Fits and Misfits in Organic Matrix Analyses: Case of the Soluble Matrices of the Nacreous Layer of Pinctada margaritifera (Mollusca)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
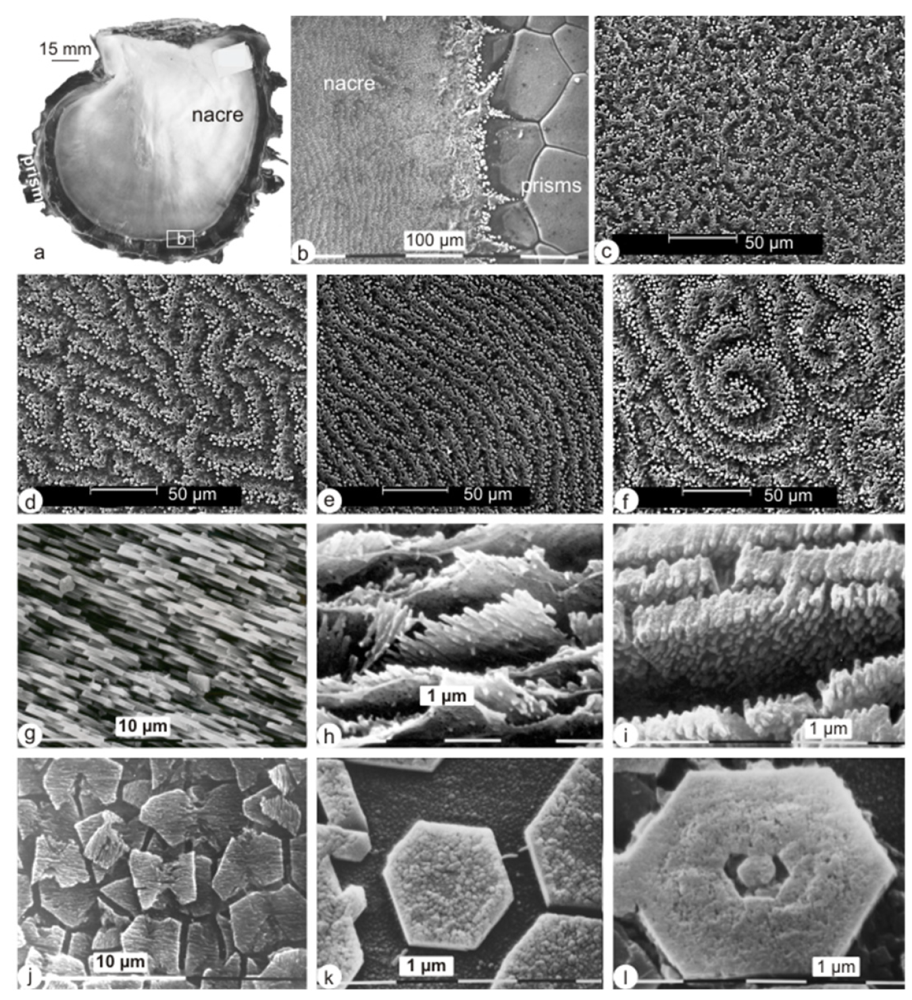
2.1. Micro- and Nanostructures
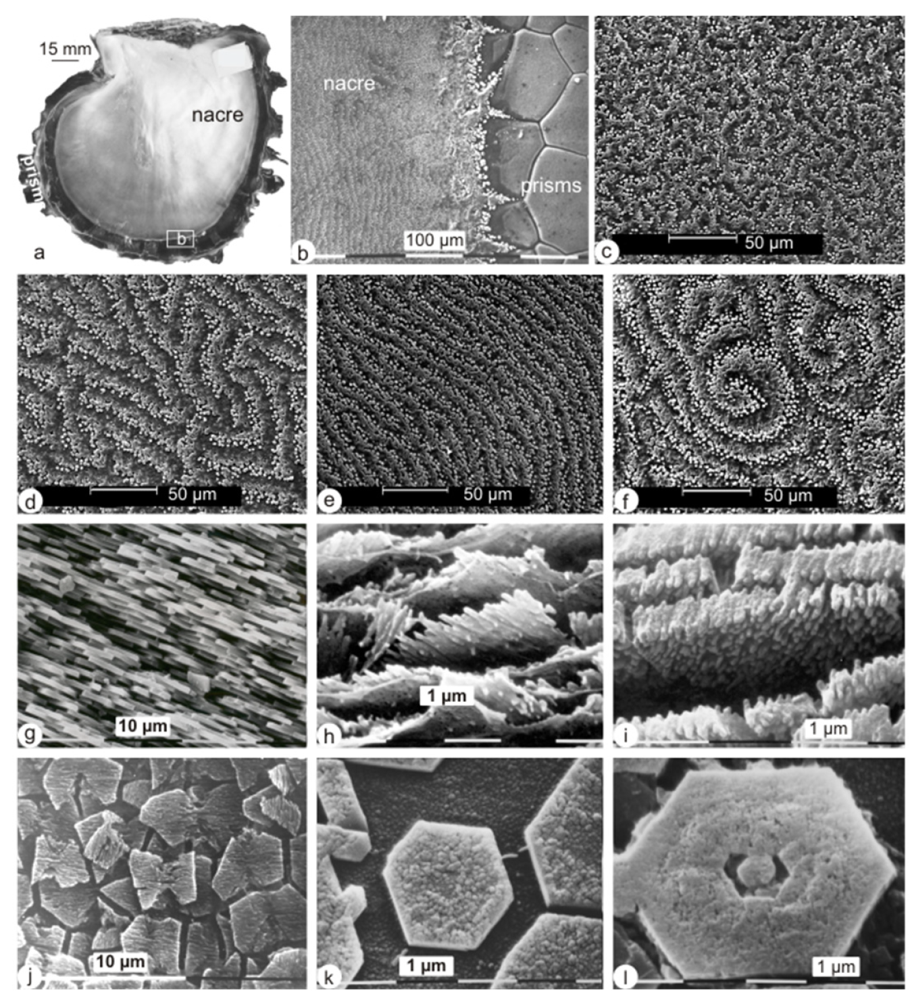

2.2. Bulk Analyses of the Soluble Organic Matrix
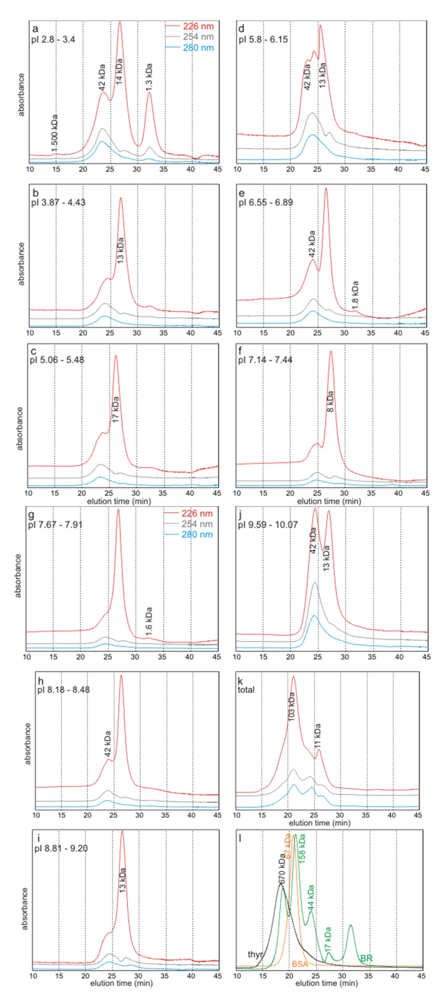
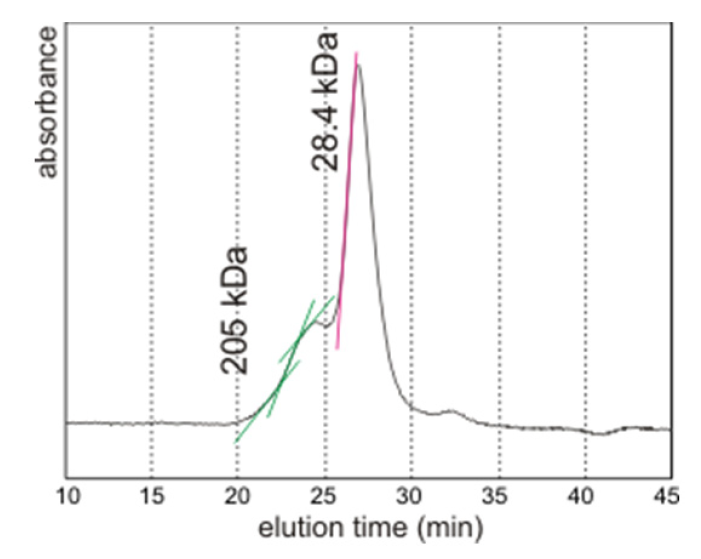
2.3. Molecular Weights Versus pI

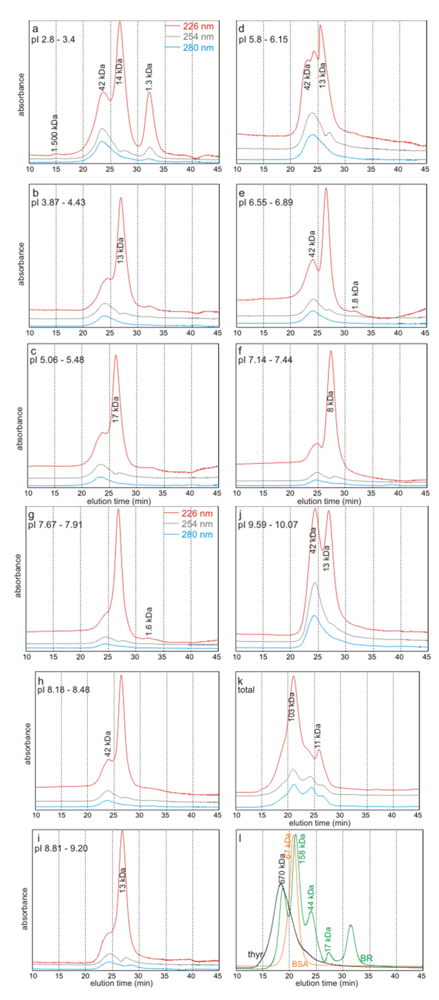
2.4. Discussion
2.4.1. Methods
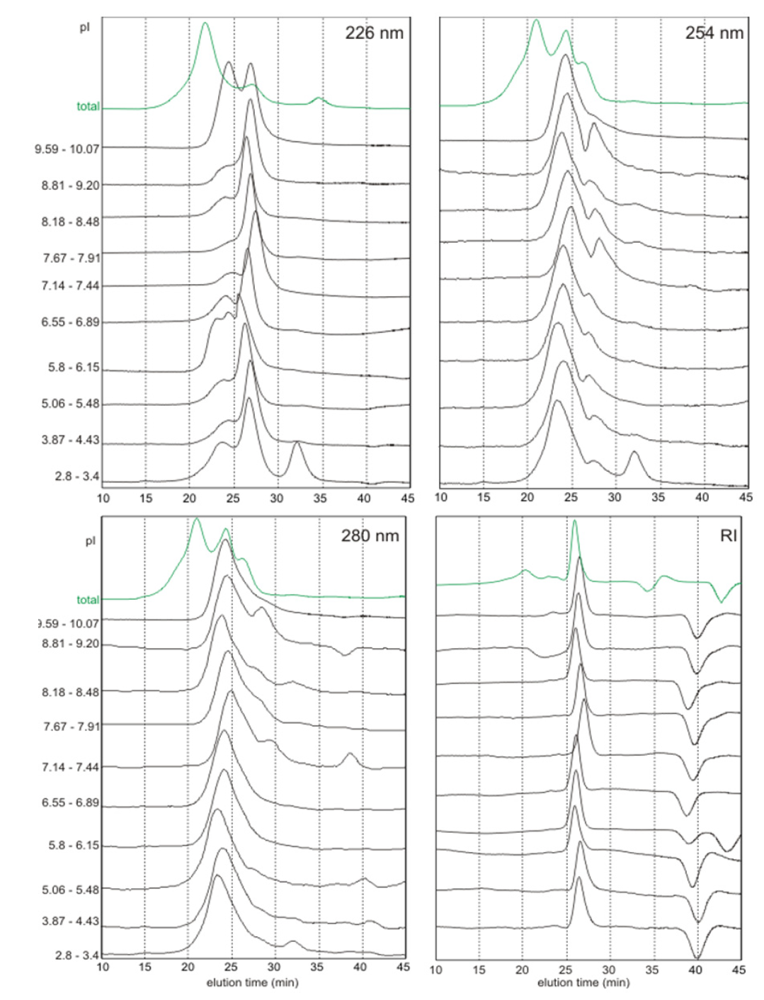
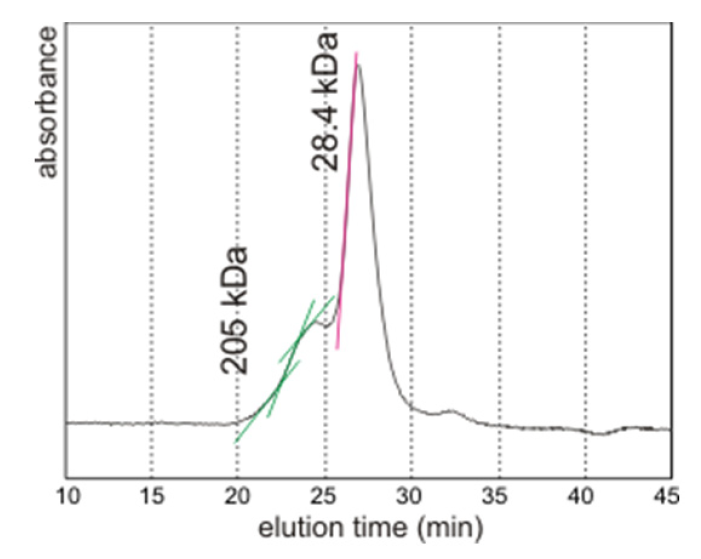

2.5. Comparison with the Known Proteins in Nacre
3. Experimental Section
3.1. Materials
3.2. Organic Matrix Extraction
3.3. Scanning Electron (SEM) and Atomic Force (AFM) Microscopes
3.4. Electrophoreses
4. Conclusions
Acknowledgements
References
- Boggild, O.B. The shell structure of the Mollusks. Det Kongelige Danske Videnskabernes Selskabs Skrifter Naturvidenskabelig Og Mathematisk Afdeling 1930, 9, 231–326. [Google Scholar]
- Hall, A.; Kennedy, W.J.; Taylor, J.D. The shell structure and mineralogy of the Bivalvia. II. Lucinacea-Clavagellacea. Bull. Br. Mus. Nat. hist. Zool. 1973, 22, 253–294. [Google Scholar]
- Kobayashi, I. Internal microstructure of the shell of Bivalve Molluscs. Am. Zool. 1969, 9, 663–672. [Google Scholar]
- Kobayashi, I. Various patterns of biomineralization and its phylogenetic significances in Bivalve mollusks. In The Mechanisms of Biomineralization in Animals and Plants; Eds., Watabe, N., Omori, M., Eds.; 1980; Tokai University Press: Tokyo, Japan. [Google Scholar]
- Wise, S.W., Jr. Microarchitecture and mode of formation of nacre (mother-of-pearl) in Pelecypods, Gastropods, and Cephalopods. Ecolog. Geol. Helv. 1970, 63, 775–797. [Google Scholar]
- Erben, H.K. Uber die Bildung und das Wachstum von Perlmutt. Biomineralization 1972, 4, 16–36. [Google Scholar]
- Mutvei, H. The nacreous layer in Mytilus, Nucula, and Unio (Bivalvia)—Crystalline composition and nucleation of nacreous tablets. Calc. Tiss. Res. 1977, 24, 11–18. [Google Scholar] [CrossRef]
- Mutvei, H. Ultrastructural characteristics of the nacre in some Gastropods. Zool. Scripta 1978, 7, 287–296. [Google Scholar] [CrossRef]
- Mutvei, H. Ultrastructural research on molluscan nacre. In. In Proceedings of the 27th International Geological Congress, Moscow, Russia, 1984.
- Grégoire, C. Topography of the organic components in mother-of-pearl. J. Biophys. Bioch. Cytol. 1957, 3, 798–808. [Google Scholar]
- Grégoire, C. Further studies on structure of the organic components in mother-of-pearl, especially in pelecypods (part 1). Bull. Inst. Roy. Sci. Nat. Belg. 1960, 36, 1–22. [Google Scholar]
- Grégoire, C. Sur la structure des matrices organiques des coquilles de Mollusques. Biol. Rev. 1967, 42, 653–688. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, H. An electron microscope study of the growing surface of nacre in two gastropod species, Turbo cornutus Tegula pfeifferi. Venus 1979, 38, 205–211. [Google Scholar]
- Nakahara, H. Calcification in gastropod nacre. In Biomineralization and Biological Metal Accumulation; Eds., de Jong, E.W., Westbroek, P., Eds.; 1983; Reidel Pub.: Dordrecht, The Netherlands. [Google Scholar]
- Itasaka, O.; Hatano, H.; Tanaka, S. Biochemical Studies on pearl. IX. Amino acid composition of conchiolin in pearl and shell. Bull. Chem. Soc. Jpn. 1960, 33, 543–545. [Google Scholar] [CrossRef]
- Voss-Foucart, F. Essais de solubilisation et de fractionnement d'une conchioline (nacre murale de Nautilus pompilus, mollusque céphalopode). Comp. Biochem. Physiol. 1968, 26, 877–880. [Google Scholar] [CrossRef]
- Voss-Foucart, F. Constituants organiques des coquilles de mollusques actuels et fossiles. Haliotis 1972, 2, 81–88. [Google Scholar]
- Weiner, S.W. Aspartic acid-rich proteins: Major components of the soluble organic matrix of mollusk shells. Calcif. Tissue Int. 1979, 29, 167–193. [Google Scholar]
- Bonucci, E. Biological Calcification. Normal and Pathological Processes in the Early Stages; 2007; p. 592. Springer-Verlag: Berlin, Germany. [Google Scholar]
- Weiner, S.; Addadi, L.; Gotliv, B.A. Mollusk shell acidic proteins: in search of individual functions. Biochemistry 2003, 4, 522–529. [Google Scholar]
- Dunca, E.; Fiebig, E.; Pfeiffer, P.; Schöne, B.R. Mutvei’s solution: An ideal agent for resolving microgrowth structures of biogenic carbonates. Paleoegeogr. Palaeoclim. Palaeoecol. 2005, 228, 149–166. [Google Scholar] [CrossRef]
- Dauphin, Y.; Farre, B. Lipids from the nacreous and prismatic layers of two Pteriomorpha Mollusk shells. Comp. Biochem. Physiol. 2009, B152, 103–109. [Google Scholar]
- Lopez, E.; Le Ny, K.; Gasser, P.; Lati, E.; Bédouet, L.; Rousseau, M. Restoration of stratum corneum with nacre lipids. Comp. Biochem. Physiol. 2006, B145, 1–9. [Google Scholar]
- Akera, S.; Horita, C.; Hasegawa, K.; Kono, M.; Hayashi, N.; Samata, T. A new matrix protein family related to the nacreous layer formation of Pinctada fucata. FEBS Lett. 1999, 462, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Giraud, M.; Lopez, E.; Milet, C.; Marin, F.; Schuller, M.J.; Bédouet, L. Soluble proteins of the nacre of the giant oyster Pinctada maxima and of the abalone Haliotis tuberculata: Extraction and partial analysis of nacre proteins. Comp. Biochem. Physiol. 2001, B128, 389–400. [Google Scholar]
- Lopez, E.; Milet, C.; Barthelemy, M.; Peduzzi, J.; Ribeiro, C.; Almeida, M.J.; Pereira-Mouries, L. Soluble silk-like organic matrix in the nacreous layer of the bivalve Pinctada maxima. Eur. J. Biochem. 2002, 269, 4994–5003. [Google Scholar] [CrossRef] [PubMed]
- Samata, T.; Hayashi, N.; Kono, M. Molecular mechanism of the nacreous layer formation in Pinctada maxima. Biochem. Biophys. Res. Com. 2000, 269, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Xie, L.; Zhang, R.; Zhang, C.; Ma, C. Extraction and purification of matrix protein from the nacre of pearl oyster Pinctada fucata. TSINGHUA Sci. Tech. 2005, 10, 499–503. [Google Scholar] [CrossRef]
- Xu, Z.F.; Zhou, W.; Wang, Y.; Chen, R.; Su, W.; Zhou, J.; Zhang, X.; Lao, Y. Characterization and in vitro mineralization function of a soluble protein complex P60 from the nacre of Pinctada fucata. Comp. Biochem. Physiol. 2007, B148, 201–208. [Google Scholar]
- Zhang, R.; Xie, L.; Ma, Z.; Yan, Z.; Liu, X.; Shangguan, J.; Gong, N. Immunolocalization of matrix proteins in nacre lamellae and their in vivo effects on aragonitic tablet growth. J. Struct. Biol. 2008, 164, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xie, L.; Yan, Z.; Li, Q.; Li, Q.; Ma, Z.; Gong, N. Characterization of calcium deposition and shell matrix protein secretion in primary mantle tissue culture from the marine pearl oyster Pinctada fucata. Biotechnology 2008, 10, 457–465. [Google Scholar]
- Samata, T.; Ishikawa, F.; Norizuki, M.; Takakura, D. Isolation and characterization of the N-linked oligosaccharides in nacrein from Pinctada fucata. Mar. Biotechnol. 2008, 10, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Westermeier, R. Electrophoresis in practice. In A Guide to Methods and Applications of DNA and Protein Separations; 1997; p. 331. VCH Verlagsgesellschaft mbH: Weinhiem, Germany. [Google Scholar]
- Dauphin, Y. Comparative studies of skeletal soluble matrices from some scleractinian corals and mollusks. Int. J. Biol. Macromol. 2001, 28, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Dauphin, Y. Comparison of the soluble matrices of the calcitic prismatic layer of Pinna nobilis (Mollusca, Bivalvia, Pteriomorpha). Comp. Biochem. Physiol. 2002, A132, 577–590. [Google Scholar]
- Mutvei, H. On the micro- and ultrastructure of the conchiolin in the nacreous layer of some recent and fossil mollusks. Stockholm Contr. Geol. 1969, XX, 1–16. [Google Scholar]
- Ristedt, H.; Crenshaw, M.A. The histochemical localization of reactive groups in septal nacre from Nautilus pompilius L. In The Mechanisms of Mineralization in the Invertebrates and Plants; Wilbur, K.M., Eds., Watabe, N., Eds.; 1976; Volume 5, pp. 355–367. The University of South Carolina Press: Columbia, SC, USA. [Google Scholar]
- Weiner, S.; Addadi, L.; Gotliv, B.A.; Nudelman, F. Mollusk shell formation: mapping the distribution of organic matrix components underlying a single aragonitic tablet in nacre. J. Struct. Biol. 2006, 153, 176–187. [Google Scholar] [CrossRef] [PubMed]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dauphin, Y.; Nouet, J. Fits and Misfits in Organic Matrix Analyses: Case of the Soluble Matrices of the Nacreous Layer of Pinctada margaritifera (Mollusca). Minerals 2012, 2, 40-54. https://doi.org/10.3390/min2010040
Dauphin Y, Nouet J. Fits and Misfits in Organic Matrix Analyses: Case of the Soluble Matrices of the Nacreous Layer of Pinctada margaritifera (Mollusca). Minerals. 2012; 2(1):40-54. https://doi.org/10.3390/min2010040
Chicago/Turabian StyleDauphin, Yannicke, and Julius Nouet. 2012. "Fits and Misfits in Organic Matrix Analyses: Case of the Soluble Matrices of the Nacreous Layer of Pinctada margaritifera (Mollusca)" Minerals 2, no. 1: 40-54. https://doi.org/10.3390/min2010040