Symmetry Adaptation of the Rotation-Vibration Theory for Linear Molecules



Abstract
:1. Introduction
2. Rotational and Vibrational Symmetry
2.1. The Groups D(M), D(EM), and D
2.2. The Point Groups D and Their Correlation with D
3. General Formulation of the Character Tables and the Irreducible Representation Transformation Matrices of the D Groups
3.1. General Structure
3.2. Irreducible Representations
3.3. Transformation Matrices
4. Symmetrization Using the TROVE Approach
Symmetrization of the Basis Set for CH Using the Coordinate TROVE Implementation
5. Numerical Example
5.1. Symmetrization
5.2. Even vs. Odd D Symmetries
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bunker, P.R.; Jensen, P. Molecular Symmetry and Spectroscopy, 2nd ed.; NRC Research Press: Ottawa, ON, Canada, 1998. [Google Scholar]
- Longuet-Higgins, H. The symmetry groups of non-rigid molecules. Mol. Phys. 1963, 6, 445–460. [Google Scholar] [CrossRef]
- Hougen, J.T. Classification of Rotational Energy Levels. II. J. Chem. Phys. 1963, 39, 358–365. [Google Scholar] [CrossRef]
- Bunker, P.; Papoušek, D. The symmetry groups of linear molecules. J. Mol. Spectrosc. 1969, 32, 419–429. [Google Scholar] [CrossRef]
- Yurchenko, S.N.; Thiel, W.; Jensen, P. Theoretical ROVibrational Energies (TROVE): A robust numerical approach to the calculation of rovibrational energies for polyatomic molecules. J. Mol. Spectrosc. 2007, 245, 126–140. [Google Scholar] [CrossRef]
- Yachmenev, A.; Yurchenko, S.N. Automatic differentiation method for numerical construction of the rotational-vibrational Hamiltonian as a power series in the curvilinear internal coordinates using the Eckart frame. J. Chem. Phys. 2015, 143, 014105. [Google Scholar] [CrossRef] [PubMed]
- Yurchenko, S.N.; Tennyson, J. ExoMol line lists IV: The rotation-vibration spectrum of methane up to 1500 K. Mon. Not. R. Astron. Soc. 2014, 440, 1649–1661. [Google Scholar] [CrossRef]
- Yurchenko, S.N.; Barber, R.J.; Yachmenev, A.; Thiel, W.; Jensen, P.; Tennyson, J. A variationally computed T = 300 K line list for NH3. J. Phys. Chem. A 2009, 113, 11845–11855. [Google Scholar] [CrossRef] [PubMed]
- Sousa-Silva, C.; Hesketh, N.; Yurchenko, S.N.; Hill, C.; Tennyson, J. High Temperature partition functions and thermodynamic data for ammonia and phosphine. J. Quant. Spectrosc. Radiat. Transf. 2014, 142, 66–74. [Google Scholar] [CrossRef]
- Sousa-Silva, C.; Al-Refaie, A.F.; Tennyson, J.; Yurchenko, S.N. ExoMol line lists-VII. The rotation-vibration spectrum of phosphine up to 1500 K. Mon. Not. R. Astron. Soc. 2015, 446, 2337–2347. [Google Scholar] [CrossRef]
- Underwood, D.S.; Yurchenko, S.N.; Tennyson, J.; Jensen, P. Rotational spectrum of SO3 and a theoretical evidence for the formation of rotational, energy level clusters in its vibrational ground state. J. Chem. Phys. 2014, 140, 244316. [Google Scholar] [CrossRef] [PubMed]
- Underwood, D.S.; Tennyson, J.; Yurchenko, S.N.; Clausen, S.; Fateev, A. ExoMol line lists XVII: A line list for hot SO3. Mon. Not. R. Astron. Soc. 2016, 462, 4300–4313. [Google Scholar] [CrossRef]
- Al-Refaie, A.F.; Yurchenko, S.N.; Yachmenev, A.; Tennyson, J. ExoMol line lists-VIII: A variationally computed line list for hot formaldehyde. Mon. Not. R. Astron. Soc. 2015, 448, 1704–1714. [Google Scholar] [CrossRef]
- Owens, A.; Yurchenko, S.N.; Yachmenev, A.; Tennyson, J.; Thiel, W. Accurate ab initio vibrational energies of methyl chloride. J. Chem. Phys. 2015, 142, 244306. [Google Scholar] [CrossRef] [PubMed]
- Owens, A.; Yurchenko, S.N.; Yachmenev, A.; Thiel, W.; Tennyson, J. ExoMol molecular line lists XXII. The rotation-vibration spectrum of silane up to 1200 K. Mon. Not. R. Astron. Soc. 2017, 471, 5025–5032. [Google Scholar] [CrossRef]
- Al-Refaie, A.F.; Ovsyannikov, R.I.; Polyansky, O.L.; Yurchenko, S.N.; Tennyson, J. A variationally calculated room temperature line-list for H2O2. J. Mol. Spectrosc. 2015, 318, 84–90. [Google Scholar] [CrossRef]
- Al-Refaie, A.F.; Polyansky, O.L.; Ovsyannikov, R.I.; Tennyson, J.; Yurchenko, S.N. ExoMol line lists XV: A hot line-list for hydrogen peroxide. Mon. Not. R. Astron. Soc. 2016, 461, 1012–1022. [Google Scholar] [CrossRef]
- Chubb, K.L.; Yachmenev, A.; Tennyson, J.; Yurchenko, S.N. TROVE: Treating linear molecule HCCH. J. Chem. Phys. 2018. submitted. [Google Scholar]
- Mant, B.P.; Yachmenev, A.; Tennyson, J.; Yurchenko, S.N. ExoMol molecular line lists-XXVII: Spectra of C2H4. Mon. Not. R. Astron. Soc. 2018. submitted for publication. [Google Scholar]
- Tennyson, J.; Yurchenko, S.N. ExoMol: Molecular line lists for exoplanet and other atmospheres. Mon. Not. R. Astron. Soc. 2012, 425, 21–33. [Google Scholar] [CrossRef]
- Tennyson, J.; Yurchenko, S.N.; Al-Refaie, A.F.; Barton, E.J.; Chubb, K.L.; Coles, P.A.; Diamantopoulou, S.; Gorman, M.N.; Hill, C.; Lam, A.Z.; et al. The ExoMol database: molecular line lists for exoplanet and other hot atmospheres. J. Mol. Spectrosc. 2016, 327, 73–94. [Google Scholar] [CrossRef]
- Yurchenko, S.N.; Yachmenev, A.; Ovsyannikov, R.I. Symmetry-Adapted Ro-vibrational Basis Functions for Variational Nuclear Motion Calculations: TROVE Approach. J. Chem. Theory Comput. 2017, 13, 4368–4381. [Google Scholar] [CrossRef] [PubMed]
- Laane, J.; Ocola, E.J. Applications of Symmetry and Group Theory for the Investigation of Molecular Vibrations. Acta Appl. Math. 2012, 118, 3–24. [Google Scholar] [CrossRef]
- Fritzsche, S. Application of point-group symmetries in chemistry and physics: A computer-algebraic approach. Int. J. Quantum Chem. 2005, 106, 98–129. [Google Scholar] [CrossRef]
- Häser, M. Molecular point-group symmetry in electronic structure calculations. J. Chem. Phys. 1991, 95, 8259–8265. [Google Scholar] [CrossRef]
- Papoušek, D.; Aliev, M.R. Molecular Vibrational-Rotational Spectra: Theory and Applications of High Resolution Infrared; Studies in Physical and Theoretical Chemistry; Elsevier: Amsterdam, The Netherlands, 1982. [Google Scholar]
- Watson, J.K.G. Vibration-rotation hamiltonian of linear molecules. Mol. Phys. 1970, 19, 465–487. [Google Scholar] [CrossRef]
- Wlodarczak, G. Linear Polyatomic Molecules: Introduction. In Linear Polyatomic Molecules; Springer: Berlin/Heidelberg, Germnay, 2012; pp. 6–24. [Google Scholar]
- Chen, C. Symmetry Adapted Analysis of Linear Molecules. J. Chin. Chem. Soc. 1973, 20, 191–202. [Google Scholar] [CrossRef]
- Hegelund, F.; Rasmussen, F.; Brodersen, S. The selection rules and the transition moment for rotation–vibrational transitions in axial molecules. J. Raman Spectrosc. 1973, 1, 433–453. [Google Scholar] [CrossRef]
- Hirano, T.; Nagashima, U.; Jensen, P. Bending wavefunctions for linear molecules. J. Mol. Spectrosc. 2018, 343, 54–61. [Google Scholar] [CrossRef]
- Chubb, K.L.; Joseph, M.; Franklin, J.; Choudhury, N.; Furtenbacher, T.; Császár, A.G.; Gaspard, G.; Oguoko, P.; Kelly, A.; Yurchenko, S.N.; et al. MARVEL analysis of the measured high-resolution spectra of C2H2. J. Quant. Spectrosc. Radiat. Transf. 2018, 204, 42–55. [Google Scholar] [CrossRef]
- Brown, J.M.; Hougen, J.T.; Huber, K.P.; Johns, J.W.C.; Kopp, I.; Lefebvre-Brion, H.; Merer, A.J.; Ramsay, D.A.; Rostas, J.; Zare, R.N. The labeling of parity doublet levels in linear molecules. J. Mol. Spectrosc. 1975, 55, 500–503. [Google Scholar] [CrossRef]
- Herman, M.; Lievin, J. Acetylene—From intensity alternation in spectra to ortho and para molecule. J. Chem. Educ. 1982, 59, 17. [Google Scholar] [CrossRef]
- Jensen, P.; Hegelund, F. Lecture Notes: Molecular Rotation-Vibration Theory; ResearchGate: Gatersleben, Germany, 2014; pp. 1–97. [Google Scholar] [CrossRef]
- Schutte, C.J.H.; Bertie, J.E.; Bunker, P.R.; Hougen, J.T.; Mills, I.M.; Watson, J.K.G.; Winnewisser, B.P. Notations and conventions in molecular spectroscopy: Part 2. Symmetry notation. Pure Appl. Chem. 1997, 69, 1641–1650. [Google Scholar] [CrossRef]
- Noumerov, B.V. A method of extrapolation of perturbations. Mon. Not. R. Astron. Soc. 1924, 84, 592–602. [Google Scholar] [CrossRef]
- Cooley, J.W. An Improved eigenvalue corrector formula for solving the Schrödinger equation for central fields. Math. Comp. 1961, 15, 363–374. [Google Scholar]
- Sørensen, G.O. A New Approach to the Hamiltonian of Nonrigid Molecules. In Large Amplitude Motion in Molecules II; Topics in Current Chemistry; Dewar, M.J.S., Ed.; Springer: Berlin/Heidelberg, Germeny, 1979; Volume 82, pp. 97–175. [Google Scholar]
- Bunker, P.R.; Jensen, P. Spherical top molecules and the molecular symmetry group. Mol. Phys. 1999, 97, 255–264. [Google Scholar] [CrossRef]
- Yurchenko, S.N.; Carvajal, M.; Jensen, P.; Lin, H.; Zheng, J.J.; Thiel, W. Rotation-vibration motion of pyramidal XY3 molecules described in the Eckart frame: Theory and application to NH3. Mol. Phys. 2005, 103, 359–378. [Google Scholar] [CrossRef]
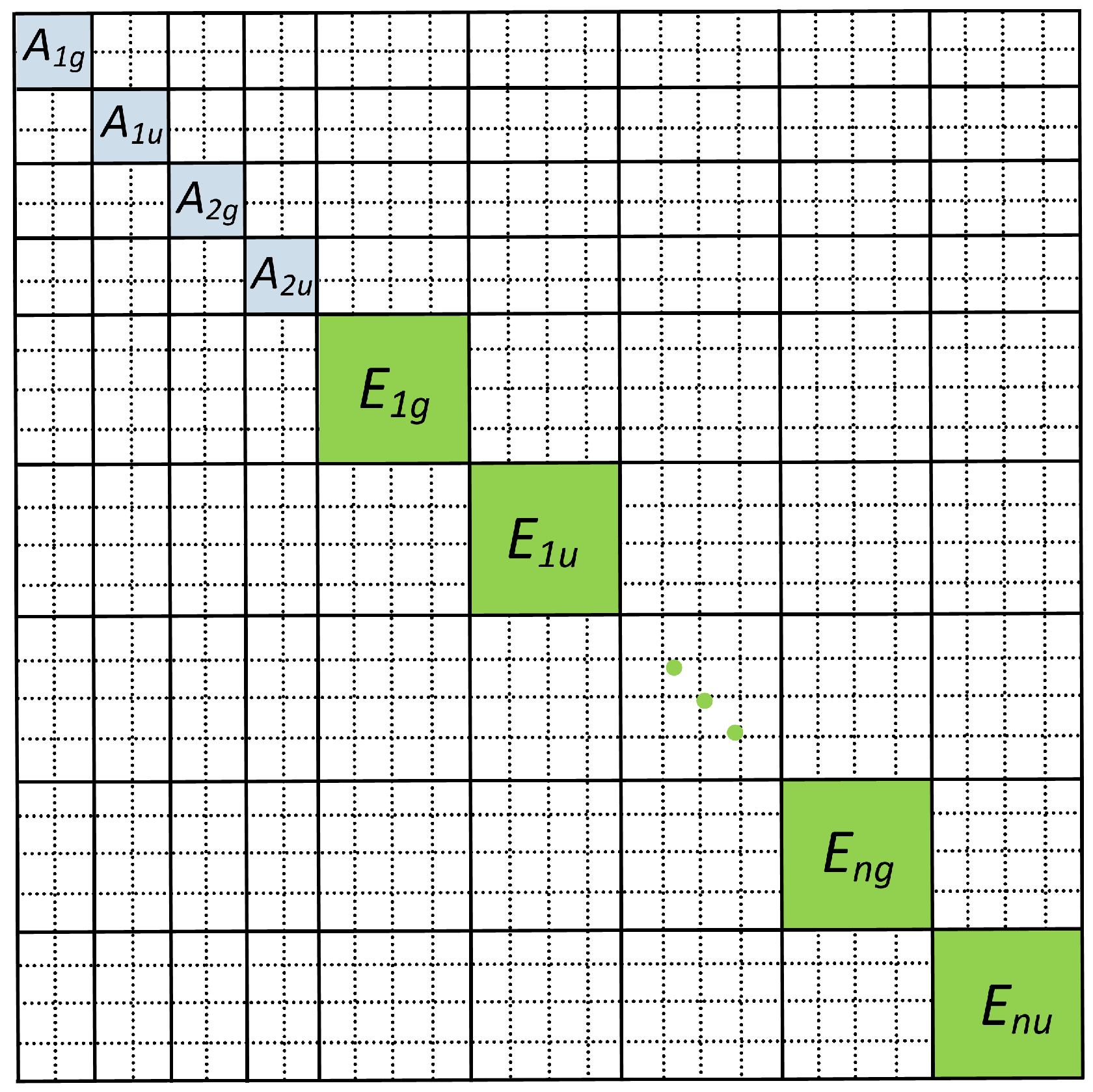
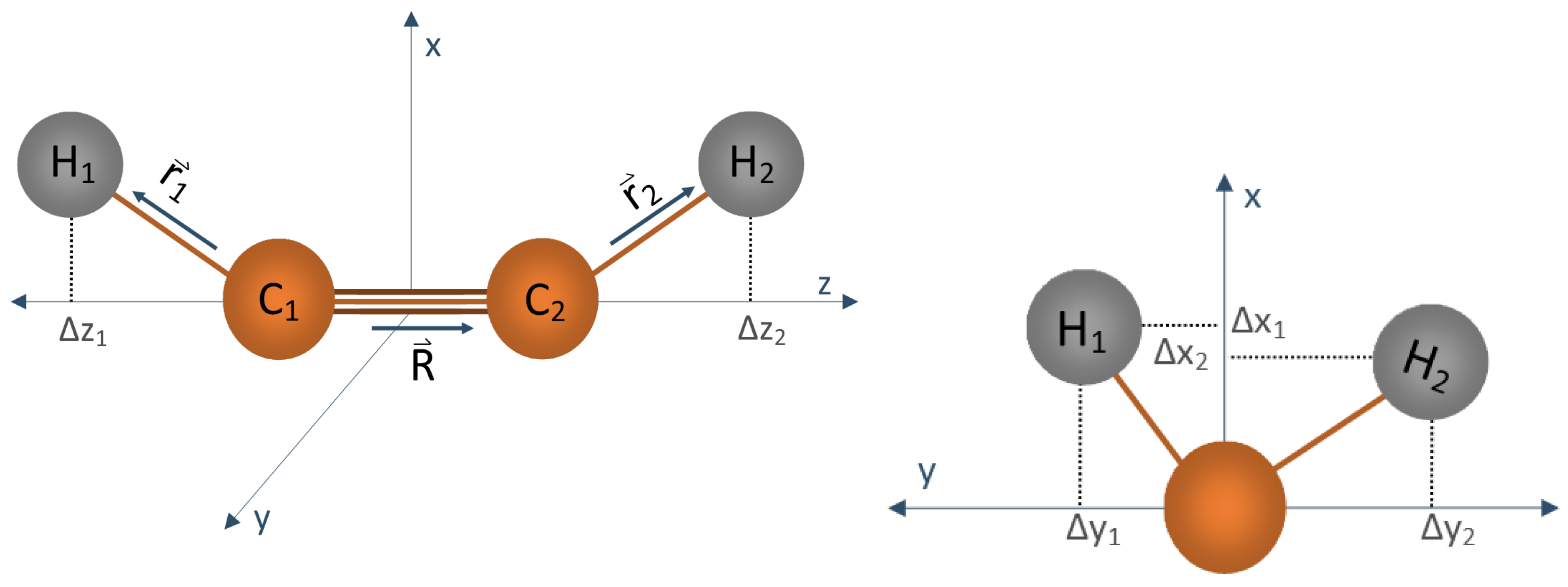

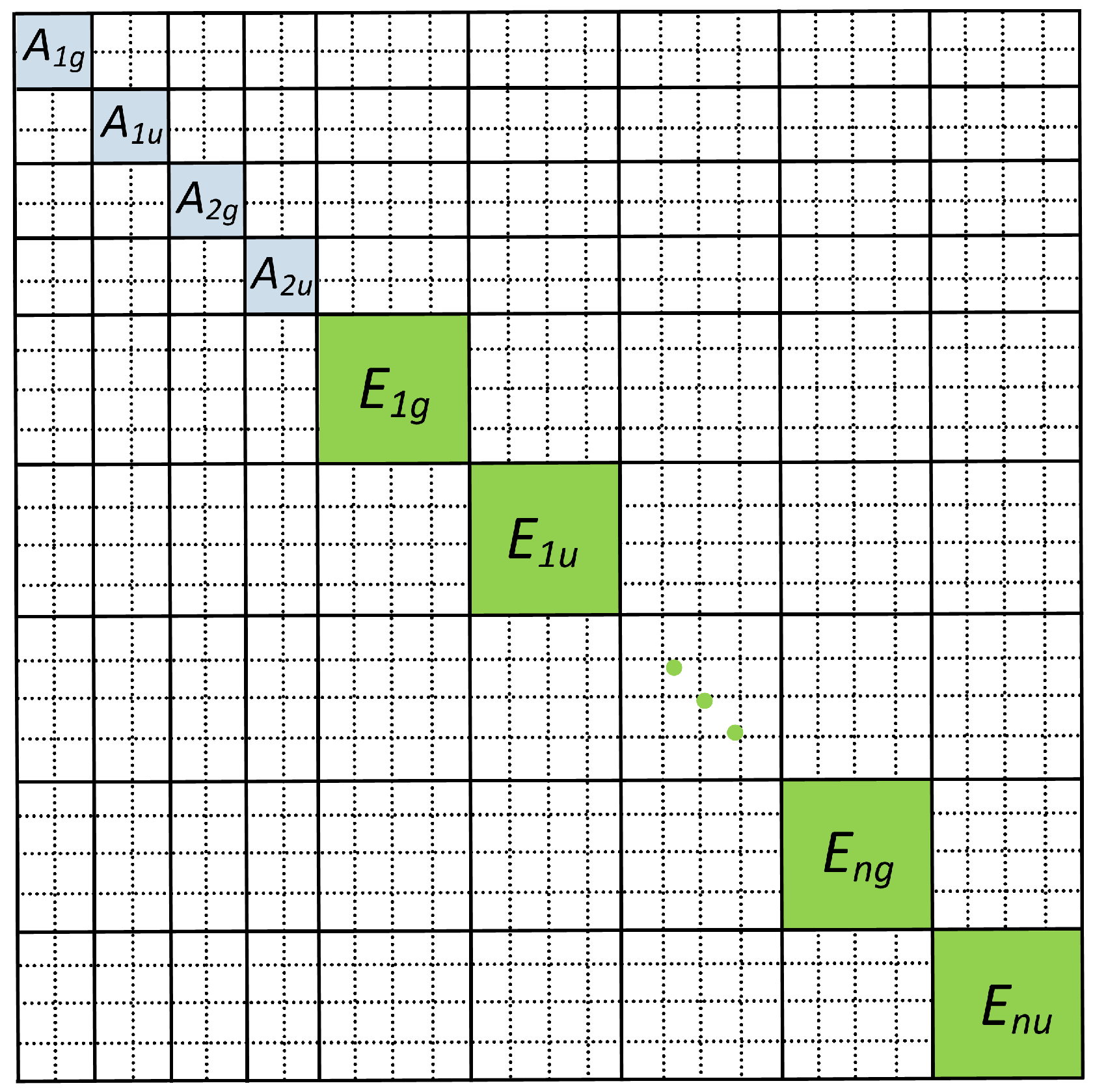{kind=link}
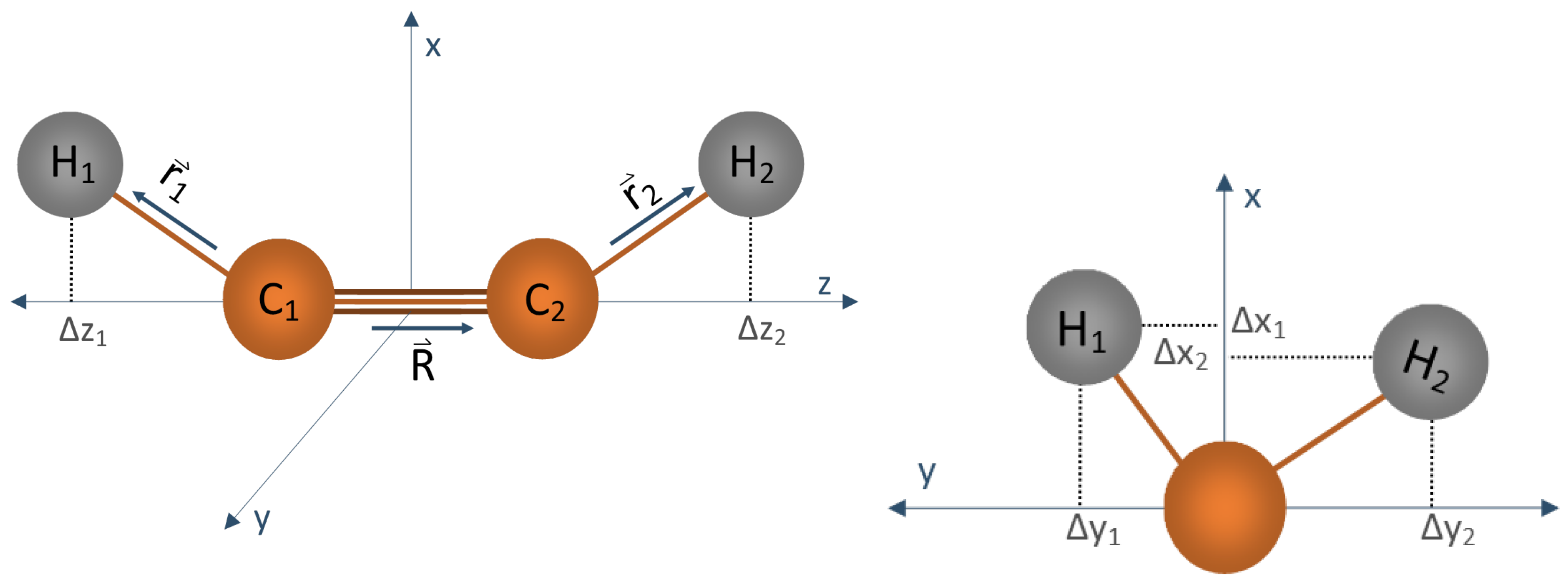{kind=link}
| D(EM): | ⋯ | ⋯ | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | ⋯ | ∞ | 1 | 2 | ⋯ | ∞ | |
| D: | E | ⋯ | i | ⋯ | ||||
| , : | 1 | 1 | ⋯ | 1 | 1 | 1 | ⋯ | 1 |
| , : | 1 | 1 | ⋯ | 1 | ⋯ | |||
| , : | 1 | 1 | ⋯ | 1 | 1 | ⋯ | ||
| , : | 1 | 1 | ⋯ | ⋯ | 1 | |||
| , : | 2 | ⋯ | 0 | 2 | ⋯ | 0 | ||
| , : | 2 | ⋯ | 0 | ⋯ | 0 | |||
| , : | 2 | ⋯ | 0 | 2 | ⋯ | 0 | ||
| , : | 2 | ⋯ | 0 | ⋯ | 0 | |||
| , : | 2 | ⋯ | 0 | 2 | ⋯ | 0 | ||
| , : | 2 | ⋯ | 0 | ⋯ | 0 | |||
| ⋮ | ⋮ | ⋮ | ⋯ | ⋮ | ⋮ | ⋮ | ⋯ | ⋮ |
| E | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 1 | 1 | ||||||
| 1 | 1 | ||||||||
| 1 | 1 | ||||||||
| 1 | 1 |
| k | ||
|---|---|---|
| 0 | (J even) | |
| (J odd) | ||
| ±1 | ||
| ±2 | ||
| ±3 | ||
| ⋮ | ⋮ |
| J odd: | f | ||||
| e | |||||
| e | |||||
| f | |||||
| J even: | e | ||||
| f | |||||
| f | |||||
| e |
| Symmetry Operation | Number of Operations | Description |
|---|---|---|
| Even n: | ||
| E | 1 | Identity |
| Rotations about the n-fold molecular axis | ||
| n | n rotations by about axes | |
| perpendicular to the molecular axis | ||
| i | 1 | Point group inversion |
| Improper rotation (see caption) | ||
| 1 | Horizontal reflection (see caption) | |
| Vertical reflection (see caption) | ||
| Diagonal reflection (see caption) | ||
| Total: | ||
| Odd n: | ||
| E | 1 | Identity |
| Rotations about the n-fold molecular axis | ||
| n | n rotations by about axes | |
| perpendicular to the molecular axis | ||
| Improper rotation (see caption) | ||
| 1 | Horizontal reflection (see caption) | |
| n | Vertical reflection (see caption) | |
| Total: |
| Point Group | |||
|---|---|---|---|
| D, n odd | |||
| D, n even | i |
| D (n even) | E | (=) | (=i) | (=) |
| 1 | −1 | 1 | 1 | |
| 1 | −1 | 1 | −1 | |
| 1 | −1 | 1 | 1 | |
| 1 | −1 | 1 | −1 | |
| 2 | 2 | 0 | ||
| 1 | −1 | −1 | 1 | |
| 1 | −1 | −1 | −1 | |
| 1 | −1 | −1 | 1 | |
| 1 | −1 | −1 | −1 | |
| 2 | −2 | 0 | ||
| D (n odd) | E | (=) | (=) | (=) |
| 1 | 1 | 1 | 1 | |
| 1 | 1 | 1 | −1 | |
| 2 | 2 | 0 | ||
| 1 | 1 | −1 | 1 | |
| 1 | 1 | −1 | −1 | |
| 2 | −2 | 0 |
| K | (even n) | (odd n) | D(EM) |
|---|---|---|---|
| 0 | |||
| >0, even | |||
| >0, odd | |||
| D (n even) | E | |||
| 1 | 1 | 1 | 1 | |
| D ( odd) | ||||
| 1 | 1 | 1 | ||
| r | ||||
|---|---|---|---|---|
| E | ||||
| i | ||||
| 1, 2, 3, …, , , …, |
| r | ||||
|---|---|---|---|---|
| E | ||||
| K | D(n even) | D(EM) | ||
| =0 | 0 | |||
| >0 | 0 | ⊕ | ⊕ | |
| >0 | ⊕ | ⊕ | ||
| >0 | = 1, 2, …, | , , , , … | ||
| D(odd) | D(EM) | |||
| =0 | 0 | J even | ||
| J odd | ||||
| >0, odd | 0 | ⊕ | ⊕ | |
| >0, even | 0 | ⊕ | ⊕ | |
| >0, odd | 1, 2, …, | |||
| >0, even | 1, 2, …, |
| Irrep | m | Transformation | |
|---|---|---|---|
| E | |||
| i | |||
| K | |||
|---|---|---|---|
| Even n | Odd n | ||
| 0 | 0 | ||
| 1 | |||
| >0, odd | 0 | ||
| 1 | |||
| >0, even | 0 | ||
| 1 | |||
| Exp. Energy (cm) [32] | Energy (cm) | J | K | D | D | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| This Work | |||||||||||
| 2.353286 | 2.356491 | 1 | 0 | 1 | |||||||
| 614.044355 | 625.810547 | 1 | 1 | 1 | |||||||
| 1232.749162 | 1283.603736 | 1 | 0 | 1 | |||||||
| 7.059822 | 7.069433 | 2 | 0 | 0 | |||||||
| 618.745653 | 630.518518 | 2 | 1 | 1 | |||||||
| 1235.874392 | 1276.518756 | 2 | 2 | 0 | |||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chubb, K.L.; Jensen, P.; Yurchenko, S.N. Symmetry Adaptation of the Rotation-Vibration Theory for Linear Molecules. Symmetry 2018, 10, 137. https://doi.org/10.3390/sym10050137
Chubb KL, Jensen P, Yurchenko SN. Symmetry Adaptation of the Rotation-Vibration Theory for Linear Molecules. Symmetry. 2018; 10(5):137. https://doi.org/10.3390/sym10050137
Chicago/Turabian StyleChubb, Katy L., Per Jensen, and Sergei N. Yurchenko. 2018. "Symmetry Adaptation of the Rotation-Vibration Theory for Linear Molecules" Symmetry 10, no. 5: 137. https://doi.org/10.3390/sym10050137
APA StyleChubb, K. L., Jensen, P., & Yurchenko, S. N. (2018). Symmetry Adaptation of the Rotation-Vibration Theory for Linear Molecules. Symmetry, 10(5), 137. https://doi.org/10.3390/sym10050137