Metalloporphyrin Symmetry in Chiral Recognition and Enantioselective Catalysis

Abstract
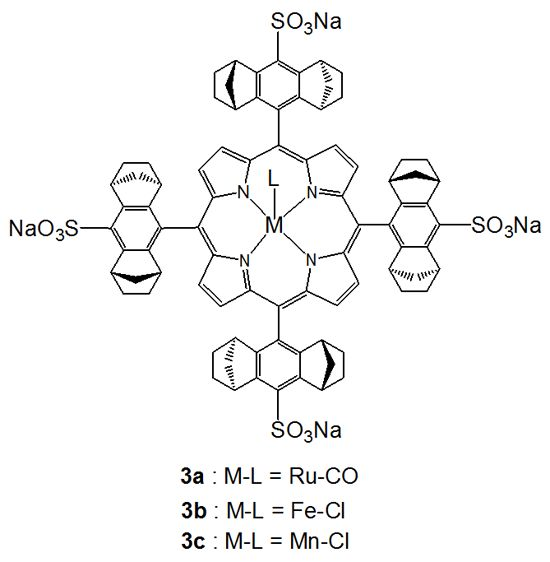
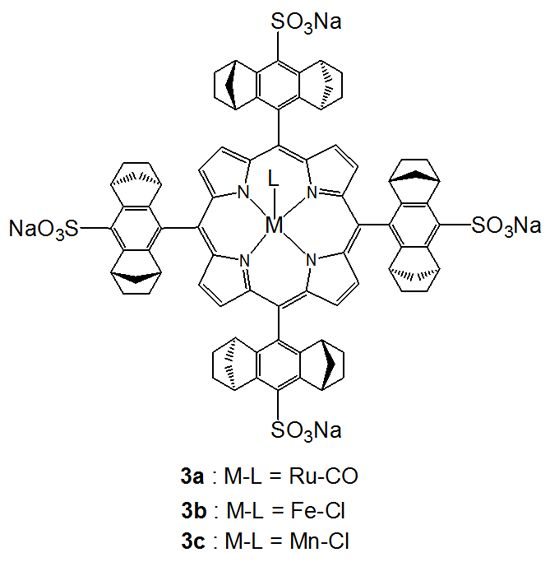
:
{kind=link}
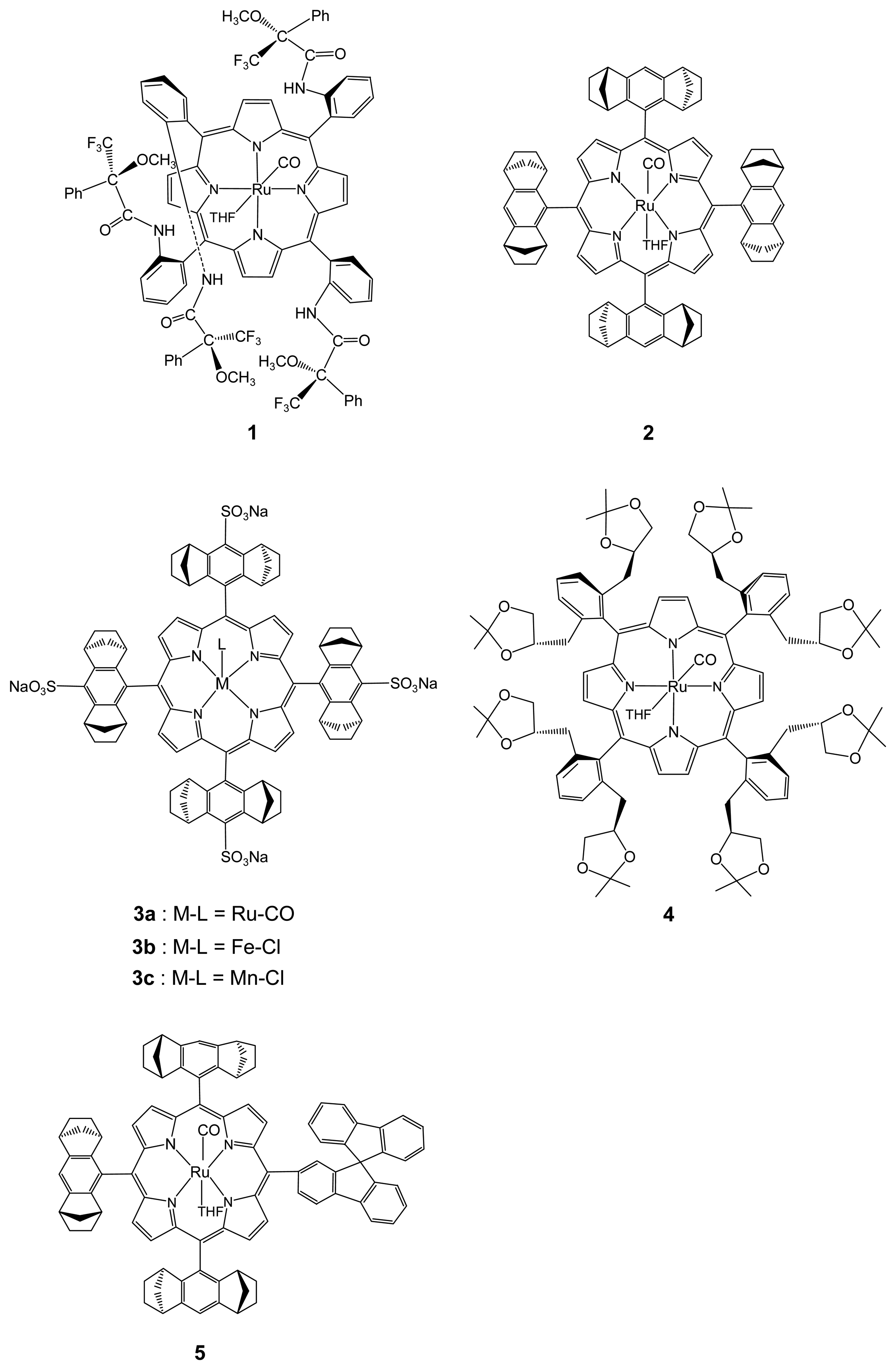{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Chiral Recognition
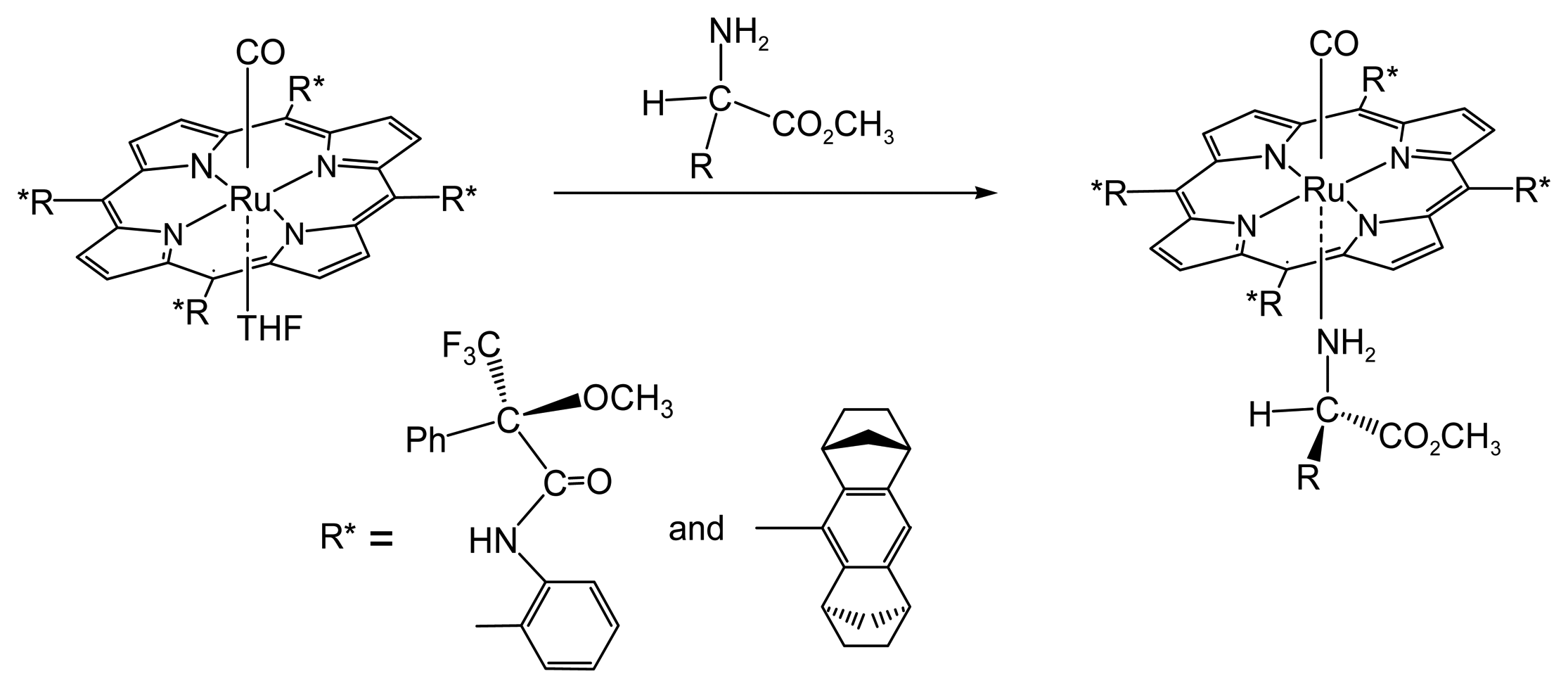
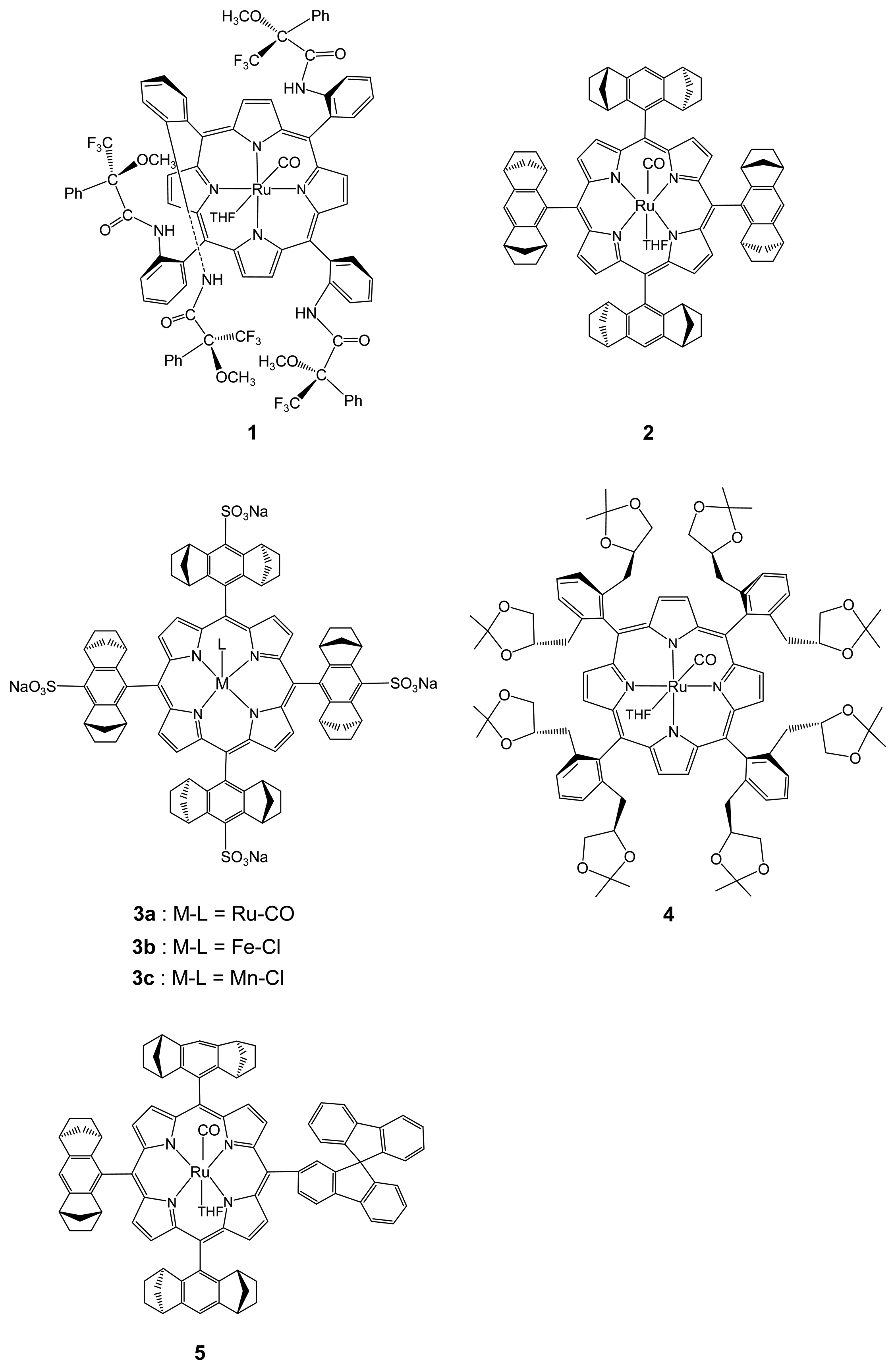
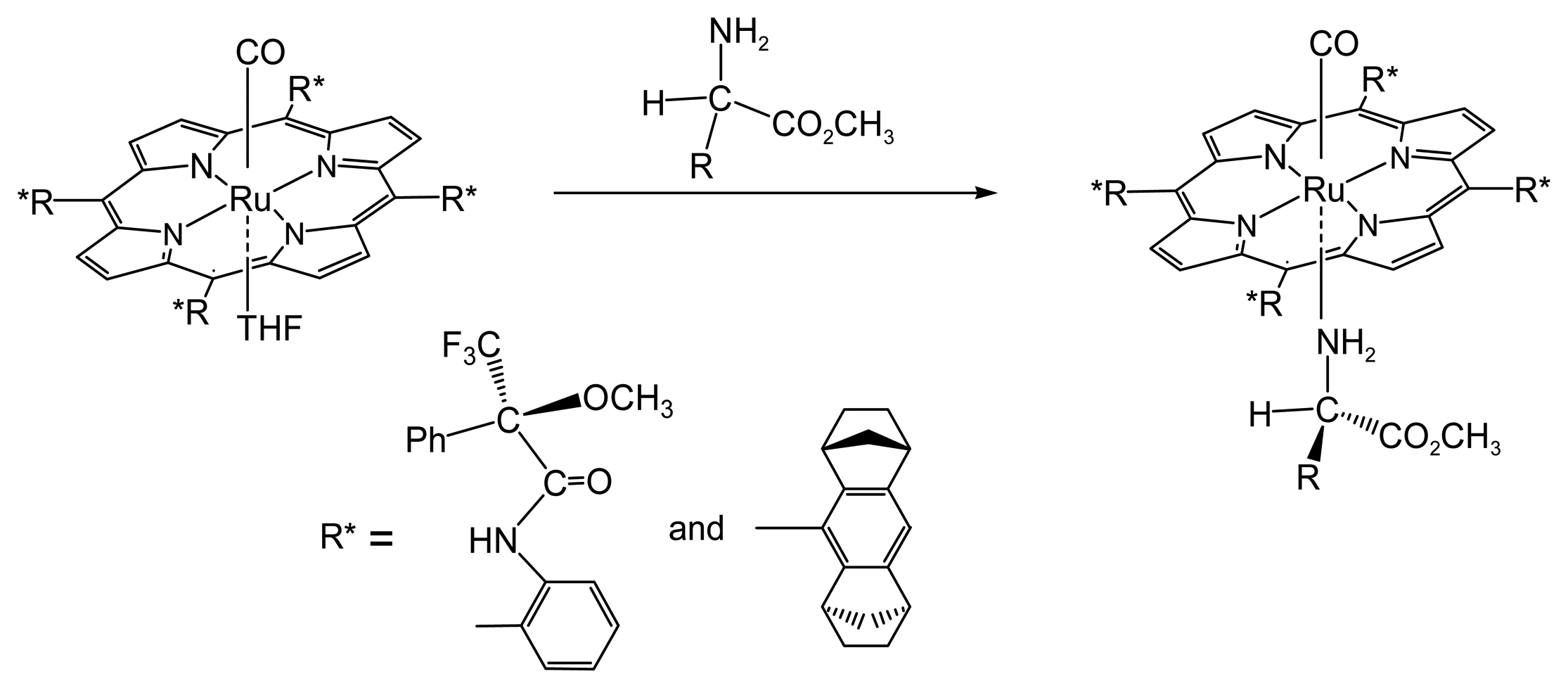
2.1.1. Chiral Recognition of Amino Acid Derivatives
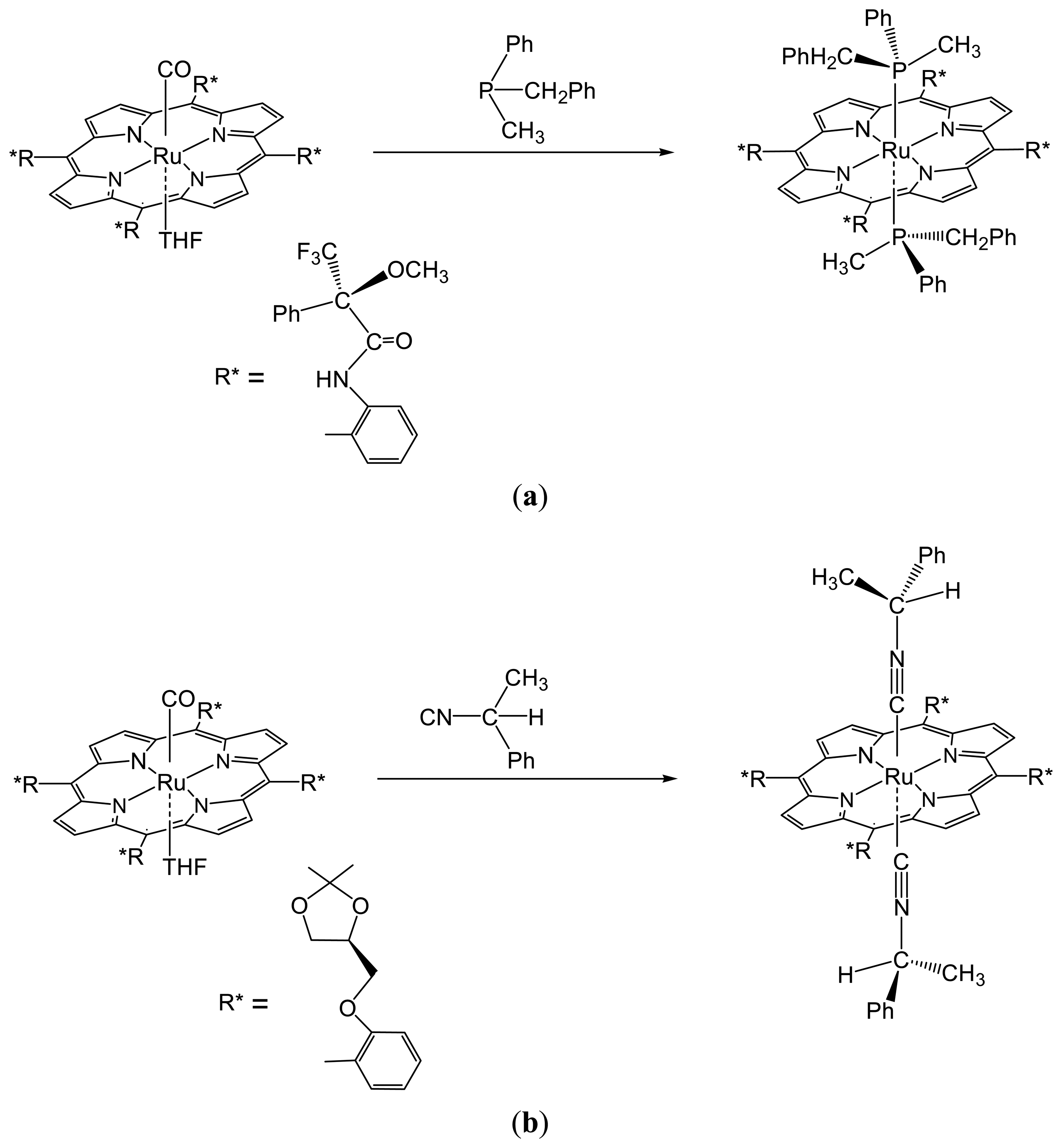
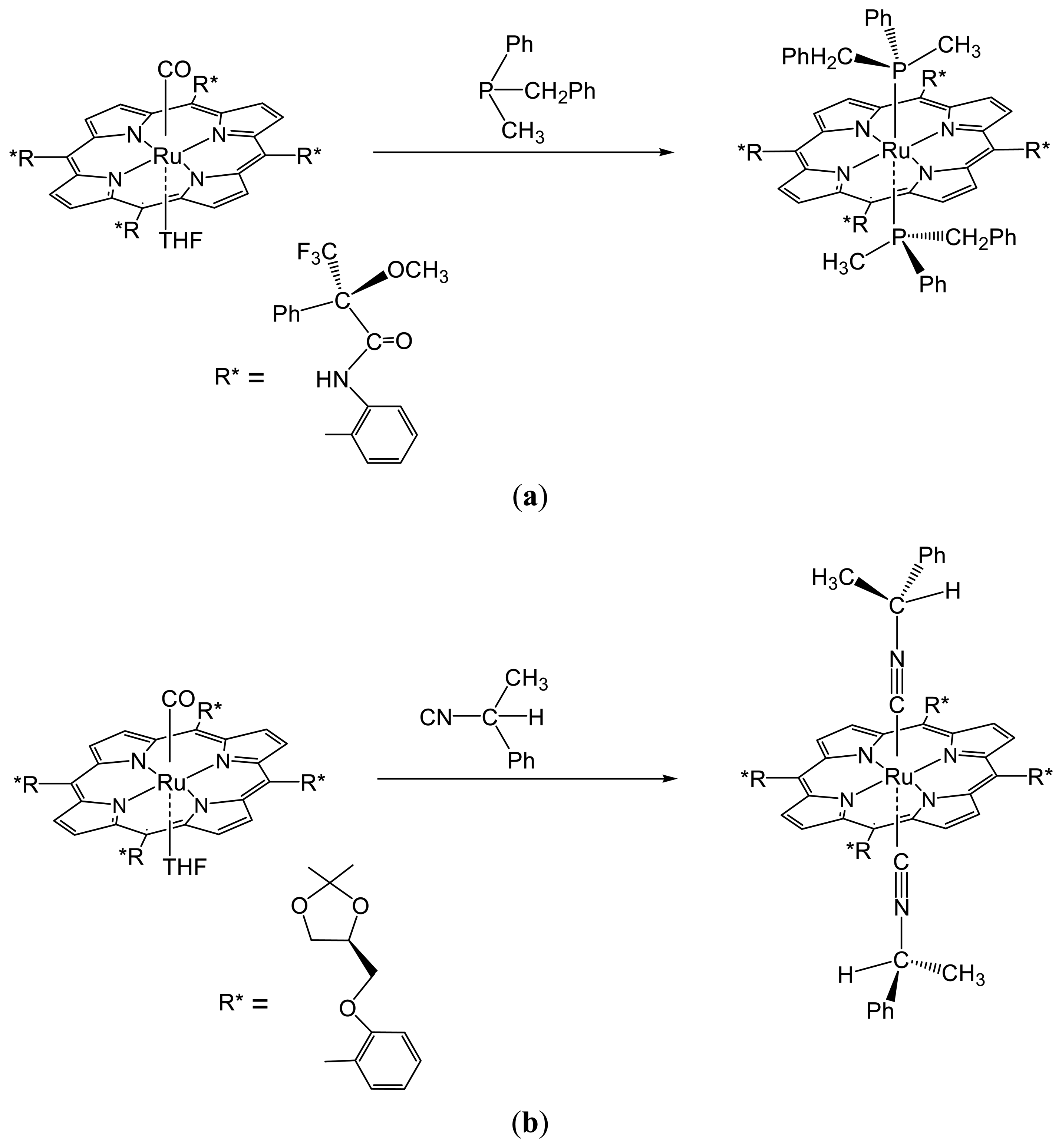
2.1.2. Chiral Recognition of Phosphines with Ruthenium Porphyrins
2.1.3. Chiral Recognition of Isocyanides with Ruthenium Porphyrins
2.2. Enantioselective Catalysis
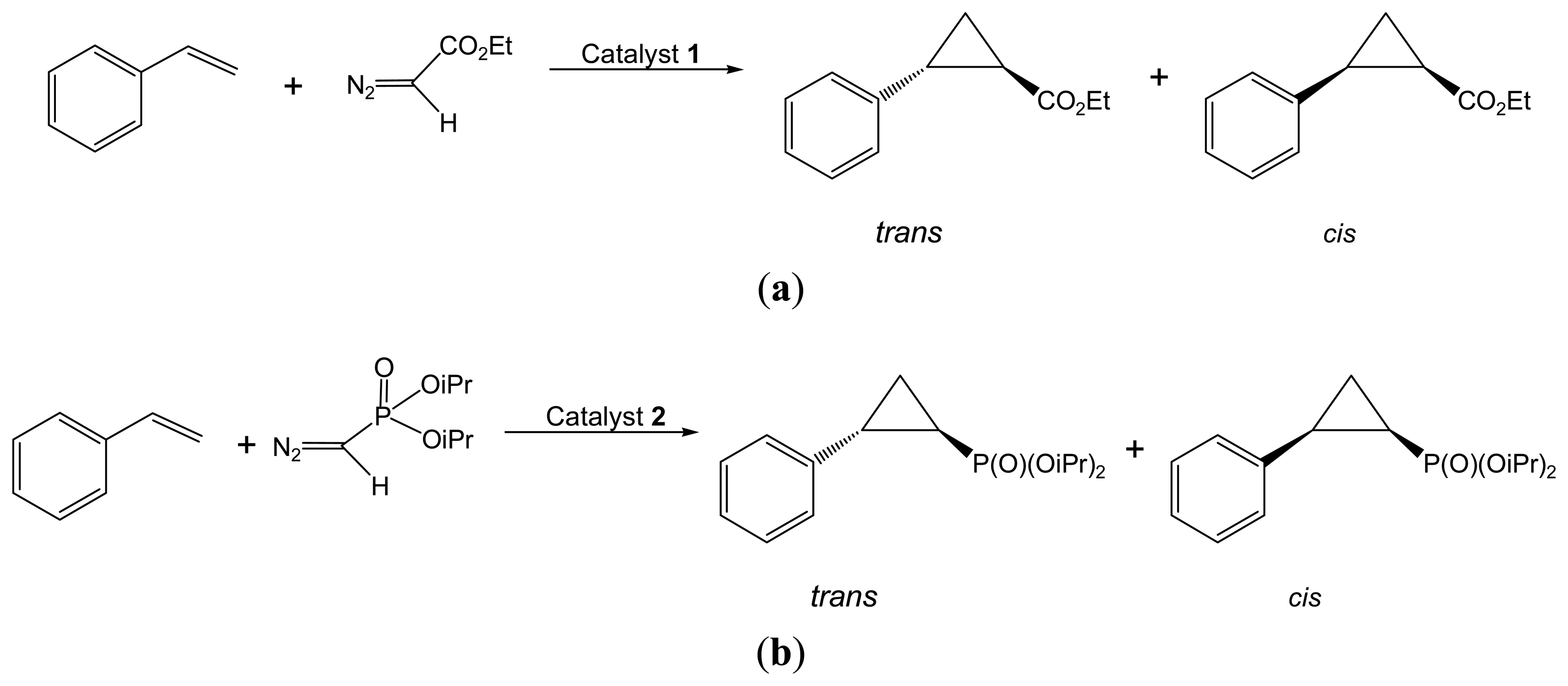
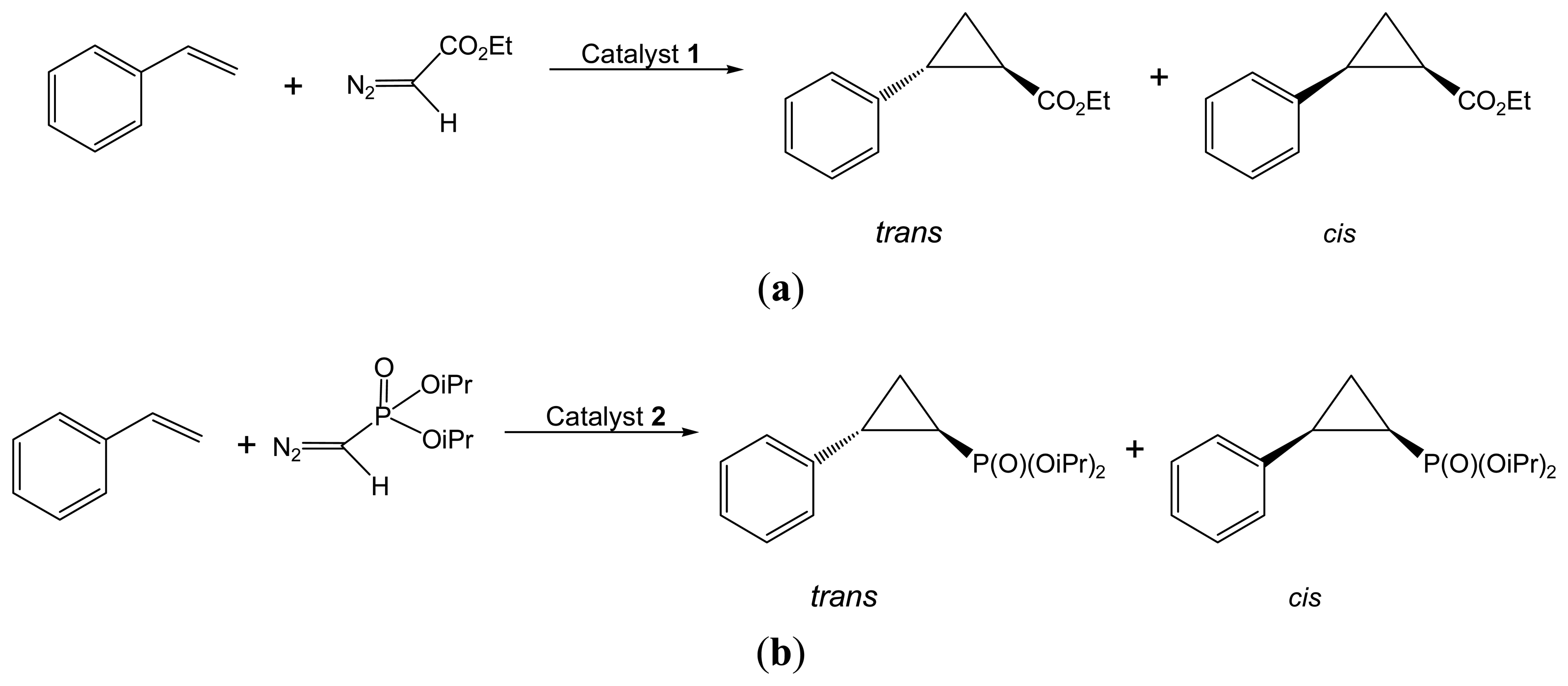
2.2.1. Asymmetric Cyclopropanation Reaction
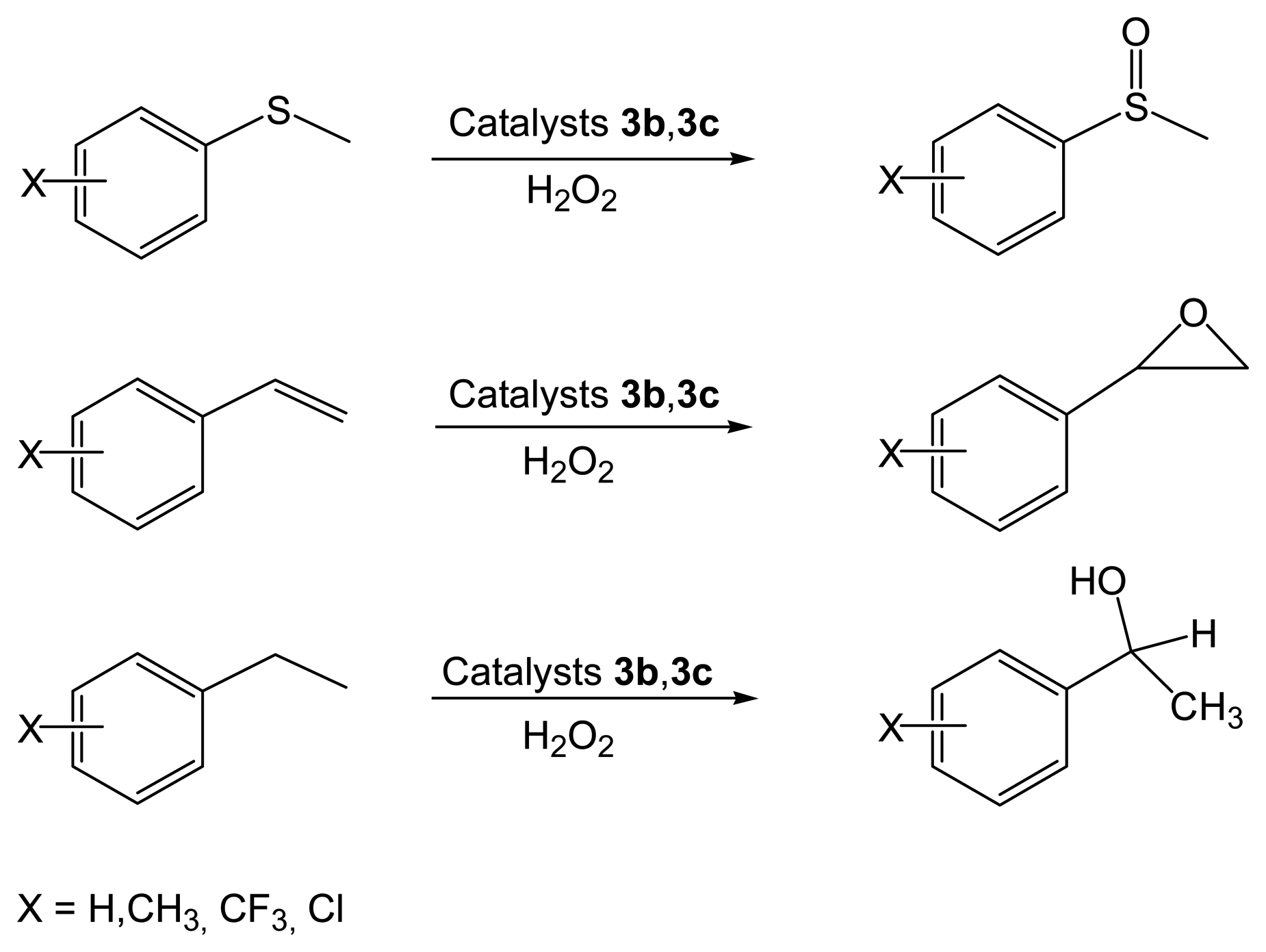
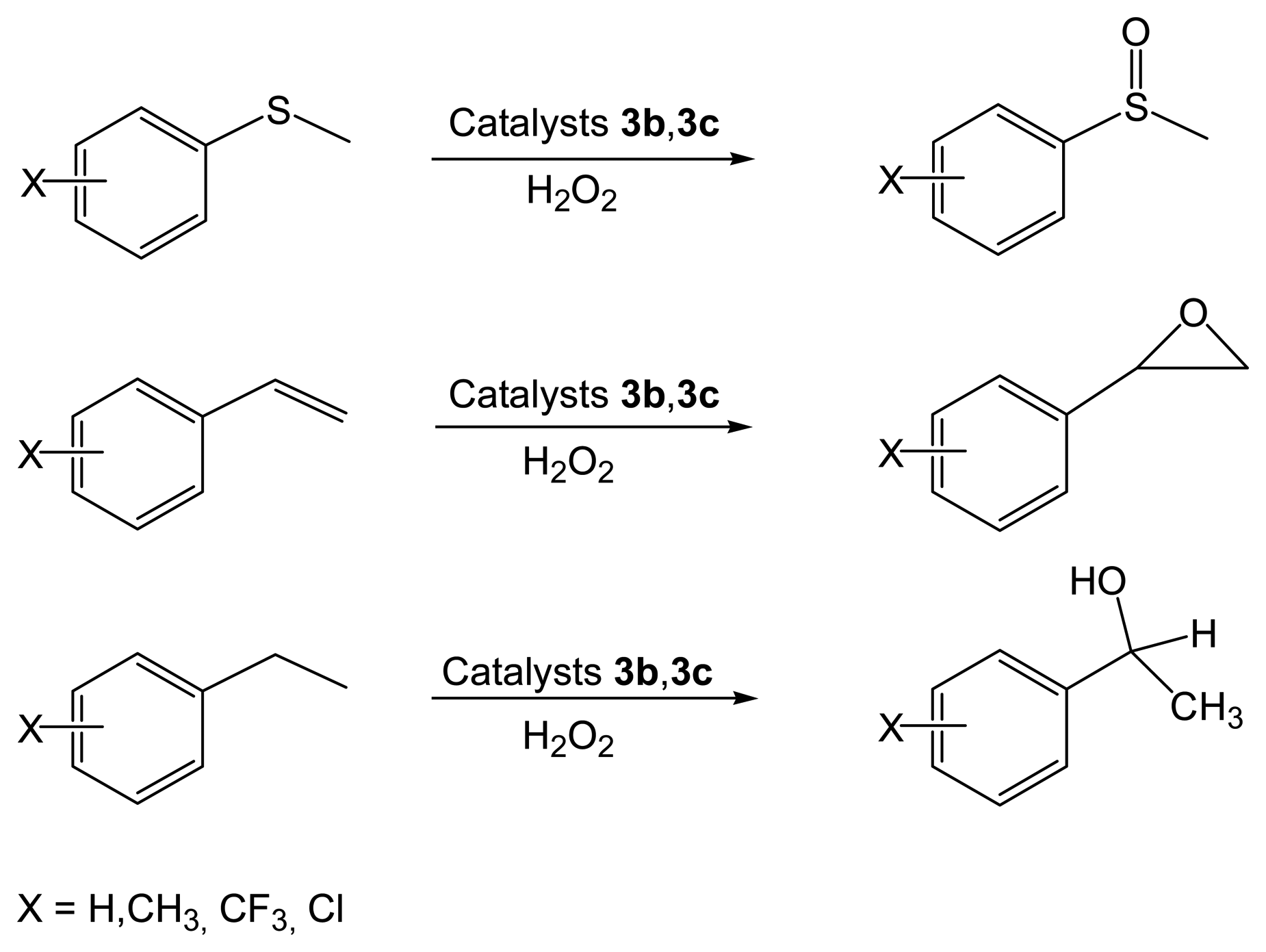
2.2.2. Asymmetric Oxidation Reactions
3. Conclusions
Conflicts of Interest
References
- Tamura, K.; Schimmel, P.R. Chiral-selective aminoacylation of an RNA minihelix: Mechanistic features and chiral suppression. Proc. Natl. Acad. Sci. USA 2006, 103, 13750–13752. [Google Scholar]
- Whitesell, J.K. C2 Symmetry and asymmetric induction. Chem. Rev. 1989, 89, 1581–1590. [Google Scholar]
- Pfaltz, A. Chiral semicorrins and related nitrogen heterocycles as ligands in asymmetric catalysis. Acc. Chem. Res. 1993, 26, 339–345. [Google Scholar]
- Moberg, C. C3 Symmetry in asymmetric catalysis and chiral recognition. Angew. Chem. Int. Ed. Engl. 1998, 37, 248–268. [Google Scholar]
- Borovkov, V.V.; Mamardashvili, N.Z.; Inoue, Y. Optically active supramolecular systems based on porphyrins. Russ. Chem. Rev. 2006, 75, 737–748. [Google Scholar]
- Meunier, B. Metalloporphyrins as versatile catalysts for oxidation reactions and oxidative DNA cleavage. Chem. Rev. 1992, 92, 1411–1456. [Google Scholar]
- Simonneaux, G.; Tagliatesta, P. Metalloporphyrin catalysts for organic synthesis. J. Porphyr Phtalocyanins 2004, 8, 1166–1171. [Google Scholar]
- Mansuy, D. A brief history of the contribution of metalloporphyrin models to cytochrome P450 chemistry and oxidation catalysis. C. R. Chim. 2007, 10, 392–413. [Google Scholar]
- Simonneaux, G.; Le Maux, P. Optically active ruthenium porphyrins: Chiral recognition and asymmetric catalysis. Coord. Chem. Rev. 2002, 228, 43–60. [Google Scholar]
- Hembury, G.A.; Borovkov, V.V.; Inoue, Y. Chirality-sensing supramolecular systems. Chem. Rev. 2008, 108, 1–73. [Google Scholar]
- Ogoshi, H.; Mizutani, T. Multifunctional and chiral porphyrins: Model receptors for chiral recognition. Acc. Chem. Res. 1998, 31, 81–89. [Google Scholar]
- Kyba, E.B.; Koga, K.; Sousa, L.R.; Siegel, M.G.; Cram, D.J. Chiral recognition in molecular complexing. J. Am. Chem. Soc. 1973, 95, 2692–2693. [Google Scholar]
- Lehn, J.M.; Simon, J.; Moradpour, A. Synthesis and properties of chiral macrotricyclic ligands. Complexation and transport of chiral molecular cations and anions. Helv. Chim. Acta 1978, 61, 2407–2418. [Google Scholar]
- Noyori, R. Chiral metal complexes as discriminating molecular catalysts. Science 1990, 248, 1194–1199. [Google Scholar]
- Groves, J.T.; Ahn, K.H. Characterization of an Oxoruthenium(IV) porphyrin complex. Inorg. Chem. 1987, 26, 3831–3833. [Google Scholar]
- Galardon, E.; Le Maux, P.; Bondon, A.; Simonneaux, G. Chiral recognition of amino esters by a ruthenium porphyrin complex: Kinetics of the exchange process determined by 1H NMR. Tetrahedron Asymmetry 1999, 10, 4203–4210. [Google Scholar]
- Morice, C.; Le Maux, P.; Simonneaux, G.; Toupet, L. Chiral recognition of amino esters by ruthenium porphyrin complexes and crystal structure of {5,10,15,20-tetrakis[o-(3,3,3-trifluoro-2-methoxy-2-phenylpropanoylamino)phenyl]porphyrin}-bis(L-valine methyl ester)ruthenium(II) (α,α,β,β isomer). Dalton Trans. 1998, 4165–4171. [Google Scholar]
- Nicolas, I.; Chevance, S.; Maux, P.L.; Simonneaux, G. Chiral recognition of amines and amino acid derivatives by optically active ruthenium Halterman porphyrins in organic solvents and water. Tetrahedron Asymmetry 2010, 21, 1788–1792. [Google Scholar]
- Eaton, S.S.; Eaton, G.R.; Holm, R.H. Inter- and intramolecular ligand exchange reactions of Ruthenium(II) carbonyl porphine complexes with nitrogen bases. J. Organomet. Chem. 1972, 39, 179–195. [Google Scholar]
- Halterman, R.L.; Jan, S.T.; Nimmons, H.L.; Standlee, D.J.; Khan, M.A. Synthesis and catalytic reactivity of D4-Symmetric dinorbornabenzene-derived metallo tetraarylporphyrins. Tetrahedron 1997, 53, 11257–11276. [Google Scholar]
- Imai, H.; Munakata, H.; Uemori, Y.; Sakura, N. Chiral Recognition of amino acids and dipeptides by a water-soluble zinc porphyrin. Inorg. Chem. 2004, 43, 1211–1213. [Google Scholar]
- Cort, A.D.; De Bernardin, P.; Schiaffino, L. A new water soluble Zn-salophen derivative as a receptor for α-aminoacids: Unexpected chiral discrimination. Chirality 2009, 21, 104–109. [Google Scholar]
- Le Maux, P.; Bahri, H.; Simonneaux, G. Molecular recognition of racemic phosphines by a chiral ruthenium porphyrin. J. Chem. Soc. Chem. Commun. 1991, 1350–1352. [Google Scholar]
- Galardon, E.; Lukas, M.; Le Maux, P.; Simonneaux, G. Synthesis and characterisation of a new chiral Ruthenium picket-fence porphyrin and its use in chiral recognition of racemic lsocyanides. Tetrahedron Lett. 1999, 40, 2753–2756. [Google Scholar]
- Ini, S.; Kapon, M.; Cohen, S.; Gross, Z. Self assembly assisted preparation of a homochiral porphyrin. Tetrahedron Asymmetry 1996, 7, 659–662. [Google Scholar]
- Knowles, W.S.; Sabacky, M.J. Catalytic asymmetric hydrogenation employing a soluble optically active Rhodium complex. Chem. Commun. 1968, 1445–1446. [Google Scholar]
- Horner, L.; Siegel, H.; Buthe, H. Asymmetric catalytic hydrogenation with an optically active phosphinerhodium complex in homogeneous solution. Angew. Chem. Int. Ed. Engl. 1968, 7, 942. [Google Scholar]
- Halpern, J.; Trost, B. Asymmetric catalysis. Proc. Natl. Acad. Sci. USA 2004, 101, 5347. [Google Scholar]
- Che, C.M.; Lo, V.K.Y.; Zhou, C.Y.; Huang, J.S. Selective functionalisation of saturated C–H bonds with metalloporphyrin catalysts. Chem. Soc. Rev. 2011, 40, 1950–1975. [Google Scholar]
- Lu, H.; Zhang, X.P. Catalytic C–H functionalization by metalloporphyrins: Recent developments and future directions. Chem. Soc. Rev. 2011, 40, 1899–1909. [Google Scholar]
- Doyle, M.P.; Forbes, D.C. Recent advances in asymmetric catalytic metal carbene transformations. Chem. Rev. 1998, 98, 911–935. [Google Scholar]
- Collman, J.P.; Rose, E.; Venburg, G.D. Reactivity of Ruthenium 5,10,15,20-tetramesitylporphyrin towards Diazoesters: Formation of olefins. J. Chem. Soc. Chem. Commun. 1993, 934–935. [Google Scholar]
- Galardon, E.; Le Maux, P.; Simonneaux, G. Cyclopropanation of alkenes with ethyl diazoacetate catalysed by Ruthenium porphyrin complexes. J. Chem. Soc. Chem. Commun. 1997, 927–928. [Google Scholar]
- Ferrand, Y.; Le Maux, P.; Simonneaux, G. Highly enantioselective synthesis of cyclopropylphosphonates catalyzed by chiral Ruthenium porphyrins. Org. Lett. 2004, 6, 3211–3214. [Google Scholar]
- Frauenkron, M.; Berkessel, A. A novel chiral Ruthenium porphyrin as highly efficient and selective catalyst for asymmetric cyclopropanations. Tetrahedron Lett. 1997, 38, 7175–7176. [Google Scholar]
- Lo, W.C.; Che, C.M.; Cheng, K.F.; Mak, T.C.W. Catalytic and asymmetric cyclopropanation of styrenes catalysed by ruthenium porphyrin and porphycene complexes. J. Chem. Soc. Chem. Commun. 1997, 1205–1206. [Google Scholar]
- Gross, Z.; Galili, N.; Simkhovich, L. Metalloporphyrin catalyzed asymmetric cyclopropanation of olefins. Tetrahedron Lett. 1999, 40, 1571–1574. [Google Scholar]
- Lane, B.S.; Burgess, K. Metal-catalyzed epoxidations of alkenes with hydrogen peroxide. Chem. Rev. 2003, 103, 2457–2474. [Google Scholar]
- De Faveri, G.; Ilyashenko, G.; Watkinson, M. Recent advances in catalytic asymmetric epoxidation using the environmentally benign oxidant hydrogen peroxide and its derivatives. Chem. Soc. Rev. 2011, 40, 1722–1760. [Google Scholar]
- Arends, I.W.C.E. Metal-catalyzed asymmetric epoxidations of terminal olefins using hydrogen peroxide as the oxidant. Angew. Chem. Int. Ed. 2006, 45, 6250–6252. [Google Scholar]
- Ferrand, Y.; Daviaud, R.; Le Maux, P.; Simonneaux, G. Catalytic asymmetric oxidation of sulfide and styrene derivatives using macroporous resins containing chiral metalloporphyrins (Fe, Ru). Tetrahedron Asymmetry 2006, 17, 952–960. [Google Scholar]
- Le Maux, P.; Simonneaux, G. First enantioselective iron-porphyrin-catalyzed sulfide oxidation with aqueous hydrogen peroxide. Chem. Commun. 2011, 47, 6957–6959. [Google Scholar]
- Le Maux, P.; Srour, H.; Simonneaux, G. Enantioselective water-soluble iron porphyrin-catalyzed epoxidation with aqueous hydrogen peroxide and hydroxylation with iodobenzene diacetate. Tetrahedron 2012, 68, 5824–5828. [Google Scholar]
- Srour, H.; Le Maux, P.; Simonneaux, G. Enantioselective Manganese-porphyrin-catalyzed epoxidation and C–H hydroxylation with hydrogen peroxide in water/methanol solutions. Inorg. Chem. 2012, 51, 5850–5856. [Google Scholar]
- Nicolas, I.; Le Maux, P.; Simonneaux, G. Synthesis of chiral water-soluble metalloporphyrins (Fe, Ru,): New catalysts for asymmetric carbene transfer in water. Tetrahedron Lett. 2008, 49, 5793–5795. [Google Scholar]
- Halterman, R.L.; Jan, S.T. Catalytic asymmetric epoxidation of unfunctionalized alkenes using the first D4-symmetric metallotetraphenylporphyrin. J. Org. Chem. 1991, 56, 5253–5254. [Google Scholar]
- Meng, L.; Cheng, Q.; Kim, C.; Gao, W.Y.; Wojtas, L.; Chen, Y.S.; Zaworotko, M.J.; Zhang, X.P.; Ma, S. Crystal engeenering of a microporous, catalytically active fcu topology MOF using a custom-designed metalloporphyrin linker. Angew. Chem. Int. Ed. 2012, 51, 10082–10085. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Simonneaux, G.; Srour, H.; Maux, P.L.; Chevance, S.; Carrie, D. Metalloporphyrin Symmetry in Chiral Recognition and Enantioselective Catalysis. Symmetry 2014, 6, 210-221. https://doi.org/10.3390/sym6020210
Simonneaux G, Srour H, Maux PL, Chevance S, Carrie D. Metalloporphyrin Symmetry in Chiral Recognition and Enantioselective Catalysis. Symmetry. 2014; 6(2):210-221. https://doi.org/10.3390/sym6020210
Chicago/Turabian StyleSimonneaux, Gérard, Hassan Srour, Paul Le Maux, Soizic Chevance, and Daniel Carrie. 2014. "Metalloporphyrin Symmetry in Chiral Recognition and Enantioselective Catalysis" Symmetry 6, no. 2: 210-221. https://doi.org/10.3390/sym6020210
APA StyleSimonneaux, G., Srour, H., Maux, P. L., Chevance, S., & Carrie, D. (2014). Metalloporphyrin Symmetry in Chiral Recognition and Enantioselective Catalysis. Symmetry, 6(2), 210-221. https://doi.org/10.3390/sym6020210