David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments

Biopharm Molecular Discovery, GlaxoSmithKline, Hertfordshire SG1 2NY, UK
*
Author to whom correspondence should be addressed.
Antibodies 2019, 8(2), 28; https://doi.org/10.3390/antib8020028
Submission received: 1 February 2019
/
Revised: 12 March 2019
/
Accepted: 2 April 2019
/
Published: 9 April 2019
(This article belongs to the Special Issue Structure and Function of Antibodies)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Since the licensing of the first monoclonal antibody therapy in 1986, monoclonal antibodies have become the largest class of biopharmaceuticals with over 80 antibodies currently approved for a variety of disease indications. The development of smaller, antigen binding antibody fragments, derived from conventional antibodies or produced recombinantly, has been growing at a fast pace. Antibody fragments can be used on their own or linked to other molecules to generate numerous possibilities for bispecific, multi-specific, multimeric, or multifunctional molecules, and to achieve a variety of biological effects. They offer several advantages over full-length monoclonal antibodies, particularly a lower cost of goods, and because of their small size they can penetrate tissues, access challenging epitopes, and have potentially reduced immunogenicity. In this review, we will discuss the structure, production, and mechanism of action of EMA/FDA-approved fragments and of those in clinical and pre-clinical development. We will also discuss current topics of interest surrounding the potential use of antibody fragments for intracellular targeting and blood–brain barrier (BBB) penetration.
1. Introduction
Since the licensing of the first monoclonal antibody (mAb) therapy, OrthocloneTM (OKT3), in 1986, the specificity, flexibility, and diversity of antibodies and antibody derivatives has led to their becoming the largest class of biopharmaceuticals [1]. Monoclonal antibodies have a history of safe use, a strong scientific basis, and a high degree of technical feasibility. As of October 2018, over 80 antibodies were marketed or approved by the EMA/FDA for multiple disease indications including cancer, inflammation/autoimmunity, transplantation, infectious and cardiovascular diseases, haematology, allergy, and ophthalmology. Over 500 more, including 2nd generation products and novel antibody formats, are currently in clinical trials around the world [1,2,3].
The modular nature of antibodies, both structurally and functionally, allows for the generation of smaller antigen binding fragments, such as fragment antigen binding (Fab), the single chain fragment variable (scFv), single-domain antibodies, and the fragment crystallizable (Fc) domain, through molecular cloning, antibody engineering, and even enzymatic methods. Antibody fragments can then be used on their own or linked to other molecules or fragments to generate bispecific, multi-specific, multimeric, or multifunctional molecules to achieve a variety of biological effects [4].
Antibody fragments can offer several advantages over the use of conventional antibodies. For example, they can be produced easily, generally using microbial expression systems, which results in faster cultivation, higher yields, and lower production costs [5]. Their small size allows access to challenging, cryptic epitopes, and tumour penetration, they have reduced immunogenicity, and the lack of Fc limits bystander activation of the immune system [6]. On the other hand, their smaller size results in faster renal excretion, which may require higher doses and/or more frequent dosing regimens in vivo unless mitigated by the addition of half-life extension moieties such as polyethylene glycol or albumin binding fragments.
In this review, we describe the history, structure, formats, mechanisms of action, and production of some of the most common antibody fragments. We also discuss some of the formats currently being tested in clinical and non-clinical settings, as well as briefly touching on future applications of this expanding class of biopharmaceuticals.
2. Antibody Fragment Formats
2.1. Fragment Variable (Fv)-Based Formats
2.1.1. Single Chain Fragment Variable (scFv)
The single chain fragment variable (scFv) was first described in 1988 by Bird et al. [7] and comprises the variable regions of the light chain (VL) and heavy chain (VH) of an antibody linked by a flexible peptide, which is most commonly glycine- and serine-rich with dispersed hydrophilic residues (Figure 1), to produce a single chain protein with an affinity for its antigen comparable to that of the parental mAb. The sequence and length of the ideal linker may differ between scFvs in order to optimise affinity for the antigen and thermostability. It is believed that the linker must span a ~3.5 nm distance between the VL and VH without disrupting the formation of the antigen binding site [8].
The scFv has several advantages over a conventional mAb. Firstly, their small size of approximately 27 kDa makes them ideal for large-scale production in microbial systems [9]. Additionally, as the VL and VH coding sequences are genetically linked in a single transcript, there is no need to balance the expression of the light chain (LC) and heavy chain (HC). This allows fragments to be produced more quickly, in higher yields, and at lower costs than full-sized mAbs, which generally require mammalian expression systems [10]. Their small size also facilitates tissue penetration and access to cryptic epitopes, making them especially useful for tumour penetration in cancer immunotherapy [11,12]. The lack of an Fc region removes the risk of bystander immune cell activation and antibody effector functions such as antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), or complement-dependent cytotoxicity (CDC), allowing the molecule to bind its target without activation of the host’s immune system [6]. This may be advantageous or disadvantageous depending on the context.
The lack of an Fc domain also brings about several disadvantages, mainly low thermostability compared to the parental mAb, a greater propensity for aggregation, therefore increasing the risk of immunogenicity, and a shorter half-life due to a lack of FcRn-mediated recycling. This can lead to the need for higher and more frequent dosing [4,13,14]. Fusion of the scFv to albumin or polyethylene glycol (PEG) can be used to improve half-life. However, such fusions can offset the advantages that an scFv holds over a mAb due to cost and the increase in size [15,16]. Examples of scFvs in clinical development include gancotamab (Merrimack Pharma), pexelizumab (Alexion), and Novartis’s brolucizumab [17].
2.1.2. Tandem scFvs
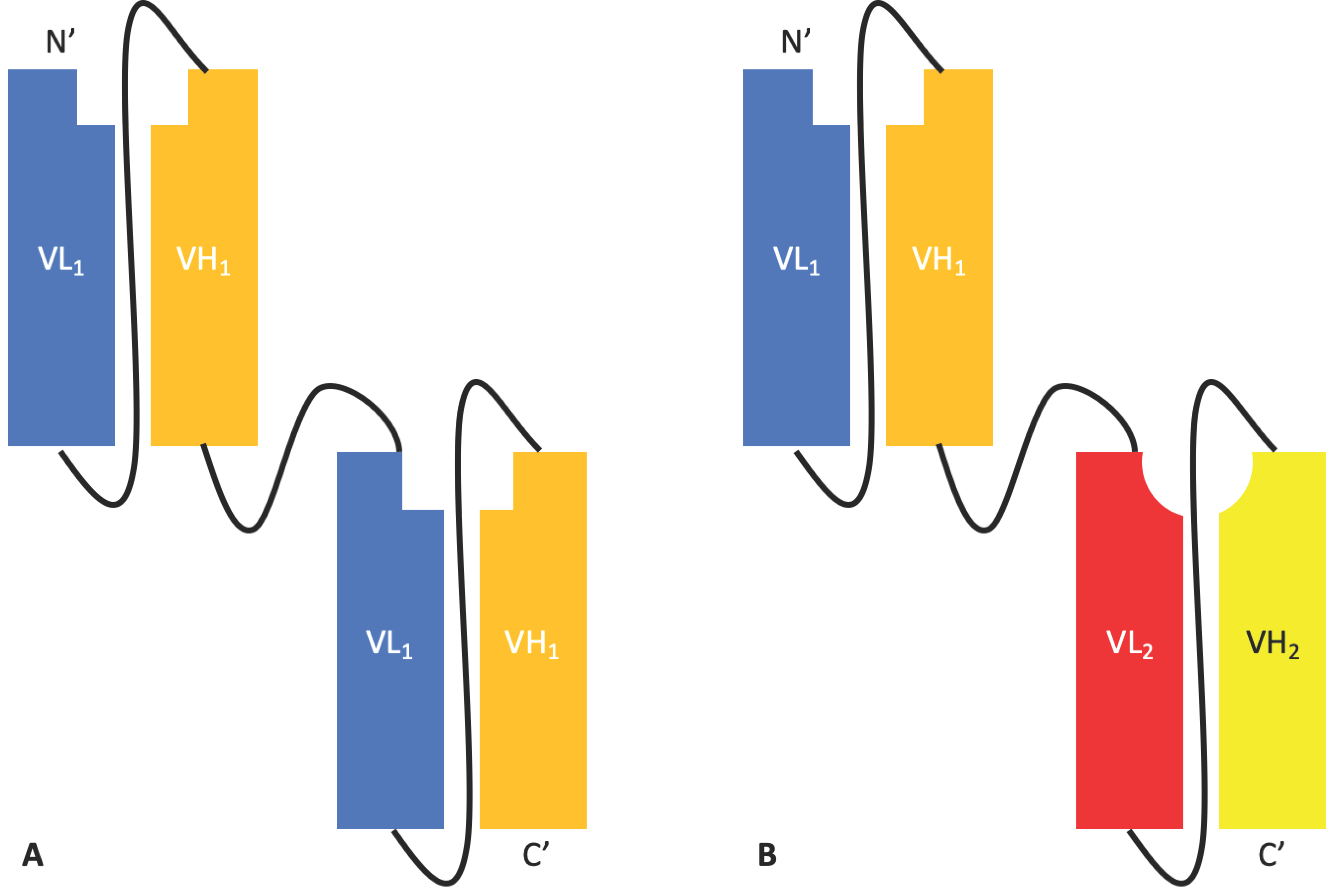
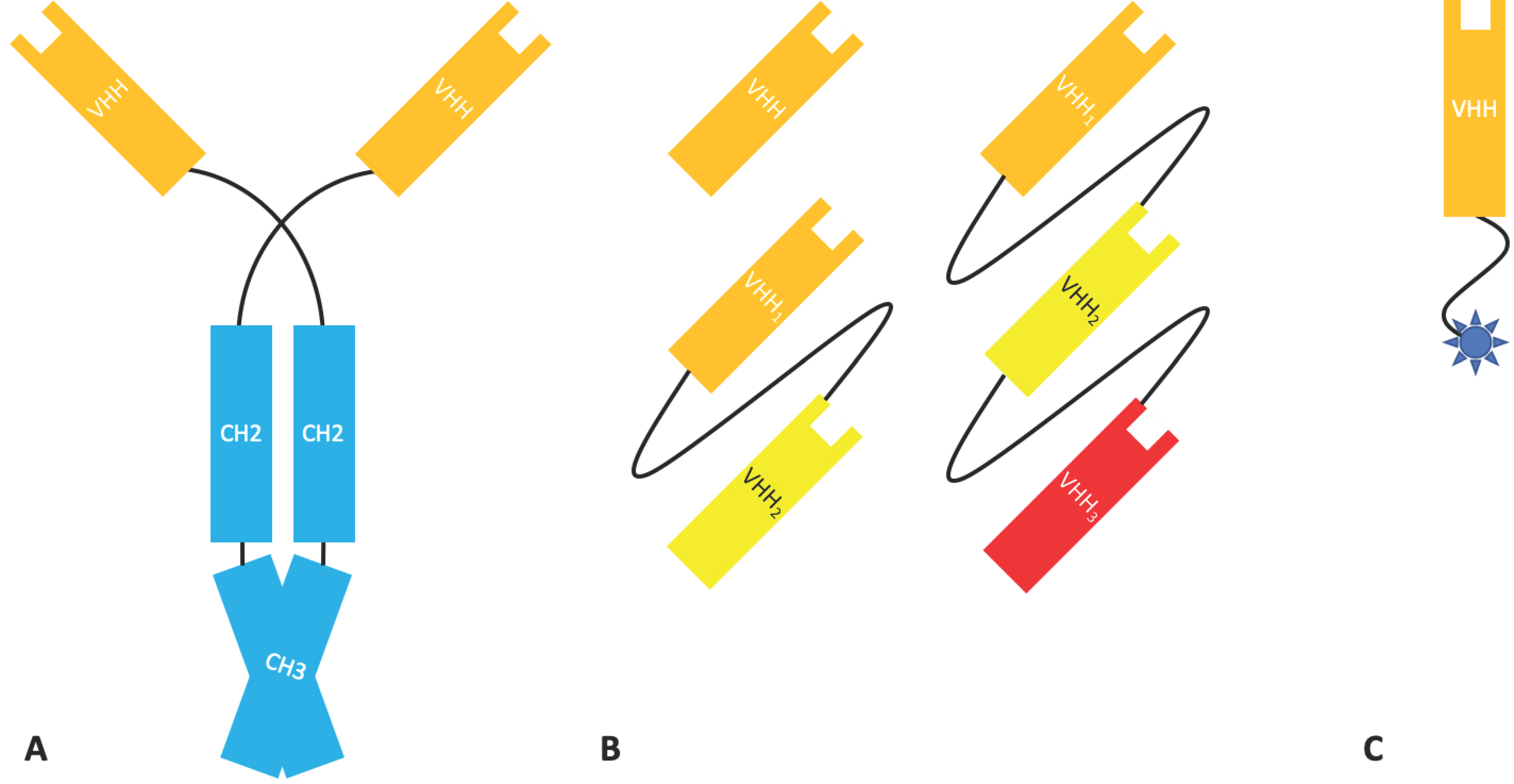
Tandem scFvs such as Micromet AG’s solitomab [17] are an adaptation of the scFv format. When two different scFvs are linked, the tandem scFv is arguably the simplest bispecific antibody platform [18]. A tandem scFv links two or more scFvs through helical peptide linkers in the orientation NH2–VL1–VH1–(linker–VL2–VH2)n–COOH, resulting in a single chain bivalent and bi-specific molecule encoded by a single gene [19] (Figure 2). These can be used to target one antigen with increased avidity, to target two distinct antigens simultaneously, or even to target albumin, thus increasing half-life [20].
2.1.3. Diabodies, DART®s, and TandAbs
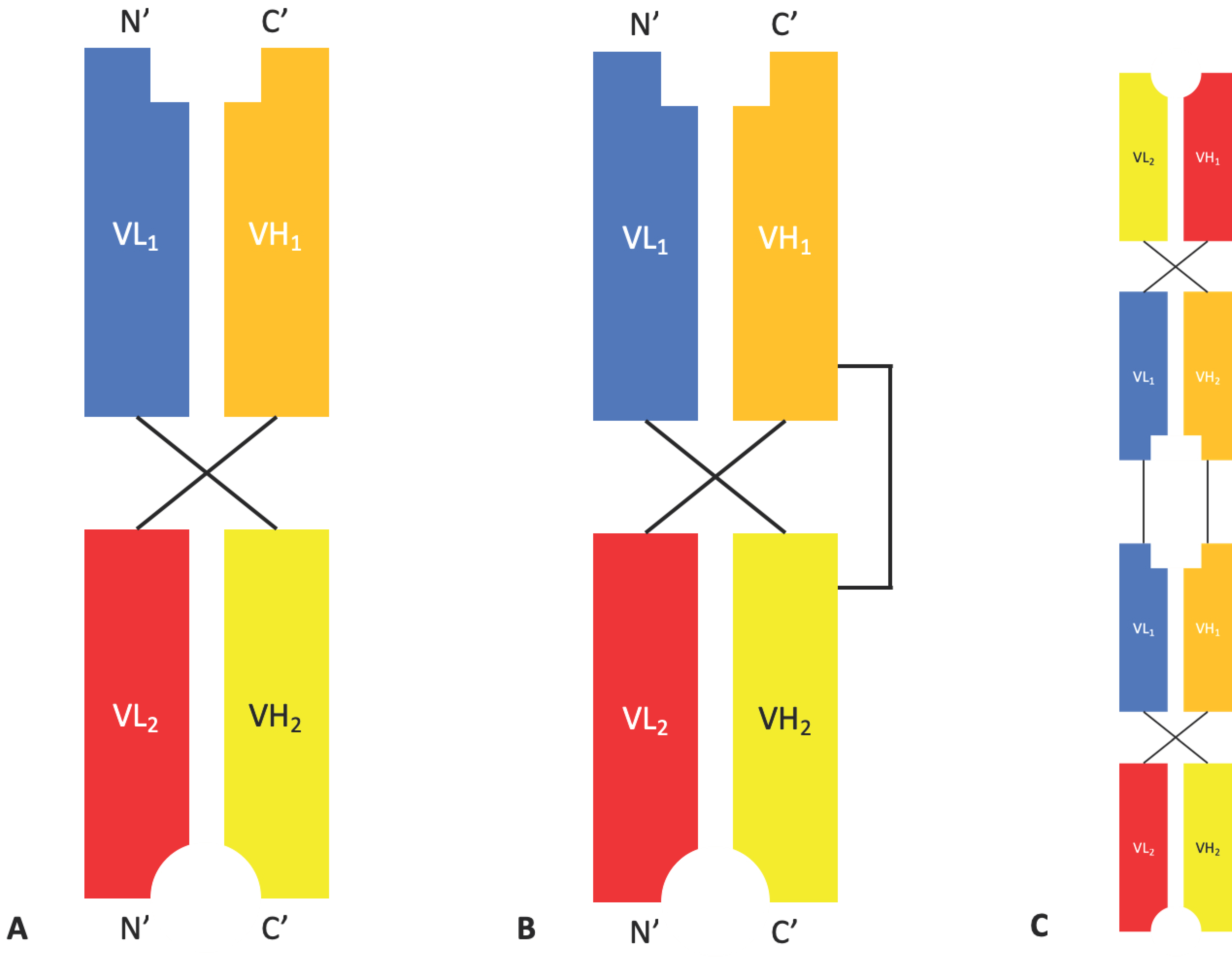
Diabodies are bivalent dimers formed from two chains, each containing a VH and a VL domain. The two domains within a chain are separated by a pentameric glycine-rich linker (G4S) that is too short to facilitate intrachain dimerization leading to two chains dimerising in a head-to-tail arrangement. By using two different chains with the same orientation, the first containing the VH of Antibody 1 and the VL of Antibody 2, and the second containing the VH of Antibody 2 and the VL of Antibody 1, bispecific bivalent dimers are produced [21] (Figure 3). A study comparing different formats of bispecific diabodies showed that not all possible diabody formats retain binding to both antigens, highlighting the importance of domain arrangement and orientation [22].
Over time, additional modifications have been made to the diabody format to further improve stability. Dual affinity re-targeting proteins (DART®s) (Figure 3) developed by MacroGenics, contain an interdomain disulphide bond for increased stability that results in a structure that is rigid and compact [23]. MacroGenics currently have four DART®s in phase I clinical trials for oncology, autoimmune disease, and HIV infection [24].
Tri- and tetra-valent molecules with a structure similar to that of a diabody can be produced by linking three or four variable domains together in a single chain. Affimed specialise in the development of TandAbs, tetravalent bispecific molecules composed of two diabodies fused in a linear fashion (Figure 3), through their proprietary ROCK® platform. There are currently five ongoing clinical trials involving a selection of T-cell and natural killer (NK) cell engagers for the treatment Hodgkin’s lymphoma and acute lymphocytic leukaemia (ALL) [25].
A major advantage of diabodies and tandem scFvs is their bivalency and ability to bring two targets into proximity. Furthermore, diabodies and tandem scFvs are bispecific-compatible formats, which has made them promising molecular formats for cancer immunotherapy, e.g., bispecific T-cell engagers (BiTE®s) [26,27].
2.1.4. Bispecific Fv Fusion Antibodies with an Fc Domain
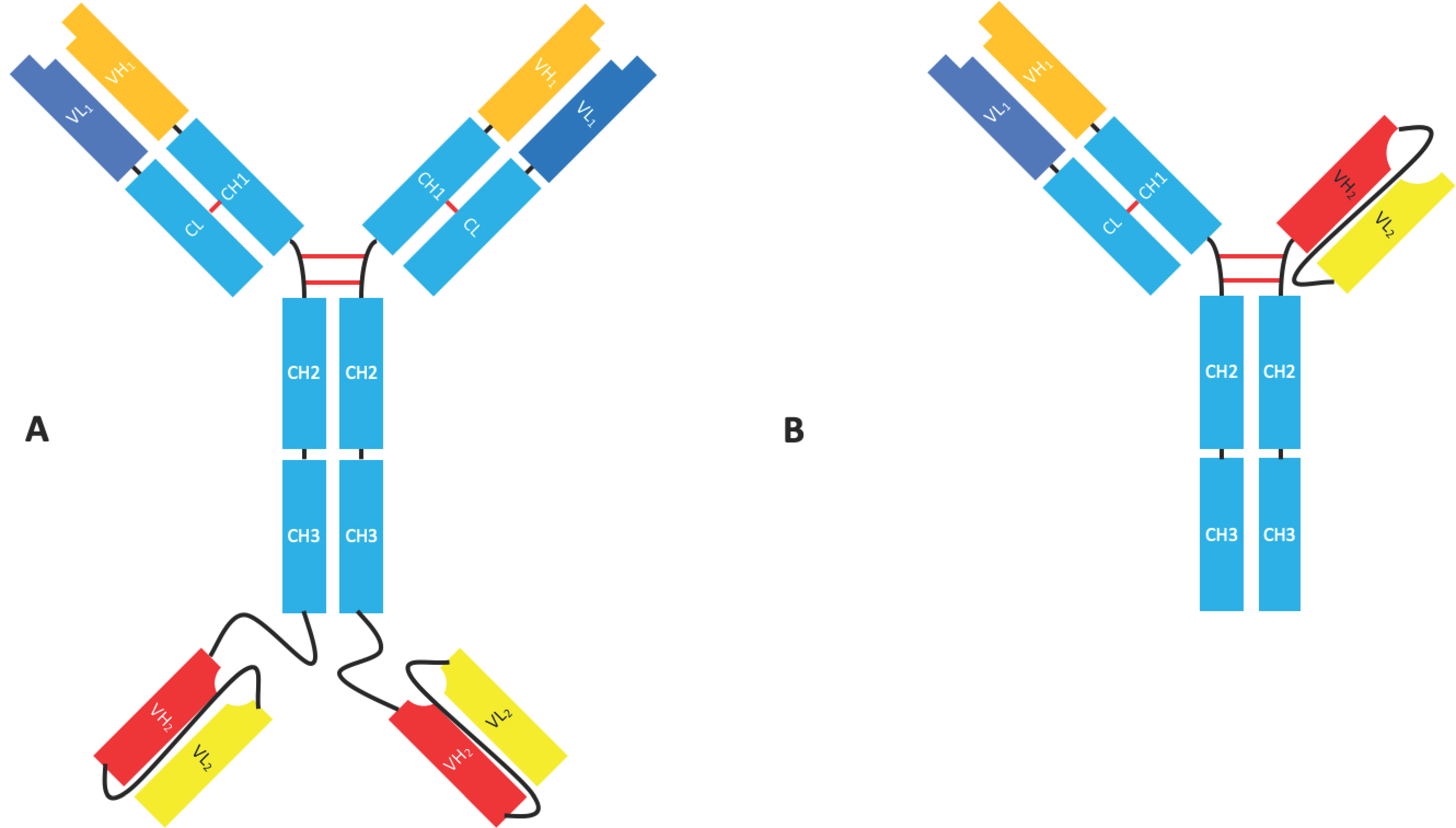
Although not fragments in their own right, there are many bispecific formats such as the IgG-scFv, which uses an scFv to target an additional epitope or antigen (Figure 4). As well as being a flexible format that allows for the production of multivalent multi-specific molecules with a modifiable effector function, these molecules can easily be produced recombinantly [18,28,29]. Istiratumab, an IgG-scFv developed by Merrimack Pharmaceuticals, is currently in clinical trials for hepatocellular carcinoma and pancreatic cancer [17]. Fab-scFv-Fc (Figure 4) is another bispecific format that uses an scFv to grant specificity for a second epitope. The format is being used in molecules such as Zymeworks’ ZW25, an anti-Her2/Her2 bispecific (biparatopic) antibody in phase I clinical trials for the treatment of Her2 expressing cancers. Xencor have four bispecific molecules with this format in phase I clinical trials for the treatment of acute myeloid leukaemia (AML), B-cell tumours, and neuroendocrine tumours [30].
2.2. Fab Based Formats
Fab & F(ab’)2 Formats
The fragment of antigen binding (Fab) (Figure 5) was the first therapeutic antibody fragment format and remains one of the most successful, laying claim to eight molecules entering clinical trials pre-1995 and comprising ~49% of all antibody fragments that have entered clinical trials [4,31]. There are currently three FDA-approved Fabs: abciximab (Reopro®), idarucizumab (Praxbind®), and ranibizumab (Leucentis®) [17].
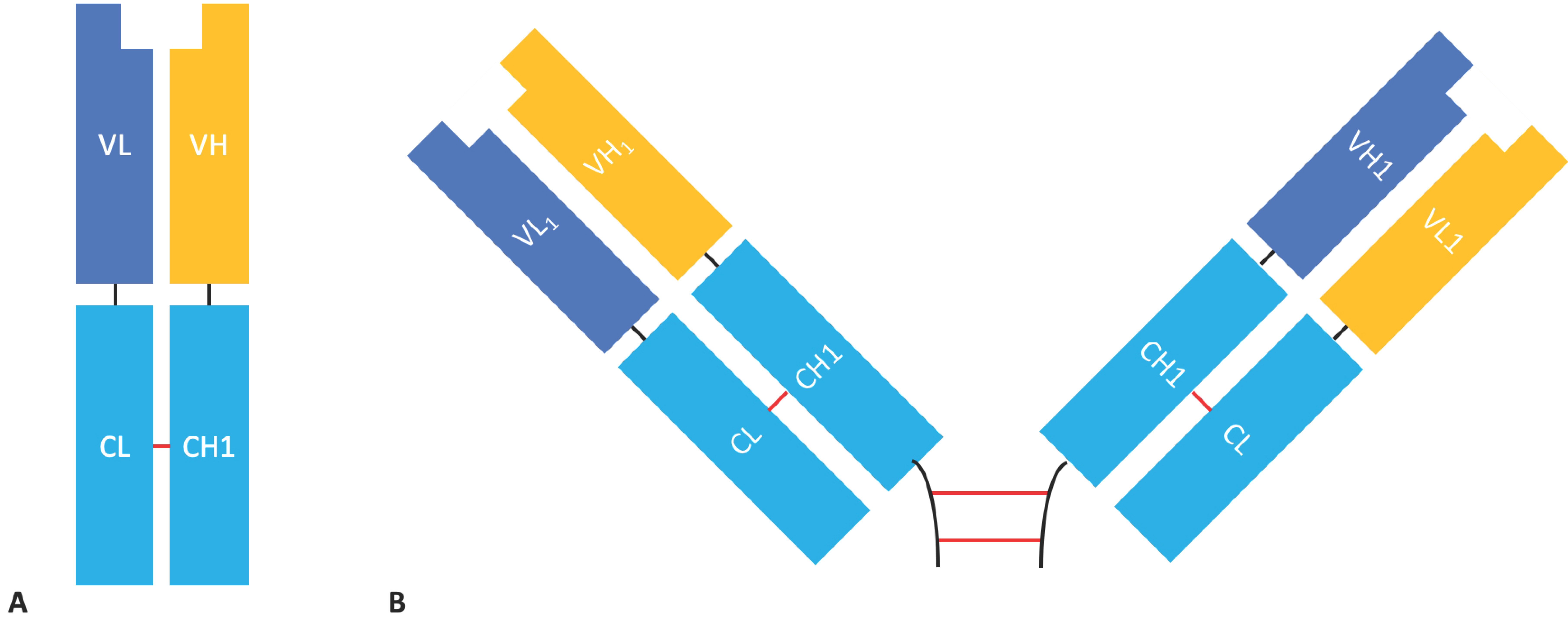
Fab fragments are composed of an antibody light chain (VL + CL domains) linked by a disulphide bond to the antibody heavy chain VH and CH1 domains; the molecular format is monovalent and monospecific and retains the parental antibody’s ability to bind its antigen with high specificity and affinity. Fabs share many of the characteristics of scFvs. Although Fabs are not as small, and therefore presumably not as good at penetrating tissue as scFvs [12], with a mass of ~50 kDa they are much smaller than mAbs (the MW of IgG is ~150 kDa). Fabs lack an Fc domain, reducing the risk of immune cell bystander activation and non-specific binding [6] and allowing for easier production at the expense of increased aggregation, lower stability, and reduced half-life [5,32]. Fab fragments are more stable than their scFv counterparts due to the mutual stabilisation that occurs between the VH/VL and CH1/CL interfaces [33]. They also have the advantage of being completely native structures and as such they avoid the time and resources required to engineer an ideal linker and are therefore less likely to be immunogenic.
F(ab’)2 fragments are composed of two Fab fragments held together by an Ig hinge region and have a molecular mass of ~110 kDa (Figure 5). F(ab’)2 fragments are bivalent, giving them increased avidity compared to Fabs [32], but their larger size may lead to reduced tissue penetration. However, their tissue penetration is still superior to that of a full-sized mAb [12]. They can be generated by the enzymatic digestion of full-length antibodies or by the expression of the recombinant F(ab’)2 in mammalian cells.
Like scFvs, Fab fragments suffer from a reduced half-life compared to their parental mAbs due to the lack of an Fc domain. This removes the possibility of FcRn-mediated recycling leading to rapid degradation of the Fab-antigen complex after absorption by macropinocytosis [34,35,36]. The half-life of Fab fragments can be extended in a similar manner to the half-life extension of scFvs, typically by conjugation to PEG or fusion to an albumin binding protein [16,37,38]. These Fab conjugate proteins have had some success with multiple molecules in clinical trials or having already received FDA approval such as certolizumab pegol (Cimzia®), a marketed PEGylated anti-TNFα Fab for rheumatoid arthritis.
2.3. Single-Domain Antibodies
2.3.1. Nanobodies
Nanobodies® (Nbs), a class of antibody fragment developed by Ablynx, are recombinantly expressed antigen binding VHH domains from heavy-chain IgG, a type of immunoglobulin found in camelids [39]. With an MW of 12–15 kDa, Nbs are one of the smallest naturally occurring antigen binding fragments. Heavy-chain IgG (Figure 6) is devoid of light chain, lacks the CH1 domain found in mammalian immunoglobulins, and therefore binds its antigen bivalently solely through two VHH domains. Isolated VHH domains retain the ability to bind their antigen and are robust under stringent conditions. Nbs can resist a wide pH range and high temperatures and have been shown to tolerate the presence of organic solvents (although these characteristics are not present in all Nbs). They are also highly soluble and, due to their small size and extended CDRH3 loop, can rapidly penetrate tissue and access cryptic epitopes [40,41,42]. Such molecules are also easy to produce recombinantly without issues relating to inter-domain interactions, such as those found in scFvs and Fab fragments [43]. Although not of human origin and frequently ‘humanised’, Nbs are rarely immunogenic due to their small size and similarities with the human VH3 gene family [43].
Fusion of Nbs that bind different epitopes allows the creation of multivalent molecules with high affinity or potency. The monomeric behaviour, good solubility, and modular nature of Nbs also allow them to be easily fused to other Nbs with different antigen specificities or covalently linked to other molecules [44].
Unfortunately, the small size of Nbs is below renal cut-off, leading to rapid renal clearance with a half-life of approximately 2 h, making them particularly unsuitable for chronic use in many therapeutic areas [43,45]. However, the fast clearance is a beneficial property for in vivo diagnostics (see Section 5.1). Strategies to extend their half-life, including PEGylation, conjugation to the Fc domain of conventional antibodies, and coupling to abundant serum proteins such as human serum albumin (HSA), apolipoprotein L1, and β-Lactamase, have been explored. Fusion of Nbs to an anti-albumin VHH has been validated in the clinic [43]. Fusion of a VHH to the Fc of IgG has also been investigated as a means to produce bispecific tetravalent molecules [46].
The favourable characteristics of VHH domains has led to excitement over their potential. With 5 Nbs currently in clinical trials, and the recent approval of caplacizumab (CabliviTM), a bivalent VHH targeting von Willebrand factor (vWF), by the EMA in October 2018 and the FDA in February 2019 for thrombotic thrombocytopenic purpura and thrombosis, this excitement is not surprising [17].
Analogous to the camelid nanobodies is the single variable new antigen receptor (V-NAR), domain antibody fragments obtained from cartilaginous fishes such as sharks (Figure 7) [47]. Whilst V-NARs share some similarities in terms of their structure and properties with domain antibodies, they are smaller (11 kDa) and more stable proteins. Contrary to mammalian variable domains, V-NAR domains have only two complementarity determining regions, CDR1 and CDR3. However, they also contain two mutation-prone regions named HV2 and HV4; the latter has been shown to contribute to antigen binding [48,49]. As V-NARs are evolutionarily derived from a non-antibody lineage, this arguably places them outside the complex and competitive antibody patent landscape. The V-NAR format is being developed by Elasmogen (UK) under the name of soloMERsTM.
2.3.2. Domain Antibodies
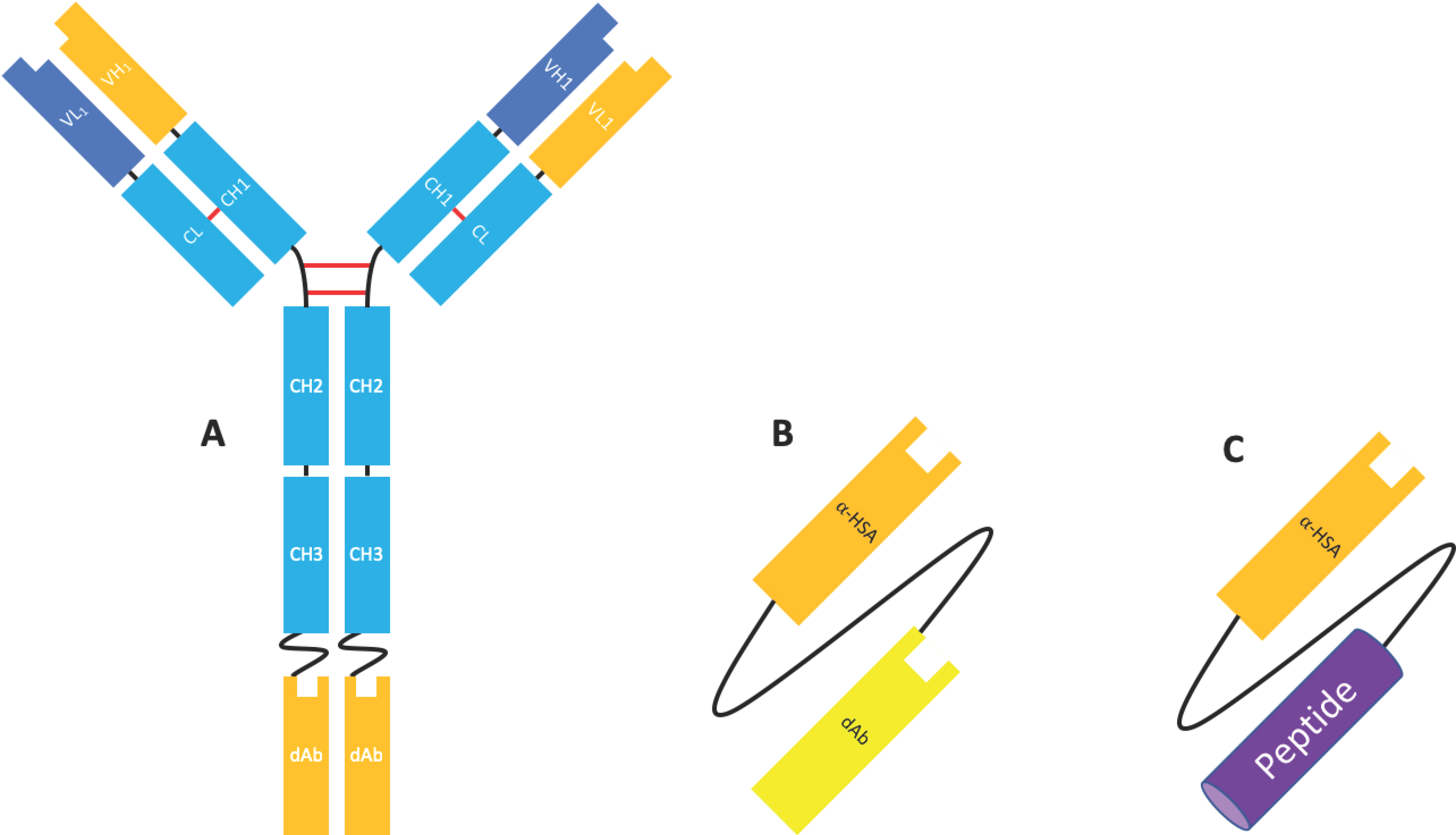
Domain antibodies (dAbs) are fully human unpaired variable domains (either VH or VL), which have been engineered to prevent dimerization whilst maintaining the specificity and affinity of a canonical antigen binding site (VH and VL). This engineering commonly involves ‘camelisation’, in which the hydrophobic residues usually found at the VH/VL interface are substituted for hydrophilic residues found in camelid VHH and in the extension of the CDRH3 [50,51]. Similar in size and structure, these molecules possess many of the advantageous properties of Nbs, including high thermostability, high solubility, and a short half-life and are amenable to conjugation/fusion and high-yield microbial expression [51]. Although naked dAbs may have some application as therapeutic molecules (see Section 4.1 and Section 4.2), dAbs have been most extensively investigated as fusion proteins to other moieties, such as full-length antibodies to enable specificity for a second antigen [18,52], an Fc domain (e.g., Placulumab), or with an anti-albumin dAb (e.g., GSK/Domantis’ AlbudAb®s) [53] (Figure 8).
Albudab®s (Figure 8) contain an anti-HSA binding domain which can increase the half-life of the species to 19 days, the half-life of HSA. GSK2374697 was an AlbudAb® developed for type 2 diabetes which phase I clinical trials were reached. It was composed of exendin-4, a GLP-1 mimetic peptide isolated from Gila monster saliva, linked to an anti-HSA dAb as a single transcript. The anti-HSA dAb extended the half-life of the peptide from 30 min to 6–10 days [53].
3. The Production of Antibody Fragments
3.1. Expression
Conventional full-length mAbs contain an N-linked glycosylation site in the CH2 domain of the heavy chain which is important for stability, preventing aggregation, and effector function [54]. Aberrant glycosylation can cause unfavourable molecular properties and be highly immunogenic. As such, the ability to produce post-translational modifications that closely resemble those that occur naturally necessitates expression in mammalian cell lines such as Chinese hamster ovary (CHO) cells, human embryonic kidney (HEK) cells, and to a lesser extent NS0 murine myeloma cells and human PER.C6TM cells [55].
Unlike full-length mAbs, antibody fragments are not generally glycosylated. Fragments are therefore amenable to production in microbial systems, allowing for faster, cheaper production in cell lines that are easier to cultivate and manipulate [10].
3.1.1. Escherichia coli
Escherichia coli was the first microbial system used for the production of biopharmaceuticals and is still used for the production of ~40% of biopharmaceuticals available today, including certolizumab pegol and ranibizumab [10,17]. As a prokaryote, E. coli’s rapid growth in inexpensive media, well understood genetics, and high manipulability make it an ideal expression system when glycosylation is not required.
Traditionally, the gene(s) of interest was placed on a self-replicating, high copy number plasmid under the control of a promoter such as the bacteriophage T7, lac operon (lac), or tryptophan (trp) promoter alongside a selectable marker. However, the greater volumetric productivity that can be achieved using this method is offset by the inhibition of cell growth and cell death caused by the metabolic burden of antibody fragment production. Recently, marker-free and plasmid-free expression systems have been developed to reduce this metabolic burden and allow for greater overall yields [10,56,57].
There are two main established approaches for the production of antibody fragments in E. coli, the first of which is to express the protein of interest in E. coli’s reducing cytoplasm. This method allows for high yields of protein, but the reducing conditions are not permissive for disulphide bond formation, and inclusion bodies are regularly formed. The protein must then be re-folded after purification which can be time-consuming, inefficient, and costly [10]. This issue can be somewhat alleviated by the co-expression of chaperones to facilitate correct protein folding [58]. The second pathway involves targeting, by fusion to an N-terminal leader peptide such as Pel B, the protein of interest to the oxidising periplasm, where disulphide bonds can readily form [59,60]. This second method does result in lower yields; therefore, if efficient re-folding of the antibody fragment is possible, cytoplasmic expression may be preferable.
Finally, due to the ability of E. coli to grow at high cell densities, antibody fragments are commonly produced in high cell density cultures grown in a stirred tank reactor using a fed-batch method [61].
3.1.2. Saccharomyces cerevisiae
Saccharomyces cerevisiae was the first yeast used as an expression system for recombinant proteins. Its high genomic stability, manipulability, and ease of cultivation have made it a strong choice for the expression of antibody fragments. Additionally, S. cerevisiae is used in a well-established production method of llama VHH fragments, consistently giving yields of hundreds of milligrams per litre [62].
There are three commonly used vector systems with S. cerevisiae. Firstly, a yeast episomal plasmid containing an origin of replication allows for gene expression with a high plasmid copy number without genomic integration [10]. Secondly, yeast centromeric plasmids containing a self-replicating sequence allow for gene expression with a single or low plasmid copy number without genomic integration [10]. Finally, yeast integrative plasmids do not contain an origin of replication but rather are incorporated into the yeast’s genome, leading to improved process quality and stability at the cost of expression levels [10,63]. However, methods have been developed to circumvent this issue such as targeted integration of the gene of interest at the ribosomal DNA locus, a highly-transcribed region [64]. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), alcohol dehydrogenase 1 (ADH1), and phosphoglycerate kinase 1 (PGK1) are promotors from S. cerevisiae’s native glycolytic pathway that are commonly used to achieve high expression levels, and methods have been developed to allow the co-expression of multiple genes on self-replicating plasmids making S. cerevisiae suitable for the expression of multi-gene fragments such as Fabs [65,66].
A major issue that plagues S. cerevisiae is endoplasmic reticulum (ER) misfolding and inefficient trafficking of the protein of interest leading to the accumulation of misfolded protein in the ER or vacuolar-like structures [10]. Although S. cerevisiae has proven to be an excellent expression system for VHH domains [41], this issue is particularly apparent with the more hydrophobic scFvs [67]; however, simultaneous overexpression of chaperones and foldases has been shown to facilitate scFv secretion [68].
3.1.3. Pichia pastoris
Pichia pastoris can be used as an alternative to S. cerevisiae and uses an integrated vector to achieve stable expression of the protein of interest.
P. pastoris metabolises methanol as its sole source of carbon and, as such, expresses large amounts of alcohol oxidase. The alcohol oxidase promoter was commonly used to express proteins of interest, but expression was hard to control. More adjustable promotors are now being investigated [70].
The genome of several strains of P. pastoris have been published online along with a genome scale metabolic model allowing for straightforward engineering and strain optimisation [71].
The preference of P. pastoris for respiratory over fermentative growth allows it to be grown to much higher cell densities than S. cerevisiae using inexpensive media, usually using a fed-batch method [72]. A P. pastoris system has been used to produce ALX-0171, a trimeric nanobody being developed for RSV infection [73], scFv-h3D6, an anti Aβ antibody fragment which is being developed for Alzheimer’s disease [9], and scFvTEG4-2c against platelet anti-αIIbβ3, for potential use as an imaging agent for atherosclerosis [74].
3.1.4. Cell-Free Expression Systems
There has recently been significant interest in cell-free production of antibody fragments. Approaches using E. coli cell lysates [75], CHO cell lysates [76], and insect cell lysates [77] have been successfully used to produce antibody fragments. Cell-free expression systems have been shown to be fast, reliable, flexible, and scalable [78]. Of note is the ability todirectly input linear DNA encoding the protein of interest rather than constructing a complex plasmid for transformation and the selection of transformed cells. Additionally, the lack of a phospholipid bilayer barrier allows forthe simple addition of resources required for the production of polypeptides, such as amino acids and ATP, and supplements, such as chaperones, the prokaryotic disulphide bond isomerase disulphide bond c (Dsbc), and oxidised/reduced glutathione rather than the co-expression or simultaneous over-expression required with cell-based systems.
Although not usually suitable for the commercial scale production of recombinant proteins, cell-free systems have been successfully used to produce functional scFvs [79] and more recently, using both prokaryotic and eukaryotic systems, functional Fabs [78,80]. Pioneered by companies like Sutro Biopharma, cell-free systems have been used to produce scFvs conjugated to moieties, such as granulocyte-monocyte colony stimulating factor (GM-CSF) and interleukin 1β-derived peptide [81] in a scalable manner, and can be used for the incorporation of non-canonical amino acids for site-directed conjugation, making cell-free expression an optimal system for protein engineering [82]. Such non-canonical amino acids are often toxic to cells or are unable to cross the cell membrane, making them difficult to incorporate whilst using cell-based systems. Although not currently a preferred method, the rise of antibody drug conjugates and improvements to cell-free expression systems may soon increase the desirability of cell-free expression systems for antibody fragment production. Sutro’s cell-free antibody production system was used to make STRO-001, a novel CD74 targeting antibody drug conjugate in phase I clinical trials for B-cell malignancies including multiple myeloma and non-Hodgkin’s lymphoma [83].
3.2. Enzymatic Cleavage
Although recombinant expression is the method most commonly used for the production of antibody fragments, fragments such as Fab, F(ab’)2, and Fc can be easily produced from their parent mAb by enzymatic cleavage using commercially available enzymes. Enzymatic cleavage can be preferable to recombinant expression for the production of certain fragment formats, such as F(ab’)2, which has a propensity to aggregate when expressed recombinantly due to the hinge region. This was originally done using papain, which cleaves just above of the hinge region to produce two Fab fragments and a hinge-CH2-CH3 fragment [84], or pepsin, which cleaves just below the hinge region to produce an F(ab’)2 and an Fc fragment [85]. Companies like Genovis now produce improved versions of these enzymes, allowing for a more efficient production of antibody fragments from the parental mAb [86].
3.3. Purification
The purification of antibody fragments is somewhat more complicated than the purification of full-sized mAbs, owing to the lack of an Fc domain which facilitates efficient purification by Protein A or Protein G affinity chromatography [87,88]. However, VH 3 family containing fragments can be purified using Protein A [89]. Currently, there are no ‘toolbox’ or generic approaches to the production of pharmaceutical antibody fragments, with the current approved fragment therapies being purified using different combinations of chromatographic and non-chromatographic techniques [90]. In theory, an antibody fragment could be captured selectively using its antigen fixed to a resin. Whilst this may be used in some cases, antigens are not always readily available and prior production, purification, and fixation of said antigen would have a significant impact on the cost of goods.
The unique nature of fragments and the lack of large conserved regions pose an additional challenge to the development of generic purification approaches. To this end, micro-fluidic approaches have been tested to quickly determine the ideal binding conditions of specific fragments to inform future purification attempts [91].
If the expression system used expresses the fragment in the cytoplasm or periplasm rather than secreting it, the fragments are first freed by lysing the cells, and proteins are then refolded if necessary. Once this has been completed, the fragment can be purified using one or more of the methods described in the following sections.
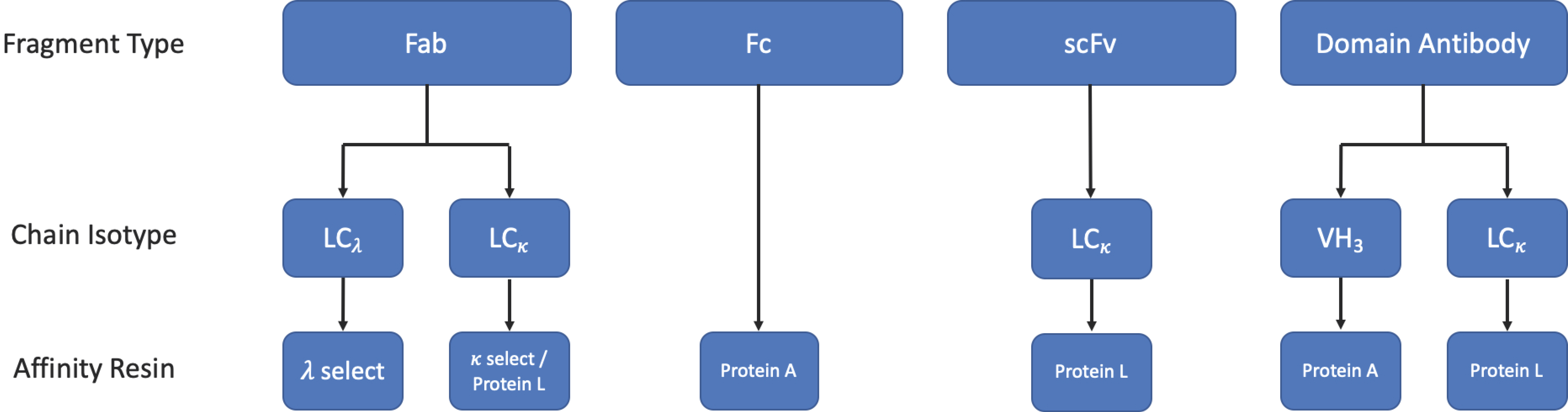
A simplified workflow for the purification of applicable fragments by affinity chromatography is shown in Figure 9.
3.3.1. Protein L Affinity Chromatography
Protein L is a cell wall-associated protein isolated from Peptostreptococcus magnus [92], which binds strongly to human Ig LCs, scFv, and Fab fragments [93]. Protein L targets a site that lies within the variable region of κ 1, 3, or 4 light chains, allowing it to capture a wide range of mAbs and fragment formats. However, should the fragment of interest be derived from a λ mAb or from the κ 2 sub-family, protein L would be unable to capture the fragment [93]. In cases such as these, commercially available κ- and λ-select resins may be suitable alternatives.
3.3.2. Affinity Tags
As antibody fragments are most commonly produced recombinantly, they can easily be generated with affinity tags such as hexa-histidine (6HIS), glutathione-S transferase (GST), or mannose binding protein (MBP) using a cleavable linker to allow purification by immobilised metal affinity chromatography (IMAC) or other affinity-based methods [94]. Such techniques would allow for the selective capture of the desired fragment regardless of the Ig germline family. However, affinity tags are not a blanket solution. Removal of the affinity tag by proteolytic cleavage may leave residual amino acids, which may cause issues with aggregation, misfolding, and immunogenicity. It is also important to consider the host’s cellular proteases when designing the linker to avoid premature cleavage and production of irrecoverable material [95,96]. Additionally, a study carried out by Das et al. showed that Protein L affinity chromatography, where applicable, was a more robust and versatile method for the purification of scFvs than IMAC using a (6HIS) tag [97].
3.3.3. Other Chromatographic Methods
As well as the affinity-based methods described above, other chromatographic methods such as size exclusion chromatography, ion exchange chromatography, and multi-modal chromatography are used to separate the desired fragment from contaminants based on size and isoelectric point [98,99]. Ion exchange chromatography usually takes the form of cation-exchange chromatography (CIEX) and has the added advantage of being able to separate charge variants [100]. In order to achieve the high purity required for pharmaceutical applications, these chromatographic methods are used in conjunction with each other, with affinity-based methods, and/or with non-chromatographic methods [90]. More recently, multi-modal approaches have been used in the purification of Fab fragments and have been shown to be superior to traditional CIEX resins due to increased salt tolerance and Fab binding [91,101].
4. Antibody Fragments in the Clinic
4.1. Oncology
The majority of antibody fragments currently being developed in the clinic are for oncological applications. In addition to the generic characteristics of antibody fragments that make them attractive as immunotherapies, e.g., their small size, which grants them superior tissue and tumour penetration compared to a conventional mAb [12], and the lack of an Fc domain that reduces non-specific activation of innate immune cells, there are many mechanisms of action that are unique to a specific format. The diversity of formats being investigated for their therapeutic potential in oncology is astounding, but the majority of fragments are reformatted as bispecific molecules combining an anti-CD3 binding moiety with an anti-tumour binding domain. For more detailed information, the reader is referred to two recent excellent reviews by Wu et al. and Velasquez et al. [102,103]. Here we will only cover the more established formats and describe the over-arching pathways that they exploit.
4.1.1. BiTE®s
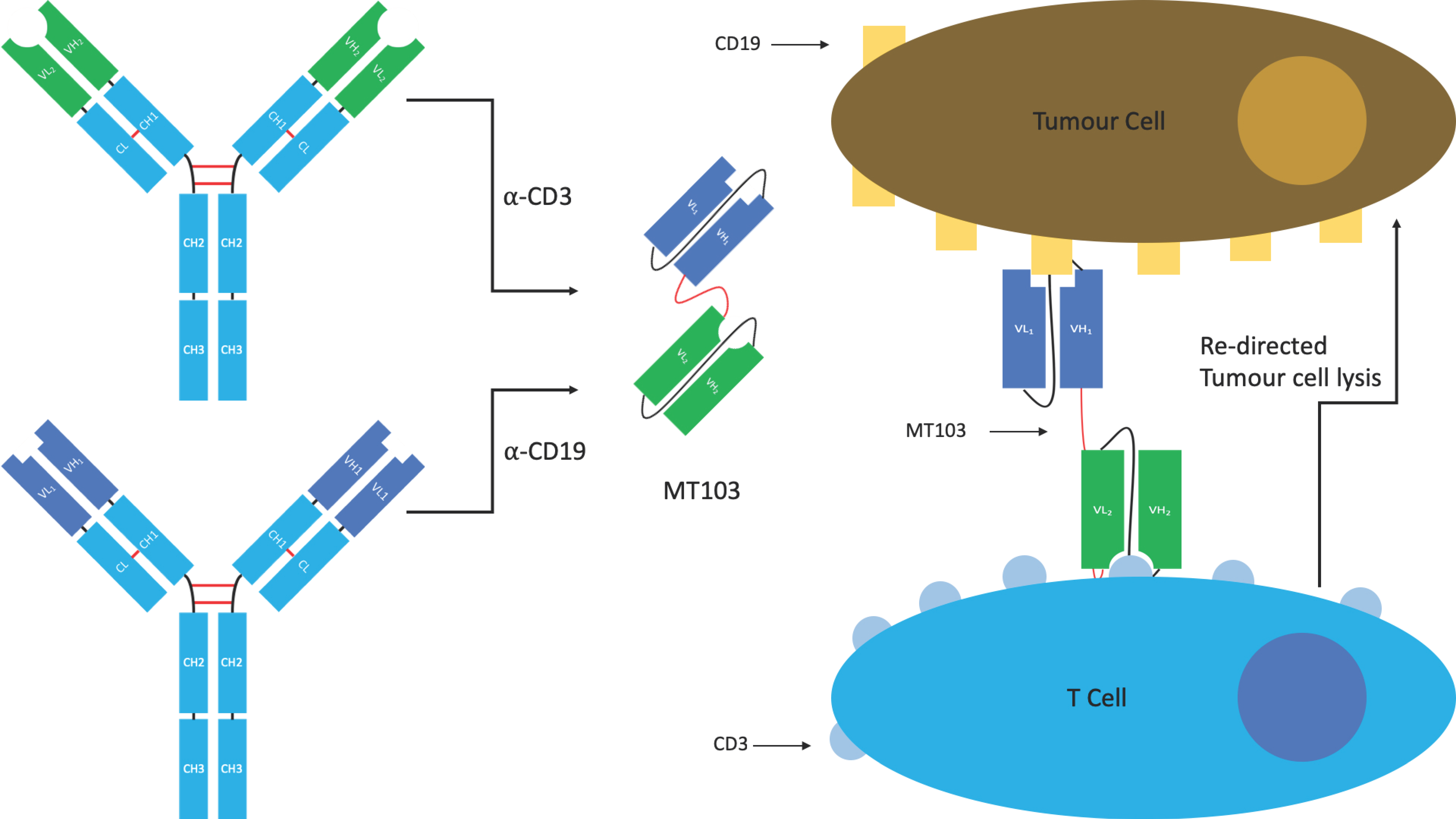
Bispecific T-cell engagers (BiTE®s) originally developed by Micromet, are a specific class of tandem scFv used to redirect cytotoxic T-cells to tumours. The induced response is highly selective for the target tumour cells, more so than can be achieved by radio- or chemotherapy. The hope is that this selectivity will lead to reduced off-target effects (Figure 10) [26].
BiTE®s contain two antigen binding sites. The first is directed against a tumour antigen, whilst the second is directed against the T-cell receptor (TCR) signalling complex CD3. Simultaneous binding of the BiTE® to CD3 and the tumour antigen (e.g., CD19) bypasses pMHC restriction and induces T-cell activation, cytokine production, the formation of cytolytic immunological synapses leading to a tumour-directed cytotoxic response, and the activation of other host immune responses [26,104]. The use of a monovalent anti-CD3 is thought to be important in limiting off-target immune activating functions that can lead to cytokine release syndrome and cytokine storm, a problem seen with some of the early anti-CD3 mAbs in the clinic such as muromonab (OKT3), which eventually led to its withdrawal.
There is currently one licenced BiTE® which received FDA approval in 2017. Blinatumomab (Blincyto®), developed by Amgen and Astellas Pharma Inc., is an anti-CD19/CD3 BiTE®, used for the treatment of Non-Hodgkin’s lymphoma and Philadelphia chromosome negative acute lymphoblastic leukaemia. It is also in clinical trials for a number of additional indications. Other tumour antigens are currently being trialled as targets for anti-CD3 containing BiTE®s including BCMA, CD33, CEA, HER2, EGFR, and EpCAM [26,102].
4.1.2. BiKEs & TriKEs
Bispecific killer cell engagers (BiKEs) and trispecific killer cell engagers (TriKEs) are bi- or tri- specific tandem scFvs used to redirect natural killer (NK) cells via an anti-CD16 scFv. They work in a similar fashion to BiTE®s. Although there are currently no BiKEs or TriKEs in clinical trials, BiKEs are currently being developed by Sanofi in collaboration with Innate Pharma using Innate Pharma’s anti-CD335 (NKp46) antibody to redirect NK cells [105].
Anti-CD16/CD33 BiKEs showed promise in an in vitro study treating myelodysplastic syndrome (MDS) [106]. Here, the anti-CD16 scFv was used to activate depleted NK cells which were redirected against myeloid-derived suppressor cells (MDSCs) expressing CD33 by the anti-CD33 scFv. The treatment was shown to reduce the immunosuppression of NK cells by MDSCs, induce MDSC cell lysis, and induce optimal MDS-NK cell function regardless of disease stage.
More recently, an anti-CD16-IL-15-anti-CD33 TriKE was also shown to overcome the cancer induced immunosuppression observed in MDS and AML [107].
4.1.3. DART®s
Developed by MacroGenics, the DART® platform has been used to produce five anti-cancer molecules, four of which entered phase I clinical trials for the treatment of AML/MDS, solid tumours, or colorectal cancer [24].
The DART® platform is compatible with several modalities. Of the five anti-cancer DARTs listed, three include an anti-CD3 binding moiety to re-direct T-cells towards cells expressing the cancer antigen complimentary to the second binding site in the same fashion as a BiTE®. The other two DARTs, which target PD-1/LAG3 or PD-1/CTLA-4, block pathways involved in T-cell inhibition leading to an enhanced T-cell response against tumour cells [108].
Although BiTE®s and DART®s can exploit the same pathways, a 2011 study comparing a CD19/CD3 bispecific molecule in DART® and BiTE® formats showed increased potency of the DART® compared to the BiTE®, which was not accompanied by an increase in non-specific T-cell activation of CD19- cell lysis in vitro [109]. The study also trialled an anti- CD19/TCR DART®, which showed activity in an in vivo xenograft mouse model and was virtually identical in vitro to the anti-CD19/CD3 DART®, describing another potential mechanism of action for the DART® platform.
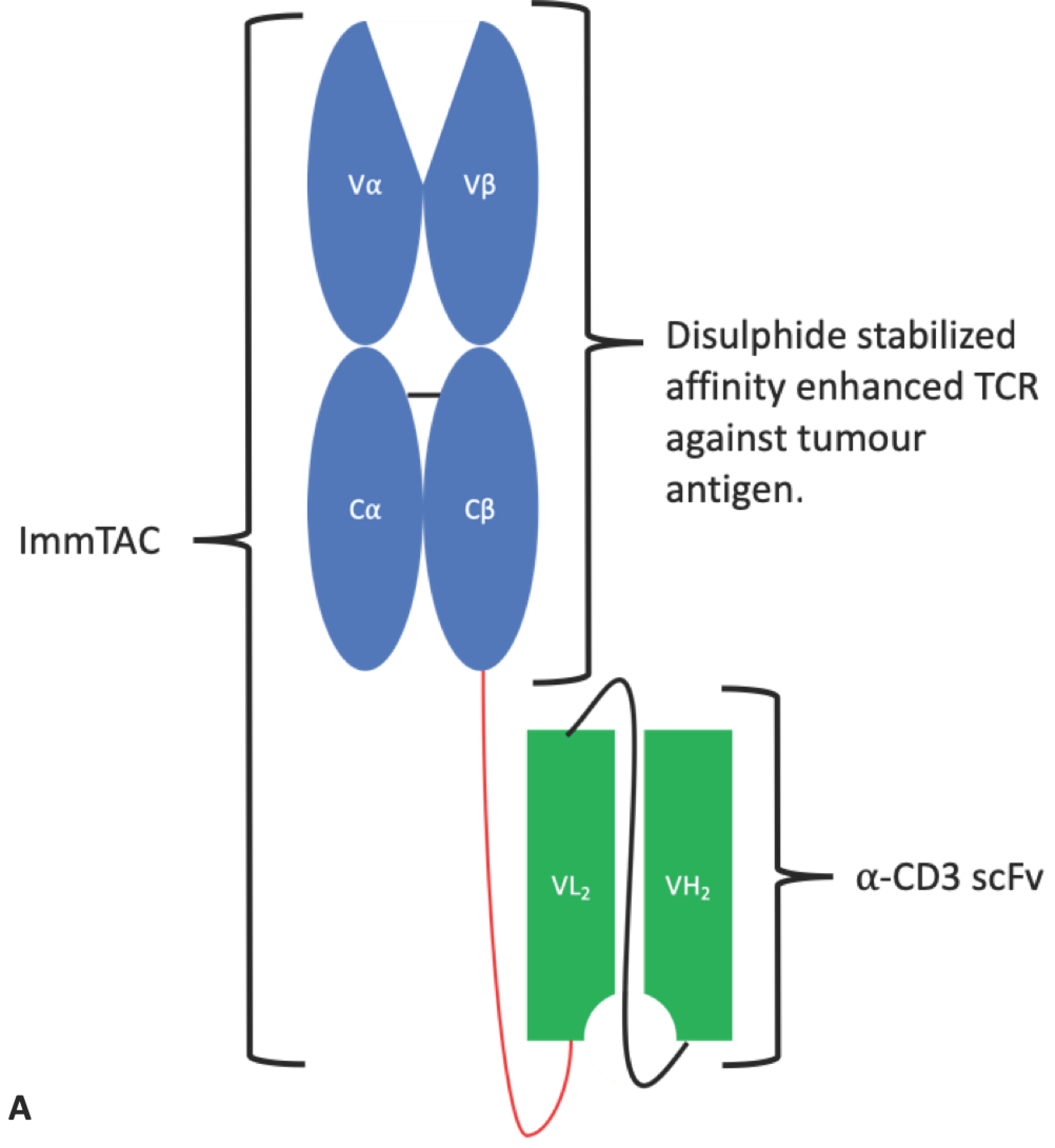
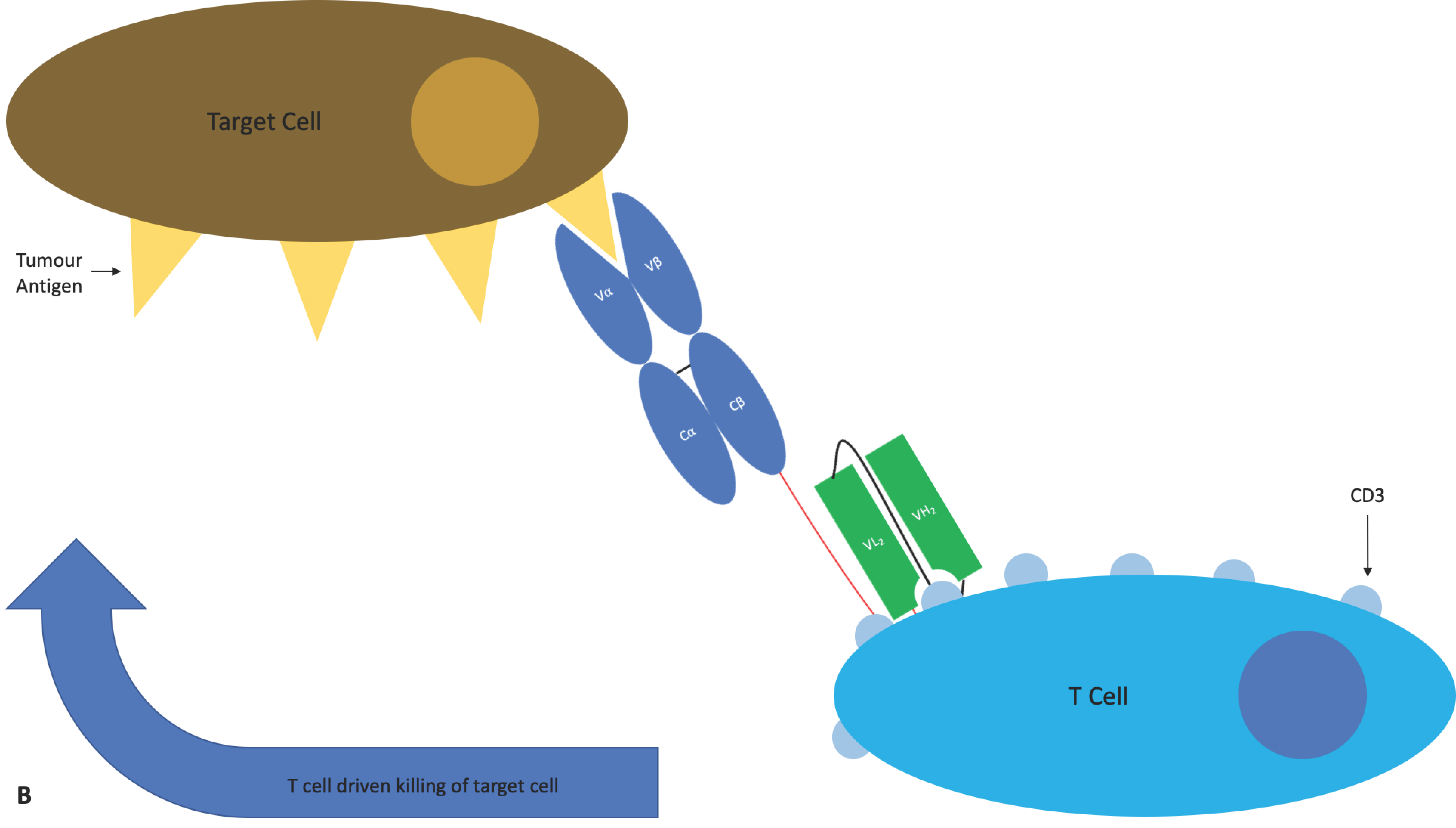
4.1.4. ImmTAC®s
The immune mobilising monoclonal T-cell receptors against cancer (ImmTAC®) is a novel scFv-TCR chimeric format developed by Immunocore. ImmTAC®s are peptide-HLA-specific, dimeric affinity-enhanced soluble TCRs containing an artificial disulphide bond, joined by a peptide linker to an anti-CD3 scFv (Figure 11). The TCR portion binds with picomolar affinity to the target cell expressing the peptide antigen in the context of MHC. The anti-CD3 portion then binds with nanomolar affinity to passing T-cells that are recruited to kill the target cell. The binding of multiple CD3 surface proteins by multiple ImmTAC®s on a single T-cell causes T-cell activation leading to an immune response against the target cell/tissue [110]. Importantly, ImmTAC®s hold the potential to overcome T-cell tolerance and the low affinity of native TCRs for cancer antigen/MHC complexes and mediate an enduring immune response [111]. An ImmTAC® targeting gp100 is being tested in two phase I trials for uveal melanoma. An ImmTAC® against NY ESO entered phase I clinical trials in 2018 for a variety of cancer indications, and IMC-C101C against melanoma associated antigen 4 is planned to enter phase I in 2019 [112].
As a side note, immune-mobilising monoclonal T-cell receptors against virus antigens (ImmTAV®s) are similar to ImmTAC®s in that they are composed of an affinity-enhanced soluble TCR linked to an anti-CD3 scFv. However, as the name would suggest, the TCR is designed to bind specific viral antigens as opposed to cancer antigens. These molecules are being investigated as a novel class of HIV therapy [113].
4.1.5. Nanobodies®
Two nanobodies reached phase I clinical trials for oncology indications: ALX-0651, which targets CXCR4 for multiple myeloma and non-Hodgkin’s lymphoma, and TAS266 (Ablynx/Novartis), which targets the death receptor DR5 for solid tumours. However, their development has not been pursued [44]. On the contrary, there are a large number of nanobodies in preclinical development for a variety of other cancers. The reader is referred to the excellent review by Steeland et al. (2016) [44].
4.1.6. Antibody Fragment-Drug Conjugates
In addition to PEGylation or fusion to albumin binding antibody fragments to improve half-life and pharmacokinetics, a wide range of effector moieties, including cellular toxins, radioisotopes, cytokines, and enzymes, have been conjugated to Fab, scFv, and Nb fragments. In 2010, there were 12 scFv and 12 Fab conjugates in clinical trials worldwide [4,40,114,115]. Citatuzumab bogatox, an anti-epithelial cell adhesion molecule (EpCAM) Fab conjugated to bouganin, developed by Viventia Biotechnologies Inc., is currently in phase I clinical trials for the treatment of solid tumours. Naptumomab estafenatox, an anti-trophoblast glycoprotein 5T4 (TBGP) that Fab fused to Staphylococcus aureus enterotoxin E, developed by Active Biotech, is currently in phase III clinical trials for renal cell carcinoma and non-small cell lung carcinoma [17].
Conjugation to cytokines is also an effective way of enhancing anti-tumour efficacy. Philogen (Switzerland) are developing a number of immunocytokines for oncology indications including Fibromun, a scFv (L19) against the tumour antigen EDB fused to TNF, Darleukin, which contains L19 scFv fused to IL-2, Teleukin, which contains a vascular targeting antibody F16 linked to IL-2, and Dodekin which contains two subunits of the immunomodulatory payload IL-12 fused to a human vascular targeting antibody in tandem diabody format [116].
Radioimmunotherapy (RIT) is the combination of radiation therapy with Ab immunotherapy and has become an attractive strategy in cancer treatment because it allows for the selective destruction of cancer cells and is less pervasive than radiotherapy. The Ab recognises and binds the surface of the primary tumour site and disseminated disease tissue and thereby delivers high doses of radiation directly to the tumour without significant damage to healthy tissue. Recent examples are the generation of radiolabelled antibodies for the radioimmunotherapy of multiple myeloma [117] and radio-iodinated anti-HER2 Nanobody® for breast cancer [118].
Although the effector moieties add another mechanism through which the antibody fragments can mediate a therapeutic effect, antibody drug conjugates are not easy to develop and optimise. Many factors need to be considered, including what antibody/fragment is used, what to conjugate, what linker/chemistry to use, and the ratio of naked to conjugated antibodies [119,120].
4.2. Autoimmune and Inflammatory Diseases
Autoimmune diseases are chronic and potentially life-threatening, and antibody therapies are extremely expensive because they usually require intensive, life-long treatment. The lower production costs of antibody fragments and potential reduced immunogenicity due to their small size renders the use of antibody fragments with half-life extension moieties as a viable alternative to full-length antibodies. Furthermore, like for cancer immunotherapies, the development of antibody fragments for the treatment of autoimmune diseases has been growing at a fast pace and there are numerous possibilities for bispecific targeting.
One of the first antibody fragments to be marketed for an autoimmune disease indication was Certolizumab pegol (Cimzia®), a pegylated Fab targeting TNF developed by UCB (Belgium), approved by the FDA for the treatment of Crohn’s disease in 2008. It has subsequently been approved for rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. Two other Fabs are in clinical trials: FR104 (OSE/Janssen) against CD28 in phase II for RA, and Dapirolizumab, an anti-CD40L Fab developed by UCB in phase II for SLE.
There are four nanobodies reported to be in clinical development: ALX-0061 (Vobarilizumab) is a monovalent Nanobody® against IL6R linked to a half-life extending Nb against HSA from Ablynx in phase II trials for RA and SLE [121,122]. ALX-0761 (Merck/Ablynx) is a bispecific Nanobody® targeting IL17A/IL17F linked to an HSA Nb in phase Ib for psoriasis [123], and Ozoralizumab or ATN-103 (Taisho) is a Nanobody® against TNF. Some success has been reported for ATN-103 in a phase II interventional long-term safety study in subjects with RA at week 48 [124]. ATN-192 is a pegylated version of ATN-103, which is in phase I clinical trials [125]. Several Nbs are also reported in pre-clinical development for autoimmune disease indications [44].
One scFv format currently being evaluated in a phase II clinical study for the treatment of RA is Dekavil or F8IL10 (Philogen). It is a fully human fusion protein composed of the vascular targeting scFv antibody F8 fused to the cytokine interleukin-10. A number of other immunocytokines fused to scFvs are also in preclinical development—the reader is referred to the Philogen website for further information [126].
MacroGenics are developing MGD-010, a DART® targeting CD32B and CD79B on B-cells for the treatment of autoimmune disorders. CD32B is a checkpoint molecule expressed on B lymphocytes that, when co-ligated with CD79B (a component of the B-cell antigen receptor complex), delivers a co-inhibitory signal that dampens B-cell activation. The intended mechanism of MGD010 is to modulate the function of human B-cells while avoiding their depletion. MGD010 completed phase I clinical trials in 2016 and has now been licensed to PreventionBio, who will evaluate the safety and efficacy of MGD-010, now called PRV-3279, in a phase Ib trial, expected to commence in the second half of 2019.
Another fragment in development for a wide variety of autoimmune disease indications is ARGX-113. This is an IgG1 Fc-fragment developed by ArGenX for the treatment of patients with high levels of circulating pathogenic IgG, found in acquired thrombotic thrombocytopenic purpura (aTTP), SLE, MS, or myasthenia gravis (MG). ARGX-113 binds to the neonatal Fc receptor (FcRn) and blocks IgG recycling, resulting in clearance of autoreactive antibodies through lysosomal degradation. ARGX-113 showed statistically significant improvement in a phase II clinical trial on patients with MG [127] and is currently being evaluated for efficacy, safety, and tolerability in a randomised, double-blind, placebo-controlled, multicentre phase III trial in patients with MG having generalised muscle weakness [128].
The last category of antibody fragments tested in clinical trials for autoimmune and inflammatory diseases are dAbs. Lulizumab pegol, a pegylated Domain Antibody® targeting CD28 developed by Bristol-Myers Squibb, was evaluated in a phase II trial in subjects with active systemic lupus erythematosus (SLE). There was no significant difference between lulizumab and placebo for the primary (BICLA response rate) or secondary endpoints at week 24, although PD activity was observed [129].
GSK has developed two inhalable anti-TNFR1 VH domain antibodies for selective antagonism of TNF in the lung interstitium for acute respiratory distress syndrome (ARDS)/acute lung inflammation (ALI). Delivery of antibody fragments directly into the lung by inhalation has great potential for treatment of inflammatory lung diseases, the advantages being rapid onset of action, reduced systemic exposure, lower doses, as well as needle-less administration. GSK1995057 was tested in a phase I clinical trial in healthy volunteers. It was not developed further because of the presence of naturally occurring, pre-existing anti-drug antibodies (ADAs) which could lead to early neutralising anti-drug-antibody responses [130]. GSK1995057 was subsequently engineered by adding a C-terminal alanine residue to render it less susceptible to ADAs. The resultant GSK2862277 was tested in a phase I trial and was well tolerated when administered both as an orally inhaled aerosol and by iv route. A phase II placebo-controlled randomised trial in patients that were undergoing esophagectomy surgery and were at risk to develop ARDS has been completed [131] with results expected in 2019.
4.3. Other Clinical Applications
While oncology and autoimmune disease are two major areas in which antibody fragments have become a prominent class of therapeutic molecules, there are several other disease areas in which, although not as dominant, fragments are being evaluated.
4.3.1. Ophthalmic Indications
Antibody fragments such as Fabs and scFvs, unlike full-length antibodies, have been shown to be able to penetrate the cornea and pass into the eye and achieve clinically useful concentrations in the anterior chamber over a reasonable time-span following topical administration [132] but to date there are no reports of this route of administration being tested in the clinic. Most are administered by direct injection into the eye (intravitreal route).
The most common eye disorder treated with antibodies or antibody fragments is age-related macular degeneration (AMD), which is the leading cause of irreversible blindness in people aged 50 years or older, in the developed world. For AMD, the antibody fragments are applied directly to the eye via the intravitreal route. Extremely high local drug concentrations can be achieved in the eye with minimal risk of systemic side effects.
Ranibizumab (Lucentis®) is an anti-angiogenic monoclonal antibody fragment targeting VEGF-A, derived from the same parental mouse antibody as bevacizumab. It was approved in 2006 for wet AMD and subsequently in 2012 and 2015 for diabetic macular oedema and diabetic retinopathy, respectively.
Lampalizumab (Roche), a Fab against complement factor D, entered phase III clinical trials for geographic atrophy, an advanced form of age-related macular degeneration [133,134] in 2014 but failed to meet primary endpoints [135].
Brolucizumab (Alcon/Novartis) is a scFv targeting VEGF that is currently in phase III for wet AMD [136,137].
A number of antibody fragments are also in preclinical development for eye indications [138,139]. Elasmogen is developing V-NARs such as ELN/21, an ICOSL G-binding soloMERTM, in preclinical development for posterior uveitis and corneal graft rejection, and ELN/12, an anti-VEGF soloMERTM for AMD [140]. Abzyme has a bivalent nanobody targeting VEGF and TfR for wet AMD.
4.3.2. Infectious Diseases
Only three full-length mAbs have been approved for the treatment of infectious diseases: Synagis® (Palivizumab) for RSV infection, Abthrax® (Raxibacumab) against anthrax, and ZinplavaTM (Bezlotoxumab) against C. difficile (although technically the latter two mAbs neutralise bacterial toxins—see below) and there are currently over 60 mAbs in various stages of clinical trials for the treatment of infectious diseases including Ebola, hepatitis B, and respiratory syncytial virus (RSV). The development of antibody fragments in infectious diseases is under-exploited, likely due to their lack of effector function. To our knowledge, there are only three molecules currently in clinical trials and no approved therapies. Afelimomab is an F(ab’)2 in phase III trials for sepsis toxic shock [17]. Rivabazumab pegol is a pegylated Fab in phase II for the treatment of chronic Pseudomonas aeruginosa infection [17]. The third is ALX-0171, a trivalent Nanobody® (VHH3) in phase III for the treatment of RSV infection [17]. ALX-0171 binds to RSV F protein. The potency of the trivalent ALX-0171 against RSV-A and RSV-B strains was found to be several thousand-fold higher than that of the monovalent nanobody. It is also the first Nanobody® treatment developed for delivery directly into the lungs, the site of RSV infection, by nebulisation [141]. Unfortunately, Sanofi decided to stop development ALX-0171 in Feb 2019 [142].
Although ALX-0171 remains the only Nb to reach clinical trials in this therapy area, there have been many in vivo and in vitro studies investigating the use of nanobodies against a wide range of bacteria, viruses, parasites, and fungi, including rotavirus, norovirus, HIV-1, Helicobacter pylori, Trypanosoma brucei, and Plasmodium falciparum. The reader is directed to two recent reviews by Steeland et al. and Wilken and McPherson [44,141].
4.3.3. Anti-Toxins and Anti-Venoms
Traditionally, the treatment for envenoming has been the transfusion of serum from immunised animals. Primarily containing IgG, Fab and F(ab’)2 fragments, much of which will not be specific for the venom, serotherapy can have several undesirable affects including IgE-mediated and non-IgE-mediated early adverse reactions, anaphylaxis and serum sickness [143]. The ability of antibody fragments to rapidly penetrate tissue, their lack of effector function, and their retained specificity and affinity for their antigen makes them promising candidates for anti-venoms. In addition, they can easily be produced recombinantly in a homogeneous form.
Although none have progressed to the clinic as yet, many Nbs have been generated that have shown the ability to neutralise toxins/venoms in in vitro and in vivo models. Venoms from Androctonus australis (Fat tailed scorpion), Hemiscorpius lepturus (Iranian scorpion), and Naja kaouthia (monocled cobra) and toxins from Clostridium difficile, Vibrio cholera, Bacillus anthracis, and E. coli, including Shiga toxins 1 and 2, have all been neutralised [44,144].
5. Non-Therapeutic Uses
Imaging & Diagnostics
The use of antibodies for molecular imaging is well established. In essence, their high affinity and specificity make them ideal for the detection of a specific surface protein in vivo or in vitro. Additionally, their large size allows for their conjugation to radioisotopes, fluorescent molecules, or even enzymes without inhibiting binding to their target [145].
However, conventional antibodies are by no means perfect for in vivo imaging. Their long half-lives and low rates of clearance necessitates a several-day waiting period to obtain an acceptable signal-to-noise ratio, exposing patients to excessive radiation from radioisotopes. In addition, potential off-target immune effects, conferred by the Fc domain, are undesirable. Antibody fragments, either produced recombinantly or by enzymatic cleavage, provide a solution to these downsides. Fragments lack an Fc domain, thus removing their immune activating potential and reducing their half-life simultaneously. This reduces the risk of disturbing the system being visualised, allows for rapid high-contrast imaging, and reduces radiation exposure [145]. In addition, their small size allows for better tissue distribution and provides more options with regard to epitope choice [12]. Nanobodies targeting CAIX and HER2 have been used for optical imaging of pre-invasive breast cancer, which requires a high tumour to background ratio [146,147].
6. Future Opportunities
6.1. Neurodegenerative Diseases
Antibody therapies have traditionally been thought to be of limited relevance in the treatment of neurodegenerative disease due to the miniscule proportion of antibodies in circulation that can cross the blood–brain barrier (BBB) [153]. The four monoclonal antibodies approved for the treatment of multiple sclerosis—natalizumab (Tysabri®), alemtuzumab (Lemtrada®), daclizumab (Zinbryta®), and ocrelizumab (Ocrevus®)—are thought to mediate their effects primarily in the periphery [154]. There are also two antibodies against alpha synuclein (PRX002/RO7046015 from Roche and BIIB-054 from Biogen) entering clinical trials for Parkinson’s disease. No antibody fragments have yet progressed into the clinic.
The causal mechanism of many neurodegenerative diseases, including Alzheimer’s and Huntington’s disease, involves aggregation of misfolded protein [155], and it has been shown that it is possible to raise antibodies that can neutralise these toxic aggregates. One possible approach to circumvent the BBB challenge is to express antibody fragments, termed intrabodies, within the cells of the brain. Although this approach may be far from reaching clinical trials, partially due to its invasive nature, it has been shown to be efficacious in some in vivo models [156]. Penetration of the BBB is discussed in Section 6.2.
6.2. Cell and Tissue Specific Antibody Delivery
Bispecific antibodies where one specificity is used to target the antibody to a specific tissue or cellular compartment and the second specificity is used to target the antigen of interest would have great advantages in limiting off-target effects due to systemic administration. Conditions in which the target of interest is located in the central nervous system (CNS) are particularly challenging, however, as most antibodies are generally unable to penetrate the BBB. Receptor-mediated transcytosis (RMT) is an example of a macromolecule transport system that is employed by cells of the BBB to supply essential proteins to the brain. This system can be utilised to deliver biologic payloads, such as antibodies, across the BBB. Increased brain penetration of therapeutic antibodies can be achieved by engineering bispecific antibodies in which one antibody binding specificity recognises a BBB receptor that undergoes RMT from the circulatory compartment into brain parenchyma, and the second binding specificity recognises a therapeutic target within the CNS. Anti-transferrin receptor (TfR)-based bispecific antibodies have previously shown promise for boosting antibody uptake in the brain [157]. Abzyme Therapeutics are now exploiting modular anti-TfR antibodies (nanobodies) and TFR-directed bispecifics capable of overcoming the blood–brain barrier for treatment of CNS disorders [158].
Some fragment formats, including F(ab’)2 and basic VHHs, have been demonstrated to cross the BBB with relative efficiency, although they may not necessarily accumulate to therapeutic concentration due to rapid clearance [159,160,161]. Engineering these formats to bind key proteins in the causal mechanism of neurodegenerative diseases will likely be challenging, but their existence shows the significant progress that has been made in recent years. In 2001, Muruganandam et al. identified two single-domain antibody fragments of camelid origin capable of crossing the BBB endothelium, FC5 and FC44, by phenotypic panning of a naive llama single-domain antibody phage display library [162]. Recently, Farrington et al. engineered FC5 as a mono- and bivalent fusion with the human Fc domain and showed up to a 30-fold enhanced apparent brain exposure (derived from serum and cerebrospinal fluid pharmacokinetic profiles) compared with control domain antibody-Fc fusions after systemic dosing in rats [163]. This study demonstrates that modular incorporation of FC5 as the BBB carrier arm in bispecific antibodies or antibody-drug conjugates may have potential use in the development of pharmacologically active biotherapeutics for CNS indications. As an alternative, Caljon et al. suggested grafting CDR loops of BBB penetrating Nbs onto an as-yet undiscovered scaffold to combine BBB penetration and high antigen specificity with the desired pharmacokinetic properties [161].
6.3. Intracellular Targeting
Antibodies have proven to be effective at modulating a wide variety of disease associated molecules belonging to different target classes, but there are still hundreds of disease-associated intracellular targets that are inaccessible to antibodies and undruggable with small molecules and that include phosphatases, E3 ubiquitin ligases, GTPases, and transcription factors. The majority of antibodies are unable to penetrate into cells. Thus, while small molecules drugs can easily penetrate cell membranes to hit intracellular targets, they often lack specificity, in particular when multiple targets have similar binding pockets. Furthermore, their small size makes them ineffective at blocking certain protein–protein interactions where large interfaces are involved. Antibodies or antibody fragments can solve the specificity problem and effectively block protein–protein interactions, but they have been largely restricted to targets in the extracellular milieu because they cannot cross the lipid bilayer. It has therefore been the ‘holy grail’ of many pharmaceutical companies to combine the targeting power of monoclonal antibodies with the cell-penetrating abilities of small molecules. Even if cell penetration can be achieved, a further complication is the need for tissue and cellular specificity to limit off-target effects, although one could argue that cell-specific action can be reached on the intracellular antigen level.
A number of different approaches have been described in the literature including transfection, cell penetrating peptides (CPPs), fragments of bacterial toxins, nanocarriers (lipid-based, polymer-based, and virus- and virus-like particle-based), and physical methods such as microinjection and electroporation and have recently been extensively reviewed [164]. Below we briefly describe some of the most promising approaches that could be applicable for the systemic delivery of antibody fragments.
A cell-penetrating, intracellular targeting antibody needs to bind to a receptor on the cell surface and trigger internalization, for example, by receptor-mediated endocytosis. Once internalised, the antibody needs to be able to escape from the endosome in order to bind to/neutralise its intracellular target in the cytosol, where the majority of potential therapeutic targets are concentrated, or in the nucleus.
A frequently used approach for intracellular delivery is the fusion of an antibody or antibody fragments including scFv, Fab, and nanobody to a CPP. Early studies showed that HIV tat and other CPPs such as the membrane translocating sequence from Kaposi fibroblast growth factor, the Antennapedia protein transduction domain, the Penetration of the Drosophila homeodomain, nona-arginine, and certain oligonucleotides could cross the plasma membrane to enter cells. However, their mechanism of intracellular delivery was unclear, and it is unknown if/how CPPs and CPP-conjugated antibody fragments are released from endocytotic vesicles, making efficient endosomal escape one of the limitations of this approach. Other limitations include the lack of optimisation for intracellular localization and the lack of cell or tissue-specific targeting. The latter may be solved by addition of a cell-targeting ligand to the antibody fragment. This was elegantly demonstrated by the authors in [165], who fused a cancer-cell specific, 17-amino acid peptide (BR2) to anti-mutated K-ras scFv, and demonstrated significant and cancer-cell-selective effects in vitro.
Most currently used CPPs seem to contain a disproportionately high number of positively charged lysine and/or arginine residues and have a high theoretical net charge. These positively charged residues facilitate interaction with negatively charged cell surface proteoglycans, such as sulfated proteoglycans like HSPG, ultimately enabling cellular uptake. These findings suggested that polycationic protein resurfacing could endow cell penetration properties, the downside being that extensive mutation of most proteins would lead to a loss of functional activity. To this end, Bruce et al. recently showed that nanobodies are amenable to cationic resurfacing [166]. Structural analysis of a GFP-binding nanobody revealed a number of solvent exposed residues that were not within the CDRs. Polycationic resurfacing of these solvent exposed residues resulted in a new protein that expressed well in E. coli, retained affinity for GFP, and penetrates mammalian cells. Analogous mutation of HER2-or β-lactamase-binding nanobodies also resulted in well-expressed nanobodies that exhibited potent cell-penetrating properties, and the majority of the internalised proteins were found to reside in the cytosol, although the mechanism of uptake remains unclear.
Probably one of the most exciting recent technology developments in the field of intracellular targeting has come from Orum Therapeutics (South Korea). Orum have developed a cell-penetrating antibody technology that uses a cell surface receptor-specific cyclic peptide fused to an antibody targeting activated K-ras that has been engineered from a naturally occurring autoimmune antibody able to penetrate into cells. The peptide, which confers cell-type specificity, is genetically linked to the light chain variable domain of the antibody. The light chain contains a sequence motif in L-CDR3, which enables the antibody to escape from acidified endosomes into the cytosol, where the heavy chain of the antibody is able to engage with and neutralise its intracellular target, activated K-ras [167]. Using this strategy, a humanised IgG1 format antibody named iMab RT11-i fused to a tumour-homing αv integrin binding RGD10 cyclic peptide was developed, and this had significant anti-tumour effects in vivo in a tumour xenograft model in mice, demonstrating the feasibility of this approach for the cytosolic delivery of antibodies. Based on this approach, it is conceivable that antibody fragments, e.g., nanobodies or domain antibodies with different targeting specificities, could be combined to achieve a similar effect.
7. Conclusions
In this review we have covered the structure of a range of antibody fragments, from the isolated domains of canonical IgG to nanobodies, their production in microbial prokaryotic and eukaryotic, cell-free systems, and their application in and outside of the clinic. We have discussed the main advantages of each fragment format with a focus on size and effector function and have highlighted the main mechanisms of action through which these fragments mediate their therapeutic effects.
Since the approval of muromonab in 1986, antibodies have rapidly expanded to become a major class of therapeutic molecule and are essential to the way we treat many diseases. Therapies have also diversified from the canonical mAb to a wide range of fragments and antibody-drug conjugate formats, which can offer context-dependent improvements to full-sized mAbs, the most common of which are discussed here. There is nothing to suggest that this trend will not continue with new formats and artificial frameworks (not discussed in this review) constantly being developed and gradually making their way into the clinic.
New formats offer exciting opportunities to expand the uses of antibodies into previously uncharted territory. For example, an orally taken antibody for the treatment of neurological diseases or antibodies against intracellular targets were once thought impossible due to an antibody’s large size and susceptibility to acidic proteases. Small, highly stable, even at low pH, nanobodies that are easy to link in series may potentially offer a way to access previously undruggable targets and tissues whilst being completely un-invasive. Fragments allow us to break central tolerance and re-target a host’s immune cells against cells displaying previously unrecognised cancer antigens in tissues too far removed from circulation for conventional antibodies to access. Fragments allow us to discover new antibodies at an astounding rate, create a vast array of multi-specific molecules, and rapidly search for indicators of disease almost anywhere in the body.
Although still not without their limitations and complications, the future of antibody-derived fragments undoubtedly looks bright. The diverse range of formats and modifications available combined with yet to be explored sequence space may allow us to overcome the challenges that we face in this modern era, from diseases of the wealthy and old aged, to the infectious and transmissible.
Funding
This research received no external funding.
Acknowledgments
Abthrax® is a registered trademark of Human Genome Science, Inc. BiTE® is a registered trademark of Micromet, Inc., Amgen; Blincyto® is a registered trademark of Amgen Inc.; CabliviTM is a registered trademark of Ablynx; Cimzia® is a registered trademark of UCB Pharma, S.A.; DART® is a registered trademark of MacroGenics; ImmTAC® and ImmTAV® are registered trademarks of Immunocore Ltd.; Lemtrada® is a registered trademark of the Genzyme Corporation; Leucentis® is a registered trademark of Novartis AG; Ocrevus® is a registered trademark of Genentech, Inc.; OrthocloneTM is a registered trademark of Johnson & Johnson; PER.C6TM is a registered trademark of Janssen Vaccines & Prevention B.V.; Praxbind® is a registered trademark of Boehringer Ingelheim; ReoPro® is a registered trademark of Eli Lilly and Company; ROCK® is a registered trademark of Affimed; SoloMERTM is a registered trademark of Elasmogen Ltd.; Synagis® is a registered trademark of the AstraZeneca group of companies; The Domain Antibody® and AlbudAb® platforms are trademarks of GlaxoSmithKline plc.; The Nanobody® platform is a registered trademark of Ablynx; Tysabri® and Zinbryta® are registered trademarks of Biogen MA Inc.; ZinplavaTM is a registered trademark of Merck Sharpe & Dohme corp.
Conflicts of Interest
The authors declare no conflicts of interest. A.B. is a complementary worker on assignment at GSK. C.A.P. is a full time employee of GSK.
Abbreviations
| 6HIS | hexa-histidine |
| ADA | anti-drug antibody |
| ADCC | antibody dependant cellular cytotoxicity |
| ADCP | antibody dependant cellular phagocytosis |
| ADH1 | alcohol dehydrogenase 1 |
| AIEX | anion exchange chromatography |
| ALI | acute lung inflammation |
| ARDS | acute respiratory distress syndrome |
| aTTP | acquired thrombotic thrombocytopenic purpura |
| ALL | acute lymphoblastic leukaemia |
| AMD | age-related macular degeneration |
| AML | acute myeloid leukaemia |
| BBB | blood–brain barrier |
| BCMA | B-cell maturation antigen |
| BiKE | bispecific natural killer cell engager |
| BiTE® | bispecific T-cell engager |
| CDC | complement-dependent cytotoxicity |
| CDR | complementarity determining region |
| CEA | carcinoembryonic antigen |
| CHO | Chinese hamster ovary |
| CIEX | cation exchange chromatography |
| CL | constant domain of immunoglobulin light chain |
| CNS | central nervous system |
| CPP | cell penetrating peptide |
| dAb | Domain antibody® |
| DART® | dual affinity re-targeting protein |
| EpCAM | epithelial cell adhesion molecule |
| ER | endoplasmic reticulum |
| Fab | fragment of antigen binding |
| Fc | fragment crystallizable |
| FcRn | neonatal fragment crystallizable receptor |
| Fv | fragment variable |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GFP | green fluorescent protein |
| GM-CSF | granulocyte-monocyte colony stimulating factor |
| GST | glutathione-S transferase |
| HAVH | human anti-VH |
| HEK | human embryonic kidney |
| HER2 | human epidermal growth factor 2 |
| HIC | hydrophobic interaction chromatography |
| HIV | human immunodeficiency virus |
| HLA | human leukocyte antigen |
| IgG | immunoglobulin gamma |
| IMAC | immobilised metal affinity chromatography |
| ImmTAC® | immune mobilising monoclonal t-cell receptors against cancer |
| ImmTAV® | immune mobilising monoclonal t-cell receptors against virus antigens |
| mAb | monoclonal antibody |
| MBP | mannose binding protein |
| MDS | myelodysplastic syndrome |
| MDSC | myeloid derived suppressor cell |
| MG | myasthenia gravis |
| MHC | major histocompatibility complex |
| MS | multiple sclerosis |
| Nb | Nanobody® |
| NK cell | Natural killer cell |
| NY ESO | New York esophageal squamous cell carcinoma |
| PD | pharmacodynamic |
| PEG | polyethylene glycol |
| PGK1 | phosphoglycerate kinase 1 |
| RMT | receptor-mediated endocytosis |
| RSV | respiratory syncytial virus |
| scFv | single chain fragment variable |
| SEC | size exclusion chromatography |
| SLE | systemic lupus erythematosus |
| TBGP | anti-trophoblast glycoprotein 5T4 |
| TCR | T-cell receptor |
| TfR | transferrin receptor |
| TNF | tumour necrosis factor |
| IL | interleukin |
| TNFR1 | tumour necrosis factor receptor 1 |
| TriKE | trispecific natural killer cell engager |
| VEGF | vascular endothelial growth factor |
| VH | variable domain of immunoglobulin heavy chain |
| VL | variable domain of immunoglobulin light chain |
| V-NAR | variable new antigen receptor |
| vWF | von Willebrand factor |
References
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. MAbs 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.; Kontermann, R.E. Bispecific Antibodies. In Handbook of Therapeutic Antibodies, 2nd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2014; ISBN 9783527682423. [Google Scholar]
- Drake, P.M.; Rabuka, D. An emerging playbook for antibody-drug conjugates: Lessons from the laboratory and clinic suggest a strategy for improving efficacy and safety. Curr. Opin. Chem. Biol. 2015, 28, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.L. Antibody fragments: Hope and hype. MAbs 2010, 2, 77–83. [Google Scholar] [CrossRef]
- Fernandes, J.C. Therapeutic application of antibody fragments in autoimmune diseases: Current state and prospects. Drug Discov. Today 2018, 23, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, R.V.; Kalinovsky, D.V.; Doronin, I.I.; Ponomarev, E.D.; Kholodenko, I. V Antibody Fragments as Potential Biopharmaceuticals for Cancer Therapy: Success and Limitations. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bird, R.E.; Hardman, K.D.; Jacobson, J.W.; Johnson, S.; Kaufman, B.M.; Lee, S.M.; Lee, T.; Pope, S.H.; Riordan, G.S.; Whitlow, M. Single-chain antigen-binding proteins. Science 1988. [Google Scholar] [CrossRef]
- Huston, J.S.; Levinson, D.; Mudgett-Hunter, M.; Tai, M.S.; Novotný, J.; Margolies, M.N.; Ridge, R.J.; Bruccoleri, R.E.; Haber, E.; Crea, R. Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1988, 85, 5879–5883. [Google Scholar] [CrossRef] [PubMed]
- Montoliu-Gaya, L.; Esquerda-Canals, G.; Bronsoms, S.; Villegas, S. Production of an anti-Aβ antibody fragment in Pichia pastoris and in vitro and in vivo validation of its therapeutic effect. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Spadiut, O.; Capone, S.; Krainer, F.; Glieder, A.; Herwig, C. Microbials for the production of monoclonal antibodies and antibody fragments. Trends Biotechnol. 2014, 32, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Yokota, T.; Milenic, D.E.; Whitlow, M.; Schlom, J. Rapid Tumor Penetration of a Single-Chain Fv and Comparison with Other Immunoglobulin Forms. Cancer Res. 1992. [Google Scholar] [CrossRef]
- Li, Z.; Krippendorff, B.F.; Sharma, S.; Walz, A.C.; Lavé, T.; Shah, D.K. Influence of molecular size on tissue distribution of antibody fragments. MAbs 2016. [Google Scholar] [CrossRef]
- Cumber, A.J.; Ward, E.S.; Winter, G.; Parnell, G.D.; Wawrzynczak, E.J. Comparative stabilities in vitro and in vivo of a recombinant mouse antibody FvCys fragment and a bisFvCys conjugate. J. Immunol. 1992, 149, 120–126. [Google Scholar]
- Sanz, L.; Cuesta, Á.M.; Compte, M.; Álvarez-Vallina, L. Antibody engineering: Facing new challenges in cancer therapy. Acta Pharmacol. Sin. 2005, 26, 641–648. [Google Scholar] [CrossRef]
- Jain, A.; Jain, S. PEGylation: An approach for drug delivery. A review. Crit. Rev. Drug Carr. Syst. 2008. [Google Scholar] [CrossRef]
- Müller, D.; Karle, A.; Meißburger, B.; Höfig, I.; Stork, R.; Kontermann, R.E. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J. Biol. Chem. 2007. [Google Scholar] [CrossRef]
- Poiron, C.; Wu, Y.; Ginestoux, C.; Ehrenmann, F.; Duroux, P.; Lefranc, M. IMGT®, the international ImMunoGeneTics information system®. Nucleic Acids Res. 2008, 37 (Suppl. S1), D1006–D1012. [Google Scholar]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.S.; Linsley, P.S.; Gayle, M.A.; Bajorath, J.; Brady, W.A.; Norris, N.A.; Fell, H.P.; Ledbetter, J.A.; Gilliland, L.K. Single-chain mono- and bispecific antibody derivatives with novel biological properties and antitumour activity from a COS cell transient expression system. Ther. Immunol. 1994, 1, 3–15. [Google Scholar] [PubMed]
- Holt, L.J.; Basran, A.; Jones, K.; Chorlton, J.; Jespers, L.S.; Brewis, N.D.; Tomlinson, I.M. Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Eng. Des. Sel. 2008. [Google Scholar] [CrossRef]
- Holliger, P. “Diabodies”: Small Bivalent and Bispecific Antibody Fragments. Proc. Natl. Acad. Sci. USA 1993. [Google Scholar] [CrossRef]
- Lu, D.; Jimenez, X.; Witte, L.; Zhu, Z. The effect of variable domain orientation and arrangement on the antigen-binding activity of a recombinant human bispecific diabody. Biochem. Biophys. Res. Commun. 2004. [Google Scholar] [CrossRef]
- Kipriyanov, S.M.; Moldenhauer, G.; Braunagel, M.; Reusch, U.; Cochlovius, B.; Le Gall, F.; Kouprianova, O.A.; Von Der Lieth, C.W.; Little, M. Effect of domain order on the activity of bacterially produced bispecific single-chain Fv antibodies. J. Mol. Biol. 2003. [Google Scholar] [CrossRef]
- MacroGenics Pipeline. Available online: https://www.macrogenics.com/pipeline/ (accessed on 16 January 2019).
- Affimed Pipeline. Available online: https://www.affimed.com/pipeline/ (accessed on 17 January 2019).
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef]
- Kipriyanov, S.M.; Moldenhauer, G.; Schuhmacher, J.; Cochlovius, B.; Von Der Lieth, C.W.; Matys, E.R.; Little, M. Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics. J. Mol. Biol. 1999. [Google Scholar] [CrossRef]
- Wu, C.; Ying, H.; Grinnell, C.; Bryant, S.; Miller, R.; Clabbers, A.; Bose, S.; McCarthy, D.; Zhu, R.R.; Santora, L.; et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007. [Google Scholar] [CrossRef]
- Metz, S.; Haas, A.K.; Daub, K.; Croasdale, R.; Stracke, J.; Lau, W.; Georges, G.; Josel, H.-P.; Dziadek, S.; Hopfner, K.-P.; et al. Bispecific digoxigenin-binding antibodies for targeted payload delivery. Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef]
- Xencor Pipeline. Available online: https://www.xencor.com/pipeline/ (accessed on 17 January 2019).
- Nelson, A.L.; Dhimolea, E.; Reichert, J.M. Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 2010, 9, 767–774. [Google Scholar] [CrossRef]
- Bazin-Redureau, M.I.; Renard, C.B.; Scherrmann, J.M.G. Pharmacokinetics of heterologous and homologous immunoglobulin G, F(ab’)2and Fab after intravenous administration in the rat. J. Pharm. Pharmacol. 1997. [Google Scholar] [CrossRef]
- Röthlisberger, D.; Honegger, A.; Plückthun, A. Domain interactions in the Fab fragment: A comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J. Mol. Biol. 2005. [Google Scholar] [CrossRef]
- Simister, N.E.; Mostov, K.E. An Fc receptor structurally related to MHC class I antigens. Nature 1989. [Google Scholar] [CrossRef]
- Ober, R.J.; Martinez, C.; Lai, X.; Zhou, J.; Ward, E.S. Exocytosis of IgG as mediated by the receptor, FcRn: An analysis at the single-molecule level. Proc. Natl. Acad. Sci. USA 2004. [Google Scholar] [CrossRef]
- Rodewald, R.; Kraehenbuhl, J.P. Receptor-mediated transport of IgG. J. Cell Biol. 1984, 99, 159s–164s. [Google Scholar] [CrossRef]
- Chapman, A.P.; Antoniw, P.; Spitali, M.; West, S.; Stephens, S.; King, D.J. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat. Biotechnol. 1999. [Google Scholar] [CrossRef]
- Schreiber, S. Certolizumab pegol for the treatment of Crohn’s disease. Ther. Adv. Gastroenterol. 2011, 357, 228–238. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hammers, R. Naturally occurring antibodies devoid of light chains. Nature 1993. [Google Scholar] [CrossRef]
- Cortez-Retamozo, V.; Backmann, N.; Senter, P.D.; Wernery, U.; De Baetselier, P.; Muyldermans, S.; Revets, H. Efficient Cancer Therapy with a Nanobody-Based Conjugate. Cancer Res. 2004. [Google Scholar] [CrossRef]
- Van Der Linden, R.H.J.; Frenken, L.G.J.; De Geus, B.; Harmsen, M.M.; Ruuls, R.C.; Stok, W.; De Ron, L.; Wilson, S.; Davis, P.; Verrips, C.T. Comparison of physical chemical properties of llama V(HH) antibody fragments and mouse monoclonal antibodies. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1999. [Google Scholar] [CrossRef]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 2006. [Google Scholar] [CrossRef]
- Harmsen, M.M.; De Haard, H.J. Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Steeland, S.; Vandenbroucke, R.E.; Libert, C. Nanobodies as therapeutics: Big opportunities for small antibodies. Drug Discov. Today 2016, 21, 1076–1113. [Google Scholar] [CrossRef]
- Cortez-Retamozo, V.; Lauwereys, M.; Hassanzadeh Gh., G.; Gobert, M.; Conrath, K.; Muyldermans, S.; De Baetselier, P.; Revets, H. Efficient tumor targeting by single-domain antibody fragments of camels. Int. J. Cancer 2002. [Google Scholar] [CrossRef]
- Shen, J.; Vil, M.D.; Jimenez, X.; Iacolina, M.; Zhang, H.; Zhu, Z. Single variable domain-IgG fusion: A novel recombinant approach to Fc domain-containing bispecific antibodies. J. Biol. Chem. 2006. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Avila, D.; Hughes, M.; Hughes, A.; McKinney, E.C.; Flajnik, M.F. A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature 1995. [Google Scholar] [CrossRef]
- Stanfield, R.L.; Dooley, H.; Verdino, P.; Flajnik, M.F.; Wilson, I.A. Maturation of Shark Single-domain (IgNAR) Antibodies: Evidence for Induced-fit Binding. J. Mol. Biol. 2007. [Google Scholar] [CrossRef]
- Zielonka, S.; Empting, M.; Grzeschik, J.; Könning, D.; Barelle, C.J.; Kolmar, H. Structural insights and biomedical potential of IgNAR scaffolds from sharks. MAbs 2015, 7, 15–25. [Google Scholar] [CrossRef]
- Davies, J.; Riechmann, L. “Camelising” human antibody fragments: NMR studies on VH domains. FEBS Lett. 1994. [Google Scholar] [CrossRef]
- Holt, L.J.; Herring, C.; Jespers, L.S.; Woolven, B.P.; Tomlinson, I.M. Domain antibodies: Proteins for therapy. Trends Biotechnol. 2003, 21, 484–490. [Google Scholar] [CrossRef]
- Scott, M.J.; Lee, J.A.; Wake, M.S.; Batt, K.V.; Wattam, T.A.; Hiles, I.D.; Batuwangala, T.D.; Ashman, C.I.; Steward, M. ‘In-Format’ screening of a novel bispecific antibody format reveals significant potency improvements relative to unformatted molecules. MAbs 2017. [Google Scholar] [CrossRef]
- O’Connor-Semmes, R.L.; Lin, J.; Hodge, R.J.; Andrews, S.; Chism, J.; Choudhury, A.; Nunez, D.J. GSK2374697, a Novel Albumin-Binding Domain Antibody (AlbudAb), Extends Systemic Exposure of Exendin-4: First Study in Humans—PK/PD and Safety. Clin. Pharmacol. Ther. 2014. [Google Scholar] [CrossRef]
- Jefferis, R. Recombinant antibody therapeutics: The impact of glycosylation on mechanisms of action. Trends Pharmacol. Sci. 2009, 30, 356–362. [Google Scholar] [CrossRef]
- Li, F.; Vijayasankaran, N.; Shen, A.; Kiss, R.; Amanullah, A. Cell culture processes for monoclonal antibody production. MAbs 2010, 2, 466–479. [Google Scholar] [CrossRef]
- Striedner, G.; Pfaffenzeller, I.; Markus, L.; Nemecek, S.; Grabherr, R.; Bayer, K. Plasmid-free T7-based Escherichia coli expression systems. Biotechnol. Bioeng. 2010. [Google Scholar] [CrossRef]
- Mairhofer, J.; Cserjan-Puschmann, M.; Striedner, G.; Nöbauer, K.; Razzazi-Fazeli, E.; Grabherr, R. Marker-free plasmids for gene therapeutic applications—Lack of antibiotic resistance gene substantially improves the manufacturing process. J. Biotechnol. 2010. [Google Scholar] [CrossRef]
- Sonoda, H.; Kumada, Y.; Katsuda, T.; Yamaji, H. Effects of cytoplasmic and periplasmic chaperones on secretory production of single-chain Fv antibody in Escherichia coli. J. Biosci. Bioeng. 2011. [Google Scholar] [CrossRef]
- Yuan, J.; Zweers, J.C.; Van Dijl, J.M.; Dalbey, R.E. Protein transport across and into cell membranes in bacteria and archaea. Cell. Mol. Life Sci. 2010, 67, 179–199. [Google Scholar] [CrossRef]
- Levy, R.; Ahluwalia, K.; Bohmann, D.J.; Giang, H.M.; Schwimmer, L.J.; Issafras, H.; Reddy, N.B.; Chan, C.; Horwitz, A.H.; Takeuchi, T. Enhancement of antibody fragment secretion into the Escherichia coli periplasm by co-expression with the peptidyl prolyl isomerase, FkpA, in the cytoplasm. J. Immunol. Methods 2013. [Google Scholar] [CrossRef]
- Jalalirad, R. Production of antibody fragment (Fab) throughout Escherichia coli fed-batch fermentation process: Changes in titre, location and form of product. Electron. J. Biotechnol. 2013. [Google Scholar] [CrossRef]
- Gorlani, A.; De Haard, H.; Verrips, T. Expression of VHHs in saccharomyces cerevisiae. Methods Mol. Biol. 2012. [Google Scholar] [CrossRef]
- Chee, M.K.; Haase, S.B. New and Redesigned pRS Plasmid Shuttle Vectors for Genetic Manipulation of Saccharomyces cerevisiae. G3 Genes Genomes Genet. 2012. [Google Scholar] [CrossRef]
- Leite, F.C.B.; dos Anjos, R.S.G.; Basilio, A.C.M.; Leal, G.F.C.; Simões, D.A.; de Morais, M.A. Construction of integrative plasmids suitable for genetic modification of industrial strains of Saccharomyces cerevisiae. Plasmid 2013. [Google Scholar] [CrossRef]
- Partow, S.; Siewers, V.; Bjørn, S.; Nielsen, J.; Maury, J. Characterization of different promoters for designing a new expression vector in Saccharomyces cerevisiae. Yeast 2010. [Google Scholar] [CrossRef]
- Maury, J.; Asadollahi, M.A.; Møller, K.; Schalk, M.; Clark, A.; Formenti, L.R.; Nielsen, J. Reconstruction of a bacterial isoprenoid biosynthetic pathway in Saccharomyces cerevisiae. FEBS Lett. 2008. [Google Scholar] [CrossRef]
- Joosten, V.; Lokman, C.; van den Hondel, C.A.M.J.J.; Punt, P.J. The production of antibody fragments and antibody fusion proteins by yeasts and filamentous fungi. Microb. Cell Fact. 2003, 2. [Google Scholar] [CrossRef]
- Xu, P.; Raden, D.; Doyle, F.J.; Robinson, A.S. Analysis of unfolded protein response during single-chain antibody expression in Saccaromyces cerevisiae reveals different roles for BiP and PDI in folding. Metab. Eng. 2005. [Google Scholar] [CrossRef]
- Ferndahl, C.; Bonander, N.; Logez, C.; Wagner, R.; Gustafsson, L.; Larsson, C.; Hedfalk, K.; Darby, R.A.J.; Bill, R.M. Increasing cell biomass in Saccharomyces cerevisiae increases recombinant protein yield: The use of a respiratory strain as a microbial cell factory. Microb. Cell Fact. 2010. [Google Scholar] [CrossRef]
- Delic, M.; Mattanovich, D.; Gasser, B. Repressible promoters—A novel tool to generate conditional mutants in Pichia pastoris. Microb. Cell Fact. 2013. [Google Scholar] [CrossRef]
- Sohn, S.B.; Graf, A.B.; Kim, T.Y.; Gasser, B.; Maurer, M.; Ferrer, P.; Mattanovich, D.; Lee, S.Y. Genome-scale metabolic model of methylotrophic yeast Pichia pastoris and its use for in silico analysis of heterologous protein production. Biotechnol. J. 2010. [Google Scholar] [CrossRef]
- Jahic, M.; Rotticci-Mulder, J.; Martinelle, M.; Hult, K.; Enfors, S.O. Modeling of growth and energy metabolism of Pichia pastoris producing a fusion protein. Bioprocess Biosyst. Eng. 2001. [Google Scholar] [CrossRef]
- Detalle, L.; Stohr, T.; Palomo, C.; Piedra, P.A.; Gilbert, B.E.; Mas, V.; Millar, A.; Power, U.F.; Stortelers, C.; Allosery, K.; et al. Generation and characterization of ALX-0171, a potent novel therapeutic nanobody for the treatment of respiratory syncytial virus infection. Antimicrob. Agents Chemother. 2016. [Google Scholar] [CrossRef]
- Vallet-Courbin, A.; Larivière, M.; Hocquellet, A.; Hemadou, A.; Parimala, S.N.; Laroche-Traineau, J.; Santarelli, X.; Clofent-Sanchez, G.; Jacobin-Valat, M.J.; Noubhani, A. A recombinant human anti-platelet SCFV antibody produced in pichia pastoris for atheroma targeting. PLoS ONE 2017. [Google Scholar] [CrossRef]
- Oh, I.S.; Lee, J.C.; Lee, M.S.; Chung, J.H.; Kim, D.M. Cell-free production of functional antibody fragments. Bioprocess Biosyst. Eng. 2010. [Google Scholar] [CrossRef]
- Stech, M.; Nikolaeva, O.; Thoring, L.; Stöcklein, W.F.M.; Wüstenhagen, D.A.; Hust, M.; Dübel, S.; Kubick, S. Cell-free synthesis of functional antibodies using a coupled in vitro transcription-Translation system based on CHO cell lysates. Sci. Rep. 2017. [Google Scholar] [CrossRef]
- Stech, M.; Hust, M.; Schulze, C.; Dübel, S.; Kubick, S. Cell-free eukaryotic systems for the production, engineering, and modification of scFv antibody fragments. Eng. Life Sci. 2014. [Google Scholar] [CrossRef]
- Stech, M.; Kubick, S. Cell-Free Synthesis Meets Antibody Production: A Review. Antibodies 2015. [Google Scholar] [CrossRef]
- Ryabova, L.A.; Desplancq, D.; Spirin, A.S.; Plückthun, A. Functional antibody production using cell-free translation: Effects of protein disulfide isomerase and chaperones. Nat. Biotechnol. 1997. [Google Scholar] [CrossRef]
- Jiang, X.; Ookubo, Y.; Fujii, I.; Nakano, H.; Yamane, T. Expression of Fab fragment of catalytic antibody 6D9 in an Escherichia coli in vitro coupled transcription/translation system. FEBS Lett. 2002. [Google Scholar] [CrossRef]
- Kanter, G.; Yang, J.; Voloshin, A.; Levy, S.; Swartz, J.R.; Levy, R. Cell-free production of scFv fusion proteins: An efficient approach for personalized lymphoma vaccines. Blood 2007. [Google Scholar] [CrossRef]
- Shimizu, Y.; Kuruma, Y.; Ying, B.W.; Umekage, S.; Ueda, T. Cell-free translation systems for protein engineering. FEBS J. 2006, 273, 4133–4140. [Google Scholar] [CrossRef] [Green Version]
- STR001—Clinical Trial: NCT03424603. Available online: https://clinicaltrials.gov/ct2/show/NCT03424603 (accessed on 15 January 2019).
- Wang, A.C.; Wang, I.Y. Cleavage sites of human IgGl immunoglobulin by papain. Immunochemistry 1977. [Google Scholar] [CrossRef]
- Jones, R.G.A.; Landon, J. Enhanced pepsin digestion: A novel process for purifying antibody F(ab′)2 fragments in high yield from serum. J. Immunol. Methods 2002. [Google Scholar] [CrossRef]
- Genovis Website. Available online: https://www.genovis.com (accessed on 17 January 2019).
- Hober, S.; Nord, K.; Linhult, M. Protein A chromatography for antibody purification. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 848, 40–47. [Google Scholar] [CrossRef]
- Grodzki, A.C.; Berenstein, E. Antibody Purification: Affinity Chromatography—Protein A and Protein G Sepharose. In Immunocytochemical Methods and Protocols; Humana Press: New York, NY, USA, 2009. [Google Scholar]
- Roben, P.W.; Salem, A.N.; Silverman, G.J. VH3 family antibodies bind domain D of staphylococcal protein A. J. Immunol. 1995, 154, 6437–6445. [Google Scholar]
- Rodrigo, G.; Gruvegård, M.; Van Alstine, J. Antibody Fragments and Their Purification by Protein L Affinity Chromatography. Antibodies 2015. [Google Scholar] [CrossRef]
- Nascimento, A.; Pinto, I.F.; Chu, V.; Aires-Barros, M.R.; Conde, J.P.; Azevedo, A.M. Studies on the purification of antibody fragments. Sep. Purif. Technol. 2018. [Google Scholar] [CrossRef]
- Björck, L. A Novel Bacterial Cell Wall Protein with Affinity for Ig L Chains. J. Immunol. 1988, 140, 1194–1197. [Google Scholar]
- De Château, M.; Nilson, B.H.K.; Erntell, M.; Myhre, E.; Magnusson, C.G.M.; Åkerström, B.; Björck, L. On the Interaction between Protein L and Immunoglobulins of Various Mammalian Species. Scand. J. Immunol. 1993. [Google Scholar] [CrossRef]
- Lichty, J.J.; Malecki, J.L.; Agnew, H.D.; Michelson-Horowitz, D.J.; Tan, S. Comparison of affinity tags for protein purification. Protein Expr. Purif. 2005. [Google Scholar] [CrossRef]
- Goel, A.; Colcher, D.; Koo, J.S.; Booth, B.J.M.; Pavlinkova, G.; Batra, S.K. Relative position of the hexahistidine tag effects binding properties of a tumor-associated single-chain Fv construct. Biochim. Biophys. Acta Gen. Subj. 2000. [Google Scholar] [CrossRef]
- Schmeisser, H.; Kontsek, P.; Esposito, D.; Gillette, W.; Schreiber, G.; Zoon, K.C. Binding Characteristics of IFN-alpha Subvariants to IFNAR2-EC and Influence of the 6-Histidine Tag. J. Interferon Cytokine Res. 2006. [Google Scholar] [CrossRef]
- Das, D.; Allen, T.M.; Suresh, M.R. Comparative evaluation of two purification methods of anti-CD19-c-myc- His6-Cys scFv. Protein Expr. Purif. 2005. [Google Scholar] [CrossRef]
- Liu, H.; Gaza-Bulseco, G.; Chumsae, C. Analysis of Reduced Monoclonal Antibodies Using Size Exclusion Chromatography Coupled with Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2009. [Google Scholar] [CrossRef]
- Ljunglöf, A.; Lacki, K.M.; Mueller, J.; Harinarayan, C.; van Reis, R.; Fahrner, R.; Van Alstine, J.M. Ion exchange chromatography of antibody fragments. Biotechnol. Bioeng. 2007. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, C.M.; Kim, K.; Yoo, J.M.; Kang, S.M.; Ha, G.S.; Park, M.K.; Choi, M.A.; Lee, D.E.; Seong, B.L. Purification of antibody fragments for the reduction of charge variants using cation exchange chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018. [Google Scholar] [CrossRef]
- Karkov, H.S.; Krogh, B.O.; Woo, J.; Parimal, S.; Ahmadian, H.; Cramer, S.M. Investigation of protein selectivity in multimodal chromatography using in silico designed Fab fragment variants. Biotechnol. Bioeng. 2015. [Google Scholar] [CrossRef]
- Wu, Z.; Cheung, N.V. T-cell engaging bispecific antibody (T-BsAb): From technology to therapeutics. Pharmacol. Ther. 2018, 182, 161–175. [Google Scholar] [CrossRef]
- Velasquez, M.P.; Bonifant, C.L.; Gottschalk, S. Redirecting T-cells to hematological malignancies with bispecific antibodies. Blood 2018, 131, 30–38. [Google Scholar] [CrossRef]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T-cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006. [Google Scholar] [CrossRef]
- Bispecific Antibodies Technology: NK Cells Engagers—Innate Pharma. Available online: https://www.innate-pharma.com/en/pipeline/bispecific-antibodies-technology-nk-cells-engagers (accessed on 18 January 2019).
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 2014. [Google Scholar] [CrossRef]
- Felices, M.; Sarhan, D.; Brandt, L.; Guldevall, K.; McElmurry, R.; Lenvik, A.; Chu, S.; Tolar, J.; Taras, E.; Spellman, S.R.; et al. CD16-IL15-CD33 Trispecific Killer Engager (TriKE) Overcomes Cancer-Induced Immune Suppression and Induces Natural Killer Cell-Mediated Control of MDS and AML Via Enhanced Killing Kinetics. Blood 2016, 128, 4291. [Google Scholar]
- Lichtenegger, F.S.; Rothe, M.; Schnorfeil, F.M.; Deiser, K.; Krupka, C.; Augsberger, C.; Schlüter, M.; Neitz, J.; Subklewe, M. Targeting LAG-3 and PD-1 to enhance T-cell activation by antigen-presenting cells. Front. Immunol. 2018. [Google Scholar] [CrossRef]
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 2011. [Google Scholar] [CrossRef]
- Oates, J.; Jakobsen, B.K. ImmTACs: Novel bi-specific agents for targeted cancer therapy. Oncoimmunology 2013, 2, e22891. [Google Scholar] [CrossRef]
- Oates, J.; Hassan, N.J.; Jakobsen, B.K. ImmTACs for targeted cancer therapy: Why, what, how, and which. Mol. Immunol. 2015, 67, 67–74. [Google Scholar] [CrossRef]
- Immunocore Pipeline. Available online: https://www.immunocore.com/pipeline (accessed on 15 January 2019).
- Zaia, J.A. A new agent in the strategy to cure AIDS. Mol. Ther. 2016, 24, 1894–1896. [Google Scholar] [CrossRef]
- Safdari, Y.; Ahmadzadeh, V. Use of Single-Chain Antibody Derivatives for Targeted Drug Delivery. Mol. Med. 2016. [Google Scholar] [CrossRef]
- Lu, Z.R.; Kopekov, P.; Kopeek, J. Polymerizable Fab’ antibody fragments for targeting of anticancer drugs. Nat. Biotechnol. 1999. [Google Scholar] [CrossRef]
- Philogen Pipeline. Available online: http://www.philogen.com/en/products/pipeline/pipeline_16.html (accessed on 17 January 2019).
- Lemaire, M.; D’Huyvetter, M.; Lahoutte, T.; Van Valckenborgh, E.; Menu, E.; De Bruyne, E.; Kronenberger, P.; Wernery, U.; Muyldermans, S.; Devoogdt, N.; et al. Imaging and radioimmunotherapy of multiple myeloma with anti-idiotypic Nanobodies. Leukemia 2014, 28, 444–447. [Google Scholar] [CrossRef]
- Pruszynski, M.; Koumarianou, E.; Vaidyanathan, G.; Revets, H.; Devoogdt, N.; Lahoutte, T.; Zalutsky, M.R. Targeting breast carcinoma with radioiodinated anti-HER2 Nanobody. Nucl. Med. Biol. 2013. [Google Scholar] [CrossRef]
- Shen, B.Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.F.; Mai, E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- ALX-0061—Clinical Trial: NCT02287922. Available online: https://clinicaltrials.gov/ct2/show/NCT02287922 (accessed on 21 January 2019).
- ALX-0061—Clinical Trial: NCT02437890. Available online: https://clinicaltrials.gov/ct2/show/NCT02437890 (accessed on 21 January 2019).
- ALX-0761—Clinical Trial: NCT02156466. Available online: https://clinicaltrials.gov/ct2/show/NCT02156466 (accessed on 21 January 2019).
- ATN-103—Clinical trial: NCT01063803. Available online: https://clinicaltrials.gov/ct2/show/NCT01063803 (accessed on 21 January 2019).
- ATN-192—Clinical Trial: NCT01284036. Available online: https://clinicaltrials.gov/ct2/show/NCT01284036 (accessed on 21 January 2019).
- Philogen Website. Available online: http://www.philogen.com/en/ (accessed on 21 January 2019).
- Ulrichts, P.; Cousin, T.; Dreier, T.; de Haard, H.; Leupin, N. Argx-113, a novel Fc-based approach for antibody-induced pathologies such as primary immune thrombocytopenia. Blood 2016, 128, 4919. [Google Scholar]
- ARGX-113—Clinical Trial NCT03669588. Available online: https://clinicaltrials.gov/ct2/show/NCT03669588 (accessed on 29 January 2019).
- Merrill, J.T.; Shevell, D.E.; Duchesne, D.; Nowak, M.; Kundu, S.; Girgis, I.G.; Hu, Y.S.; Nadler, S.G.; Banerjee, S.; Throup, J. An Anti-CD28 Domain Antibody, Lulizumab, in Systemic Lupus Erythematosus: Results of a Phase II Study. In Arthritis & Rheumatology; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar]
- Cordy, J.C.; Morley, P.J.; Wright, T.J.; Birchler, M.A.; Lewis, A.P.; Emmins, R.; Chen, Y.Z.; Powley, W.M.; Bareille, P.J.; Wilson, R.; et al. Specificity of human anti-variable heavy (VH) chain autoantibodies and impact on the design and clinical testing of a VH domain antibody antagonist of tumour necrosis factor-α receptor 1. Clin. Exp. Immunol. 2015. [Google Scholar] [CrossRef]
- GSK286227—Clinical Trial: NCT02221037. Available online: https://clinicaltrials.gov/ct2/show/NCT02221037 (accessed on 29 January 2019).
- Thiel, M.A.; Coster, D.J.; Standfield, S.D.; Brereton, H.M.; Mavrangelos, C.; Zola, H.; Taylor, S.; Yusim, A.; Williams, K.A. Penetration of engineered antibody fragments into the eye. Clin. Exp. Immunol. 2002. [Google Scholar] [CrossRef]
- Lampalizumab—Clinical Trial: NCT02247531. Available online: https://clinicaltrials.gov/ct2/show/NCT02247531 (accessed on 24 January 2019).
- Lampalizumab—Clinical Trial: NCT02247479. Available online: https://clinicaltrials.gov/ct2/show/NCT02247479 (accessed on 24 January 2019).
- Holz, F.G.; Sadda, S.R.; Busbee, B.; Chew, E.Y.; Mitchell, P.; Tufail, A.; Brittain, C.; Ferrara, D.; Gray, S.; Honigberg, L.; et al. Efficacy and safety of lampalizumab for geographic atrophy due to age-related macular degeneration: Chroma and spectri phase 3 randomized clinical trials. JAMA Ophthalmol. 2018. [Google Scholar] [CrossRef]
- RTH 258—Clinical Trial: NCT02307682. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=1&cad=rja&uact=8&ved=2ahUKEwiNsqmMi5PgAhUS2qQKHbi1BZoQFjAAegQICRAB&url=https%3A%2F%2Fclinicaltrials.gov%2Fct2%2Fshow%2FNCT02307682&usg=AOvVaw2oDAJKgexjOb5qZHcOVqJy (accessed on 24 January 2018).
- RTH 258—Clinical Trial: NCT02434328. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=1&cad=rja&uact=8&ved=2ahUKEwiMjoKbi5PgAhVO3KQKHa2cDKoQFjAAegQIChAB&url=https%3A%2F%2Fclinicaltrials.gov%2Fct2%2Fshow%2FNCT02434328&usg=AOvVaw1Uobk9ivR8iGx_t6gl-Lec (accessed on 24 January 2019).
- Camacho-Villegas, T.A.; Mata-González, M.T.; García-Ubbelohd, W.; Núñez-García, L.; Elosua, C.; Paniagua-Solis, J.F.; Licea-Navarro, A.F. Intraocular penetration of a vNAR: In vivo and in vitro VEGF165 neutralization. Mar. Drugs 2018. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, M.; Johnson, K.; Steven, J.; Barelle, C.J.; Porter, A. Therapeutic potential of shark anti-ICOSL VNAR domains is exemplified in a murine model of autoimmune non-infectious uveitis. Front. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Elasmogen Website. Available online: www.elasmogen.com (accessed on 24 January 2019).
- Wilken, L.; McPherson, A. Application of camelid heavy-chain variable domains (VHHs) in prevention and treatment of bacterial and viral infections. Int. Rev. Immunol. 2018, 37, 69–76. [Google Scholar] [CrossRef] [PubMed]
- AdisInsight ALX-0171. Available online: https://adisinsight.springer.com/drugs/800035341 (accessed on 24 January 2019).
- Laustsen, A.H.; María Gutiérrez, J.; Knudsen, C.; Johansen, K.H.; Bermúdez-Méndez, E.; Cerni, F.A.; Jürgensen, J.A.; Ledsgaard, L.; Martos-Esteban, A.; Øhlenschlæger, M.; et al. Pros and cons of different therapeutic antibody formats for recombinant antivenom development. Toxicon 2018. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.M.; Mukherjee, J.; Leysath, C.E.; Debatis, M.; Ofori, K.; Baldwin, K.; Boucher, C.; Peters, R.; Beamer, G.; Sheoran, A.; et al. A single VHH-based toxin-neutralizing agent and an effector antibody protect mice against challenge with Shiga toxins 1 and 2. Infect. Immun. 2013. [Google Scholar] [CrossRef]
- Freise, A.C.; Wu, A.M. In vivo imaging with antibodies and engineered fragments. Mol. Immunol. 2015, 67, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Brussel, A.S.A.; Adams, A.; Oliveira, S.; Dorresteijn, B.; El Khattabi, M.; Vermeulen, J.F.; van der Wall, E.; Mali, W.P.T.M.; Derksen, P.W.B.; van Diest, P.J.; et al. Hypoxia-Targeting Fluorescent Nanobodies for Optical Molecular Imaging of Pre-Invasive Breast Cancer. Mol. Imaging Biol. 2016. [Google Scholar] [CrossRef]
- Kijanka, M.M.; van Brussel, A.S.A.; van der Wall, E.; Mali, W.P.T.M.; van Diest, P.J.; van Bergen en Henegouwen, P.M.P.; Oliveira, S. Optical imaging of pre-invasive breast cancer with a combination of VHHs targeting CAIX and HER2 increases contrast and facilitates tumour characterization. EJNMMI Res. 2016. [Google Scholar] [CrossRef]
- Viola-Villegas, N.T.; Sevak, K.K.; Carlin, S.D.; Doran, M.G.; Evans, H.W.; Bartlett, D.W.; Wu, A.M.; Lewis, J.S. Noninvasive imaging of PSMA in prostate tumors with89Zr-Labeled huJ591 engineered antibody fragments: The faster alternatives. Mol. Pharm. 2014. [Google Scholar] [CrossRef]
- Raubitschek, A.A.; Tsai, S.-W.; Shively, J.E.; Yazaki, P.J.; Williams, L.E.; Ikle’, D.N.; Wu, A.M.; Wong, J.Y.C. Tumor Targeting of Radiometal Labeled Anti-CEA Recombinant T84.66 Diabody and T84.66 Minibody: Comparison to Radioiodinated Fragments. Bioconjug. Chem. 2002. [Google Scholar] [CrossRef]
- Maier, J.; Traenkle, B.; Rothbauer, U. Real-time analysis of epithelial-mesenchymal transition using fluorescent single-domain antibodies. Sci. Rep. 2015. [Google Scholar] [CrossRef]
- Broisat, A.; Hernot, S.; Toczek, J.; De Vos, J.; Riou, L.M.; Martin, S.; Ahmadi, M.; Thielens, N.; Wernery, U.; Caveliers, V.; et al. Nanobodies targeting mouse/human VCAM1 for the nuclear imaging of atherosclerotic lesions. Circ. Res. 2012. [Google Scholar] [CrossRef]
- Broisat, A.; Toczek, J.; Dumas, L.S.; Ahmadi, M.; Bacot, S.; Perret, P.; Slimani, L.; Barone-Rochette, G.; Soubies, A.; Devoogdt, N.; et al. 99mTc-cAbVCAM1-5 Imaging Is a Sensitive and Reproducible Tool for the Detection of Inflamed Atherosclerotic Lesions in Mice. J. Nucl. Med. 2014. [Google Scholar] [CrossRef]
- Yu, Y.J.; Watts, R.J. Developing Therapeutic Antibodies for Neurodegenerative Disease. Neurotherapeutics 2013, 10, 459–472. [Google Scholar] [CrossRef] [Green Version]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the “high-hanging fruit”. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004. [Google Scholar] [CrossRef]
- Messer, A.; Lynch, S.M.; Butler, D.C. Developing intrabodies for the therapeutic suppression of neurodegenerative pathology. Expert Opin. Biol. Ther. 2009. [Google Scholar] [CrossRef]
- Pardridge, W.M. Targeted delivery of protein and gene medicines through the blood-brain barrier. Clin. Pharmacol. Ther. 2015, 97, 347–361. [Google Scholar] [CrossRef]
- Abzyme Website. Available online: www. abzymetx.com (accessed on 21 January 2019).
- Li, T.; Bourgeois, J.P.; Celli, S.; Glacial, F.; Le Sourd, A.M.; Mecheri, S.; Weksler, B.; Romero, I.; Couraud, P.O.; Rougeon, F.; et al. Cell-penetrating anti-GFAP VHH and corresponding fluorescent fusion protein VHH-GFP spontaneously cross the blood-brain barrier and specifically recognize astrocytes: Application to brain imaging. FASEB J. 2012. [Google Scholar] [CrossRef]
- Poduslo, J.F.; Ramakrishnan, M.; Holasek, S.S.; Ramirez-Alvarado, M.; Kandimalla, K.K.; Gilles, E.J.; Curran, G.L.; Wengenack, T.M. In vivo targeting of antibody fragments to the nervous system for Alzheimer’s disease immunotherapy and molecular imaging of amyloid plaques. J. Neurochem. 2007. [Google Scholar] [CrossRef]
- Caljon, G.; Caveliers, V.; Lahoutte, T.; Stijlemans, B.; Ghassabeh, G.H.; Van Den Abbeele, J.; Smolders, I.; De Baetselier, P.; Michotte, Y.; Muyldermans, S.; et al. Using microdialysis to analyse the passage of monovalent nanobodies through the blood-brain barrier. Br. J. Pharmacol. 2012. [Google Scholar] [CrossRef]
- Muruganandam, A.; Tanha, J.; Narang, S.; Stanimirovic, D. Selection of phage-displayed llama single-domain antibodies that transmigrate across human blood-brain barrier endothelium. FASEB J. 2002. [Google Scholar] [CrossRef]
- Farrington, G.K.; Caram-Salas, N.; Haqqani, A.S.; Brunette, E.; Eldredge, J.; Pepinsky, B.; Antognetti, G.; Baumann, E.; Ding, W.; Garber, E.; et al. A novel platform for engineering blood-brain barrier-crossing bispecific biologics. FASEB J. 2014. [Google Scholar] [CrossRef]
- Slastnikova, T.A.; Ulasov, A.V.; Rosenkranz, A.A.; Sobolev, A.S. Targeted intracellular delivery of antibodies: The state of the art. Front. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Lim, K.J.; Sung, B.H.; Shin, J.R.; Lee, Y.W.; Kim, D.J.; Yang, K.S.; Kim, S.C. A Cancer Specific Cell-Penetrating Peptide, BR2, for the Efficient Delivery of an scFv into Cancer Cells. PLoS ONE 2013. [Google Scholar] [CrossRef]
- Bruce, V.J.; Lopez-Islas, M.; McNaughton, B.R. Resurfaced cell-penetrating nanobodies: A potentially general scaffold for intracellularly targeted protein discovery. Protein Sci. 2016. [Google Scholar] [CrossRef]
- Shin, S.M.; Choi, D.K.; Jung, K.; Bae, J.; Kim, J.S.; Park, S.W.; Song, K.H.; Kim, Y.S. Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic administration. Nat. Commun. 2017. [Google Scholar] [CrossRef]
Figure 1.
The single chain fragment variable format. The C-terminus of the light chain (VL) is linked the N-terminus of the heavy chain (VH) by a flexible glycine- and serine- rich linker.
Figure 1.
The single chain fragment variable format. The C-terminus of the light chain (VL) is linked the N-terminus of the heavy chain (VH) by a flexible glycine- and serine- rich linker.

Figure 2.
The tandem scFv platform. (A) A monospecific bivalent tandem scFv composed of two identical scFvs joined by a helical linker. (B) A bispecific bivalent scFv composed of two different scFvs joined by a helical linker.
Figure 2.
The tandem scFv platform. (A) A monospecific bivalent tandem scFv composed of two identical scFvs joined by a helical linker. (B) A bispecific bivalent scFv composed of two different scFvs joined by a helical linker.

Figure 3.
The structures of diabody, DART®, and TandAb fragments. (A) A bispecific diabody composed of two different chains, each containing a VL and VH from different antibodies, in a head-to-tail arrangement. (B) A bispecific dual affinity re-targeting (DART®) protein containing two distinct polypeptide chains held together by non-covalent interactions and a disulphide bond. (C) A TandAb composed of two diabodies linked in a linear arrangement to produce a tetravalent bispecific molecule.
Figure 3.
The structures of diabody, DART®, and TandAb fragments. (A) A bispecific diabody composed of two different chains, each containing a VL and VH from different antibodies, in a head-to-tail arrangement. (B) A bispecific dual affinity re-targeting (DART®) protein containing two distinct polypeptide chains held together by non-covalent interactions and a disulphide bond. (C) A TandAb composed of two diabodies linked in a linear arrangement to produce a tetravalent bispecific molecule.

Figure 4.
scFv fusion bispecific formats with an Fc domain. (A) IgG-scFv. Canonical IgG with a scFv fused to the C-terminus of the CH3 domain to produce a tetravalent bispecific molecule. (B) Fab-scFv-Fc IgG with one IgG Fab arm exchanged for a scFv.
Figure 4.
scFv fusion bispecific formats with an Fc domain. (A) IgG-scFv. Canonical IgG with a scFv fused to the C-terminus of the CH3 domain to produce a tetravalent bispecific molecule. (B) Fab-scFv-Fc IgG with one IgG Fab arm exchanged for a scFv.

Figure 5.
The structure of Fab and F(ab’)2 fragments. (A) Fab fragment composed of an LC (containing VL and CL) linked to an Fd (containing VH and CH1) by a disulphide bond between the CL and CH1 domains. (B) F(ab’)2 fragment composed of two Fab fragments joined by an IgG hinge region.
Figure 5.
The structure of Fab and F(ab’)2 fragments. (A) Fab fragment composed of an LC (containing VL and CL) linked to an Fd (containing VH and CH1) by a disulphide bond between the CL and CH1 domains. (B) F(ab’)2 fragment composed of two Fab fragments joined by an IgG hinge region.

Figure 6.
Camelid heavy-chain IgG and Nanobody® fragments. (A) The structure of heavy-chain IgG, composed of two heavy chains, each containing a VHH domain, a CH2 domain, and a CH3 domain. (B) Mono-, bi-, and tri-valent Nb formats with each VHH having a different antigen specificity. (C) A Nanobody® drug conjugate.
Figure 6.
Camelid heavy-chain IgG and Nanobody® fragments. (A) The structure of heavy-chain IgG, composed of two heavy chains, each containing a VHH domain, a CH2 domain, and a CH3 domain. (B) Mono-, bi-, and tri-valent Nb formats with each VHH having a different antigen specificity. (C) A Nanobody® drug conjugate.

Figure 7.
Shark heavy chain antibody (Ig-NAR), a dimer of heavy chains containing five constant domains and the antigen binding variable nucleotide antigen receptor (V-NAR).
Figure 7.
Shark heavy chain antibody (Ig-NAR), a dimer of heavy chains containing five constant domains and the antigen binding variable nucleotide antigen receptor (V-NAR).

Figure 8.
The uses of domain antibodies (dAbs). (A) IgG-dAb (also called a mAb-dAb). IgG with a dAb fused to the C terminus of each heavy chain to produce a bispecific tetravalent molecule. (B) Tandem dAb with an anti-HSA domain. The dAb against the target of interest is linked to an anti-HSA dAb to improve half-life. (C) AlbuDab®. A peptide linked to an anti-HSA dAb to improve the peptide’s half-life.
Figure 8.
The uses of domain antibodies (dAbs). (A) IgG-dAb (also called a mAb-dAb). IgG with a dAb fused to the C terminus of each heavy chain to produce a bispecific tetravalent molecule. (B) Tandem dAb with an anti-HSA domain. The dAb against the target of interest is linked to an anti-HSA dAb to improve half-life. (C) AlbuDab®. A peptide linked to an anti-HSA dAb to improve the peptide’s half-life.

Figure 9.
Simplified purification workflow for different fragment formats. A series of stages that can be used to purify common antibody fragments based on the presence of certain domains. The workflow typically includes affinity chromatography (where applicable) followed by multi-modal polishing stages, which may include size exclusion chromatography (SEC), cation (CIEX) or anion (AIEX) exchange, and hydrophobic interaction chromatography (HIC). Antibody fragments can also be purified using cation exchange chromatography (CIEX) or immobilised metal affinity chromatography (IMAC) as the initial capture step.
Figure 9.
Simplified purification workflow for different fragment formats. A series of stages that can be used to purify common antibody fragments based on the presence of certain domains. The workflow typically includes affinity chromatography (where applicable) followed by multi-modal polishing stages, which may include size exclusion chromatography (SEC), cation (CIEX) or anion (AIEX) exchange, and hydrophobic interaction chromatography (HIC). Antibody fragments can also be purified using cation exchange chromatography (CIEX) or immobilised metal affinity chromatography (IMAC) as the initial capture step.

Figure 10.
The structure and mechanism of action of MT103, an -CD3/-CD19 bispecific T-cell engager (BiTE®). The antigen binding site of each parental antibody is isolated and converted into an scFv format. The two scFvs are then joined by a flexible peptide linker to produce a bispecific moiety. The anti-CD19 scFv binds to tumour cells whilst the anti-CD3 scFv will bind passing T-cells, re-directing them to attack the tumour cell.
Figure 10.
The structure and mechanism of action of MT103, an -CD3/-CD19 bispecific T-cell engager (BiTE®). The antigen binding site of each parental antibody is isolated and converted into an scFv format. The two scFvs are then joined by a flexible peptide linker to produce a bispecific moiety. The anti-CD19 scFv binds to tumour cells whilst the anti-CD3 scFv will bind passing T-cells, re-directing them to attack the tumour cell.

Figure 11.
The structure and mechanism of action of ImmTAC®s. (A) The structure of an ImmTAC®, comprising an -CD3 scFv linked to a disulphide-stabilised, affinity-enhanced soluble T-cell receptor. (B) The T-cell receptor binds its target with picomolar affinity causing the ImmTAC® to cluster on the target cell. The anti-CD3 scFv then recruits passing T-cells by binding CD3 with nanomolar affinity. The clustering of CD3 on the T-cell leads to activation and re-direction of the T-cell to produce an immune response against the target cell.
Figure 11.
The structure and mechanism of action of ImmTAC®s. (A) The structure of an ImmTAC®, comprising an -CD3 scFv linked to a disulphide-stabilised, affinity-enhanced soluble T-cell receptor. (B) The T-cell receptor binds its target with picomolar affinity causing the ImmTAC® to cluster on the target cell. The anti-CD3 scFv then recruits passing T-cells by binding CD3 with nanomolar affinity. The clustering of CD3 on the T-cell leads to activation and re-direction of the T-cell to produce an immune response against the target cell.


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bates, A.; Power, C.A. David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments. Antibodies 2019, 8, 28. https://doi.org/10.3390/antib8020028
AMA Style
Bates A, Power CA. David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments. Antibodies. 2019; 8(2):28. https://doi.org/10.3390/antib8020028
Chicago/Turabian StyleBates, Adam, and Christine A. Power. 2019. "David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments" Antibodies 8, no. 2: 28. https://doi.org/10.3390/antib8020028
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.