Molecular Diagnostic of Solid Tumor Using a Next Generation Sequencing Custom-Designed Multi-Gene Panel

 , , ,
, , ,
Abstract
:1. Introduction
2. Material and Methods
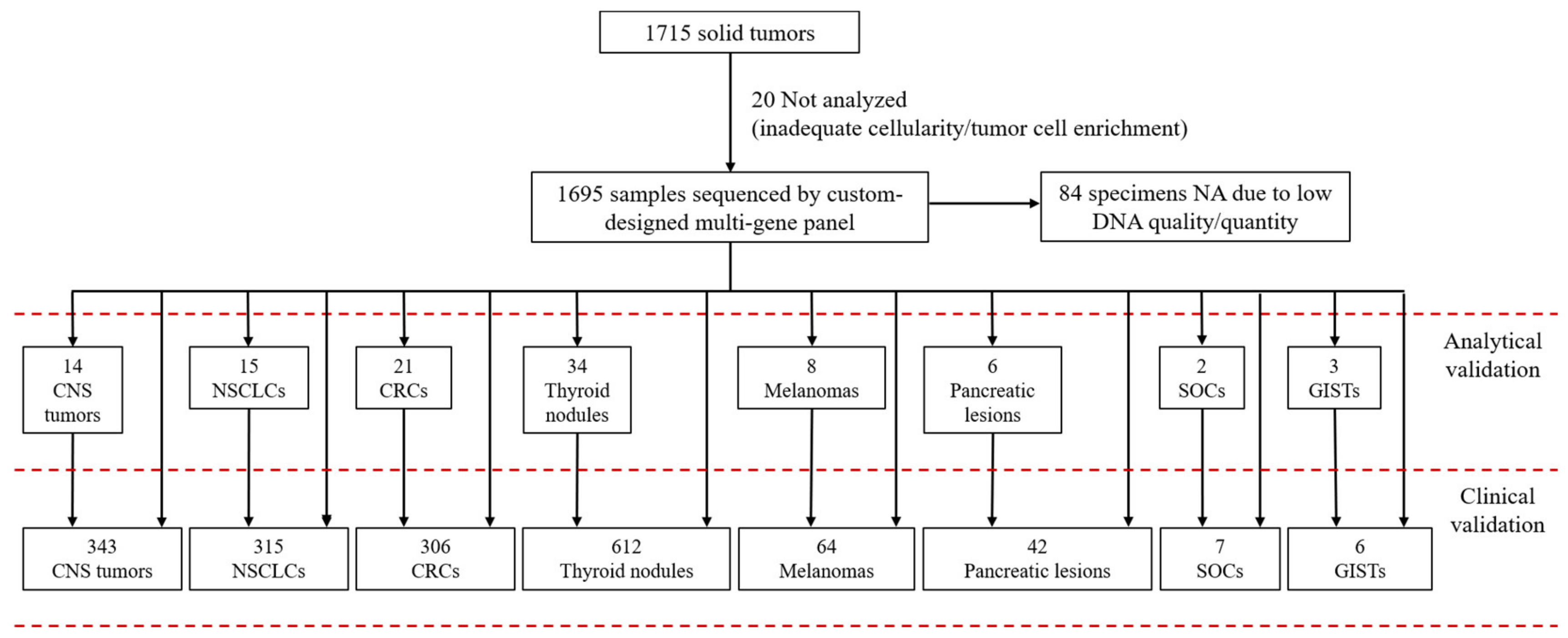
2.1. Case Selection
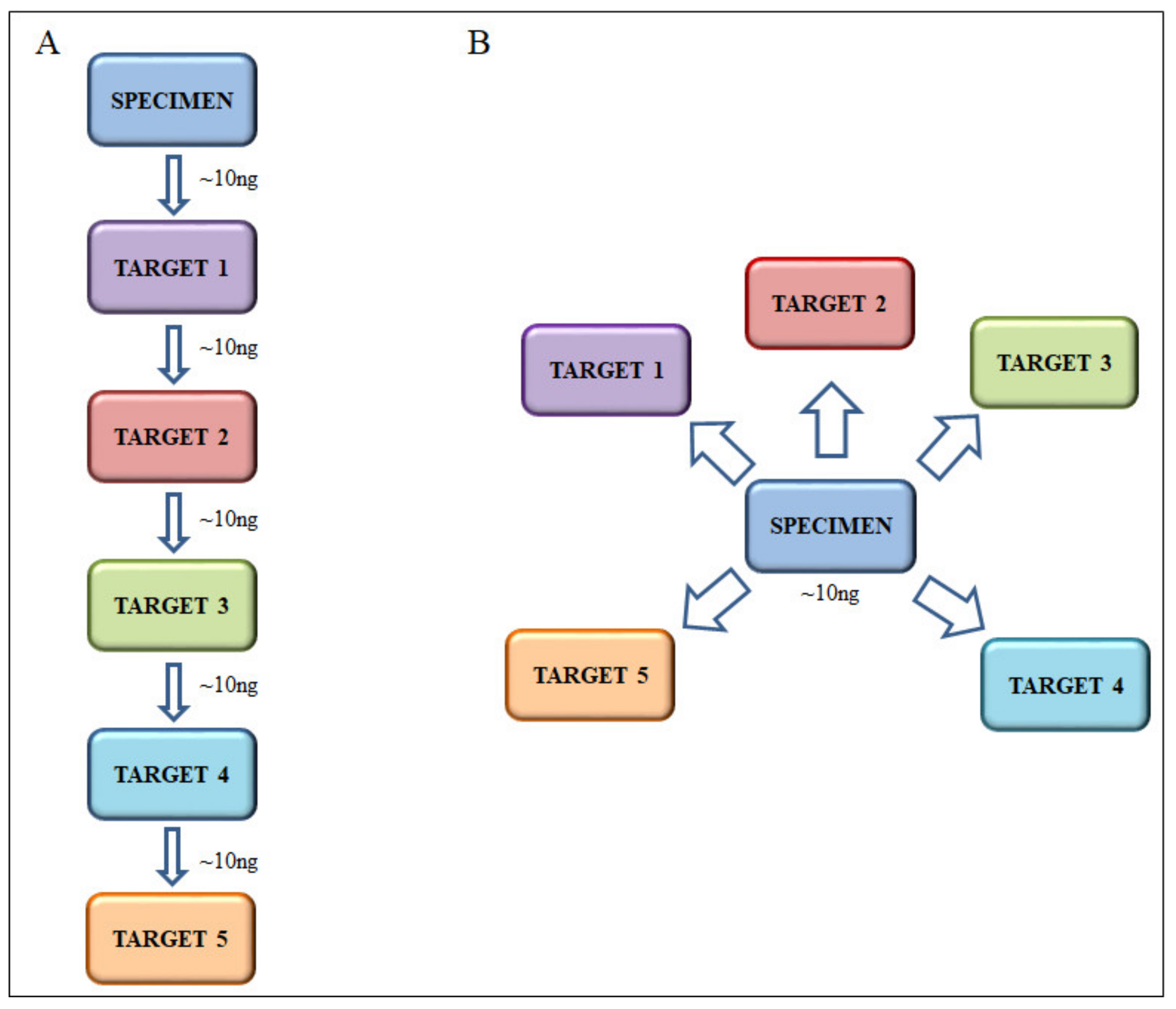
2.2. Custom-Designed Multi-Gene Panel
2.3. Pre-Analytical Evaluation
2.4. DNA Extraction and Next Generation Sequencing
3. Results
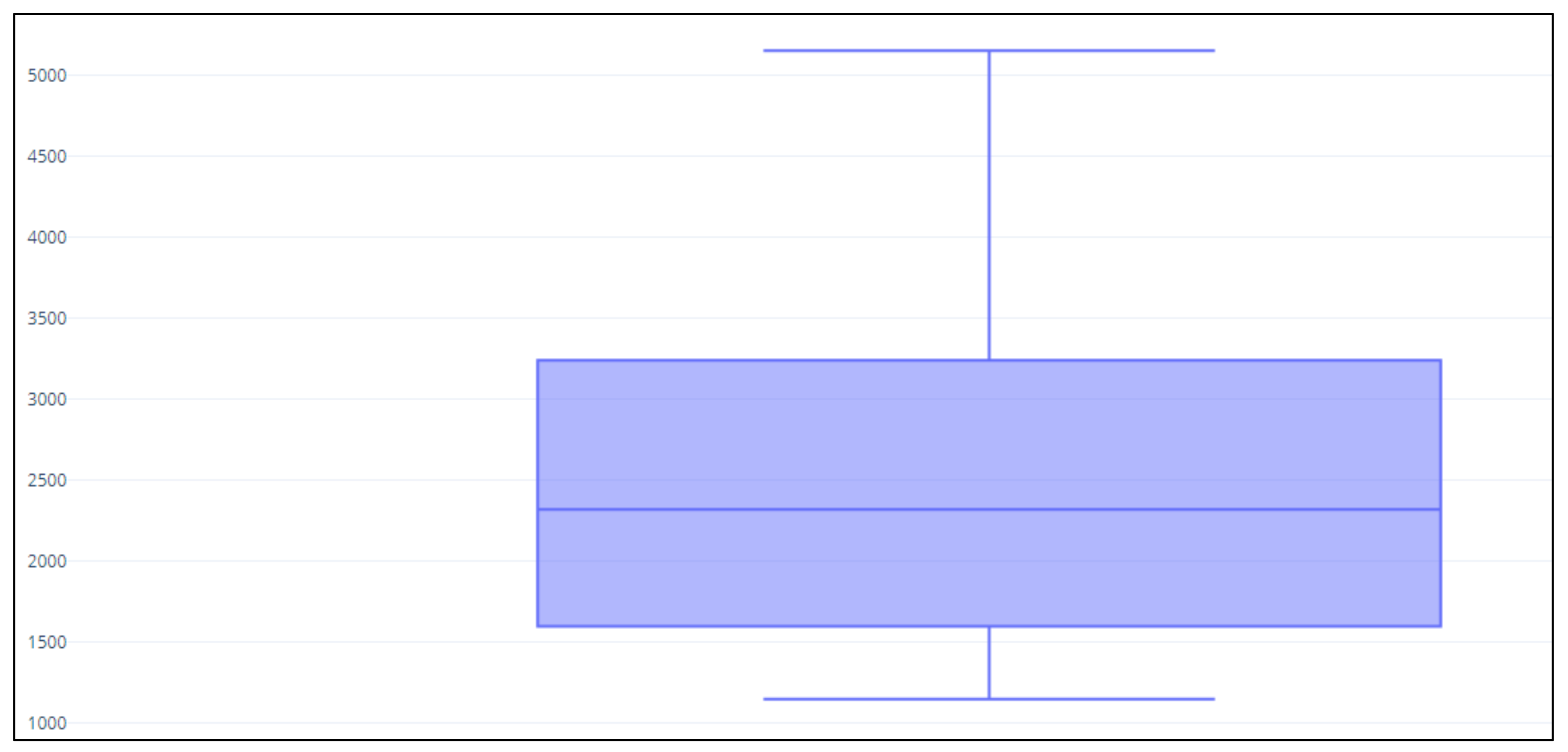
3.1. Statistical Measures of Performance
3.2. Custom-Designed Multi-Gene Panel Analytical Sensitivity
3.3. Custom-Designed Multi-Gene Panel Analytical Validation
3.4. Prospective Analysis of Routine Clinical Samples (Clinical Validation)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Forbes, S.A.; Beare, D.; Bindal, N.; Bamford, S.; Ward, S.; Cole, C.G.; Jia, M.; Kok, C.; Boutselakis, H.; De, T.; et al. COSMIC: High-Resolution Cancer Genetics Using the Catalogue of Somatic Mutations in Cancer. Curr. Protoc. Hum. Genet. 2016, 91, 10–11. [Google Scholar] [CrossRef]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, A.P.G. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducreux, M.; Cuhna, A.S.; Caramella, C.; Hollebecque, A.; Burtin, P.; Goere, D.; Seufferlein, T.; Haustermans, K.; Van Laethem, J.L.; Conroy, T.; et al. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26 (Suppl. 5), v56–v68. [Google Scholar] [CrossRef]
- Dummer, R.; Hauschild, A.; Lindenblatt, N.; Pentheroudakis, G.; Keilholz, U.; Committee, E.G. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26 (Suppl. 5), v126–v132. [Google Scholar] [CrossRef]
- Garbe, C.; Peris, K.; Hauschild, A.; Saiag, P.; Middleton, M.; Bastholt, L.; Grob, J.J.; Malvehy, J.; Newton-Bishop, J.; Stratigos, A.J.; et al. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline—Update 2016. Eur. J. Cancer 2016, 63, 201–217. [Google Scholar] [CrossRef]
- Casali, P.G.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.; Brodowicz, T.; et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv68–iv78. [Google Scholar] [CrossRef]
- Poveda, A.; Garcia Del Muro, X.; Lopez-Guerrero, J.A.; Cubedo, R.; Martinez, V.; Romero, I.; Serrano, C.; Valverde, C.; Martin-Broto, J. GEIS guidelines for gastrointestinal sarcomas (GIST). Cancer Treat. Rev. 2017, 55, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Lindeman, N.I.; Cagle, P.T.; Beasley, M.B.; Chitale, D.A.; Dacic, S.; Giaccone, G.; Jenkins, R.B.; Kwiatkowski, D.J.; Saldivar, J.S.; Squire, J.; et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: Guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J. Mol. Diagn. 2013, 15, 415–453. [Google Scholar] [CrossRef] [Green Version]
- Wells, S.A., Jr.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.P.; Treece, A.L.; Lindeman, N.I.; Vasalos, P.; Shan, M.; Jennings, L.J.; Rimm, D.L. Worldwide Frequency of Commonly Detected EGFR Mutations. Arch. Pathol. Lab. Med. 2018, 142, 163–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinoni, M.; Marucci, G.; de Biase, D.; Rubboli, G.; Volpi, L.; Riguzzi, P.; Marliani, F.; Toni, F.; Naldi, I.; Bisulli, F.; et al. BRAF V600E mutation in neocortical posterior temporal epileptogenic gangliogliomas. J. Clin. Neurosci. 2015, 22, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; de Biase, D.; Zoli, M.; Faustini-Fustini, M.; Bacci, A.; Pasquini, E.; Visani, M.; Mazzatenta, D.; Frank, G.; Tallini, G. Targeted BRAF and CTNNB1 next-generation sequencing allows proper classification of nonadenomatous lesions of the sellar region in samples with limiting amounts of lesional cells. Pituitary 2015, 18, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.D.; Lee, S.E.; Oh, D.Y.; Yu, D.B.; Jeong, H.M.; Kim, J.; Hong, S.; Jung, H.S.; Oh, E.; Song, J.Y.; et al. MET Exon 14 Skipping Mutations in Lung Adenocarcinoma: Clinicopathologic Implications and Prognostic Values. J. Thorac. Oncol. 2017, 12, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, A.R.; Hamilton, S.R.; Allegra, C.J.; Grody, W.; Cushman-Vokoun, A.M.; Funkhouser, W.K.; Kopetz, S.E.; Lieu, C.; Lindor, N.M.; Minsky, B.D.; et al. Molecular Biomarkers for the Evaluation of Colorectal Cancer: Guideline Summary From the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and American Society of Clinical Oncology. J. Oncol. Pract. 2017, 13, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, G.; de Biase, D.; Sancisi, V.; Ragazzi, M.; Acquaviva, G.; Pession, A.; Piana, S.; Tallini, G.; Ciarrocchi, A. Deep sequencing of KIT, MET, PIK3CA, and PTEN hotspots in papillary thyroid carcinomas with distant metastases. Endocr. Relat. Cancer 2014, 21, L23–L26. [Google Scholar] [CrossRef] [Green Version]
- Acquaviva, G.; Visani, M.; Repaci, A.; Rhoden, K.J.; de Biase, D.; Pession, A.; Giovanni, T. Molecular pathology of thyroid tumours of follicular cells: A review of genetic alterations and their clinicopathological relevance. Histopathology 2018, 72, 6–31. [Google Scholar] [CrossRef]
- Ibrahimpasic, T.; Xu, B.; Landa, I.; Dogan, S.; Middha, S.; Seshan, V.; Deraje, S.; Carlson, D.L.; Migliacci, J.; Knauf, J.A.; et al. Genomic Alterations in Fatal Forms of Non-Anaplastic Thyroid Cancer: Identification of MED12 and RBM10 as Novel Thyroid Cancer Genes Associated with Tumor Virulence. Clin. Cancer Res. 2017, 23, 5970–5980. [Google Scholar] [CrossRef] [Green Version]
- Karunamurthy, A.; Panebianco, F.; Hsiao, S.J.; Vorhauer, J.; Nikiforova, M.N.; Chiosea, S.; Nikiforov, Y.E. Prevalence and phenotypic correlations of EIF1AX mutations in thyroid nodules. Endocr. Relat. Cancer 2016, 23, 295–301. [Google Scholar] [CrossRef] [Green Version]
- De Biase, D.; Gandolfi, G.; Ragazzi, M.; Eszlinger, M.; Sancisi, V.; Gugnoni, M.; Visani, M.; Pession, A.; Casadei, G.; Durante, C.; et al. TERT Promoter Mutations in Papillary Thyroid Microcarcinomas. Thyroid 2015, 25, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- De Biase, D.; Torricelli, F.; Ragazzi, M.; Donati, B.; Kuhn, E.; Visani, M.; Acquaviva, G.; Pession, A.; Tallini, G.; Piana, S.; et al. Not the same thing: Metastatic PTCs have a different background than ATCs. Endocr. Connect. 2018, 7, 1370–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandolfi, G.; Ragazzi, M.; de Biase, D.; Visani, M.; Zanetti, E.; Torricelli, F.; Sancisi, V.; Gugnoni, M.; Manzotti, G.; Braglia, L.; et al. Genome-wide profiling identifies the THYT1 signature as a distinctive feature of widely metastatic Papillary Thyroid Carcinomas. Oncotarget 2018, 9, 1813–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Kim, Y.; Choi, J.W.; Kim, Y.S. KRAS, GNAS, and RNF43 mutations in intraductal papillary mucinous neoplasm of the pancreas: A meta-analysis. Springerplus 2016, 5, 1172. [Google Scholar] [CrossRef] [Green Version]
- De Biase, D.; Visani, M.; Acquaviva, G.; Fornelli, A.; Masetti, M.; Fabbri, C.; Pession, A.; Tallini, G. The Role of Next-Generation Sequencing in the Cytologic Diagnosis of Pancreatic Lesions. Arch. Pathol. Lab. Med. 2018, 142, 458–464. [Google Scholar] [CrossRef] [Green Version]
- Altimari, A.; de Biase, D.; De Maglio, G.; Gruppioni, E.; Capizzi, E.; Degiovanni, A.; D’Errico, A.; Pession, A.; Pizzolitto, S.; Fiorentino, M.; et al. 454 next generation-sequencing outperforms allele-specific PCR, Sanger sequencing, and pyrosequencing for routine KRAS mutation analysis of formalin-fixed, paraffin-embedded samples. OncoTargets Ther. 2013, 6, 1057–1064. [Google Scholar] [CrossRef] [Green Version]
- De Biase, D.; Visani, M.; Malapelle, U.; Simonato, F.; Cesari, V.; Bellevicine, C.; Pession, A.; Troncone, G.; Fassina, A.; Tallini, G. Next-generation sequencing of lung cancer EGFR exons 18–21 allows effective molecular diagnosis of small routine samples (cytology and biopsy). PLoS ONE 2013, 8, e83607. [Google Scholar] [CrossRef] [Green Version]
- Masetti, M.; Acquaviva, G.; Visani, M.; Tallini, G.; Fornelli, A.; Ragazzi, M.; Vasuri, F.; Grifoni, D.; Di Giacomo, S.; Fiorino, S.; et al. Long-term survivors of pancreatic adenocarcinoma show low rates of genetic alterations in KRAS, TP53 and SMAD4. Cancer Biomark. 2018, 21, 323–334. [Google Scholar] [CrossRef]
- Ragazzi, M.; Tamagnini, I.; Bisagni, A.; Cavazza, A.; Pagano, M.; Baldi, L.; Boni, C.; Cantile, F.; Barbieri, F.; Nicoli, D.; et al. Diamond: Immunohistochemistry versus sequencing in EGFR analysis of lung adenocarcinomas. J. Clin. Pathol. 2016, 69, 440–447. [Google Scholar] [CrossRef]
- Malapelle, U.; Mayo-de-Las-Casas, C.; Molina-Vila, M.A.; Rosell, R.; Savic, S.; Bihl, M.; Bubendorf, L.; Salto-Tellez, M.; de Biase, D.; Tallini, G.; et al. Consistency and reproducibility of next-generation sequencing and other multigene mutational assays: A worldwide ring trial study on quantitative cytological molecular reference specimens. Cancer Cytopathol. 2017, 125, 615–626. [Google Scholar] [CrossRef]
- Pisapia, P.; Malapelle, U.; Roma, G.; Saddar, S.; Zheng, Q.; Pepe, F.; Bruzzese, D.; Vigliar, E.; Bellevicine, C.; Luthra, R.; et al. Consistency and reproducibility of next-generation sequencing in cytopathology: A second worldwide ring trial study on improved cytological molecular reference specimens. Cancer Cytopathol. 2019, 127, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Visani, M.; Acquaviva, G.; Marucci, G.; Paccapelo, A.; Mura, A.; Franceschi, E.; Grifoni, D.; Pession, A.; Tallini, G.; Brandes, A.A.; et al. Non-canonical IDH1 and IDH2 mutations: A clonal and relevant event in an Italian cohort of gliomas classified according to the 2016 World Health Organization (WHO) criteria. J. Neurooncol. 2017, 135, 245–254. [Google Scholar] [CrossRef] [PubMed]
- El Osta, B.; Behera, M.; Kim, S.; Berry, L.D.; Sica, G.; Pillai, R.N.; Owonikoko, T.K.; Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J.; et al. Characteristics and Outcomes of Patients with Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. 2019, 14, 876–889. [Google Scholar] [CrossRef]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular epidemiology of EGFR and KRAS mutations in 3026 lung adenocarcinomas: Higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Biase, D.; Visani, M.; Baccarini, P.; Polifemo, A.M.; Maimone, A.; Fornelli, A.; Giuliani, A.; Zanini, N.; Fabbri, C.; Pession, A.; et al. Next generation sequencing improves the accuracy of KRAS mutation analysis in endoscopic ultrasound fine needle aspiration pancreatic lesions. PLoS ONE 2014, 9, e87651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Biase, D.; de Luca, C.; Gragnano, G.; Visani, M.; Bellevicine, C.; Malapelle, U.; Tallini, G.; Troncone, G. Fully automated PCR detection of KRAS mutations on pancreatic endoscopic ultrasound fine-needle aspirates. J. Clin. Pathol. 2016. [Google Scholar] [CrossRef]
- Kim, S.W.; Lee, J.I.; Kim, J.W.; Ki, C.S.; Oh, Y.L.; Choi, Y.L.; Shin, J.H.; Kim, H.K.; Jang, H.W.; Chung, J.H. BRAFV600E mutation analysis in fine-needle aspiration cytology specimens for evaluation of thyroid nodule: A large series in a BRAFV600E-prevalent population. J. Clin. Endocrinol. Metab. 2010, 95, 3693–3700. [Google Scholar] [CrossRef] [Green Version]
- Hemalatha, R.; Pai, R.; Manipadam, M.T.; Rebekah, G.; Cherian, A.J.; Abraham, D.T.; Rajaratnam, S.; Thomas, N.; Ramakant, P.; Jacob, P.M. Presurgical Screening of Fine Needle Aspirates from Thyroid Nodules for BRAF Mutations: A Prospective Single Center Experience. Indian J. Endocrinol. Metab. 2018, 22, 785–792. [Google Scholar] [CrossRef]
- Radkay, L.A.; Chiosea, S.I.; Seethala, R.R.; Hodak, S.P.; LeBeau, S.O.; Yip, L.; McCoy, K.L.; Carty, S.E.; Schoedel, K.E.; Nikiforova, M.N.; et al. Thyroid nodules with KRAS mutations are different from nodules with NRAS and HRAS mutations with regard to cytopathologic and histopathologic outcome characteristics. Cancer Cytopathol. 2014, 122, 873–882. [Google Scholar] [CrossRef]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J. Mol. Diagn. 2015, 17, 251–264. [Google Scholar] [CrossRef]
- Malapelle, U.; Pepe, F.; Pisapia, P.; Sgariglia, R.; Nacchio, M.; De Luca, C.; Lacalamita, R.; Tommasi, S.; Pinto, R.; Palomba, G.; et al. Harmonization of Next-Generation Sequencing Procedure in Italian Laboratories: A Multi-Institutional Evaluation of the SiRe(R) Panel. Front. Oncol. 2020, 10, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Censi, S.; Cavedon, E.; Bertazza, L.; Galuppini, F.; Watutantrige-Fernando, S.; De Lazzari, P.; Nacamulli, D.; Pennelli, G.; Fassina, A.; Iacobone, M.; et al. Frequency and Significance of Ras, Tert Promoter, and Braf Mutations in Cytologically Indeterminate Thyroid Nodules: A Monocentric Case Series at a Tertiary-Level Endocrinology Unit. Front. Endocrinol. 2017, 8, 273. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Khan, H.A.; Irfan, O.; Samad, A.; Mirza, Y.; Awan, M.S. Correlation of TP53 Overexpression and Clinical Parameters with Five-Year Survival in Oral Squamous Cell Carcinoma Patients. Cureus 2017, 9, e1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Tested (Number of Samples) (Total Samples: n = 53) | 454 GS-Junior Results | MiSeq Results |
|---|---|---|
| KRAS (n = 19) | 3 KRAS p.G12D | 3 KRAS p.G12D |
| 2 KRAS p.G13D | 2 KRAS p.G13D | |
| 1 KRAS p.Q61K | 1 KRAS p.Q61K | |
| 1 KRAS p.Q61R | 1 KRAS p.Q61R | |
| 12 WT | 12 WT | |
| NRAS (n = 11) | 2 NRAS p.Q61R | 2 NRAS p.Q61R |
| 9 WT | 9 WT | |
| BRAF (n = 6) | 1 BRAF p.V600E | 1 BRAF p.V600E |
| 1 BRAF p.V600K | 1 BRAF p.V600K | |
| 4 WT | 4 WT | |
| HRAS (n = 4) | 1 HRAS p.Q61R | 1 HRAS p.Q61R |
| 3 WT | 3 WT | |
| EGFR (n = 6) | 1 EGFR p.E746_A750delELREA | 1 EGFR p.E746_A750delELREA |
| 2 EGFR p.L858R | 2 EGFR p.L858R | |
| 3 WT | 3 WT | |
| IDH1/IDH2 (n = 5) | 3 IDH1 p.R132H | 3 IDH1 p.R132H |
| 1 IDH1 p.R132S | 1 IDH1 p.R132S | |
| 1 WT | 1 WT | |
| c-kit/PDGFRa (n = 1) | 1 c-Kit p.V599D | 1 c-Kit p.V599D |
| RET (n = 1) | 1 WT | 1 WT |
| Mutational Status MiSeq | % Mutated Allele | Mutational Status GeneStudio S5 | % Mutated Allele |
|---|---|---|---|
| IDH1 p.R132C | 23 | IDH1 p.R132C | 24 |
| KRAS p.G12S | 43 | KRAS p.G12S | 42 |
| KRAS p.G12R | 14 | KRAS p.G12R | 13 |
| SMAD4 p.E526K | 22 | SMAD4 p.E526K | 19 |
| TP53 p.R175H | 31 | TP53 p.R175H | 32 |
| TP53 p.R306Ter | 48 | TP53 p.R306Ter | 51 |
| c-Kit p.W557_K558del | 51 | c-Kit p.W557_K558del | 44 |
| c-Kit p.A502_Y503dup | 41 | c-Kit p.A502_Y503dup | 32 |
| BRAF p.V600E | 29 | BRAF p.V600E | 23 |
| EGFR p.E746_A750delELREA | 65 | EGFR p.E746_A750delELREA | 48 |
| EGFR E746_S752delELREATSinsV | 49 | EGFR p.E746_S752delELREATSinsV | 51 |
| H3F3A p.K28M | 90 | H3F3A p.K28M | 88 |
| CTNNB1 p.S37C | 35 | CTNNB1 p.S37C | 40 |
| RET p.E768D | 51 | RET p.E768D | 51 |
| 16 Samples without mutations | / | 16 WT | / |
| Gene Tested (Number of Samples) (Total Samples: n = 20) | Results-Analysis 1 | Results-Analysis 2 | ||||
|---|---|---|---|---|---|---|
| Mutational Status | Mutated Reads % | Coverage (Reads) ^ | Mutational Status | Mutated Reads % | Coverage (Reads) ^ | |
| BRAF (n = 4) | 1 p.V600E | 15% | 3800× | 1 p.V600E | 7% | 3000× |
| 1 p.V600E | 32% | 3400× | 1 p.V600E | 18% | 1400× | |
| WT | 0% | 500× | WT | 0% | 700× | |
| WT | 0% | 2600× | WT | 0% | 3000× | |
| KRAS/NRAS/BRAF (n = 2) | WT | 0% | 3600–4500× | WT | 0% | 2029–5500× |
| WT | 0% | 1250–2500× | WT | 0% | 2300–3500× | |
| BRAF/KRAS/HRAS/NRAS (n = 5) | KRAS p.G12V | 13% | 620× | KRAS p.G12V | 9% | 1000× |
| HRAS p.Q61R | 36% | 1100× | HRAS p.Q61R | 39% | 3850× | |
| HRAS p.Q61R | 35% | 3200× | HRAS p.Q61R | 27% | 700× | |
| WT | 0% | 3600–4700× | WT | 0% | 2300–2560× | |
| WT | 0% | 1400–3100× | WT | 0% | 700–1800× | |
| EGFR/KRAS (n = 4) * | 1 EGFR p.E746_A750delELREA * | 44% | 5250× | 1 EGFR p.E746_A750delELREA* | 47% | 5900× |
| 1 EGFR p.T790M * | 40% | 1050× | 1 EGFR p.T790M * | 34% | 900× | |
| 1 EGFR p.L858R | 39% | 550× | 1 EGFR p.L858R | 39% | 900× | |
| WT | 0% | 1750–5130× | WT | 0% | 1700–2400× | |
| WT | 0% | 800–1100× | WT | 0% | 600–1200× | |
| KRAS (n = 2) | KRAS p.G12C | 58% | 550× | KRAS p.G12C | 49% | 500× |
| KRAS p.G12C | 17% | 6500× | KRAS p.G12C | 25% | 3000× | |
| IDH1/IDH2 (n = 3) | 1 IDH1 p.R132C | 11% | 2500× | 1 IDH1 p.R132C | 23% | 4700× |
| WT | 0% | 1500–2200× | WT | 0% | 1300–2050× | |
| WT | 0% | 2300–3000× | WT | 0% | 1800–3500× | |
| Type and Number of Samples | Genes Evaluated (Number of Samples per Gene) | Frequency of Mutation (Number of Samples per Gene) |
|---|---|---|
| CNS/intracranial tumors | ||
| Brain neoplasms (n = 341) | IDH1 (321) | 22.1 |
| IDH2 (321) | 1.9 | |
| H3F3A (12) | 16.7 | |
| BRAF (14) | 14.3 | |
| Sellar lesions (n = 2) | CTNNB1 (2) | 100.0 |
| BRAF (2) | / | |
| NA: 20 (5.9%) | ||
| NSCLCs (n = 315) | EGFR (306) | 14.1 |
| KRAS (306) | 38.9 | |
| MET (50) | / | |
| NA: 9 (2.8%) | ||
| CRCs (n = 306) | KRAS (298) | 44.6 |
| NRAS (298) | 5.4 | |
| BRAF (205) | 15.1 | |
| NA: 8 (2.6%) | ||
| Thyroid nodules (n = 612) | BRAF (568) | 19.2 |
| KRAS (481) | 3.3 | |
| HRAS (481) | 2.5 | |
| NRAS (481) | 8.5 | |
| TERT (123) | 9.8 | |
| PIK3CA (83) | 8.4 | |
| TP53 (67) | 7.5 | |
| EIF1AX (67) | / | |
| MED12 (67) | / | |
| PTEN (37) | / | |
| RET (9) | 55.6 | |
| NA: 43 (7.0%) | ||
| Melanomas (n = 64) | BRAF (63) | 41.3 |
| NRAS (56) | 21.4 | |
| c-Kit (56) | 1.8 | |
| NA: 1 (1.6%) | ||
| Pancreatic lesions (n = 42) | KRAS (39) | 46.2 |
| GNAS (6) | 33.3 | |
| RNF43 (6) | / | |
| SMAD4 (6) | / | |
| NA: 3 (7.1%) | ||
| SOCs (n = 7) | TP53 (7) | 71.4 |
| GISTs (n = 6) | c-Kit (6) | 66.7 |
| PDFGRα (6) | 16.7 | |
| BRAF (6) | / |
| Sample Type | Gene | Frequency in Our Series | Frequency Reported in the Literature | References |
|---|---|---|---|---|
| Brain neoplasms | IDH1 | 22.1 | 30–39 | [1,32] |
| IDH2 | 1.9 | 1–2.8 | [1,32] | |
| NSCLCs | EGFR | 14.1 | 13.9–22.2 | [4,12] |
| KRAS | 38.9 | 29.7–40 | [4,12,34] | |
| CRCs | KRAS | 44.6 | 34–44.2 | [1,4] |
| NRAS | 5.4 | 4–4.5 | [1,4] | |
| BRAF | 15.1 | 10–11.2 | [1,4] | |
| Thyroid nodules | BRAF | 19.2 | 13.7–22 * | [1,37,38] |
| HRAS | 2.5 | 4 | [1,39] | |
| KRAS | 3.3 | 2 | [1,39] | |
| NRAS | 8.5 | 8 | [1,39] | |
| PIK3CA | 8.4 | 4 | [1] | |
| TP53 | 7.5 | 11 | [1] | |
| TERT | 9.8 | 11 | [1] | |
| Melanomas | BRAF | 41.3 | 36–44 | [1,4] |
| NRAS | 21.4 | 17–23.4 | [1,4] | |
| c-Kit | 1.8 | 6.6–8 | [1,4] | |
| Pancreatic lesions | KRAS | 46.2 | ~45–55 | [1,35] |
| Panel (Manufacturer) | Targets | Type of Starting Material | Minimum Amount of Input DNA (Recommended Yeld) | Multiplatform (i.e., NGS from Different Company) | Samples × run (for at least 500–1000× Coverage) |
|---|---|---|---|---|---|
| SOPHiA Solid Tumor Solution (Sophia Genetics) | 42 | Fresh/Frozen | 10–50 ng | Yes (ThermoFisher and Illumina) | 12–24 * |
| FFPE | |||||
| Oncomine Focus Assay (ThermoFisher Scientific) | 52 ^ | Fresh/Frozen | 10 ng | No (IonTorrent) | 8–16 * |
| FFPE | |||||
| Oncomine Comprehensive Assay v3 (ThermoFisher Scientific) | 161 ^ | Fresh/Frozen | 30 ng (10 ng per pool) | No (IonTorrent) | 8 ° |
| FFPE | |||||
| Human Actionable Solid Tumor Panel (Qiagen) | 22 | Fresh/Frozen | 10–40 ng (fresh)/40–250 ng (FFPE) | Yes (ThermoFisher and Illumina) | 24–32 * |
| FFPE | |||||
| GeneRead QIAact AIT DNA UMI Panel (Qiagen) | 30 | Fresh/Frozen | 40–160 ng | No (Qiagen) | 8 |
| FFPE | |||||
| Myriapod® NGS 56G Onco panel (Diatech) | 56 | Fresh/Frozen | 10–25 ng | Yes (ThermoFisher and Illumina) | 8–16 * |
| FFPE | |||||
| Custom-designed multi-gene panel of this study | 22 | Fresh/Frozen | 10–50 ng | Yes (Illumina and ThermoFisher) | 32–40 |
| FFPE |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Biase, D.; Acquaviva, G.; Visani, M.; Sanza, V.; Argento, C.M.; De Leo, A.; Maloberti, T.; Pession, A.; Tallini, G. Molecular Diagnostic of Solid Tumor Using a Next Generation Sequencing Custom-Designed Multi-Gene Panel. Diagnostics 2020, 10, 250. https://doi.org/10.3390/diagnostics10040250
de Biase D, Acquaviva G, Visani M, Sanza V, Argento CM, De Leo A, Maloberti T, Pession A, Tallini G. Molecular Diagnostic of Solid Tumor Using a Next Generation Sequencing Custom-Designed Multi-Gene Panel. Diagnostics. 2020; 10(4):250. https://doi.org/10.3390/diagnostics10040250
Chicago/Turabian Stylede Biase, Dario, Giorgia Acquaviva, Michela Visani, Viviana Sanza, Chiara M. Argento, Antonio De Leo, Thais Maloberti, Annalisa Pession, and Giovanni Tallini. 2020. "Molecular Diagnostic of Solid Tumor Using a Next Generation Sequencing Custom-Designed Multi-Gene Panel" Diagnostics 10, no. 4: 250. https://doi.org/10.3390/diagnostics10040250