Small-Format Drug Conjugates: A Viable Alternative to ADCs for Solid Tumours?

 ,
,
Abstract
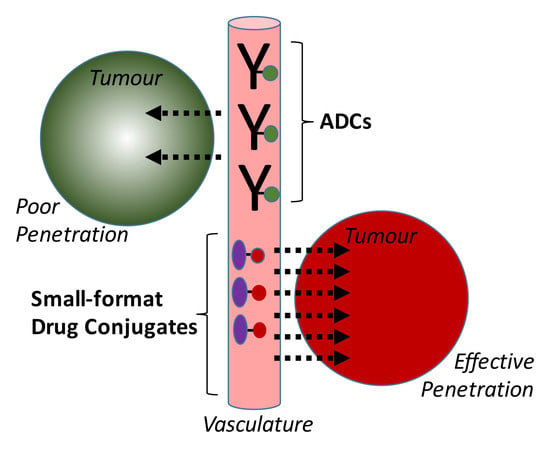
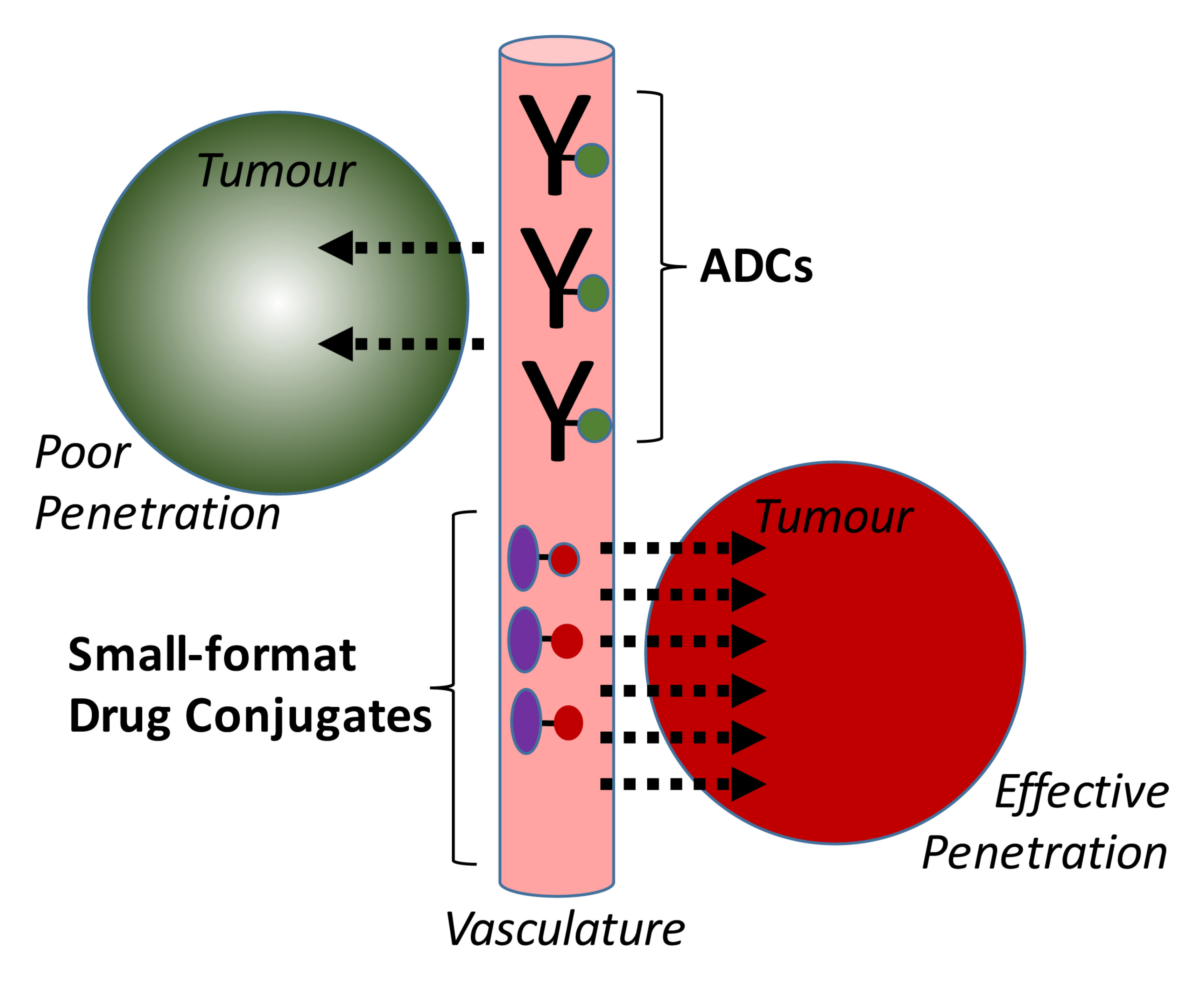
:
1. Introduction
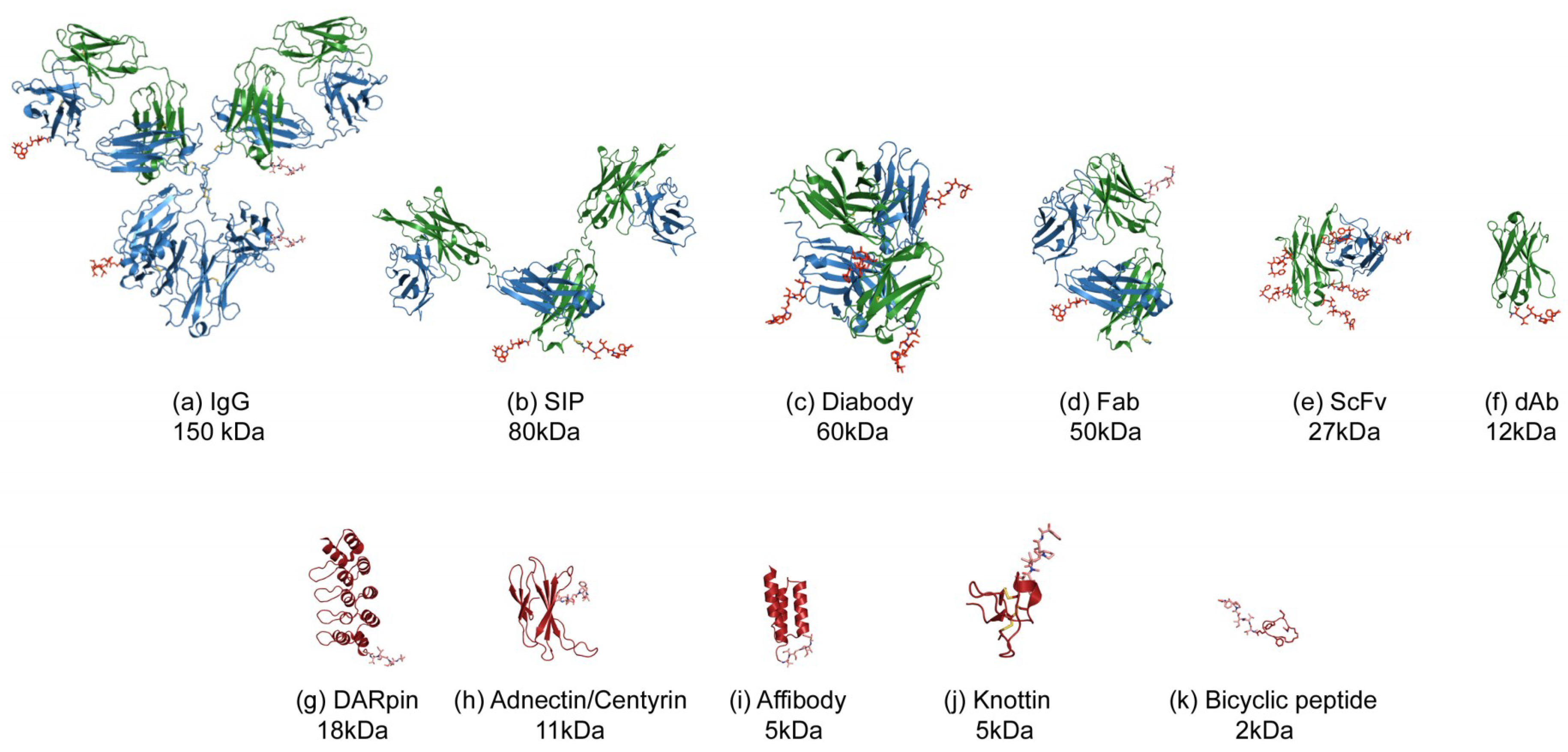
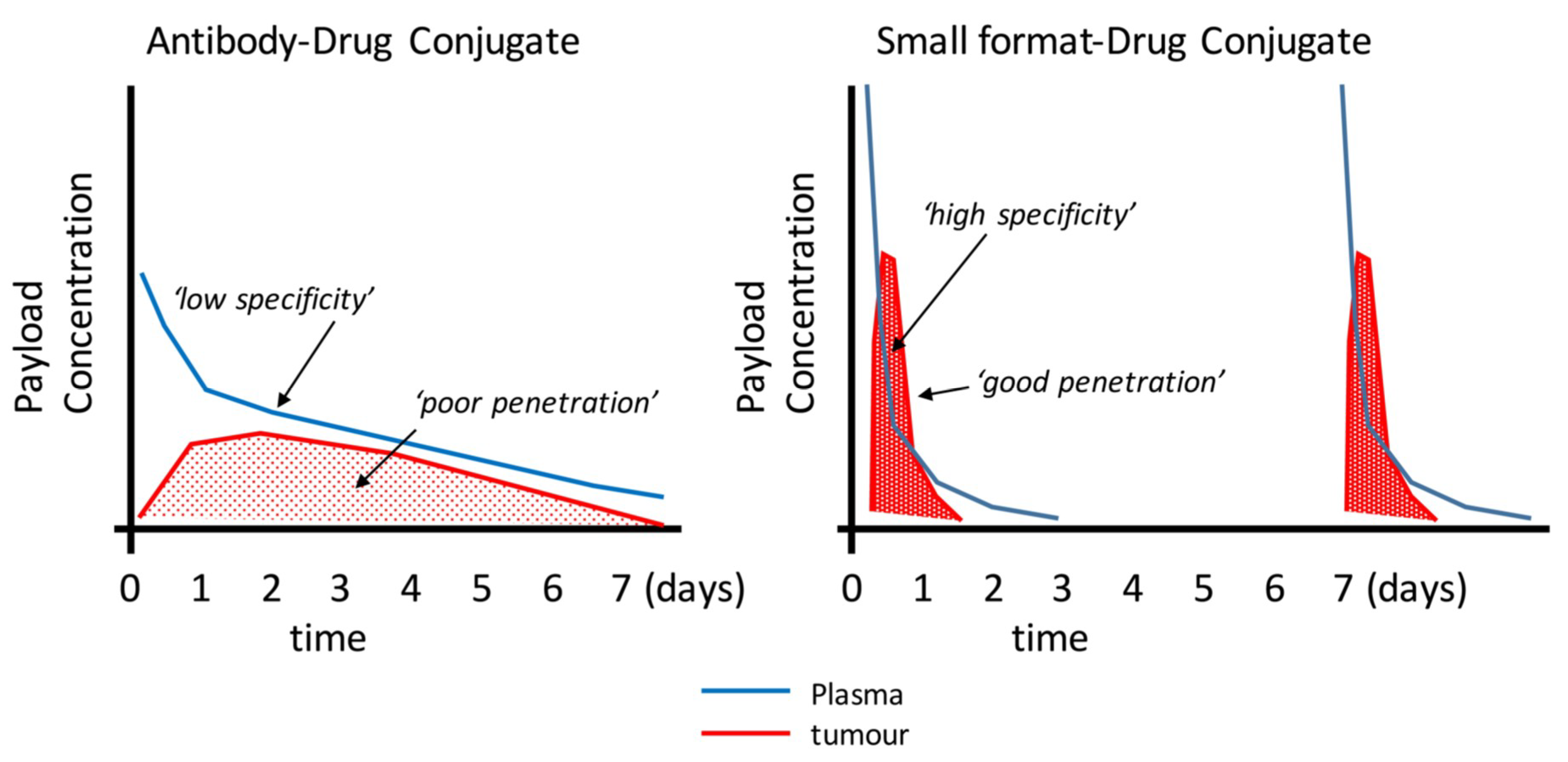
2. Uptake and Tumour Penetration of Smaller Binding Formats
3. Antibody Fragment–Drug Conjugates
3.1. Fab–Drug Conjugates
3.2. Diabody–Drug Conjugates
3.3. SIP–Drug Conjugates
3.4. scFv–Drug Conjugates
3.5. Domain Antibody–Drug Conjugates
4. Scaffold–Drug Conjugates (SDCs)
4.1. Affibody–Drug Conjugates
4.2. Fibronectin Type III–Drug Conjugates
4.3. Cystine Knot–Drug Conjugates
4.4. DARPin Drug Conjugates
4.5. Abdurin–Drug Conjugates
5. Peptide–Drug Conjugates
5.1. Pentarin–Drug Conjugates
5.2. ‘Bicycle’–Drug Conjugates
5.3. RGD Peptide–Drug Conjugates
6. Small-Molecule–Drug Conjugates (SMDCs)
6.1. CA-IX Ligand–Drug Conjugates
6.2. PSMA Ligand–Drug Conjugates
6.3. Folate Ligand–Drug Conjugates
6.4. Phosphatidylserine Ligand–Drug Conjugates
6.5. Asialoglycoprotein Receptor Ligand–Drug Conjugates
7. Discussion and Conclusions
Conflicts of Interest
References
- Available online: https://clinicaltrials.gov (accessed on 5 January 2018).
- Available online: https://pharma.globaldata.com/ (accessed on 2 November 2017).
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.beacon-intelligence.com (accessed on 2 February 2017).
- Lambert, J.M.; Morris, C.Q. Antibody-Drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Adv. Ther. 2017, 34, 1015–1035. [Google Scholar] [CrossRef] [PubMed]
- Beckman, R.A.; Weiner, L.M.; Davis, H.M. Antibody constructs in cancer therapy Protein engineering strategies to improve exposure in solid tumors. Cancer 2007, 109, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Deonarain, M.P.; Yahioglu, G.; Stamati, I.; Marklew, J. Emerging formats for next-generation antibody drug conjugates. Expert Opin. Drug Discov. 2015, 10, 463–481. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Krippendorff, B.F.; Sharma, S.; Walz, A.C.; Lavé, T.; Shah, D.K. Influence of molecular size on tissue distribution of antibody fragments. MAbs 2016, 8, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Xenaki, K.T.; Oliveira, S.; van Bergen En Henegouwen, P.M.P. Antibody or Antibody Fragments: Implications for Molecular Imaging and Targeted Therapy of Solid Tumors. Front. Immunol. 2017, 8, 1287. [Google Scholar] [CrossRef] [PubMed]
- Freise, A.C.; Wu, A.M. In vivo imaging with antibodies and engineered fragments. Mol. Immunol. 2015, 67, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.; Phipps, C.; Edginton, A.; Blay, J. Pharmacokinetic Considerations for Antibody-Drug Conjugates against Cancer. Pharm. Res. 2017, 34, 2579–2595. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.J.; Shaletsky, E.; McPerson, A. Crystallographic structure of an intact IgG1 monoclonal antibody. J. Mol. Biol. 1998, 275, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Terrabube, V.; Tam, A.; Stochaj, W.; Fennell, B.; Lin, L.; Stahl, M.; LaVallie, E.; Somers, W.; Finlay, W.; et al. Optimization of a SCFV-BASED BIOTHERAPEUTIC by CDR Side-Chain Clash Repair. 2018. Available online: http://www.jbc.org/content/suppl/2015/10/29/M115.688010.DC1/jbc.M115.688010-1.pdf (accessed on 5 January 2018).
- Mosyak, L.; Root, A. Development of PF-06671008, a Highly Potent Anti-P-cadherin/Anti-CD3 Bispecific DART Molecule with Extended Half-Life for the Treatment of Cancer. Antibodies 2016, 5, 6. [Google Scholar]
- Bloch, Y.; Bouchareychas, L.; Merceron, R.; Składanowska, K.; Van den Bossche, L.; Detry, S.; Govindarajan, S.; Elewaut, D.; Haerynck, F.; Dullaers, M.; et al. Structural Activation of Pro-inflammatory Human Cytokine IL-23 by Cognate IL-23 Receptor Enables Recruitment of the Shared Receptor IL-12Rβ1. Immunity 2018, 48, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.B.; Smits, C.; Wong, A.S.W.; Stock, D.; Christie, M.; Sandin, S.; Stewart, A.G. Cryo-EM analysis of a domain antibody bound rotary ATPase complex. J. Struct. Biol. 2017, 197, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Seeger, M.A.; Zbinden, R.; Flütsch, A.; Gutte, P.G.; Engeler, S.; Roschitzki-Voser, H.; Grütter, M.G. Design, construction, and characterization of a second-generation DARP in library with reduced hydrophobicity. Protein Sci. 2013, 22, 1239–1257. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, V.; Krystek, S.R., Jr.; Bush, A.; Wei, A.; Emanuel, S.L.; Das Gupta, R.; Janjua, A.; Cheng, L.; Murdock, M.; Abramczyk, B.; et al. Structures of adnectin/protein complexes reveal an expanded binding footprint. Structure 2012, 20, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Högbom, M.; Eklund, M.; Nygren, P.A.; Nordlund, P. Structural basis for recognition by an in vitro evolved affibody. Proc. Natl. Acad. Sci. USA 2003, 100, 3191–3196. [Google Scholar] [CrossRef] [PubMed]
- Krätzner, R.; Debreczeni, J.E.; Pape, T.; Schneider, T.R.; Wentzel, A.; Olmar, H.; Sheldrick, G.M.; Uson, I. Structure of Ecballium elaterium trypsin inhibitor II (EETI-II): A rigid molecular scaffold. Acta Crystallogr. D 2005, 61, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Di Bonaventura, I.; Jin, X.; Visini, R.; Probst, D.; Javor, S.; Gan, B.H.; Michau, D.G.; Natalello, A.; Doglia, S.M.; Köhler, T.; et al. Chemical space guided discovery of antimicrobial bridged bicyclic peptides against Pseudomonas aeruginosa and its biofilms. Chem. Sci. 2017, 8, 6784–6798. [Google Scholar] [CrossRef] [PubMed]
- Waight, A.B.; Bargsten, K.; Doronina, S.; Steinmetz, M.O.; Sussman, D.; Prota, A.E. Structural Basis of Microtubule Destabilization by Potent Auristatin Anti-Mitotics. PLoS ONE 2016, 11, e0160890. [Google Scholar] [CrossRef] [PubMed]
- Drake, P.M.; Rabuka, D. Recent Developments in ADC Technology: Preclinical Studies Signal Future Clinical Trends. BioDrugs 2017, 31, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.P.; Schier, R.; McCall, A.M.; Simmons, H.H.; Horak, E.M.; Alpaugh, R.K.; Marks, J.D.; Weiner, L.M. High affinity restricts the localization and tumor penetration of single-chain Fv antibody molecules. Cancer Res. 2001, 61, 4750–4755. [Google Scholar] [PubMed]
- Beckman, R.A.; von Roemeling, R.; Scott, A.M. Monoclonal antibody dose determination and biodistribution into solid tumors. Ther. Deliv. 2011, 2, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Thurber, G.M.; Wittrup, K.D. A mechanistic compartmental model for total antibody uptake in tumors. J. Theor. Biol. 2012, 314, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Orcutt, K.D.; Adams, G.P.; Wu, A.M.; Silva, M.D.; Harwell, C.; Hoppin, J.; Matsumura, M.; Kotsuma, M.; Greenberg, J.; Scott, A.M.; et al. Molecular Simulation of Receptor Occupancy and Tumor Penetration of an Antibody and Smaller Scaffolds: Application to Molecular Imaging. Mol. Imaging Biol. 2017, 19, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Saggar, J.K.; Yu, M.; Wang, M.; Tannock, I.F. Mechanisms of Drug Resistance Related to the Microenvironment of Solid Tumors and Possible Strategies to Inhibit Them. Cancer J. 2015, 21, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance. Adv. Drug Deliv. Rev. 2008, 60, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug penetration into solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Vasalou, C.; Helmlinger, G.; Gomes, B. A mechanistic tumor penetration model to guide antibody drug conjugate design. PLoS ONE 2015, 10, e0118977. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, S.I.; Lou, J.; Shaller, C.C.; Tang, Y.; Klein-Szanto, J.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res. 2011, 71, 2250–2259. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Delivery of molecular and cellular medicine to solid tumors. Adv. Drug Deliv. Rev. 2001, 46, 149–168. [Google Scholar] [CrossRef]
- Yokota, T.; Milenic, D.E.; Whitlow, M.; Schlom, J. Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res. 1992, 52, 3402–3408. [Google Scholar] [PubMed]
- Dennis, M.S.; Jin, H.; Dugger, D. Imaging tumors with an albumin-binding Fab, a novel tumor-targeting agent. Cancer Res. 2007, 67, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Freeman, D.J.; McDorman, K.; Ogbagabriel, S.; Kozlosky, C.; Yang, B.B.; Doshi, S.; Perez-Ruxio, J.J.; Fanslow, W.; Starnes, C.; Radinsky, R. Tumor penetration and epidermal growth factor receptor saturation by panitumumab correlate with antitumor activity in a preclinical model of human cancer. Mol. Cancer 2012, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- Cilliers, C.; Menezes, B.; Nessler, I.; Linderman, J.; Thurber, G. Improved Tumor Penetration and Single-Cell Targeting of Antibody Drug Conjugates Increases Anticancer Efficacy and Host Survival. Cancer Res. 2018, 78, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, V.; Maruani, A.; Caddick, S. Recent advances in the construction of antibody-drug conjugates. Nat. Chem. 2016, 8, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, J.; Zhao, P. In vitro efficacy of immuno-chemotherapy with anti-EGFR human Fab-Taxol conjugate on A431 epidermoid carcinoma cells. Cancer Biol. Ther. 2007, 6, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ding, G.; Gao, Q.; Sun, J.; Zhang, Q.; Du, L.; Qiu, Z.; Wang, C.; Zheng, F.; Sun, B.; et al. A human anti-c-Met Fab fragment conjugated with doxorubicin as targeted chemotherapy for hepatocellular carcinoma. PLoS ONE 2013, 8, e63093. [Google Scholar] [CrossRef] [PubMed]
- Badescu, G.; Bryant, P.; Bird, M.; Henseleit, K.; Swierkosz, J.; Parekh, V.; Tommasi, R.; Pawlisz, E.; Jurlewicz, K.; Farys, M.; et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjug. Chem. 2014, 25, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Wang, X.; Zheng, F.; Wang, C.; Tang, Q.; Tang, X.; Xu, N.; Zhang, H.; Zhang, D.; Xiong, L.; et al. The tumor-inhibitory effectiveness of a novel anti-Trop2 Fab conjugate in pancreatic cancer. Oncotarget 2016, 7, 24810–24823. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhang, J.; Zhang, Y.; Ma, G.; Su, Z. Specific Conjugation of the Hinge Region for Homogeneous Preparation of Antibody Fragment-Drug Conjugate: A Case Study for Doxorubicin-PEG-anti-CD20 Fab’ Synthesis. Bioconjug. Chem. 2016, 27, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Puthenveetil, S.; Loganzo, F.; He, H.; Dirico, K.; Green, M.; Teske, J.; Musto, S.; Clark, T.; Rago, B.; Koehn, F.; et al. Natural Product Splicing Inhibitors: A New Class of Antibody-Drug Conjugate (ADC) Payloads. Bioconjug. Chem. 2016, 27, 1880–1888. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Axup, J.Y.; Lawson, B.R.; Yun, H.; Tardif, V.; Choi, S.H.; Zhou, Q.; Dubrovska, A.; Biroc, S.L.; Marsden, R.; et al. Bispecific small molecule-antibody conjugate targeting prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 17796–17801. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; McDonagh, C.F.; Westendorf, L.; Brown, L.L.; Sussman, D.; Feist, T.; Lyon, R.; Alley, S.C.; Okeley, N.M.; Zhang, X.; et al. Anti-CD30 diabody-drug conjugates with potent antitumor activity. Mol. Cancer Ther. 2008, 7, 2486–2497. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.avipep.com.au/technology/background-on-avibodies/ (accessed on 5 January 2018).
- Bernardes, G.J.; Casi, G.; Trüssel, S.; Hartmann, I.; Schwager, K.; Scheuermann, J.; Neri, D. A traceless vascular-targeting antibody-drug conjugate for cancer therapy. Angew. Chem. Int. Ed. Engl. 2012, 51, 941–944. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.; Hartmann, I.; Perrino, E.; Casi, G.; Brighton, S.; Jelesarov, I.; Bernardes, G.J.L.; Neri, D. Spacer length shapes drug release and therapeutic efficacy of traceless disulfide-linked ADCs targeting the tumor neovasculature. Chem. Sci. 2013, 4, 297–302. [Google Scholar] [CrossRef]
- Perrino, E.; Steiner, M.; Krall, N.; Bernardes, G.J.; Pretto, F.; Casi, G.; Neri, D. Curative properties of noninternalizing antibody-drug conjugates based on maytansinoids. Cancer Res. 2014, 74, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Gébleux, R.; Stringhini, M.; Casanova, R.; Soltermann, A.; Neri, D. Non-internalizing antibody-drug conjugates display potent anti-cancer activity upon proteolytic release of monomethyl auristatin E in the subendothelial extracellular matrix. Int. J. Cancer 2017, 140, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Casi, G.; Huguenin-Dezot, N.; Zuberbühler, K.; Scheuermann, J.; Neri, D. Site-specific traceless coupling of potent cytotoxic drugs to recombinant antibodies for pharmacodelivery. J. Am. Chem. Soc. 2012, 134, 5887–5892. [Google Scholar] [CrossRef] [PubMed]
- Gébleux, R.; Wulhfard, S.; Casi, G.; Neri, D. Antibody Format and Drug Release Rate Determine the Therapeutic Activity of Noninternalizing Antibody-Drug Conjugates. Mol. Cancer Ther. 2015, 14, 2606–2612. [Google Scholar] [CrossRef] [PubMed]
- List, T.; Casi, G.; Neri, D. A chemically defined trifunctional antibody-cytokine-drug conjugate with potent antitumor activity. Mol. Cancer Ther. 2014, 13, 2641–2652. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, Y.H.; Li, Y.H.; Jiang, Y.; Xie, P.L.; Zhou, G.H.; Li, G.C. Anti-hepatoma human single-chain Fv antibody and adriamycin conjugates with potent antitumor activity. Int. Immunopharmacol. 2014, 18, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Kuimova, M.K.; Bhatti, M.; Deonarain, M.; Yahioglu, G.; Levitt, J.A.; Stamati, I.; Suhling, K.; Phillips, D. Fluorescence characterisation of multiply-loaded anti-HER2 single chain Fv-photosensitizer conjugates suitable for photodynamic therapy. Photochem. Photobiol. Sci. 2007, 6, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, M.; Yahioglu, G.; Milgrom, L.R.; Garcia-Maya, M.; Chester, K.A.; Deonarain, M.P. Targeted photodynamic therapy with multiply-loaded recombinant antibody fragments. Int. J. Cancer 2008, 122, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Hauler, F.; Dziunycz, P.; Schwager, K.; Soltermann, A.; Pretto, F.; Alonso, C.; Hofbauer, G.F.; Boyle, R.W.; Neri, D. A chemically modified antibody mediates complete eradication of tumours by selective disruption of tumour blood vessels. Br. J. Cancer 2011, 104, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Bauerschlag, D.; Meinhold-Heerlein, I.; Maass, N.; Bleilevens, A.; Bräutigam, K.A.; Rawashdeh, W.; Di Fiore, S.; Haugg, A.M.; Gremse, F.; Steitz, J.; et al. Detection and Specific Elimination of EGFR(+) Ovarian Cancer Cells Using a Near Infrared Photoimmunotheranostic Approach. Pharm. Res. 2017, 34, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Pye, H.; Butt, M.A.; Reinert, H.W.; Maruani, A.; Nunes, J.P.; Marklew, J.S.; Qurashi, M.; Funnell, L.; May, A.; Stamati, I.; et al. A HER2 selective theranostic agent for surgical resection guidance and photodynamic therapy. Photochem. Photobiol. Sci. 2016, 15, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Deonarain, M.P.; Yahioglu, G.; Stamati, I.; Saouros, S.; Kapadnis, P.D. Biological Materials and Uses Thereof. PCT Patent WO 2,016,046,574, 31 March 2016. [Google Scholar]
- Deonarain, M.P. Fragment Antibody Fragment Drug Conjugates (FDCs): A unique drug class or just smaller ADCs? In Proceedings of the Protein Engineering Summit (PEGS) Conference, Lisbon, Portugal, 13–17 November 2017. [Google Scholar]
- Rapid Optimal Assembly of Building Blocks to Create Humabody® Products. Available online: https://www.crescendobiologics.com/humabody/humabody-therapeutics/ (accessed on 5 January 2018).
- Vazquez-Lombardi, R.; Phan, T.G.; Zimmermann, C.; Lowe, D.; Jermutus, L.; Christ, D. Challenges and opportunities for non-antibody scaffold drugs. Drug Discov. Today 2015, 20, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Non-Antibody Protein Scaffolds: Drugs and Diagnostics Market, 2017-2030. Available online: https://www.rootsanalysis.com/reports/view_document/non-antibody-protein-scaffolds-drugs-and-diagnostics-market-2017-2030/181.html (accessed on 31 January 2018).
- Ståhl, S.; Gräslund, T.; Eriksson Karlström, A.; Frejd, F.Y.; Nygren, P.Å.; Löfblom, J. Affibody Molecules in Biotechnological and Medical Applications. Trends Biotechnol. 2017, 35, 691–712. [Google Scholar] [CrossRef] [PubMed]
- Frejd, F.Y.; Kim, K.T. Affibody molecules as engineered protein drugs. Exp. Mol. Med. 2017, 24, e306. [Google Scholar] [CrossRef] [PubMed]
- Ghanemi, M.; Pourshohod, A.; Ghaffari, M.A.; Kheirollah, A.; Amin, M.; Zeinali, M.; Jamalan, M. Specific targeting of HER2-positive head and neck squamous cell carcinoma line HN5 by Idarubicin-ZHER2 affibody conjugate. Curr. Cancer Drug Targets 2017. [Google Scholar] [CrossRef] [PubMed]
- Sochaj-Gregorczyk, A.M.; Serwotka-Suszczak, A.M.; Otlewski, J. A Novel Affibody-Auristatin E Conjugate With a Potent and Selective Activity against HER2+ Cell Lines. J. Immunother. 2016, 39, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Sochaj-Gregorczyk, A.M.; Ludzia, P.; Kozdrowska, E.; Jakimowicz, P.; Sokolowska-Wedzina, A.; Otlewski, J. Design and In Vitro Evaluation of a Cytotoxic Conjugate Based on the Anti-HER2 Affibody Fused to the Fc Fragment of IgG1. Int. J. Mol. Sci. 2017, 18, 1688. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.D.; Cardoso, R.M.; Lin, T.; Spinka-Doms, T.; Klein, D.; Jacobs, S.A.; Dudkin, V.; Gilliland, G.; O’Neil, K.T. Engineering a targeted delivery platform using Centyrins. Protein Eng. Des. Sel. 2016, 29, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, S.M.; Dudkin, V.Y.; Goldberg, S.; Klein, D.; Yi, F.; Singhal, S.; O’Neil, K.T.; Low, P.S. Evaluation of a Centyrin-Based Near-Infrared Probe for Fluorescence-Guided Surgery of Epidermal Growth Factor Receptor Positive Tumors. Bioconjug. Chem. 2017, 28, 2865–2873. [Google Scholar] [CrossRef] [PubMed]
- Kintzing, J.R.; Cochran, J.R. Engineered knottin peptides as diagnostics, therapeutics and drug delivery vehicles. Curr. Opin. Chem. Biol. 2016, 34, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Cox, N.; Kintzing, J.R.; Smith, M.; Grant, G.A.; Cochran, J.R. Integrin-Targeting Knottin Peptide-Drug Conjugates Are Potent Inhibitors of Tumor Cell Proliferation. Angew. Chem. Int. Ed. Engl. 2016, 55, 9894–9897. [Google Scholar] [CrossRef] [PubMed]
- Currier, N.V.; Ackerman, S.E.; Kintzing, J.R.; Chen, R.; Filsinger Interrante, M.; Steiner, A.; Sato, A.K.; Cochran, J.R. Targeted Drug Delivery with an Integrin-Binding Knottin-Fc-MMAF Conjugate Produced by Cell-Free Protein Synthesis. Mol. Cancer Ther. 2016, 15, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.cyclogenix.com (accessed on 5 January 2018).
- Plückthun, A. Designed ankyrin repeat proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Frey, R.; Zangemeister-Wittke, U.; Plückthun, A. Orthogonal assembly of a designed ankyrin repeat protein-cytotoxin conjugate with a clickable serum albumin module for half-life extension. Bioconjug. Chem. 2013, 24, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Ullman, C.; Mathonet, P.; Oleksy, A.; Diamandakis, A.; Tomei, L.; Demartis, A.; Nardi, C.; Sambucini, S.; Missineo, A.; Alt, K.; et al. High Affinity Binders to EphA2 Isolated from Abdurin Scaffold Libraries; Characterization, Binding and Tumor Targeting. PLoS ONE 2015, 10, e0135278. [Google Scholar] [CrossRef] [PubMed]
- Peretti, S. Abdurin-Drug Conjugates: A New Generation of Targeted Therapeutics. In Proceedings of the Protein Engineering Summit (PEGS) Conference, Lisbon, Portugal, 13–17 November 2017. [Google Scholar]
- White, B.H.; Bazinet, P.; Whalen, K. Discovery of PEN-221, an SSTR2-targeting maytansinoid conjugate with potent activity in vitro and in vivo. In Proceedings of the American Association for Cancer Research Annual Conference, Washington, DC, USA, 1–5 April 2017. [Google Scholar]
- PEN221 Clinical Trial. Available online: https://clinicaltrials.gov/ct2/show/NCT02936323 (accessed on 5 January 2018).
- PEN 866 Clinical Trial. Available online: https://clinicaltrials.gov/ct2/show/NCT03221400 (accessed on 5 January 2018).
- Deyle, K.; Kong, X.D.; Heinis, C. Phage Selection of Cyclic Peptides for Application in Research and Drug Development. Acc. Chem. Res. 2017, 50, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Heinis, C.; Rutherford, T.; Freund, S.; Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Teufel, D.; Eder, M.; Pavan, S. Bicyclic Peptides for Positron Emission Tomography (PET) Imaging of MT1-MMP Expressing Tumors. In Proceedings of the American Association for Cancer Research Annual Conference, Washington, DC, USA, 1–5 April 2017. [Google Scholar]
- Harrison, H.; Bennett, G.; Blakeley, D. BT1718, a novel bicyclic peptide-maytansinoid conjugate targeting MT1-MMP for the treatment of solid tumours: Design of bicyclic peptide and linker selection. In Proceedings of the American Association for Cancer Research Annual Conference, Washington, DC, USA, 1–5 April 2017. [Google Scholar]
- Katsamakas, S.; Chatzisideri, T.; Thysiadis, S.; Sarli, V. RGD-mediated delivery of small-molecule drugs. Future Med. Chem. 2017, 9, 579–604. [Google Scholar] [CrossRef] [PubMed]
- Krall, N.; Pretto, F.; Decurtins, W.; Bernardes, G.J.; Supuran, C.T.; Neri, D. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew. Chem. Int. Ed. Engl. 2014, 53, 4231–4235. [Google Scholar] [CrossRef] [PubMed]
- Krall, N.; Pretto, F.; Neri, D. A bivalent small molecule-drug conjugate directed against carbonic anhydrase IX can elicit complete tumour regression in mice. Chem. Sci. 2014, 5, 3640–3644. [Google Scholar] [CrossRef]
- Cazzamalli, S.; Dal Corso, A.; Widmayer, F.; Neri, D. Chemically Defined Antibody and Small Molecule-Drug Conjugates for in Vivo Tumor Targeting Applications: A Comparative Analysis. J. Am. Chem. Soc. 2018, 140, 1617–1621. [Google Scholar] [CrossRef] [PubMed]
- Kularatne, S.A.; Wang, K.; Santhapuram, H.K.; Low, P.S. Prostate-specific membrane antigen targeted imaging and therapy of prostate cancer using a PSMA inhibitor as a homing ligand. Mol. Pharm. 2009, 6, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Vogelzang, N.J.; Sartor, O.; Armour, A.; Groaning, M. 793PD Phase 1 study of the PSMA-targeted small-molecule drug conjugate EC1169 in patients with metastatic castrate-resistant prostate cancer (mCRPC). Ann. Oncol. 2017, 28 (Suppl. 5), 370. [Google Scholar] [CrossRef]
- Kumar, A.; Mastren, T.; Wang, B.; Hsieh, J.T.; Hao, G.; Sun, X. Design of a Small-Molecule Drug Conjugate for Prostate Cancer Targeted Theranostics. Bioconjug. Chem. 2016, 27, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Vergote, I.; Leamon, C.P. Vintafolide: A novel targeted therapy for the treatment of folate receptor expressing tumors. Ther. Adv. Med. Oncol. 2015, 7, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Maurer, A.H.; Elsinga, P.; Fanti, S.; Nguyen, B.; Oyen, W.J.; Weber, W.A. Imaging the folate receptor on cancer cells with 99mTc-etarfolatide: Properties, clinical use, and future potential of folate receptor imaging. J. Nucl. Med. 2014, 55, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Vallabhajosula, S.; Nikolopoulou, A.; Babich, J.W.; Osborne, J.R.; Tagawa, S.T.; Lipai, I.; Solnes, L.; Maresca, K.P.; Armor, T.; Joyal, J.L.; et al. 99mTc-labeled small-molecule inhibitors of prostate-specific membrane antigen: Pharmacokinetics and biodistribution studies in healthy subjects and patients with metastatic prostate cancer. J. Nucl. Med. 2014, 55, 1791–1798. [Google Scholar] [PubMed]
- Huang, B.; Otis, J.; Joice, M.; Kotlyar, A.; Thomas, T.P. PSMA-targeted stably linked “dendrimer-glutamate urea-methotrexate” as a prostate cancer therapeutic. Biomacromolecules 2014, 15, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.W.; Shia, K.S.; Wu, C.H.; Liu, K.L.; Yeh, Y.C.; Lo, C.F.; Chen, C.T.; Chen, Y.Y.; Yeh, T.K.; Chen, W.H.; et al. Targeting Tumor Associated Phosphatidylserine with New Zinc Dipicolylamine-Based Drug Conjugates. Bioconjug. Chem. 2017, 28, 1878–1892. [Google Scholar] [CrossRef] [PubMed]
- Ivanenkov, Y.A.; Majouga, A.G.; Petrov, R.A.; Petrov, S.A.; Kovalev, S.V.; Maklakova, S.Y.; Yamansarov, E.Y.; Saltykova, I.V.; Deyneka, E.V.; Filkov, G.I.; et al. Synthesis and biological evaluation of novel doxorubicin-containing ASGP-R-targeted drug-conjugates. Bioorg. Med. Chem. Lett. 2017, 28, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. Ablynx nanobody fragments go places antibodies cannot. Nat. Biotechnol. 2017, 35, 1115–1117. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, E.; Sachdev, D.; Mita, M. Aldoxorubicin for the treatment of soft tissue sarcoma. Expert Opin. Investig. Drugs 2017, 26, 1175–1179. [Google Scholar] [CrossRef] [PubMed]
- Lobocki, M.; Zakrzewska, M.; Szlachcic, A.; Krzyscik, M.A.; Sokolowska-Wedzina, A.; Otlewski, J. High-Yield Site-Specific Conjugation of Fibroblast Growth Factor 1 with Monomethylauristatin E via Cysteine Flanked by Basic Residues. Bioconjug. Chem. 2017, 28, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Masters, J.C.; Nickens, D.J.; Xuan, D.; Shazer, R.L.; Amantea, M. Clinical toxicity of antibody drug conjugates: A meta-analysis of payloads. Investig. New Drugs 2018, 36, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, M.J.M.; Ryan, P.M.; Zheng, B.; Afif-Rider, S.; Yu, X.Q.; Gunsior, M.; Zhong, H.; Harper, J.; Bezabeh, B.; Vashishy, K.; et al. Fractionated Dosing Improves Preclinical Therapeutic Index of Pyrrolobenzodiazepine-Containing Antibody Drug Conjugates. Clin. Cancer Res. 2017, 23, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.affilogic.com/nanofitin-drug-conjugates (accessed on 5 January 2018).
- Available online: https://www.avacta.com/pipeline (accessed on 5 January 2018).
- Erickson, H.K.; Lewis Phillips, G.D.; Leipold, D.D.; Provenzano, C.A.; Mai, E.; Johnson, H.A.; Gunter, B.; Audette, C.A.; Gupta, M.; Pinkas, J.; et al. The effect of differentlinkers on target cell catabolism and pharmacokinetics/ pharmacodynamics of trastuzumab maytansinoid conjugates. Mol. Cancer Ther. 2012, 11, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
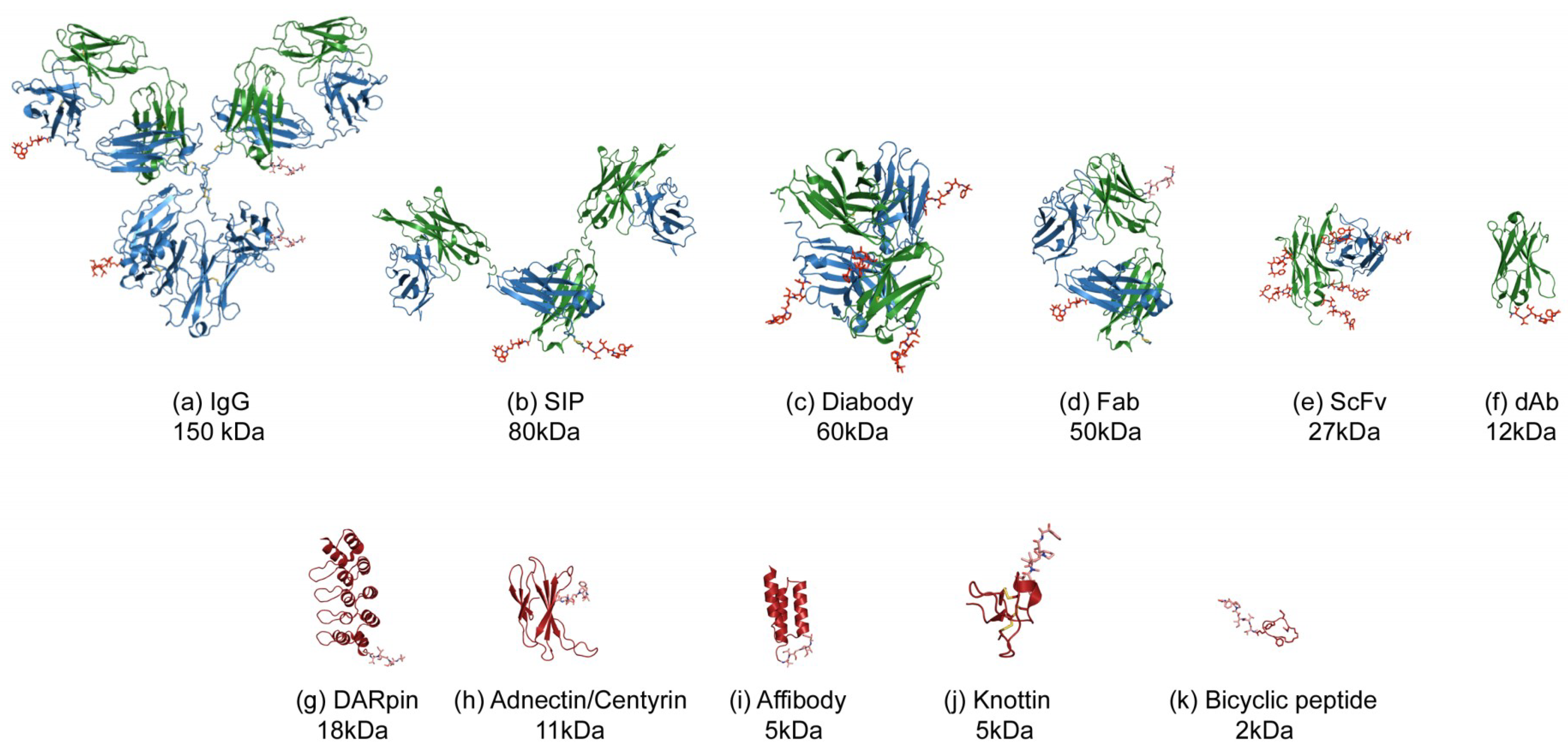
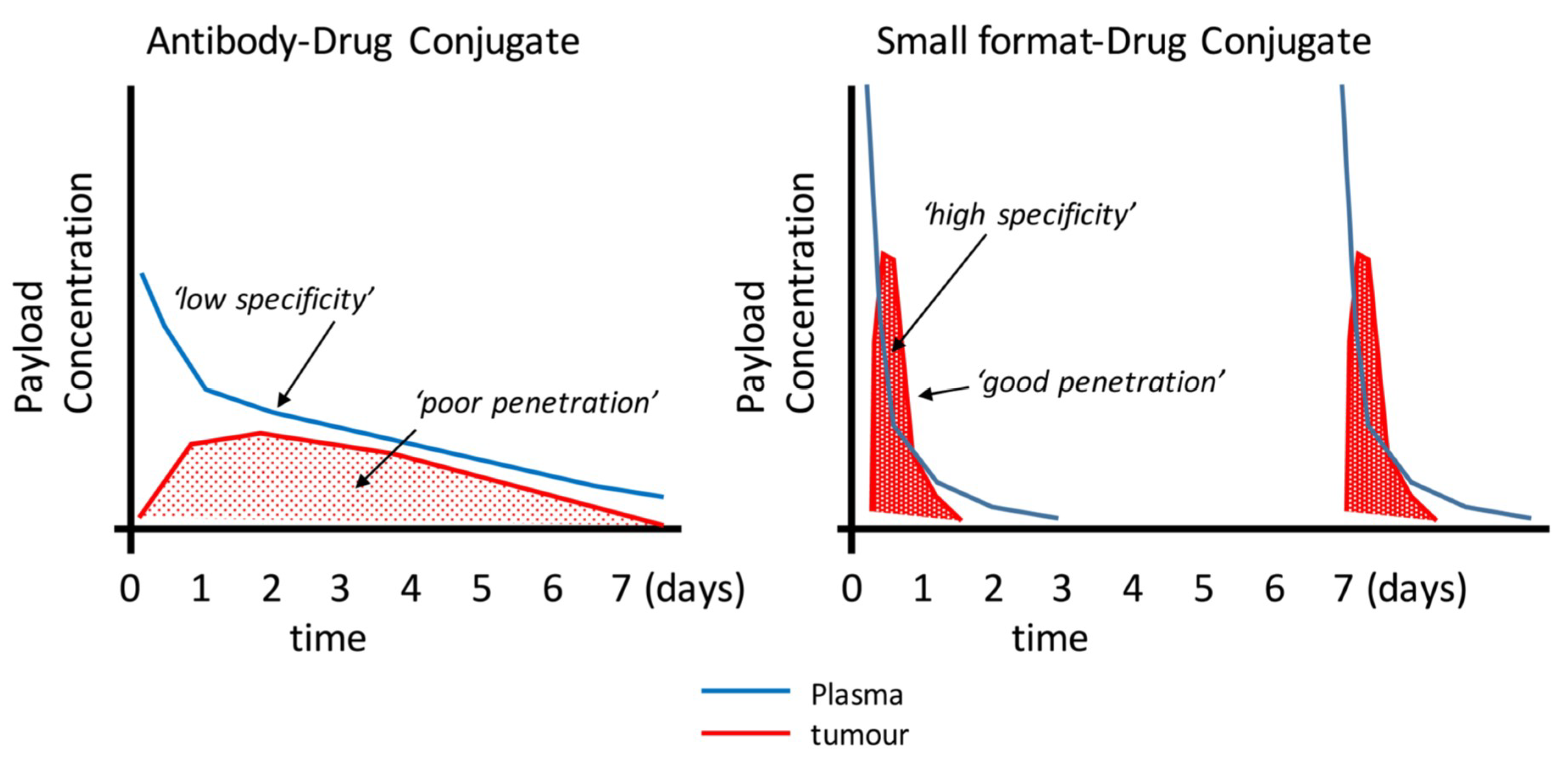

{kind=link}
{kind=link}
{kind=link}
| Small Format Carrier Size (kDa) | Small Format Carrier | Target | Payload | Status [Reference] | Commercial Organisation (If Any) |
|---|---|---|---|---|---|
| ~0.2 | N-Acetyl-galactosamine | Asialo-glycoprotein receptor | doxorubicin, paclitaxel | Pre-clinical R&D [101] | |
| ~0.2 | Acetazolamide | Carbonic anhydrase IX | DM1, duocarmycin | Pre-clinical R&D [90,91,92] | Philogen SA |
| ~0.3 | DUPA | PSMA | Tubulysin | Pre-clinical R&D [94] | Endocyte Inc. |
| ~0.4 | Folate | Folate receptor | Vinca alkaloid, Tubulysin | Pre-clinical R&D [96,97] | Endocyte Inc. |
| ~0.7 | Zinc dipicolylamine | Phosphatidyl-serine | SN38 | Pre-clinical R&D [100] | |
| ~1 | RGD-based peptides | Intergrins | Various: doxorubicin, vcMMAE, paclitaxel | Pre-clinical R&D [89] | |
| 1.5–2 | Bicycle (bicyclic peptides) | Matrix metallo-protease 14 | DM1 | BT1718 is in Phase 1/2a clinical trials [86,87,88] | Bicycle Therapeutics Ltd. |
| ~3–5 | Pentarin | Somatostatin receptor | DM1 | PEN-221 is in Phase 1/2a clinical trials [82,83,84] | Tarveda Inc. |
| ~3.5–5 | Cystine knots | Integrin, Matrix metallo-protease 2 | Gemcitabine, MMAF, Cis-platin | ||
| 5–6.5 | Affibody | HER2 | Idarubicin, vcMMAE | Pre-clinical R&D [67,68,69] | Affibody |
| 7 | Nanofitin (sac 7d) | ND * | ND * | Pre-clinical R&D [108] | Affilogic |
| ~10–11 | Centyrin | EGFR | vcMMAF | Pre-clinical R&D [72] | Janssen R&D LLP |
| ~12 | VH (like) domains | ND * | ND * | Pre-clinical R&D | Crescendo Biologics Ltd. |
| 12–14 | Affimer | ND * | ND * | Pre-clinical R&D [109] | Avacta PLC |
| ~15–18 | DARPIn | EpCAM | MMAF | Pre-clinical R&D [78,79] | Molecular Partners AG |
| ~15 | Abdurin | ephA2 | vcMMAE | Pre-clinical R&D [80] | IRBM |
| ~25–27 | Single-chain Fv | Various: HER2, CEA, PLAP, Fibronectin, Tenascin-C | Adriamycin, photosensitisers, Infra-red dyes, MMAF, vcMMAE, doxorubicin, cemadotin, dolostatin-10 | Pre-clinical R&D [56,57,58,59,60,61,62,63] | Antikor Biopharma Ltd.; Philogen SA |
| ~55–60 | Diabody | CD30 | MMAF | Pre-clinical R&D [47,48] | Seattle Genetics Inc. Avipep |
| ~50 | Fab | Various: TROP2, CD20, HER2 | Doxorubicin, vcMMAE, Auristatin | Pre-clinical R&D [40,41,42,43,44,45] | Abzena |
| ~80 | SIP-Small immunoprotein | Fibronectin, Tenascin-C | Cemadotin, DM1 | Pre-clinical R&D [49,50,51,52,53,54] | Philogen SA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deonarain, M.P.; Yahioglu, G.; Stamati, I.; Pomowski, A.; Clarke, J.; Edwards, B.M.; Diez-Posada, S.; Stewart, A.C. Small-Format Drug Conjugates: A Viable Alternative to ADCs for Solid Tumours? Antibodies 2018, 7, 16. https://doi.org/10.3390/antib7020016
Deonarain MP, Yahioglu G, Stamati I, Pomowski A, Clarke J, Edwards BM, Diez-Posada S, Stewart AC. Small-Format Drug Conjugates: A Viable Alternative to ADCs for Solid Tumours? Antibodies. 2018; 7(2):16. https://doi.org/10.3390/antib7020016
Chicago/Turabian StyleDeonarain, Mahendra P., Gokhan Yahioglu, Ioanna Stamati, Anja Pomowski, James Clarke, Bryan M. Edwards, Soraya Diez-Posada, and Ashleigh C. Stewart. 2018. "Small-Format Drug Conjugates: A Viable Alternative to ADCs for Solid Tumours?" Antibodies 7, no. 2: 16. https://doi.org/10.3390/antib7020016