Interaction of Natural Organic Matter with Layered Minerals: Recent Developments in Computational Methods at the Nanoscale


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction to Natural Organic Matter-Mineral Systems
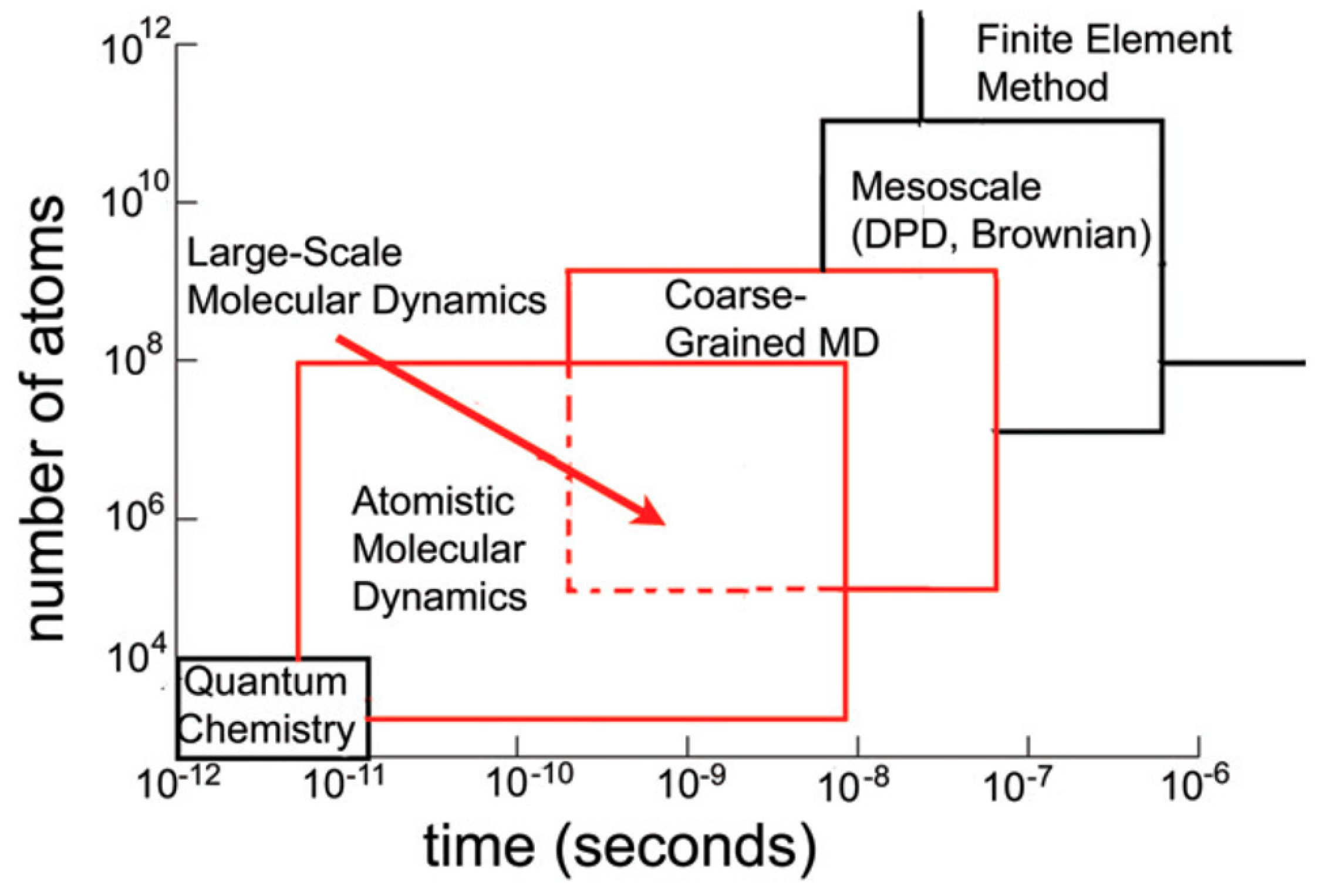
2. Introduction to Molecular Simulation Methods

3. Simulation Studies of Organic-Mineral Structures

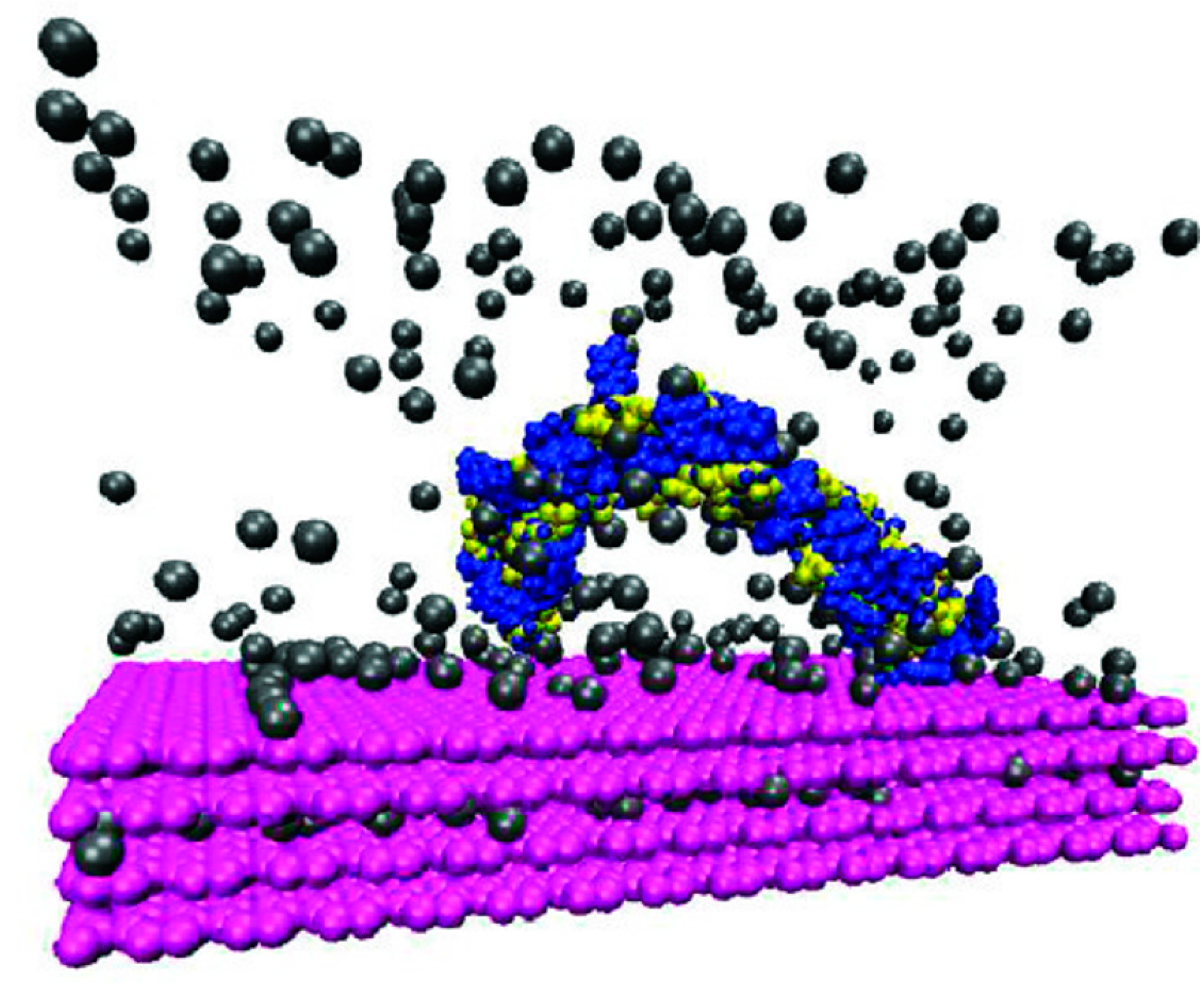
4. Experimental Studies of Natural Organic Matter-Mineral Structures
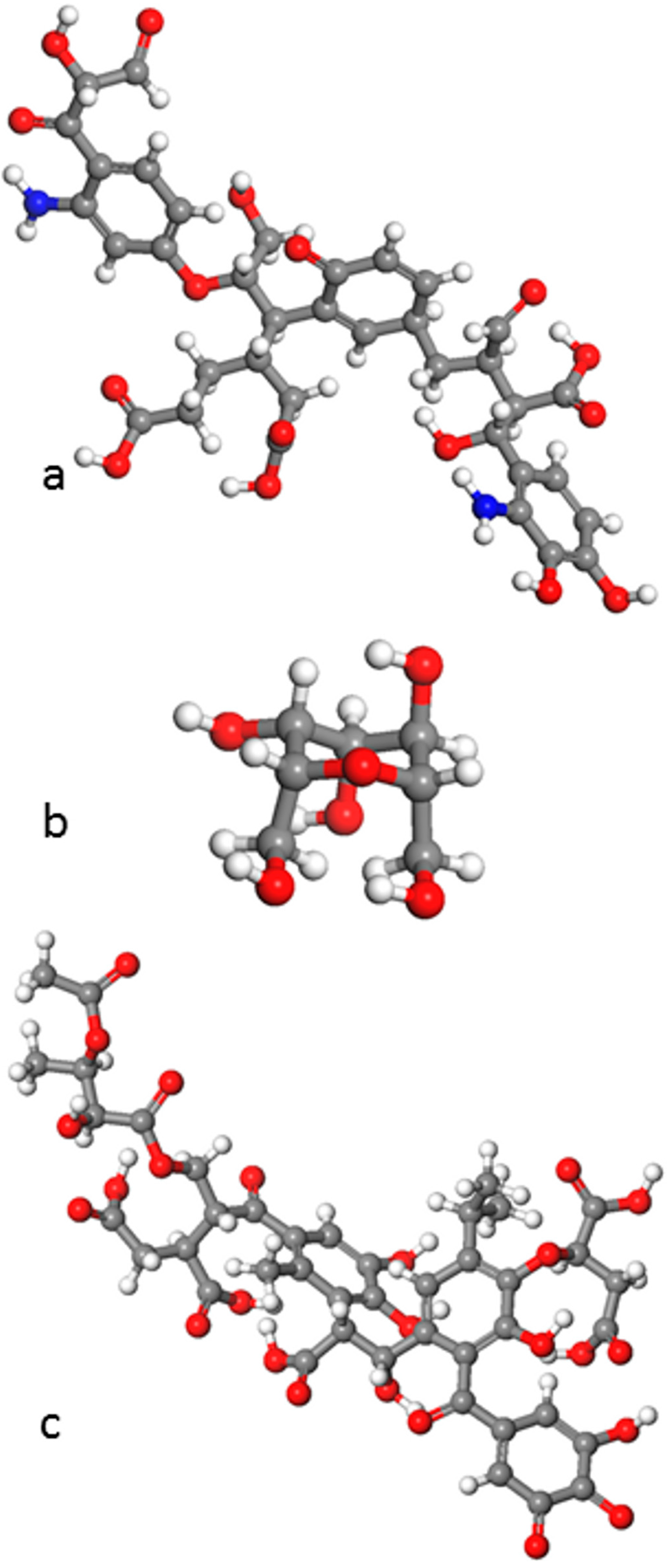
5. Simulations of Natural Organic Matter (NOM) and NOM-Mineral Interfaces
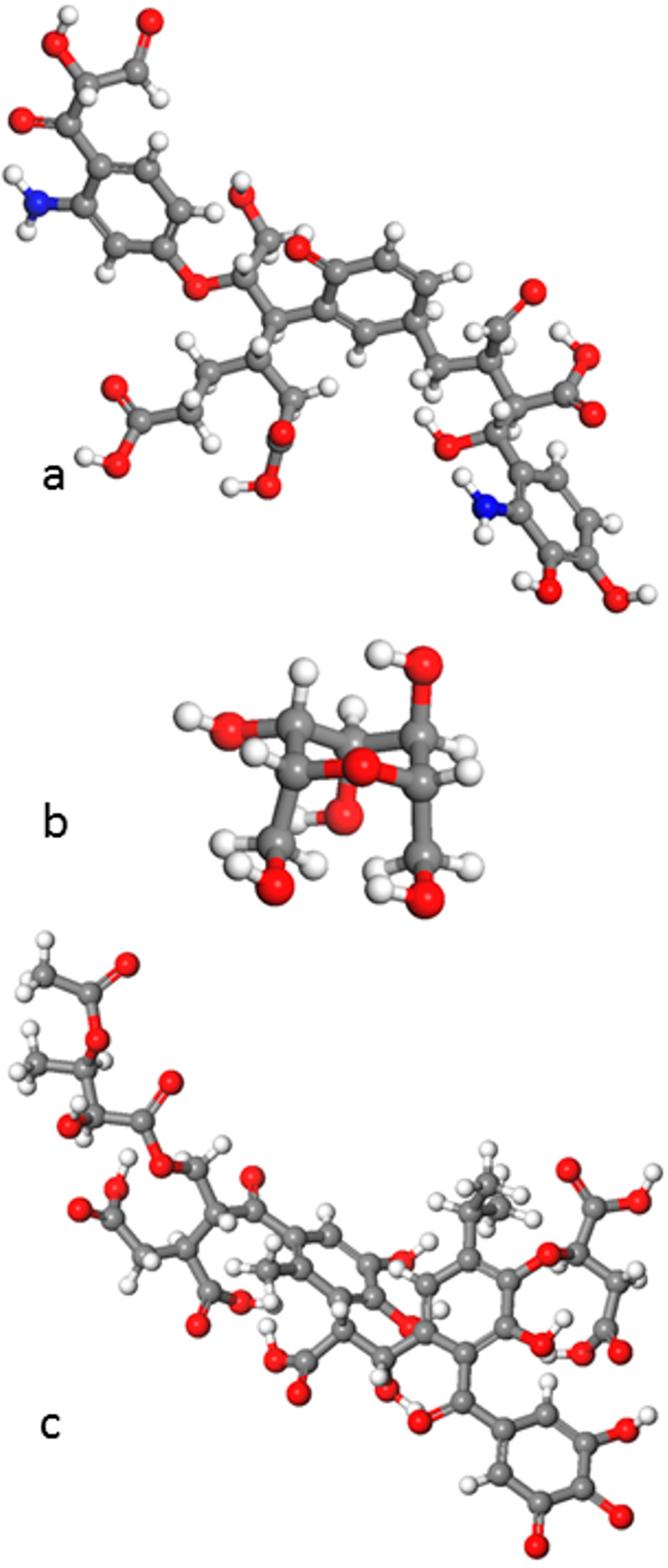
6. Redox Active Mineral-Natural Organic Matter—A Future Challenge
6.1. Role of Iron Based Minerals
6.2. Role of Manganese Oxides
- Charging of any amide groups present in the organic contaminant resulting in electrostatic attraction to the oxide surface [140];
- Increased sorption of neutral phenolic groups;
- Decrease of the negative charge on MnO2;
- Increased redox potential of the MnO2/Mn2+ couple [145]; and
- Enhanced removal of Mn2+ from the oxide surface, exposing new reactive sites [146].
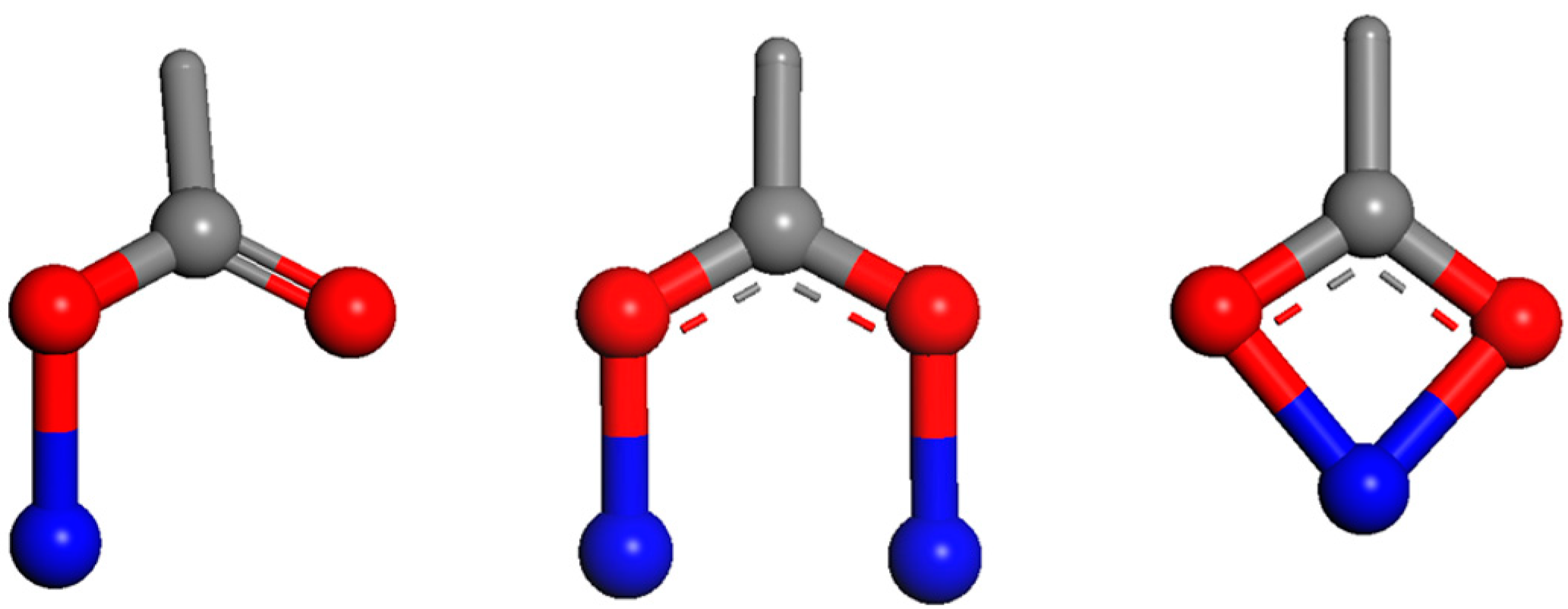
7. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Senesi, N.; Xing, B.; Huang, P.M. Biophysico-Chemical Processes Involving Natural Nonliving Organic Matter in Environmental Systems; John Wiley and Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Keil, R.G.; Mayer, L.M. Mineral matrices and organic matter. In Treatise on Geochemistry, 2nd ed.; Holland, H.D., Turekian, K.K., Eds.; Elsevier: Oxford, UK, 2014; pp. 337–359. [Google Scholar]
- Kennedy, M.J.; Wagner, T. Clay mineral continental amplifier for marine carbon sequestration in a greenhouse ocean. Proc. Natl. Acad. Sci. USA 2011, 108, 9776–9781. [Google Scholar] [CrossRef]
- Polubesova, T.; Chefetz, B. DOM-affected transformation of contaminants on mineral surfaces: A review. Crit. Rev. Env. Sci. Technol. 2014, 44, 223–254. [Google Scholar] [CrossRef]
- Matilainen, A.; Vepsalainen, M.; Sillanpaa, M. Natural organic matter removal by coagulation during drinking water treatment: A review. Adv. Colloid Interface Sci. 2010, 159, 189–197. [Google Scholar] [CrossRef]
- Al-Amoudi, A.S. Factors affecting natural organic matter (NOM) and scaling fouling in NF membranes: A review. Desalination 2010, 259, 1–10. [Google Scholar] [CrossRef]
- Jansen, S.A.; Malaty, M.; Nwabara, S.; Johnson, E.; Ghabbour, E.; Davies, G.; Varnum, J.M. Structural modeling in humic acids. Mater. Sci. Eng. C Bio. S. 1996, 4, 175–179. [Google Scholar] [CrossRef]
- Snoeyink, V.L.; Jenkins, D. Water Chemistry; John Wiley and Sons: New York, NY, USA, 1980. [Google Scholar]
- Hedges, J.I.; Eglinton, G.; Hatcher, P.G.; Kirchman, D.L.; Arnosti, C.; Derenne, S.; Evershed, R.P.; Kogel-Knabner, I.; de Leeuw, J.W.; Littke, R.; et al. The molecularly-uncharacterized component of nonliving organic matter in natural environments. Org. Geochem. 2000, 31, 945–958. [Google Scholar] [CrossRef]
- Burdige, D.J. Preservation of organic matter in marine sediments: Controls, mechanisms, and an imbalance in sediment organic carbon budgets? Che. Rev. 2007, 107, 467–485. [Google Scholar] [CrossRef]
- Von Lutzow, M.; Kogel-Knabner, I.; Ekschmitt, K.; Matzner, E.; Guggenberger, G.; Marschner, B.; Flessa, H. Stabilization of organic matter in temperate soils: Mechanisms and their relevance under different soil conditions—A review. Eur. J. Soil Sci. 2006, 57, 426–445. [Google Scholar] [CrossRef]
- Weber, W.J., Jr.; McGinley, P.M.; Katz, L.E. A distributed reactivity model for sorption by soils and sediments. 1. Conceptual basis and equilibrium assessments. Environ. Sci. Technol. 1992, 26, 1955–1962. [Google Scholar] [CrossRef]
- Vandenbroucke, M.; Largeau, C. Kerogen origin, evolution and structure. Org. Geochem. 2007, 38, 719–833. [Google Scholar] [CrossRef]
- Collins, M.J.; Bishop, A.N.; Farrimond, P. Sorption by mineral surfaces—Rebirth of the classical condensation pathway for kerogen formation. Geochim. Cosmochim. Acta 1995, 59, 2387–2391. [Google Scholar] [CrossRef]
- Brown, G.E.; Calas, G. Mineral-aqueous solution interfaces and their impact on the environment. Geochem. Perspect. 2012, 1, 483–742. [Google Scholar] [CrossRef]
- Geatches, D.L.; Clark, S.J.; Greenwell, H.C. Role of clay minerals in oil-forming reactions. J. Phys. Chem. A 2010, 114, 3569–3575. [Google Scholar] [CrossRef]
- Wu, L.M.; Zhou, C.H.; Keeling, J.; Tong, D.S.; Yu, W.H. Towards an understanding of the role of clay minerals in crude oil formation, migration and accumulation. Earth Sci. Rev. 2012, 115, 373–386. [Google Scholar] [CrossRef]
- Chen, B.; Evans, J.R.G.; Greenwell, H.C.; Boulet, P.; Coveney, P.V.; Bowden, A.A.; Whiting, A. A critical appraisal of polymer-clay nanocomposites. Chem. Soc. Rev. 2008, 37, 568–594. [Google Scholar] [CrossRef]
- Sels, B.F.; de vos, D.E.; Jacobs, P.A. Hydrotalcite-like anionic clays in catalytic organic reactions. Catal. Rev. Sci. Eng. 2001, 43, 443–488. [Google Scholar] [CrossRef]
- Debecker, D.P.; Gaigneaux, E.M.; Busca, G. Exploring, tuning, and exploiting the basicity of hydrotalcites for applications in heterogeneous catalysis. Chem. Eur. J. 2009, 15, 3920–3935. [Google Scholar] [CrossRef]
- Suter, J.L.; Anderson, R.L.; Greenwell, H.C.; Coveney, P.V. Recent advances in large-scale atomistic and coarse-grained molecular dynamics simulation of clay minerals. J. Mater. Chem. 2009, 19, 2482–2493. [Google Scholar] [CrossRef]
- Geysermans, P.; Noguera, C. Advances in atomistic simulations of mineral surfaces. J. Mater. Chem. 2009, 19, 7807–7821. [Google Scholar] [CrossRef]
- Greenwell, H.C.; Jones, W.; Coveney, P.V.; Stackhouse, S. On the application of computer simulation techniques to anionic and cationic clays: A materials chemistry perspective. J. Mater. Chem. 2006, 16, 708–723. [Google Scholar] [CrossRef]
- Anderson, R.L.; Ratcliffe, I.; Greenwell, H.C.; Williams, P.A.; Cliffe, S.; Coveney, P.V. Clay swelling—A challenge in the oilfield. Earth Sci. Rev. 2010, 98, 201–216. [Google Scholar] [CrossRef]
- Cygan, R.T.; Greathouse, J.A.; Heinz, H.; Kalinichev, A.G. Molecular models and simulations of layered minerals. J. Mater. Chem. 2009, 19, 2470–2481. [Google Scholar] [CrossRef]
- Lyubartsev, A.; Tu, Y.Q.; Laaksonen, A. Hierarchical multiscale modelling scheme from first principles to mesoscale. J. Comput. Theor. Nanosci. 2009, 6, 951–959. [Google Scholar] [CrossRef]
- Moras, G.; Choudhury, R.; Kermode, J.R.; CsÁnyi, G.; Payne, M.C.; de Vita, A. Hybrid quantum/classical modeling of material systems: The “learn on the fly” molecular dynamics scheme. In Trends in Computational Nanomechanics: Transcending Length and Time Scales; Dumitrica, T., Ed.; Springer Netherlands: Dordrecth, The Netherlands, 2010; Volume 9, pp. 1–23. [Google Scholar]
- Boulet, P.; Greenwell, H.C.; Stackhouse, S.; Coveney, P.V. Recent advances in understanding the structure and reactivity of clays using electronic structure calculations. J. Mol. Struct. 2006, 762, 33–48. [Google Scholar] [CrossRef]
- Larentzos, J.P.; Greathouse, J.A.; Cygan, R.T. An ab initio and classical molecular dynamics investigation of the structural and vibrational properties of talc and pyrophyllite. J. Phys. Chem. C 2007, 111, 12752–12759. [Google Scholar] [CrossRef]
- Ortega-Castro, J.; Hernandez-Haro, N.; Hernandez-Laguna, A.; Sainz-Diaz, C.I. DFT calculation of cystallographic properties of dioctahedral 2:1 phyllosilicates. Clay Miner. 2008, 43, 351–361. [Google Scholar] [CrossRef]
- Berghout, A.; Tunega, D.; Zaoui, A. Density functional theory (DFT) study of the hydration steps of Na+/Mg2+/Ca2+/Sr2+/Ba2+-exchanged montmorillonites. Clays Clay Miner. 2010, 58, 174–187. [Google Scholar] [CrossRef]
- Voora, V.K.; Al-Saidi, W.A.; Jordan, K.D. Density functional theory study of pyrophyllite and M-montmorillonites (M = Li, Na, K, Mg, and Ca): Role of dispersion interactions. J. Phys. Chem. A 2011, 115, 9695–9703. [Google Scholar]
- Gerzabek, M.H.; Aquino, A.J.A.; Haberhauer, G.; Tunega, D.; Lischka, H. Molecular modelling-opportunities for soil research. Bodenkultur 2001, 52, 133–146. [Google Scholar]
- Aquino, A.J.A.; Tunega, D.; Gerzabek, M.H.; Lischka, H. Modeling catalytic effects of clay mineral surfaces on peptide bond formation. J. Phys. Chem. B 2004, 108, 10120–10130. [Google Scholar] [CrossRef]
- Tunega, D.; Gerzabek, M.H.; Haberhauer, G.; Totsche, K.U.; Lischka, H. Model study on sorption of polycyclic aromatic hydrocarbons to goethite. J. Colloid Interface Sci. 2009, 330, 244–249. [Google Scholar]
- Liu, X.D.; Lu, X.C.; Wang, R.C.; Zhou, H.Q.; Xu, S.J. Surface complexes of acetate on edge surfaces of 2:1 type phyllosilicate: Insights from density functional theory calculation. Geochim. Cosmochim. Acta 2008, 72, 5896–5907. [Google Scholar] [CrossRef]
- Geatches, D.L.; Jacquet, A.; Clark, S.J.; Greenwell, H.C. Monomer adsorption on kaolinite: Modeling the essential ingredients. J. Phys. Chem. C 2012, 116, 22365–22374. [Google Scholar] [CrossRef]
- Aquino, A.J.A.; Tunega, D.; Haberhauer, G.; Gerzabek, M.H.; Lischka, H. Adsorption of organic substances on broken clay surfaces: A quantum chemical study. J. Comput. Chem. 2003, 24, 1853–1863. [Google Scholar] [CrossRef]
- Kwon, K.D.; Vadillo-Rodriguez, V.; Logan, B.E.; Kubicki, J.D. Interactions of biopolymers with silica surfaces: Force measurements and electronic structure calculation studies. Geochim. Cosmochim. Acta 2006, 70, 3803–3819. [Google Scholar] [CrossRef]
- Lee, B.H.; Lee, S.K. Effect of lattice topology on the adsorption of benzyl alcohol on kaolinite surfaces: Quantum chemical calculations of geometry optimization, binding energy, and NMR chemical shielding. Am. Mineral. 2009, 94, 1392–1404. [Google Scholar]
- Geatches, D.L.; Clark, S.J.; Greenwell, H.C. DFT plus U investigation of the catalytic properties of ferruginous clay. Am. Mineral. 2013, 98, 132–140. [Google Scholar]
- Dauber-Osguthorpe, P.; Roberts, V.A.; Osguthorpe, D.J.; Wolff, J.; Genest, M.; Hagler, A.T. Structure and energetics of ligand binding to proteins: Escherichia coli dihydrofolate reductase-trimethoprim, a drug-receptor system. Proteins Struct. Funct. Genet. 1988, 4, 31–47. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tiradorives, J. The OPLS potential functions for proteins. Energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM—A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; TiradoRives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Cygan, R.T.; Liang, J.-J.; Kalinichev, A.G. Molecular models of hydroxide, oxyhydroxide, and clay phases and the development of a general force field. J. Phys. Chem. B 2004, 108, 1255–1266. [Google Scholar] [CrossRef]
- Teppen, B.J.; Rasmussen, K.R.; Bertsch, P.M.; Miller, D.M.; Schafer, L. Molecular dynamics modeling of clay minerals. 1. Gibbsite, kaolinite, pyrophillite, and beidellite. J. Phys. Chem. B 1997, 101, 1579–1587. [Google Scholar] [CrossRef]
- Heinz, H.; Lin, T.J.; Mishra, R.K.; Emami, F.S. Thermodynamically consistent force fields for the assembly of inorganic, organic, and biological nanostructures: The INTERFACE force field. Langmuir 2013, 29, 1754–1765. [Google Scholar] [CrossRef]
- Pitman, M.C.; van Duin, A.C.T. Dynamics of confined reactive water in smectite clay-zeolite composites. J. Am. Chem. Soc. 2012, 134, 3042–3053. [Google Scholar] [CrossRef]
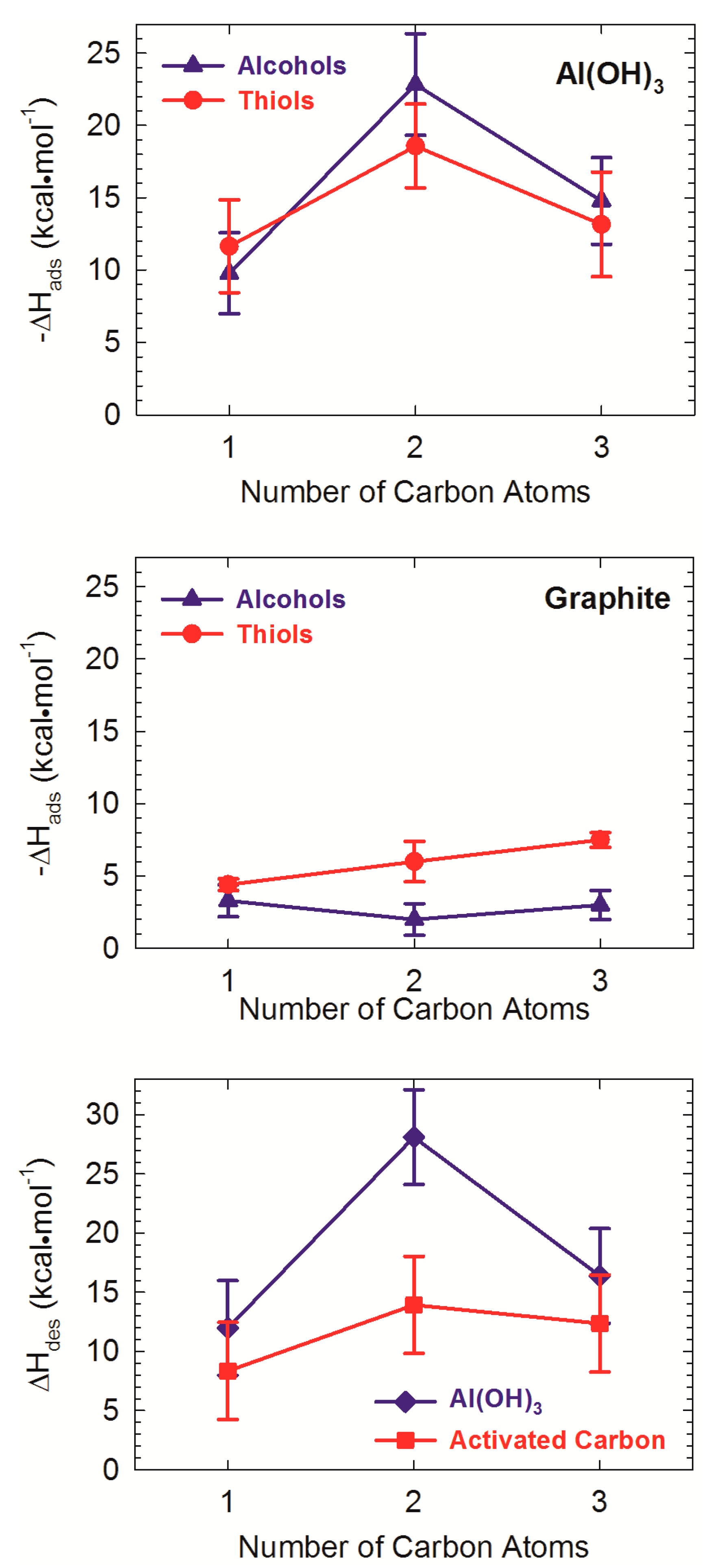
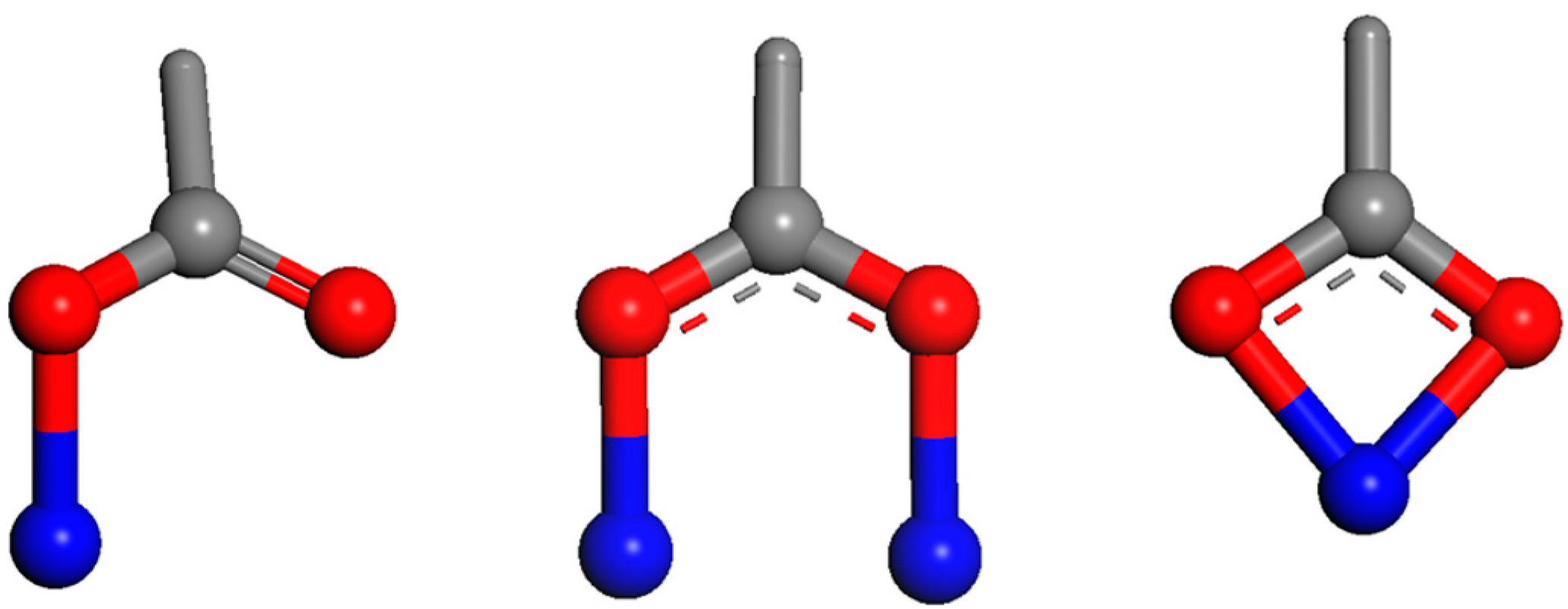
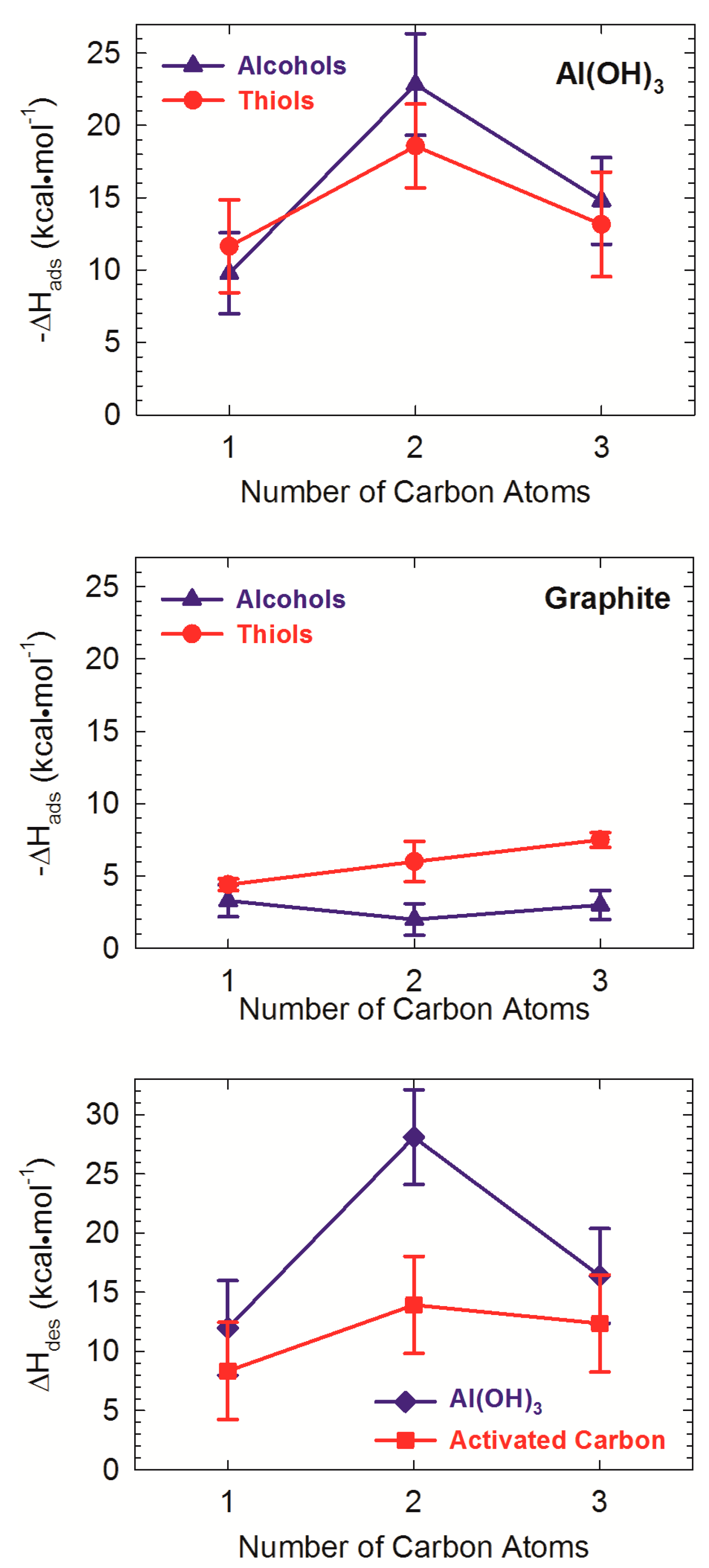
- Greathouse, J.A.; Hart, D.B.; Ochs, M.E. Alcohol and thiol adsorption on (oxy)hydroxide and carbon surfaces: Molecular dynamics simulation and desorption experiments. J. Phys. Chem. C 2012, 116, 26756–26764. [Google Scholar] [CrossRef]
- Veteska, M.; Pospisil, M.; Melanova, K.; Benes, L.; Zima, V. Structure analysis of hydrotalcite intercalated with pyrenetetrasulfonate; experiments and molecular modelling. J. Mol. Model. 2008, 14, 1119–1129. [Google Scholar] [CrossRef]
- Kovar, P.; Pospisil, M.; Kafunkova, E.; Lang, K.; Kovanda, F. Mg-Al layered double hydroxide intercalated with porphyrin anions: Molecular simulations and experiments. J. Mol. Model. 2010, 16, 223–233. [Google Scholar] [CrossRef]
- Praus, P.; Veteska, M.; Pospisil, M. Adsorption of phenol and aniline on natural and organically modified montmorillonite: Experiment and molecular modelling. Mol. Simul. 2011, 37, 964–974. [Google Scholar] [CrossRef]
- Liu, C.; Li, H.; Johnston, C.T.; Boyd, S.A.; Teppen, B.J. Relating clay structural factors to dioxin adsorption by smectites: Molecular dynamics simulations. Soil Sci. Soc. Am. J. 2012, 76, 110–120. [Google Scholar] [CrossRef]
- Greenwell, H.C.; Bowden, A.A.; Chen, B.Q.; Boulet, P.; Evans, J.R.G.; Coveney, P.V.; Whiting, A. Intercalation and in situ polymerization of poly (alkylene oxide) derivatives within M+-montmorillonite (M = Li, Na, K). J. Mater. Chem. 2006, 16, 1082–1094. [Google Scholar] [CrossRef]
- Anderson, R.L.; Greenwell, H.C.; Suter, J.L.; Coveney, P.V.; Thyveetil, M.A. Determining materials properties of natural composites using molecular simulation. J. Mater. Chem. 2009, 19, 7251–7262. [Google Scholar] [CrossRef]
- Kulhankova, L.; Tokarsky, J.; Peikertova, P.; Kutlakova, K.M.; Ivanek, L.; Capkova, P. Montmorillonite intercalated by conducting polyanilines. J. Phys. Chem. Solids 2012, 73, 1530–1533. [Google Scholar] [CrossRef]
- Kumar, P.P.; Kalinichev, A.G.; Kirkpatrick, R.J. Hydration, swelling, interlayer structure, and hydrogen bonding in organolayered double hydroxides: Insights from molecular dynamics simulation of citrate-intercalated hydrotalcite. J. Phys. Chem. B 2006, 110, 3841–3844. [Google Scholar] [CrossRef]
- Kumar, P.P.; Kalinichev, A.G.; Kirkpatrick, R.J. Molecular dynamics simulation of the energetics and structure of layered double hydroxides intercalated with carboxylic acids. J. Phys. Chem. C 2007, 111, 13517–13523. [Google Scholar] [CrossRef]
- Kalinichev, A.G.; Kumar, P.P.; Kirkpatrick, R.J. Molecular dynamics computer simulations of the effects of hydrogen bonding on the properties of layered double hydroxides intercalated with organic acids. Philos. Mag. 2010, 90, 2475–2488. [Google Scholar] [CrossRef]
- Thyveetil, M.A.; Coveney, P.V.; Greenwell, H.C.; Suter, J.L. Computer simulation study of the structural stability and materials properties of DNA-intercalated layered double hydroxides. J. Am. Chem. Soc. 2008, 130, 4742–4756. [Google Scholar] [CrossRef]
- Thyveetil, M.A.; Coveney, P.V.; Greenwell, H.C.; Suter, J.L. Role of host layer flexibility in DNA guest intercalation revealed by computer simulation of layered nanomaterials. J. Am. Chem. Soc. 2008, 130, 12485–12495. [Google Scholar] [CrossRef]
- Swadling, J.B.; Coveney, P.V.; Greenwell, H.C. Clay minerals mediate folding and regioselective interactions of RNA: A large-scale atomistic simulation study. J. Am. Chem. Soc. 2010, 132, 13750–13764. [Google Scholar]
- Swadling, J.B.; Coveney, P.V.; Greenwell, H.C. Stability of free and mineral-protected nucleic acids: Implications for the RNA world. Geochim. Cosmochim. Acta 2012, 83, 360–378. [Google Scholar] [CrossRef]
- Swadling, J.B.; Suter, J.L.; Greenwell, H.C.; Coveney, P.V. Influence of surface chemistry and charge on mineral-RNA interactions. Langmuir 2013, 29, 1573–1583. [Google Scholar] [CrossRef]
- Lee, S.S.; Fenter, P.; Park, C.; Nagy, K.L. Fulvic acid sorption on muscovite mica as a function of pH and time using in situ X-ray reflectivity. Langmuir 2008, 24, 7817–7829. [Google Scholar] [CrossRef]
- Lee, S.S.; Park, C.; Fenter, P.; Sturchio, N.C.; Nagy, K.L. Competitive adsorption of strontium and fulvic acid at the muscovite-solution interface observed with resonant anomalous X-ray reflectivity. Geochim. Cosmochim. Acta 2010, 74, 1762–1776. [Google Scholar] [CrossRef]
- Zhang, L.C.; Luo, L.; Zhang, S.Z. Integrated investigations on the adsorption mechanisms of fulvic and humic acids on three clay minerals. Colloids Surf. A 2012, 406, 84–90. [Google Scholar] [CrossRef]
- Ha, J.Y.; Yoon, T.H.; Wang, Y.G.; Musgrave, C.B.; Brown, G.E. Adsorption of organic matter at mineral/water interfaces: 7. ATR-FTIR and quantum chemical study of lactate interactions with hematite nanoparticles. Langmui 2008, 24, 6683–6692. [Google Scholar] [CrossRef]
- Yoon, T.H.; Johnson, S.B.; Brown, G.E. Adsorption of organic matter at mineral/water interfaces. IV. Adsorption of humic substances at boehmite/water interfaces and impact on boehmite dissolution. Langmuir 2005, 21, 5002–5012. [Google Scholar] [CrossRef]
- Conte, P.; Abbate, C.; Baglieri, A.; Negre, M.; De Pasquale, C.; Alonzo, G.; Gennari, M. Adsorption of dissolved organic matter on clay minerals as assessed by infra-red, CPMAS C-13 NMR spectroscopy and low field T-1 NMR relaxometry. Org. Geochem. 2011, 42, 972–977. [Google Scholar] [CrossRef]
- Kang, S.; Xing, B. Humic acid fractionation upon sequential adsorption onto goethite. Langmuir 2008, 24, 2525–2531. [Google Scholar] [CrossRef]
- Spagnuolo, M.; Jacobson, A.R.; Baveye, P. Electron paramagnetic resonance analysis of the distribution of a hydrophobic spin probe in suspensions of humic acids, hectorite, and aluminum hydroxide-humate-hectorite complexes. Environ. Toxicol. Chem. 2005, 24, 2435–2444. [Google Scholar] [CrossRef]
- Kaiser, K.; Guggenberger, G.; Haumaier, L.; Zech, W. Dissolved organic matter sorption on subsoils and minerals studied by C-13-NMR and drift spectroscopy. Eur. J. Soil Sci. 1997, 48, 301–310. [Google Scholar] [CrossRef]
- Evanko, C.R.; Dzombak, D.A. Influence of structural features on sorption of NOM-analogue organic acids to goethite. Environ. Sci. Technol. 1998, 32, 2846–2855. [Google Scholar] [CrossRef]
- Ali, M.A.; Dzombak, D.A. Competitive sorption of simple organic acids and sulfate on goethite. Environ. Sci. Technol. 1996, 30, 1061–1071. [Google Scholar] [CrossRef]
- Maillard, L.C. Formation of humic matters by the effect of polypeptides on sugars. C. R. Hebd. Seances Acad. Sci. 1913, 156, 1159–1160. [Google Scholar]
- Chorover, J.; Amistadi, M.K. Reaction of forest floor organic matter at goethite, birnessite and smectite surfaces. Geochim. Cosmochim. Acta 2001, 65, 95–109. [Google Scholar] [CrossRef]
- Jokic, A.; Frenkel, A.I.; Vairavamurthy, M.A.; Huang, P.M. Birnessite catalysis of the Maillard reaction: Its significance in natural humification. Geophys. Res. Lett. 2001, 28, 3899–3902. [Google Scholar] [CrossRef]
- Keil, R.G.; Montlucon, D.B.; Prahl, F.G.; Hedges, J.I. Sorptive preservation of labile organic-matter in marine-sediments. Nature 1994, 370, 549–552. [Google Scholar] [CrossRef]
- Janot, N.; Reiller, P.E.; Zheng, X.; Croue, J.-P.; Benedetti, M.F. Characterization of humic acid reactivity modifications due to adsorption onto alpha-Al2O3. Water Res. 2012, 46, 731–740. [Google Scholar] [CrossRef]
- Schnitzer, M.; Kodama, H. Interactions between organic and inorganic components in particle-size fractions separated from 4 soils. Soil Sci. Soc. Am. J. 1992, 56, 1099–1105. [Google Scholar] [CrossRef]
- Saidy, A.R.; Smernik, R.J.; Baldock, J.A.; Kaiser, K.; Sanderman, J. The sorption of organic carbon onto differing clay minerals in the presence and absence of hydrous iron oxide. Geoderma 2013, 209, 15–21. [Google Scholar]
- Lalonde, K.; Mucci, A.; Ouellet, A.; Gelinas, Y. Preservation of organic matter in sediments promoted by iron. Nature 2012, 483, 198–200. [Google Scholar]
- Wiseman, C.L.S.; Puttmann, W. Soil organic carbon and its sorptive preservation in central germany. Eur. J. Soil Sci. 2005, 56, 65–76. [Google Scholar] [CrossRef]
- Davies, G.; Fataftah, A.; Cherkasskiy, A.; Ghabbour, E.A.; Radwan, A.; Jansen, S.A.; Kolla, S.; Paciolla, M.D.; Sein, L.T.; Buermann, W.; et al. Tight metal binding by humic acids and its role in biomineralization. J. Chem. Soc. Dalton Trans. 1997, 4047–4060. [Google Scholar]
- Kunhi Mouvenchery, Y.; Kucerik, J.; Diehl, D.; Schaumann, G.E. Cation-mediated cross-linking in natural organic matter: A review. Rev. Env. Sci. Biotechnol. 2012, 11, 41–54. [Google Scholar]
- Schaumann, G.E.; Thiele-Bruhn, S. Molecular modeling of soil organic matter: Squaring the circle? Geoderma 2011, 166, 1–14. [Google Scholar] [CrossRef]
- Perry, T.D.; Cygan, R.T.; Mitchell, R. Molecular models of alginic acid: Interactions with calcium ions and calcite surfaces. Geochim. Cosmochim. Acta 2006, 70, 3508–3532. [Google Scholar] [CrossRef]
- Sutton, R.; Sposito, G. Molecular simulation of humic substance-Ca-montmorillonite complexes. Geochim. Cosmochim. Acta 2006, 70, 3566–3581. [Google Scholar] [CrossRef]
- Sutton, R.; Sposito, G. Molecular structure in soil humic substances: The new view. Environ. Sci. Technol. 2005, 39, 9009–9015. [Google Scholar] [CrossRef]
- Aquino, A.J.A.; Tunega, D.; Pasalic, H.; Haberhauer, G.; Gerzabek, M.H.; Lischka, H. The thermodynamic stability of hydrogen bonded and cation bridged complexes of humic acid models—A theoretical study. Chem. Phys. 2008, 349, 69–76. [Google Scholar] [CrossRef]
- Aquino, A.J.A.; Tunega, D.; Schaumann, G.E.; Haberhauer, G.; Gerzabek, M.H.; Lischka, H. Stabilizing capacity of water bridges in nanopore segments of humic substances: A theoretical investigation. J. Phys. Chem. C 2009, 113, 16468–16475. [Google Scholar] [CrossRef]
- Aquino, A.J.A.; Tunega, D.; Schaumann, G.E.; Haberhauer, G.; Gerzabek, M.H.; Lischka, H. The functionality of cation bridges for binding polar groups in soil aggregates. Int. J. Quantum Chem. 2011, 111, 1531–1542. [Google Scholar] [CrossRef]
- Aquino, A.J.A.; Tunega, D.; Pasalic, H.; Schaumann, G.E.; Haberhauer, G.; Gerzabek, M.H.; Lischka, H. Molecular dynamics simulations of water molecule-bridges in polar domains of humic acids. Environ. Sci. Technol. 2011, 45, 8411–8419. [Google Scholar] [CrossRef]
- Aquino, A.J.A.; Tunega, D.; Pasalic, H.; Schaumann, G.E.; Haberhauer, G.; Gerzabek, M.H.; Lischka, H. Study of solvent effect on the stability of water bridge-linked carboxyl groups in humic acid models. Geoderma 2011, 169, 20–26. [Google Scholar] [CrossRef]
- Kalinichev, A.G. Molecular models of natural organic matter and its colloidal aggregation in aqueous solutions: Challenges and opportunities for computer simulations. Pure Appl. Chem. 2013, 85, 149–158. [Google Scholar]
- Sein, L.T.; Varnum, J.M.; Jansen, S.A. Conformational modeling of a new building block of humic acid: Approaches to the lowest energy conformer. Environ. Sci. Technol. 1999, 33, 546–552. [Google Scholar] [CrossRef]
- Xu, X.; Kalinichev, A.G.; Kirkpatrick, R.J. 133Cs and 35Cl NMR spectroscopy and molecular dynamics modeling of Cs+ and Cl− complexation with natural organic matter. Geochim. Cosmochim. Acta 2006, 70, 4319–4331. [Google Scholar] [CrossRef]
- Kalinichev, A.G.; Kirkpatrick, R.J. Molecular dynamics simulation of cationic complexation with natural organic matter. Eur. J. Soil Sci. 2007, 58, 909–917. [Google Scholar] [CrossRef]
- Ahn, W.Y.; Kalinichev, A.G.; Clark, M.M. Effects of background cations on the fouling of polyethersulfone membranes by natural organic matter: Experimental and molecular modeling study. J. Membr. Sci. 2008, 309, 128–140. [Google Scholar] [CrossRef]
- Schulten, H.R.; Schnitzer, M. Chemical model structures for soil organic matter and soils. Soil Sci. 1997, 162, 115–130. [Google Scholar] [CrossRef]
- Schulten, H.R.; Schnitzer, M. A state-of-the-art structural concept for humic substances. Naturwissenschaften 1993, 80, 29–30. [Google Scholar]
- Shevchenko, S.M.; Bailey, G.W.; Akim, L.G. The conformational dynamics of humic polyanions in model organic and organo-mineral aggregates. J. Mol. Struct. 1999, 460, 179–190. [Google Scholar] [CrossRef]
- Shevchenko, S.M.; Bailey, G.W. Modeling sorption of soil organic matter on mineral surfaces: Wood-derived polymers on mica. Supramol. Sci. 1998, 5, 143–157. [Google Scholar] [CrossRef]
- Leenheer, J.A.; Brown, G.K.; MacCarthy, P.; Cabaniss, S.E. Models of metal binding structures in fulvic acid from the Suwannee River, Georgia. Environ. Sci. Technol. 1998, 32, 2410–2416. [Google Scholar] [CrossRef]
- Iskrenova-Tchoukova, E.; Kalinichev, A.G.; Kirkpatrick, R.J. Metal cation complexation with natural organic matter in aqueous solutions: Molecular dynamics simulations and potentials of mean force. Langmuir 2010, 26, 15909–15919. [Google Scholar] [CrossRef]
- Kalinichev, A.G.; Iskrenova-Tchoukova, E.; Ahn, W.-Y.; Clark, M.M.; Kirkpatrick, R.J. Effects of Ca2+ on supramolecular aggregation of natural organic matter in aqueous solutions: A comparison of molecular modeling approaches. Geoderma 2011, 169, 27–32. [Google Scholar] [CrossRef]
- Kirishima, A.; Tanaka, K.; Niibori, Y.; Tochiyama, O. Complex formation of calcium with humic acid and polyacrylic acid. Radiochim. Acta 2002, 90, 555–561. [Google Scholar]
- Plaschke, M.; Rothe, J.; Armbruster, M.K.; Denecke, M.A.; Naber, A.; Geckeis, H. Humic acid metal cation interaction studied by spectromicroscopy techniques in combination with quantum chemical calculations. J. Synchrotron Radiat. 2010, 17, 158–165. [Google Scholar] [CrossRef]
- Roger, G.M.; Durand-Vidal, S.; Bernard, O.; Meriguet, G.; Altmann, S.; Turq, P. Characterization of humic substances and polyacrylic acid: A high precision conductimetry study. Colloids Surf. A 2010, 356, 51–57. [Google Scholar] [CrossRef]
- Crea, F.; Giacalone, A.; Gianguzza, A.; Piazzese, D.; Sammartano, S. Modelling of natural and synthetic polyelectrolyte interactions in natural waters by using SIT, Pitzer and ion pairing approaches. Mar. Chem. 2006, 99, 93–105. [Google Scholar] [CrossRef]
- Crea, F.; de Stefano, C.; Gianguzza, A.; Pettignano, A.; Piazzese, D.; Sammartano, S. Acid-base properties of synthetic and natural polyelectrolytes: Experimental results and models for the dependence on different aqueous media. J. Chem. Eng. Data 2009, 54, 589–605. [Google Scholar] [CrossRef]
- Lovley, D.R. Dissimilatory Fe(III) and Mn(IV) reduction. Microbiol. Rev. 1991, 55, 259–287. [Google Scholar]
- Gu, B.H.; Schmitt, J.; Chen, Z.H.; Liang, L.Y.; McCarthy, J.F. Adsorption and desorption of natural organic-matter on iron-oxide - mechanisms and models. Environ. Sci. Technol. 1994, 28, 38–46. [Google Scholar] [CrossRef]
- Kaiser, K.; Guggenberger, G. The role of DOM sorption to mineral surfaces in the preservation of organic matter in soils. Org. Geochem. 2000, 31, 711–725. [Google Scholar] [CrossRef]
- Geatches, D.L.; Clark, S.J.; Greenwell, H.C. Iron reduction in nontronite-type clay minerals: Modelling a complex system. Geochim. Cosmochim. Acta 2012, 81, 13–27. [Google Scholar] [CrossRef]
- Alexandrov, V.; Neumann, A.; Scherer, M.M.; Rosso, K.M. Electron exchange and conduction in nontronite from first-principles. J. Phys. Chem. C 2013, 117, 2032–2040. [Google Scholar]
- Alexandrov, V.; Rosso, K.M. Insights into the mechanism of Fe(II) adsorption and oxidation at Fe-clay mineral surfaces from first-principles calculations. J. Phys. Chem. C 2013, 117, 22880–22886. [Google Scholar] [CrossRef]
- Wander, M.C.F.; Rosso, K.M.; Schoonen, M.A.A. Structure and charge hopping dynamics in green rust. J. Phys. Chem. C 2007, 111, 11414–11423. [Google Scholar] [CrossRef]
- Qiang, L.H.; Li, Z.F.; Zhao, T.Q.; Zhong, S.L.; Wang, H.Y.; Cui, X.J. Atomic-scale interactions of the interface between chitosan and Fe3O4. Colloids Surf. A 2013, 419, 125–132. [Google Scholar] [CrossRef]
- Aryanpour, M.; van Duin, A.C.T.; Kubicki, J.D. Development of a reactive force field for iron-oxyhydroxide systems. J. Phys. Chem. A 2010, 114, 6298–6307. [Google Scholar] [CrossRef]
- Dismukes, G.C. The metal centers of the photosynthetic oxygen-evolving complex. Photochem. Photobiol. 1986, 43, 99–115. [Google Scholar] [CrossRef]
- Burns, R.G.; Burns, V.M. Mineralogy. In Marine Manganese Deposits; Glasby, G.P., Ed.; Elsevier: Amsterdam, The Netherlands, 1977; pp. 185–248. [Google Scholar]
- O’Reilly, S.E.; Hochella, M.F. Lead sorption efficiencies of natural and synthetic Mn and Fe-oxides. Geochim. Cosmochim. Acta 2003, 67, 4471–4487. [Google Scholar] [CrossRef]
- Sposito, G. The Chemistry of Soils, 2nd ed.; Oxford University Press: Oxford, UK, 2008. [Google Scholar]
- Sparks, D.L. Environmental Soil Chemistry, 2nd ed.; Academic Press: San Diego, CA, USA, 2003. [Google Scholar]
- Villalobos, M.; Lanson, B.; Manceau, A.; Toner, B.; Sposito, G. Structural model for the biogenic Mn oxide produced by pseudomonas putida. Am. Mineral. 2006, 91, 489–502. [Google Scholar] [CrossRef]
- Essington, M.E. Soil and Water Chemistry: An Integrative Approach; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Post, J.E. Manganese oxide minerals: Crystal structures and economic and environmental significance. Proc. Natl. Acad. Sci. USA 1999, 96, 3447–3454. [Google Scholar] [CrossRef]
- Gasparatos, D. Sequestration of heavy metals from soil with Fe-Mn concretions and nodules. Environ. Chem. Lett. 2013, 11, 1–9. [Google Scholar]
- Cygan, R.T.; Post, J.E.; Heaney, P.J.; Kubicki, J.D. Molecular models of birnessite and related hydrated layered minerals. Am. Mineral. 2012, 97, 1505–1514. [Google Scholar]
- Kwon, K.D.; Refson, K.; Sposito, G. Surface complexation of Pb(II) by hexagonal birnessite nanoparticles. Geochim. Cosmochim. Acta 2010, 74, 6731–6740. [Google Scholar]
- Kwon, K.D.; Refson, K.; Sposito, G. On the role of Mn(IV) vacancies in the photoreductive dissolution of hexagonal birnessite. Geochim. Cosmochim. Acta 2009, 73, 4142–4150. [Google Scholar] [CrossRef]
- Kwon, K.D.; Refson, K.; Sposito, G. Understanding the trends in transition metal sorption by vacancy sites in birnessite. Geochim. Cosmochim. Acta 2013, 101, 222–232. [Google Scholar] [CrossRef]
- Pena, J.; Kwon, K.D.; Refson, K.; Bargar, J.R.; Sposito, G. Mechanisms of nickel sorption by a bacteriogenic birnessite. Geochim. Cosmochim. Acta 2010, 74, 3076–3089. [Google Scholar] [CrossRef]
- Kwon, K.D.; Refson, K.; Sposito, G. Zinc surface complexes on birnessite: A density functional theory study. Geochim. Cosmochim. Acta 2009, 73, 1273–1284. [Google Scholar] [CrossRef]
- Tebo, B.M.; Bargar, J.R.; Clement, B.G.; Dick, G.J.; Murray, K.J.; Parker, D.; Verity, R.; Webb, S.M. Biogenic manganese oxides: Properties and mechanisms of formation. Annu. Rev. Earth Planet. Sci. 2004, 32, 287–328. [Google Scholar] [CrossRef]
- Madison, A.S.; Tebo, B.M.; Mucci, A.; Sundby, B.; Luther, G.W. Abundant porewater Mn(III) is a major component of the sedimentary redox system. Science 2013, 341, 875–878. [Google Scholar] [CrossRef]
- Laha, S.; Luthy, R.G. Oxidation of aniline and other primary aromatic-amines by manganese-dioxide. Environ. Sci. Technol. 1990, 24, 363–373. [Google Scholar] [CrossRef]
- Stone, A.T. Reductive dissolution of manganese(III/IV) oxides by substituted phenols. Environ. Sci. Technol. 1987, 21, 979–988. [Google Scholar] [CrossRef]
- Ulrich, H.J.; Stone, A.T. Oxidation of chlorophenols adsorbed to manganese oxide surfaces. Environ. Sci. Technol. 1989, 23, 421–428. [Google Scholar] [CrossRef]
- Zhang, H.C.; Huang, C.H. Oxidative transformation of triclosan and chlorophene by manganese oxides. Environ. Sci. Technol. 2003, 37, 2421–2430. [Google Scholar] [CrossRef]
- Zhang, H.C.; Huang, C.H. Reactivity and transformation of antibacterial N-oxides in the presence of manganese oxide. Environ. Sci. Technol. 2005, 39, 593–601. [Google Scholar] [CrossRef]
- Zhang, H.C.; Huang, C.H. Oxidative transformation of fluoroquinolone antibacterial agents and structurally related amines by manganese oxide. Environ. Sci. Technol. 2005, 39, 4474–4483. [Google Scholar] [CrossRef]
- Klausen, J.; Haderlein, S.B.; Schwarzenbach, R.P. Oxidation of substituted anilines by aqueous MnO2: Effect of co-solutes on initial and quasi-steady-state kinetics. Environ. Sci. Technol. 1997, 31, 2642–2649. [Google Scholar] [CrossRef]
- Banerjee, D.; Nesbitt, H.W. XPS study of dissolution of birnessite by humate with constraints on reaction mechanism. Geochim. Cosmochim. Acta 2001, 65, 1703–1714. [Google Scholar] [CrossRef]
- Banerjee, D.; Nesbitt, H.W. XPS study of reductive dissolution of birnessite by oxalate: Rates and mechanistic aspects of dissolution and redox processes. Geochim. Cosmochim. Acta 1999, 63, 3025–3038. [Google Scholar] [CrossRef]
- Durand, J.P.; Senanayake, S.D.; Suib, S.L.; Mullins, D.R. Reaction of formic acid over amorphous manganese oxide catalytic systems: An in situ study. J. Phys. Chem. C 2010, 114, 20000–20006. [Google Scholar]
- Reiller, P.E. Modelling metal-humic substances-surface systems: Reasons for success, failure and possible routes for peace of mind. Miner. Mag. 2012, 76, 2643–2658. [Google Scholar] [CrossRef]
- Wang, N.-H.; Lo, S.-L. Preparation, characterization and adsorption performance of cetyltrimethylammonium modified birnessite. Appl. Surf. Sci. 2014, 299, 123–130. [Google Scholar] [CrossRef]
- Myeongjin, K.; Myeongyeol, Y.; Youngjae, Y.; Jooheon, K. Capacitance behavior of composites for supercapacitor applications prepared with different durations of graphene/nanoneedle MnO2 reduction. Microelectron. Reliab. 2014, 54, 587–594. [Google Scholar] [CrossRef]
- Ahmed, K.A.M.; Huang, K. Synthesis, characterization and catalytic activity of birnessite type potassium manganese oxide nanotubes and nanorods. Mater. Chem. Phys. 2012, 133, 605–610. [Google Scholar] [CrossRef]
- Xia, H.; Wang, Y.; Lin, J.; Lu, L. Hydrothermal synthesis of MnO2/CNT nanocomposite with a CNT core/porous MnO2 sheath hierarchy architecture for supercapacitors. Nanoscale Res. Lett. 2012, 7, 1–10. [Google Scholar] [CrossRef]
- Jaramillo-Botero, A.; Nielsen, R.; Abrol, R.; Su, J.; Pascal, T.; Mueller, J.; Goddard, W.A. First-principles-based multiscale, multiparadigm molecular mechanics and dynamics methods for describing complex chemical processes. In Multiscale Molecular Methods in Applied Chemistry; Kirchner, B., Vrabec, J., Eds.; Springer-Verlag: Berlin, Germany, 2012; Volume 307, pp. 1–42. [Google Scholar]
- Abrams, C.; Bussi, G. Enhanced sampling in molecular dynamics using metadynamics, replica-exchange, and temperature-acceleration. Entropy 2014, 16, 163–199. [Google Scholar] [CrossRef]
- Stamatakis, M.; Vlachos, D.G. Unraveling the complexity of catalytic reactions via kinetic Monte Carlo simulation: Current status and frontiers. ACS Catal. 2012, 2, 2648–2663. [Google Scholar] [CrossRef]
- Liu, S.Y.; Kleber, M.; Takahashi, L.K.; Nico, P.; Keiluweit, M.; Ahmed, M. Synchrotron-based mass spectrometry to investigate the molecular properties of mineral-organic associations. Anal. Chem. 2013, 85, 6100–6106. [Google Scholar] [CrossRef]
- Smalley, M. Clay Swelling and Colloid Stability; Taylor and Francis Group: Boca Raton, FL, USA, 2006. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Greathouse, J.A.; Johnson, K.L.; Greenwell, H.C. Interaction of Natural Organic Matter with Layered Minerals: Recent Developments in Computational Methods at the Nanoscale. Minerals 2014, 4, 519-540. https://doi.org/10.3390/min4020519
Greathouse JA, Johnson KL, Greenwell HC. Interaction of Natural Organic Matter with Layered Minerals: Recent Developments in Computational Methods at the Nanoscale. Minerals. 2014; 4(2):519-540. https://doi.org/10.3390/min4020519
Chicago/Turabian StyleGreathouse, Jeffery A., Karen L. Johnson, and H. Christopher Greenwell. 2014. "Interaction of Natural Organic Matter with Layered Minerals: Recent Developments in Computational Methods at the Nanoscale" Minerals 4, no. 2: 519-540. https://doi.org/10.3390/min4020519
APA StyleGreathouse, J. A., Johnson, K. L., & Greenwell, H. C. (2014). Interaction of Natural Organic Matter with Layered Minerals: Recent Developments in Computational Methods at the Nanoscale. Minerals, 4(2), 519-540. https://doi.org/10.3390/min4020519