1. Introduction
Inflammatory bowel disease (IBD) is a chronic intestinal disease that is multifactorial and polygenic, resulting from the dysregulation of the mucosal immune response and intestinal microflora [
1]. Recent genome-wide association studies of IBD, including both Crohn’s disease (CD) and ulcerative colitis (UC), have led to the identification of a number of susceptibility genes or loci associated with CD and UC [
2,
3]. Over 200 distinct susceptibility loci for IBD were identified, with some contributing to a unique risk and others contributing to a combined risk for CD and UC [
2,
4]. In fact, approximately 70% of the genes are shared between CD and UC, thereby highlighting a significant genetic overlap in these disease entities [
2]. These results strengthen earlier suggestions that CD and UC are, at the molecular level, two related yet different forms of chronic intestinal disease. However, UC and CD are heterogeneous, with marked differences in disease location, behavior, clinical presentation, and response to treatment [
5,
6,
7].
Emerging challenges have now moved from gene identification to functional understanding. RNA sequencing (RNA-seq) is a technique that can examine the quantity and sequences of RNA in a sample using next-generation sequencing (NGS). It analyzes the transcriptome, which is key for connecting the information in our genome with its functional protein expression [
8]. With high-throughput omics data such as proteomics, epigenomics, metabolomics, and transcriptomics, powerful tools such as artificial intelligence, including machine learning and deep learning, are emerging to unravel novel mechanistic insights and help address unmet clinical needs in IBD, including diagnosis, disease risk assessment, and prognosis [
9].
As previous GWAS emphasized a substantial overlap in the genetic risk loci between UC and CD [
2], and previous RNA-seq transcription profiles in the specific colon segment also reflected a “common IBD” signature with a major overlap between the UC and CD [
10], the need for a new method to discriminate between UC and CD using high-throughput omics data emerged.
In this study, we profiled gene expression in inflammatory or normal mucosal samples obtained from patients with IBD using RNA-seq with the aim of differentiating between UC and CD using a machine learning model.
2. Materials and Methods
2.1. Study Population
In our study, we utilized two cohorts of patients. Patients with CD were included from the IMPACT (identification of the mechanism of the occurrence and progression of CD through integrated analysis on both genetic and environmental factors) study cohort, which is a prospective multicenter study established in Korea in 2017 [
11,
12]. A total of 16 university hospitals participated in this study, and clinical data and biological specimens (including blood, stool, and tissue specimens) of CD patients, who were newly diagnosed or followed-up within the institutions, were collected. Patients with UC were included from the UC multiomics study cohort, which is a prospective multicenter study established in Korea in 2020. A total of 14 university hospitals participated in this study, and collected clinical data and biological specimens (including blood, stool, tissue, and saliva specimens) of UC patients. Ethical approval of the present study was provided by the institutional review boards of Kangbuk Samsung Hospital (KBSMC 2016-07-029, KBSMC 2020-05-021) and each center. Written consent was obtained from all participants after the nature and possible consequences of the studies were explained.
2.2. Sample Collection
RNA-seq data were collected from the colonoscopic tissue samples of patients with CD and UC. Patients previously diagnosed with UC or CD underwent colonoscopy as part of their routine medical care. Biopsy was performed from the most severe inflammatory lesion or normal lesion. Colon tissue was assessed as inflamed or normal by the performing endoscopist based on endoscopic findings at the time of collection. Normal tissue was obtained mostly from the terminal ileum of CD patients and the rectum of UC patients when the patient’s colonoscopic findings showed endoscopic remission.
2.3. RNA Extraction, Library Construction, and Sequencing
Colon tissue for RNA-seq analysis was transferred to individual tubes containing RNA, later (Invitrogen, Waltham, MA, USA, AM7021), stored at 4 °C overnight, and then stored at −80 °C until processing. The samples were added to TRI-Reagent (Molecular Research Center, Inc., Cincinnati, OH, USA, TR118) for lysis of the tissue. The samples were homogenized, and chloroform was added to the samples containing TRI-Reagent. The samples were vortexed and centrifuged, and 70% ethanol was added to the supernatant. Total RNA was treated with the RNase-free DNase Kit (Invitrogen, AM1906) to remove contaminating DNA from the RNA preparations. Total RNA was re-suspended in 30 μL of RNA/DNA-free water.
The quality and quantity of the extracted total RNA were analyzed using an ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and a 2100 Agilent Bioanalyzer (Agilent Technologies, Waldbronn, Germany) was used to estimate the RNA integrity number (RIN) score. Samples were only used in subsequent analyses if the RNA integrity number (RIN) was greater than 5.5.
Approximately 1 μg of total RNA was used for library construction with the Illumina TruSeq Stranded Total RNA Library Prep Kit (San Diego, CA, USA). Next, paired-end sequencing was performed using the Illumina NovaSeq 6000 System, according to the manufacturer’s instructions, yielding 101-bp paired-end reads. All primary RNA sequencing data were deposited in the SRA database under the accession number PRJNA774132.
2.4. RNA Sequencing Data Analysis
The sequenced reads were mapped to the human reference genome GRCh38 using HISAT2 [
13]. The gene-level expression was quantified with featureCounts [
14] from the Subread package using the Ensembl gene model (Release 100). The read counts were processed using the Bioconductor R package edgeR [
15]. Specifically, the filtration of genes with very low counts across all samples and the calculation of sample-specific effective library sizes for normalization were performed with the default options of edgeR. For the analysis of differentially expressed genes, the negative binomial generalized linear model was fitted to each gene with naïve Bayesian dispersion and a design matrix of two factors (disease and inflammatory status), followed by a quasi-likelihood F-test comparing all four combinations: inflammatory vs. normal of each disease, as well as CD vs. UC of inflammatory or normal tissues. In addition, edgeR was used to calculate the log2-transformed normalized counts, which were input into the principal component analysis (PCA), heatmap drawing, and machine learning algorithms. The Bioconductor R package mixOmics [
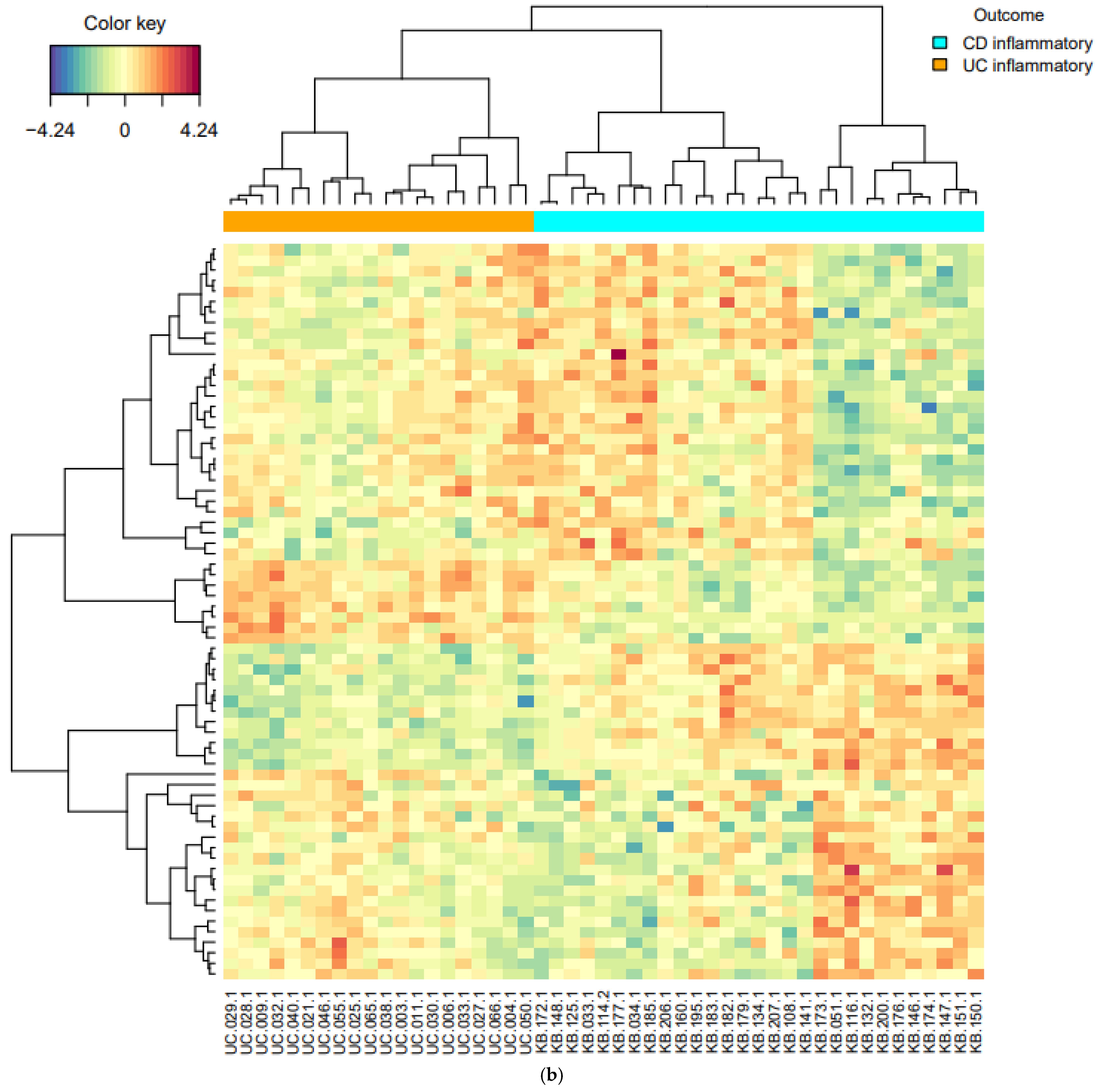
16] was used to draw the heatmaps.
2.5. Machine Learning for Prediction Model Development
The Bioconductor R package mixOmics was used for PCA and prediction model development. We applied partial least-squares discriminant analysis (PLS-DA), which is computationally efficient with rich graphical outputs, in two stages: first classifying CD normal vs. UC normal, followed by pruning strong classifiers and classifying inflammatory CD vs. inflammatory UC. The strong classifiers of normal samples were identified by calculating the loading vector of each gene along the direction connecting the class means. From the sorted list of the vector lengths, we tested three trenches that were cut into 1–3 quartiles. We confirmed that the lower quartiles, that is, those with more pruning, showed poorer performance in classifying normal CD versus normal UC.
While pruning more genes in this way worsens the classification performance of inflammatory CD vs. inflammatory UC slightly, we pruned half of the genes and used the rest in the subsequent classification of inflammatory CD vs. inflammatory UC.
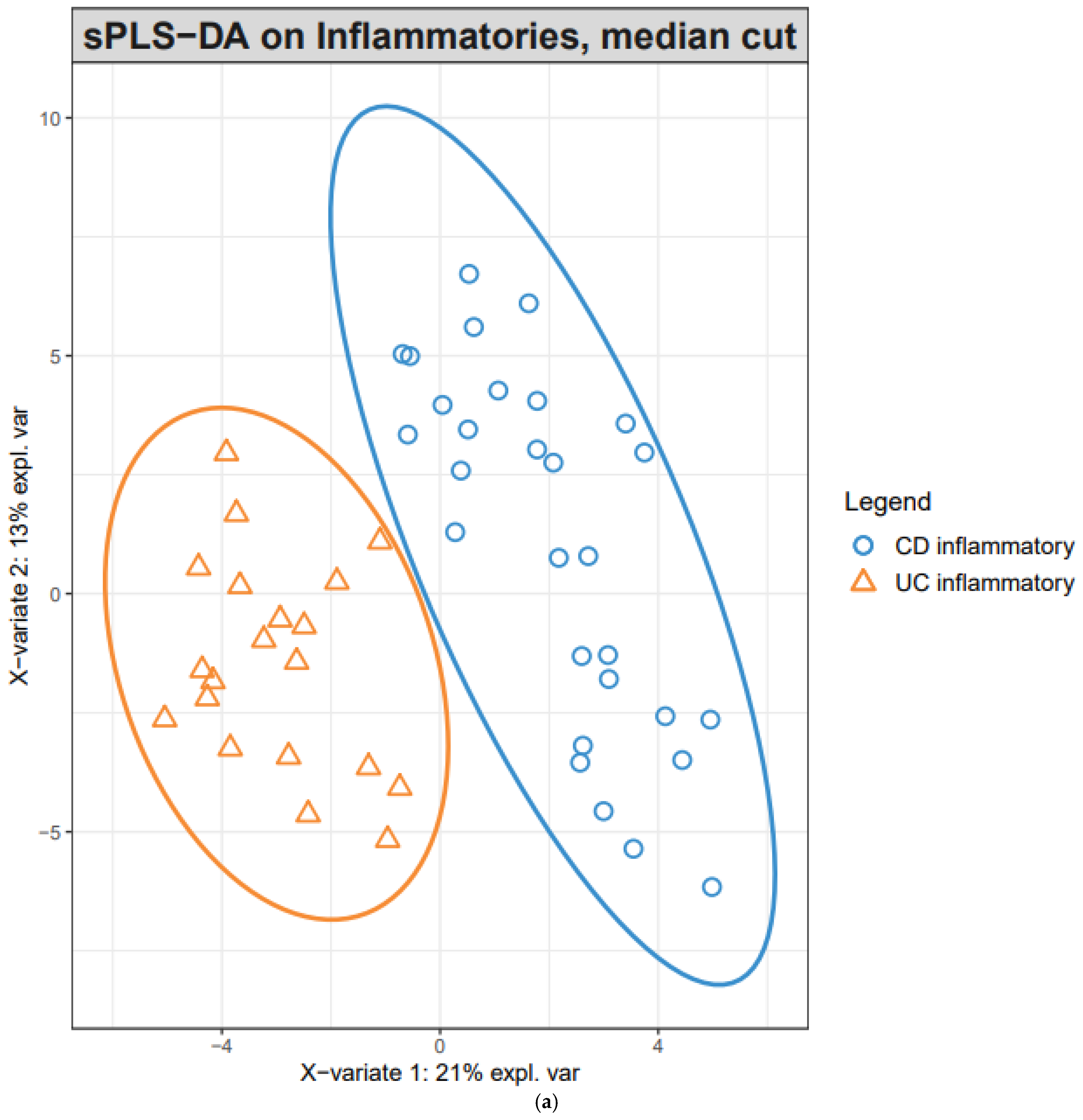
The classification performance of the PLS-DA model was assessed by a five-fold cross-validation (5-CV), which was repeated 10 times with random splits. The optimum number of components was determined from the trend of the overall error rate of 5-CV. A sparse PLS-DA model was developed for the classification of inflammatory CD vs. inflammatory UC. sPLS-DA enables the selection of the most predictive or discriminative features in the data to help classify the samples. sPLS-DA tuned the number of genes from 1 to 300 for each of the components suggested by the previous PLS-DA. The performance of this process was also evaluated using 5-CV, which was repeated 100 times with random splits. The final model was then defined using the optimum number of genes for each of the optimum number of components.
4. Discussion
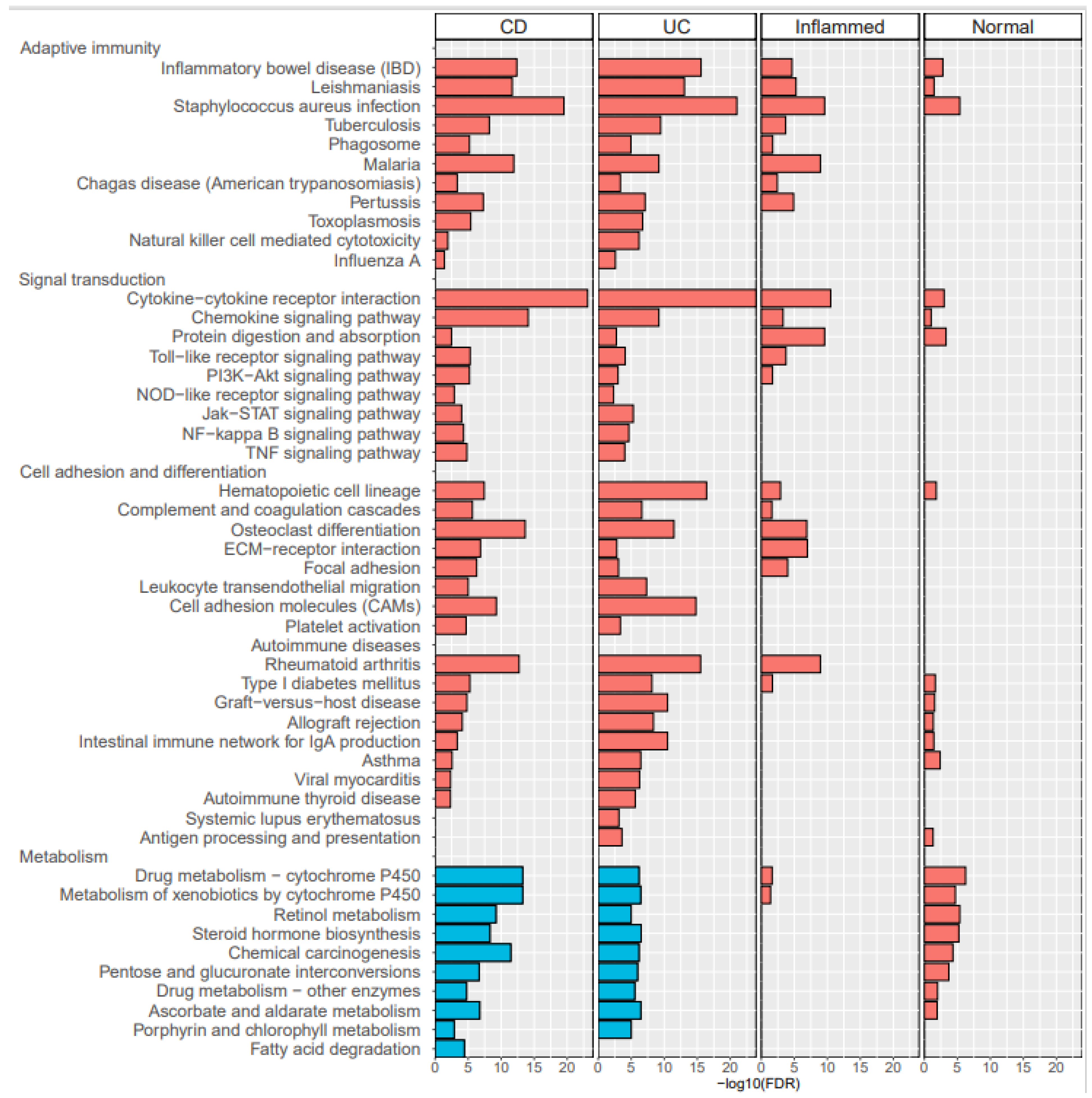
In this study, initial unsupervised analysis of mucosal RNA-seq profiles revealed highly inflammation-specific signatures and suggested disease-associated alterations in gene expression. Given the supervised learning of DEGs, common pathways in inflammatory samples are associated with IBD pathogenesis. In addition, pathways of upregulated genes in CD inflammation, compared with UC inflammation, suggested CD-specific pathogenesis. In the supervised classification model for distinguishing CD from UC, sPLS-DA revealed two components (20 and 50 genes), and inflammatory CD and inflammatory UC were well-differentiated within these two components.
Numerous previous studies led to the identification of a number of susceptibility genes or loci associated with CD and UC. Recent GWASs identified a total of 163 IBD susceptibility loci, which focus the attention on positional candidate genes involved in immunoregulation and microbial homeostasis [
2]. However, GWAS emphasized a substantial overlap in the genetic risk loci between UC and CD. To further characterize the molecular pathways underlying disease development, omics approaches such as proteomics, epigenomics, metabolomics, and transcriptomics emerged to unravel novel mechanistic insights and help address unmet clinical needs in the IBD field. Among the omics data, we used RNA-seq data and transciptomics to examine molecular pathways and to differentiate between UC and CD.
A few studies analyzed RNA-seq data in the mucosal tissues of patients with IBD. Noble et al. [
18] performed a genome-wide expression study with 67 patients with UC and 31 healthy controls (HC). When expression signals were compared between 35 inflamed and 22 non-inflamed UC biopsies, 700 genes showed a fold change greater than 1.5. In our study, 1498 genes were upregulated (>2-fold) in inflammatory UC compared with normal UC mucosa, and while some genes overlapped with Noble’s results, the list of notably upregulated genes was different. Holgersen et al. [
19] quantified the expression of 115 selected genes that were known to be linked to IBD. They investigated the inflamed mucosal tissues of 9 CD, 10 UC, and 15 HC. Although they compared inflammatory CD or UC tissue with HC, notably upregulated genes in CD, such as
IL1B,
CXCL1, and
CXCL2, were also present in our list of the genes upregulated in inflammatory CD compared with normal CD.
Howell et al. [
10] elaborated on these findings. They obtained mucosal biopsies collected from children (median age, 13 years) newly diagnosed with IBD (43 with CD, 23 with UC) and 30 HC. They investigated gut-segment-specific differences and disease-specific alterations considering inflammation status in the transcription profiles of intestinal epithelial cells (IECs). In addition to a number of genes that were found to be differentially expressed between the IBD and control samples (e.g.,
DEFA5,
DEFA6,
LYZ,
PLA2G2A,
CD40, and
CD44), ileal IECs revealed CD-specific changes in gene expression when compared with either controls or UC. However, the changes observed in the sigmoid colon reflected a “common IBD” signature, with a major overlap between the UC and CD signatures. To overcome this issue, we applied PLS-DA, a machine learning analysis to distinguish CD from UC.
Using PLS-DA we found that two components, 20 and 50 genes, can effectively discriminate CD from UC, regardless of the gut segment. Interestingly, most genes included in the first and second components did not overlap with the DEGs of inflammatory CD compared with inflammatory UC. For example, among the first 20 components, only one gene, complement C1q B chain (C1QB), was included in the DEG list of inflammatory CD vs. inflammatory UC, and the remaining 19 genes were not included. This suggests that a better discernment of the combination of genes and components specific to CD was achieved by PLS-DA.
The present result, with a relatively large IBD cohort, allowed us to gain a valuable insight into the differences in gene expression in mucosal tissue along the disease phenotype and inflammation status. In addition, as it is frequently challenging to differentiate CD from UC for those with less severe disease in real-world practice, the two components revealed by our machine learning analysis could be of clinical value. The ability to use endoscopic mucosal biopsies rather than resection samples will allow investigators to assess a larger range of patients, encompassing those with mild to moderate disease.
However, this study has several limitations. First, the predictive ability of this model was not validated in an independent cohort. Thus, further research with a multicenter IBD cohort is ongoing. Second, although UC was more likely to reveal the diseases processes resulting in the inflammation of the regional mucosa, and previous studies showed that cluster analysis presented differences in gene expression between gut segments [
18], we could not assess the relative contribution of diagnosis and inflammation to the observed variance within each data layer via the gut segments. However, before classifying inflammatory CD vs. inflammatory UC, we first classified CD normal vs. UC normal, as most of the normal samples in CD and UC were collected from the terminal ileum and rectum, respectively. Thus, in the final sPLS-DA model, the effect of the gut segment may have been minimal. Third, most genes included in the first and second components did not overlap with the DEGs of inflammatory CD compared with inflammatory UC, which was associated with the pathogenesis of CD. Thus, the mechanism of how the combination of genes can discriminate CD and UC and the role of each gene in the component needs to be further studied.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}