Supramolecular Chirality in Porphyrin Chemistry

Abstract
: Supramolecular chirality, being an intelligent combination of supramolecular chemistry and chiral science, plays a decisive role in the functioning of various natural assemblies and has attracted much attention from the scientific community, due to different applications in modern technologies, medicine, pharmacology, catalysis and biomimetic research. Porphyrin molecules are of particular interest to study this phenomenon owing to their unique spectral, physico-chemical and synthetic properties. This review highlights the most important types of chiral porphyrin structures by using the best-suited representative examples, which are frequently used in the area of supramolecular chirality.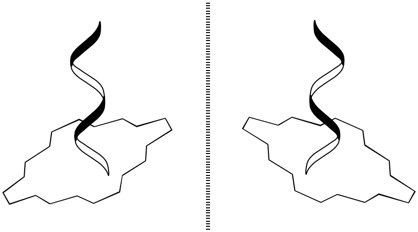
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
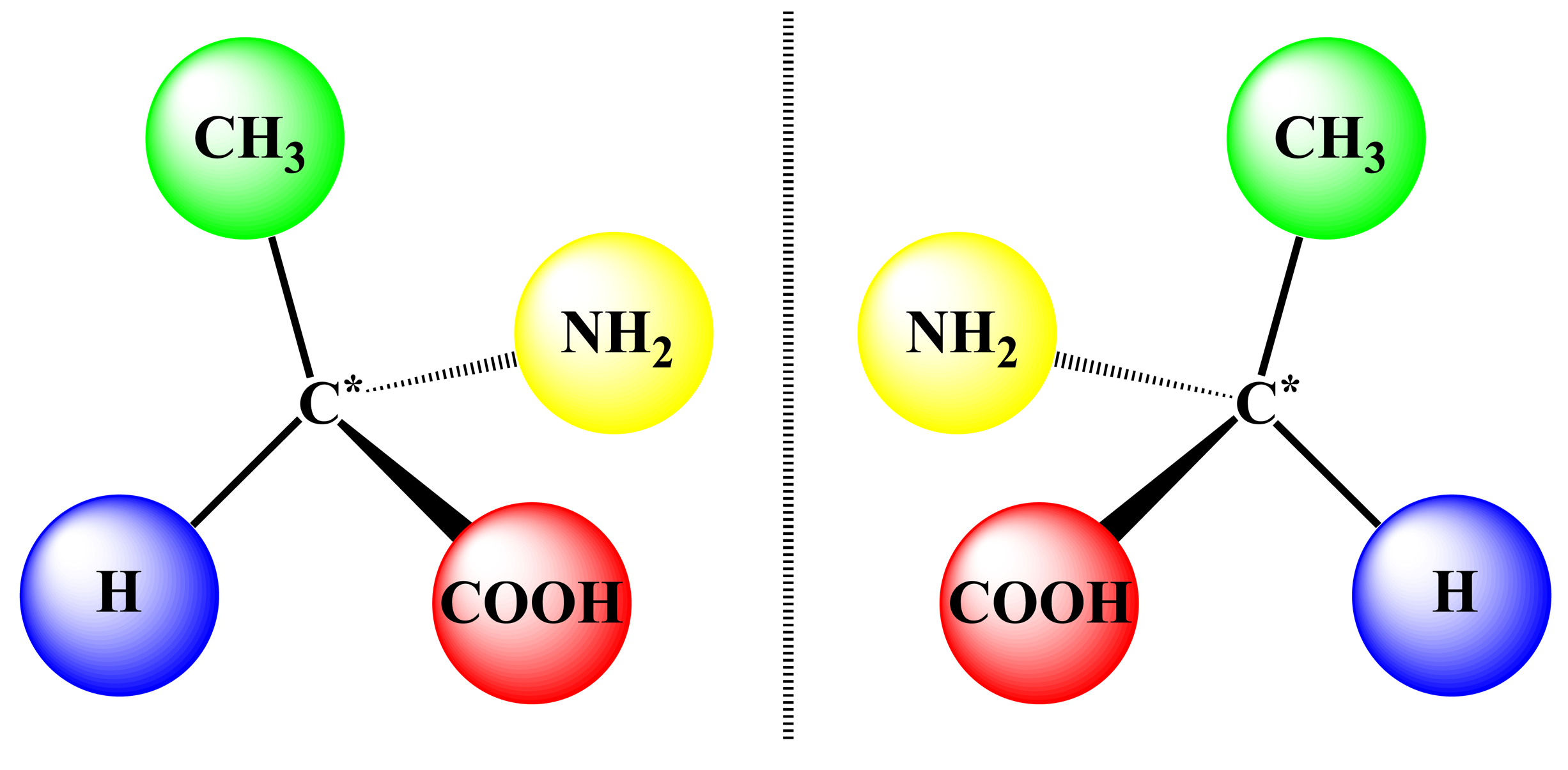
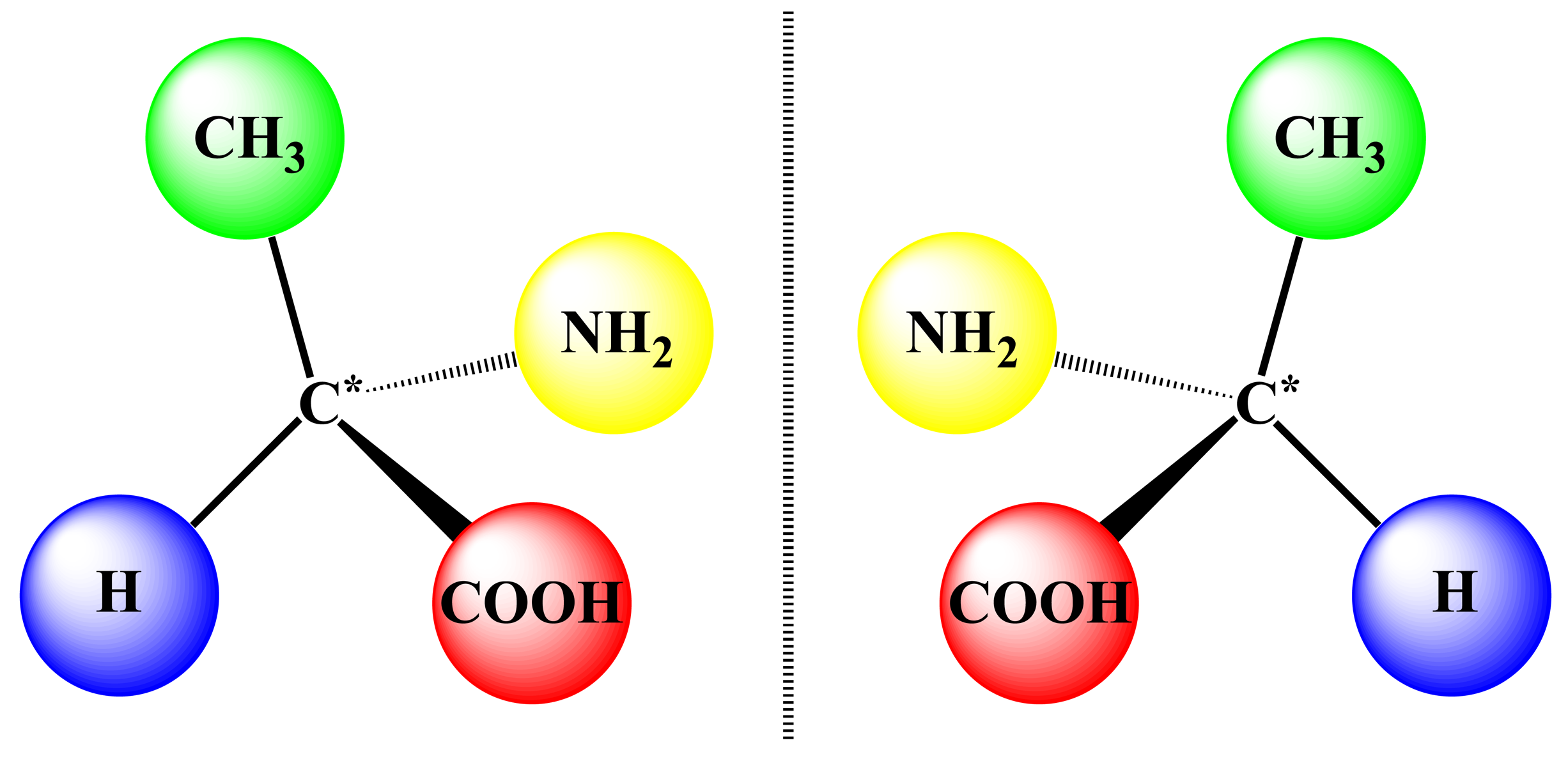
Chirality is one of the most fundamental principles of nature and describes the ability of any object to exist as a pair of non-superimposable mirror images, which are termed enantiomers (Figure 1). In the case of supramolecular chirality, the processes associated with the chirality phenomena are driven by various types of noncovalent interactions between the components of these systems. In this respect, the chiral properties of porphyrinoids are of particular interest, due to their direct relevance to many vital biological processes, such as oxygen transport, electron transfer, enzyme functioning and photosynthesis [1]. Furthermore, these compounds have turned out to be particularly well suited for investigating different chiral processes and, particularly, supramolecular chirality, because of their specific and highly appropriate spectral, physico-chemical and synthetic characteristics, facile handling and superior propensity to form various supramolecular assemblies [2]. Furthermore, this kind of molecular and supramolecular system, so far, has attracted considerable attention of the scientific community on account of the wide applicability in different fields of fundamental and applied sciences and modern technologies lying behind the judicious design of various chiroptical devices and sensors, molecular switches and machines, enantioselective materials and catalysts, as well documented in numerous reviews discussing these topics to a greater or lesser extent [3–15]. In general, chirality in the porphyrin-based supramolecular systems may be generated either via the intrinsic chiral modification of achiral porphyrinoids, by employing naturally occurring chiral pigments or via the external chiral field. In the case of dimeric and multimeric porphyrinoids, the asymmetry induction can be additionally achieved by a chiral linkage. Taking into account the vast number of publications in this area, only the most illustrative examples of each type of structural organization are overviewed in this review article. From the chirality point of view, the rigidly fixed bis- and multi-porphyrinoid systems exhibit a more straightforward relationship between their structures and properties, thus being more suitable systems for comprehensive rationalization, and therefore, their discussion should come first.
2. Chirality and Supramolecular Chirality of Covalently Fixed Rigid Architectures
Generally, in chemistry, the chirality phenomenon most conventionally relates to molecules and/or (supra-) molecular systems, the intrinsic components of which (atoms or functional groups) are asymmetrically arranged in three spatial dimensions around a center, axis or plane. In this regard, the rigidly linked structures provide direct access to the chiral architectures. In the case of bis- and multi-porphyrinoids, this has come about through the introduction of stereogenic element(s) either into the macrocycle directly or into the connecting linkage. Besides, there is another type of multiporphyrin structure, which is based upon the asymmetrical spatial fixation of macrocycles, whilst neither porphyrin units nor covalent linkages themselves have stereogenic centers. Typically, the conformational stability of the overall structure may be ensured by two covalent bridges on the opposite sides of the porphyrin ring or by a single rigid linkage. At first, bis-porphyrinoid structures on the basis of chirally modified macrocycles connected by achiral covalent bridges are discussed.
2.1. Chirality Introduced via Chiral Porphyrinoids
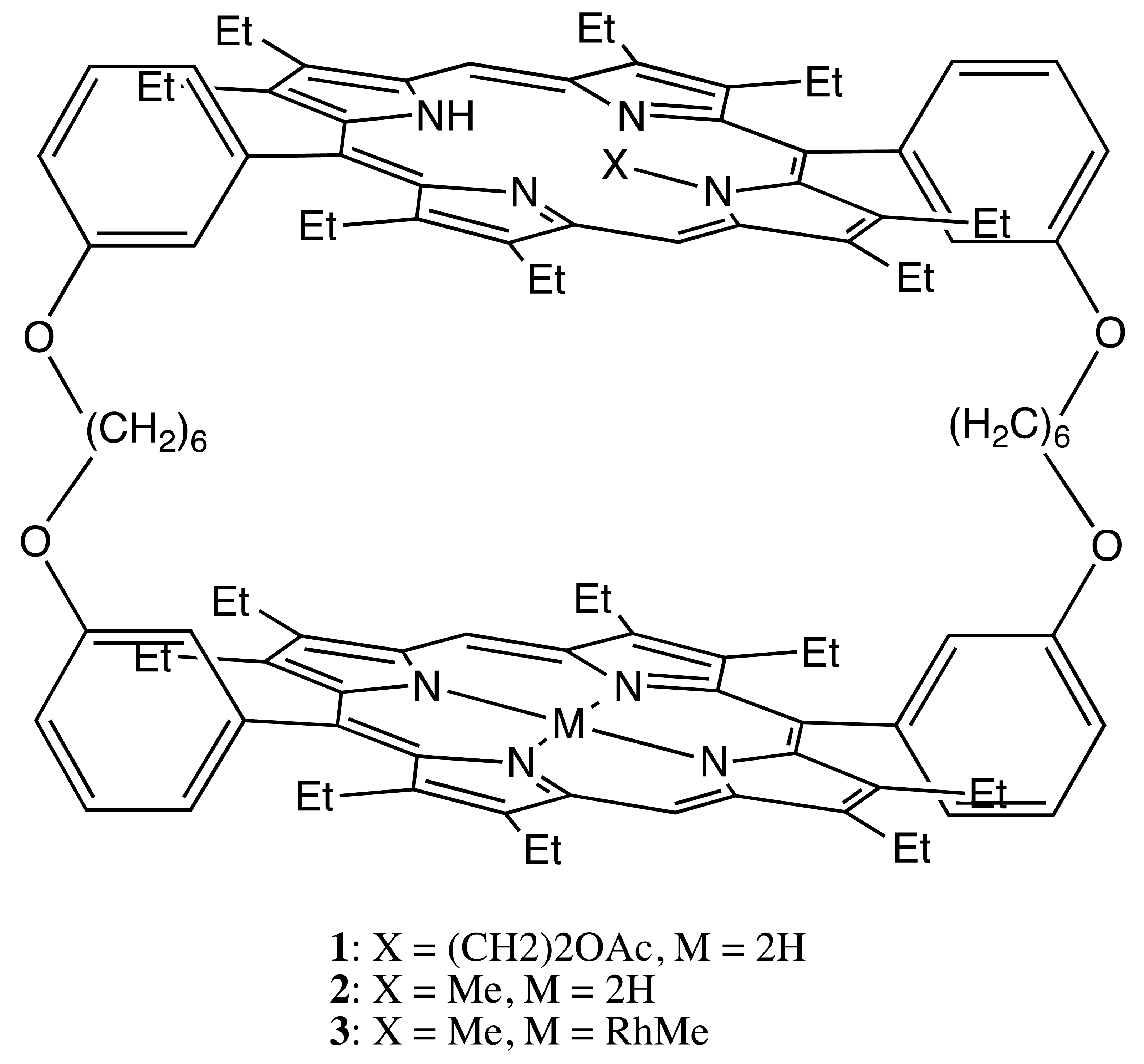
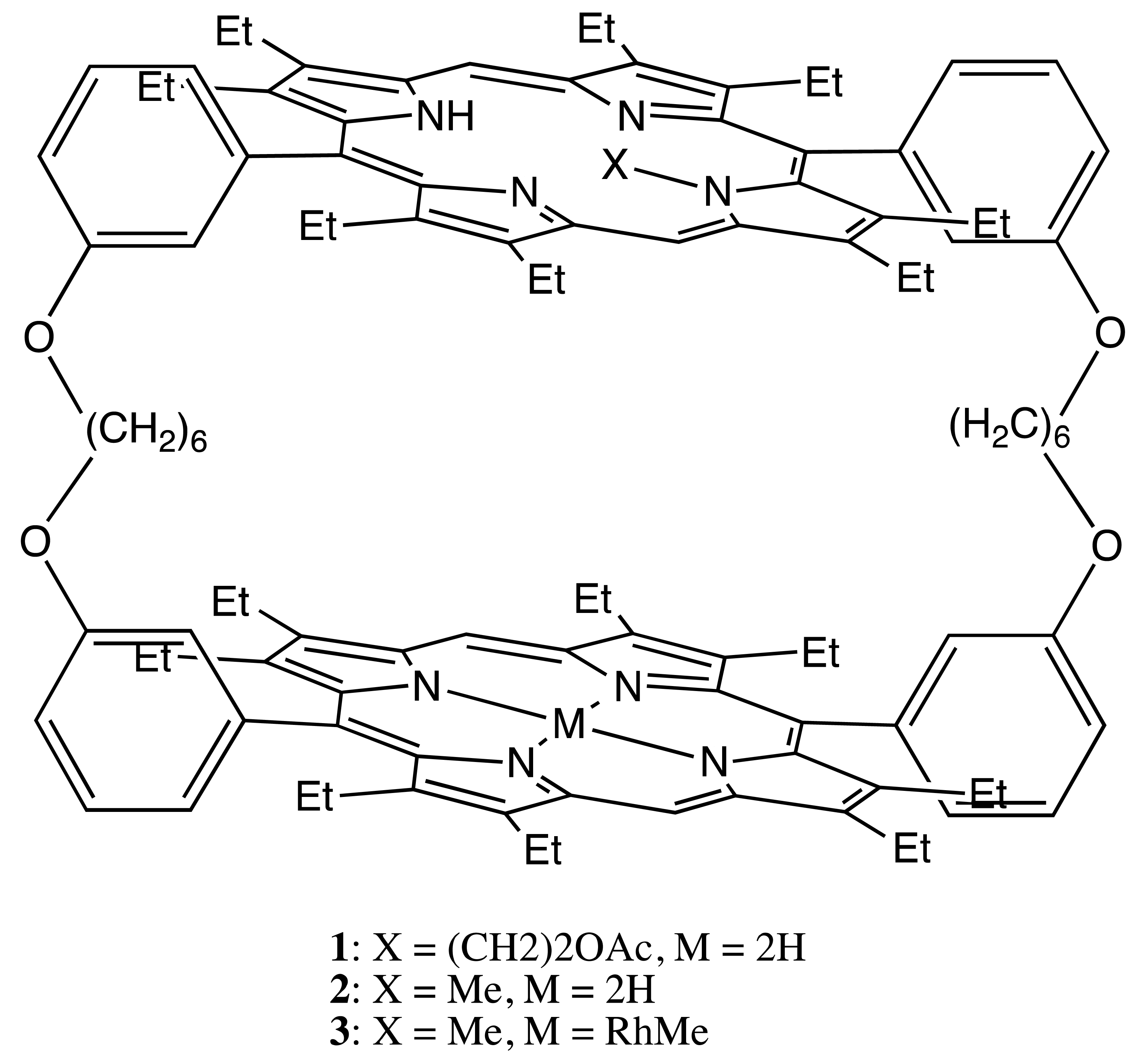
One of the most representative examples of the first structural type is a series of doubly-strapped bis-porphyrins, 1–3 (Figure 2) [16,17]. In this case, chirality was introduced via the alkylation of one of the pyrrole nitrogens, making the alkylated porphyrin asymmetrical, whilst the rigidity of whole structure prevented the inversion process. The mirror image circular dichroism (CD) spectra were obtained for the corresponding enantiomers. The supramolecular chirality of 1–3 was explored upon the noncovalent interaction with the appropriate host molecules. Particularly, the interporphyrin cavity of these bis-porphyrins was designed to accommodate spheroidal fullerenes, especially C76, whilst the chiral properties were applied for discriminating the enantiomers of C76 (by 3) and enantioselective extraction (by 1), resulting in 7% enantiomeric excess (ee) in a single procedure.
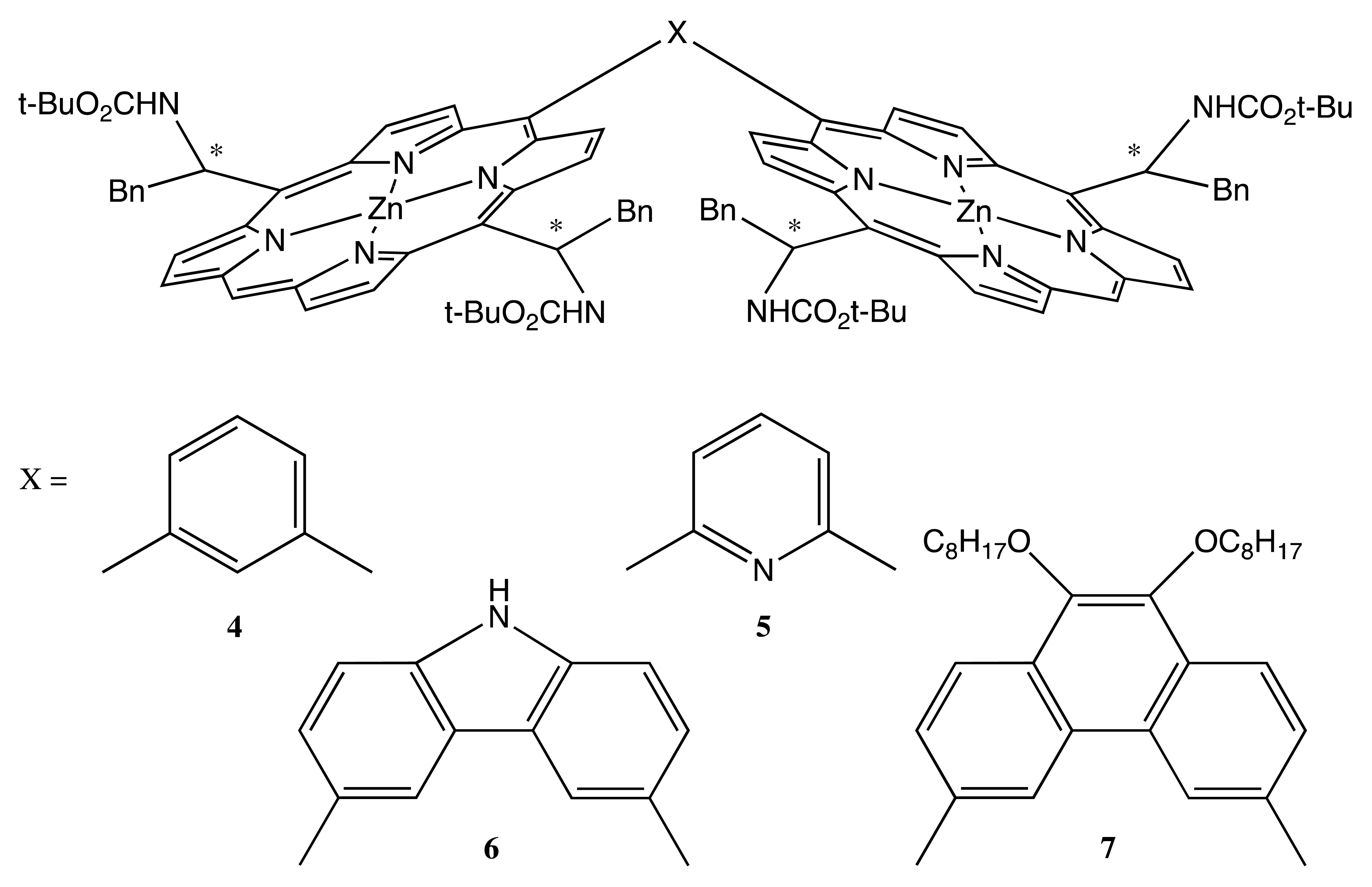
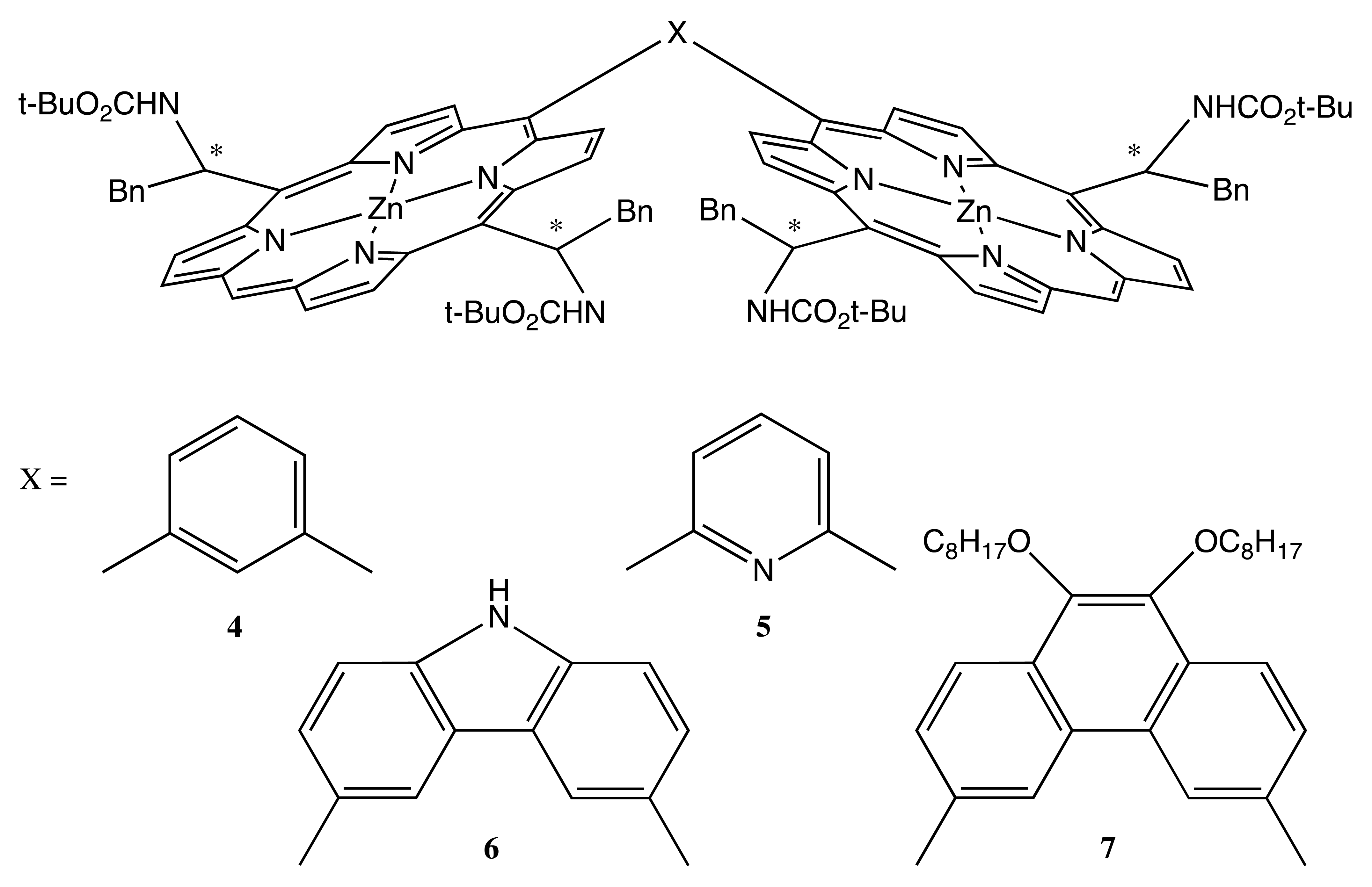
Another kind of covalent linkage was employed for the preparation of fixed bis-porphyrins, 4–7 (Figure 3) [18–21]. The chiral modification was carried out at two meso positions of each porphyrin ring, thus yielding four stereogenic centers correspondingly. Since the synthesis was based on enantiopure phenylalaninal, the chirality of 4–7 was predetermined as R or S exclusively. Two porphyrin moieties were rigidly connected by various aromatic bridges, resulting in the different spatial arrangement of macrocycles. In this case, supramolecular chirality was a result of noncovalent interaction with single-walled carbon nanotubes (SWCN). The judicious combination of chirality and the molecular geometry of these bis-porphyrins allowed the size selective discrimination of left- and right-handed SWCN, with the ee value of extracted SWCN being as high as 67% (in the case of 7).
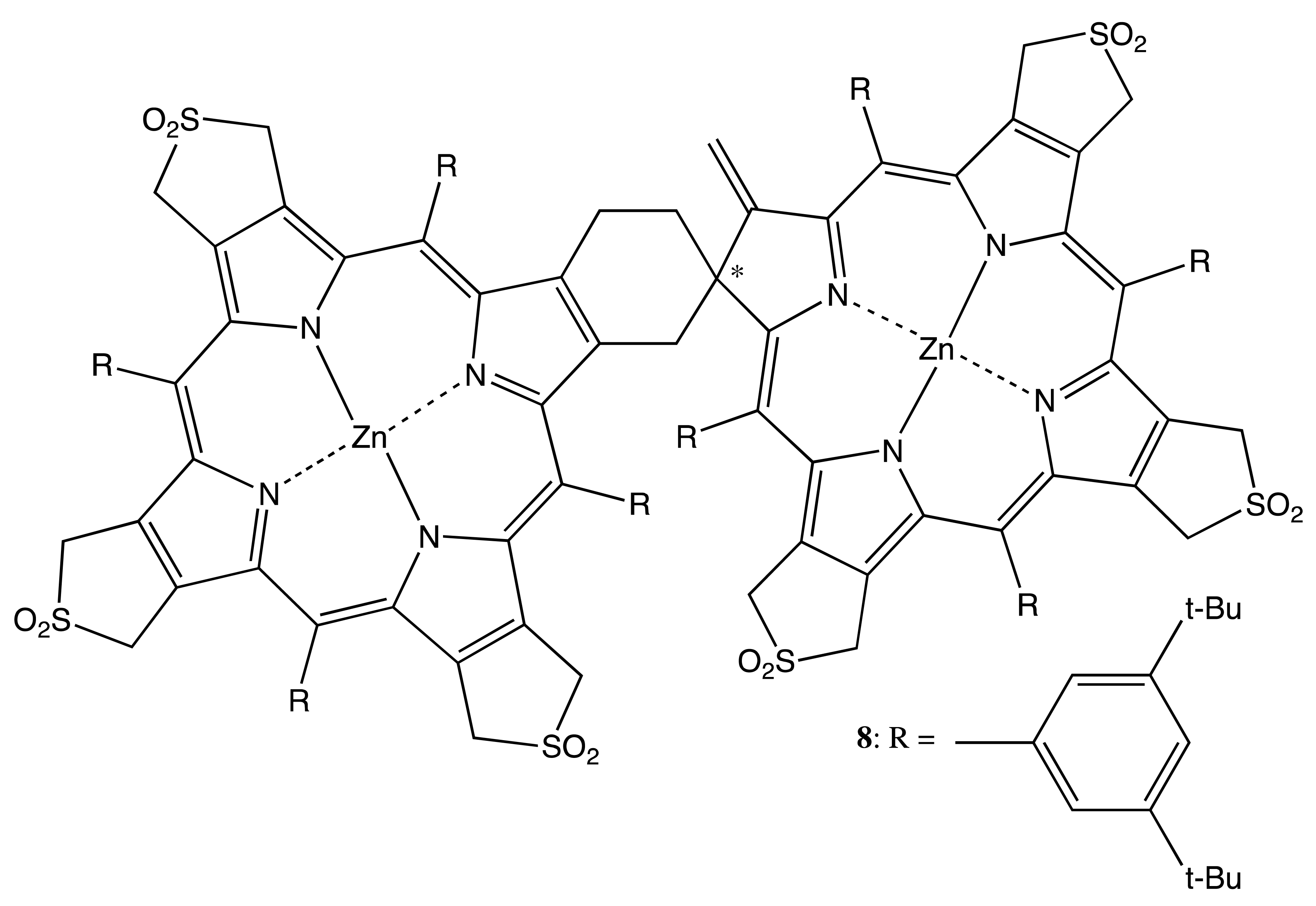
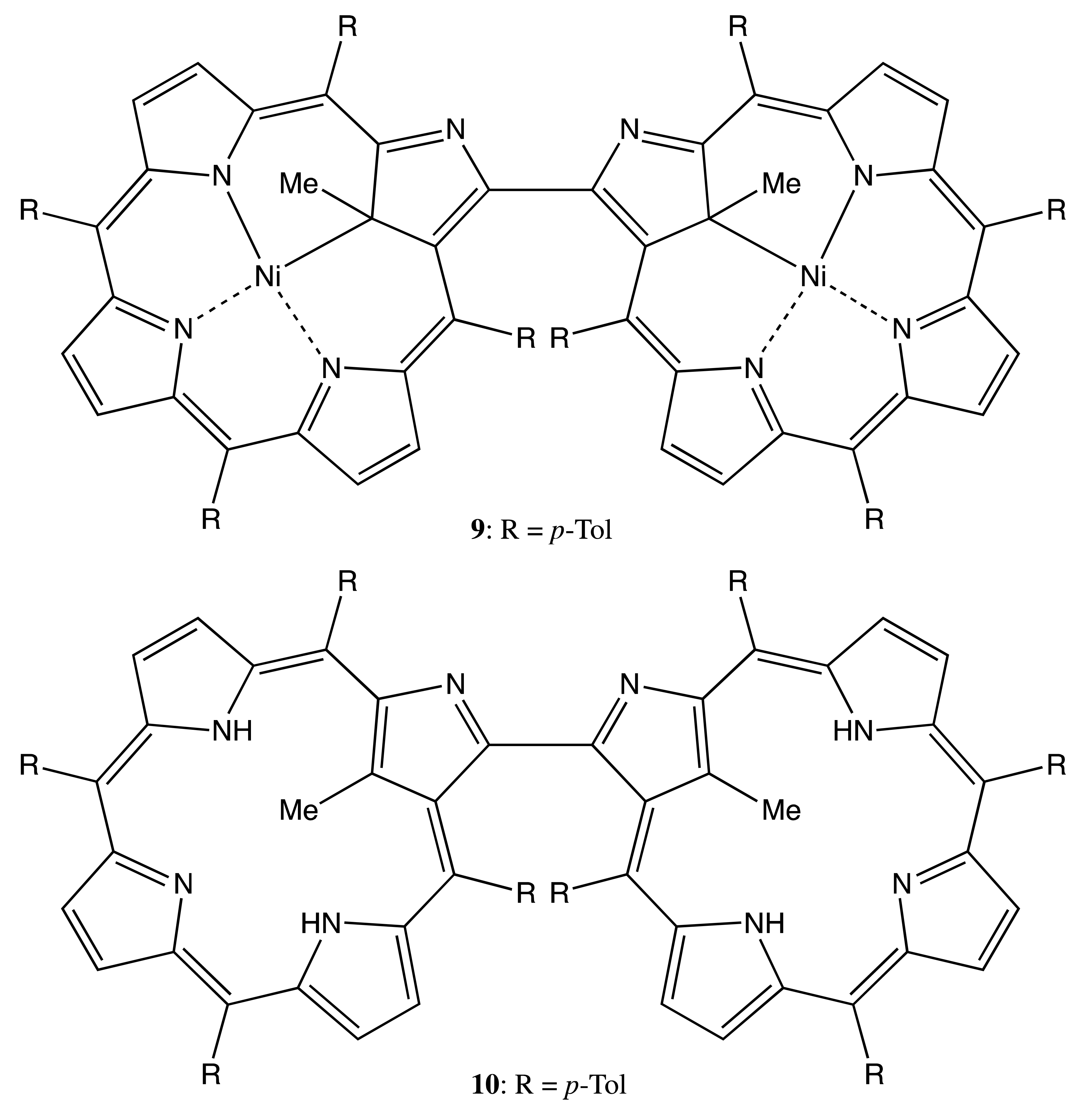
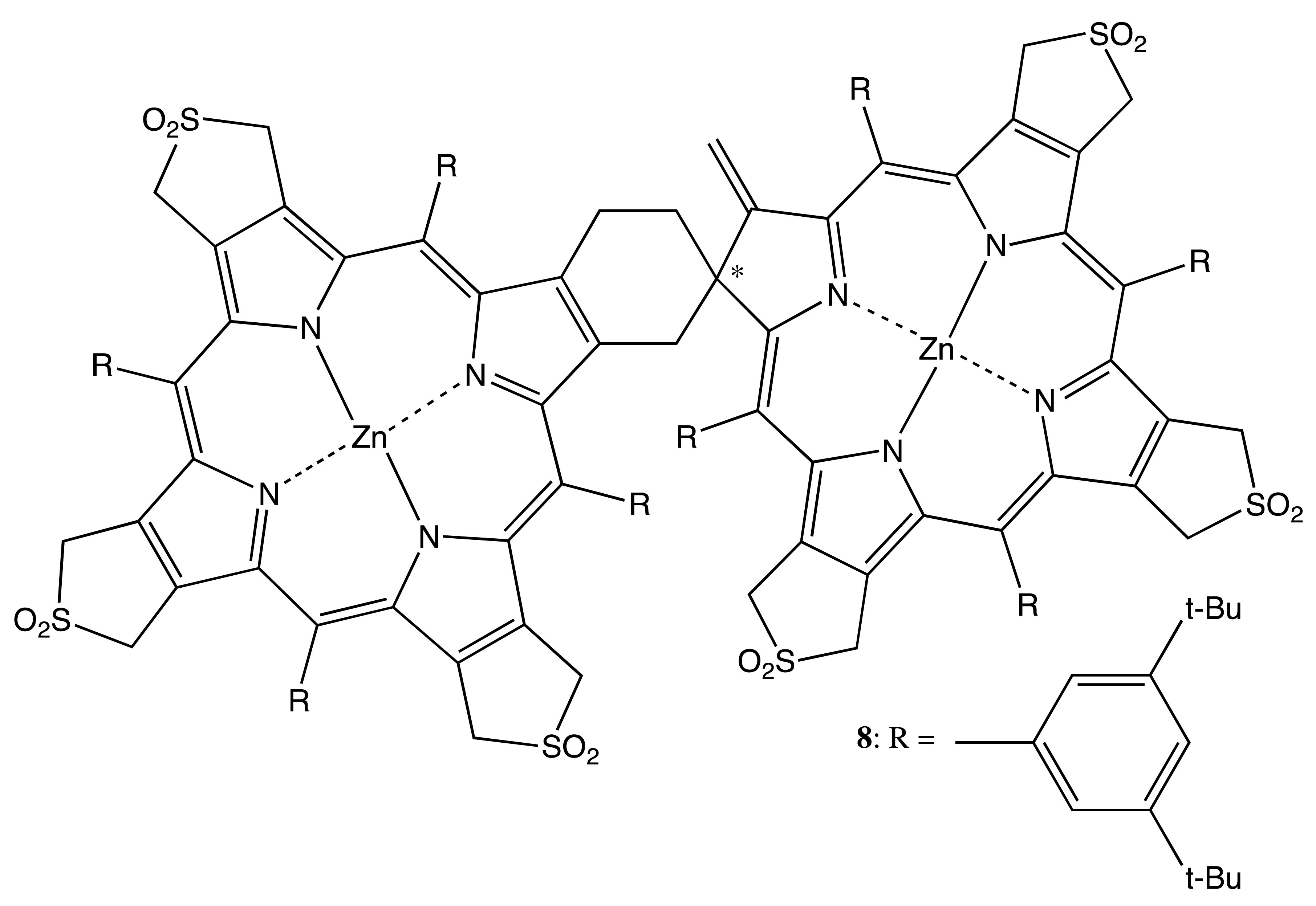
Chiral dimeric structures can also be obtained by the direct connection of porphyrin and chlorin structures, which is an intrinsically chiral macrocycle, or two chiral porphyrinoids. This type of linkage and the spatially bulky peripheral aromatic substituents are able to provide the conformational rigidity of the overall geometry (Figure 4). Hence, the thermal self-cycloaddition of symmetrical (tetra-β,β′-sulfolenoporphyrinato)zinc with the subsequent extrusion of SO2 yields the racemic chlorin-porphyrin complex, 8, the chiral property of which has not been explored yet, because the corresponding enantiomers have not been resolved [22]. However, in the case of β,β′-bonded chiral porphyrinoids, the corresponding enantiomers were successfully obtained [23]. In particular, the C21 methylation of 3,3′-bis(N-confused nickel porphyrin) resulted in the corresponding di-methylated derivative, 9. The asymmetry was derived as from the two homochiral subunits and from the rigid chiral conformation, which allowed memorizing chirality in the configurationally stable free base bis-porphyrinoid, 10, obtained upon demetalation of 9. Other examples of conformational chirality in bis-porphyrinoids will be discussed in the corresponding topic below. The chiral properties of 9 and 10 were characterized by the corresponding CD spectra exhibiting a typical bisignate pattern of exciton couplets in the region of porphyrinoid absorption, whilst the supramolecular chirality features are yet to be explored.
2.2. Chirality Introduced via Chiral Linkage
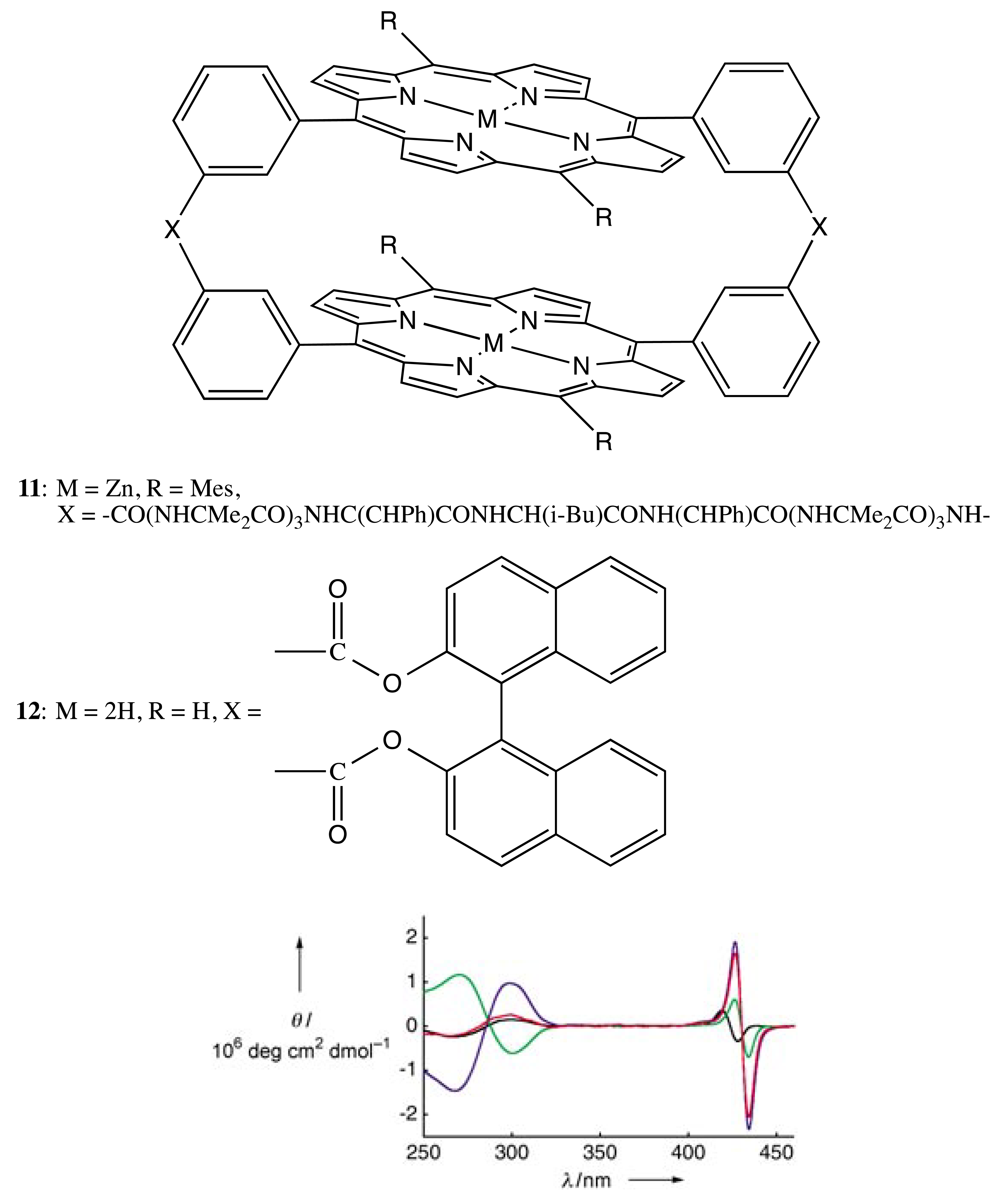
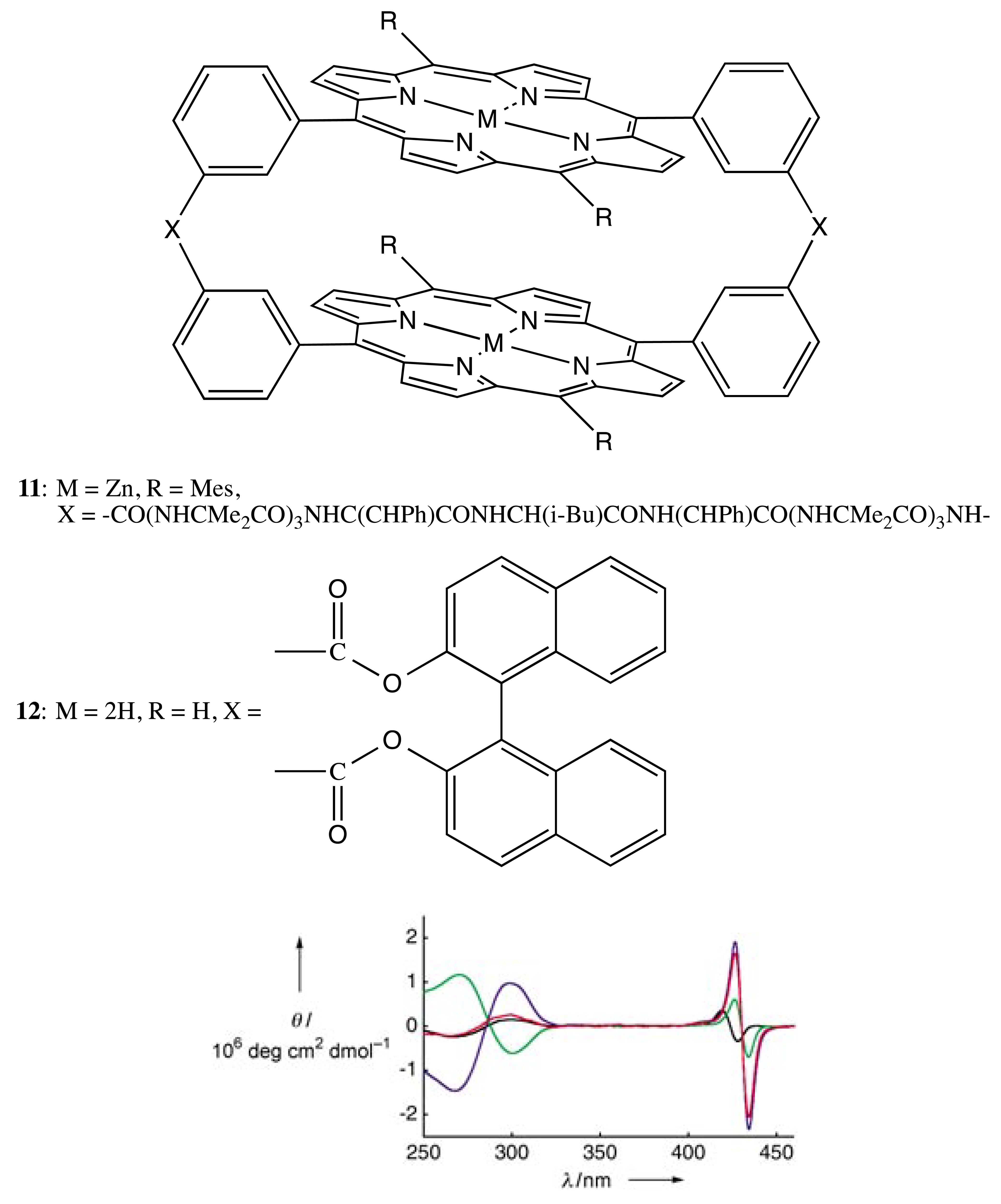
Besides the asymmetry generated by chiral macrocycles itself, the chirality of bis- and multi-porphyrins may be produced via a rigid chiral linkage. In this case, both the double and single rigid bridges are also commonly employed. For example, two chiral straps containing the leucine residue brought about two porphyrin moieties in the bis-porphyrin, 11 (Figure 5), into the helical conformation. This spatial arrangement was evidenced by the characteristic exciton couplet in the corresponding CD spectra of porphyrin absorption [24]. Essentially, the l- and d-leucine derivatives resulted in the right- and left-handed orientation, respectively. The supramolecular chirality property of 11 was employed for optical resolution of a series of bidentate artificial oligopeptides (OPs) with an ee value as high as 80%. Upon formation of the favorable host-guest complex, the CD signal of the porphyrin chromophore was significantly enhanced, apparently due to the directional stabilization of the twisted geometry of bis-porphyrin by the helicity of the bound oligopeptide (Figure 5).
As another example of the double chiral bridges, the 1,1′-substituted binaphthyl (BNP) moiety, which is inherently chiral, was employed as one of the frequently used structural motifs to link two (or more) porphyrinoids. Hence, two BNP fragments were applied to fix two porphyrin subunits in the bis-porphyrin, 12, in a chiral fashion (Figure 5) [25,26]. Whilst the chiroptical properties have not been studied, the supramolecular chirality functionality of 12 was applied for the chiral discrimination of electron-deficient aromatic compounds bearing a dinitrophenyl group.
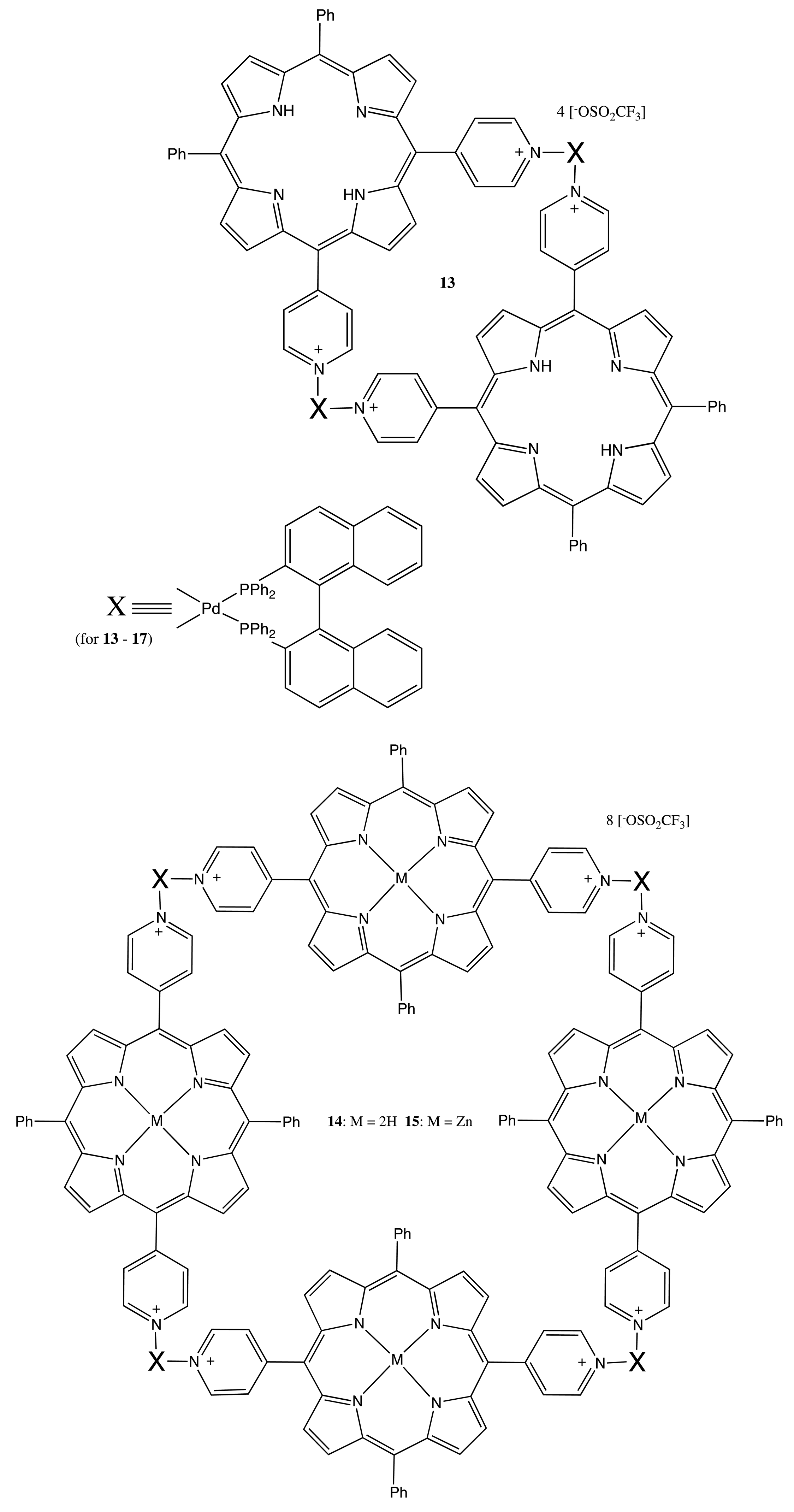
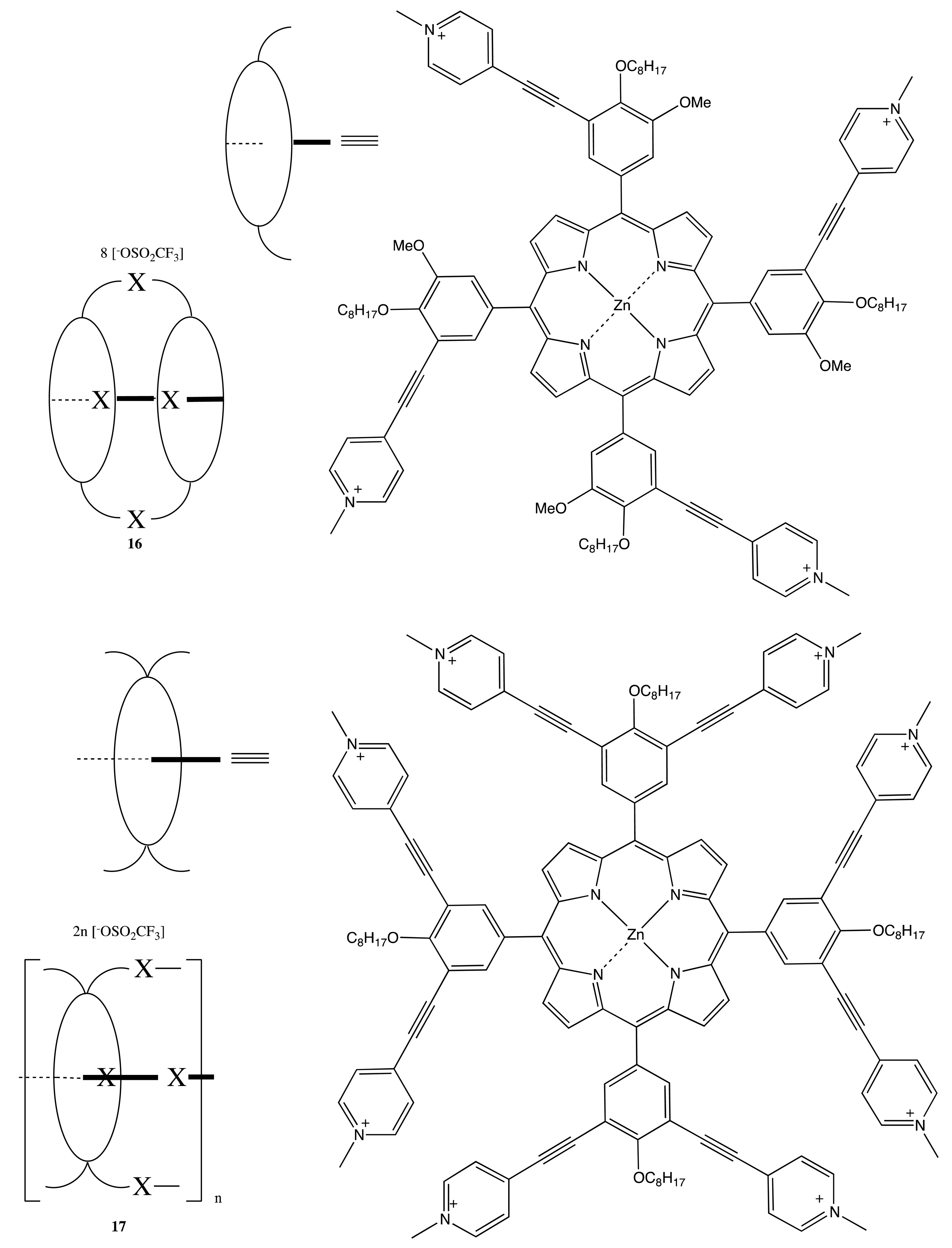
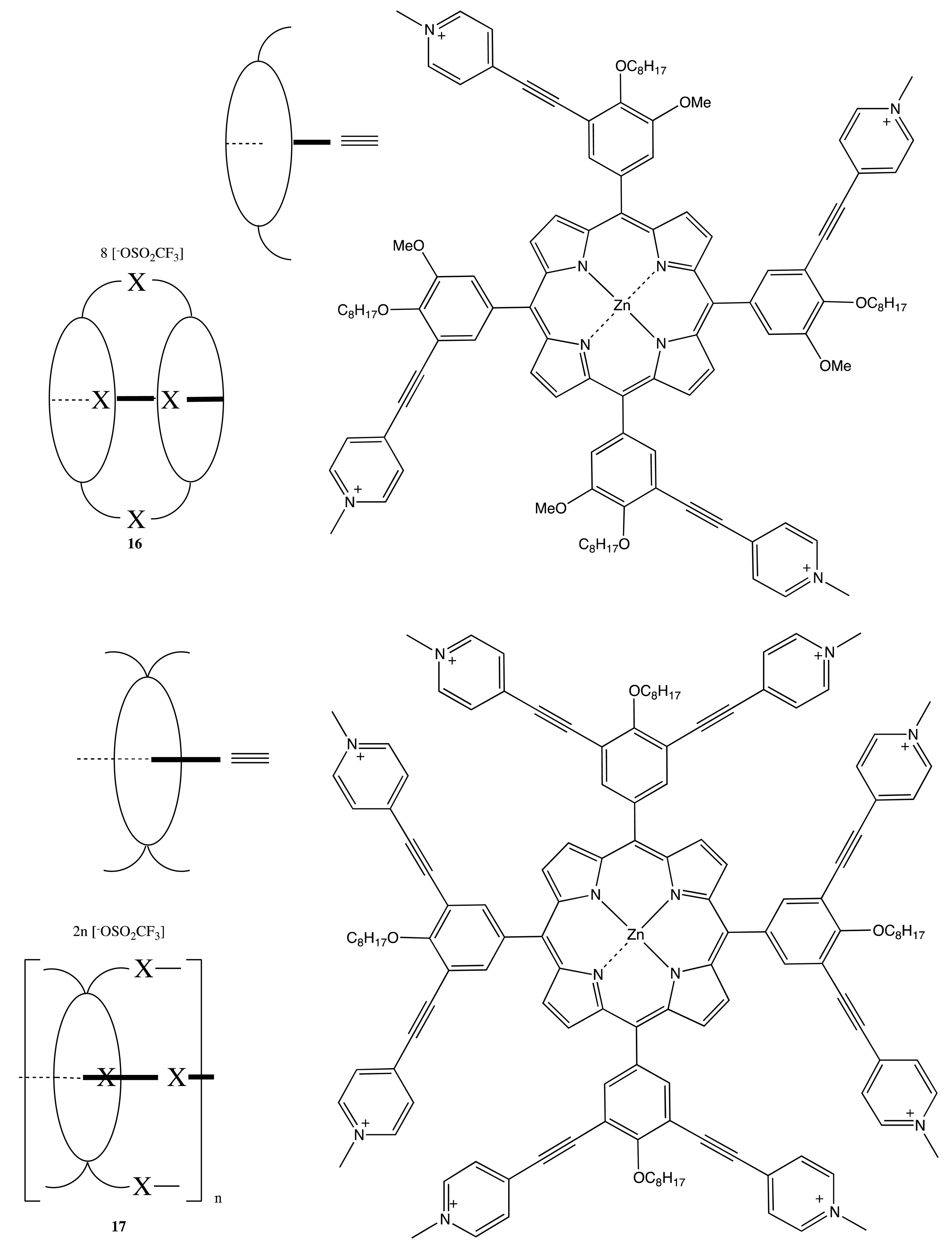
Analogously, the BNP chiral unit was used to produce the corresponding bis-, tetra- and poly-porphyrins upon self-assembly of the pyridyl-substituted monomeric porphyrins with the bisphosphine-coordinated Pd(II) complexes of BNP to yield the optically active bis- and multi-porphyrins, 13–17 (Figure 6) [27,28]. Interestingly, the chiroptical properties of these systems were profoundly diverse and dependent upon the corresponding structural organization. Hence, the tetrameric porphyrin, 14, exhibited a strong bisignate CD signal in the region of the porphyrin Soret band, being as high as 1800 cm−1·M−1, whilst no CD signal was detected in the case of the Zn complex, 15, and only a moderate monosignate CD profile was observed in the case of the dimer, 13. This remarkable behavior of these systems was attributed to the effective multiporphyrin exciton coupling in 14, the high symmetry of 15 and the coplanar arrangement of two porphyrins in 13. The asymmetry of BNP in the face-to-face oriented 16 and 17 forced the macrocycles to adopt a twisted conformation, resulting in the unidirectional (clockwise or anticlockwise) coupling of porphyrin electronic transitions. For example, the (R)-enantiomer induced the left-handed helical structure, resulting in a negative exciton couplet in the region of Soret absorption, whilst the multiporphyrin coupling in 17 considerably increased the intensity of the CD signal.
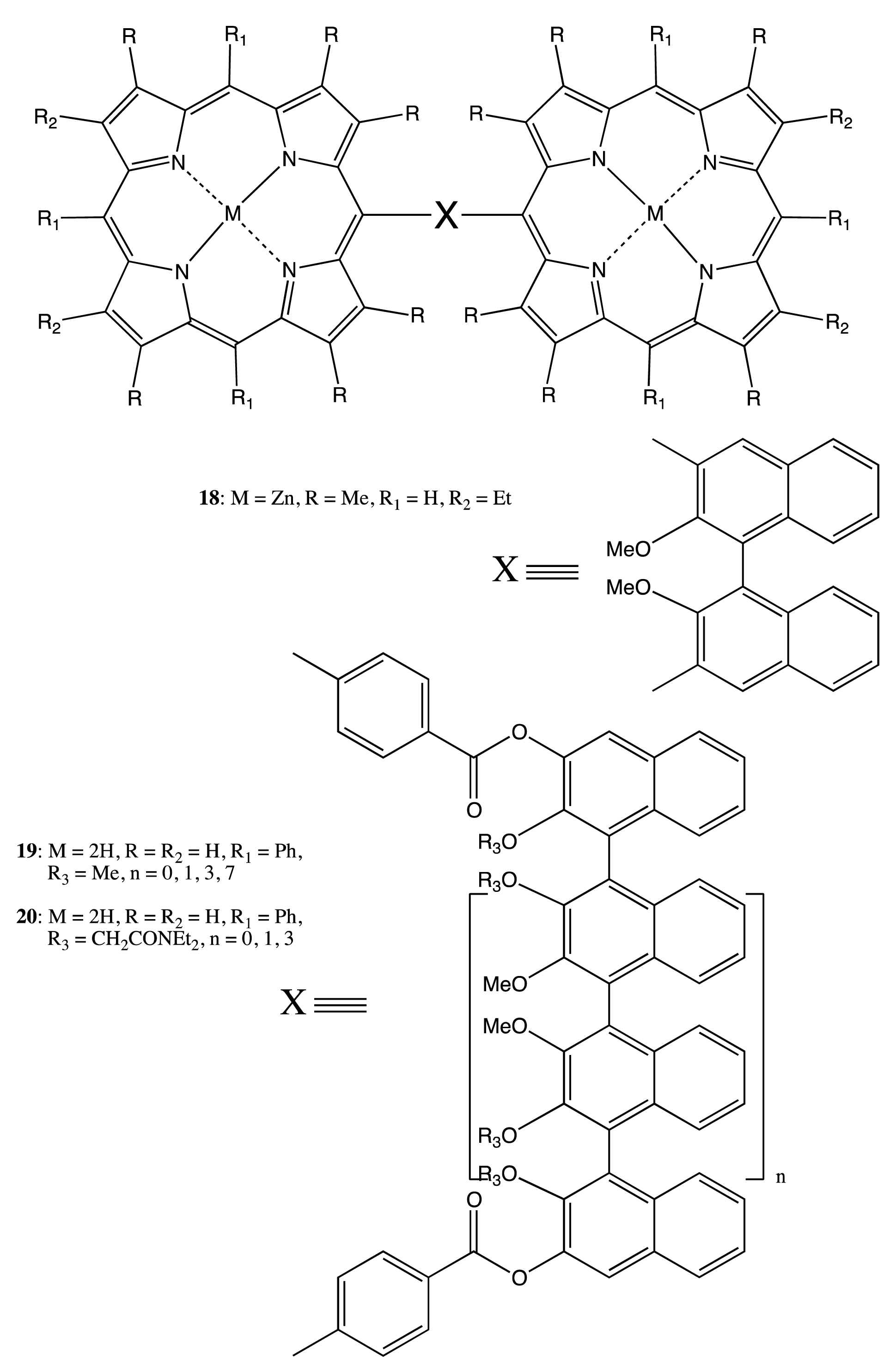
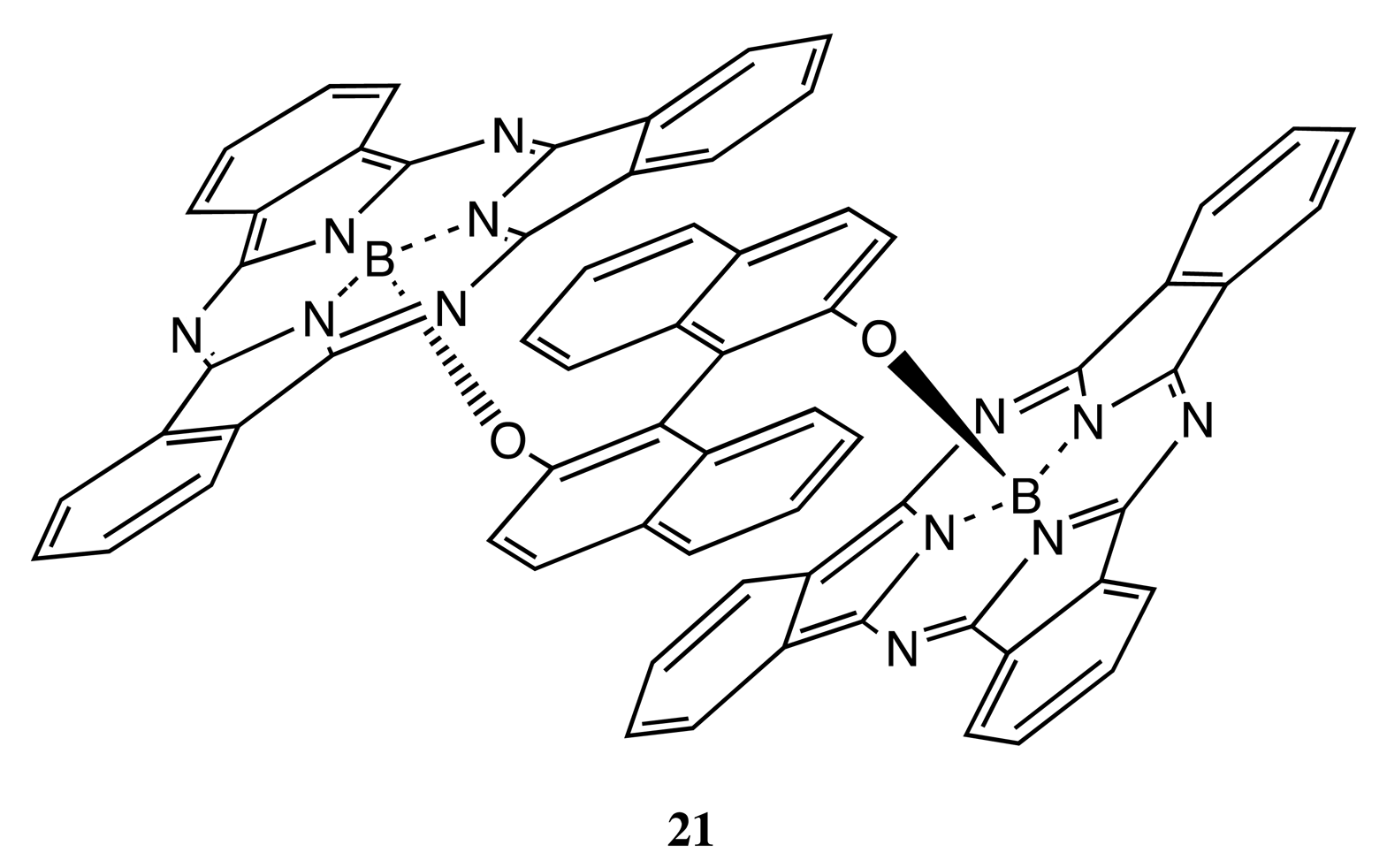
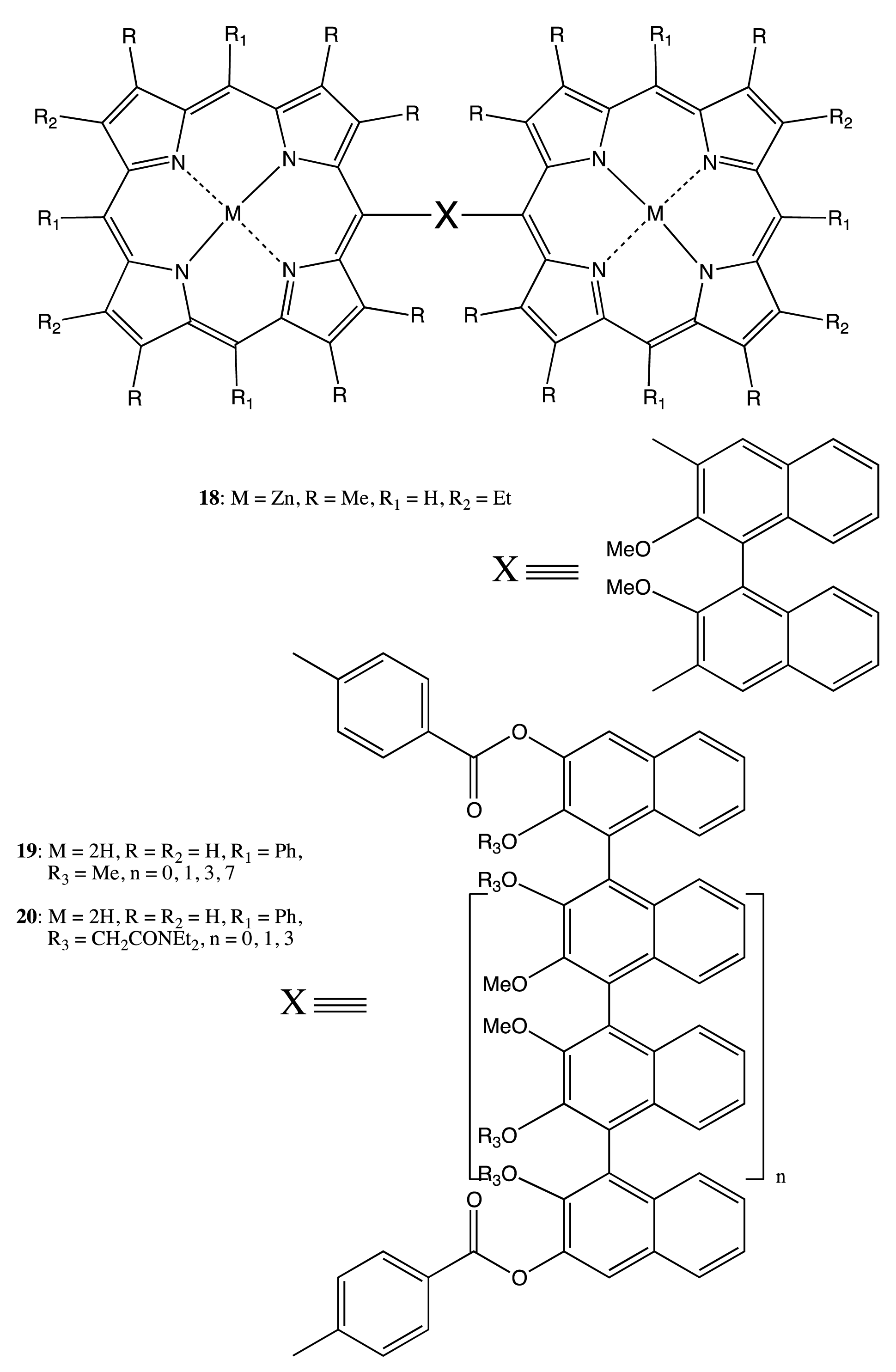
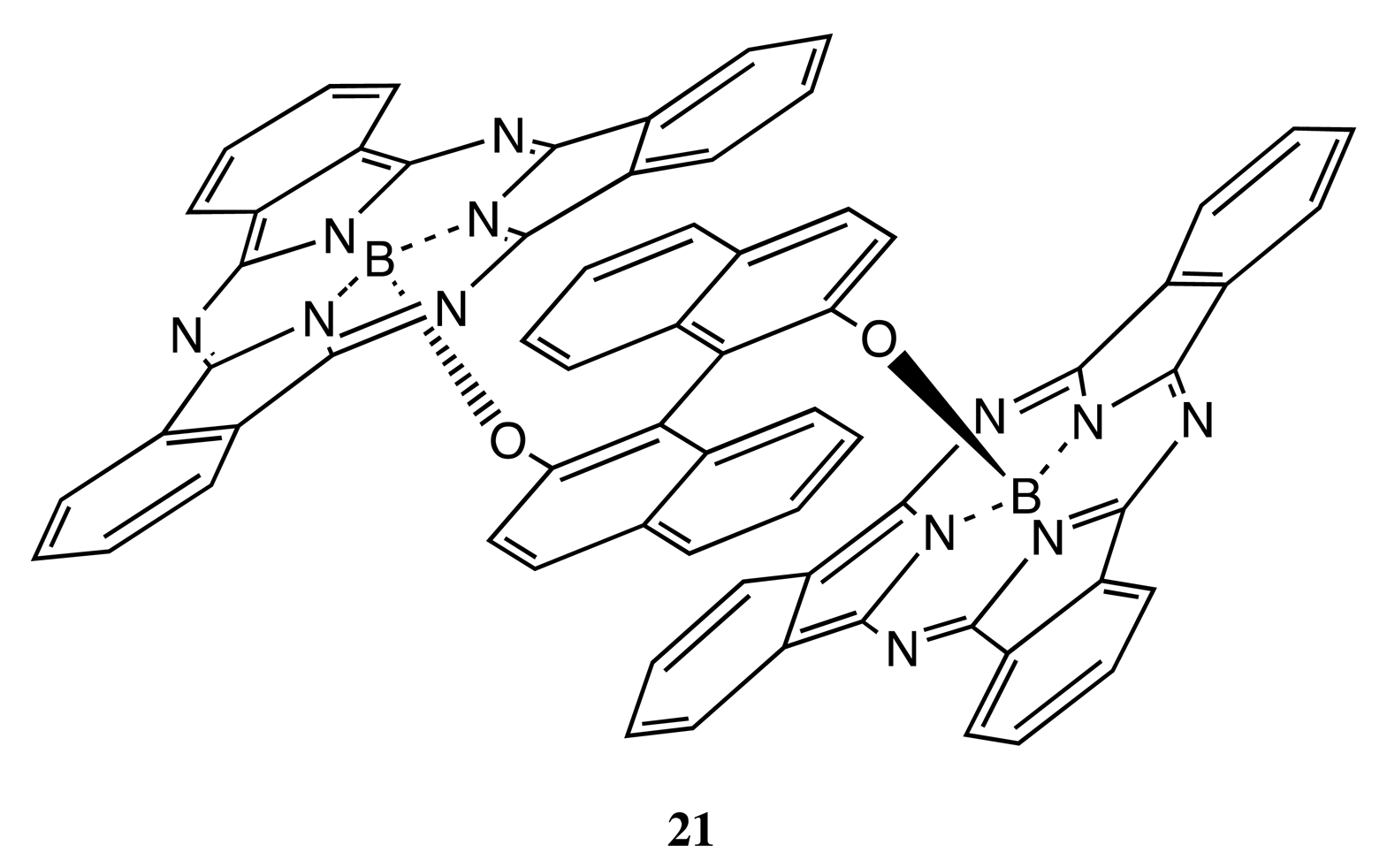
Owing to the considerable rigidity of the BNP fragment, this structural pattern was often employed for the design of bis- and multi-porphyrinoids connecting by a single covalent bridge, as well. By using this construction approach, the enantiopure bis-porphyrin, 18 (Figure 7), has been synthesized, and its supramolecular chirality properties have been demonstrated in the appreciable chiral discrimination towards diamine molecules [29,30]. The fixed distance between two porphyrins in 18 ensured the best enantioselectivity for the lysine derivatives being as high as 11–12 times, as determined by comparison of the corresponding association constants. The host-guest complex optimization revealed that the selectivity mechanism was based upon steric repulsion occurring between the methoxy groups of the BNP moiety and the amide group of amino acid derivatives in the favorable diastereomeric pair. Furthermore, the chiroptical response of 18 was greatly enhanced upon interaction with the appropriate diamines, resulting in a CD amplitude enhancement of up to 1788 cm−1·M−1 in the case of 1,8-diaminooctane, apparently due to additional fixation of the bis-porphyrin conformation by a diamine guest. In addition, the BNP-based linkage was used to sense a long-range exciton coupling in CD. Hence, a series of the BNP fixed bis-porphyrins, 19, 20 (Figure 7), with a certain spacer length between the porphyrin units has been synthesized, and a bisignate CD signal has been clearly detected in the case of 19 (n = 7), having an interchromophoric distance of over 66 Å [31]. However, the supramolecular chirality properties of 19, 20 have yet to be investigated. Besides the porphyrins, the porphyrinoid structures have also been linked by the BNP moiety. Thus, subphthalocyanine moieties have been successfully connected by the BNP bridge in 21 (Figure 7) to generate an intense CD signal in the region of low energy Q transitions. The complicated shape of which was rationalized in terms of the excitonic interactions by analyzing the band deconvolution results [32]. The asymmetrical CD profile was derived from the chirality transfer from the BNP unit to the porphyrinoid moieties.
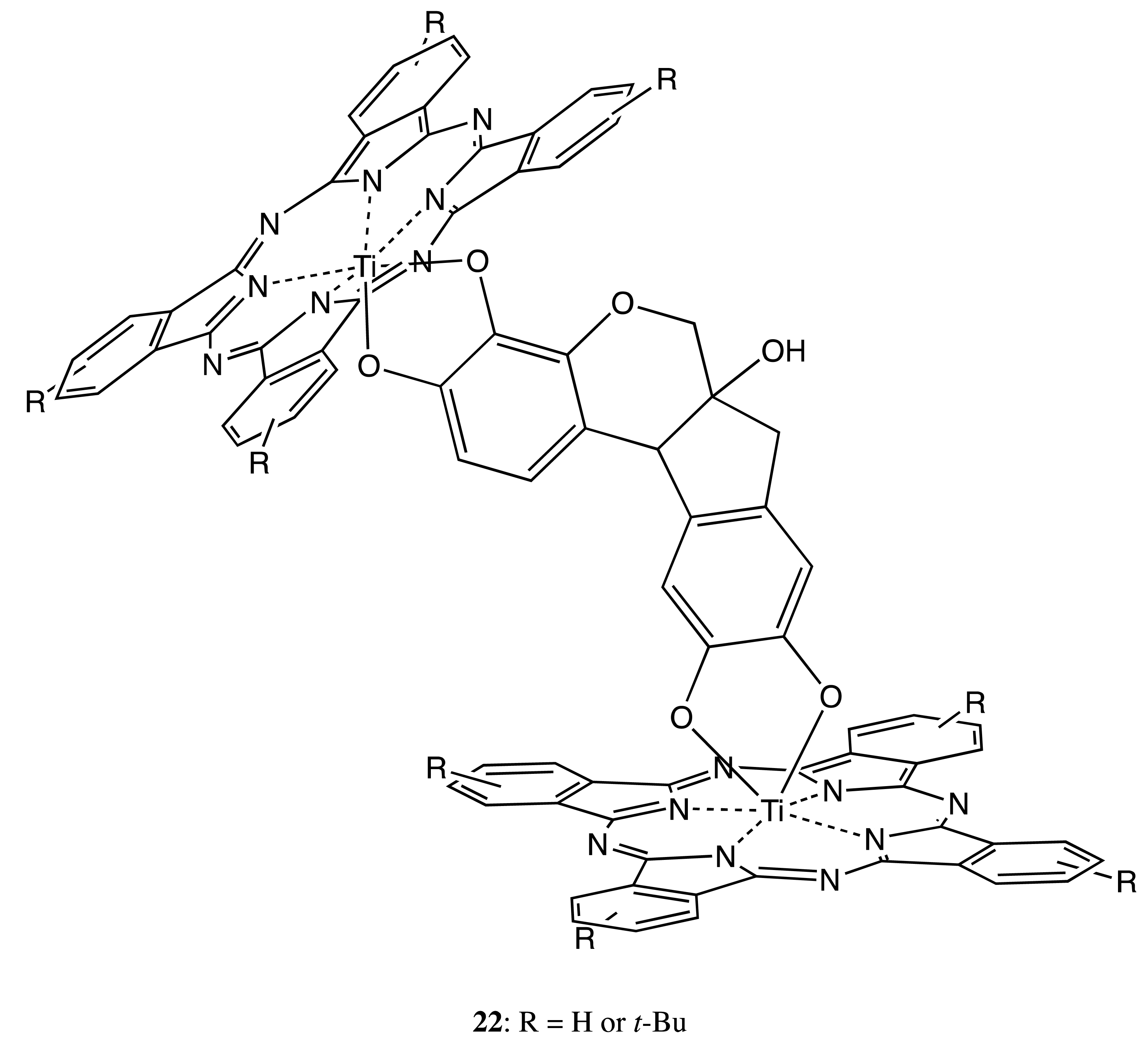
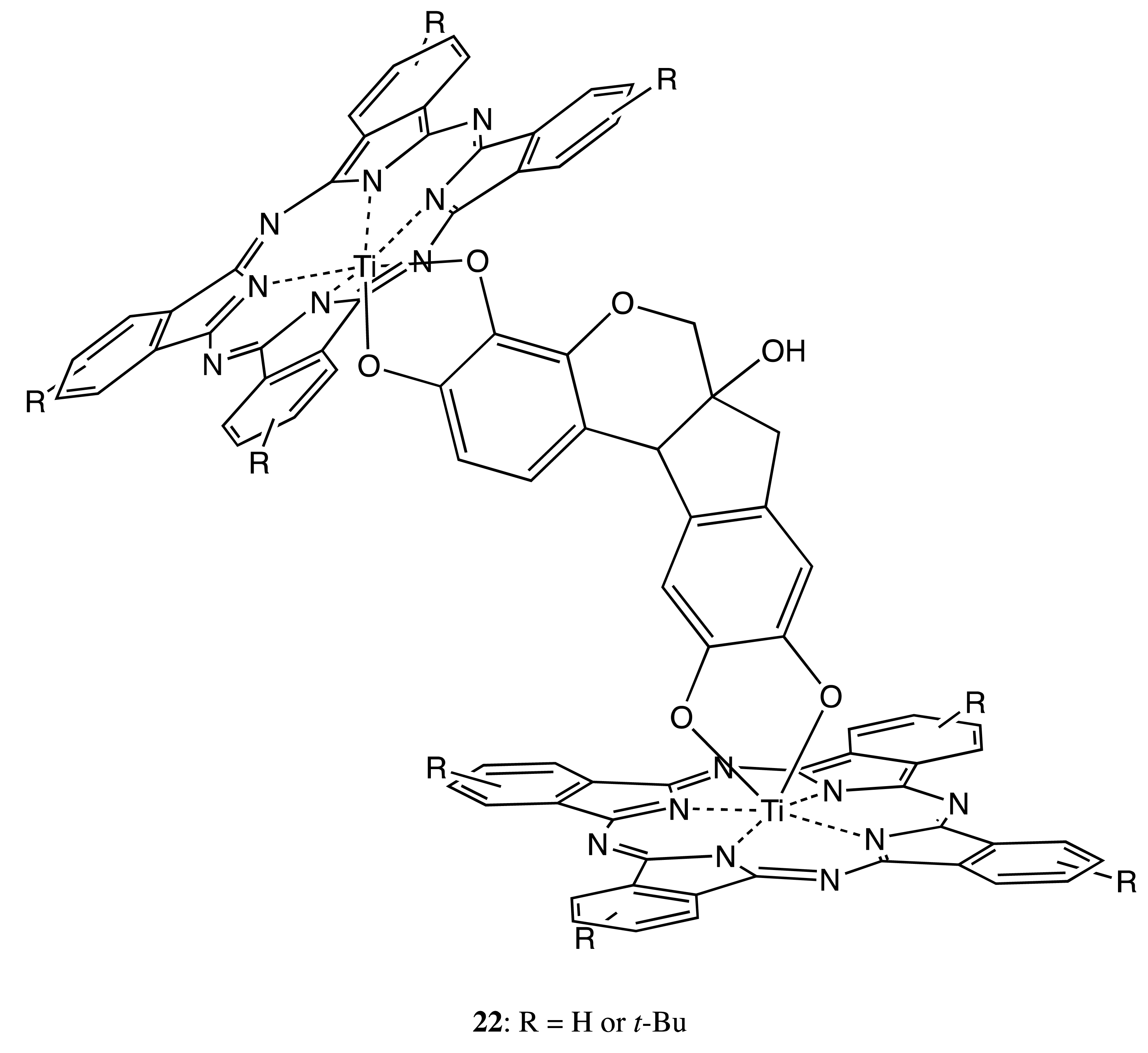
Different structural types of rigidly linked bridges have also been successfully employed, as shown in the following chiral bis-porphyrinoids. Hence, the hematoxylin fragment was applied to fix a pair of phthalocyanines in 22 to produce the corresponding chiral orientation (Figure 8) [33]. The chiroptical outcome consisted of an intense negative-to-positive bisignate CD signal in the Q-band region and several weak signals in the Soret band area as a result of the excitonic interaction between two porphyrinoids. The induced optical activity was unambiguously confirmed by ab initio geometry optimization combined with a Kuhn–Kirkwood coupled-oscillator mechanism, which additionally allowed the determination of the absolute configuration of hematoxylin as the (6aS,11bR)-form.
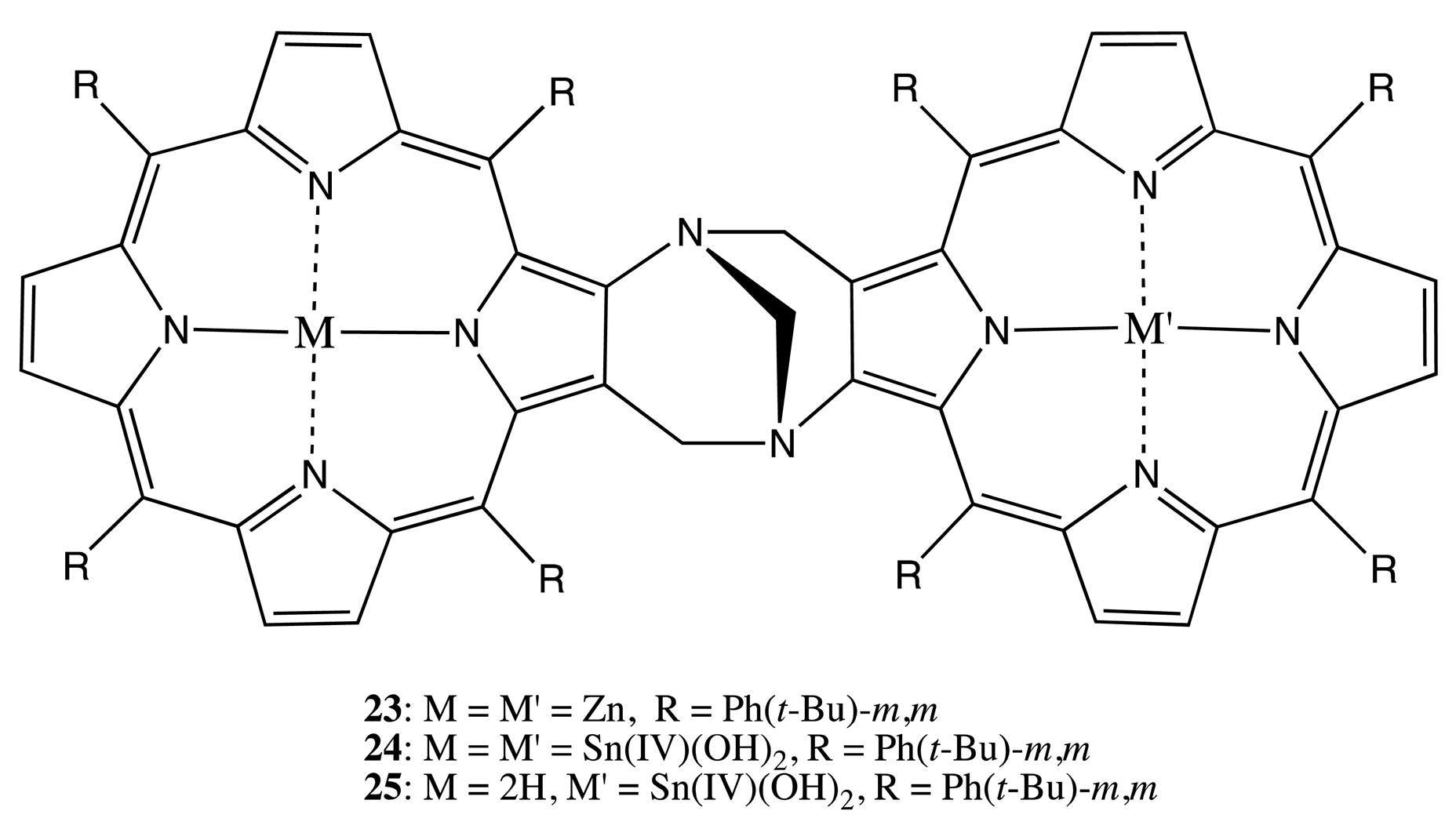
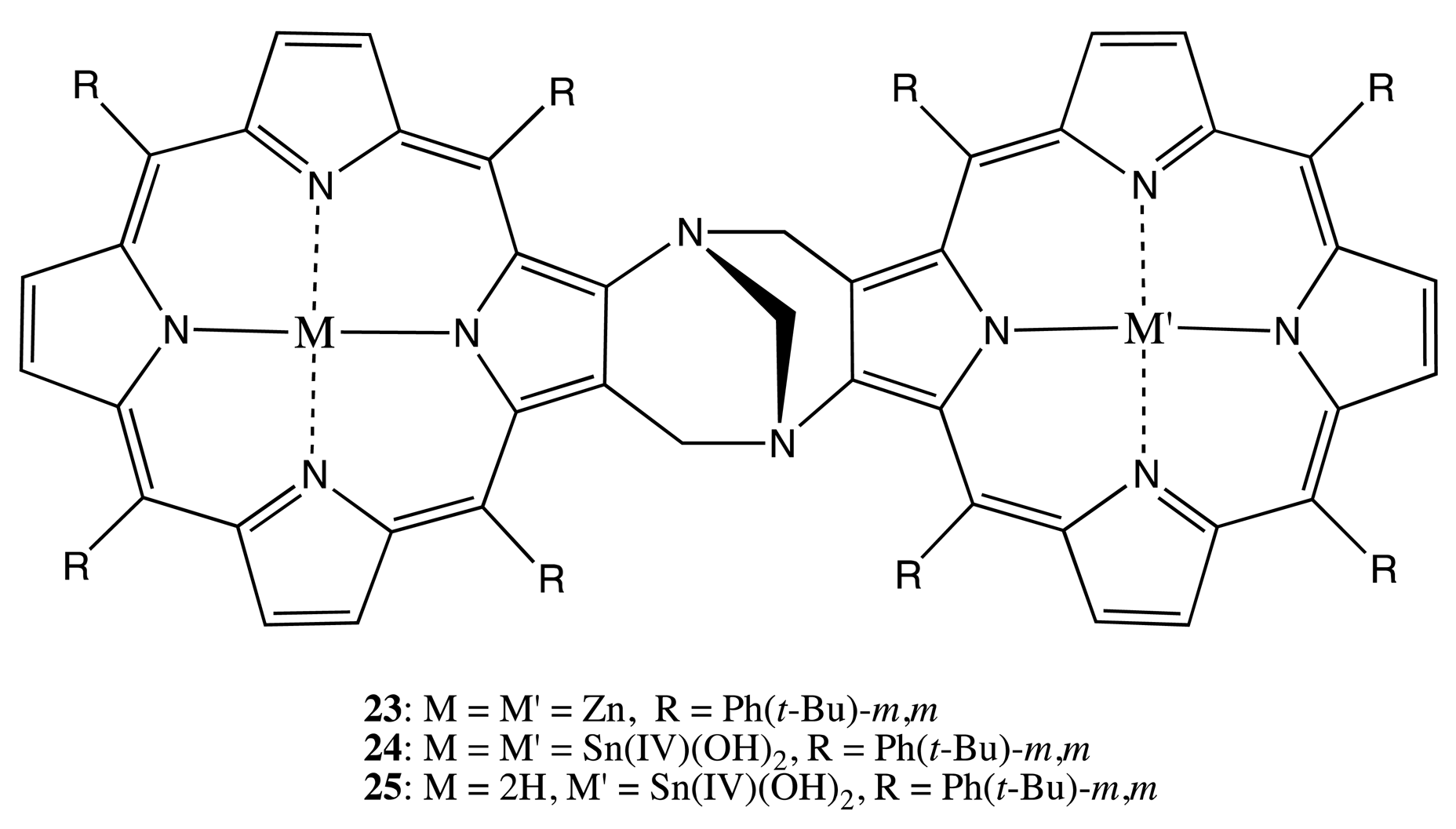
Tröger's base was employed as another suitable rigid linkage, which is inherently chiral and consequently used to bind two porphyrin units in the bis-porphyrins, 23–25 (Figure 9) [34,35]. The resolved enantiomers exhibited a bisignate CD pattern with the Cotton effects of high molar ellipticities (up to 819 cm−1·M−1 at 425 nm, a high energy Cotton effect). The supramolecular chiral properties of 23 were applied to obtain 80%–86% and 48% ee for the binding of histidine and lysine esters, respectively. This enantioselectivity is due to the ditopic interaction of two nitrogen sites on the guest with the two zinc centers of bis-porphyrin, leading to the corresponding tweezer structure [34]. Furthermore, the corresponding tin complexes, 24, 25, were used to rationalize the mechanism accounting for the high selectivity for the binding of dicarboxylic acids in 24, which binds just ≤1 equivalent of dicarboxylic acid quantitatively within the chiral cavity [35].
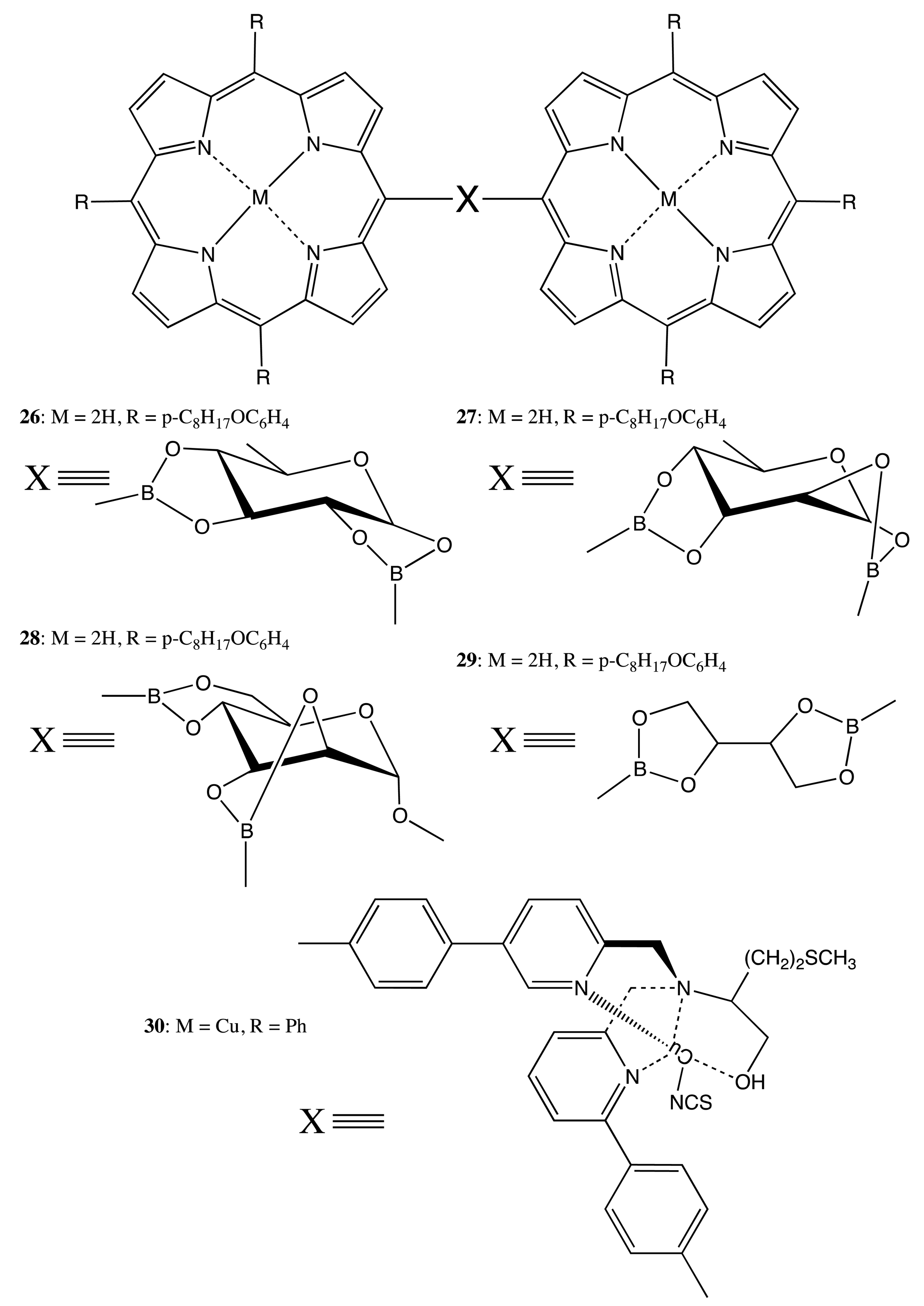
Saccharides also appeared to be a very effective template for the formation of the fixed bis-porphyrins, 26–29 (Figure 10) [36]. Hence, upon applying the supramolecular chirality approach, the boronic acid-appended porphyrin produces the corresponding bis-porphyrin adduct upon mixing with monosaccharides. In turn, this results in the effective chirality information transfer from the chiral saccharide bridge to the achiral porphyrin moieties. The exciton coupling CD spectra were observed in all the cases, whilst the chiroptical properties were strongly affected by the different spatial orientation of two porphyrins, which was governed by the structure of the sugar derivatives. For example, in the case of α-methyl-d-mannopyranoside bridged bis-porphyrin, 28, the highest optical activity (−264 cm−1·M−1 at 427 nm) was attained.
A rather rigid linkage between two porphyrins was proposed on the basis of a chiral tripodal ligand [37]. The supramolecular dimerization of chiral porphyrins assisted by the complexation with Cu(II) ions in the presence of NH4NCS resulted in fixing the geometry of 30 (Figure 10). Subsequently, the conformational stability of 30 induced a strong exciton coupled response in the region of Soret transition of the CD spectrum, whilst no CD signal was observed for the corresponding monomeric porphyrin.
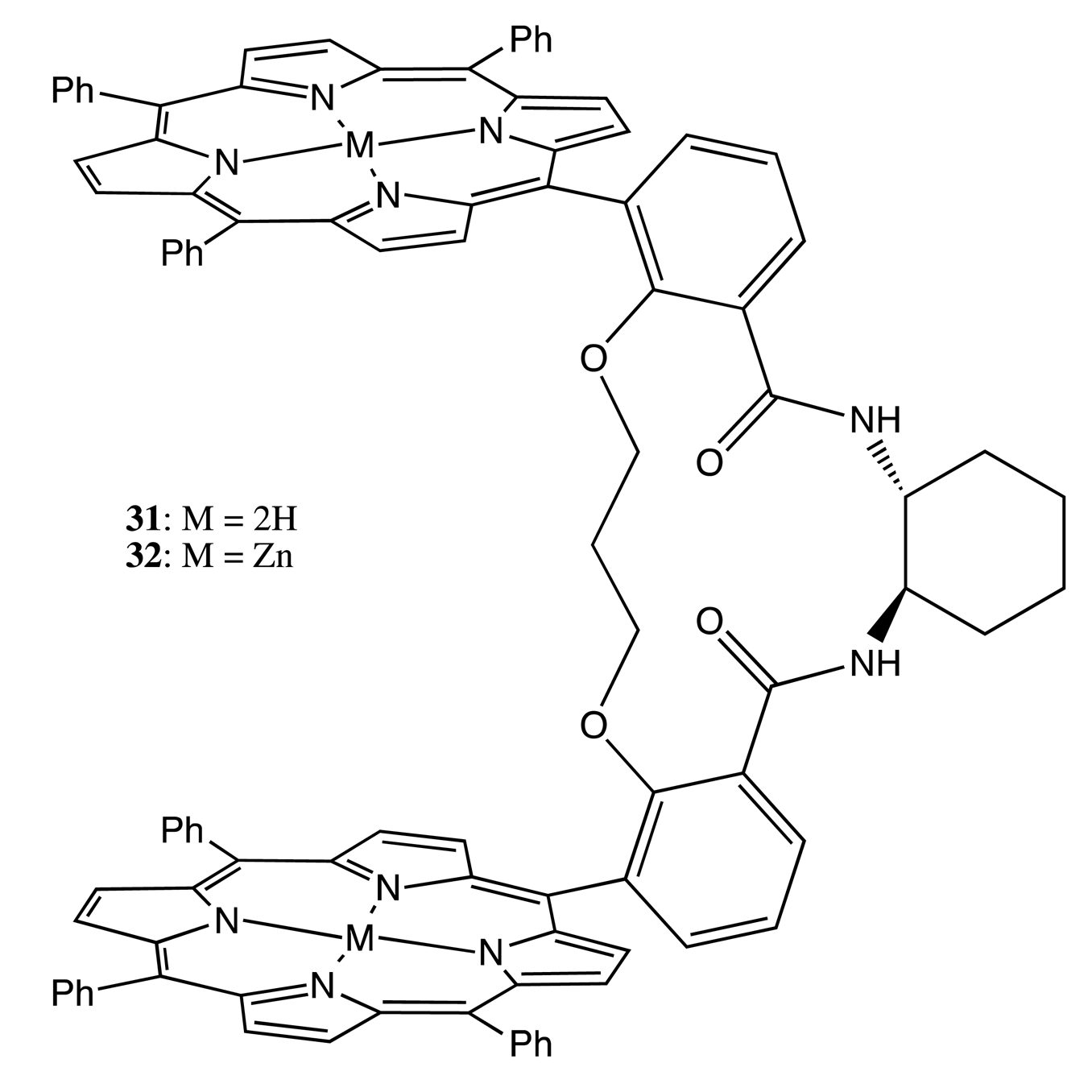
An alternative approach to fix two porphyrin subunits lies in the additional linkage. For example, two porphyrins in the bis-porphyrins, 31, 32, were linked by a chiral diaminocyclohexane (DACH) structure and by an achiral dioxopropane bridge (Figure 11) [38]. The resulting double strapping considerably increased the chiroptical outcome in comparison to the single bonded bis-porphyrin, owing to the enhanced rigidity of the system. Furthermore, the pre-organized cavity allowed the selective binding of diamines of various lengths, which then was able to modulate the CD signal as a result of the supramolecular chirality function. The maximal CD intensity was observed for the complexation of diaminohexane arising from the appropriate length of this diamine to form an additional (third) linkage between two porphyrins, thus further stabilizing the geometry of 32.
2.3. Conformational Chirality
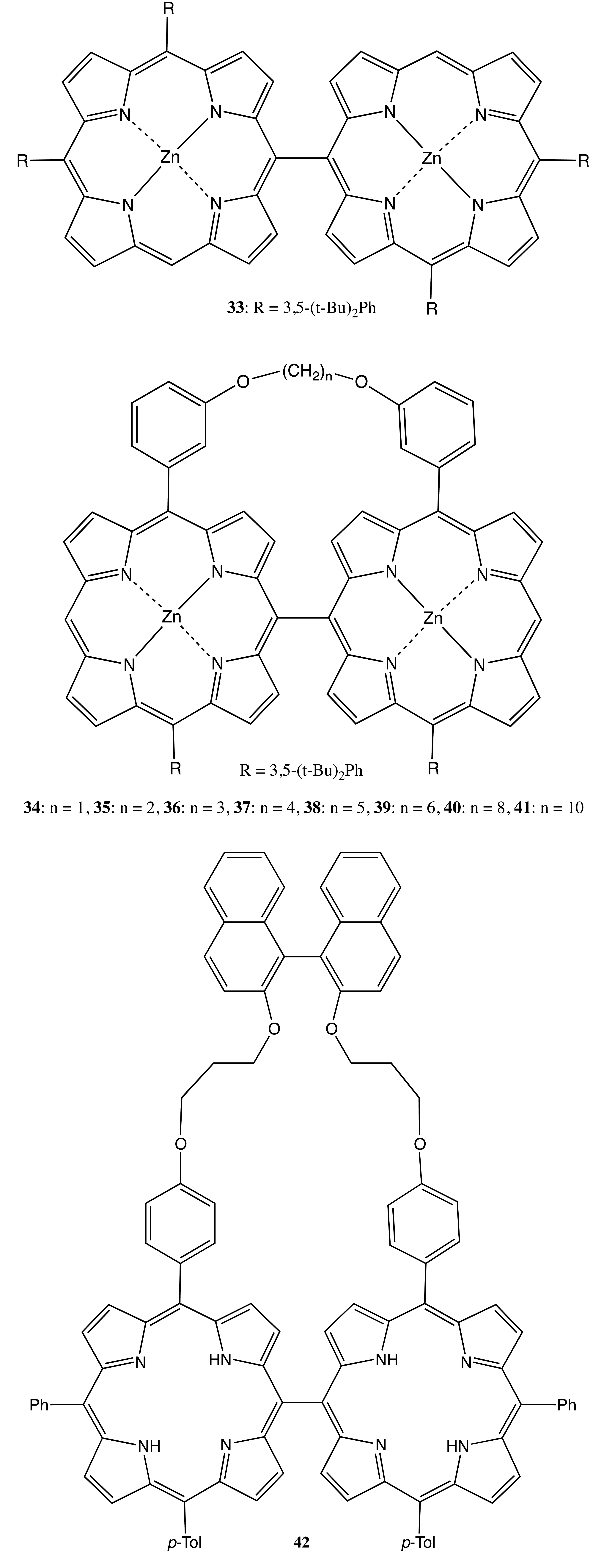
The last type of rigidly fixed chiral bis- and multi-porphyrins discussed in this subchapter is based upon the conformational chirality; that is, when neither the porphyrins themselves nor the linkage between the macrocycles have a stereogenic center, but the overall fixed spatial arrangement of two (or more) porphyrins yields the non-superimposable mirror images. The key point of these systems is the hindered interconversion between the separated enantiomers resulting in the stabilization of the chiral structure. For example, a direct C–C bond between two porphyrins at the corresponding meso- or beta-positions may be a suitable target for this approach if the rotation around the bond is blocked, for example, by bulky substituents or an additional linkage. Hence, a simple meso-meso linked bis-porphyrin, 33, was optically separated into two enantiomers through chiral high performance liquid chromatography (HPLC) (Figure 12) [39]. However, whilst the 180° rotation around the binding axis was prohibited, the overall geometry was not completely fixed, leaving room for several existing conformers, resulting in a rather moderate CD outcome (within ±20 cm−1·M−1). The situation with the chiroptical activity was considerably improved upon employing an additional strapping between the two porphyrins in the case of the bis-porphyrins, 34–41 (Figure 12) [40]. This was a result of a severe constraint imposed by the alkyl bridge, yielding further conformational fixation. The largest CD signal (up to ±2211 cm−1·M−1 at 420 nm) was observed for 37, due to the optimal dihedral angle between the interacting electronic transitions of the porphyrin subunits secured by the alkyl strap. Replacement of the achiral alkyl bridge with a chiral one, such as the dinaphthyl derivative in 42 (Figure 12), allowed the optical resolution to be considerably simplified by avoiding chiral HPLC. This facilitation is due to the formation of a diastereomeric mixture, whilst the optical activity of 42 is almost the same as in the case of 37 [41]. However, the supramolecular chirality properties of these bis-porphyrins have yet to be explored.
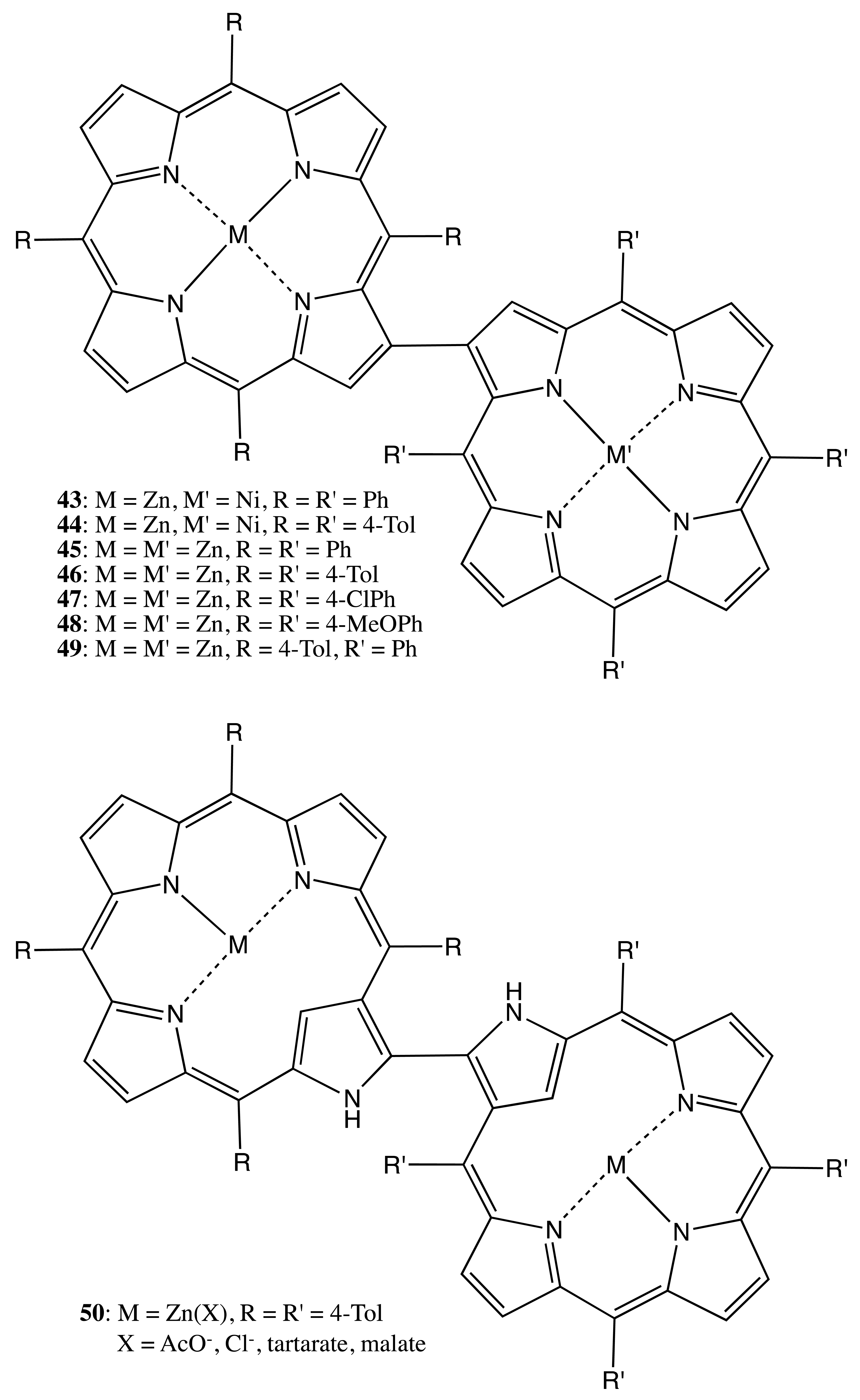
The beta-beta linkage between two porphyrins was also successfully employed to fix the overall conformation in a series of bis-porphyrins, 43–49 (Figure 13), and it resolved the corresponding enantiomers [42]. The obtained stereochemical assignments were in agreement with the exciton chirality method [43], whilst the rotational stability about the porphyrin-porphyrin axis was found to be dependent upon the nature of central metal ion, with the Gibbs free energy being as high as 115 kJ mol−1 (in the case of 45), as determined by the HPLC-UV measurements. Besides conventional porphyrins, this type of linkage was also applied to connect N-confused porphyrins to obtain the corresponding dimers, 50 (Figure 13) [44]. Apparently, the rotational barrier in the case of achiral ligands (AcO− and Cl−) was rather low, thus resulting in a non-separable racemic mixture of 50. However, the chiral tartrate and malate anions induced optical activity in the region of the porphyrin Soret band absorption, due to the preferential stereoisomer formation as a result of the supramolecular chirality transfer. The chiral structures were also stabilized either via the intermolecular self-assembly assisted by an Ag cation [44] or via the formation of an intramolecular helical arrangement of two porphyrinoids, which could then be optically separated [45].
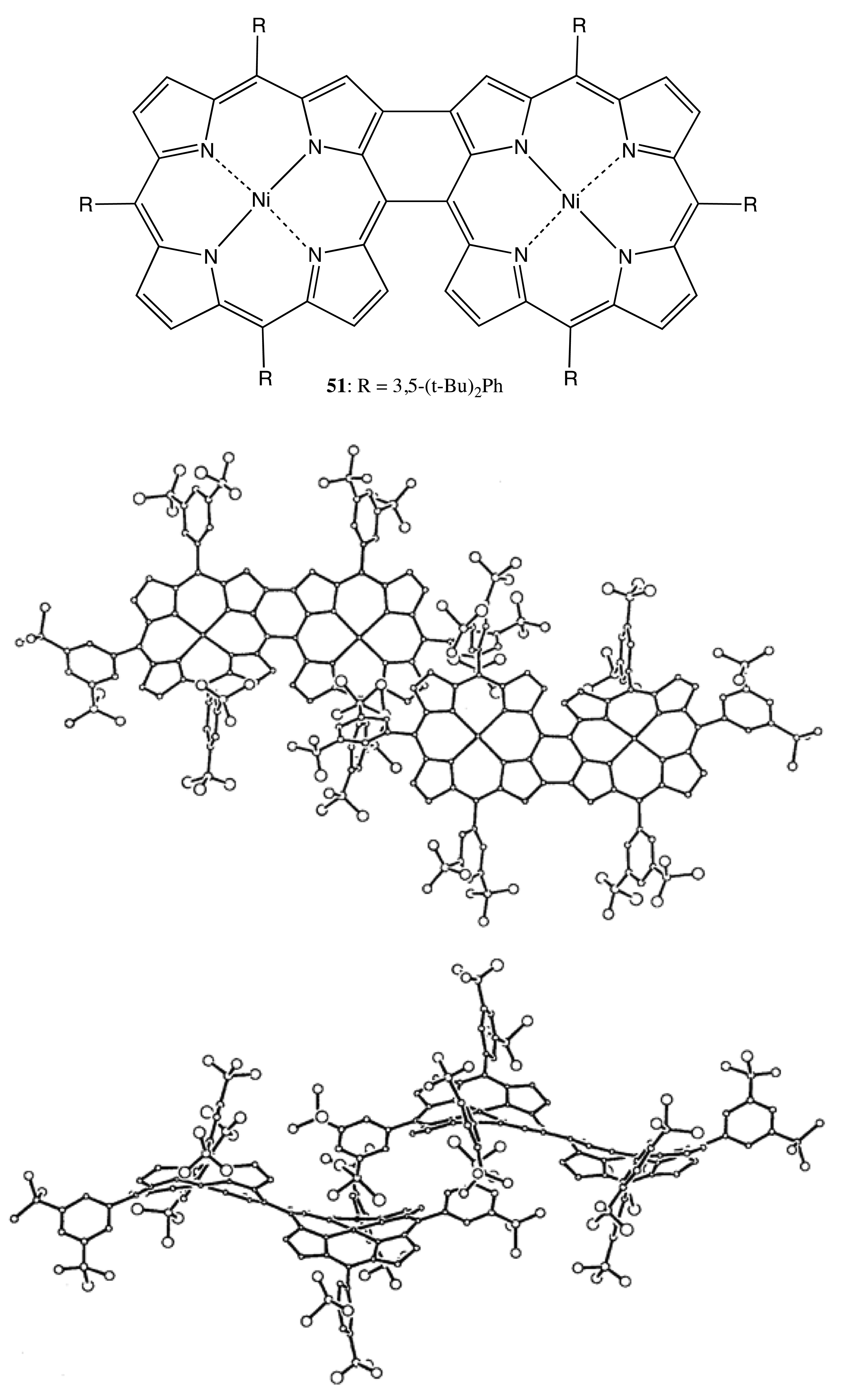
Another approach to fix asymmetry in the bis-porphyrin structures was used in the case of 51, where the porphyrin moieties were covalently linked by a meso-meso and a beta-beta bond (Figure 14) [46]. This double strapping resulted in the opposite ruffling of two macrocycles in 51, thus producing a helical structure with nearly C2 symmetry. The steric constraints of this bis-porphyrin allowed the optical resolution of the corresponding enantiomers, which exhibited mirror image CD spectra with a Cotton effect intensity of up to ±200 cm−1·M−1 at 416 nm and ΔG‡ = 111 kJ mol−1. However, the supramolecular chirality properties of this bis-porphyrin have yet to be investigated.
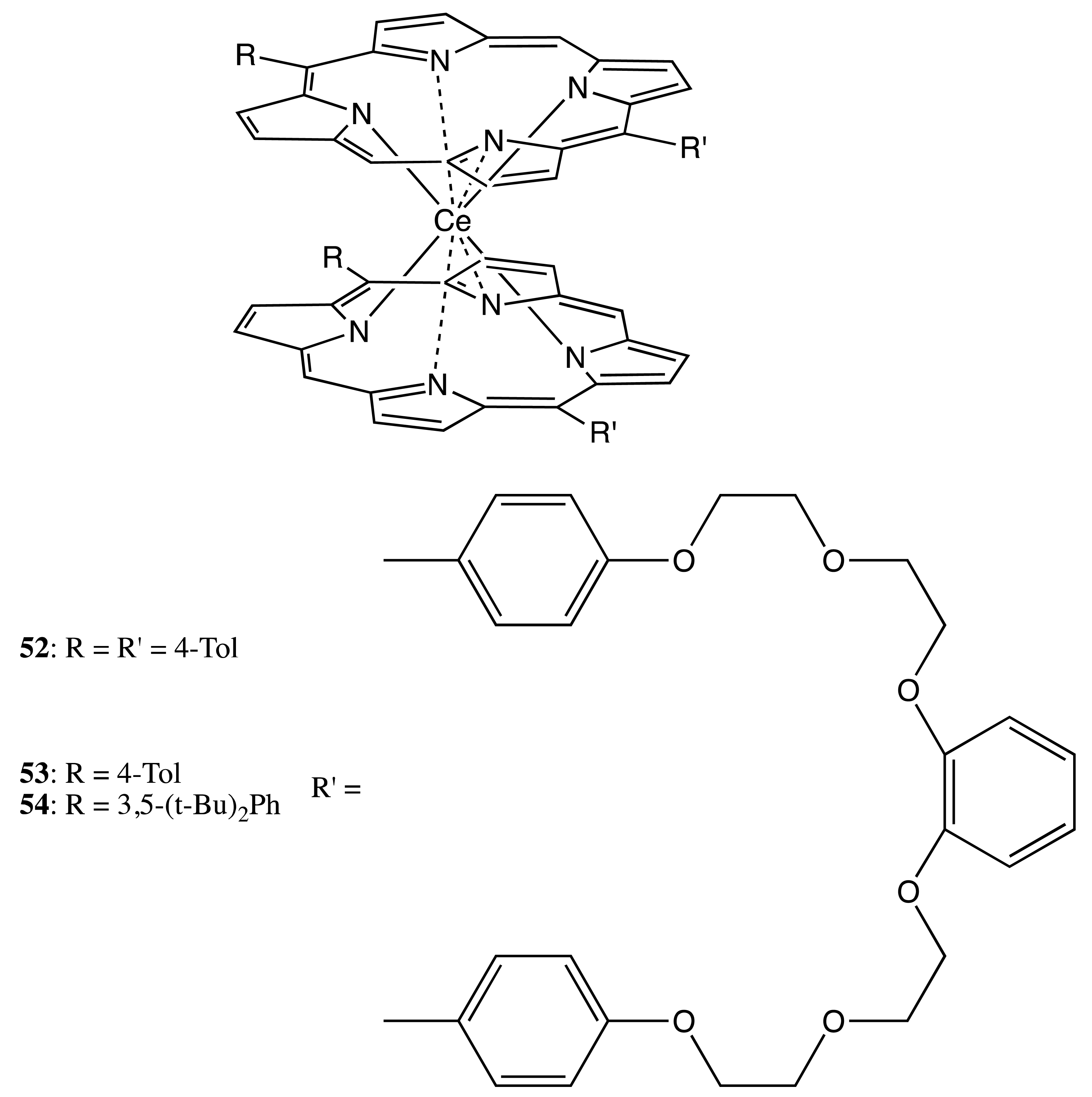
A different type of binding was used for the last example in this category of bis-porphyrinoids. Particularly, the cerium-bonded bis-porphyrin, 52 (Figure 15), possessed a D2 symmetry group that resulted in a chiral structure. However the corresponding enantiomers have not been optically resolved, due to the fast rotation of the porphyrin sub-units [47]. In order to reduce this rotation and, thus, to stabilize the corresponding antipodes, an additional oligoether strapping at the meso positions of the macrocycles was introduced to give the corresponding bis-porphyrins, 53 and 54 (Figure 15) [48]. This modification sufficiently hindered the rotation process to allow the separation of the enantiomers of 54, which are rotationally oscillating around the metal center with a rate constant of 0.39 × 10−3 s−1, eventually resulting in racemization.
3. Chirality and Supramolecular Chirality of Flexibly-Linked Structures
A different and more structurally diverse category of bis-porphyrinoids is based upon the flexible covalent linkages. Due to the conformational instability, the chiral properties of such systems are usually less pronounced, and some additional factors are generally needed to enhance the asymmetry performance. In this respect, the supramolecular chemistry approach plays a particularly important role in the chirality function. Additionally, there are some advantages of these bis-porphyrinoids from the applicability point of view. For example, these systems are more sensitive to the external chiral field, thus making it suitable for the purposes of chirality sensing and supramolecular chirality. Firstly, chiral porphyrinoids with achiral linkage are to be discussed.
3.1. Chiral Porphyrinoids with Achiral Linkage
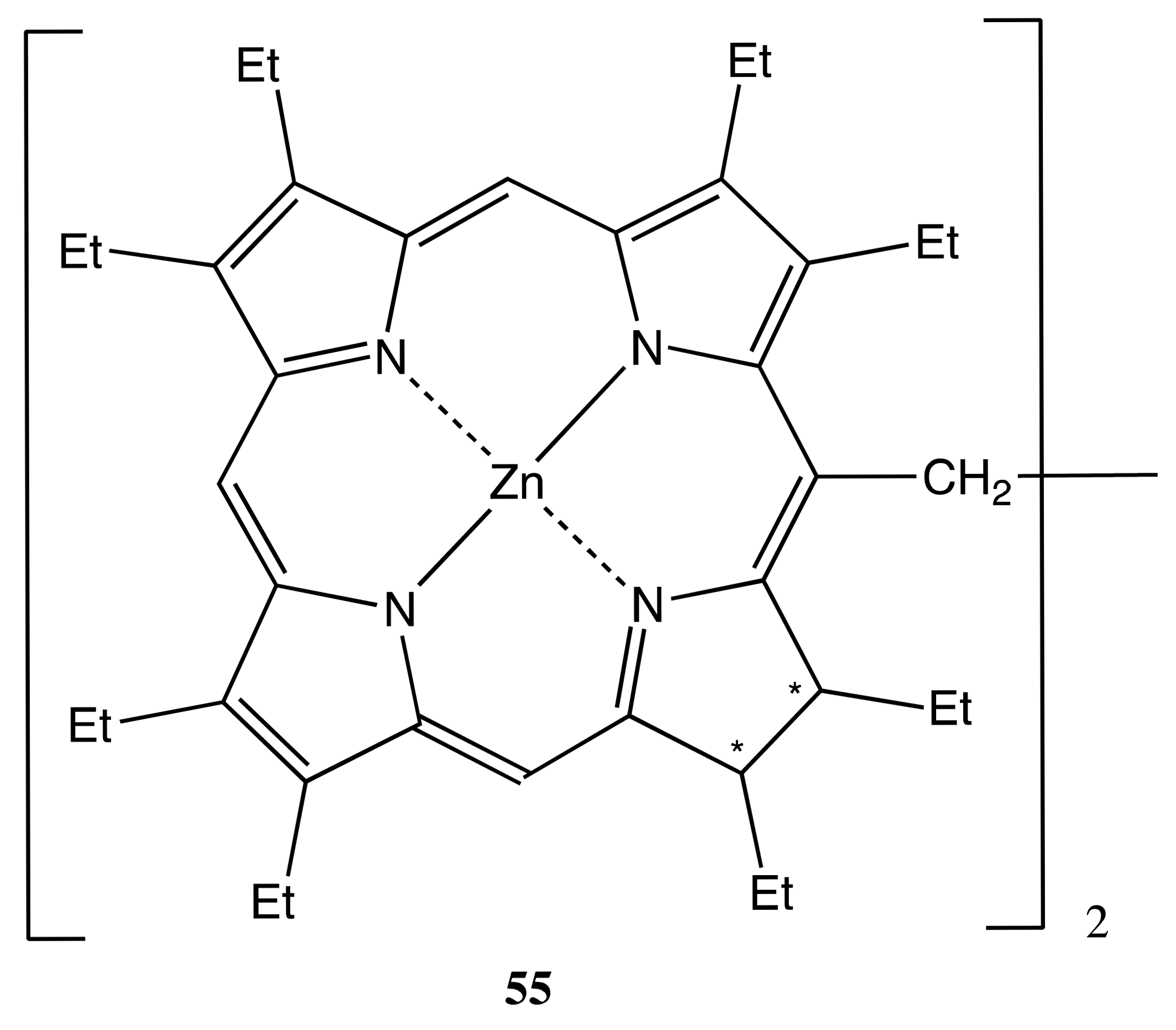
This type of bis- or multi-porphyrinoid consists of two or more macrocycles connected by a flexible covalent linkage. Such a kind of binding allows conformational freedom to a greater or lesser extent, which directly affects the chiral properties of the whole system. Whilst the primary chirality is produced by the chiral macrocycles, the overall chiral features can be further increased or modulated by additional fixation of the entire conformation via various ways, including the supramolecular chemistry methods. Thus, the first example to be dealt with in this category is based upon the bis-chlorin, 55 (Figure 16), the two chlorin subunits of which are connected by the semi-flexible ethane bridge, resulting in four stereogenic centers in total [49]. Surprisingly, only two (R,R/R,R and S,S/S,S) enantiomers were obtained by a chiral HPLC with an enantiomeric excess of up to >88%. Despite the relative flexibility of the ethane linkage, the geometry of 55 was stabilized by minimizing the total dipole moment. This was a result of the dipole-dipole interaction between two chlorine moieties yielding in the V-shaped conformation with the reduced pyrrole rings of two chlorin moieties located at the closest spatial position to each other. In contrast to the porphyrin chromophores, their mirror image CD spectra exhibited substantial optical activity in the region of both B and Q electronic transitions, with the corresponding amplitudes being ±163 and ±135 cm−1·M−1, respectively. The B and Q signals were of opposite CD signs, owing to the different spatial orientation of the coupling transitions. Upon supramolecular interaction with chiral guests, the CD signals of enantiopure 55 were reduced due to the induced conformational changes in the bis-chlorin. Furthermore, the chiroptical responses were significantly different in the case of corresponding antipodal guests [50,51]. Therefore, these distinct supramolecular chirality properties make it possible to apply 55 for the purposes of chiral recognition. This ability utilizes a new principle of enantioselectivity, which is based upon only a two-point host-guest interaction mode in combination with the coupling electronic transitions of the chromophoric host, the relative orientation of which is totally controlled by the guest stereochemistry.
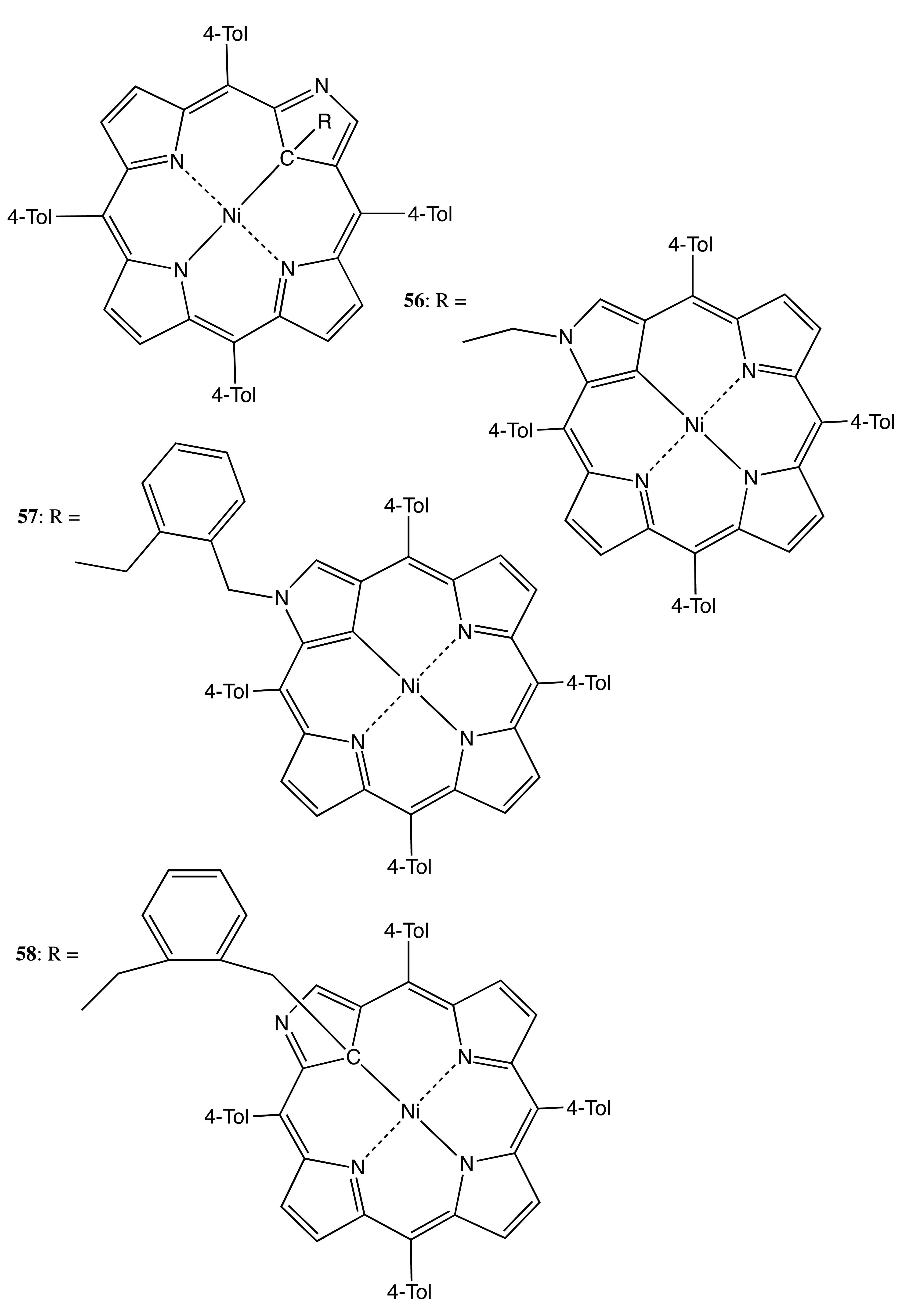
Bis-porphyrinoids on the basis of C21-alkylated N-confused porphyrin served as other examples in this category [52]. Hence, the methylene- and o-xylene-linked bis-porphyrinoids, 56–58 (Figure 17), the binding modes of which involved one or two C21 atoms, were obtained and optically resolved. The chiroptical outcome of 56–58 consisted of multiple Cotton effects of rather modest intensities (up to ±30 cm−1·M−1) in the region of the porphyrinoids' absorption. The observed moderate optical activity was a result of the conformational freedom of these systems, whilst the supramolecular chirality properties of 56–58 have not yet been explored.
3.2. Achiral Porphyrinoids with Chiral Linkage
This category is more plentiful and consists of achiral porphyrinoids linked to each other via a chiral linkage. In these types of porphyrin arrangements, a great variety of different structurally organized systems can be observed. The largest number is based upon the peripherally substituted porphyrinoids at the meso- or beta-positions. For example, two tetraphenylporphyrin derivatives were connected via enantiopure trans-1,2-dithiane-4,5-diol to yield the chiral bis-porphyrin, 59 (Figure 18) [53]. This induced optical activity in the region of porphyrin absorption with Cotton effect intensities of up to –455 cm−1·M−1 at 440 nm. The supramolecular chirality properties of 59 were expressed in the considerable decrease of the CD signal in the presence of enantiopure limonene down to –297 cm−1·M−1 and –237 cm−1·M−1 in the case of S- and R-enantiomers, correspondingly. This noticeable difference between the enantiomers arose from the chiral recognition properties of 59, which were further observed in the solid state upon deposition onto gold electrodes of quartz crystal microbalances and exposure to a guest containing gas phase as the first step for developing nanogravimetric sensors.
A similar structural approach was used in constructing the bis-porphyrin, 60, where two porphyrins were linked by a flexible chiral covalent bridge (Figure 18) [54]. The supramolecular chirality properties of 60 have been observed upon interaction with diamines. Hence, whilst the CD couplet of 60 itself was rather small, due to the existence of numerous conformations, the presence of diamines amplified the optical activity from 10 to 20 times. This effect was a result of the geometry stabilization due to the bidentate binding mode, forming a rigid tweezer structure. Furthermore, the nonequivalent interaction with antipodes as a consequence of the chiral recognition ability allowed 60 to be used as a chiral shift reagent.
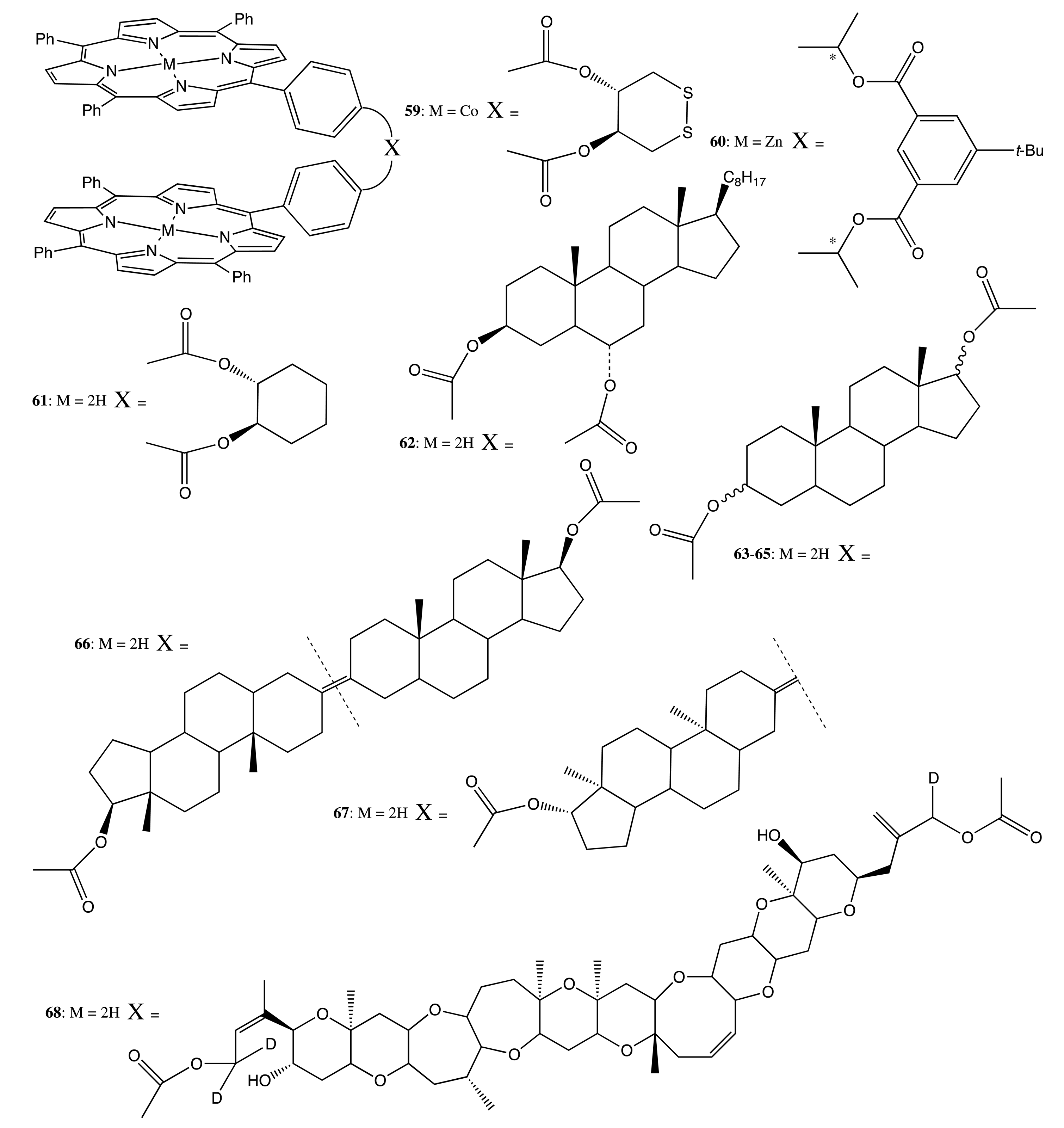
A more rigid structural motif on the basis of a steroidal skeleton was used to connect two porphyrin moieties in the bis-porphyrins, 61–68 (Figure 18) [55]. This type of linkage resulted in a considerably stronger chiroptical outcome, with the CD amplitude being as high as +675 cm−1·M−1 in the case of 62, whilst the interporphyrin exciton coupling was dependent on the distance and orientation between the porphyrin subunits detectable over a distance of 59 Å, as in the case of 68. The observed high sensitivity was suggested to apply to the stereochemical assignment of steroidal diols. However, the supramolecular chirality properties of these bis-porphyrins have not been investigated so far.
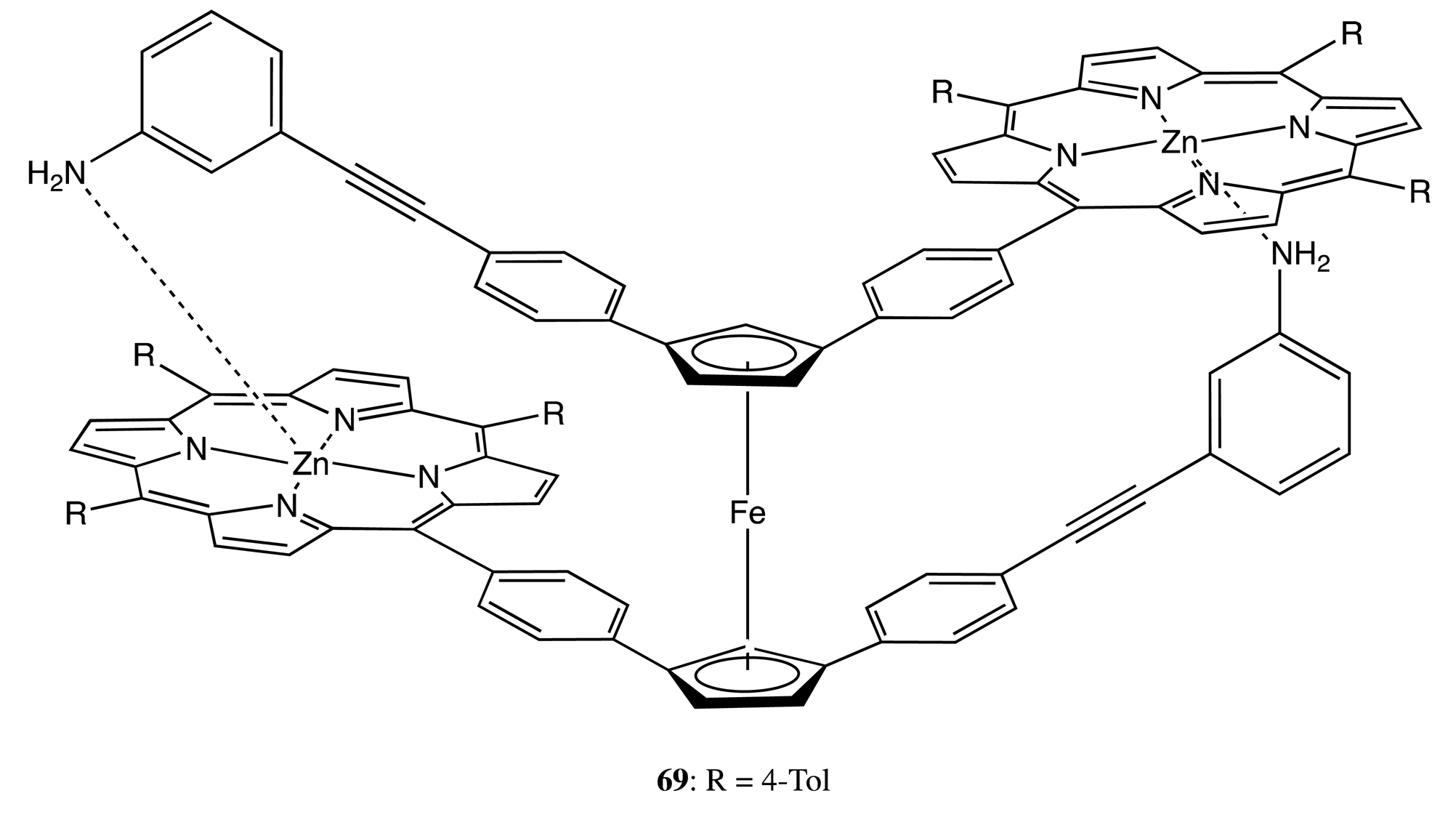
Another type of conformational flexibility, the rotational freedom, was realized in the bis-porphyrin, 69 (Figure 19) [56]. The principles of supramolecular chirality in 69 have been applied as follows. Two porphyrin moieties were coupled via a ferrocene unit using it as a stirring module. The rotary motion allowed the formation of two Zn–N coordination bonds between the porphyrin and aniline units, hence producing a noticeable optical activity in the regions of ferrocene and porphyrin absorptions. Interaction with the cis- and trans-forms of 1,2-bispyridylethylene resulted in the corresponding modulation of CD signals upon formation of the 1:1 and 1:2 intermolecular host-guest complexes. However, the subsequent photochemical cis-to-trans isomerization switched back to the self-locked state of 69, due to the weaker binding affinity of the trans-isomer.
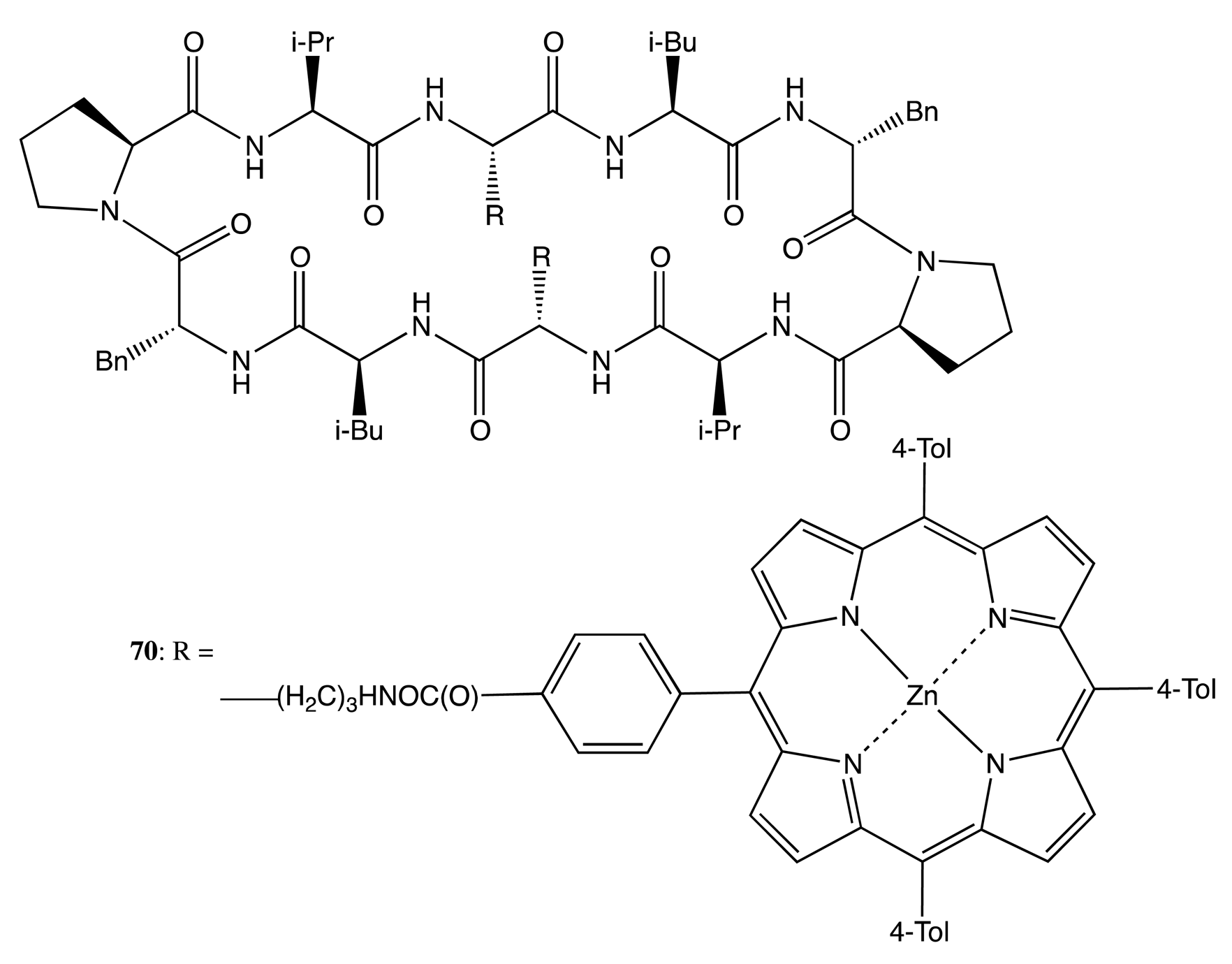
Peptides have also been widely used as a chiral backbones in linking porphyrin moieties. For instance, a pair of porphyrins was bound to the antiparallel beta-sheet cyclic decapeptide, Gramicidin S, to yield the bis-porphyrin, 70 (Figure 20) [57]. Owing to the asymmetrical orientation of porphyrin fragments and the left-twisted arrangement of polypeptide, a relatively strong negative couplet was observed in all cases. For the dizinc complex, 70, according to the supramolecular chirality concept that the solvent is an active part of the overall system and may directly affect the chiral properties, the intensity of the CD signal depended on the media used, with the amplitude being as high as −675 cm−1·M−1 in the case of toluene. This solvent-controlled variability reflected the conformational flexibility of porphyrin moieties. However, upon interaction with 4,4′-dipyridyl, the CD profile was considerably changed to exhibit a complex profile, with the major positive peak at 424 nm, the intensity of which was strongly diminished to 23.7 cm−1·M−1 as a result of the almost parallel mutual orientation of two porphyrins.
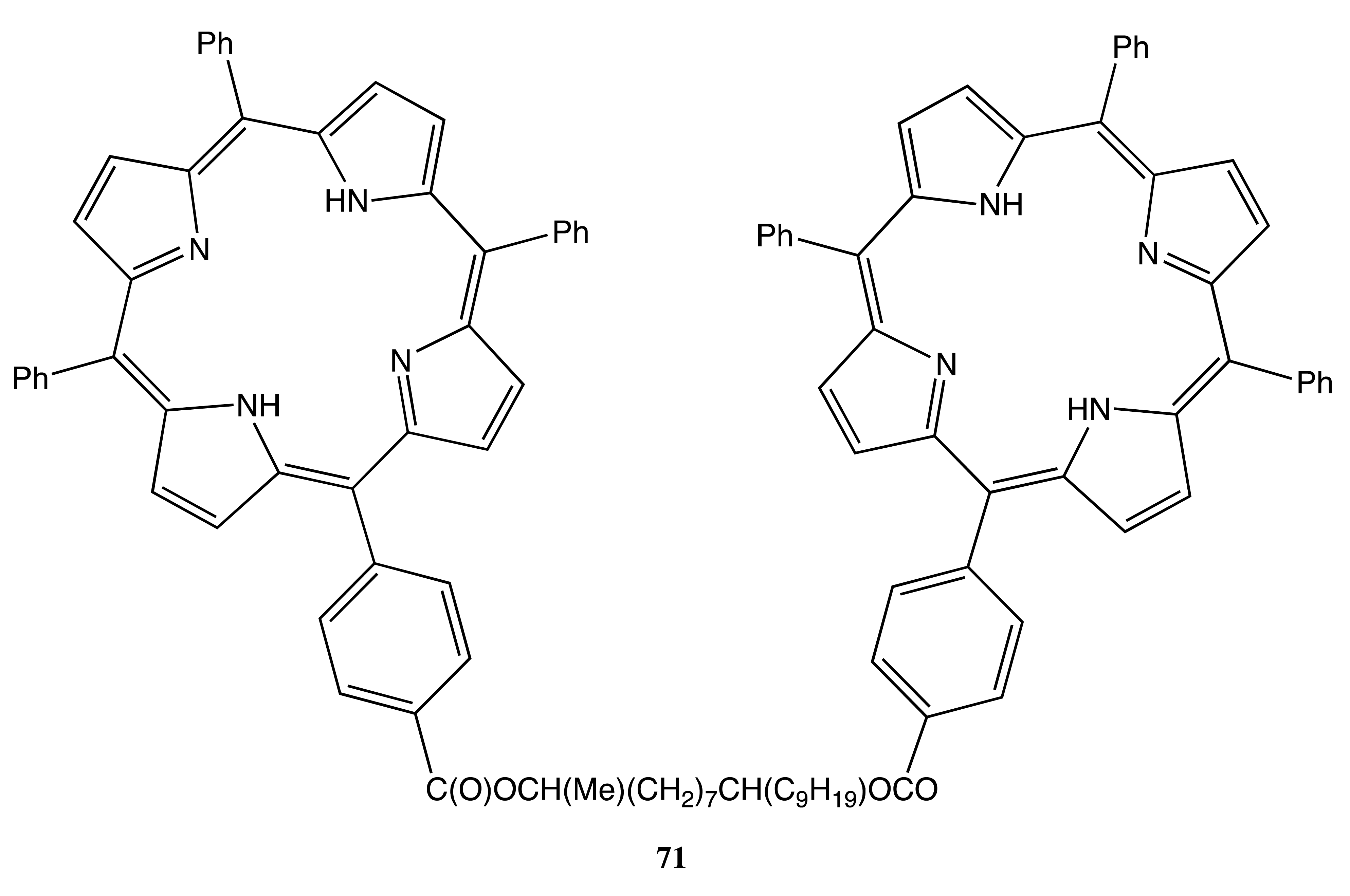
The wide range of applications of bis-porphyrin systems was nicely demonstrated by two porphyrins connected to the chiral derivatives of 1,9-glycol to yield the corresponding structure, 71 (Figure 21) [58]. This bis-porphyrin was applied for the determination of the absolute configuration of caminoside A. Whilst the conformational freedom of 71 resulted in no chiroptical response in the solution phase, the supramolecular chirality approach using a liposome environment as the ordering media to fix the spatial orientation of two porphyrin moieties successfully induced a noticeable bisignate CD signal of 22 cm−1·M−1. Further exciton coupling analysis combined with the comparative studies of reference bis-porphyrins led to the assignment of the C10 configuration as R.
Multi-porphyrin subunits are frequently attached to an enantiopure polymeric backbone in order to transfer the asymmetric properties from the chiral matrix to the achiral chromophores. In particular, about 100 porphyrin moieties have been selectively linked to cellulose at the C(6)-O position to form the corresponding porphyrin-containing polymers, 72 and 73 (Figure 22) [59]. The spatial structure of cellulose dictated the unidirectional helical arrangement of porphyrins, hence inducing the exciton coupling CD signal in the region of porphyrin Soret band with an amplitude well above −400 cm−1·M−1 in the case of 73. Interestingly, the chiroptical outcome of 72 was reversibly off-on switched upon the one-electron oxidation and subsequent reduction, thus offering the CD read-out of an electrochemically-induced information input.
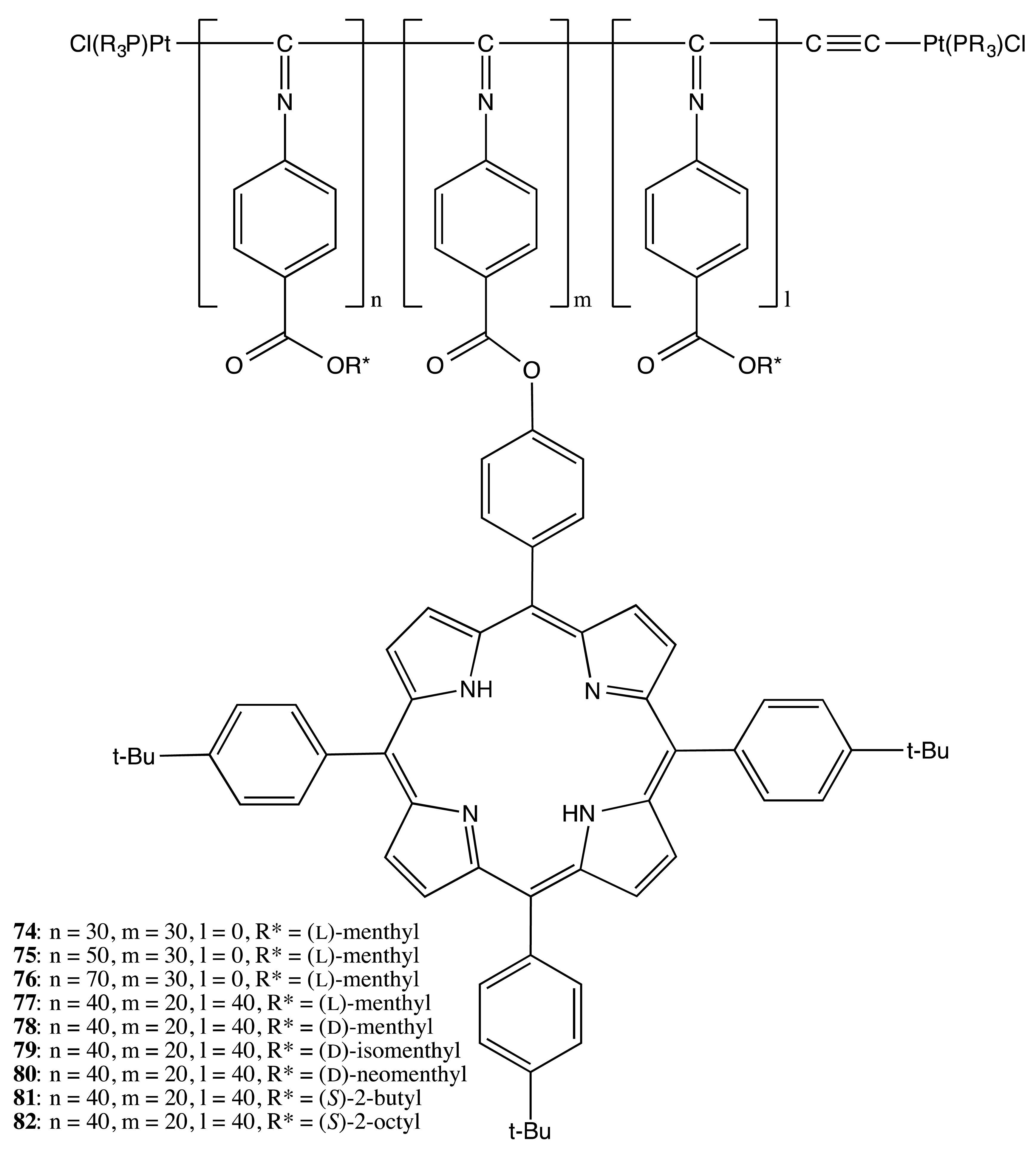
Another type of the chiral polymer matrix used in the preparation of multi-porphyrins was based on artificial helical polyisocyanides containing enantiopure groups. Hence, copolymerization of the corresponding blocks containing different chiral and porphyrin moieties resulted in the optically active polymers of various structures, 74–82 (Figure 23) [60]. The helical sense of the polymer was transferred to the porphyrins' spatial arrangement, thus inducing an intense CD signal in the region of the porphyrin Soret band, the sign of which was unambiguously governed by the handedness of the chiral backbone. In particular, the positive sign was indicative of a right-handed helix, whilst the negative sign implied a left-handed helix.
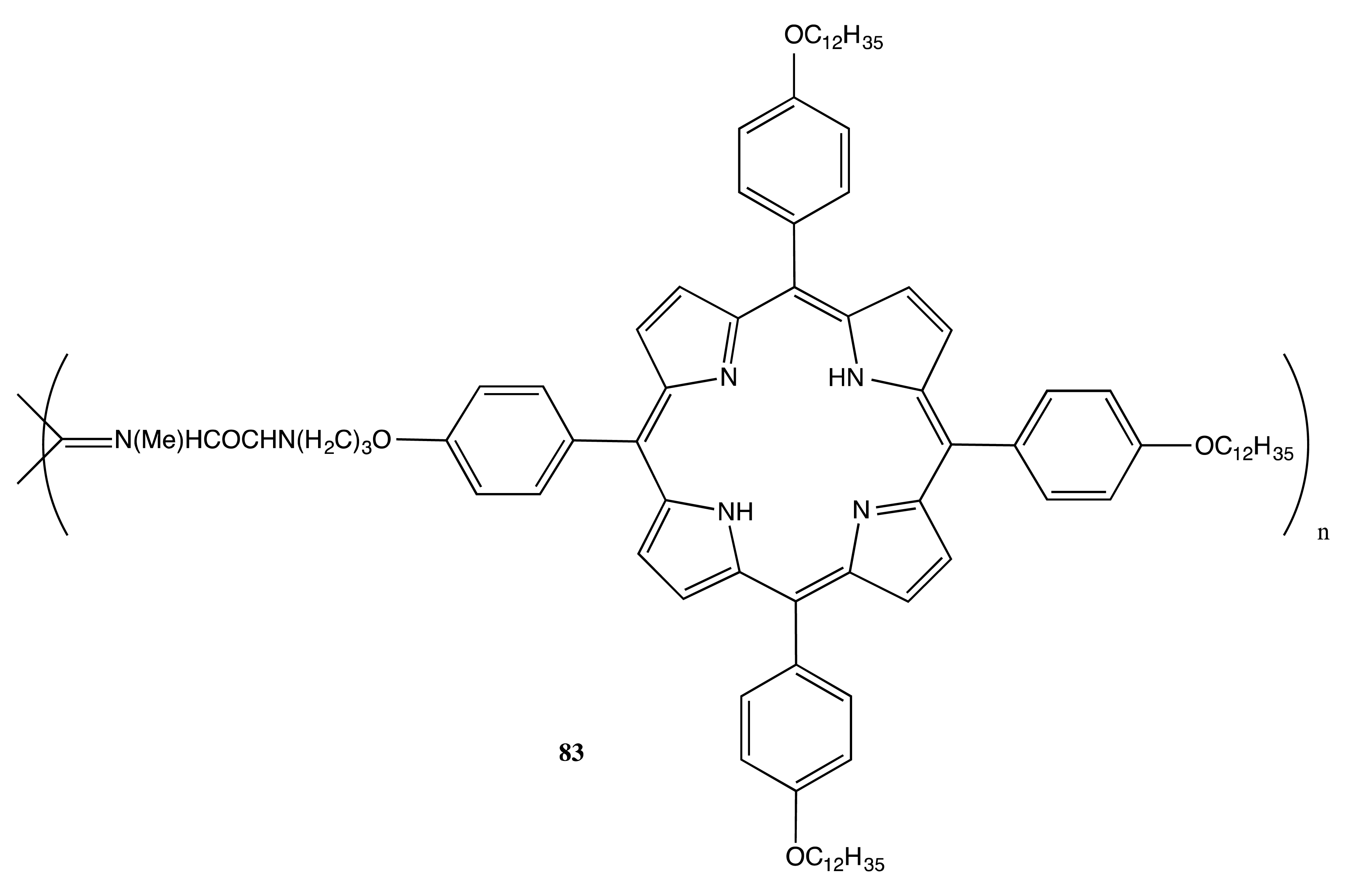
A similar approach was employed in the construction of helical polymer-anchored porphyrin nanorods [61]. Hence, the Ni(II) catalyzed polymerization of chiral porphyrin containing isocyanide monomer resulted in the corresponding polymer, 83 (Figure 24), the porphyrin moieties in which were arranged in four well-defined stacks that ran parallel to the polymer axis. One stack contained at least 25 interacting porphyrin units, which were 4.2 Å apart from each other with a slip angle of 30° and an overall average length of 87 nm. In this geometry, the backbone chirality was effectively transferred to the porphyrin chromophores, thus inducing an exceptionally strong CD couplet with an amplitude of over −3500 cm−1·M−1 in the region of the porphyrin Soret band.
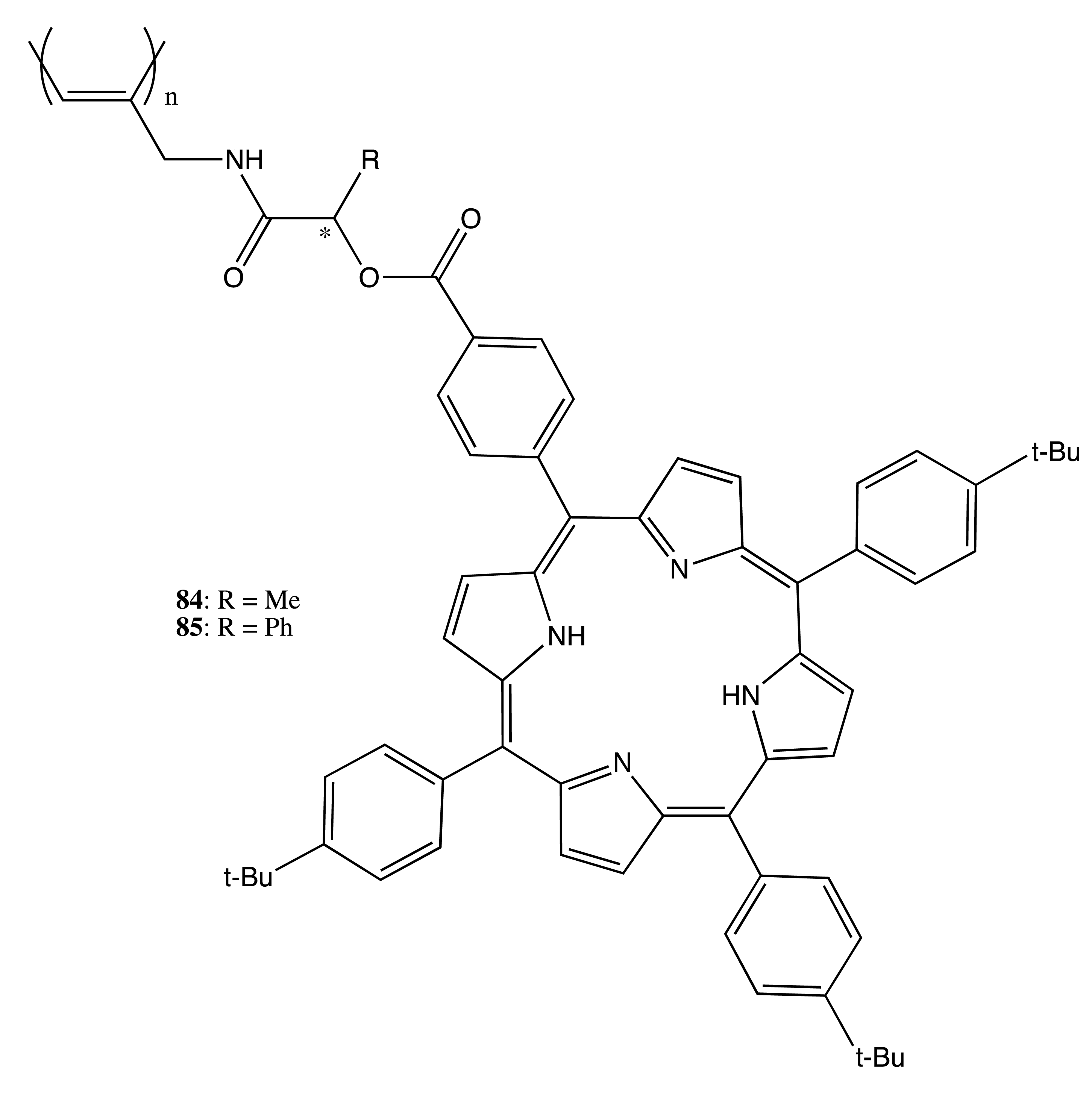
A different polymeric matrix was used to obtain chiral poly(N-propargylamides)-bearing porphyrin groups, 84 and 85 (Figure 25) [62]. The molecular weights of 84 and 85 were determined as 17.000 and 24.000, with the cis contents being 85% and 78%, correspondingly, hence inducing fairly stereoregular structures and high optical activity in the region of the porphyrin Soret band. The direction of CD exciton coupling revealed the right-handed relative arrangement of porphyrin units on the left-handed helical polyacetylene.
3.3. Achiral Porphyrinoids with Achiral Linkage
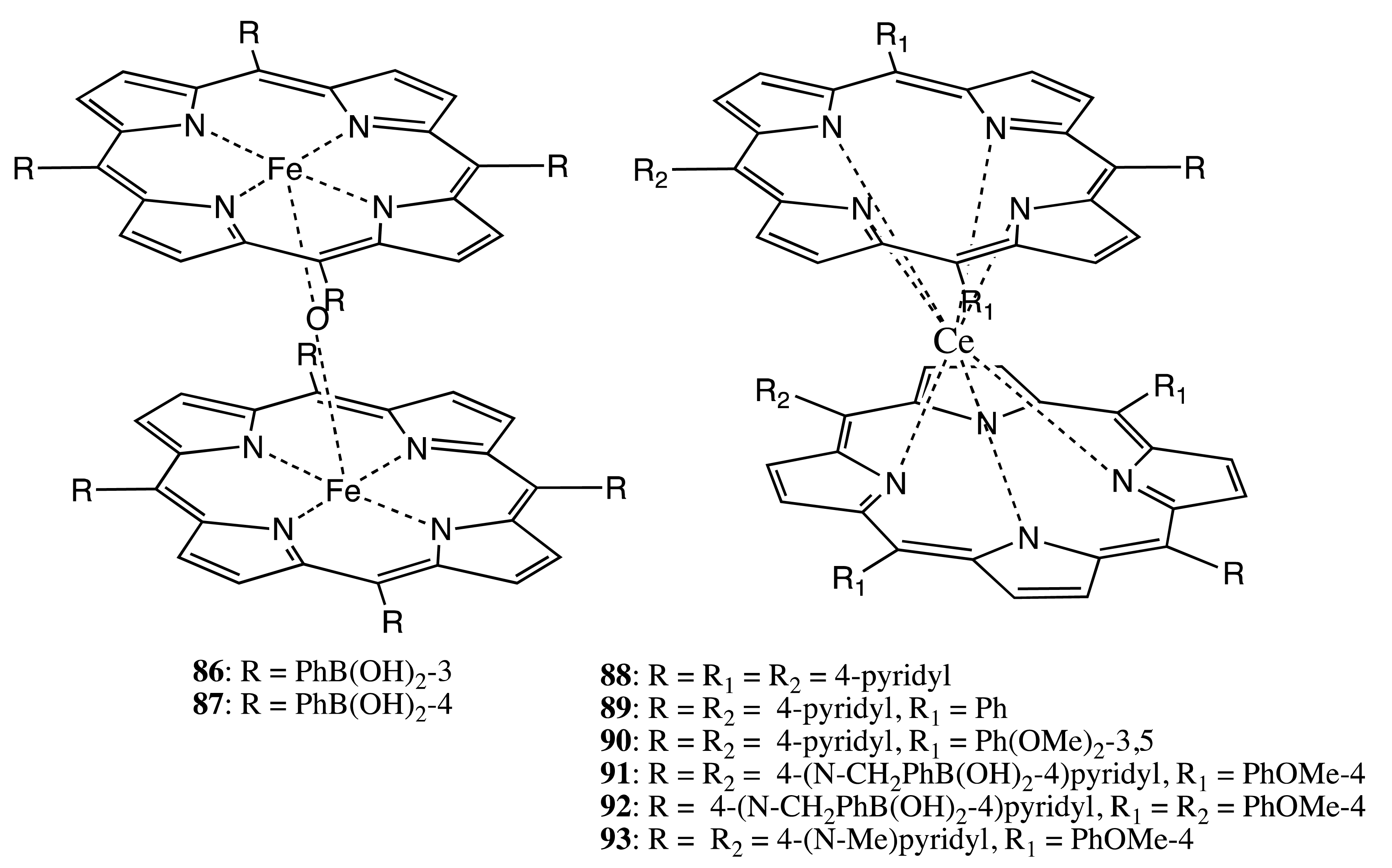
The last structural type in this category includes achiral porphyrinoids linked through a flexible achiral bridge. These systems are most frequently used for supramolecular chirality purposes. This is because both the chromophore and linkage units are inherently achiral, and an additional chiral influence involving noncovalent interactions is needed to induce asymmetry to these structures, also making it highly sensitive to the chirality modulation. Therefore, most often, these systems are applied in chiral sensing and the determination of the absolute configuration. One of the first examples was based upon the double-decker architecture, in which two porphyrins were bound through the central metal ion core as μ-oxo dimers in the case of 86, 87, and Ce complexes in the case of 88–93 (Figure 26) [63–68]. The rotational freedom around the central axis of these bis-porphyrins was used to sense the chirality of various enantiopure compounds upon the supramolecular interaction of a bidentate guest with two anchor binding groups of the neighboring porphyrins. This generated a unidirectional twist either in the right- or in the left-handed direction, depending upon the guest's stereochemistry, and subsequently induced appreciable optical activity in the region of the porphyrin Soret band, owing to the interchromophoric exciton coupling. Hence, these systems were successfully applied for sensing the chirality of saccharides (in the case of 86, 87, 91, 92), dicarboxylic acids (in the case of 88–90), dianions (in the case of 93) and memorizing the chiral information (in the case of 90). The durability of chiral memory was estimated to last for three days at 0 °C, one year at –37 °C and 1.9 × 106 years at −100 °C. The host-guest binding occurred in a highly cooperative manner, exhibiting a positive allosteric effect, whilst the CD response was strongly affected by the guest's structure [63,64], solvent [68] and pH value [67]. Besides, the effect of supramolecular chirality switching controlled by the number of sugar units was observed for 91 upon interaction with maltooligosaccharide guests.
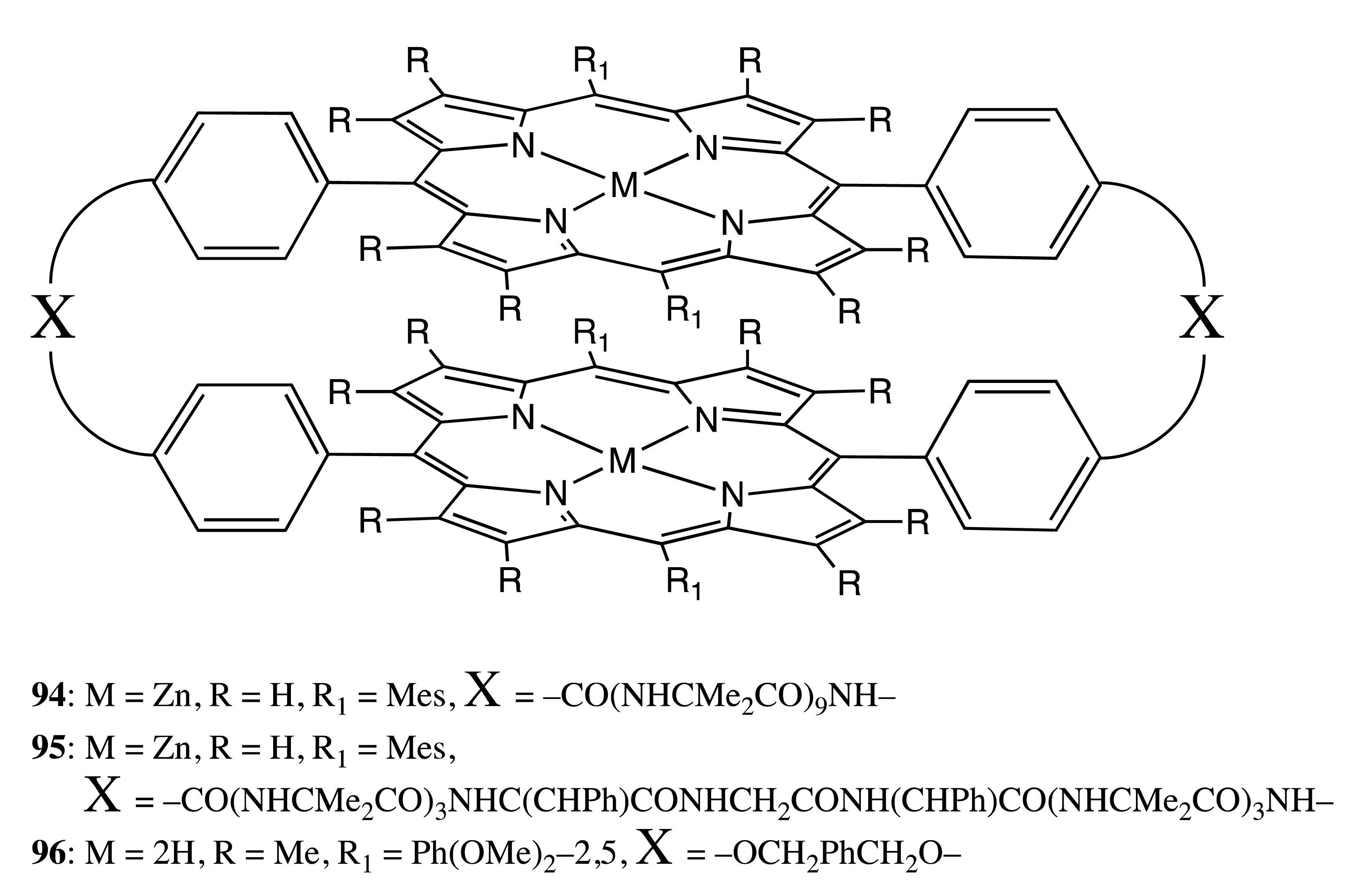
The face-to-face spatial arrangement of bis-porphyrins could be also ensured by two covalent linkages located at the opposite sides of porphyrin cycles, as in the structures, 94–96 (Figure 27) [69,70]. Owing to a certain semi-flexibility and sufficiently long length of the covalent bridges, 94 and 95 showed a high specificity toward the bis-pyridyl-substituted chiral guests of appropriate length. The corresponding supramolecular host-guest interactions resulted in the stable 1:1 inclusion complexes with the association constants of up to 2.3 × 106 M−1. The chiral information of the guests effectively transferred to the bis-porphyrin hosts, thus inducing a strong exciton coupling signal in the region of the Soret band. Bis-porphyrin, 96, with a shorter linkage was employed for the complexation with smaller guests, such as mandelic acid, exhibiting significant amplification (more than seven times) of the CD outcome (with the amplitude of 260 cm−1·M−1) in comparison to the corresponding monomer, due to the translation of nonplanar chirality from the porphyrin ring into the helical chirality of the entire supramolecular assembly.
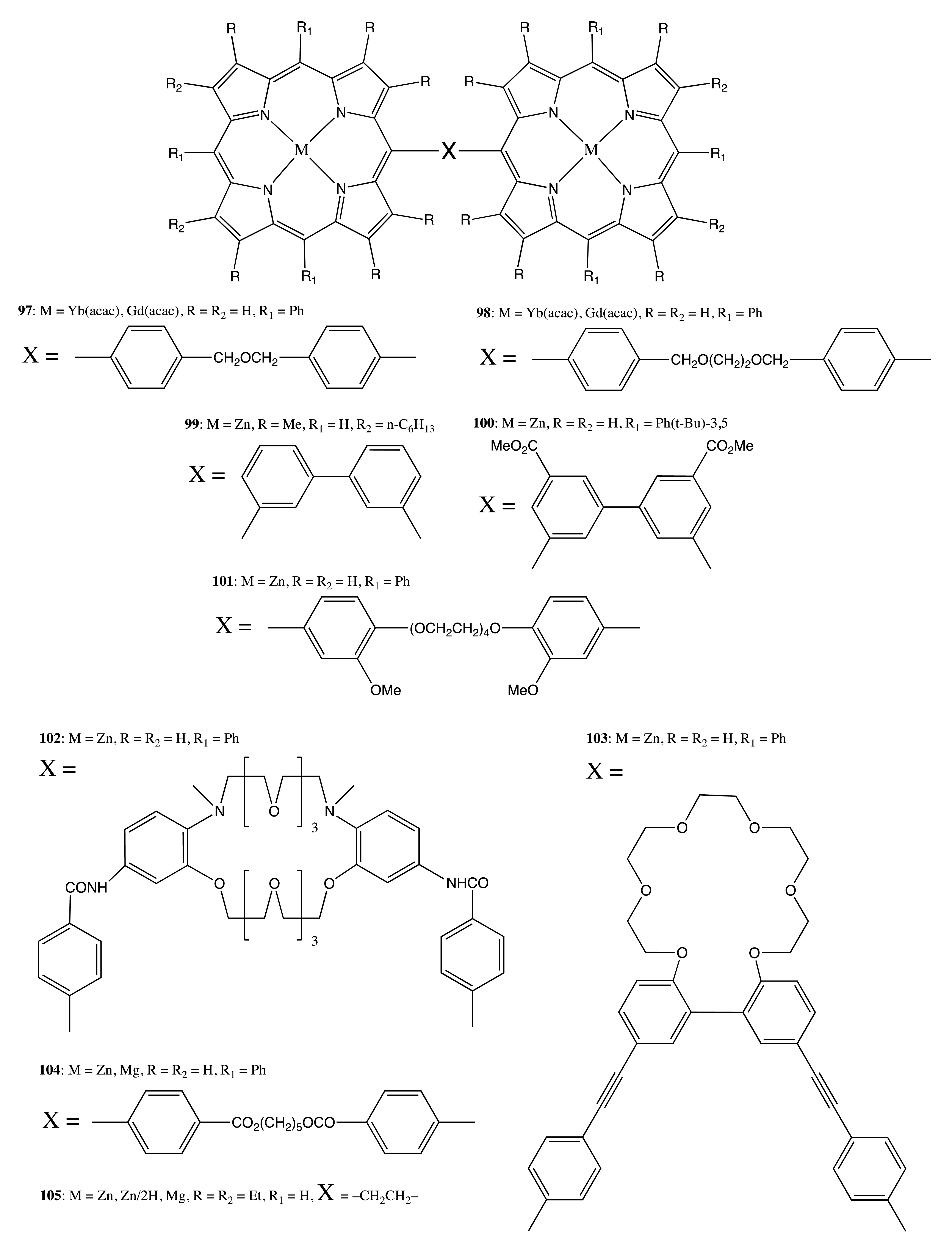
Bis-porphyrins linked by a single covalent bridge are another structural motif in this category of porphyrin architectures. In this case, the overall rigidity of the system is decreased, resulting in the co-existence of a number of conformations, and hence, the additional complexity from the chirality point of view. Thus, for successful supramolecular chirality generation, the asymmetry inductor should be able to stabilize the spatial organization of the whole assembly. For example, a series of the lanthanide bis-porphyrins, 97 and 98 (Figure 28), was synthesized for binding cysteine polyions and exhibited complicated CD profiles in the region of the Soret band upon noncovalent interaction with chiral guests [71]. The CD shape was greatly affected by the mode of binding and the size of the guest. Cysteine was efficiently extracted from an aqueous solution by the Yb complex of 97 in comparison to other guests tested. This resulted in the greater CD outcome of the corresponding 1:1 complex. In the case of longer homocysteine, the bis-porphyrin, 98, was more effective.
The chiroptical properties of more rigidly bound bis-porphyrins, 99 and 100 (Figure 28), generated by supramolecular interactions with chiral diamines, were also strongly influenced by the bulkiness of the guest, the number of stereogenic centers, the porphyrin structure and temperature [30]. The greatest induced CD amplitude of 1340 cm−1·M−1 was further increased 1.5 times upon lowering the temperature down to −45 °C, due to the stabilization of the chiral conformation, whilst the association constant for the best host-guest pair was determined to be as high as 2.4 × 106 M−1.
The crown ether linkage provided a certain degree of rigidity for the bis-porphyrin structure, particularly via specific complexation with alkali metal ions. For example, the corresponding chiroptical properties of the 1:1 supramolecular tweezer complex between the bis-porphyrin, 101 (Figure 28), and DACH were precisely modulated by the addition of Na ions and monitored by the intensity of the Cotton effect at 436 nm, −415 and −620 cm−1·M−1 without and in the presence of Na+, respectively [72]. Furthermore, the association constant was also controlled in the same way, increasing from 2.6 × 105 M−1 to 4.5 × 105 M−1, correspondingly. Similarly, the CD amplitude and binding affinity were greatly enhanced for the bis-porphyrin, 102 (Figure 28), upon interaction with the N-alkyl substituted DACH in the presence of larger alkali ions [73,74]. The best-fitted K+ ion exhibited the greatest chirality amplification effect, increasing the intensity from 770 cm−1·M−1 up to almost 1000 cm−1·M−1. Additionally, this bis-porphyrin was able to perform an extraction and chirality probing of unprotected amino acids as a result of the amphiphilic property of the crown spacer, where encapsulated K+ assisted the formation of the corresponding host-guest complex. The crown ether moiety provided an additional functionality of the host-guest complex that holds a chiral memory function. Particularly, the induced CD signal of a supramolecular tweezer system consisted of the bis-porphyrin, 103 (Figure 28), and the chiral diamine was effectively preserved upon replacement of the latter with an achiral diamine in the presence of Ba2+ ions, which played the role of the chiral conformation stabilizer [75]. The chiral memory on 103 was stable with just a small decrease of the CD intensity over one day.
In the case of flexible bound porphyrins, the corresponding bis-porphyrin, 104 (Figure 28), required a bidentate enantiopure ligand to adopt the supramolecular chiral tweezer conformation [76,77]. Hence, the interaction of 104 with various chiral diamines resulted in the induction of a CD couplet in the region of porphyrin absorption. The CD intensities were greatly varied depending upon the guest structure, media, hydrogen bonding and other factors, reaching a value of up to 1078 cm−1·M−1. The chirogenic mechanism included the stereospecific differentiation of the substituents' relative bulkiness at the asymmetry center, thus forcing two porphyrins to follow the guest's stereochemistry, which was reflected by the CD sign. This property was used for the absolute configuration determination of different bidentate compounds.
The more rigidly ethane-linked bis-porphyrin, 105 (Figure 28), was able to form the corresponding supramolecular chiral structures upon interaction, as with bidentate and with monodentate guests [5,15,78–86]. In the case of enantiopure monoamines, the induced CD amplitude was in the range of 7.2–129.7 cm−1·M−1. This chirogenic activity was a result of noncovalent interactions between 105 and a monodentate guest leading to the formation of the stable 1:2 host-guest complex in a cooperative manner with the Gibbs free energy varying from −6.7 to −8.4 kcal mol−1 for primary amines, as determined by absorption spectroscopy. The competitive repulsive interactions between the two most bulky substituents at the guest's stereogenic center and the ethyl groups of the neighboring porphyrin ring generated a unidirectional screw structure in the extended anti-conformation of 105. The chirality sign unequivocally correlated with the induced helicity, thus allowing the straightforward determination of the absolute configuration of monodentate guests and the investigation of various external and internal controlling factors. In contrast, the interaction with bidentate guests resulted in the formation of extremely stable supramolecular 1:1 tweezer complexes possessing a significantly enhanced optical activity. For example, in the case of enantiopure DACH, the Gibbs free energy was determined to be as great as −9.5 kcal mol−1, whilst the CD amplitude was as high as 590 cm−1·M−1. This was a result of the increased structural rigidity and optimal geometry for the process of chirality transfer experienced by the tweezer complexes in comparison to the anti-species, owing to the concerted nature of the two coordination bonds in the 1:1 complex, rather than the dynamic single coordination bonds of the 1:2 anti-complexes.
4. Supramolecular Chirality of Self-Assembled Monomeric Units
Besides covalently linked bis- and multi-porphyrins, simple monomeric porphyrins are also able to demonstrate a cooperative supramolecular chirality upon various noncovalent interactions. However, for effective chirality generation, the external binding matrix or the self-organization ability combined with the corresponding asymmetry field are needed. As in the case of covalently linked systems, there are two major classes of chromophores: chiral and achiral porphyrinoids involved in various chiroptical effects and supramolecular processes.
4.1. Chiral Porphyrinoids
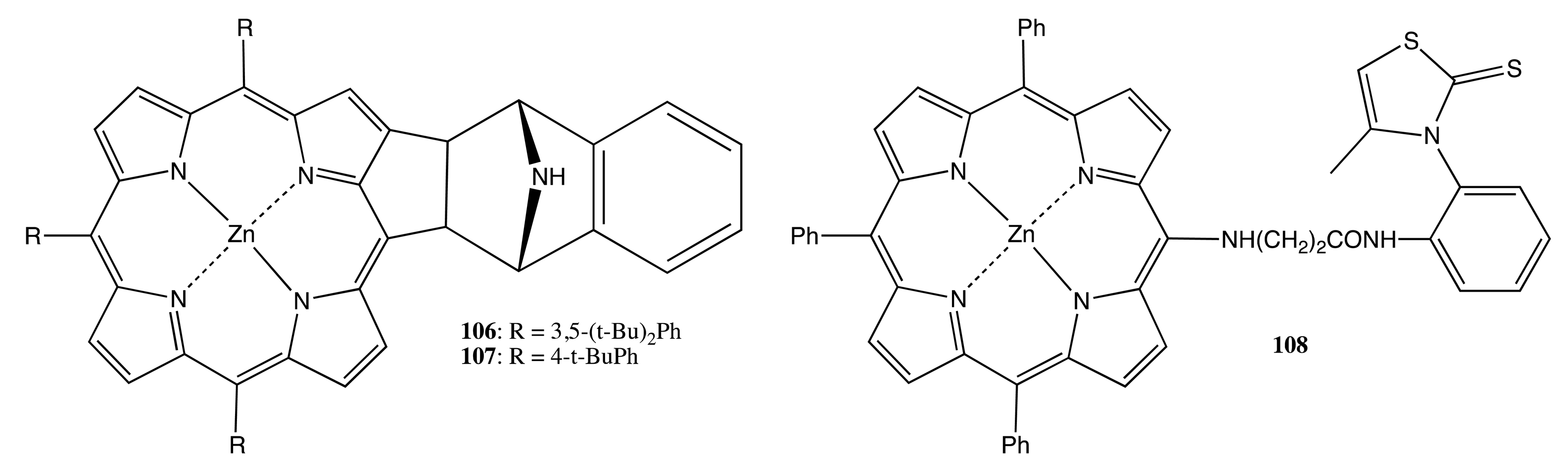
Although this type of porphyrinoids possesses its own center(s) of asymmetry, which is frequently used for chiral recognition, enantio-differentiating catalysis, medical purposes and various biomimetic studies, generally, the chiroptical outcome itself is rather moderate. In order to amplify the chiral properties, two or more porphyrinoids should be brought together in close proximity by means of different supramolecular interactions. For example, the simplest self-assembly via the dimerization process was achieved for the chiral porphyrins, 106 and 107 (Figure 29) [87]. The complimentary coordination of enantiopure 106 resulted in a homodimeric structure with the association constant of 1.2 × 107 M−1, whilst a mixture of 106 and 107 produced the corresponding heterodimer, reflecting the enhanced stability of the hetero assembly in comparison to the homo structure.
More complicated supramolecular self-assembly behavior was observed for the enantiopure porphyrin, 108 (Figure 29) [88]. Whilst the monomeric species was essentially CD silent, the corresponding self-coordinated complex exhibited a noticeable CD signal in the region of Soret absorption consisting of three Cotton effects as a result of the interporphyrin exciton coupling.
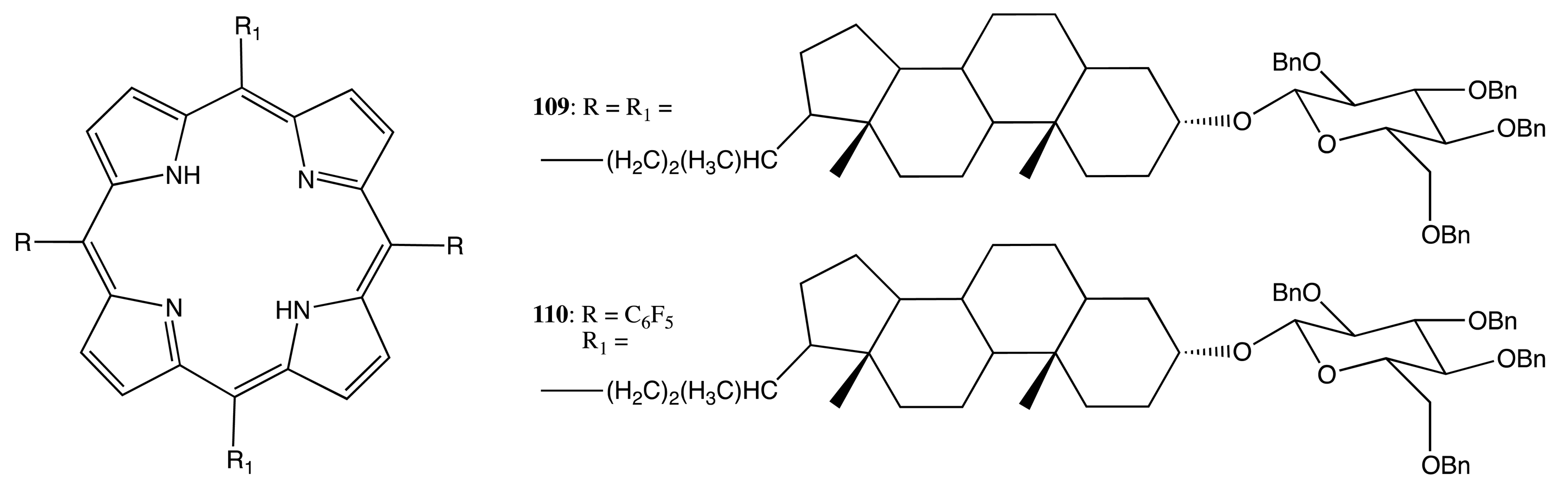
A different type of supramolecular self-assembly was employed for the chiral porphyrins, 109, 110 (Figure 30) [89]. Whilst the monomeric species showed a rather weak monosignate Cotton effect (less than 10 cm−1·M−1), the solvent-driven aggregated state induced an appreciable CD outcome consisting of several moderate exciton couplets (in the case of 109) or a single negative bisignated signal (in the case of 110) in the region of the Soret band with an amplitude of up to 450 cm−1·M−1. The difference in the chiroptical behavior was governed by the morphology of aggregated species and dictated by the arrangement of glycosylated substituents.
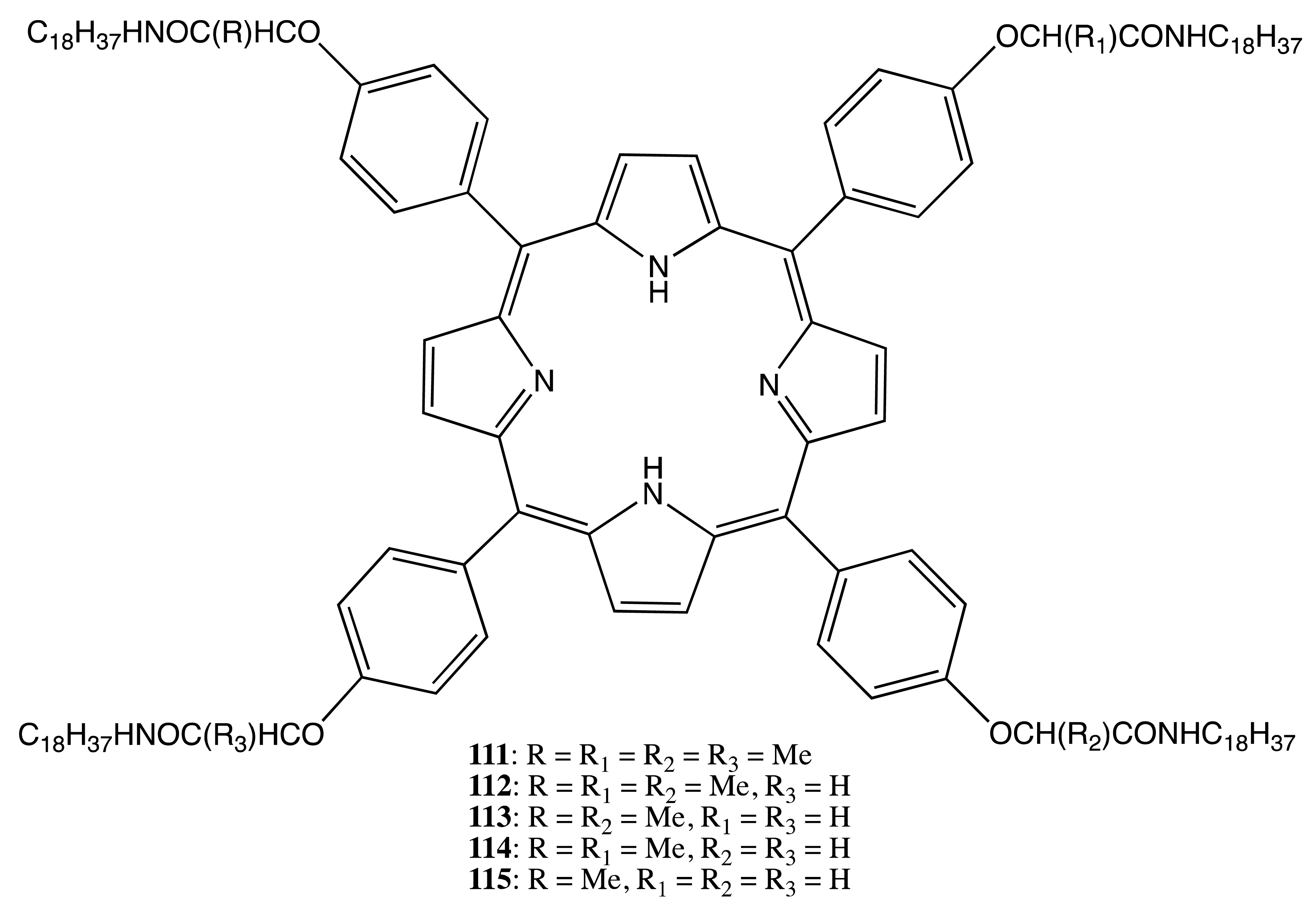
Similarly, the porphyrins, 111–115 (Figure 31), possessing from one to four stereogenic centers formed the corresponding hydrogen bonded chiral H-aggregates [90]. The resulting CD exciton couplings were dependent upon the number of chiral moieties and noticeably enhanced upon lowering the temperature. The degree of structural chirality of the monolayers increased almost linearly with the number of stereogenic centers, whilst the helical handedness was the same for the same absolute configuration of asymmetric carbon.
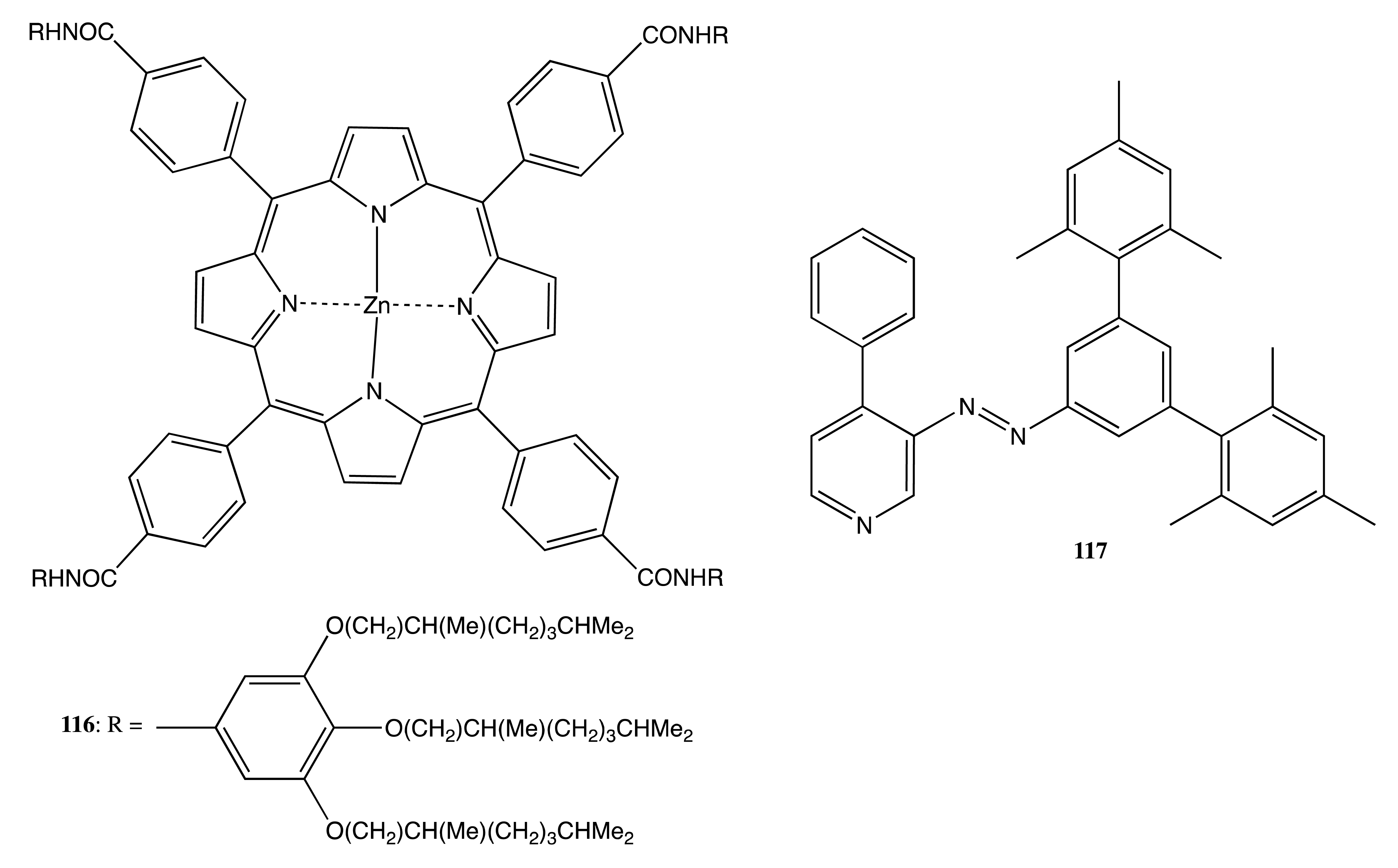
A programmed photocontrol of supramolecular chiral self-assembly was undertaken in the case of the optically active porphyrin, 116 (Figure 32) [91]. In particular, a complex of 116 with 117 dissociated upon the trans-to-cis photoisomerization of 117, owing to the steric hindrance imposed by the bulky mesityl groups in the cis-conformation. The removal of the ligand resulted in the subsequent self-assembly of 116 governed by four intermolecular hydrogen bonds to produce the corresponding helical cofacial structures. Simultaneously, the CD signal intensity in the region of Soret absorption increased up to 81% relative to the reference solution of 116 in the absence of 117.
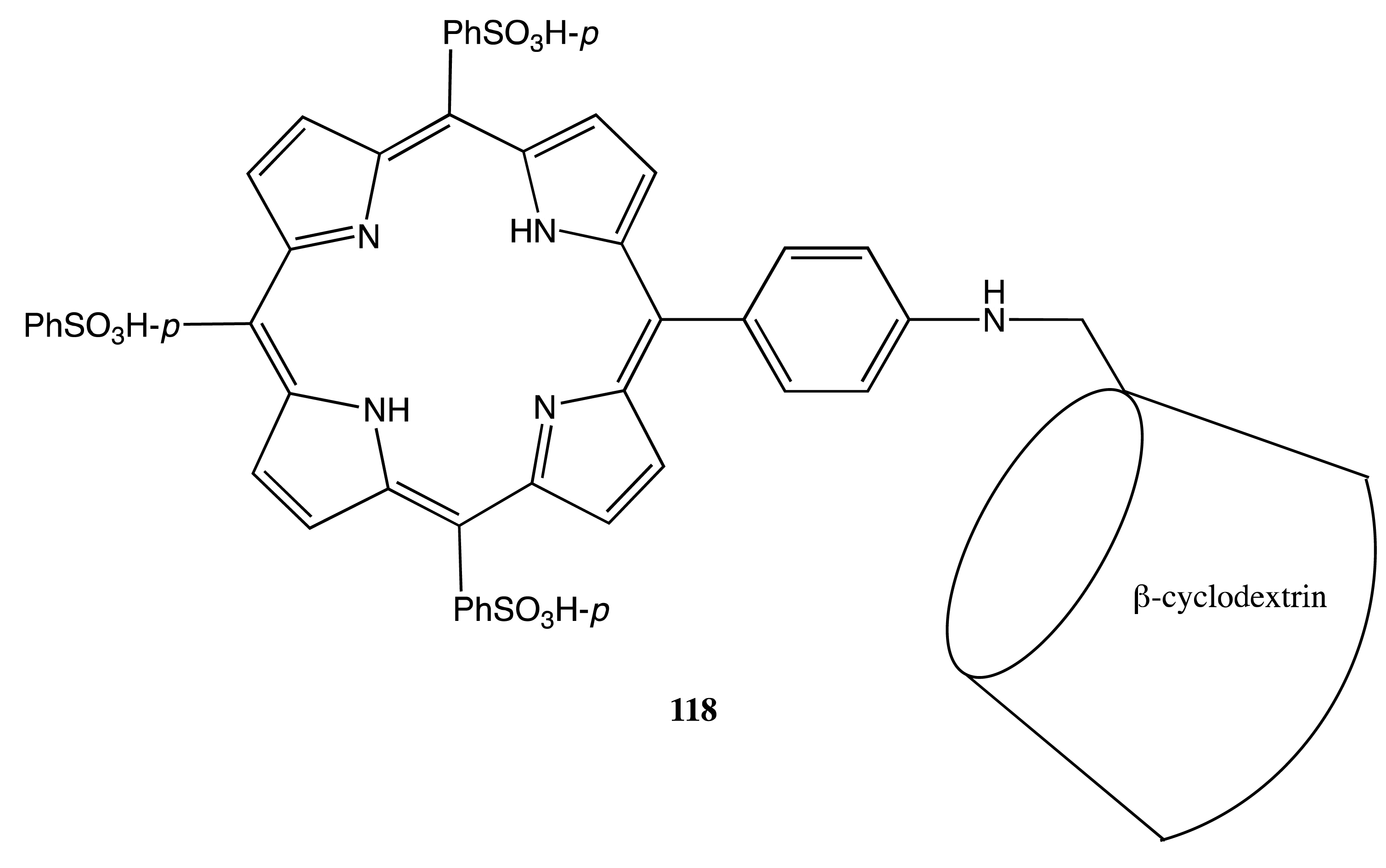
Another example of the supramolecular self-organization external control via pH modulation was demonstrated by the porphyrin-cyclodextrin conjugate, 118 (Figure 33) [92]. Thus, a strong negative CD exciton couplet in the Soret band was observed under basic conditions (pH 8.5), due to intermolecular interactions upon the inclusion of the porphyrin moiety of one molecule into the cyclodextrin cavity of another molecule. In the case of acidic conditions (pH 3.5), a positive bisignate CD signal of a complicated profile arose from the split absorption bands and was detected as a result of electrostatic interactions between the porphyrins in a zwitterionic form.
4.2. Achiral Porphyrinoids
Achiral monomeric porphyrinoids as bis- and multi-porphyrins described above require an external asymmetry field to induce supramolecular chirality. However, in general, this field should possess not only the asymmetry properties, but also a specific ability to organize structurally the corresponding monomeric units. For example, a simple zinc octaethylporphyrin, which exhibited a negligible optical activity upon interaction with monodentate chiral amines in solution [79], developed a strong CD signal in the presence of enantiopure 1-cyclohexylethylamine in a solid glassy KBr matrix [93]. Interestingly, the chirogenic process of the formation of highly stable chiral J-aggregates was time-dependent with rate constants of 0.026–0.030 h−1. The supramolecular helicity was controlled by the absolute configuration of the ligand, whilst the induced chirality was a result of the interporphyrin excitonic interactions in the asymmetrically orientated assembly. The chirality transfer process was very efficient, as documented by a remarkably large anisotropy (g) factor at the wavelength corresponding to the first Cotton effect (g = 0.015), which is 10–100 times greater than the g factors of allowed π–π transitions.
A unidirectional vortex can also serve as a supramolecular chiral and organizing force for the monomeric porphyrins [94–96]. Hence, 5,10,15,20-tetrakis(4-sulfophenyl)porphyrin (TPPS) and related derivatives formed chiral J-aggregates upon the vortex motion and the corresponding self-assembly process caused by the intermolecular association between the positively charged pyrrolic nitrogen and the negatively charged sulfonato groups. Without stirring, the left- and right-handed aggregates were obtained in equal amounts, whilst upon stirring, the porphyrins were arranged with a unidirectional helical orientation. The supramolecular helicity was dependent upon the vortex direction with a probability of about 85%. The clockwise and counterclockwise direction produced the right- and left-handed chirality, respectively, which corresponded to a strong positive and negative CD couplet in the porphyrin absorption region, as a result of the interporphyrin exciton coupling. The mechanism was based upon the following assumption. The chiral vortex acted at the mesoscale level upon the kinetically controlled growth of the supramolecular assembly and spontaneous symmetry-breaking processes in the diffusion-limited generation of the high-molecular-weight homo-associates that, in turn, was further supported by the direct visualization of 3D homochiral helices using atomic force microscopy.
Owing to the negatively charged peripheral substituents, TPPS is also frequently used as a suitable monomeric porphyrin for structural and supramolecular chiral organization on the positively charged polypeptide matrix. For example, a chiral template on the basis of poly(Glu-Val-Lys-Val) was used to study the asymmetry generation in the porphyrin aggregates of TPPS [97]. The porphyrin-polypeptide electrostatic interactions induced two negative, well-resolved CD couplets and a negative monosignate Cotton effect in the region of Soret and Q absorption, respectively, due to the formation of optically active aggregates. The chiroptical properties were controlled by the corresponding host-guest ratio, pH and ionic strength. The CD amplitudes were enhanced upon decreasing the pH value, whilst the chirality sign could be even switched at a high salt concentration. This indicated a spatial rearrangement of the porphyrin coupling electronic transitions within the aggregates via modulation of the porphyrin-polypeptide electrostatic interactions.
Polynucleotides (DNA and RNA) are another type of natural supramolecular chiral matrix, which is often applied to arrange monomeric porphyrins in an asymmetrical manner. However, the phosphate residues of DNA/RNA produce negative charges that require cationic porphyrins as suitable counterparts for the effective self-assembly process. In general, there are three major binding modes for the porphyrin-DNA/RNA complexes: intercalation between the base pairs, binding to the major or minor grooves and outside stacking, depending upon the porphyrin and polynucleotide structures and media [98–101]. The chiroptical responses from these types of supramolecular complexation were also well established, reflecting the orientation of porphyrin electronic transitions in relation to the chirally arranged bases of DNA/RNA and interporphyrin coupling within the porphyrin aggregates stacked in a chiral fashion along the double helix. Particularly, the intercalation mode induces a negative CD signal, whilst the external binding results in a positive CD signal of larger intensity. In the case of the aggregation mode, the chiroptical outcome consists of a bisignate exciton couplet in the region of the porphyrin Soret band.
5. Conclusions
In conclusion, using these several representative examples of the different structural types of monomeric, dimeric and multimeric porphyrinoids, it was demonstrated that the supramolecular chirality properties are strongly depended upon the spatial arrangement of chromophoric units and the judicious choice of covalent or non-covalent linkages. The origin of chirality is another highly important factor to control the overall asymmetry of a (supra-) molecular system. In particular, the chiral properties of rigidly fixed systems are mainly predetermined by the rigidity of the covalent bridge, whilst in the case of flexibly linked systems, one of the major factors is an additional stabilizing component, which fixes the overall geometry of the supramolecular assembly. For monomeric porphyrinoids, the external stabilizing matrix (chiral or achiral) is a key element to control the supramolecular chirality of a system. Taking into account these general principles and specific factors of porphyrin-based (supra-) molecular systems, the corresponding chiral properties can be effectively controlled and modulated, which is vitally important for developing efficient chiroptical sensors, biomimetic processes, asymmetric catalysts and medical drugs.
Acknowledgments
I warmly thank all the collaborators and co-workers and, particularly, Yoshihisa Inoue, Reiko Kuroda, Ludovico Valli, Hiroyuki Higuchi, Michiya Fujiki and many others who have greatly contributed to my research in this area.
Conflicts of Interest
The author declares no conflict of interest.
References
- Voet, D.; Voet, J.G. Biochemistry, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1995. [Google Scholar]
- Kadish, K.M.; Smith, K.M.; Guilard, R. The Porphyrin Handbook; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Escarcega-Bobadilla, M.V.; Kleij, A.W. Artificial chirogenesis: A gateway to new opportunities in material science and catalysis. Chem. Sci. 2012, 3, 2421–2428. [Google Scholar]
- Dai, Z.; Lee, J.; Zhang, W. Chiroptical switches: Applications in sensing and catalysis. Molecules 2012, 17, 1247–1277. [Google Scholar]
- Borovkov, V. Effective supramolecular chirogenesis in ethane-bridged bis-porphyrinoids. Symmetry 2010, 2, 184–200. [Google Scholar]
- Borovkov, V.; Inoue, Y. A versatile bisporphyrinoid motif for supramolecular chirogenesis. Eur. J. Org. Chem. 2009, 2009, 189–197. [Google Scholar]
- Hembury, G.A.; Borovkov, V.V.; Inoue, Y. Chirality sensing supramolecular systems. Chem. Rev. 2008, 108, 1–73. [Google Scholar]
- Mamardashvili, N.Z.; Borovkov, V.V.; Mamardashvili, G.M.; Inoue, Y.; Koifman, O.I. Complexation of porphyrins with ions and organic molecules. In Chemical Processes with Participation of Biological and Related Compounds. Biophysical and Chemical Aspects of Porphyrins, Pigments, Drugs, Biodegradable Polymers and Nanofibers; Lomova, T.N., Zaikov, G.E., Eds.; Koninklijke Brill: Leiden, The Netherlands, 2008; pp. 117–168. [Google Scholar]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar]
- Borovkov, V.V.; Mamardashvili, N.Z.; Inoue, Y. Optically active supramolecular systems on the basis of porphyrins. Russ. Chem. Rev. 2006, 75, 820–832. [Google Scholar]
- Borovkov, V.V.; Inoue, Y. Supramolecular chirogenesis in host-guest systems containing porphyrinoids. Top. Curr. Chem. 2006, 265, 89–146. [Google Scholar]
- Balaban, T.S.; Tamiaki, H.; Holzwarth, A.R. Chlorins programmed for self-assembly. Top. Curr. Chem. 2005, 258, 1–38. [Google Scholar]
- Balaban, T.S. Tailoring porphyrins and chlorins for self-assembly in biomimetic artificial antenna systems. Acc. Chem. Res. 2005, 38, 612–623. [Google Scholar]
- Miyake, H.; Tsukube, H. Helix architecture and helicity switching via dynamic metal coordination chemistry. Supramol. Chem. 2005, 17, 53–59. [Google Scholar]
- Borovkov, V.V.; Hembury, G.A.; Inoue, Y. Origin, control, and application of supramolecular chirogenesis in bis-porphyrin based systems. Acc. Chem. Res. 2004, 37, 449–459. [Google Scholar]
- Shoji, Y.; Tashiro, K.; Aida, T. Sensing of chiral fullerenes by a cyclic host with an asymmetrically distorted π-electronic component. J. Am. Chem. Soc. 2006, 128, 10690–10691. [Google Scholar]
- Shoji, Y.; Tashiro, K.; Aida, T. One-pot enantioselective extraction of chiral fullerene C76 using a cyclic host carrying an asymmetrically distorted, highly π-basic porphyrin module. J. Am. Chem. Soc. 2010, 132, 5928–5929. [Google Scholar]
- Peng, X.; Komatsu, N.; Bhattacharya, S.; Shimawaki, T.; Aonuma, S.; Kimura, T.; Osuka, A. Optically active single-walled carbon nanotubes. Nat. Nanotechnol. 2007, 2, 361–365. [Google Scholar]
- Peng, X.; Komatsu, N.; Kimura, T.; Osuka, A. Improved optical enrichment of SWNTs through extraction with chiral nano-tweezers of 2,6-pyridylene-bridged diporphyrins. J. Am. Chem. Soc. 2007, 129, 15947–15953. [Google Scholar]
- Peng, X.; Komatsu, N.; Kimura, T.; Osuka, A. Simultaneous enrichments of optical purity and (n, m) abundance of SWNTs through extraction with 3,6-carbazolylene-bridged chiral diporphyrin nanotweezers. ACS Nano 2008, 2, 2045–2050. [Google Scholar]
- Wang, F.; Matsuda, K.; Rahman, A.F.M.M.; Peng, X.; Kimura, T.; Komatsu, N. Simultaneous discrimination of handedness and diameter of single-walled carbon nanotubes (SWNTs) with chiral diporphyrin nanotweezers leading to enrichment of single enantiomer of (6,5)-SWNTs. J. Am. Chem. Soc. 2010, 132, 10876–10881. [Google Scholar]
- Banala, S.; Sintic, P.; Kräutler, R. A functionalized di-zinc-porphyrin-chlorin-type spirodimer from rapid self-[4+2]-cycloaddition of a conjugated zinc-porphyrin-β,β′-diene. Helv. Chim. Acta 2012, 95, 211–220. [Google Scholar]
- Chmielewski, P.J.; Maciolek, J. Diastereoselective methylation of bis(N-confused porphyrinatonickel(II): Access to configurationally stable chiral bis(porphyrinoid) and non-symmetric dimers. Chem. Commun. 2012, 48, 428–430. [Google Scholar]
- Guo, Y.-M.; Oike, H.; Saeki, N.; Aida, T. One-pot optical resolution of oligopeptide through artificial peptide bundling. Angew. Chem. Int. Ed. 2004, 43, 4915–4918. [Google Scholar]
- Ema, T.; Ura, N.; Eguchi, K.; Ise, Y.; Sakai, T. Chiral porphyrin dimer with a macrocyclic cavity for intercalation of aromatic guests. Chem. Commun. 2011, 47, 6090–6092. [Google Scholar]
- Ema, T.; Ura, N.; Eguchi, K.; Sakai, T. Molecular recognition of chiral diporphyrin receptor with a macrocyclic cavity for intercalation of aromatic compounds. Bull. Chem. Soc. Jpn. 2012, 85, 101–109. [Google Scholar]
- Fan, J.; Whiteford, J.A.; Olenyuk, B.; Levin, M.D.; Stang, P.J.; Fleischer, E.B. Self-Assembly of porphyrin arrays via coordination to transition metal bisphosphine complexes and the unique spectral properties of the product metallacyclic ensembles. J. Am. Chem. Soc. 1999, 121, 2741–2752. [Google Scholar]
- Ayabe, M.; Yamashita, K.; Sada, K.; Shinkai, S.; Ikeda, A.; Sakamoto, S.; Yamaguchi, K. Construction of monomeric and polymeric porphyrin compartments by a Pd(II)–pyridine interaction and their chiral twisting by a BINAP ligand. J. Org. Chem. 2003, 68, 1059–1066. [Google Scholar]
- Hayashi, T.; Nonoguchi, M.; Aya, T.; Ogoshi, H. Molecular recognition of α,ω-diamines by metalloporphyrin dimer. Tetrahedron Lett. 1997, 38, 1603–1606. [Google Scholar]
- Hayashi, T.; Aya, T.; Nonoguchi, M.; Mizutani, T.; Hisaeda, Y.; Kitagawa, S.; Ogoshi, H. Chiral recognition and chiral sensing using zinc porphyrin dimers. Tetrahedron 2002, 58, 2803–2811. [Google Scholar]
- Tsubaki, K.; Takaishi, K.; Tanaka, H.; Miura, M.; Kawabata, T. Long-range exciton-coupled circular dichroism: Application for determination of the absolute configuration of oligonaphthalenes. Org. Lett. 2006, 8, 2587–2590. [Google Scholar]
- Fukuda, T.; Olmstead, M.M.; Durfee, W.S.; Kobayashi, N. An optically-active subphthalocyanine dimer. Chem. Commun. 2003, 1256–1257. [Google Scholar]
- Muranaka, A.; Okuda, M.; Kobayashi, N.; Somers, K.; Ceulemans, A. Recognition of chiral catechols using oxo-titanium phthalocyanine. J. Am. Chem. Soc. 2004, 126, 4596–4604. [Google Scholar]
- Crossley, M.J.; Mackay, L.G.; Try, A.C. Enantioselective recognition of histidine and lysine esters by porphyrin chiral clefts and detection of amino acid conformations in the bound state. J. Chem. Soc. Chem. Commun. 1995, 1925–1927. [Google Scholar]
- Brotherhood, P.R.; Wu, R.A.-S.; Turner, P.; Crossley, M.J. Cavity effect amplification in the recognition of dicarboxylic acids by initial ditopic H-bond formation followed by kinetic trapping. Chem. Commun. 2007, 225–227. [Google Scholar]
- Takeuchi, M.; Chin, Y.; Imada, T.; Shinkai, S. Sugar-boronic acid interactions in the formation of novel chiral porphyrin dimers with various porphyrin-porphyrin angles. J. Chem. Soc. Chem. Commun. 1996, 1867–1868. [Google Scholar]
- Holmes, A.E.; Das, D.; Canary, J.W. Chelation-enhanced circular dichroism of tripodal bisporphyrin ligands. J. Am. Chem. Soc. 2007, 129, 1506–1507. [Google Scholar]
- Ema, T.; Misawa, S.; Nemugaki, S.; Sakai, T.; Utaka, M. New optically active diporphyrin having a chiral cyclophane as a spacer. Chem. Lett. 1997, 487–488. [Google Scholar]
- Nakamura, Y.; Hwang, I.-W.; Aratani, N.; Ahn, T.K.; Ko, D.M.; Takagi, A.; Kawai, T.; Matsumoto, T.; Kim, D.; Osuka, A. Directly meso-meso linked porphyrin rings: Synthesis, characterization, and efficient excitation energy hopping. J. Am. Chem. Soc. 2005, 127, 236–246. [Google Scholar]
- Yoshida, N.; Ishizuka, T.; Osuka, A.; Jeong, D.H.; Cho, H.S.; Kim, D.; Matsuzaki, Y.; Nogami, A.; Tanaka, K. Fine tuning of photophysical properties of meso-meso-linked ZnII-diporphyrins by dihedral angle control. Chem. Eur. J. 2003, 9, 58–75. [Google Scholar]
- Ouyang, Q.; Zhu, Y.-Z.; Li, Y.-C.; Wei, H.-B.; Zheng, J.-Y. Diastereoselective synthesis of chiral diporphyrins via intramolecular meso-meso oxidative coupling. J. Org. Chem. 2009, 74, 3164–3167. [Google Scholar]
- Bringmann, G.; Götz, D.C.G.; Gulder, T.A.M.; Gehrke, T.H.; Bruhn, T.; Kupfer, T.; Radacki, K.; Braunschweig, H.; Heckmann, A.; Lambert, C. Axially chiral β,β′-bisporphyrins: synthesis and configurational stability tuned by the central metals. J. Am. Chem. Soc. 2008, 130, 17812–17825. [Google Scholar]
- Harada, N.; Nakanishi, K. Circular Dichroism Spectroscopy Exciton Coupling in Organic Stereochemistry; University Science Books: Mill Valley, CA, USA, 1983. [Google Scholar]
- Siczek, M.; Chmielewski, P.J. Synthesis, characterization, and chirality of dimeric N-confused porphyrin-zinc complexes: Toward the enantioselective synthesis of bis(porphyrinoid) systems. Angew. Chem. Int. Ed. 2007, 46, 7432–7436. [Google Scholar]
- Chmielewski, P.J.; Durlej, B.; Siczek, M.; Szterenberg, L. Helical bis(N-confused porphyrins) with subunits fused by double orthometalation with platinum: Adaptability of an apparently rigid system. Angew. Chem. Int. Ed. 2009, 48, 8736–8739. [Google Scholar]
- Tsuda, A.; Furuta, H.; Osuka, A. Syntheses, structural characterizations, and optical and electrochemical properties of directly fused diporphyrins. J. Am. Chem. Soc. 2001, 123, 10304–10321. [Google Scholar]
- Tashiro, K.; Konishi, K.; Aida, T. Enantiomeric resolution of chiral metallobis(porphyrin)s: Studies on rotatability of electronically coupled porphyrin ligands. Angew. Chem. Int. Ed. Engl. 1997, 36, 856–858. [Google Scholar]
- Tashiro, K.; Fujiwara, T.; Konishi, K.; Aida, T. Rotational oscillation of two interlocked porphyrins in cerium bis(5,15-diarylporphyrinate) double-deckers. Chem. Commun. 1998, 1121–1122. [Google Scholar]
- Borovkov, V.V.; Muranaka, A.; Hembury, G.A.; Origane, Y.; Ponomarev, G.V.; Kobayashi, N.; Inoue, Y. Chiral bis-chlorin: Effective enantiomer resolution and absolute configuration determination. Org. Lett. 2005, 7, 1015–1018. [Google Scholar]
- Borovkov, V.V.; Hembury, G.A.; Inoue, Y. Supramolecular chirogenesis with bis-chlorin versus bis-porphyrin hosts: Peculiarities of chirality induction and modulation of optical activity. J. Org. Chem. 2005, 70, 8743–8754. [Google Scholar]
- Borovkov, V.V.; Inoue, Y. Supramolecular chiral recognition by bischlorins: A two-point interaction mode combined with the host's conformational modulation controlled by the guest's stereochemistry and bulkiness. Org. Lett. 2006, 8, 2337–2340. [Google Scholar]
- Chmielewski, P.J.; Szterenberg, L.; Siczek, M. Enantiomer resolution of intrinsically chiral C21-alkylated N-confused porphyrin complexes. Chem. Eur. J. 2011, 17, 1009–1020. [Google Scholar]
- Paolesse, R.; Monti, D.; La Monica, L.; Venanzi, M.; Froiio, A.; Nardis, S.; Di Natale, C.; Martinelli, E.; D'Amico, A. Preparation and self-assembly of chiral porphyrin diads on the gold electrodes of quartz crystal microbalances: A novel potential approach to the development of enantioselective chemical sensors. Chem. Eur. J. 2002, 8, 2476–2483. [Google Scholar]
- Ema, T.; Ouchi, N.; Doi, T.; Korenaga, T.; Sakai, T. Highly sensitive chiral shift reagent bearing two zinc porphyrins. Org. Lett. 2005, 7, 3985–3988. [Google Scholar]
- Matile, S.; Berova, N.; Nakanishi, K.; Fleischhauer, J.; Woody, R.W. Structural studies by exciton coupled circular dichroism over a large distance: Porphyrin derivatives of steroids, dimeric steroids, and brevetoxin. B. J. Am. Chem. Soc. 1996, 118, 5198–5206. [Google Scholar]
- Muraoka, T.; Kinbara, K.; Aida, T. A self-locking molecule operative with a photoresponsive key. J. Am. Chem. Soc. 2006, 128, 11600–11605. [Google Scholar]
- Arai, T.; Araki, K.; Maruo, N.; Sumida, Y.; Korosue, C.; Fukuma, K.; Kato, T.; Nishino, N. Spectroscopic characterization of an assembled pair of free-base and zinc porphyrins linked by the cyclic β-sheet peptide Gramicidin S. New. J. Chem. 2004, 28, 1151–1159. [Google Scholar]
- MacMillan, J.B.; Linington, R.G.; Andersen, R.J.; Molinski, T.F. Stereochemical assignment in acyclic lipids across long distance by circular dichroism: Absolute stereochemistry of the aglycone of caminoside A. Angew. Chem. Int. Ed. 2004, 43, 5946–5951. [Google Scholar]
- Redl, F.X.; Lutz, M.; Daub, J. Chemistry of porphyrin-appended cellulose strands with a helical structure: Spectroscopy, electrochemistry, and in situ circular dichroism spectroelectrochemistry. Chem. Eur. J. 2001, 7, 5350–5358. [Google Scholar]
- Takei, F.; Hayashi, H.; Onitsuka, K.; Kobayashi, N.; Takahashi, S. Helical chiral polyisocyanides possessing porphyrin pendants: Determination of helicity by exciton-coupled circular dichroism. Angew. Chem. Int. Ed. 2001, 40, 4092–4094. [Google Scholar]
- De Witte, P.A.J.; Castriciano, M.; Cornelissen, J.J.L.M.; Scolaro, L.M.; Nolte, R.J.M.; Rowan, A.E. Helical polymer-anchored porphyrin nanorods. Chem. Eur. J. 2003, 9, 1775–1781. [Google Scholar]
- Tabei, J.; Shiotsuki, M.; Sanda, F.; Masuda, T. Determination of helical Sense of poly(N-propargylamides) by exciton-coupled circular dichroism. Macromolecules 2005, 38, 9448–9454. [Google Scholar]
- Takeuchi, M.; Imada, T.; Shinkai, S. Molecular design of highly selective and sensitive sugar tweezers from boronic-acid-appended m-oxo-bis[porphinatoiron (III)]s. Bull. Chem. Soc. Jpn. 1998, 71, 1117–1123. [Google Scholar]
- Takeuchi, M.; Imada, T.; Shinkai, S. A strong positive allosteric effect in the molecular recognition of dicarboxylic acids by a cerium(IV) bis[tetrakis(4-pyridyl)-porphyrinate] double decker. Angew. Chem. Int. Ed. 1998, 37, 2096–2099. [Google Scholar]
- Sugasaki, A.; Ikeda, M.; Takeuchi, M.; Robertson, A.; Shinkai, S. Efficient chirality transcription utilizing a cerium(IV) double decker porphyrin: A prototype for development of a molecular memory system. J. Chem. Soc. Perkin Trans. 1999, 1, 3259–3264. [Google Scholar]
- Sugasaki, A.; Ikeda, M.; Takeuchi, M.; Shinkai, S. Novel oligosaccharide binding to CeIV bis(porphyrinate) double decker: Effective amplification of a binding signal through positive homotropic allosterism. Angew. Chem. Int. Ed. 2000, 39, 3839–3842. [Google Scholar]
- Sugasaki, A.; Sugiyasu, K.; Ikeda, M.; Takeuchi, M.; Shinkai, S. First successful molecular design of an artificial Lewis oligosaccharide binding system utilizing positive homotropic allosterism. J. Am. Chem. Soc. 2001, 123, 10239–10244. [Google Scholar]
- Yamamoto, M.; Sugasaki, A.; Ikeda, M.; Takeuchi, M.; Frimat, K.; James, T.D.; Shinkai, S. Efficient anion binding to cerium(IV) bis(porphyrinate) double decker utilizing positive homotropic allosterism. Chem. Lett. 2001, 6, 520–521. [Google Scholar]
- Guo, Y.-M.; Oike, H.; Aida, T. Chiroptical transcription of helical information through supramolecular harmonization with dynamic helices. J. Am. Chem. Soc. 2004, 126, 716–717. [Google Scholar]
- Mizuno, Y.; Aida, T. Nonlinear amplification of circular dichroism activity upon cyclodimerization of a chiral saddle-shaped porphyrin. Chem. Commun. 2003, 20–21. [Google Scholar]
- Tsukube, H.; Tameshige, N.; Shinoda, S.; Unno, S.; Tamiaki, H. A specific receptor of biological cystine polyion: Distance-selective extraction and efficient chirality sensing with an ytterbium porphyrinate tweezer. Chem. Commun. 2002, 2574–2575. [Google Scholar]
- Monti, D.; La Monica, L.; Scipioni, A.; Mancini, G. Effect of the inclusion of sodium cations on the binding properties of a switchable diporphyrin receptor. New J. Chem. 2001, 25, 780–782. [Google Scholar]
- Kubo, Y.; Ishii, Y.; Yoshizawa, T.; Tokita, S. Effective cation-assisted chirality induction using a dibenzo-diaza-30-crown-10 with bis(zinc(II) porphyrin) units. Chem. Commun. 2004, 1394–1395. [Google Scholar]
- Ishii, Y.; Yoshizawa, T.; Kubo, Y. Dibenzodiaza-30-crown-10-appended bis(zinc porphyrin) tweezers: Synthesis and crown-assisted chiroptical behaviour. Org. Biomol. Chem. 2007, 5, 1210–1217. [Google Scholar]
- Kubo, Y.; Ohno, T.; Yamanaka, J.-I.; Tokita, S.; Iida, T.; Ishimaru, Y.T. Chirality-transfer control using a heterotopic zinc(II) porphyrin dimer. J. Am. Chem. Soc. 2001, 123, 12700–12701. [Google Scholar]
- Kurtan, T.; Nesnas, N.; Li, Y.-Q.; Huang, X.; Nakanishi, K.; Berova, N. Chiral recognition by CD-sensitive dimeric zinc porphyrin host. 1. Chiroptical protocol for absolute configurational assignments of monoalcohols and primary monoamines. J. Am. Chem. Soc. 2001, 123, 5962–5973. [Google Scholar]
- Huang, X.; Fujioka, N.; Pescitelli, G.; Koehn, F.E.; Williamson, R.T.; Nakanishi, K.; Berova, N. Absolute configurational assignments of secondary amines by CD-sensitive dimeric zinc porphyrin host. J. Am. Chem. Soc. 2002, 124, 10320–10335. [Google Scholar]
- Borovkov, V.V.; Lintuluoto, J.M.; Fujiki, M.; Inoue, Y. Temperature effect on supramolecular chirality induction in bis (zinc porphyrin). J. Am. Chem. Soc. 2000, 122, 4403–4407. [Google Scholar]
- Borovkov, V.V.; Lintuluoto, J.M.; Inoue, Y. Supramolecular chirogenesis in zinc porphyrins: Mechanism, role of guest structure, and application for the absolute configuration determination. J. Am. Chem. Soc. 2001, 123, 2979–2989. [Google Scholar]
- Borovkov, V.V.; Lintuluoto, J.M.; Sugeta, H.; Fujiki, M.; Arakawa, R.; Inoue, Y. Supramolecular chirogenesis in zinc porphyrins: equilibria, binding properties, and thermodynamics. J. Am. Chem. Soc. 2002, 124, 2993–3006. [Google Scholar]
- Borovkov, V.V.; Lintuluoto, J.M.; Sugiura, M.; Inoue, Y.; Kuroda, R. Remarkable stability and enhanced optical activity of a chiral supramolecular bis-porphyrin tweezer in both solution and solid state. J. Am. Chem. Soc. 2002, 124, 11282–11283. [Google Scholar]
- Lintuluoto, J.M.; Borovkov, V.V.; Inoue, Y. Direct determination of absolute configuration of monoalcohols by bis(magnesium porphyrin). J. Am. Chem. Soc. 2002, 124, 13676–13677. [Google Scholar]
- Borovkov, V.V.; Lintuluoto, J.M.; Hembury, G.A.; Sugiura, M.; Arakawa, R.; Inoue, Y. Supramolecular chirogenesis in zinc porphyrins: Interaction with bidentate ligands, formation of tweezer structures, and the origin of enhanced optical activity. J. Org. Chem. 2003, 68, 7176–7192. [Google Scholar]
- Borovkov, V.V.; Hembury, G.A.; Inoue, Y. The origin of solvent-controlled supramolecular chirality switching in a bis(zinc porphyrin) system. Angew. Chem. Int. Ed. 2003, 42, 5310–5314. [Google Scholar]
- Borovkov, V.V.; Fujii, I.; Muranaka, A.; Hembury, G.A.; Tanaka, T.; Ceulemans, A.; Kobayashi, N.; Inoue, Y. Rationalization of supramolecular chirality in a bis-porphyrin system. Angew. Chem. Int. Ed. 2004, 43, 5481–5485. [Google Scholar]
- Bhyrappa, P.; Borovkov, V.V.; Inoue, Y. Supramolecular chirogenesis in bis-porphyrins: Interaction with chiral acids and application for the absolute configuration assignment. Org. Lett. 2007, 9, 433–435. [Google Scholar]
- Mizumura, M.; Shinokubo, H.; Osuka, A. Synthesis of chiral porphyrins through Pd-catalyzed [3+2]annulation and heterochiral self-assembly. Angew. Chem. 2008, 120, 5458–5461. [Google Scholar]
- Chappaz-Gillot, C.; Canard, G.; Andreoli, F.; Vanthuyne, N.; Giorgi, M.; Naubron, J.-V.; Monnier, V.; Rosas, R.; Roussel, C.; Balaban, T.S. Atropisomeric chiral probes to study the supramolecular organization in porphyrin self-assemblies. Eur. J. Org. Chem. 2012, 2012, 6526–6536. [Google Scholar]
- Zelenka, K.; Trnka, T.; Tislerova, I.; Monti, D.; Cinti, S.; Naitana, M.L.; Schiaffino, L.; Vananzi, M.; Laguzzi, G.; Luvidi, L.; et al. Spectroscopic, morphological, and mechanistic investigation of the solvent-promoted aggregation of porphyrins modified in meso-positions by glucosylated steroids. Chem. Eur. J. 2011, 17, 13743–13753. [Google Scholar]
- Iavicoli, P.; Xu, H.; Feldborg, L.N.; Linares, M.; Paradinas, M.; Stafström, S.; Ocal, C.; Nieto-Ortega, B.; Casado, J.; Navarrete, J.T.L.; et al. Tuning the supramolecular chirality of one- and two-dimensional aggregates with the number of stereogenic centers in the component porphyrins. Am. Chem. Soc. 2010, 132, 9350–9362. [Google Scholar]
- Hirose, T.; Helmich, F.; Meijer, E.W. Photocontrol over cooperative porphyrin self-assembly with phenylazopyridine ligands. Angew. Chem. Int. Ed. 2013, 51, 304–309. [Google Scholar]
- Carofiglio, T.; Fornasier, R.; Lucchini, V.; Simonato, L.; Tonellato, U. Synthesis, characterization, and supramolecular properties of a hydrophilic porphyrin−β-cyclodextrin conjugate. J. Org. Chem. 2000, 65, 9013–9021. [Google Scholar]
- Borovkov, V.V.; Harada, T.; Hembury, G.A.; Inoue, Y.; Kuroda, R. Solid-state supramolecular chirogenesis: High optical activity and gradual development of zinc octaethylporphyrin aggregates. Angew. Chem. Int. Ed. 2003, 42, 1746–1749. [Google Scholar]
- Ribó, J.M.; Crusats, J.; Sagues, F.; Claret, J.; Rubires, R. Chiral sign induction by vortices during the formation of mesophases in stirred solutions. Science 2001, 292, 2063–2066. [Google Scholar]
- Rubires, R.; Ferrera, J.-A.; Ribó, J.M. Stirring effects on the spontaneous formation of chirality in the homoassociation of diprotonated meso-tetraphenylsulfonato porphyrins. Chem. Eur. J. 2001, 7, 436–446. [Google Scholar]
- Crusats, J.; Claret, J.; Diez-Perez, I.; El-Hachemi, Z.; Garcia-Ortega, H.; Rubires, R.; Sagues, F.; Ribó, J.M. Chiral shape and enantioselective growth of colloidal particles of self-assembled meso-tetra(phenyl and 4-sulfonatophenyl)porphyrins. Chem. Commun. 2003, 1588–1589. [Google Scholar]
- Fukushima, Y. Salt effect on the interaction of 22,24-diprotonated 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrin with a β-sheet structure of a zwitterionic polypeptide. Bull. Chem. Soc. Jpn. 1996, 69, 1719–1726. [Google Scholar]
- Allenmark, S. Induced circular dichroism by chiral molecular interaction. Chirality 2003, 15, 409–422. [Google Scholar]
- Pasternack, R.F. Circular dichroism and the interactions of water soluble porphyrins with DNA—A minireview. Chirality 2003, 15, 329–332. [Google Scholar]
- McMillin, D.R.; McNett, K.M. Photoprocesses of copper complexes that bind to DNA. Chem. Rev. 1998, 98, 1201–1220. [Google Scholar]
- Fiel, R.J.; Howard, J.C.; Mark, E.H.; Datta Gupta, N. Interaction of DNA with a porphyrin ligand: Evidence for intercalation. Nucleic Acids Res. 1979, 6, 3093–3118. [Google Scholar]



© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Borovkov, V. Supramolecular Chirality in Porphyrin Chemistry. Symmetry 2014, 6, 256-294. https://doi.org/10.3390/sym6020256
Borovkov V. Supramolecular Chirality in Porphyrin Chemistry. Symmetry. 2014; 6(2):256-294. https://doi.org/10.3390/sym6020256
Chicago/Turabian StyleBorovkov, Victor. 2014. "Supramolecular Chirality in Porphyrin Chemistry" Symmetry 6, no. 2: 256-294. https://doi.org/10.3390/sym6020256