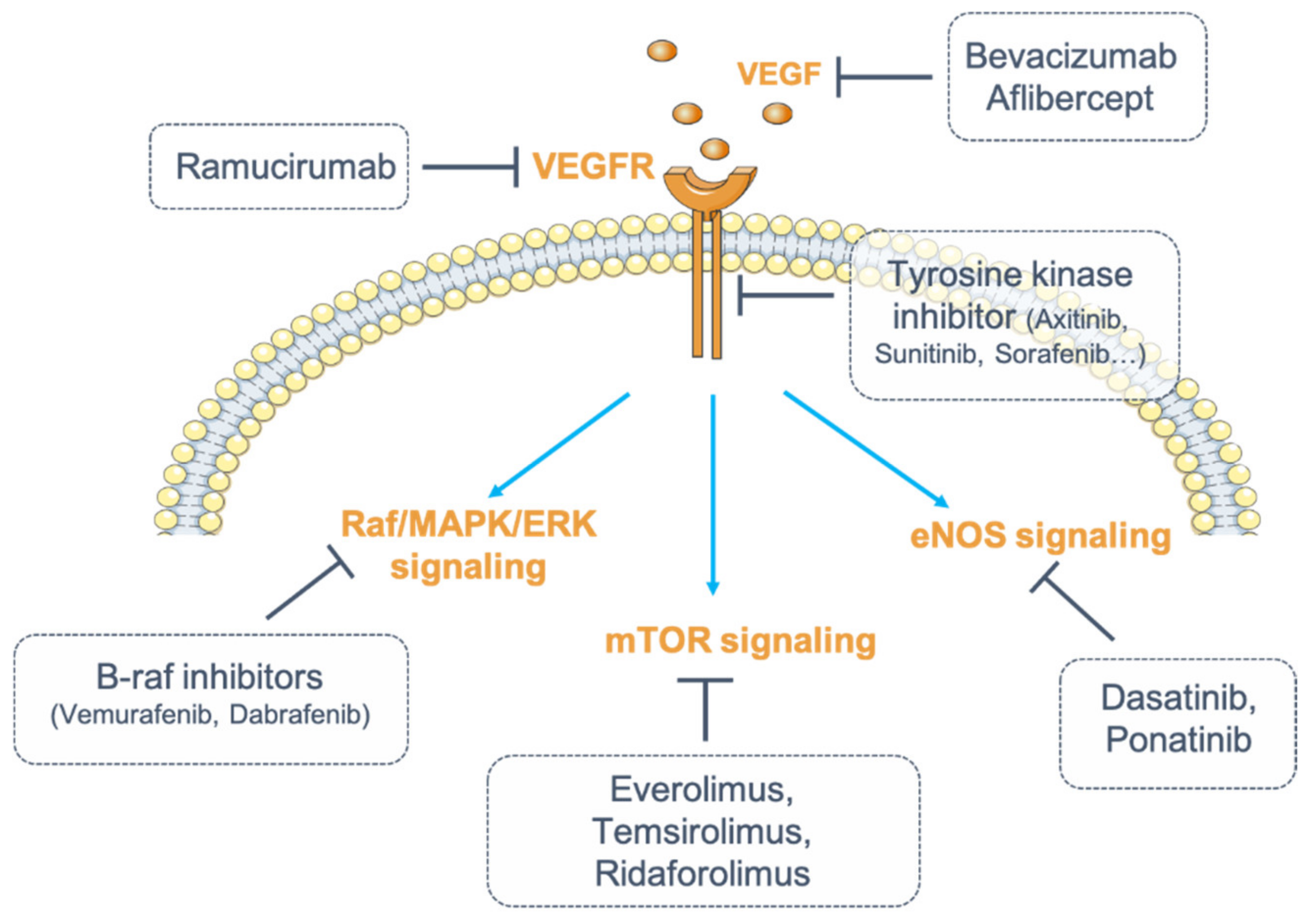
3. Anti-Angiogenic Drugs
3.1. Anti-VEGF mAb and Tyrosine Kinase Inhibitors
3.2. Other VEGF Signaling Inhibitors
3.2.1. RAF/MAPK/ERK Pathway
3.2.2. eNOS Pathway
3.2.3. mTOR Pathway
3.3. Current and Future Anti-Angiogenic Therapies
4. Adverse Effects of Anti-Angiogenic Therapy
4.1. Hypertension
4.2. Proteinuria
4.3. Kidney Dysfunction
4.4. Electrolyte Disorders
4.5. Cell Damages
4.5.1. Cytoskeleton and Focal Adhesion Architecture
4.5.2. Tubular Cell Apoptosis
4.5.3. Autophagy and mTOR Signaling
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carmeliet, P.; Jain, R.K. Angiogenesis in Cancer and Other Diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Ferrara, N.; Kerbel, R.S. Angiogenesis as a Therapeutic Target. Nature 2005, 438, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.; Mirza, M.M.; Ganti, A.K.; Tendulkar, K. Renal Toxicities of Targeted Therapies. Target. Oncol. 2015, 10, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Semeniuk-Wojtaś, A.; Lubas, A.; Stec, R.; Szczylik, C.; Niemczyk, S. Influence of Tyrosine Kinase Inhibitors on Hypertension and Nephrotoxicity in Metastatic Renal Cell Cancer Patients. Int. J. Mol. Sci. 2016, 17, 2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Feng, L.-J.; Teng, F.; Li, Y.-H.; Zhang, X.; Ran, Y.-G. Incidence and Risk of Proteinuria Associated with Newly Approved Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitors in Cancer Patients: An up-to-Date Meta-Analysis of Randomized Controlled Trials. Expert Rev. Clin. Pharmacol. 2020, 13, 311–320. [Google Scholar] [CrossRef]
- Calizo, R.C.; Bhattacharya, S.; van Hasselt, J.G.C.; Wei, C.; Wong, J.S.; Wiener, R.J.; Ge, X.; Wong, N.J.; Lee, J.-J.; Cuttitta, C.M.; et al. Disruption of Podocyte Cytoskeletal Biomechanics by Dasatinib Leads to Nephrotoxicity. Nat. Commun. 2019, 10, 2061. [Google Scholar] [CrossRef] [Green Version]
- Izzedine, H.; Mangier, M.; Ory, V.; Zhang, S.-Y.; Sendeyo, K.; Bouachi, K.; Audard, V.; Péchoux, C.; Soria, J.C.; Massard, C.; et al. Expression Patterns of RelA and C-Mip Are Associated with Different Glomerular Diseases Following Anti-VEGF Therapy. Kidney Int. 2014, 85, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Ivy, S.P.; Wick, J.Y.; Kaufman, B.M. An Overview of Small-Molecule Inhibitors of VEGFR Signaling. Nat. Rev. Clin. Oncol. 2009, 6, 569–579. [Google Scholar] [CrossRef]
- Adams, R.H.; Alitalo, K. Molecular Regulation of Angiogenesis and Lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor Angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, K.; Wada, J.; Sato, Y. Targeting Angiogenesis and Lymphangiogenesis in Kidney Disease. Nat. Rev. Nephrol. 2020, 16, 289–303. [Google Scholar] [CrossRef]
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. VEGF Inhibition and Renal Thrombotic Microangiopathy. N. Engl. J. Med. 2008, 358, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Sivaskandarajah, G.A.; Jeansson, M.; Maezawa, Y.; Eremina, V.; Baelde, H.J.; Quaggin, S.E. Vegfa Protects the Glomerular Microvasculature in Diabetes. Diabetes 2012, 61, 2958–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sison, K.; Eremina, V.; Baelde, H.; Min, W.; Hirashima, M.; Fantus, I.G.; Quaggin, S.E. Glomerular Structure and Function Require Paracrine, Not Autocrine, VEGF-VEGFR-2 Signaling. J. Am. Soc. Nephrol. JASN 2010, 21, 1691–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yue, Z.; Wu, J.; Liu, T.; Mo, Y.; Jiang, X.; Sun, L. The Accumulation of VEGFA in the Glomerular Basement Membrane and Its Relationship with Podocyte Injury and Proteinuria in Alport Syndrome. PLoS ONE 2015, 10, e0135648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veron, D.; Reidy, K.J.; Bertuccio, C.; Teichman, J.; Villegas, G.; Jimenez, J.; Shen, W.; Kopp, J.B.; Thomas, D.B.; Tufro, A. Overexpression of VEGF-A in Podocytes of Adult Mice Causes Glomerular Disease. Kidney Int. 2010, 77, 989–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller-Deile, J.; Worthmann, K.; Saleem, M.; Tossidou, I.; Haller, H.; Schiffer, M. The Balance of Autocrine VEGF-A and VEGF-C Determines Podocyte Survival. Am. J. Physiol. Renal Physiol. 2009, 297, F1656–F1667. [Google Scholar] [CrossRef]
- Ku, C.-H.; White, K.E.; Dei Cas, A.; Hayward, A.; Webster, Z.; Bilous, R.; Marshall, S.; Viberti, G.; Gnudi, L. Inducible Overexpression of SFlt-1 in Podocytes Ameliorates Glomerulopathy in Diabetic Mice. Diabetes 2008, 57, 2824–2833. [Google Scholar] [CrossRef] [Green Version]
- Bertuccio, C.; Veron, D.; Aggarwal, P.K.; Holzman, L.; Tufro, A. Vascular Endothelial Growth Factor Receptor 2 Direct Interaction with Nephrin Links VEGF-A Signals to Actin in Kidney Podocytes. J. Biol. Chem. 2011, 286, 39933–39944. [Google Scholar] [CrossRef] [Green Version]
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.-P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-Specific Alterations of VEGF-A Expression Lead to Distinct Congenital and Acquired Renal Diseases. J. Clin. Invest. 2003, 111, 707–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phipps, E.A.; Thadhani, R.; Benzing, T.; Karumanchi, S.A. Pre-Eclampsia: Pathogenesis, Novel Diagnostics and Therapies. Nat. Rev. Nephrol. 2019, 15, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.E.; Min, J.-Y.; Merchan, J.; Lim, K.-H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess Placental Soluble Fms-like Tyrosine Kinase 1 (SFlt1) May Contribute to Endothelial Dysfunction, Hypertension, and Proteinuria in Preeclampsia. J. Clin. Invest. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisler, H.; Llurba, E.; Chantraine, F.; Vatish, M.; Staff, A.C.; Sennström, M.; Olovsson, M.; Brennecke, S.P.; Stepan, H.; Allegranza, D.; et al. Predictive Value of the SFlt-1:PlGF Ratio in Women with Suspected Preeclampsia. N. Engl. J. Med. 2016, 374, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Henao, D.E.; Saleem, M.A.; Cadavid, A.P. Glomerular Disturbances in Preeclampsia: Disruption between Glomerular Endothelium and Podocyte Symbiosis. Hypertens. Pregnancy 2010, 29, 10–20. [Google Scholar] [CrossRef]
- Henao, D.E.; Saleem, M.A. Proteinuria in Preeclampsia from a Podocyte Injury Perspective. Curr. Hypertens. Rep. 2013, 15, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Tanabe, K.; Croker, B.P.; Johnson, R.J.; Grant, M.B.; Kosugi, T.; Li, Q. Endothelial Dysfunction as a Potential Contributor in Diabetic Nephropathy. Nat. Rev. Nephrol. 2011, 7, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Schrijvers, B.F.; Flyvbjerg, A.; De Vriese, A.S. The Role of Vascular Endothelial Growth Factor (VEGF) in Renal Pathophysiology. Kidney Int. 2004, 65, 2003–2017. [Google Scholar] [CrossRef] [Green Version]
- Guan, F.; Villegas, G.; Teichman, J.; Mundel, P.; Tufro, A. Autocrine VEGF-A System in Podocytes Regulates Podocin and Its Interaction with CD2AP. Am. J. Physiol. Renal Physiol. 2006, 291, F422–F428. [Google Scholar] [CrossRef]
- Ollero, M.; Sahali, D. Inhibition of the VEGF Signalling Pathway and Glomerular Disorders. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2015, 30, 1449–1455. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Ronca, R.; Benkheil, M.; Mitola, S.; Struyf, S.; Liekens, S. Tumor Angiogenesis Revisited: Regulators and Clinical Implications. Med. Res. Rev. 2017, 37, 1231–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-Inducible Factor 1 Is a Basic-Helix-Loop-Helix-PAS Heterodimer Regulated by Cellular O2 Tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J. Tumor Angiogenesis: Therapeutic Implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada, C.C.; Maldonado, A.; Mallipattu, S.K. Therapeutic Inhibition of VEGF Signaling and Associated Nephrotoxicities. J. Am. Soc. Nephrol. JASN 2019, 30, 187–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Li, A.; Yi, M.; Yu, S.; Zhang, M.; Wu, K. Recent Advances on Anti-Angiogenesis Receptor Tyrosine Kinase Inhibitors in Cancer Therapy. J. Hematol. Oncol. 2019, 12, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, R.M.; Barsoum, M.; Arman, F.; Selamet, U.; Hasnain, H.; Kurtz, I. Nephrotoxicity Induced by Intravitreal Vascular Endothelial Growth Factor Inhibitors: Emerging Evidence. Kidney Int. 2019, 96, 572–580. [Google Scholar] [CrossRef]
- Shye, M.; Hanna, R.M.; Patel, S.S.; Tram-Tran, N.; Hou, J.; Mccannel, C.; Khalid, M.; Hanna, M.; Abdelnour, L.; Kurtz, I. Worsening Proteinuria and Renal Function after Intravitreal Vascular Endothelial Growth Factor Blockade for Diabetic Proliferative Retinopathy. Clin. Kidney J. 2020, 13, 969–980. [Google Scholar] [CrossRef]
- Tewari, K.S.; Sill, M.W.; Penson, R.T.; Huang, H.; Ramondetta, L.M.; Landrum, L.M.; Oaknin, A.; Reid, T.J.; Leitao, M.M.; Michael, H.E.; et al. Bevacizumab for Advanced Cervical Cancer: Final Overall Survival and Adverse Event Analysis of a Randomised, Controlled, Open-Label, Phase 3 Trial (Gynecologic Oncology Group 240). Lancet 2017, 390, 1654–1663. [Google Scholar] [CrossRef] [Green Version]
- Gridelli, C.; de Castro Carpeno, J.; Dingemans, A.-M.C.; Griesinger, F.; Grossi, F.; Langer, C.; Ohe, Y.; Syrigos, K.; Thatcher, N.; Das-Gupta, A.; et al. Safety and Efficacy of Bevacizumab Plus Standard-of-Care Treatment Beyond Disease Progression in Patients With Advanced Non-Small Cell Lung Cancer: The AvaALL Randomized Clinical Trial. JAMA Oncol. 2018, 4, e183486. [Google Scholar] [CrossRef]
- Coleman, R.L.; Brady, M.F.; Herzog, T.J.; Sabbatini, P.; Armstrong, D.K.; Walker, J.L.; Kim, B.-G.; Fujiwara, K.; Tewari, K.S.; O’Malley, D.M.; et al. Bevacizumab and Paclitaxel-Carboplatin Chemotherapy and Secondary Cytoreduction in Recurrent, Platinum-Sensitive Ovarian Cancer (NRG Oncology/Gynecologic Oncology Group Study GOG-0213): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2017, 18, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Saito, H.; Fukuhara, T.; Furuya, N.; Watanabe, K.; Sugawara, S.; Iwasawa, S.; Tsunezuka, Y.; Yamaguchi, O.; Okada, M.; Yoshimori, K.; et al. Erlotinib plus Bevacizumab versus Erlotinib Alone in Patients with EGFR-Positive Advanced Non-Squamous Non-Small-Cell Lung Cancer (NEJ026): Interim Analysis of an Open-Label, Randomised, Multicentre, Phase 3 Trial. Lancet Oncol. 2019, 20, 625–635. [Google Scholar] [CrossRef]
- Carvalho, B.; Lopes, R.G.; Linhares, P.; Costa, A.; Caeiro, C.; Fernandes, A.C.; Tavares, N.; Osório, L.; Vaz, R. Hypertension and Proteinuria as Clinical Biomarkers of Response to Bevacizumab in Glioblastoma Patients. J. Neurooncol. 2020, 147, 109–116. [Google Scholar] [CrossRef]
- Wu, S.; Kim, C.; Baer, L.; Zhu, X. Bevacizumab Increases Risk for Severe Proteinuria in Cancer Patients. J. Am. Soc. Nephrol. JASN 2010, 21, 1381–1389. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Wang, X.; Xu, T.; Xu, X.; Liu, Z. Bevacizumab Significantly Increases the Risks of Hypertension and Proteinuria in Cancer Patients: A Systematic Review and Comprehensive Meta-Analysis. Oncotarget 2017, 8, 51492–51506. [Google Scholar] [CrossRef] [Green Version]
- Al-Samkari, H.; Kasthuri, R.S.; Parambil, J.G.; Albitar, H.A.; Almodallal, Y.A.; Vázquez, C.; Serra, M.M.; Dupuis-Girod, S.; Wilsen, C.B.; McWilliams, J.P.; et al. An International, Multicenter Study of Intravenous Bevacizumab for Bleeding in Hereditary Hemorrhagic Telangiectasia: The InHIBIT-Bleed Study. Haematologica 2020. [Google Scholar] [CrossRef]
- Kreisl, T.N.; Zhang, W.; Odia, Y.; Shih, J.H.; Butman, J.A.; Hammoud, D.; Iwamoto, F.M.; Sul, J.; Fine, H.A. A Phase II Trial of Single-Agent Bevacizumab in Patients with Recurrent Anaplastic Glioma. Neuro Oncol. 2011, 13, 1143–1150. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, M.; Thareja, N.; Benjamin, M.; Akhondi, A.; Mitchell, G.D. Tyrosine Kinase Inhibitor-Induced Hypertension. Curr. Oncol. Rep. 2018, 20, 65. [Google Scholar] [CrossRef]
- Zhang, Z.-F.; Wang, T.; Liu, L.-H.; Guo, H.-Q. Risks of Proteinuria Associated with Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitors in Cancer Patients: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e90135. [Google Scholar] [CrossRef]
- Bellini, E.; Pia, A.; Brizzi, M.P.; Tampellini, M.; Torta, M.; Terzolo, M.; Dogliotti, L.; Berruti, A. Sorafenib May Induce Hypophosphatemia through a Fibroblast Growth Factor-23 (FGF23)-Independent Mechanism. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2011, 22, 988–990. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in Advanced Clear-Cell Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Lalami, Y.; Garcia, C.; Flamen, P.; Ameye, L.; Paesmans, M.; Awada, A. Phase II Trial Evaluating the Efficacy of Sorafenib (BAY 43-9006) and Correlating Early Fluorodeoxyglucose Positron Emission Tomography–CT Response to Outcome in Patients with Recurrent and/or Metastatic Head and Neck Cancer. Head Neck 2016, 38, 347–354. [Google Scholar] [CrossRef]
- Kelley, R.K.; Nimeiri, H.S.; Munster, P.N.; Vergo, M.T.; Huang, Y.; Li, C.-M.; Hwang, J.; Mulcahy, M.F.; Yeh, B.M.; Kuhn, P.; et al. Temsirolimus Combined with Sorafenib in Hepatocellular Carcinoma: A Phase I Dose-Finding Trial with Pharmacokinetic and Biomarker Correlates. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, 1900–1907. [Google Scholar] [CrossRef] [PubMed]
- Terashima, T.; Yamashita, T.; Takata, N.; Takeda, Y.; Kido, H.; Iida, N.; Kitahara, M.; Shimakami, T.; Takatori, H.; Arai, K.; et al. Safety and Efficacy of Sorafenib Followed by Regorafenib or Lenvatinib in Patients with Hepatocellular Carcinoma. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Grignani, G.; Palmerini, E.; Ferraresi, V.; D’Ambrosio, L.; Bertulli, R.; Asaftei, S.D.; Tamburini, A.; Pignochino, Y.; Sangiolo, D.; Marchesi, E.; et al. Sorafenib and Everolimus for Patients with Unresectable High-Grade Osteosarcoma Progressing after Standard Treatment: A Non-Randomised Phase 2 Clinical Trial. Lancet Oncol. 2015, 16, 98–107. [Google Scholar] [CrossRef]
- Gounder, M.M.; Mahoney, M.R.; Van Tine, B.A.; Ravi, V.; Attia, S.; Deshpande, H.A.; Gupta, A.A.; Milhem, M.M.; Conry, R.M.; Movva, S.; et al. Sorafenib for Advanced and Refractory Desmoid Tumors. N. Engl. J. Med. 2018, 379, 2417–2428. [Google Scholar] [CrossRef]
- Ueda, T.; Uemura, H.; Tomita, Y.; Tsukamoto, T.; Kanayama, H.; Shinohara, N.; Tarazi, J.; Chen, C.; Kim, S.; Ozono, S.; et al. Efficacy and Safety of Axitinib versus Sorafenib in Metastatic Renal Cell Carcinoma: Subgroup Analysis of Japanese Patients from the Global Randomized Phase 3 AXIS Trial. Jpn. J. Clin. Oncol. 2013, 43, 616–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Arén Frontera, O.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; et al. Nivolumab plus Ipilimumab versus Sunitinib in First-Line Treatment for Advanced Renal Cell Carcinoma: Extended Follow-up of Efficacy and Safety Results from a Randomised, Controlled, Phase 3 Trial. Lancet Oncol. 2019, 20, 1370–1385. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Fujita, T.; Hirayama, T.; Ishii, D.; Matsumoto, K.; Yoshida, K.; Iwamura, M. Efficacy and Safety of Sunitinib in Elderly Patients with Advanced Renal Cell Carcinoma. Mol. Clin. Oncol. 2018, 9, 394–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Zhang, J.; Shen, K.; Hao, J.; Feng, Y.; Yuan, C.; Zhu, Y.; Ma, X. Efficacy and Safety of Perioperative Appliance of Sunitinib in Patients with Metastatic or Advanced Renal Cell Carcinoma: A Systematic Review and Meta-Analysis. Medicine 2019, 98, e15424. [Google Scholar] [CrossRef]
- Zhu, X.; Stergiopoulos, K.; Wu, S. Risk of Hypertension and Renal Dysfunction with an Angiogenesis Inhibitor Sunitinib: Systematic Review and Meta-Analysis. Acta Oncol. 2009, 48, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Ravaud, A.; Motzer, R.J.; Pandha, H.S.; George, D.J.; Pantuck, A.J.; Patel, A.; Chang, Y.-H.; Escudier, B.; Donskov, F.; Magheli, A.; et al. Adjuvant Sunitinib in High-Risk Renal-Cell Carcinoma after Nephrectomy. N. Engl. J. Med. 2016, 375, 2246–2254. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus Bevacizumab versus Sunitinib in Patients with Previously Untreated Metastatic Renal Cell Carcinoma (IMmotion151): A Multicentre, Open-Label, Phase 3, Randomised Controlled Trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kakutani, S.; Sato, Y.; Hanashi, A.; Kinoshita, Y.; Ishikawa, A. Drug Review: Pazopanib. Jpn. J. Clin. Oncol. 2018, 48, 503–513. [Google Scholar] [CrossRef]
- Toulmonde, M.; Pulido, M.; Ray-Coquard, I.; Andre, T.; Isambert, N.; Chevreau, C.; Penel, N.; Bompas, E.; Saada, E.; Bertucci, F.; et al. Pazopanib or Methotrexate-Vinblastine Combination Chemotherapy in Adult Patients with Progressive Desmoid Tumours (DESMOPAZ): A Non-Comparative, Randomised, Open-Label, Multicentre, Phase 2 Study. Lancet Oncol. 2019, 20, 1263–1272. [Google Scholar] [CrossRef]
- Motzer, R.J.; Haas, N.B.; Donskov, F.; Gross-Goupil, M.; Varlamov, S.; Kopyltsov, E.; Lee, J.L.; Melichar, B.; Rini, B.I.; Choueiri, T.K.; et al. Randomized Phase III Trial of Adjuvant Pazopanib Versus Placebo after Nephrectomy in Patients with Localized or Locally Advanced Renal Cell Carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 3916–3923. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; de Souza, P.; Merchan, J.R.; et al. Pazopanib versus Sunitinib in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef] [Green Version]
- Van der Graaf, W.T.A.; Blay, J.-Y.; Chawla, S.P.; Kim, D.-W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for Metastatic Soft-Tissue Sarcoma (PALETTE): A Randomised, Double-Blind, Placebo-Controlled Phase 3 Trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in Locally Advanced or Metastatic Renal Cell Carcinoma: Results of a Randomized Phase III Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Berardi, R.; Santoni, M.; Rinaldi, S.; Nunzi, E.; Smerilli, A.; Caramanti, M.; Morgese, F.; Torniai, M.; Savini, A.; Fiordoliva, I.; et al. Risk of Hyponatraemia in Cancer Patients Treated with Targeted Therapies: A Systematic Review and Meta-Analysis of Clinical Trials. PLoS ONE 2016, 11, e0152079. [Google Scholar] [CrossRef] [PubMed]
- Leboulleux, S.; Bastholt, L.; Krause, T.; de la Fouchardiere, C.; Tennvall, J.; Awada, A.; Gómez, J.M.; Bonichon, F.; Leenhardt, L.; Soufflet, C.; et al. Vandetanib in Locally Advanced or Metastatic Differentiated Thyroid Cancer: A Randomised, Double-Blind, Phase 2 Trial. Lancet Oncol. 2012, 13, 897–905. [Google Scholar] [CrossRef]
- Wells, S.A.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in Patients with Locally Advanced or Metastatic Medullary Thyroid Cancer: A Randomized, Double-Blind Phase III Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoh, K.; Seto, T.; Satouchi, M.; Nishio, M.; Yamamoto, N.; Murakami, H.; Nogami, N.; Matsumoto, S.; Kohno, T.; Tsuta, K.; et al. Vandetanib in Patients with Previously Treated RET-Rearranged Advanced Non-Small-Cell Lung Cancer (LURET): An Open-Label, Multicentre Phase 2 Trial. Lancet Respir. Med. 2017, 5, 42–50. [Google Scholar] [CrossRef]
- Hu, M.I.; Elisei, R.; Dedecjus, M.; Popovtzer, A.; Druce, M.; Kapiteijn, E.; Pacini, F.; Locati, L.; Krajewska, J.; Weiss, R.; et al. Safety and Efficacy of Two Starting Doses of Vandetanib in Advanced Medullary Thyroid Cancer. Endocr. Relat. Cancer 2019, 26, 241–250. [Google Scholar] [CrossRef]
- Lee, J.S.; Hirsh, V.; Park, K.; Qin, S.; Blajman, C.R.; Perng, R.-P.; Chen, Y.-M.; Emerson, L.; Langmuir, P.; Manegold, C. Vandetanib Versus Placebo in Patients with Advanced Non-Small-Cell Lung Cancer after Prior Therapy with an Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor: A Randomized, Double-Blind Phase III Trial (ZEPHYR). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 1114–1121. [Google Scholar] [CrossRef]
- Thornton, K.; Kim, G.; Maher, V.E.; Chattopadhyay, S.; Tang, S.; Moon, Y.J.; Song, P.; Marathe, A.; Balakrishnan, S.; Zhu, H.; et al. Vandetanib for the Treatment of Symptomatic or Progressive Medullary Thyroid Cancer in Patients with Unresectable Locally Advanced or Metastatic Disease: U.S. Food and Drug Administration Drug Approval Summary. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 3722–3730. [Google Scholar] [CrossRef] [Green Version]
- Rini, B.I.; Escudier, B.; Tomczak, P.; Kaprin, A.; Szczylik, C.; Hutson, T.E.; Michaelson, M.D.; Gorbunova, V.A.; Gore, M.E.; Rusakov, I.G.; et al. Comparative Effectiveness of Axitinib versus Sorafenib in Advanced Renal Cell Carcinoma (AXIS): A Randomised Phase 3 Trial. Lancet 2011, 378, 1931–1939. [Google Scholar] [CrossRef]
- Gross-Goupil, M.; Kwon, T.G.; Eto, M.; Ye, D.; Miyake, H.; Seo, S.I.; Byun, S.-S.; Lee, J.L.; Master, V.; Jin, J.; et al. Axitinib versus Placebo as an Adjuvant Treatment of Renal Cell Carcinoma: Results from the Phase III, Randomized ATLAS Trial. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 2371–2378. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; Tomczak, P.; Hutson, T.E.; Michaelson, M.D.; Negrier, S.; Oudard, S.; Gore, M.E.; Tarazi, J.; Hariharan, S.; et al. Axitinib versus Sorafenib as Second-Line Treatment for Advanced Renal Cell Carcinoma: Overall Survival Analysis and Updated Results from a Randomised Phase 3 Trial. Lancet Oncol. 2013, 14, 552–562. [Google Scholar] [CrossRef]
- Hutson, T.E.; Lesovoy, V.; Al-Shukri, S.; Stus, V.P.; Lipatov, O.N.; Bair, A.H.; Rosbrook, B.; Chen, C.; Kim, S.; Vogelzang, N.J. Axitinib versus Sorafenib as First-Line Therapy in Patients with Metastatic Renal-Cell Carcinoma: A Randomised Open-Label Phase 3 Trial. Lancet Oncol. 2013, 14, 1287–1294. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Martinelli, E.; Cascinu, S.; Sobrero, A.; Banzi, M.; Seitz, J.-F.; Barone, C.; Ychou, M.; Peeters, M.; Brenner, B.; et al. Regorafenib for Patients with Metastatic Colorectal Cancer Who Progressed After Standard Therapy: Results of the Large, Single-Arm, Open-Label Phase IIIb CONSIGN Study. Oncologist 2019, 24, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Xu, R.-H.; Qin, S.; Pan, H.; Bai, Y.; Chi, Y.; Wang, L.; Bi, F.; Cheng, Y.; Liu, T.; et al. Regorafenib in Chinese Patients with Metastatic Colorectal Cancer: Subgroup Analysis of the Phase 3 CONCUR Trial. J. Gastroenterol. Hepatol. 2020, 35, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib Monotherapy for Previously Treated Metastatic Colorectal Cancer (CORRECT): An International, Multicentre, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Apolo, A.B.; Nadal, R.; Tomita, Y.; Davarpanah, N.N.; Cordes, L.M.; Steinberg, S.M.; Cao, L.; Parnes, H.L.; Costello, R.; Merino, M.J.; et al. Cabozantinib in Patients with Platinum-Refractory Metastatic Urothelial Carcinoma: An Open-Label, Single-Centre, Phase 2 Trial. Lancet Oncol. 2020, 21, 1099–1109. [Google Scholar] [CrossRef]
- Drilon, A.; Rekhtman, N.; Arcila, M.; Wang, L.; Ni, A.; Albano, M.; Van Voorthuysen, M.; Somwar, R.; Smith, R.S.; Montecalvo, J.; et al. Cabozantinib in Patients with Advanced RET-Rearranged Non-Small-Cell Lung Cancer: An Open-Label, Single-Centre, Phase 2, Single-Arm Trial. Lancet Oncol. 2016, 17, 1653–1660. [Google Scholar] [CrossRef] [Green Version]
- Italiano, A.; Mir, O.; Mathoulin-Pelissier, S.; Penel, N.; Piperno-Neumann, S.; Bompas, E.; Chevreau, C.; Duffaud, F.; Entz-Werlé, N.; Saada, E.; et al. Cabozantinib in Patients with Advanced Ewing Sarcoma or Osteosarcoma (CABONE): A Multicentre, Single-Arm, Phase 2 Trial. Lancet Oncol. 2020, 21, 446–455. [Google Scholar] [CrossRef]
- Xu, J.; Higgins, M.J.; Tolaney, S.M.; Come, S.E.; Smith, M.R.; Fornier, M.; Mahmood, U.; Baselga, J.; Yeap, B.Y.; Chabner, B.A.; et al. A Phase II Trial of Cabozantinib in Hormone Receptor-Positive Breast Cancer with Bone Metastases. Oncologist 2020, 25, 652–660. [Google Scholar] [CrossRef]
- Okamoto, I.; Miyazaki, M.; Takeda, M.; Terashima, M.; Azuma, K.; Hayashi, H.; Kaneda, H.; Kurata, T.; Tsurutani, J.; Seto, T.; et al. Tolerability of Nintedanib (BIBF 1120) in Combination with Docetaxel: A Phase 1 Study in Japanese Patients with Previously Treated Non-Small-Cell Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2015, 10, 346–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, H.; Iwasaki, H.; Takasaki, H.; Suganuma, N.; Sakai, R.; Masudo, K.; Nakayama, H.; Rino, Y.; Masuda, M. Efficacy and Tolerability of Initial Low-Dose Lenvatinib to Treat Differentiated Thyroid Cancer. Medicine 2019, 98, e14774. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.; Shields, J.; Passero, V. Tyrosine Kinase Inhibitor-Associated Syndrome of Inappropriate Secretion of Anti-Diuretic Hormone. J. Oncol. Pharm. Pract. Off. Publ. Int. Soc. Oncol. Pharm. Pract. 2016, 22, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Lo Russo, P.; MacPherson, I.R.J.; Wang, D.; Morgan, J.A.; Brunton, V.G.; Paliwal, P.; Agrawal, S.; Voi, M.; Evans, T.R.J. Phase I Dose-Escalation and Pharmacokinetic Study of Dasatinib in Patients with Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 6232–6240. [Google Scholar] [CrossRef] [Green Version]
- Lipton, J.H.; Chuah, C.; Guerci-Bresler, A.; Rosti, G.; Simpson, D.; Assouline, S.; Etienne, G.; Nicolini, F.E.; le Coutre, P.; Clark, R.E.; et al. Ponatinib versus Imatinib for Newly Diagnosed Chronic Myeloid Leukaemia: An International, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2016, 17, 612–621. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kim, D.-W.; Pinilla-Ibarz, J.; le Coutre, P.D.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. Ponatinib Efficacy and Safety in Philadelphia Chromosome-Positive Leukemia: Final 5-Year Results of the Phase 2 PACE Trial. Blood 2018, 132, 393–404. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kim, D.-W.; Pinilla-Ibarz, J.; le Coutre, P.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. A Phase 2 Trial of Ponatinib in Philadelphia Chromosome-Positive Leukemias. N. Engl. J. Med. 2013, 369, 1783–1796. [Google Scholar] [CrossRef] [Green Version]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [Green Version]
- Perico, L.; Mandalà, M.; Schieppati, A.; Carrara, C.; Rizzo, P.; Conti, S.; Longaretti, L.; Benigni, A.; Remuzzi, G. BRAF Signaling Pathway Inhibition, Podocyte Injury, and Nephrotic Syndrome. Am. J. Kidney Dis. 2017, 70, 145–150. [Google Scholar] [CrossRef]
- Chaib, H.; Hoskins, B.E.; Ashraf, S.; Goyal, M.; Wiggins, R.C.; Hildebrandt, F. Identification of BRAF as a New Interactor of PLCepsilon1, the Protein Mutated in Nephrotic Syndrome Type 3. Am. J. Physiol. Renal Physiol. 2008, 294, F93–F99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanchoo, R.; Jhaveri, K.D.; Deray, G.; Launay-Vacher, V. Renal Effects of BRAF Inhibitors: A Systematic Review by the Cancer and the Kidney International Network. Clin. Kidney J. 2016, 9, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Feliers, D.; Chen, X.; Akis, N.; Choudhury, G.G.; Madaio, M.; Kasinath, B.S. VEGF Regulation of Endothelial Nitric Oxide Synthase in Glomerular Endothelial Cells. Kidney Int. 2005, 68, 1648–1659. [Google Scholar] [CrossRef] [Green Version]
- Quilot, F.-M.; Georges, M.; Favrolt, N.; Beltramo, G.; Foignot, C.; Grandvuillemin, A.; Montani, D.; Bonniaud, P.; Camus, P. Pulmonary Hypertension Associated with Ponatinib Therapy. Eur. Respir. J. 2016, 47, 676–679. [Google Scholar] [CrossRef] [Green Version]
- Moslehi, J.J. Cardiovascular Toxic Effects of Targeted Cancer Therapies. N. Engl. J. Med. 2016, 375, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Neves, K.B.; Montezano, A.C.; Lang, N.N.; Touyz, R.M. Vascular Toxicity Associated with Anti-Angiogenic Drugs. Clin. Sci. Lond. Engl. 1979 2020, 134, 2503–2520. [Google Scholar] [CrossRef] [PubMed]
- Moslehi, J.J.; Deininger, M. Tyrosine Kinase Inhibitor-Associated Cardiovascular Toxicity in Chronic Myeloid Leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 4210–4218. [Google Scholar] [CrossRef]
- Guba, M.; von Breitenbuch, P.; Steinbauer, M.; Koehl, G.; Flegel, S.; Hornung, M.; Bruns, C.J.; Zuelke, C.; Farkas, S.; Anthuber, M.; et al. Rapamycin Inhibits Primary and Metastatic Tumor Growth by Antiangiogenesis: Involvement of Vascular Endothelial Growth Factor. Nat. Med. 2002, 8, 128–135. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/MTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting MTOR for Cancer Therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Magaway, C.; Kim, E.; Jacinto, E. Targeting MTOR and Metabolism in Cancer: Lessons and Innovations. Cells 2019, 8, 1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comunanza, V.; Bussolino, F. Therapy for Cancer: Strategy of Combining Anti-Angiogenic and Target Therapies. Front. Cell Dev. Biol. 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination Cancer Immunotherapy and New Immunomodulatory Targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor Endothelium FasL Establishes a Selective Immune Barrier Promoting Tolerance in Tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Kudo, M. Systemic Therapy for Hepatocellular Carcinoma: Latest Advances. Cancers 2018, 10, 412. [Google Scholar] [CrossRef] [Green Version]
- Rini, B.I.; Cohen, D.P.; Lu, D.R.; Chen, I.; Hariharan, S.; Gore, M.E.; Figlin, R.A.; Baum, M.S.; Motzer, R.J. Hypertension as a Biomarker of Efficacy in Patients with Metastatic Renal Cell Carcinoma Treated with Sunitinib. J. Natl. Cancer Inst. 2011, 103, 763–773. [Google Scholar] [CrossRef] [Green Version]
- Hamnvik, O.-P.R.; Choueiri, T.K.; Turchin, A.; McKay, R.R.; Goyal, L.; Davis, M.; Kaymakcalan, M.D.; Williams, J.S. Clinical Risk Factors for the Development of Hypertension in Patients Treated with Inhibitors of the VEGF Signaling Pathway. Cancer 2015, 121, 311–319. [Google Scholar] [CrossRef]
- Zhu, X.; Wu, S.; Dahut, W.L.; Parikh, C.R. Risks of Proteinuria and Hypertension with Bevacizumab, an Antibody against Vascular Endothelial Growth Factor: Systematic Review and Meta-Analysis. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2007, 49, 186–193. [Google Scholar] [CrossRef]
- Facemire, C.S.; Nixon, A.B.; Griffiths, R.; Hurwitz, H.; Coffman, T.M. Vascular Endothelial Growth Factor Receptor 2 Controls Blood Pressure by Regulating Nitric Oxide Synthase Expression. Hypertension 2009, 54, 652–658. [Google Scholar] [CrossRef] [Green Version]
- Hayman, S.R.; Leung, N.; Grande, J.P.; Garovic, V.D. VEGF Inhibition, Hypertension, and Renal Toxicity. Curr. Oncol. Rep. 2012, 14, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Zou, A.P.; Cowley, A.W. Role of Nitric Oxide in the Control of Renal Function and Salt Sensitivity. Curr. Hypertens. Rep. 1999, 1, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Witte, J.; Lampe, J.; Koenen, A.; Urbaneck, I.; Steinbach, A.; Rettig, R.; Grisk, O. The Role of Distal Tubule and Collecting Duct Sodium Reabsorption in Sunitinib-Induced Hypertension. J. Hypertens. 2018, 36, 892–903. [Google Scholar] [CrossRef] [PubMed]
- Grisk, O.; Koenen, A.; Meissner, T.; Donner, A.; Braun, D.; Steinbach, A.; Glöckl, G.; Zimmermann, U.; Evert, K.; Evert, M.; et al. Rho Kinase Inhibition Mitigates Sunitinib-Induced Rise in Arterial Pressure and Renal Vascular Resistance but Not Increased Renal Sodium Reabsorption. J. Hypertens. 2014, 32, 2199–2210, discussion 2110. [Google Scholar] [CrossRef] [PubMed]
- Kanellis, J.; Fraser, S.; Katerelos, M.; Power, D.A. Vascular Endothelial Growth Factor Is a Survival Factor for Renal Tubular Epithelial Cells. Am. J. Physiol. Renal Physiol. 2000, 278, F905–F915. [Google Scholar] [CrossRef]
- De Jesus-Gonzalez, N.; Robinson, E.; Penchev, R.; von Mehren, M.; Heinrich, M.C.; Tap, W.; Wang, Q.; Demetri, G.; George, S.; Humphreys, B.D. Regorafenib Induces Rapid and Reversible Changes in Plasma Nitric Oxide and Endothelin-1. Am. J. Hypertens. 2012, 25, 1118–1123. [Google Scholar] [CrossRef] [Green Version]
- Kappers, M.H.W.; van Esch, J.H.M.; Sluiter, W.; Sleijfer, S.; Danser, A.H.J.; van den Meiracker, A.H. Hypertension Induced by the Tyrosine Kinase Inhibitor Sunitinib Is Associated with Increased Circulating Endothelin-1 Levels. Hypertension 2010, 56, 675–681. [Google Scholar] [CrossRef] [Green Version]
- Kappers, M.H.W.; Smedts, F.M.M.; Horn, T.; van Esch, J.H.M.; Sleijfer, S.; Leijten, F.; Wesseling, S.; Strevens, H.; Jan Danser, A.H.; van den Meiracker, A.H. The Vascular Endothelial Growth Factor Receptor Inhibitor Sunitinib Causes a Preeclampsia-like Syndrome with Activation of the Endothelin System. Hypertension 2011, 58, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Lankhorst, S.; Baelde, H.J.; Kappers, M.H.W.; Smedts, F.M.M.; Hansen, A.; Clahsen-van Groningen, M.C.; Sleijfer, S.; Mathijssen, R.H.J.; Danser, A.H.J.; van den Meiracker, A.H. Greater Sensitivity of Blood Pressure Than Renal Toxicity to Tyrosine Kinase Receptor Inhibition With Sunitinib. Hypertension 2015, 66, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Barton, M. Reversal of Proteinuric Renal Disease and the Emerging Role of Endothelin. Nat. Clin. Pract. Nephrol. 2008, 4, 490–501. [Google Scholar] [CrossRef]
- Buelli, S.; Rosanò, L.; Gagliardini, E.; Corna, D.; Longaretti, L.; Pezzotta, A.; Perico, L.; Conti, S.; Rizzo, P.; Novelli, R.; et al. β-Arrestin-1 Drives Endothelin-1-Mediated Podocyte Activation and Sustains Renal Injury. J. Am. Soc. Nephrol. JASN 2014, 25, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Lankhorst, S.; Danser, A.H.J.; van den Meiracker, A.H. Endothelin-1 and Antiangiogenesis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R230–R234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, J.; Ingram, D.A.; Haneline, L.S. Oxidative Stress Impairs Endothelial Progenitor Cell Function. Antioxid. Redox Signal. 2008, 10, 1895–1907. [Google Scholar] [CrossRef] [PubMed]
- Baffert, F.; Le, T.; Sennino, B.; Thurston, G.; Kuo, C.J.; Hu-Lowe, D.; McDonald, D.M. Cellular Changes in Normal Blood Capillaries Undergoing Regression after Inhibition of VEGF Signaling. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H547–H559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamba, T.; Tam, B.Y.Y.; Hashizume, H.; Haskell, A.; Sennino, B.; Mancuso, M.R.; Norberg, S.M.; O’Brien, S.M.; Davis, R.B.; Gowen, L.C.; et al. VEGF-Dependent Plasticity of Fenestrated Capillaries in the Normal Adult Microvasculature. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H560–H576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mourad, J.-J.; des Guetz, G.; Debbabi, H.; Levy, B.I. Blood Pressure Rise Following Angiogenesis Inhibition by Bevacizumab. A Crucial Role for Microcirculation. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2008, 19, 927–934. [Google Scholar] [CrossRef]
- Qin, C.; Cao, Q.; Li, P.; Wang, S.; Wang, J.; Wang, M.; Chu, H.; Zhou, L.; Li, X.; Ye, D.; et al. The Influence of Genetic Variants of Sorafenib on Clinical Outcomes and Toxic Effects in Patients with Advanced Renal Cell Carcinoma. Sci. Rep. 2016, 6, 20089. [Google Scholar] [CrossRef] [Green Version]
- Eechoute, K.; Van der Veldt, A.A.M.; Oosting, S.; Kappers, M.H.W.; Wessels, J.A.M.; Gelderblom, H.; Guchelaar, H.-J.; Reyners, A.K.L.; Van Herpen, C.M.L.; Haanen, J.B.; et al. Polymorphisms in Endothelial Nitric Oxide Synthase (ENOS) and Vascular Endothelial Growth Factor (VEGF) Predict Sunitinib-Induced Hypertension. Clin. Pharmacol. Ther. 2012, 92, 503–510. [Google Scholar] [CrossRef]
- Kim, J.J.; Vaziri, S.A.J.; Rini, B.I.; Elson, P.; Garcia, J.A.; Wirka, R.; Dreicer, R.; Ganapathi, M.K.; Ganapathi, R. Association of VEGF and VEGFR2 Single Nucleotide Polymorphisms with Hypertension and Clinical Outcome in Metastatic Clear Cell Renal Cell Carcinoma Patients Treated with Sunitinib. Cancer 2012, 118, 1946–1954. [Google Scholar] [CrossRef]
- Schneider, B.P.; Wang, M.; Radovich, M.; Sledge, G.W.; Badve, S.; Thor, A.; Flockhart, D.A.; Hancock, B.; Davidson, N.; Gralow, J.; et al. Association of Vascular Endothelial Growth Factor and Vascular Endothelial Growth Factor Receptor-2 Genetic Polymorphisms with Outcome in a Trial of Paclitaxel Compared with Paclitaxel plus Bevacizumab in Advanced Breast Cancer: ECOG 2100. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 4672–4678. [Google Scholar] [CrossRef]
- Sibertin-Blanc, C.; Mancini, J.; Fabre, A.; Lagarde, A.; Del Grande, J.; Levy, N.; Seitz, J.-F.; Olschwang, S.; Dahan, L. Vascular Endothelial Growth Factor A c.*237C>T Polymorphism Is Associated with Bevacizumab Efficacy and Related Hypertension in Metastatic Colorectal Cancer. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2015, 47, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Gampenrieder, S.P.; Hufnagl, C.; Brechelmacher, S.; Huemer, F.; Hackl, H.; Rinnerthaler, G.; Romeder, F.; Monzo Fuentes, C.; Morre, P.; Hauser-Kronberger, C.; et al. Endothelin-1 Genetic Polymorphism as Predictive Marker for Bevacizumab in Metastatic Breast Cancer. Pharmacogenom. J. 2017, 17, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Haworth, L.; Sherry, R.M.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Steinberg, S.M.; Chen, H.X.; Rosenberg, S.A. A Randomized Trial of Bevacizumab, an Anti-Vascular Endothelial Growth Factor Antibody, for Metastatic Renal Cancer. N. Engl. J. Med. 2003, 349, 427–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzedine, H.; Massard, C.; Spano, J.P.; Goldwasser, F.; Khayat, D.; Soria, J.C. VEGF Signalling Inhibition-Induced Proteinuria: Mechanisms, Significance and Management. Eur. J. Cancer 2010, 46, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tryggvason, K.; Patrakka, J.; Wartiovaara, J. Hereditary Proteinuria Syndromes and Mechanisms of Proteinuria. N. Engl. J. Med. 2006, 354, 1387–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Person, F.; Rinschen, M.M.; Brix, S.R.; Wulf, S.; de las Mercedes Noriega, M.; Fehrle, W.; Schmitz, J.; Schwarz, A.; Ivanyi, P.; Steinmetz, O.M.; et al. Bevacizumab-Associated Glomerular Microangiopathy. Mod. Pathol. 2019, 32, 684–700. [Google Scholar] [CrossRef]
- Verheul, H.M.W.; Pinedo, H.M. Possible Molecular Mechanisms Involved in the Toxicity of Angiogenesis Inhibition. Nat. Rev. Cancer 2007, 7, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Francia, G.; Viloria-Petit, A.; Hicklin, D.J.; du Manoir, J.; Rak, J.; Kerbel, R.S. In Vitro Procoagulant Activity Induced in Endothelial Cells by Chemotherapy and Antiangiogenic Drug Combinations: Modulation by Lower-Dose Chemotherapy. Cancer Res. 2005, 65, 5365–5373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keir, L.; Coward, R.J.M. Advances in Our Understanding of the Pathogenesis of Glomerular Thrombotic Microangiopathy. Pediatr. Nephrol. 2011, 26, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Brocklebank, V.; Wood, K.M.; Kavanagh, D. Thrombotic Microangiopathy and the Kidney. Clin. J. Am. Soc. Nephrol. CJASN 2018, 13, 300–317. [Google Scholar] [CrossRef] [Green Version]
- Keir, L.S.; Firth, R.; Aponik, L.; Feitelberg, D.; Sakimoto, S.; Aguilar, E.; Welsh, G.I.; Richards, A.; Usui, Y.; Satchell, S.C.; et al. VEGF Regulates Local Inhibitory Complement Proteins in the Eye and Kidney. J. Clin. Invest. 2017, 127, 199–214. [Google Scholar] [CrossRef] [Green Version]
- D’Agati, V.D.; Kaskel, F.J.; Falk, R.J. Focal Segmental Glomerulosclerosis. N. Engl. J. Med. 2011, 365, 2398–2411. [Google Scholar] [CrossRef] [Green Version]
- De Vriese, A.S.; Sethi, S.; Nath, K.A.; Glassock, R.J.; Fervenza, F.C. Differentiating Primary, Genetic, and Secondary FSGS in Adults: A Clinicopathologic Approach. J. Am. Soc. Nephrol. JASN 2018, 29, 759–774. [Google Scholar] [CrossRef] [Green Version]
- New, L.A.; Martin, C.E.; Scott, R.P.; Platt, M.J.; Keyvani Chahi, A.; Stringer, C.D.; Lu, P.; Samborska, B.; Eremina, V.; Takano, T.; et al. Nephrin Tyrosine Phosphorylation Is Required to Stabilize and Restore Podocyte Foot Process Architecture. J. Am. Soc. Nephrol. JASN 2016, 27, 2422–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, M.; Schwarz, K.; Kriz, W.; Miettinen, A.; Reiser, J.; Mundel, P.; Holthöfer, H. Involvement of Lipid Rafts in Nephrin Phosphorylation and Organization of the Glomerular Slit Diaphragm. Am. J. Pathol. 2001, 159, 1069–1077. [Google Scholar] [CrossRef] [Green Version]
- Hara, A.; Wada, T.; Furuichi, K.; Sakai, N.; Kawachi, H.; Shimizu, F.; Shibuya, M.; Matsushima, K.; Yokoyama, H.; Egashira, K.; et al. Blockade of VEGF Accelerates Proteinuria, via Decrease in Nephrin Expression in Rat Crescentic Glomerulonephritis. Kidney Int. 2006, 69, 1986–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrasse, M. Anti-VEGF Therapy Induces Proteinuria through Endothelial Disorganization Leading to Nephrin Decrease in Podocytes. Int. J. Immunother. Cancer Res. 2015, 021–028. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.-Y.; Kamal, M.; Dahan, K.; Pawlak, A.; Ory, V.; Desvaux, D.; Audard, V.; Candelier, M.; BenMohamed, F.; Mohamed, F.B.; et al. C-Mip Impairs Podocyte Proximal Signaling and Induces Heavy Proteinuria. Sci. Signal. 2010, 3, ra39. [Google Scholar] [CrossRef] [Green Version]
- Launay-Vacher, V.; Oudard, S.; Janus, N.; Gligorov, J.; Pourrat, X.; Rixe, O.; Morere, J.-F.; Beuzeboc, P.; Deray, G. Renal Insufficiency and Cancer Medications (IRMA) Study Group Prevalence of Renal Insufficiency in Cancer Patients and Implications for Anticancer Drug Management: The Renal Insufficiency and Anticancer Medications (IRMA) Study. Cancer 2007, 110, 1376–1384. [Google Scholar] [CrossRef]
- Rosner, M.H.; Perazella, M.A. Acute Kidney Injury in Patients with Cancer. N. Engl. J. Med. 2017, 376, 1770–1781. [Google Scholar] [CrossRef]
- Porta, C.; Bamias, A.; Danesh, F.R.; Dębska-Ślizień, A.; Gallieni, M.; Gertz, M.A.; Kielstein, J.T.; Tesarova, P.; Wong, G.; Cheung, M.; et al. KDIGO Controversies Conference on Onco-Nephrology: Understanding Kidney Impairment and Solid-Organ Malignancies, and Managing Kidney Cancer. Kidney Int. 2020, 98, 1108–1119. [Google Scholar] [CrossRef]
- Lameire, N. Nephrotoxicity of Recent Anti-Cancer Agents. Clin. Kidney J. 2014, 7, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Peng, H.; Qi, X.; Wu, M.; Zhao, X. Targeted Therapies in Gynecological Cancers: A Comprehensive Review of Clinical Evidence. Signal Transduct. Target. Ther. 2020, 5, 137. [Google Scholar] [CrossRef] [PubMed]
- Bear, H.D.; Tang, G.; Rastogi, P.; Geyer, C.E.; Robidoux, A.; Atkins, J.N.; Baez-Diaz, L.; Brufsky, A.M.; Mehta, R.S.; Fehrenbacher, L.; et al. Bevacizumab Added to Neoadjuvant Chemotherapy for Breast Cancer. N. Engl. J. Med. 2012, 366, 310–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, J.-C.; Mauguen, A.; Reck, M.; Sandler, A.B.; Saijo, N.; Johnson, D.H.; Burcoveanu, D.; Fukuoka, M.; Besse, B.; Pignon, J.-P.; et al. Systematic Review and Meta-Analysis of Randomised, Phase II/III Trials Adding Bevacizumab to Platinum-Based Chemotherapy as First-Line Treatment in Patients with Advanced Non-Small-Cell Lung Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Krishna, N.; Ravi, P.; Meyer, C.P.; Becker, A.; Dalela, D.; Sood, A.; Chun, F.K.-H.; Kibel, A.S.; Menon, M.; et al. Trends of Acute Kidney Injury after Radical or Partial Nephrectomy for Renal Cell Carcinoma. Urol. Oncol. 2016, 34, 293.e1. [Google Scholar] [CrossRef]
- Li, L.; Lau, W.L.; Rhee, C.M.; Harley, K.; Kovesdy, C.P.; Sim, J.J.; Jacobsen, S.; Chang, A.; Landman, J.; Kalantar-Zadeh, K. Risk of Chronic Kidney Disease after Cancer Nephrectomy. Nat. Rev. Nephrol. 2014, 10, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.-J. Chronic Kidney Disease. Nat. Rev. Dis. Primer 2017, 3, 17088. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Piscitani, L.; Sirolli, V.; Di Liberato, L.; Morroni, M.; Bonomini, M. Nephrotoxicity Associated with Novel Anticancer Agents (Aflibercept, Dasatinib, Nivolumab): Case Series and Nephrological Considerations. Int. J. Mol. Sci. 2020, 21, 4878. [Google Scholar] [CrossRef]
- Soo, J.Y.-C.; Jansen, J.; Masereeuw, R.; Little, M.H. Advances in Predictive in Vitro Models of Drug-Induced Nephrotoxicity. Nat. Rev. Nephrol. 2018, 14, 378–393. [Google Scholar] [CrossRef]
- Nakada, T.; Kudo, T.; Kume, T.; Kusuhara, H.; Ito, K. Estimation of Changes in Serum Creatinine and Creatinine Clearance Caused by Renal Transporter Inhibition in Healthy Subjects. Drug Metab. Pharmacokinet. 2019, 34, 233–238. [Google Scholar] [CrossRef]
- Shen, H.; Yang, Z.; Zhao, W.; Zhang, Y.; Rodrigues, A.D. Assessment of Vandetanib as an Inhibitor of Various Human Renal Transporters: Inhibition of Multidrug and Toxin Extrusion as a Possible Mechanism Leading to Decreased Cisplatin and Creatinine Clearance. Drug Metab. Dispos. Biol. Fate Chem. 2013, 41, 2095–2103. [Google Scholar] [CrossRef] [Green Version]
- Tanihara, Y.; Masuda, S.; Inui, K.-I. European Journal of Pharmaceutical Sciences: PHASCI-D-20-01517R2Inhibitory Effects of Vandetanib on Creatinine Transport via Renal Organic Cation Transporter OCT2. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2020, 105666. [Google Scholar] [CrossRef]
- Mir, O.; Coriat, R.; Boudou-Rouquette, P.; Durand, J.P.; Goldwasser, F. Sorafenib-Induced Diarrhea and Hypophosphatemia: Mechanisms and Therapeutic Implications. Ann. Oncol. 2012, 23, 280–281. [Google Scholar] [CrossRef]
- Xiao, J.; Wang, J.; Yuan, L.; Hao, L.; Wang, D. Study on the Mechanism and Intervention Strategy of Sunitinib Induced Nephrotoxicity. Eur. J. Pharmacol. 2019, 864, 172709. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Xu, L.; Shi, Y.; Zhuang, S. Podocyte Autophagy: A Potential Therapeutic Target to Prevent the Progression of Diabetic Nephropathy. J. Diabetes Res. 2017, 2017, 3560238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoni, M.; Amantini, C.; Morelli, M.B.; Liberati, S.; Farfariello, V.; Nabissi, M.; Bonfili, L.; Eleuteri, A.M.; Mozzicafreddo, M.; Burattini, L.; et al. Pazopanib and Sunitinib Trigger Autophagic and Non-Autophagic Death of Bladder Tumour Cells. Br. J. Cancer 2013, 109, 1040–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milano, V.; Piao, Y.; LaFortune, T.; de Groot, J. Dasatinib-Induced Autophagy Is Enhanced in Combination with Temozolomide in Glioma. Mol. Cancer Ther. 2009, 8, 394–406. [Google Scholar] [CrossRef] [Green Version]
- Tai, W.-T.; Shiau, C.-W.; Chen, H.-L.; Liu, C.-Y.; Lin, C.-S.; Cheng, A.-L.; Chen, P.-J.; Chen, K.-F. Mcl-1-Dependent Activation of Beclin 1 Mediates Autophagic Cell Death Induced by Sorafenib and SC-59 in Hepatocellular Carcinoma Cells. Cell Death Dis. 2013, 4, e485. [Google Scholar] [CrossRef]
- Bork, T.; Liang, W.; Yamahara, K.; Lee, P.; Tian, Z.; Liu, S.; Schell, C.; Thedieck, K.; Hartleben, B.; Patel, K.; et al. Podocytes Maintain High Basal Levels of Autophagy Independent of Mtor Signaling. Autophagy 2020, 16, 1932–1948. [Google Scholar] [CrossRef] [Green Version]
- Gödel, M.; Hartleben, B.; Herbach, N.; Liu, S.; Zschiedrich, S.; Lu, S.; Debreczeni-Mór, A.; Lindenmeyer, M.T.; Rastaldi, M.-P.; Hartleben, G.; et al. Role of MTOR in Podocyte Function and Diabetic Nephropathy in Humans and Mice. J. Clin. Investig. 2011, 121, 2197–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinà, D.P.; Onay, T.; Paltoo, A.; Li, C.; Maezawa, Y.; De Arteaga, J.; Jurisicova, A.; Quaggin, S.E. Inhibition of MTOR Disrupts Autophagic Flux in Podocytes. J. Am. Soc. Nephrol. JASN 2012, 23, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenoir, O.; Jasiek, M.; Hénique, C.; Guyonnet, L.; Hartleben, B.; Bork, T.; Chipont, A.; Flosseau, K.; Bensaada, I.; Schmitt, A.; et al. Endothelial Cell and Podocyte Autophagy Synergistically Protect from Diabetes-Induced Glomerulosclerosis. Autophagy 2015, 11, 1130–1145. [Google Scholar] [CrossRef] [PubMed]
- Halimi, J.-M.; Azizi, M.; Bobrie, G.; Bouché, O.; Deray, G.; des Guetz, G.; Lecomte, T.; Levy, B.; Mourad, J.-J.; Nochy, D.; et al. Vascular and renal effects of anti-angiogenic therapy. Nephrol. Ther. 2008, 4, 602–615. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drugs | Molecular Targets | Tumor Targets | Adverse Events (Incidence) | Electrolytic Disorders | References |
|---|---|---|---|---|---|
| Monoclonal antibodies | |||||
| Bevacizumab | VEGF | CRC, NSCLC, RCC, GBM, epithelial ovarian cancer, primary peritoneal cancer, cervical cancer, fallopian cancer, glioblastoma, ocular diseases | HTN (23–41%), Proteinuria (2–32%) | Hypophosphatemia, Hyponatremia | [38,39,40,41,42,43,44,45,46,47,48] |
| Ranibizumab | VEGF | Ocular diseases | HTN, Proteinuria | - | [38,39] |
| Ramucirumab | VEGFR2 | CRC, NSCLC, GC | HTN, Proteinuria | - | [38,39] |
| Recombinant fusion protein | |||||
| Aflibercept | VEGF | CRC, ocular diseases | HTN, Proteinuria | - | [38,39] |
| Multitargeted TKI | |||||
| Sorafenib | VEGFRs, PDGFRs, RAF, c-Kit, FLT3, Ret | RCC, HCC, DTC | HTN (17–55%), Proteinuria (10%) | Hypophosphataemia (16–85%), Hyponatremia (39%) | [49,50,51,52,53,54,55,56,57,58] |
| Sunitinib | VEGFRs, PDGFRs, FLT3, CSF1R, Ret | RCC, GIST, pNETs | HTN (22–60%), Proteinuria (10–65%) | Hypophosphatemia, Hyponatremia | [59,60,61,62,63,64,65,66] |
| Pazopanib | VEGFRs, PDGFRs, FGFR1, c-Kit | RCC, STS | HTN (40–52%), Proteinuria (13.5–18%) | Hypophosphatemia (34%), Hypocalcemia (33%), Hyponatremia (31%), Hypomagnesemia (11%) | [49,50,67,68,69,70,71,72,73] |
| Vandetanib | VEGFRs, EGFR, Ret | MTC | HTN (23.5–84%), Proteinuria (5.6–26%) | Hypomagnesemia (10–40%)Hypocalcemia (4–29%)Hypokaliemia (4–17%) | [5,50,74,75,76,77,78,79] |
| Axitinib | VEGFRs, PDGFRs, c-Kit | RCC | HTN (40–64%), Proteinuria (4.6–23%) | Hyponatremia, Hypophosphatemia (13%), Hypocalcemia (39%) | [5,49,50,80,81,82,83] |
| Regorafenib | VEGFRs, PDGFRs, FGFRs, Tie2, c-Kit, Ret, RAF | GIST, CRC, HCC | HTN (13–59%), Proteinuria (7–9.4%) | Hypophosphataemia (5–18%) | [5,49,50,55,84,85,86] |
| Cabozantib | VEGFRs, c-Met, AXL, c-Kit, FLT3, Ret | MTC, RCC | HTN (7–16), Proteinuria (6%) | Hypophosphatemia (4–8%) | [5,87,88,89,90,91] |
| Nintedanib | VEGFRs, PDGFRs, FGFRs, SRC | IPF, NSCLC | HTN, Proteinuria | - | [92] |
| Lenvatinib | VEGFRs, FGFRs, PDGFRa, Ret, c-Kit | DTC, RCC, HCC | HTN (45–100%), Proteinuria (26.9–100%) | Hypophosphatemia (45%) | [5,55,93] |
| Dasatinib | BCR-ABL, SRC, LCK, YES, FYN, c-Kit, VEGFR, PDGFR | CML, Ph+ ALL | Proteinuria | Hyponatremia | [94,95] |
| Ponatinib | VEGFRs, BCR-ABL, FLT3, Ret, c-Kit, FGFRs, PDGFR | CML, Ph+ ALL | HTN (9–32%) | - | [96,97,98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Wynsberghe, M.; Flejeo, J.; Sakhi, H.; Ollero, M.; Sahali, D.; Izzedine, H.; Henique, C. Nephrotoxicity of Anti-Angiogenic Therapies. Diagnostics 2021, 11, 640. https://doi.org/10.3390/diagnostics11040640
Van Wynsberghe M, Flejeo J, Sakhi H, Ollero M, Sahali D, Izzedine H, Henique C. Nephrotoxicity of Anti-Angiogenic Therapies. Diagnostics. 2021; 11(4):640. https://doi.org/10.3390/diagnostics11040640
Chicago/Turabian StyleVan Wynsberghe, Margaux, Joanne Flejeo, Hamza Sakhi, Mario Ollero, Dil Sahali, Hassan Izzedine, and Carole Henique. 2021. "Nephrotoxicity of Anti-Angiogenic Therapies" Diagnostics 11, no. 4: 640. https://doi.org/10.3390/diagnostics11040640