
Organocatalytic Asymmetric α-Chlorination of 1,3-Dicarbonyl Compounds Catalyzed by 2-Aminobenzimidazole Derivatives



Abstract
: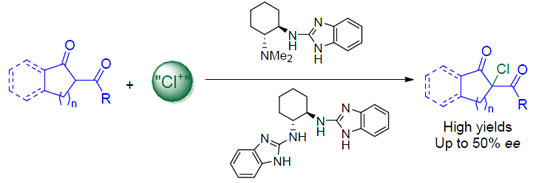
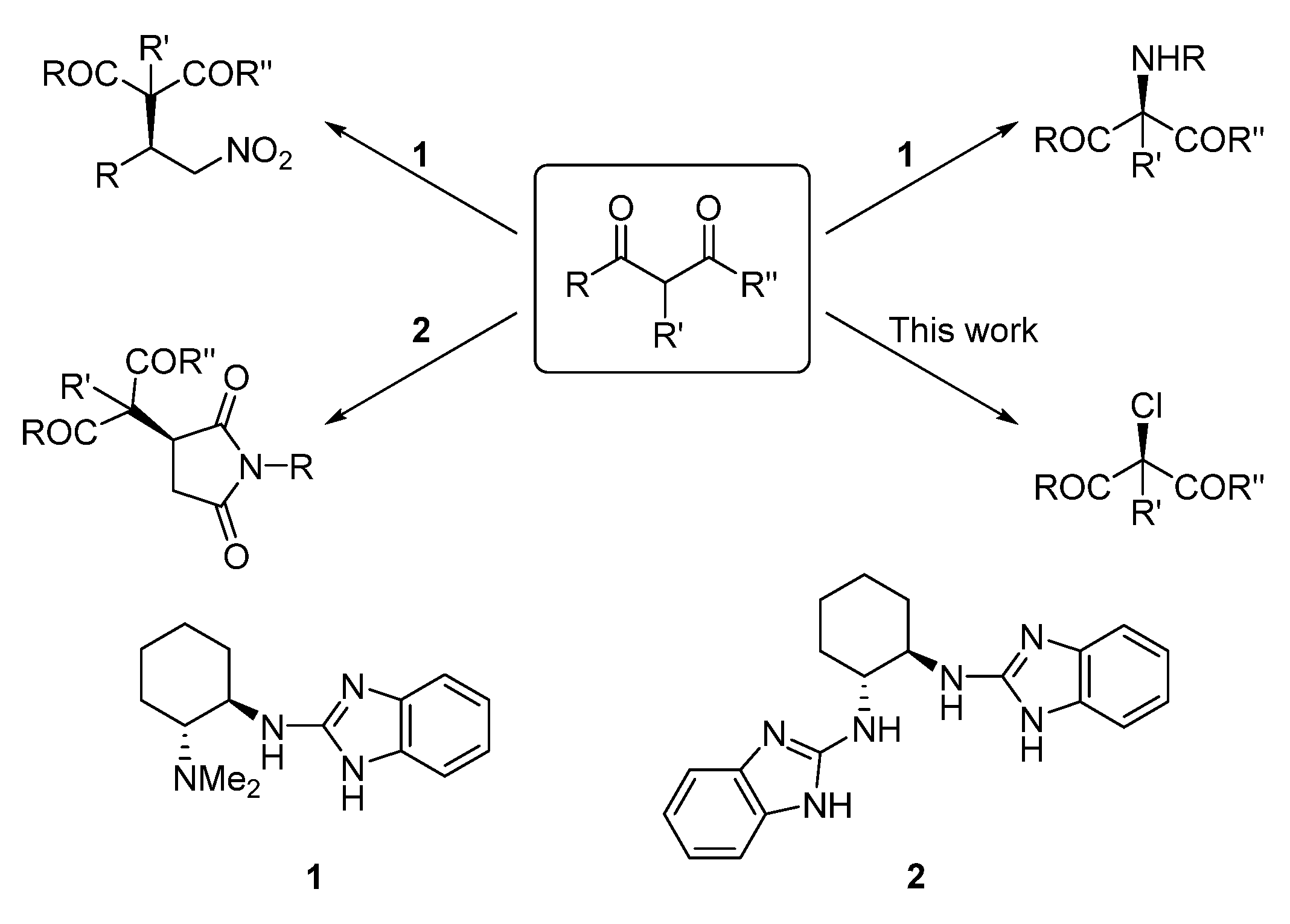
1. Introduction
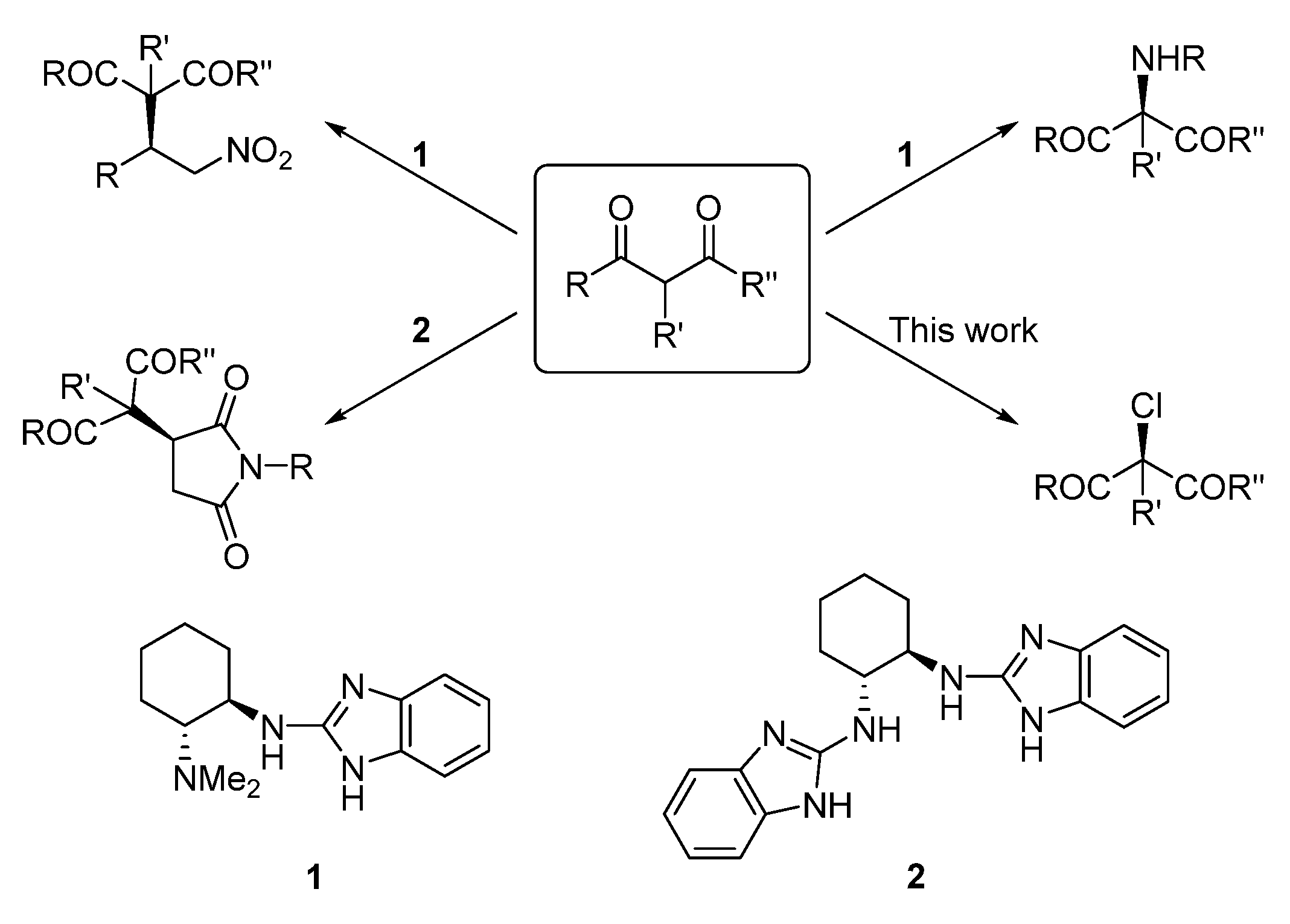
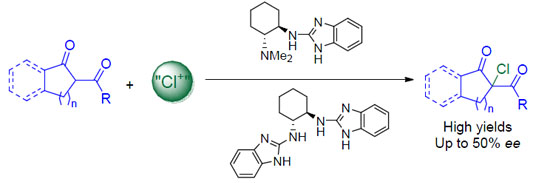
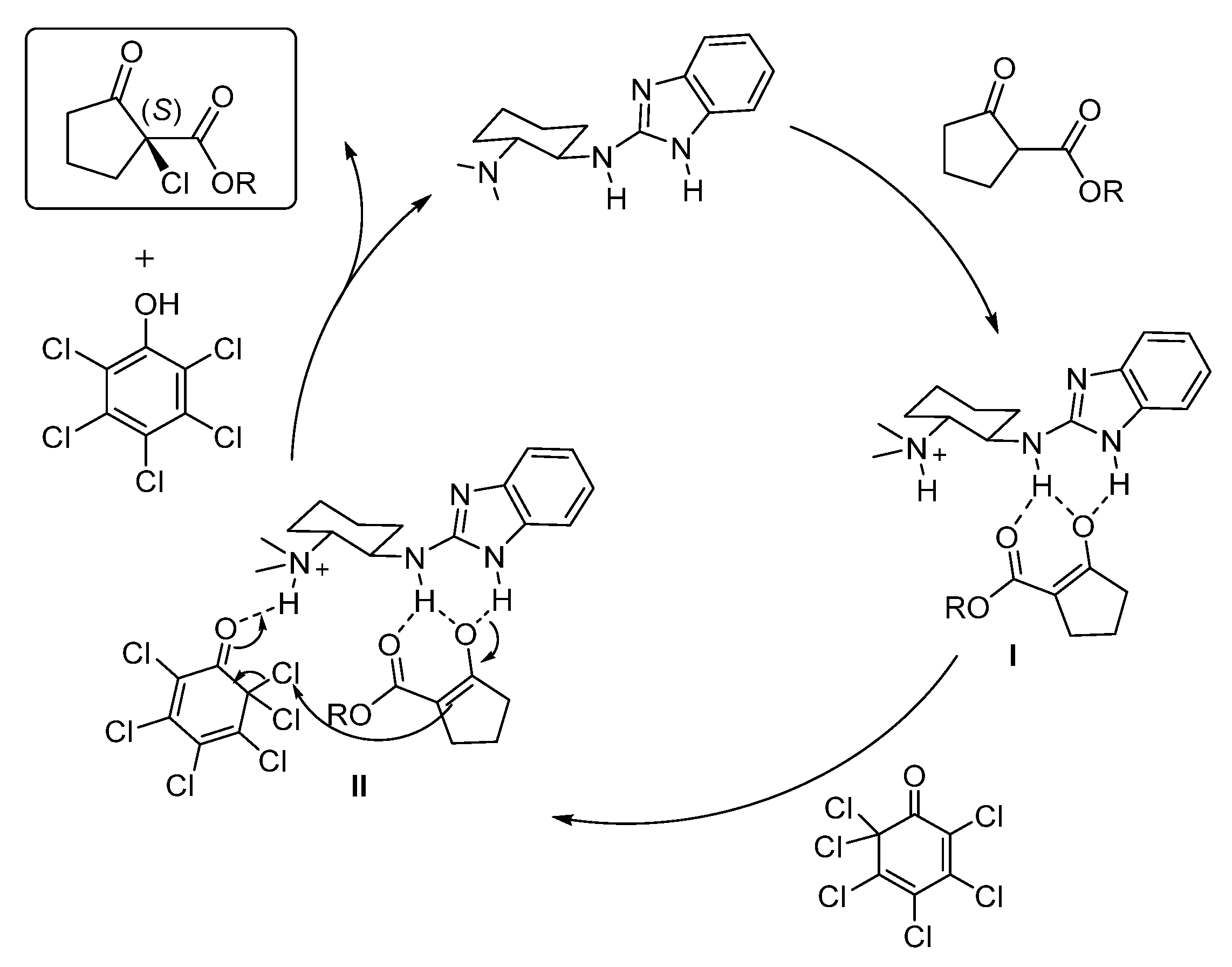
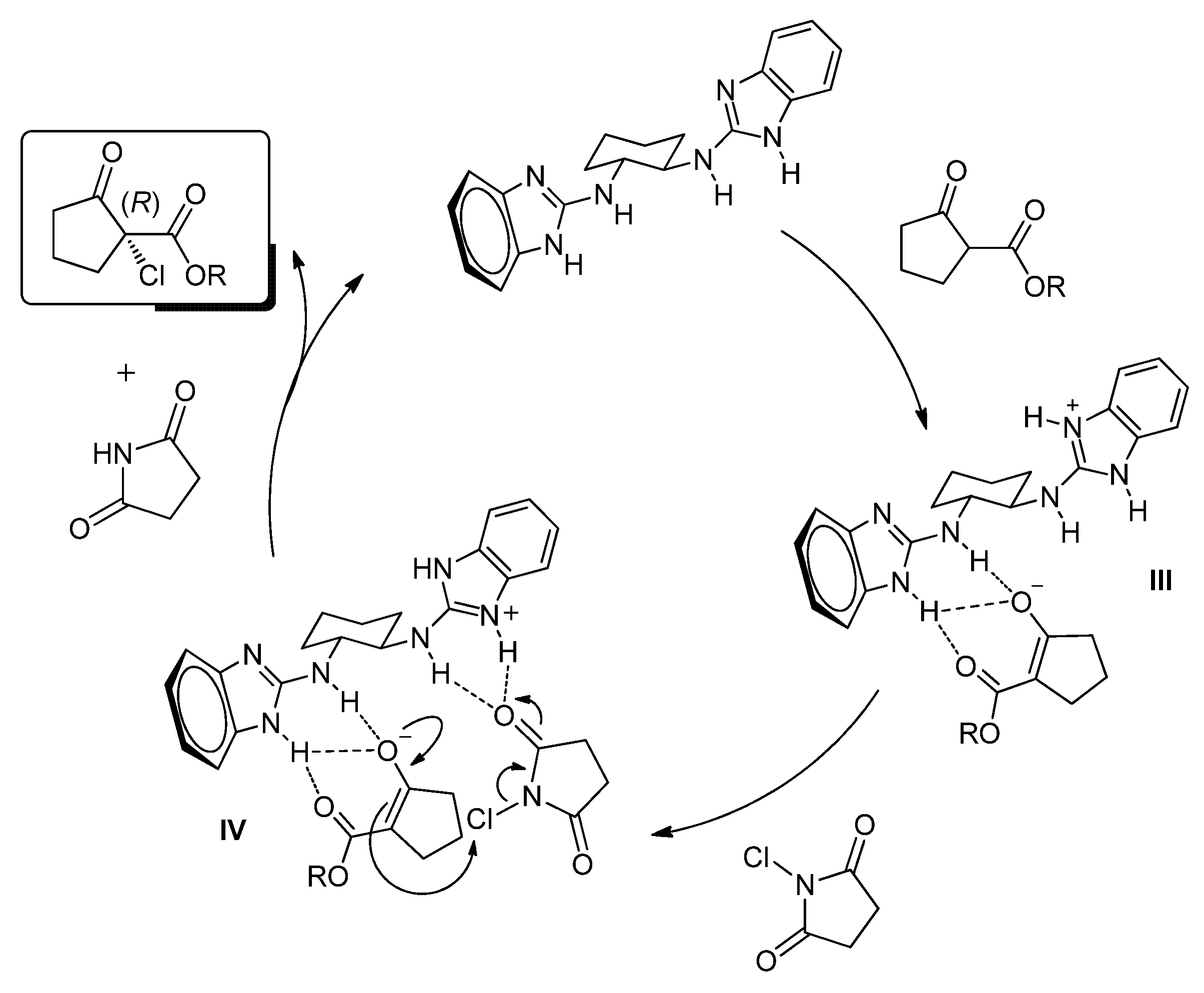
2. Results and Discussion


| Entry | Catalyst | T (°C) | Conv. (%) a | ee (%) a |
|---|---|---|---|---|
| 1 | 1 | 20 | 99 | 5 |
| 2 | 1 | −50 | 99 | 9 |
| 3 | 2 | −50 | 99 (95) b | 40 |
| 4 | 2 | −78 | 99 | 40 |
| 5 | 3 | −50 | 99 | 12 |
| Entry | Catalyst (mol%) | Cocatalyst (mol%) | Chlorine Source | T (°C) | Conv. (%) a | ee (%) a |
|---|---|---|---|---|---|---|
| 1 | 1 (10) | – | 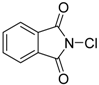 | −50 | 99 | 7 |
| 2 | 2 (10) | – | −50 | 99 | 40 | |
| 3 | 1 (10) | – | 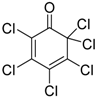 | −50 | 99 | 31 |
| 4 | 2 (10) | – | −50 | 99 | 10 | |
| 5 | 1 (10) | – | 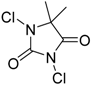 | −50 | 99 | 8 |
| 6 | 2 (10) | – | −50 | 99 | 14 | |
| 7 | 1 (10) | – | 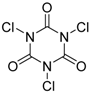 | −50 | 99 | 8 |
| 8 | 2 (10) | – | −50 | 99 | 1 | |
| 9 | 1 (10) | – | 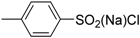 | −50 | <5 | – |
| 10 | 2 (10) | – | −50 | <5 | – | |
| 11 | 1 (10) | – |  | −78 | 99 | 31 |
| 12 | 1 (5) | – | −50 | 99 | 27 | |
| 13 | 1 (20) | – | −50 | 99 | 31 | |
| 14 | 2 (20) | – | 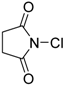 | −50 | 99 | 47 |
| 15 | 2 (20) | TFA (20) | −50 | 99 | 5 | |
| 16 | 2 (20) | NaHCO3 (100) | −50 | 99 | 28 | |
| 17 | 2 (10) | TEA (10) | −50 | 99 | 2 |
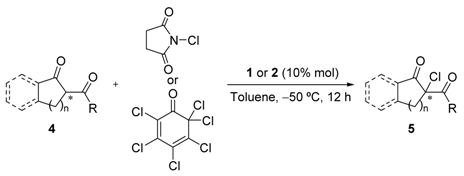
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Nucleophile | No. | Method | Product | |||
|---|---|---|---|---|---|---|---|
| Structure | No. | Yield (%) b | ee (%) c | ||||
| 1 |  | 4a | A | 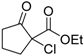 | 5a | 99 | 31 |
| 2 | 4a | B | 5a | 99 | 40 | ||
| 3 | 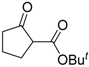 | 4b | A | 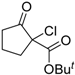 | 5b | 97 | 21 |
| 4 | 4b | B | 5b | 95 | 47 | ||
| 5 | 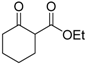 | 4c | A |  | 5c | 93 | 39 |
| 6 | 4c | B | 5c | 92 | <5 | ||
| 7 |  | 4d | A | 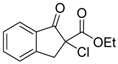 | 5d | 98 | 39 |
| 8 | 4d | B | 5d | 96 | 21 | ||
| 9 |  | 4e | A | 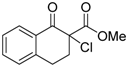 | 5e | 98 | 40 |
| 10 | 4e | B | 5e | 98 | 15 | ||
| 11 |  | 4f | A |  | 5f | 97 | 50 |
| 12 | 4f | B | 5f | 99 | 34 | ||
| 13 | 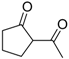 | 4g | A | 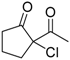 | 5g | 99 | <5 |
| 14 | 4g | B | 5g | 99 | 5 | ||
| 15 | 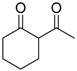 | 4h | A |  | 5h | 98 | 10 |
| 16 | 4h | B | 5h | 98 | 5 | ||
| 17 | 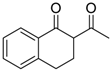 | 4i | A | 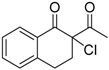 | 5i | 96 | 14 |
| 18 | 4i | B | 5i | 94 | 8 | ||

3. Experimental Section
3.1. General Remarks
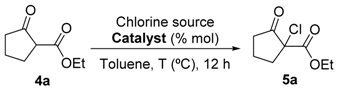
3.2. General Procedure for the Asymmetric Chlorination of 1,3-Dicarbonyl Compounds
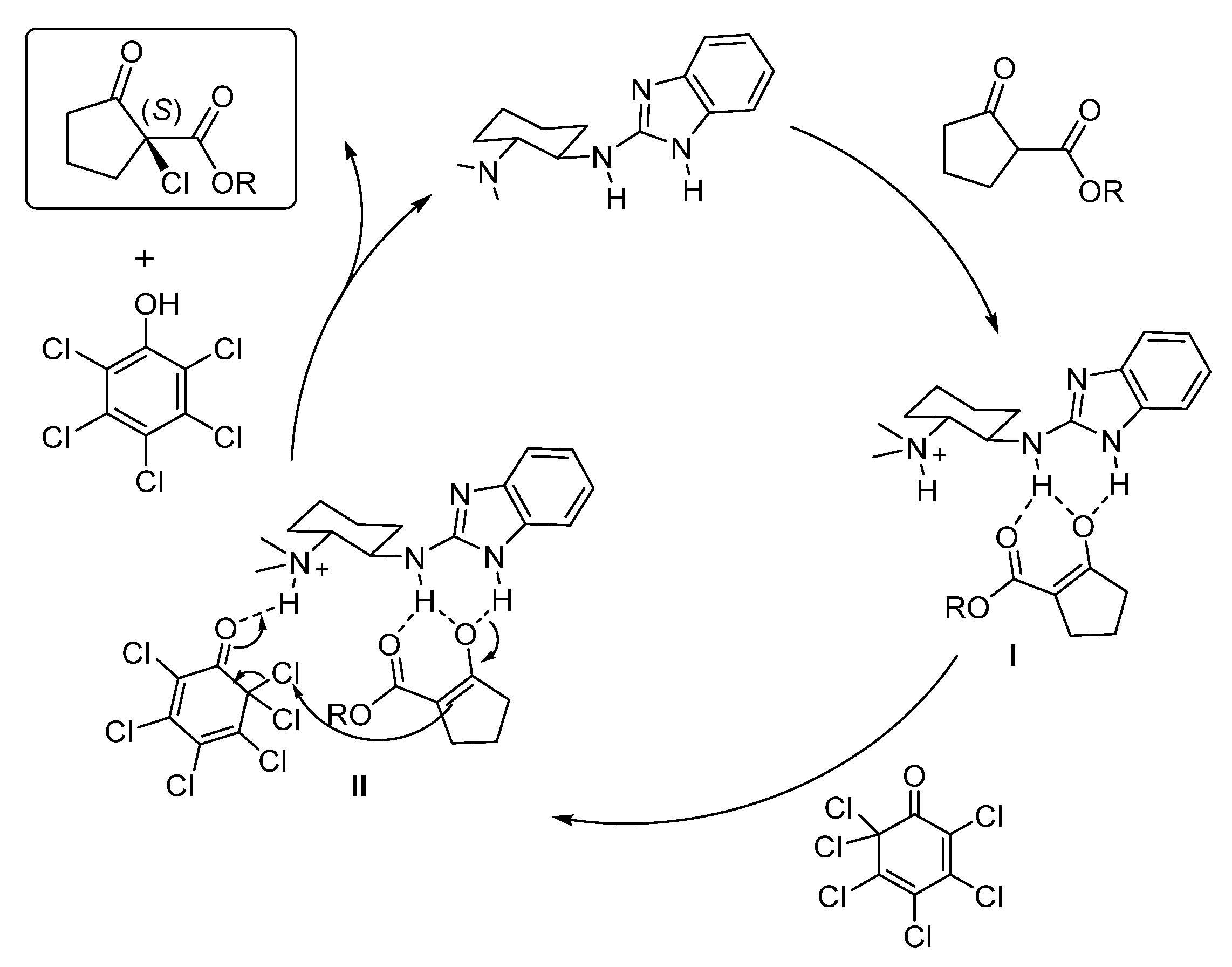
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Thomas, G. Medicinal Chemistry: An Introduction; John Wiley & Sons: New York, NY, USA, 2000. [Google Scholar]
- De Kimpe, N.; Verhé, R. The Chemistry of α-Haloketones, α-Haloaldehydes, and α-Haloimines; John Wiley & Sons: New York, NY, USA, 1990. [Google Scholar]
- Cornil, J.; Guérinot, A.; Cossy, J. Linchpin dienes: Key building-blocks in the synthesis of polyenic frameworks. Org. Biomol. Chem. 2015, 13, 4129–4142. [Google Scholar] [CrossRef] [PubMed]
- St. Denis, J.D.; He, Z.; Yudin, A.K. Amphoteric α-boryl aldehyde linchpins in the synthesis of heterocycles. ACS Catal. 2015, 5, 5373–5379. [Google Scholar] [CrossRef]
- Marigo, M.; Jørgensen, K.A. Organocatalytic direct asymmetric α-heteroatom functionalization of aldehydes and ketones. Chem. Commun. 2006, 2015, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Kano, T.; Maruoka, K. Organocatalyzed direct asymmetric α-halogenation of carbonyl compounds. Org. Biomol. Chem. 2009, 7, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Shibatomi, K.; Narayama, A. Catalytic enantioselective α-chlorination of carbonyl compounds. Asian J. Org. Chem. 2013, 2, 812–823. [Google Scholar] [CrossRef]
- Dalko, P.I. Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- De Figueiredo, R.M.; Christmann, M. Organocatalytic Synthesis of Drugs and Bioactive Natural Products. Eur. J. Org. Chem. 2007, 2007, 2575–2600. [Google Scholar] [CrossRef]
- Marqués-López, E.; Herrera, R.P.; Christmann, M. Asymmetric organocatalysis in total synthesis—A trial by fire. Nat. Prod. Rep. 2010, 27, 1138–1167. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.; Cabrera, S. Applications of asymmetric organocatalysis in medicinal chemistry. Chem. Soc. Rev. 2013, 42, 774–793. [Google Scholar] [CrossRef] [PubMed]
- Brochu, M.P.; Brown, S.P.; MacMillan, D.W.C. Direct and enantioselective organocatalytic α-chlorination of aldehydes. J. Am. Chem. Soc. 2004, 126, 4108–4109. [Google Scholar] [CrossRef] [PubMed]
- France, S.; Wack, H.; Taggi, A.E.; Hafez, A.M.; Wagerle, T.R.; Shah, M.H.; Dusich, C.L.; Lectka, T. Catalytic, Asymmetric α-chlorination of acid halides. J. Am. Chem. Soc. 2004, 126, 4245–4255. [Google Scholar] [CrossRef] [PubMed]
- Marigo, M.; Bachmann, S.; Halland, N.; Braunton, A.; Jørgensen, K.A. Highly enantioselective direct organocatalytic α-chlorination of ketones. Angew. Chem. Int. Ed. 2004, 43, 5507–5510. [Google Scholar] [CrossRef] [PubMed]
- Bergeron-Briek, M.; Teoh, T.; Britton, R. A tandem organocatalytic α-chlorination-aldol reaction thet proceeds with dynamic kinetic resolution: A powerful tool for carbohydrate synthesis. Org. Lett. 2013, 15, 3554–3557. [Google Scholar] [CrossRef] [PubMed]
- Halland, N.; Braunton, A.; Bachmann, S.; Marigo, M.; Jørgensen, K.A. Direct Organocatalytic Asymmetric α-Chlorination of Aldehydes. J. Am. Chem. Soc. 2004, 126, 4790–4791. [Google Scholar] [CrossRef] [PubMed]
- Fadeyi, O.O.; Schulte, M.L.; Lindsley, C.W. General Access to Chiral N-Alkyl Terminal Aziridines via Organocatalysis. Org. Lett. 2010, 12, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cai, C.; Curran, D.P.; Zhang, W. Enantioselective α-Chlorination of Aldehydes with Recyclable Fluorous (S)-Pyrrolidine-Thiourea Bifunctional Organocatalyst. Synlett 2010, 3, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Winter, P.; Swatschek, J.; Willout, M.; Radtke, L.; Olbrisch, T.; Schäfer, A.; Christmann, M. Transforming Terpene-derived Aldehydes into 1,2-Epoxides via Asymmetric α-chlorination: Subsequent epoxide opening with carbon nucleophiles. Chem. Commun. 2011, 47, 12200–12202. [Google Scholar] [CrossRef] [PubMed]
- Johannes, M.; Brimble, M.A. Synthesis of an Azido Precursor to (2S,5R)-5-Hydroxylysine Using an Asymmetric Organocatalytic Chlorination/Reduction Sequence. J. Org. Chem. 2013, 78, 12809–12813. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, G.; Bosco, M.; Carlone, A.; Locatelli, M.; Melchiorre, P.; Sambri, L. Organocatalytic Asymmetric α-Halogenation of 1,3-Dicarbonyl Compounds. Angew. Chem. Int. Ed. 2005, 44, 6219–6222. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Wang, W.; Shen, K.; Wang, J.; Hu, X.; Lin, L.; Liu, X.; Feng, X. Highly α-Chlorination of Cyclic β-Ketoesters Catalyzed by N,N′-Dioxide Using NCS as the Chlorice Source. Chem. Commun. 2010, 46, 1250–1252. [Google Scholar] [CrossRef] [PubMed]
- Etayo, P.; Badorrey, R.; Díaz-de-Villegas, M.D.; Gálvez, J.A. Chral Amino Diol Derivatives as New Modular Organocatalysts for the Enantioselective α-Chlorination of Cyclic β-Keto Esters. Adv. Synth. Catal. 2010, 352, 3329–3338. [Google Scholar] [CrossRef]
- Shirakawa, S.; Tokuda, T.; Kasai, A.; Maruoka, K. Design of Chiral Bifunctional Quaternary Phosphonium Bromide Catalysts Possessing an Amide Moiety. Org. Lett. 2013, 15, 3350–3353. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, F.; Meng, Q.; Gao, Z. Enantioselective α-Chlorination of β-Oxo Esters Catalyzed by Chiral Diterpenoid Alkaloid Derivatives. Tetrahedron Asymmetry 2014, 25, 1215–1220. [Google Scholar] [CrossRef]
- Novacek, J.; Monkowius, U.; Himmelsbach, M.; Waser, M. Asymmetric α-Chlorination of β-Ketoesters Using Bifunctional Ammonium Salt Catalysis. Monatsh. Chem. 2015. [Google Scholar] [CrossRef]
- Yin, Q.; Wang, S.-G.; Liang, X.-W.; Gao, D.W.; Zheng, J. You, S.-L. Organocatalytic Asymmetric Chlorinative Dearomatization of Naphthols. Chem. Sci. 2015, 6, 4179–4183. [Google Scholar] [CrossRef]
- Zhao, M.-X.; Zhang, Z.-W.; Chen, M.-X.; Tang, W.-H.; Shi, M. Cinchona Alkaloid Catalyzed Enantioselective Chlorination of 3-Aryloxindoles. Eur. J. Org. Chem. 2011, 2011, 3001–3008. [Google Scholar] [CrossRef]
- Gao, X.; Han, J.; Wang, L. Design of Highly Stable Iminophosphoranes as Recyclable Organocatalysts: Application to Asymmetric Chlorinations of Oxindoles. Org. Lett. 2015, 17, 4596–4599. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.Y.; Wasa, M.; Jacobsen, E.N. Enantioselective Synthesis of α-Chloro Esters by non-Covalent Catalysis. Tetrahedron Lett. 2015, 56, 3428–3430. [Google Scholar] [CrossRef] [PubMed]
- Almaşi, D.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Chiral 2-Aminobenzimidazoles as Recoverable Organocatalysts for the Addition of 1,3-Dicarbonyl Compounds to Nitroalkenes. J. Org. Chem. 2009, 74, 6163–6168. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Torres, E.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Conjugate Addition of 1,3-Dicarbonyl Compounds to Maleimides Using a Chiral C2-Symmetric Bis(2-aminobenzimidazole) as Recyclable Organocatalyst. Org. Lett. 2011, 13, 6106–6109. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Torres, E.; Alonso, D.A.; Gómez-Bengoa, E.; Nájera, C. Enantioselective Synthesis of Succinimides by Michael Addition of 1,3-Dicarbonyl Compounds to Maleimides Catalyzed by a Chiral Bis(2-aminobenzimidazole) Organocatalyst. Eur. J. Org. Chem. 2013, 2013, 1434–1440. [Google Scholar] [CrossRef]
- Trillo, P.; Gómez-Martínez, M.; Alonso, D.A.; Baeza, A. 2-Aminobenzimidazole Organocatalyzed Asymmetric Amination of Cyclic 1,3-Dicarbonyl Compounds. Synlett 2015, 26, 95–100. [Google Scholar]
- Duan, X.-H.; Mayr, H. Electrophilicities of α-Chlorinating Agents Used in Organocatalysis. Org. Lett. 2010, 12, 2238–2241. [Google Scholar] [CrossRef] [PubMed]
- We have also corroborated an important contribution of the non-chiral background reaction for methods A (85% conversion by GC after 12 h) and B (94% conversion by GC after 12 h).
- Since only a 7% improvement of ee was obtained using 20 mol% of catalyst loading, we decided to carry out the substrate scope with a 10 mol% of the corresponding chiral catalyst.
- Northrup, A.B.; MacMillan, D.W.C. Two-Step Synthesis of Carbohydrates by Selective Aldol Reactions. Science 2004, 305, 1752–1755. [Google Scholar] [CrossRef] [PubMed]
- Flores-Ferrándiz, J.; Fiser, B.; Gómez-Bengoa, E.; Chinchilla, R. Solvent-Induced Reversal of Enantioselectivity in the Synthesis of Succinimides by the Addition of Aldehydes to Maleimides Catalysed by Carbamate-Monoprotected 1,2-Diamines. Eur. J. Org. Chem. 2015, 2015, 1218–1225. [Google Scholar] [CrossRef]
- Moteki, S.A.; Han, J.; Arimitsu, S.; Akakura, M.; Nakayama, K.; Maruoka, K. An Achiral-Acid-Induced Switch in the Enantioselectivity of a Chiral cis-Diamine-Based Organocatalyst for Asymmetric Aldol and Mannich Reactions. Angew. Chem. Int. Ed. 2012, 51, 1187–1190. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.T.; Luparia, M.; Audisio, D.; Maulide, N. Dual Catalysis Becomes Diastereodivergent. Angew. Chem. Int. Ed. 2013, 52, 13149–13152. [Google Scholar] [CrossRef] [PubMed]
- Krautwald, S.; Sarlah, D.; Schafroth, M.A.; Carreira, E.M. Enantio- and Diastereodivergent Dual Catalysis: α-Allylation of Branched Aldehydes. Science 2013, 340, 1065–1068. [Google Scholar] [CrossRef] [PubMed]
- Halland, N.; Lie, M.A.; Kjærsgaard, A.; Marigo, M.; Schiøtt, B.; Jørgensen, K.A. Mechanistic Investigation of the 2,5-Diphenylpyrrolidine-Catalyzed Enantioselective α-Chlorination of Aldehydes. Chem. Eur. J. 2005, 11, 7083–7090. [Google Scholar] [CrossRef] [PubMed]
- Marquez, C.A.; Fabbretti, F.; Metzger, J.O. Electrospray Ionization Mass Spectrometric Study on the Direct Organocatalytic α-Halogenation of Aldehydes. Angew. Chem. Int. Ed. 2007, 46, 6915–6917. [Google Scholar] [CrossRef] [PubMed]
- Hintermann, L.; Togni, A. Catalytic Enantioselective Chlorination and Bromination of β-Keto Esters. Helv. Chim. Acta 2000, 83, 2425–2435. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez, D.S.; Baeza, A.; Alonso, D.A. Organocatalytic Asymmetric α-Chlorination of 1,3-Dicarbonyl Compounds Catalyzed by 2-Aminobenzimidazole Derivatives. Symmetry 2016, 8, 3. https://doi.org/10.3390/sym8010003
Sánchez DS, Baeza A, Alonso DA. Organocatalytic Asymmetric α-Chlorination of 1,3-Dicarbonyl Compounds Catalyzed by 2-Aminobenzimidazole Derivatives. Symmetry. 2016; 8(1):3. https://doi.org/10.3390/sym8010003
Chicago/Turabian StyleSánchez, Daniel Serrano, Alejandro Baeza, and Diego A. Alonso. 2016. "Organocatalytic Asymmetric α-Chlorination of 1,3-Dicarbonyl Compounds Catalyzed by 2-Aminobenzimidazole Derivatives" Symmetry 8, no. 1: 3. https://doi.org/10.3390/sym8010003