IgG Antibody 3D Structures and Dynamics

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods for Characterizing Basic Antibody Structure
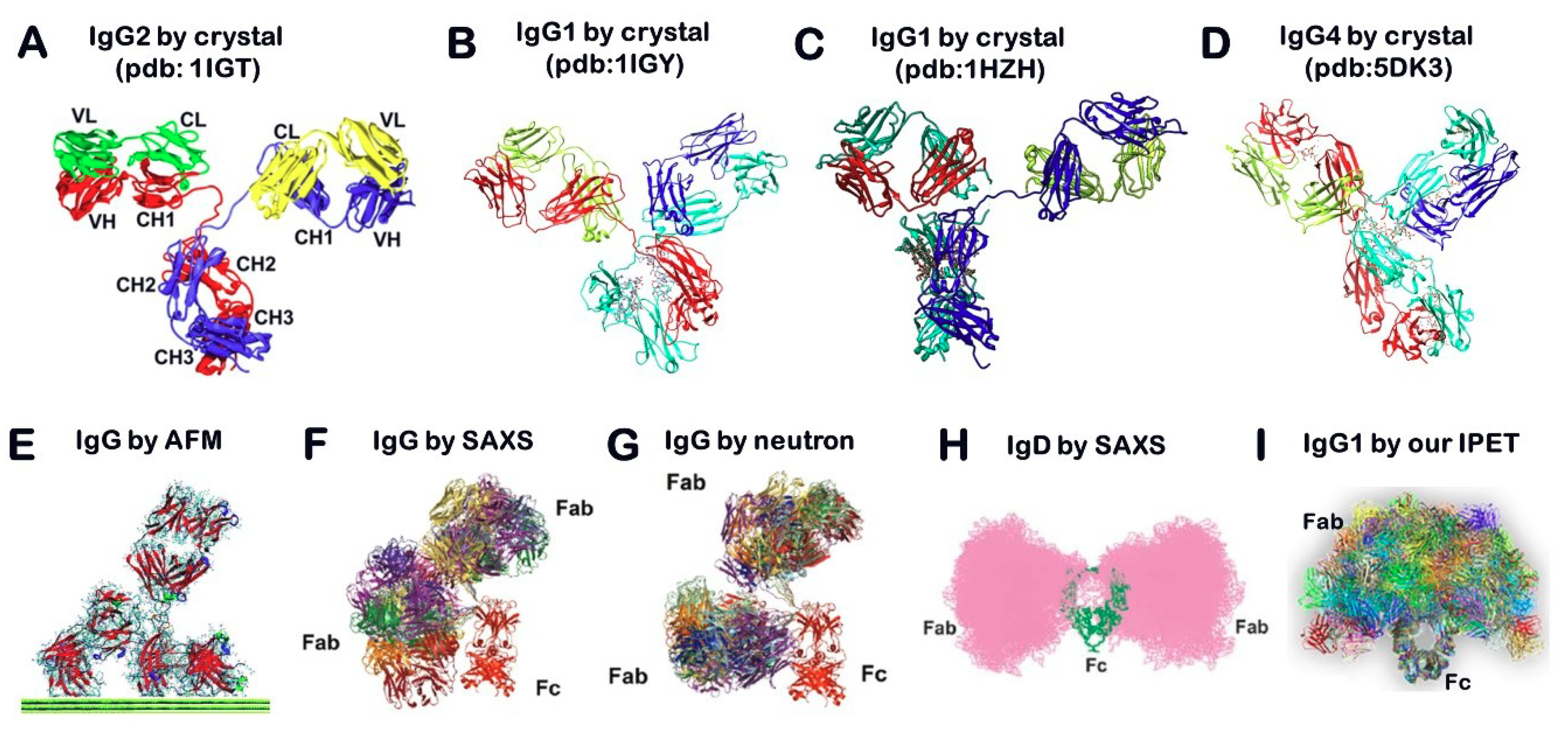
2.1. X-ray Crystallography
2.2. Atomic Force Microscopy (AFM)
2.3. Small Angle Scattering (SAS) Method
2.4. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.5. Transmission Electron Microscopy (TEM) Method for Imaging and 3D Reconstruction of Proteins
2.6. Negative-Staining (NS) and Positive-Staining (PS) Transmission Electron Microscopy (TEM) for Imaging Proteins
2.7. Optimized Negative-Staining (OpNS) for Imaging Protein
2.8. Cryo-Electron Microscopy (cryo-EM) for Imaging Protein
2.9. Cryo-Negative-Staining (cryo-NS) and Cryo-Positive Staining (cryo-PS) for Imaging Protein
2.10. Electron Crystallography for 3D Reconstruction of Protein
2.11. Single-Particle 3D Reconstruction for Averaging 3D Structure of Protein
2.12. Single-Particle Electron Tomography (ET) for Subvolume, Averaged 3D Structure of Proteins
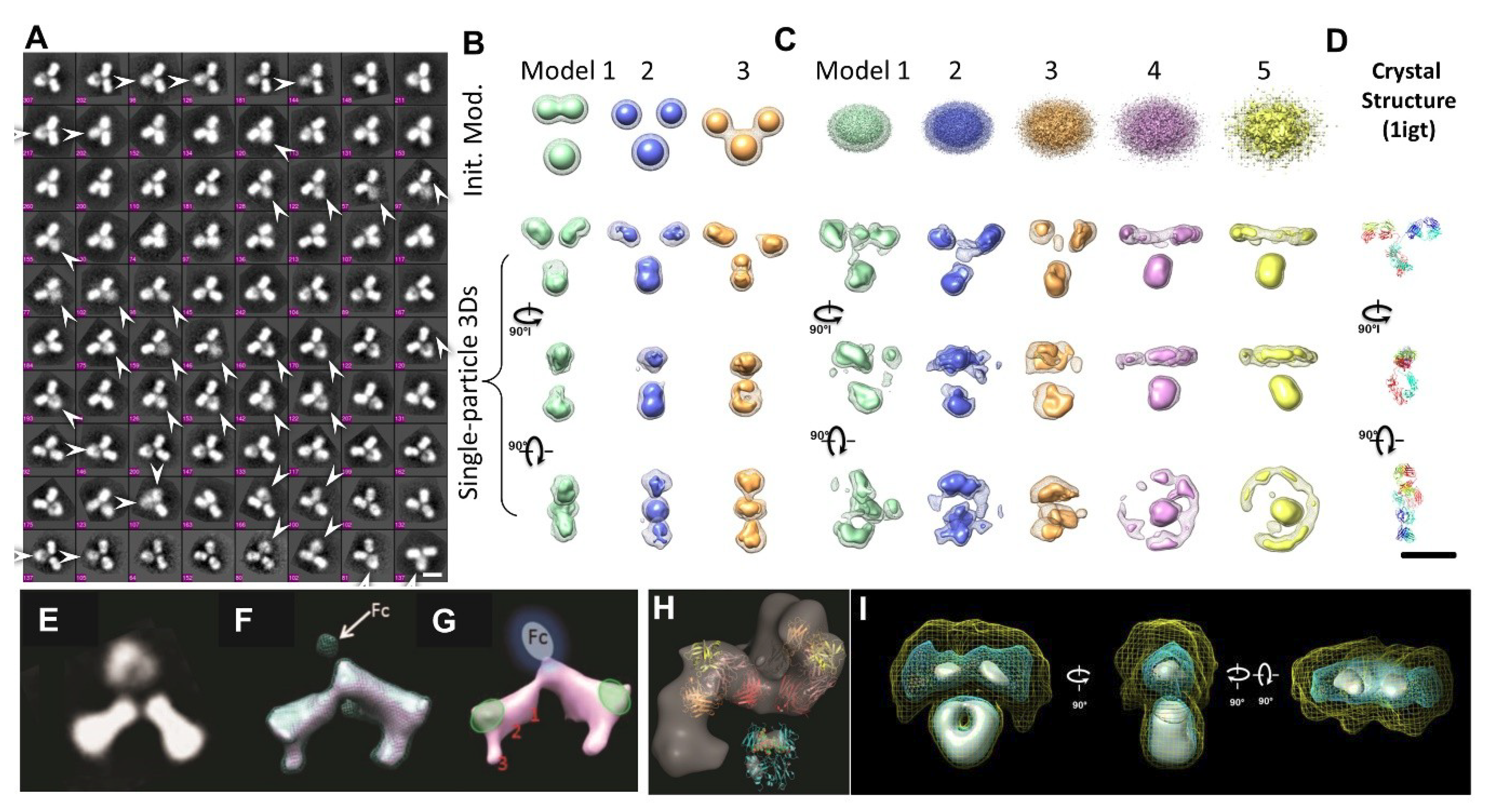
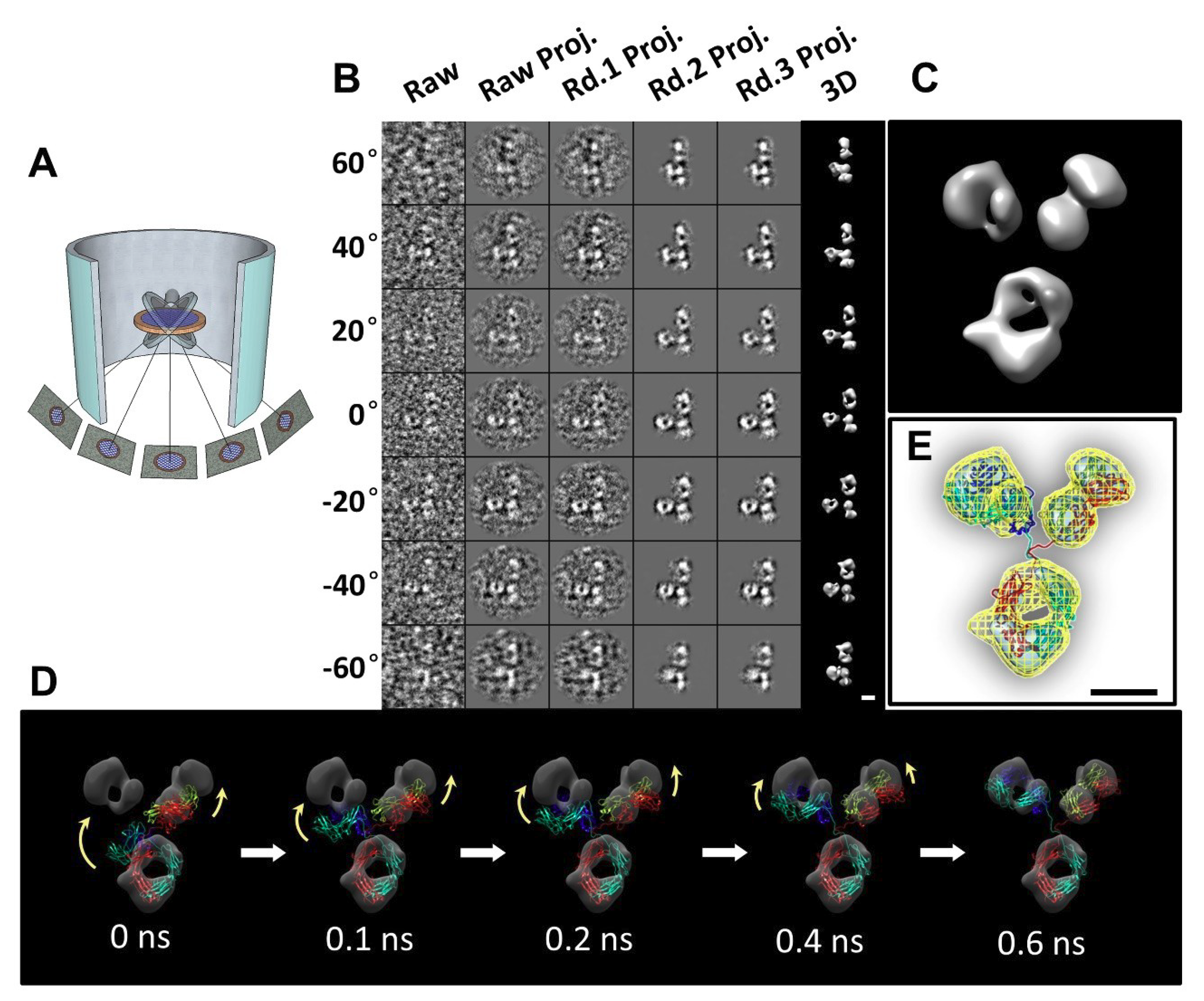
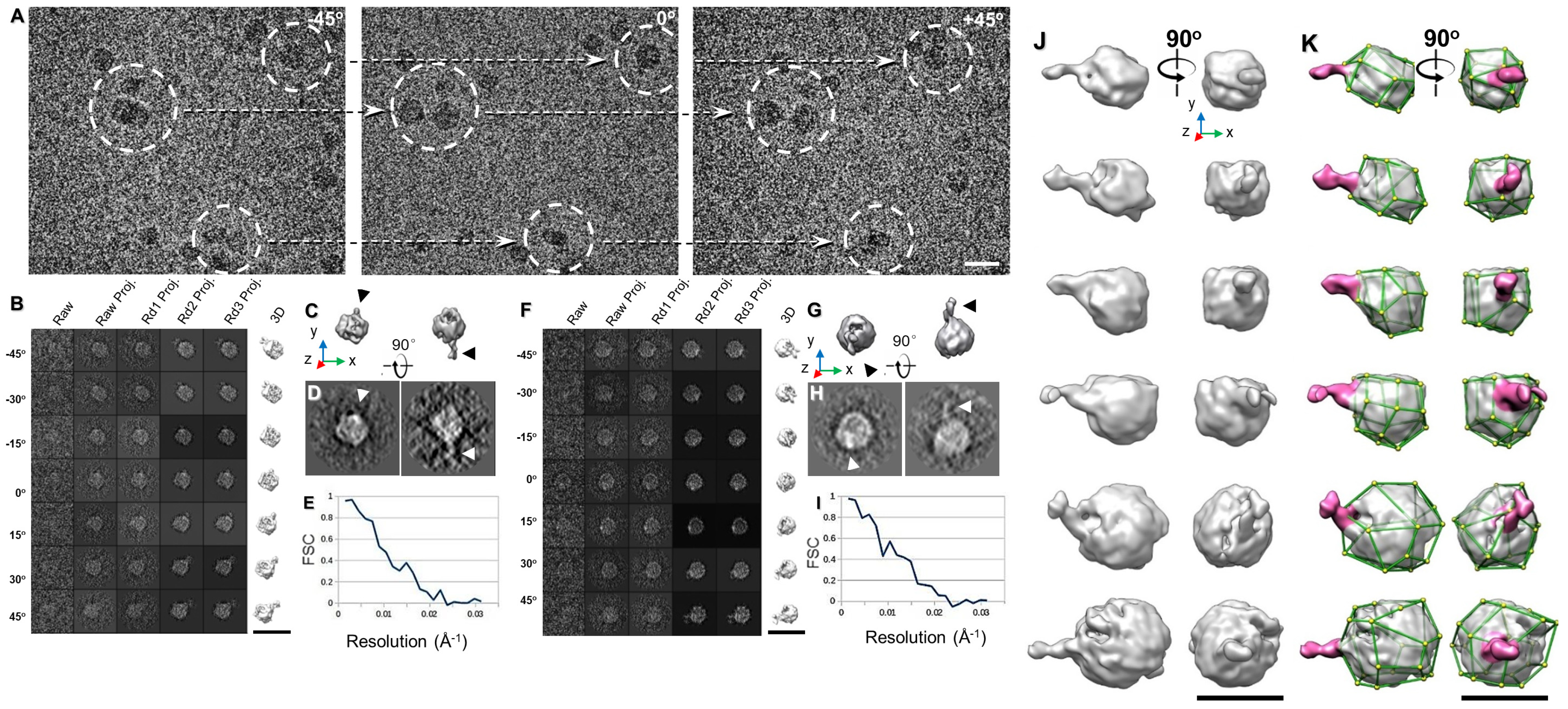
2.13. Individual-Particle Electron Tomography (IPET) for Single Molecule 3D Protein Structure
3. Applications of IPET 3D Reconstruction Approach on Antibody Studying
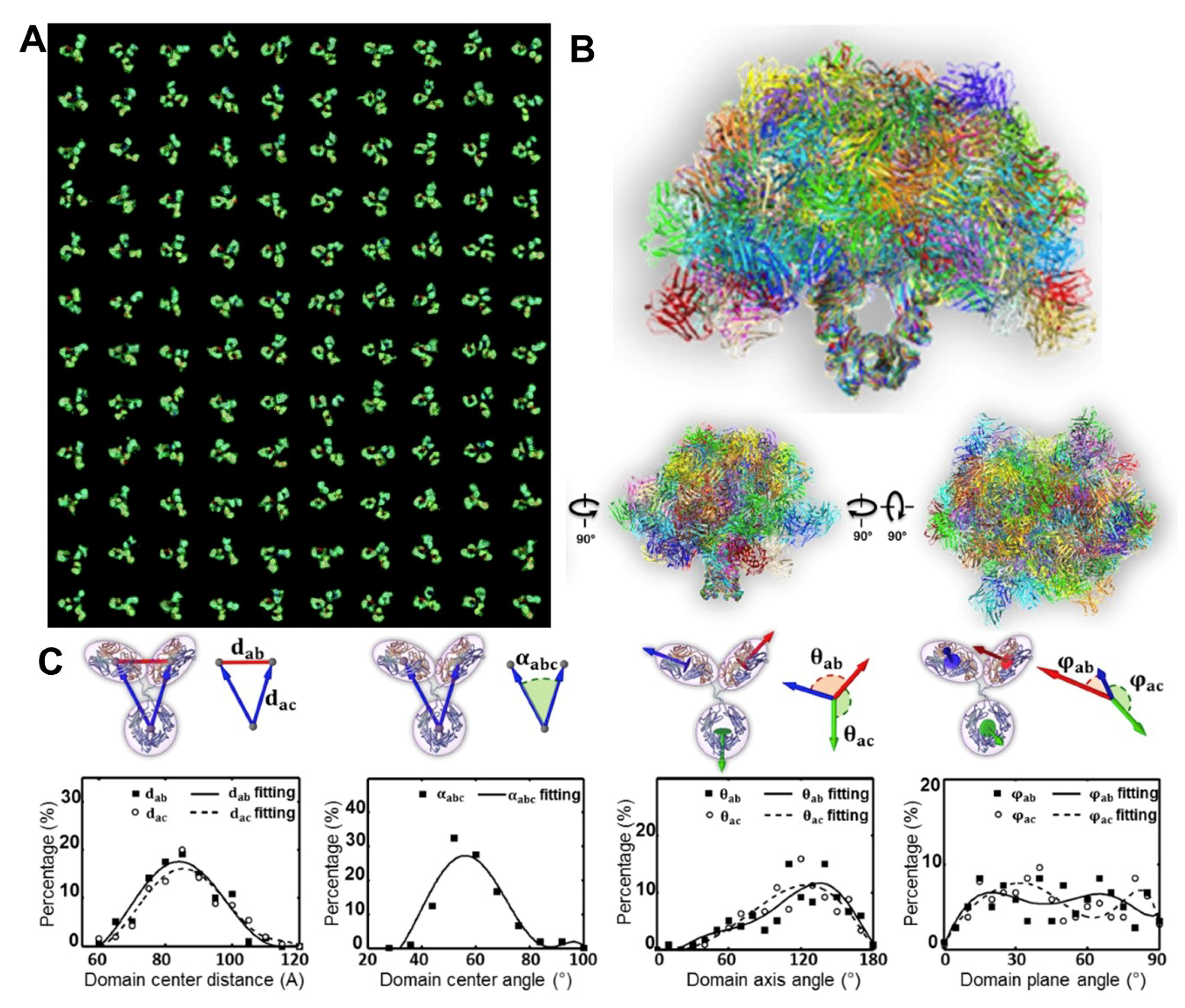
3.1. 3D Structure Variety of IgG1 Antibody by IPET
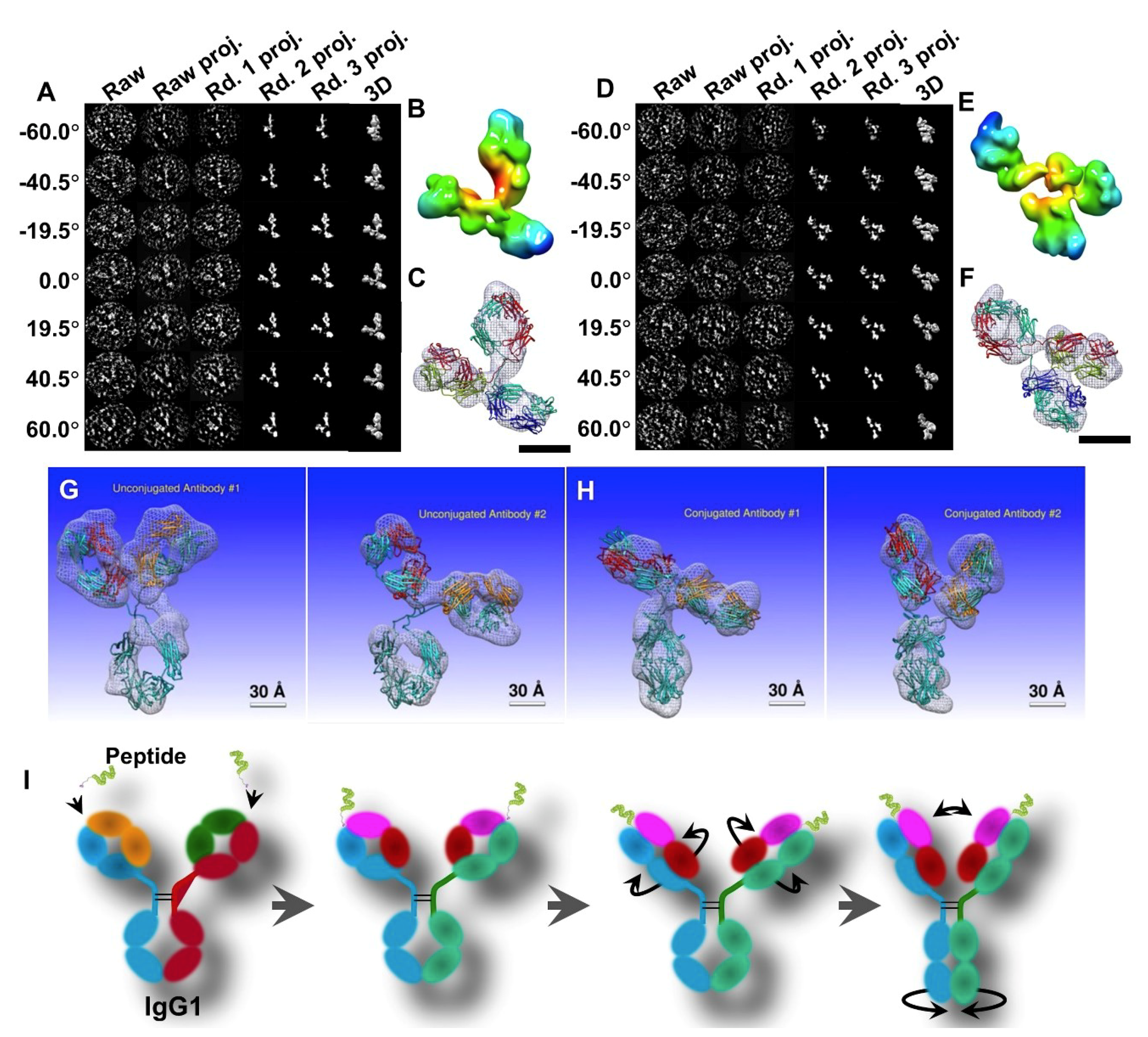
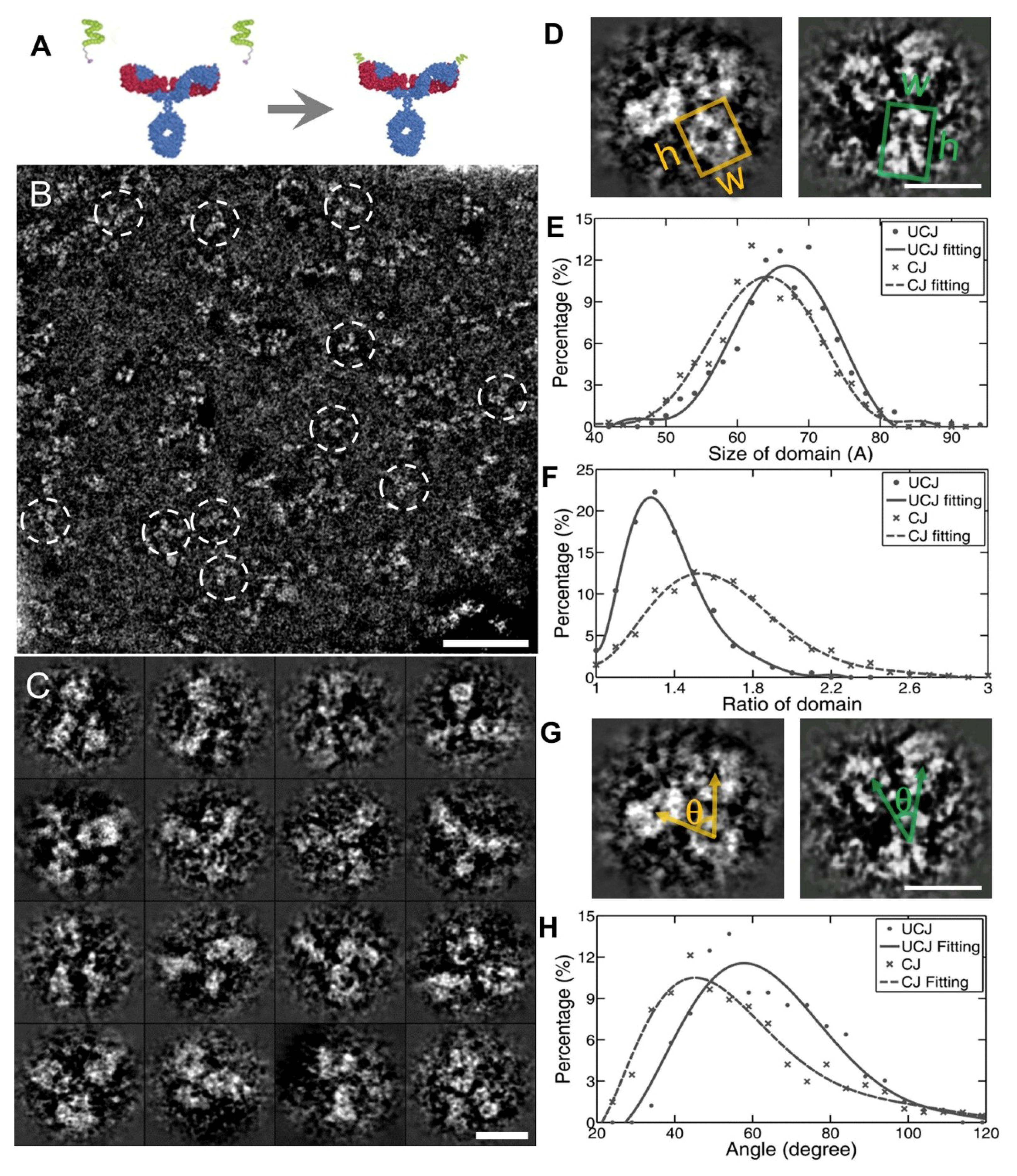
3.2. 3D Structure and Conformational Changes of Antibody Drug Conjugates (ADCs) by IPET
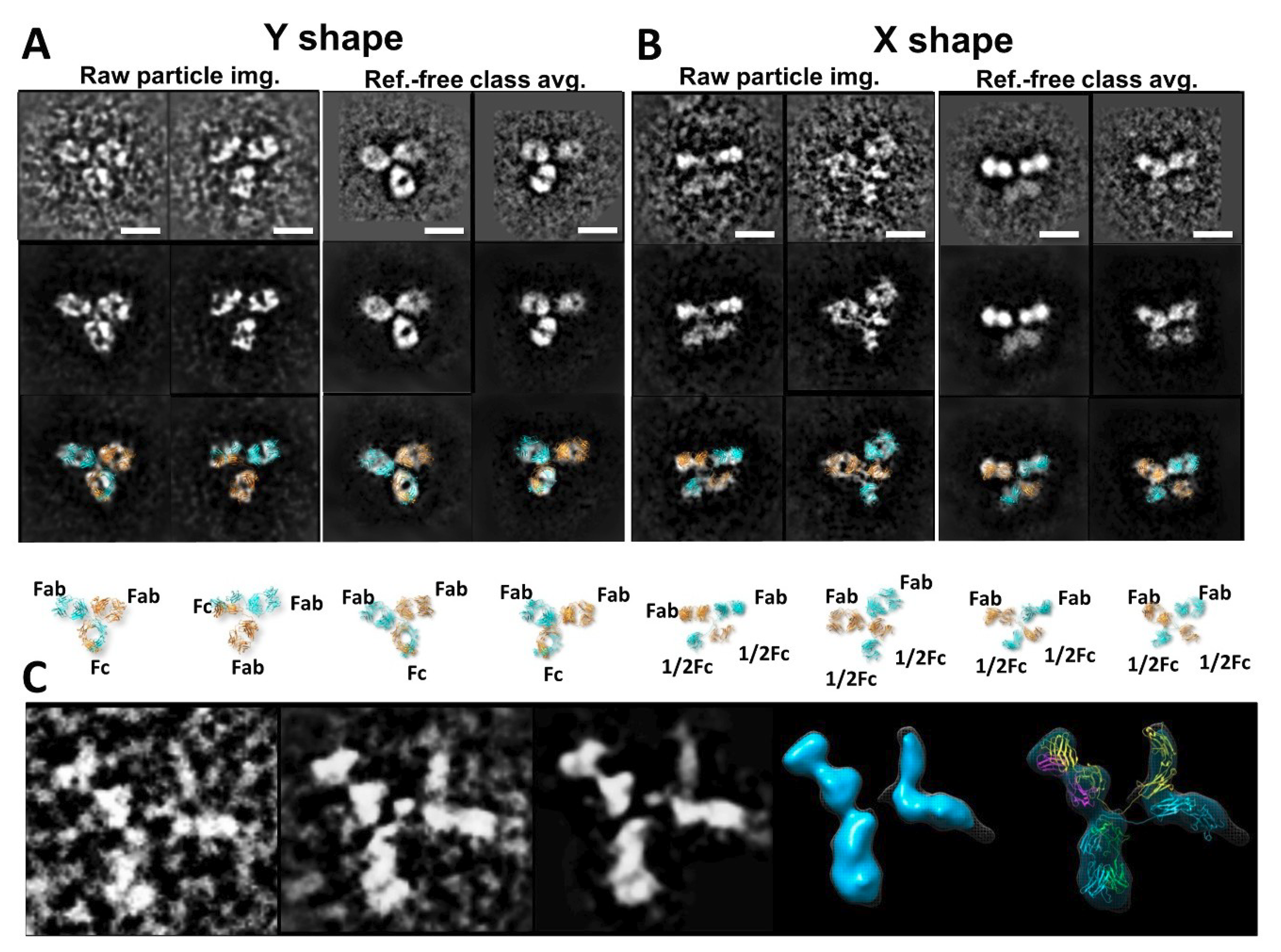
3.3. Bispecific Antibody Structure Variety by IPET
4. Future
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 3D | three-dimensional |
| EM | electron microscopy |
| ET | electron tomography |
| IgG | Immunoglobulin-G molecule |
| IPET | individual-particle electron tomography |
| NS | negative-staining |
| OpNS | optimized negative-staining |
| cryo-EM | cryo-electron microscopy |
References
- Abbas, A.K.; Lichtman, A.H.H.; Pillai, S. Basic Immunology: Functions and Disorders of the Immune System; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Schur, P.H. IgG subclasses. A historical perspective. Monogr. Allergy 1988, 23, 1–11. [Google Scholar] [PubMed]
- Campbell, A.M. Monoclonal Antibody Technology: The Production and Characterization of Roadent and Human Hybridomas; Elsevier: New York, NY, USA, 1984; Volume 13. [Google Scholar]
- Harris, L.J.; Larson, S.B.; Hasel, K.W.; Day, J.; Greenwood, A.; McPherson, A. The three-dimensional structure of an intact monoclonal antibody for canine lymphoma. Nature 1992, 360, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, A.; Beard, L.J.; Feldman, R.G. IgG subclass distribution of antibodies to bacterial and viral antigens. Pediatr. Infect. Dis. J. 1990, 9, S16–S24. [Google Scholar] [CrossRef] [PubMed]
- Gall, W.E.; Edelman, G.M. The covalent structure of a human gamma G-immunoglobulin. X. Intrachain disulfide bonds. Biochemistry 1970, 9, 3188–3196. [Google Scholar] [CrossRef] [PubMed]
- Dreker, L.; Schwarz, J.; Reichel, W.; Hilschmann, N. [Rule of antibody structure. the primary structure of a monoclonal IgG1 immunoglobulin (myeloma protein Nie), I: Purification and characterization of the protein, the L- and H-chains, the cyanogenbromide cleavage products, and the disulfide bridges (author’s transl)]. Hoppe-Seyler's Z. Physiol. Chem. 1976, 357, 1515–1540. [Google Scholar] [PubMed]
- Salonen, E.M.; Hovi, T.; Meurman, O.; Vesikari, T.; Vaheri, A. Kinetics of specific IgA, IgD, IgE, IgG, and IgM antibody responses in rubella. J. Med. Virol. 1985, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Parekh, R.B.; Dwek, R.A.; Sutton, B.J.; Fernandes, D.L.; Leung, A.; Stanworth, D.; Rademacher, T.W.; Mizuochi, T.; Taniguchi, T.; Matsuta, K.; et al. Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature 1985, 316, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, W.C.; Frazier-Jessen, M.R.; Wu, W.J.; Weir, A.; Hartsough, M.; Keegan, P.; Fuchs, C. Development and regulation of monoclonal antibody products: Challenges and opportunities. Cancer Metast. Rev. 2005, 24, 569–584. [Google Scholar] [CrossRef] [PubMed]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Hillan, K.J.; Novotny, W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem. Biophys. Res. Commun. 2005, 333, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Scapin, G.; Yang, X.; Prosise, W.W.; McCoy, M.; Reichert, P.; Johnston, J.M.; Kashi, R.S.; Strickland, C. Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat. Struct. Mol. Biol. 2015, 22, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Getts, D.R.; Getts, M.T.; McCarthy, D.P.; Chastain, E.M.; Miller, S.D. Have we overestimated the benefit of human(ized) antibodies? Mabs 2010, 2, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.T.; Aboody, K.S.; Najbauer, J. Strategies for enhancing antibody delivery to the brain. Biochim. Biophys. Acta 2011, 1816, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Vilhena, J.G.; Dumitru, A.C.; Herruzo, E.T.; Mendieta-Moreno, J.I.; Garcia, R.; Serena, P.A.; Perez, R. Adsorption orientations and immunological recognition of antibodies on graphene. Nanoscale 2016, 8, 13463–13475. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.S.; Martin, J.H. x ray crystallography. Mol. Pathol. MP 2000, 53, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.J.; Larson, S.B.; Hasel, K.W.; McPherson, A. Refined structure of an intact IgG2a monoclonal antibody. Biochemistry 1997, 36, 1581–1597. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.J.; Skaletsky, E.; McPherson, A. Crystallographic structure of an intact IgG1 monoclonal antibody. J. Mol. Biol. 1998, 275, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Saphire, E.O.; Parren, P.W.; Pantophlet, R.; Zwick, M.B.; Morris, G.M.; Rudd, P.M.; Dwek, R.A.; Stanfield, R.L.; Burton, D.R.; Wilson, I.A. Crystal structure of a neutralizing human IGG against HIV-1: A template for vaccine design. Science 2001, 293, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.; Lyubchenko, Y.; Muller, D. Atomic force microscopy: A powerful tool to observe biomolecules at work. Trends Cell Biol. 1999, 9, 77–80. [Google Scholar] [CrossRef]
- Preiner, J.; Kodera, N.; Tang, J.; Ebner, A.; Brameshuber, M.; Blaas, D.; Gelbmann, N.; Gruber, H.J.; Ando, T.; Hinterdorfer, P. IgGs are made for walking on bacterial and viral surfaces. Nat. Commun. 2014, 5, 4934. [Google Scholar] [CrossRef] [PubMed]
- Rayner, L.E.; Kadkhodayi-Kholghi, N.; Heenan, R.K.; Gor, J.; Dalby, P.A.; Perkins, S.J. The solution structure of rabbit IgG accounts for its interactions with the Fc receptor and complement C1q and its conformational stability. J. Mol. Biol. 2013, 425, 506–523. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Almogren, A.; Furtado, P.B.; Chowdhury, B.; Kerr, M.A.; Perkins, S.J. Semi-extended solution structure of human myeloma immunoglobulin D determined by constrained X-ray scattering. J. Mol. Biol. 2005, 353, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Paterson, Y.; Englander, S.W.; Roder, H. An antibody binding site on cytochrome c defined by hydrogen exchange and two-dimensional NMR. Science 1990, 249, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Kheddo, P.; Cliff, M.J.; Uddin, S.; van der Walle, C.F.; Golovanov, A.P. Characterizing monoclonal antibody formulations in arginine glutamate solutions using (1)H NMR spectroscopy. Mabs 2016, 8, 1245–1258. [Google Scholar] [CrossRef] [PubMed]
- Rosen, O.; Anglister, J. Epitope mapping of antibody-antigen complexes by nuclear magnetic resonance spectroscopy. Methods Mol. Biol. 2009, 524, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Abbott, W.M.; Damschroder, M.M.; Lowe, D.C. Current approaches to fine mapping of antigen-antibody interactions. Immunology 2014, 142, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Miyazawa, M.; Sakakura, M.; Terasawa, H.; Takahashi, H.; Shimada, I. Determination of the interface of a large protein complex by transferred cross-saturation measurements. J. Mol. Biol. 2002, 318, 245–249. [Google Scholar] [CrossRef]
- Zhang, L.; Ren, G. IPET and FETR: Experimental approach for studying molecular structure dynamics by cryo-electron tomography of a single-molecule structure. PLoS ONE 2012, 7, e30249. [Google Scholar] [CrossRef] [PubMed]
- Horne, R.W.; Wildy, P. An historical account of the development and applications of the negative staining technique to the electron microscopy of viruses. J. Microsc. 1979, 117, 103–122. [Google Scholar] [CrossRef] [PubMed]
- Farrant, J.L. An electron microscopic study of ferritin. Biochim. Biophys. Acta 1954, 13, 569–576. [Google Scholar] [CrossRef]
- Hall, C.E. Electron densitometry of stained virus particles. J. Biophys. Biochem. Cytol. 1955, 1, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Forte, T.M.; Nordhausen, R.W. Electron microscopy of negatively stained lipoproteins. Methods Enzymol. 1986, 128, 442–457. [Google Scholar] [PubMed]
- Colliex, C.; Cowley, J.M.; Dudarev, S.L.; Fink, M.; Gjonnes, J.; Hilderbrandt, R.; Howie, A.; Lynch, D.F.; Peng, L.M.; Ren, G.; et al. Electron Diffraction. In International Tables For Crystallography, 4 Ed.; Prince, E., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2006; Volume C, pp. 259–429. [Google Scholar]
- Ren, G.; Zuo, J.M.; Peng, L.-M. Accurate Measurements of Crystal Structure Factors Using a FEG Electron Microscope Using Digital Micrographs. Micron 1997, 28, 459–467. [Google Scholar] [CrossRef]
- Peng, L.M.; Ren, G.; Dudarev, S.L.; Whelan, M.J. Robust parameterization of elastic and absorptive electron atomic scattering factors. Acta Crystallogr. A 1996, 52, 257–276. [Google Scholar] [CrossRef]
- Peng, L.M.; Ren, G.; Dudarev, S.L.; Whelan, M.J. Debye-Waller factors and absorptive scattering factors of elemental crystals. Acta Crystallogr. A 1996, 52, 456–470. [Google Scholar] [CrossRef]
- Stouthamer, A.H.; Daems, W.T.; Eigner, J. Electron microscope studies of bacteriophage adsorption with negative and positive staining. Virology 1963, 20, 246–250. [Google Scholar] [CrossRef]
- Zobel, C.R.; Roe, J.Y. Positive staining of protein molecules for electron microscopy. Biochim. Biophys. Acta 1967, 133, 157–161. [Google Scholar] [CrossRef]
- Sander, B.; Golas, M.M. Visualization of bionanostructures using transmission electron microscopical techniques. Microsc. Res. Tech. 2011, 74, 642–663. [Google Scholar] [CrossRef] [PubMed]
- Melchior, V.; Hollingshead, C.J.; Cahoon, M.E. Stacking in Lipid Vesicle Tubulin Mixtures Is an Artifact of Negative Staining. J. Cell Biol. 1980, 86, 881–884. [Google Scholar] [CrossRef] [PubMed]
- De Carlo, S.; Harris, J.R. Negative staining and cryo-negative staining of macromolecules and viruses for TEM. Micron 2011, 42, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Allan, V.; Vale, R. Movement of Membrane Tubules Along Microtubules in-Vitro - Evidence for Specialized Sites of Motor Attachment. J. Cell Sci. 1994, 107, 1885–1897. [Google Scholar] [PubMed]
- Catte, A.; Patterson, J.C.; Jones, M.K.; Jerome, W.G.; Bashtovyy, D.; Su, Z.; Gu, F.; Chen, J.; Aliste, M.P.; Harvey, S.C.; et al. Novel changes in discoidal high density lipoprotein morphology: A molecular dynamics study. Biophys. J. 2006, 90, 4345–4360. [Google Scholar] [CrossRef] [PubMed]
- Forester, G.P.; Tall, A.R.; Bisgaier, C.L.; Glickman, R.M. Rat intestine secretes spherical high density lipoproteins. J. Biol. Chem. 1983, 258, 5938–5943. [Google Scholar] [PubMed]
- Gantz, D.L.; Walsh, M.T.; Small, D.M. Morphology of sodium deoxycholate-solubilized apolipoprotein B-100 using negative stain and vitreous ice electron microscopy. J. Lipid Res. 2000, 41, 1464–1472. [Google Scholar] [PubMed]
- Pentikainen, M.O.; Lehtonen, E.M.; Kovanen, P.T. Aggregation and fusion of modified low density lipoprotein. J. Lipid Res. 1996, 37, 2638–2649. [Google Scholar] [PubMed]
- Tall, A.R.; Green, P.H.; Glickman, R.M.; Riley, J.W. Metabolic fate of chylomicron phospholipids and apoproteins in the rat. J. Clin. Investig. 1979, 64, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Song, J.; Newhouse, Y.; Zhang, S.; Weisgraber, K.H.; Ren, G. An optimized negative-staining protocol of electron microscopy for apoE4 POPC lipoprotein. J. Lipid Res. 2010, 51, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Song, J.; Cavigiolio, G.; Ishida, B.Y.; Zhang, S.; Kane, J.P.; Weisgraber, K.H.; Oda, M.N.; Rye, K.A.; Pownall, H.J.; et al. Morphology and structure of lipoproteins revealed by an optimized negative-staining protocol of electron microscopy. J. Lipid Res. 2011, 52, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Gong, E.L.; Nichols, A.V.; Weisgraber, K.H.; Forte, T.M.; Shore, V.G.; Blanche, P.J. Discoidal complexes containing apolipoprotein E and their transformation by lecithin-cholesterol acyltransferase. Biochim. Biophys. Acta 1989, 1006, 317–328. [Google Scholar] [CrossRef]
- Schneeweis, L.A.; Koppaka, V.; Lund-Katz, S.; Phillips, M.C.; Axelsen, P.H. Structural analysis of lipoprotein E particles. Biochemistry 2005, 44, 12525–12534. [Google Scholar] [CrossRef] [PubMed]
- Raussens, V.; Drury, J.; Forte, T.M.; Choy, N.; Goormaghtigh, E.; Ruysschaert, J.M.; Narayanaswami, V. Orientation and mode of lipid-binding interaction of human apolipoprotein E C-terminal domain. Biochem. J. 2005, 387, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kan, H.Y.; Lavrentiadou, S.; Krieger, M.; Zannis, V. Reconstituted discoidal ApoE-phospholipid particles are ligands for the scavenger receptor BI. The amino-terminal 1-165 domain of ApoE suffices for receptor binding. J. Biol. Chem. 2002, 277, 21149–21157. [Google Scholar] [CrossRef] [PubMed]
- Innerarity, T.L.; Pitas, R.E.; Mahley, R.W. Binding of arginine-rich (E) apoprotein after recombination with phospholipid vesicles to the low density lipoprotein receptors of fibroblasts. J. Biol. Chem. 1979, 254, 4186–4190. [Google Scholar] [PubMed]
- Lu, B.; Morrow, J.A.; Weisgraber, K.H. Conformational reorganization of the four-helix bundle of human apolipoprotein E in binding to phospholipid. J. Biol. Chem. 2000, 275, 20775–20781. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yan, F.; Zhang, S.; Lei, D.; Charles, M.A.; Cavigiolio, G.; Oda, M.; Krauss, R.M.; Weisgraber, K.H.; Rye, K.A.; et al. Structural basis of transfer between lipoproteins by cholesteryl ester transfer protein. Nat. Chem. Biol. 2012, 8, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, L.; Tong, H.; Peng, B.; Rames, M.J.; Zhang, S.; Ren, G. 3D Structural Fluctuation of IgG1 Antibody Revealed by Individual Particle Electron Tomography. Sci. Rep.-UK 2015, 5, 9803. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Zhang, L.; Kaspar, A.; Rames, M.J.; Huang, L.; Woodnutt, G.; Ren, G. Peptide-conjugation induced conformational changes in human IgG1 observed by optimized negative-staining and individual-particle electron tomography. Sci. Rep.-UK 2013, 3, 1089. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Li, C.; Lei, M.; Lundin, V.; Lee, H.Y.; Ninonuevo, M.; Lin, K.; Han, G.; Sandoval, W.; Lei, D.; et al. Structural and Functional Characterization of a Hole-Hole Homodimer Variant in a "Knob-Into-Hole" Bispecific Antibody. Anal. Chem. 2017, 89, 13494–13501. [Google Scholar] [CrossRef] [PubMed]
- Rames, M.; Yu, Y.; Ren, G. Optimized negative staining: A high-throughput protocol for examining small and asymmetric protein structure by electron microscopy. J. Vis. Exp. 2014, e51087. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Reddy, M.V.; Liu, J.; Kalichava, A.; Liu, J.; Zhang, L.; Chen, F.; Wang, Y.; Holthauzen, L.M.; White, M.A.; et al. Molecular Architecture of Contactin-associated Protein-like 2 (CNTNAP2) and Its Interaction with Contactin 2 (CNTN2). J. Biol. Chem. 2016, 291, 24133–24147. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Wang, Y.; Chen, F.; Tong, H.; Reddy, M.V.; Luo, L.; Seshadrinathan, S.; Zhang, L.; Holthauzen, L.M.; Craig, A.M.; et al. Calsyntenin-3 molecular architecture and interaction with neurexin 1alpha. J. Biol. Chem. 2014, 289, 34530–34542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lei, D.; Smith, J.M.; Zhang, M.; Tong, H.; Zhang, X.; Lu, Z.; Liu, J.; Alivisatos, A.P.; Ren, G. Three-dimensional structural dynamics and fluctuations of DNA-nanogold conjugates by individual-particle electron tomography. Nat. Commun. 2016, 7, 11083. [Google Scholar] [CrossRef] [PubMed]
- Lei, D.; Marras, A.E.; Liu, J.; Huang, C.; Zhou, L.; Castro, C.E.; Su, H.; Ren, G. 3D Structural Dynamics of DNA Origami Bennett Linkages Using Individual-Particle Electron Tomography. Nat. Commun. 2018, in press. [Google Scholar] [CrossRef]
- Zhang, M.; Charles, R.; Tong, H.; Zhang, L.; Patel, M.; Wang, F.; Rames, M.J.; Ren, A.; Rye, K.A.; Qiu, X.; et al. HDL surface lipids mediate CETP binding as revealed by electron microscopy and molecular dynamics simulation. Sci. Rep.-UK 2015, 5, 8741. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Kuang, Y.L.; Lei, D.; Zhai, X.; Zhang, M.; Krauss, R.M.; Ren, G. Polyhedral 3D structure of human plasma very low density lipoproteins by individual particle cryo-electron tomography1. J. Lipid Res. 2016, 57, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Segrest, J.P.; Jones, M.K.; Catte, A.; Manchekar, M.; Datta, G.; Zhang, L.; Zhang, R.; Li, L.; Patterson, J.C.; Palgunachari, M.N.; et al. Surface Density-Induced Pleating of a Lipid Monolayer Drives Nascent High-Density Lipoprotein Assembly. Structure 2015, 23, 1214–1226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tong, H.; Garewal, M.; Ren, G. Optimized negative-staining electron microscopy for lipoprotein studies. Biochim. Biophys. Acta 2013, 1830, 2150–2159. [Google Scholar] [CrossRef] [PubMed]
- Cressey, D.; Callaway, E. Cryo-electron microscopy wins chemistry Nobel. Nature 2017, 550, 167. [Google Scholar] [CrossRef] [PubMed]
- Merk, A.; Bartesaghi, A.; Banerjee, S.; Falconieri, V.; Rao, P.; Davis, M.I.; Pragani, R.; Boxer, M.B.; Earl, L.A.; Milne, J.L.S.; et al. Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery. Cell 2016, 165, 1698–1707. [Google Scholar] [CrossRef] [PubMed]
- Dubochet, J.; Lepault, J.; Freeman, R.; Berriman, J.A.; Homo, J.C. Electron microscopy of frozen water and aqueous solutions. J. Microsc. 1982, 128, 219–237. [Google Scholar] [CrossRef]
- Li, X.; Mooney, P.; Zheng, S.; Booth, C.R.; Braunfeld, M.B.; Gubbens, S.; Agard, D.A.; Cheng, Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 2013, 10, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Adrian, M.; Dubochet, J.; Fuller, S.D.; Harris, J.R. Cryo-negative staining. Micron 1998, 29, 145–160. [Google Scholar] [CrossRef]
- Kartha, G. Combination of multiple isomorphous replacement and anomalous dispersion data for protein structure determination. 3. Refinement of heavy atom positions by the least-squares method. Acta Crystallogr. 1965, 19, 883–885. [Google Scholar] [CrossRef] [PubMed]
- Kartha, G.; Parthasarathy, R. Combination of Multiple Isomorphous Replacement and Anomalous Dispersion Data for Protein Structure Determination. I. Determination of Heavy-Atom Positions in Protein Derivatives. Acta Crystallogr. 1965, 18, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Kartha, G.; Parthasarathy, R. Combination of Multiple Isomorphous Replacement and Anomalous Dispersion Data for Protein Structure Determination. Ii. Correlation of the Heavy-Atom Positions in Different Isomorphous Protein Crystals. Acta Crystallogr. 1965, 18, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Scheres, S.H. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012, 180, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Grigorieff, N. FREALIGN: High-resolution refinement of single particle structures. J. Struct. Biol. 2007, 157, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Vieth, M.; Timm, D.E.; Humblet, C.; Schneidman-Duhovny, D.; Chemmama, I.E.; Sali, A.; Zeng, W.; Lu, J.; Liu, L. Reconstruction of 3D structures of MET antibodies from electron microscopy 2D class averages. PLoS ONE 2017, 12, e0175758. [Google Scholar] [CrossRef] [PubMed]
- Correia, I.; Sung, J.; Burton, R.; Jakob, C.G.; Carragher, B.; Ghayur, T.; Radziejewski, C. The structure of dual-variable-domain immunoglobulin molecules alone and bound to antigen. Mabs 2013, 5, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Grigorieff, N.; Penczek, P.A.; Walz, T. A primer to single-particle cryo-electron microscopy. Cell 2015, 161, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Milne, J.L.; Subramaniam, S. Cryo-electron tomography of bacteria: Progress, challenges and future prospects. Nat. Rev. Microbiol. 2009, 7, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Koning, R.I.; Koster, A.J. Cryo-electron tomography in biology and medicine. Ann. Anat. 2009, 191, 427–445. [Google Scholar] [CrossRef] [PubMed]
- Frey, T.G.; Perkins, G.A.; Ellisman, M.H. Electron tomography of membrane-bound cellular organelles. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 199–224. [Google Scholar] [CrossRef] [PubMed]
- Galaz-Montoya, J.G.; Ludtke, S.J. The advent of structural biology in situ by single particle cryo-electron tomography. Biophys. Rep. 2017, 3, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Kosinski, J.; Mosalaganti, S.; von Appen, A.; Teimer, R.; DiGuilio, A.L.; Wan, W.; Bui, K.H.; Hagen, W.J.; Briggs, J.A.; Glavy, J.S.; et al. Molecular architecture of the inner ring scaffold of the human nuclear pore complex. Science 2016, 352, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Bartesaghi, A.; Lecumberry, F.; Sapiro, G.; Subramaniam, S. Protein secondary structure determination by constrained single-particle cryo-electron tomography. Structure 2012, 20, 2003–2013. [Google Scholar] [CrossRef] [PubMed]
- Turonova, B.; Schur, F.K.M.; Wan, W.; Briggs, J.A.G. Efficient 3D-CTF correction for cryo-electron tomography using NovaCTF improves subtomogram averaging resolution to 3.4A. J. Struct. Biol. 2017, 199, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, H.; Zhang, L.; Rames, M.; Zhang, M.; Yu, Y.; Peng, B.; Celis, C.D.; Xu, A.; Zou, Q.; et al. Fully Mechanically Controlled Automated Electron Microscopic Tomography. Sci. Rep.-UK 2016, 6, 29231. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.M.; Park, J.; Ercius, P.; Kim, K.; Hellebusch, D.J.; Crommie, M.F.; Lee, J.Y.; Zettl, A.; Alivisatos, A.P. High-resolution EM of colloidal nanocrystal growth using graphene liquid cells. Science 2012, 336, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Leedom, T.A.; Do, J.; Huang, H.; Lai, J.; Johnson, K.; Osothprarop, T.F.; Rizzo, J.D.; Doppalapudi, V.R.; Bradshaw, C.W.; et al. Antitumor efficacy of a thrombospondin 1 mimetic CovX-body. Transl. Oncol. 2011, 4, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Said, N.; Gahoual, R.; Kuhn, L.; Beck, A.; Francois, Y.N.; Leize-Wagner, E. Structural characterization of antibody drug conjugate by a combination of intact, middle-up and bottom-up techniques using sheathless capillary electrophoresis—Tandem mass spectrometry as nanoESI infusion platform and separation method. Anal. Chim. Acta 2016, 918, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Debaene, F.; Boeuf, A.; Wagner-Rousset, E.; Colas, O.; Ayoub, D.; Corvaia, N.; Van Dorsselaer, A.; Beck, A.; Cianferani, S. Innovative native MS methodologies for antibody drug conjugate characterization: High resolution native MS and IM-MS for average DAR and DAR distribution assessment. Anal. Chem. 2014, 86, 10674–10683. [Google Scholar] [CrossRef] [PubMed]

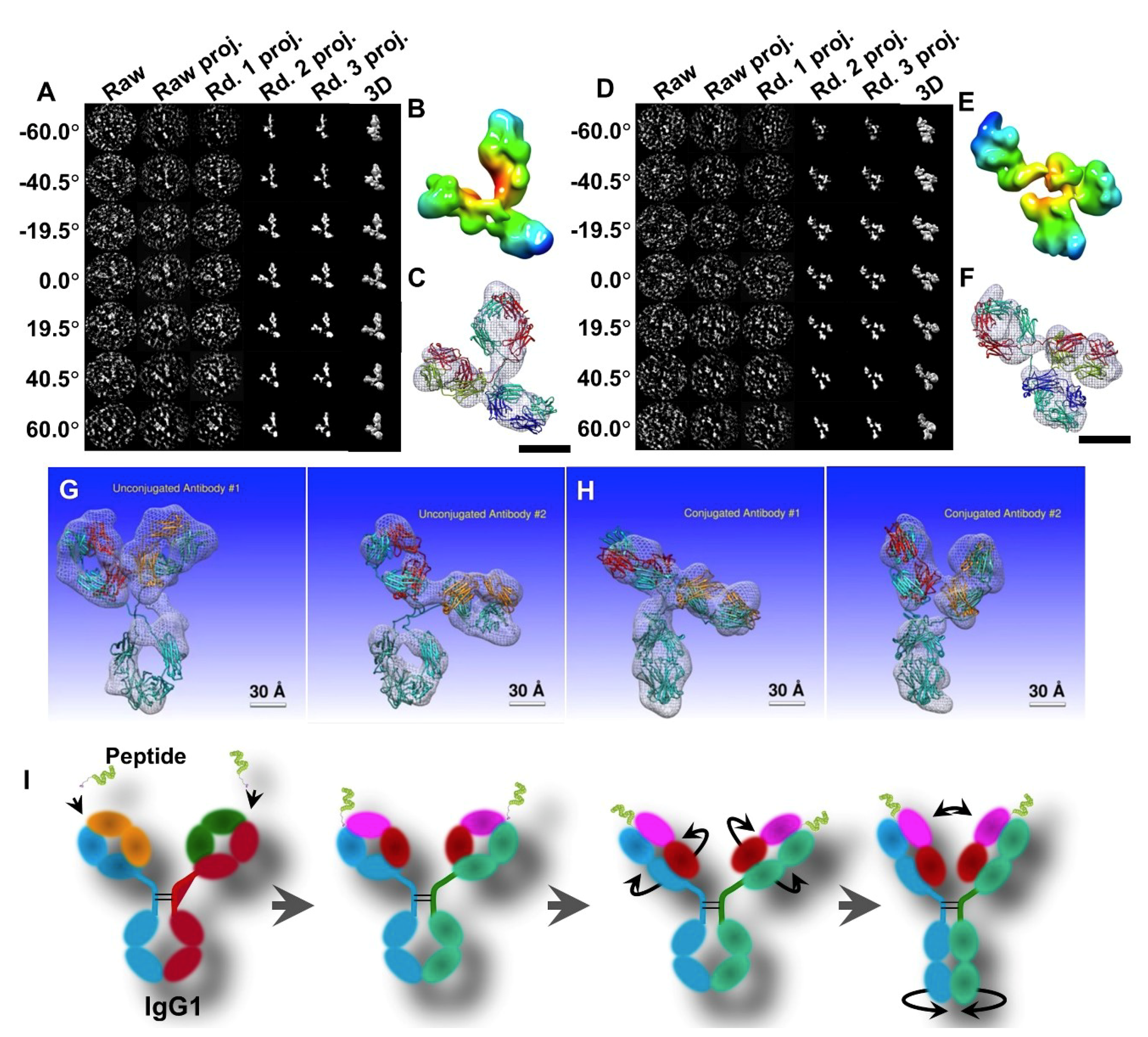
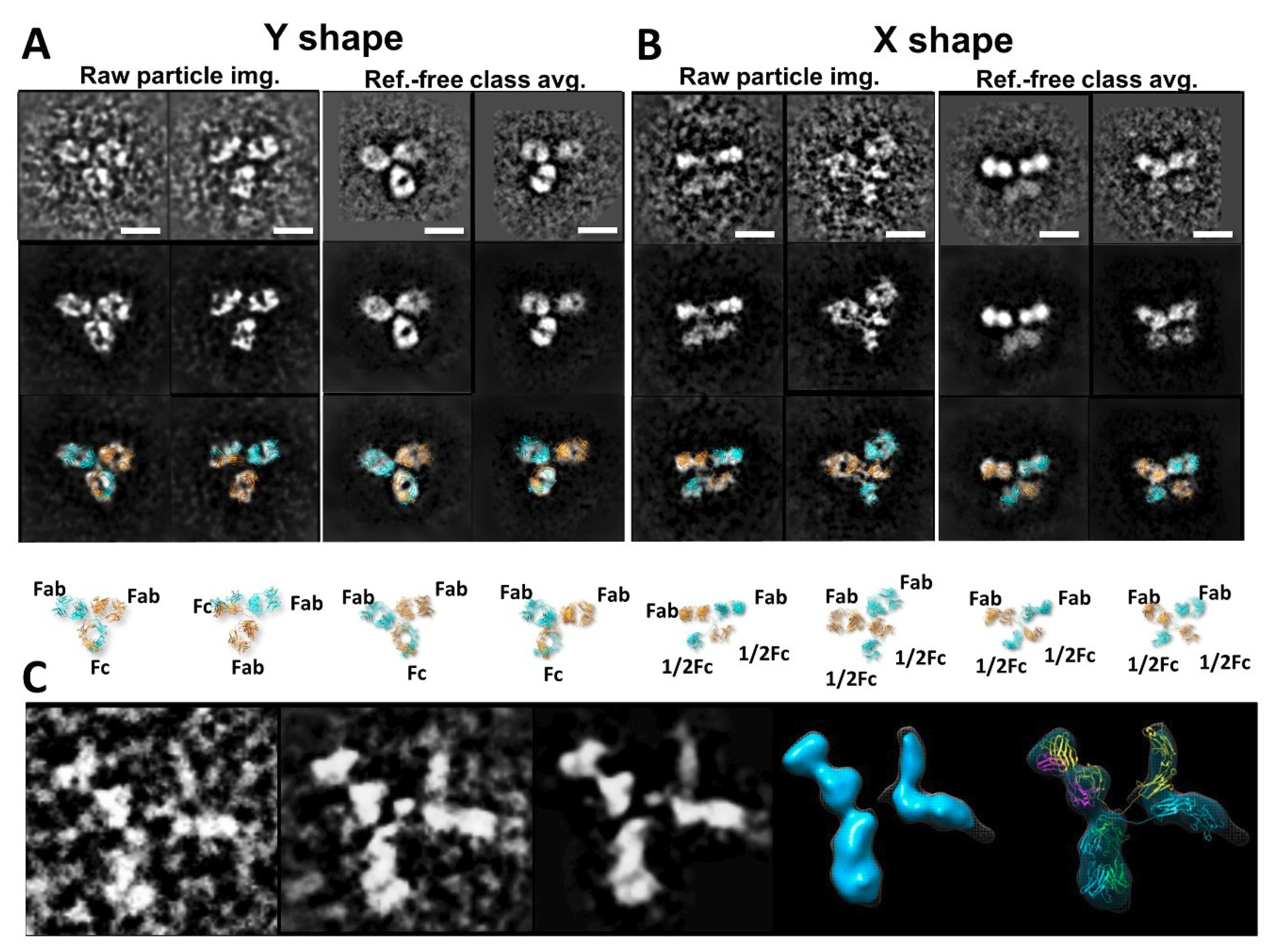
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jay, J.W.; Bray, B.; Qi, Y.; Igbinigie, E.; Wu, H.; Li, J.; Ren, G. IgG Antibody 3D Structures and Dynamics. Antibodies 2018, 7, 18. https://doi.org/10.3390/antib7020018
Jay JW, Bray B, Qi Y, Igbinigie E, Wu H, Li J, Ren G. IgG Antibody 3D Structures and Dynamics. Antibodies. 2018; 7(2):18. https://doi.org/10.3390/antib7020018
Chicago/Turabian StyleJay, Jacob White, Brinkley Bray, Yaozhi Qi, Eseosaserea Igbinigie, Hao Wu, Jinping Li, and Gang Ren. 2018. "IgG Antibody 3D Structures and Dynamics" Antibodies 7, no. 2: 18. https://doi.org/10.3390/antib7020018