Immunotherapy of B-Cell Lymphoma with an Engineered Bispecific Antibody Targeting CD19 and CD5

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Hybridomas
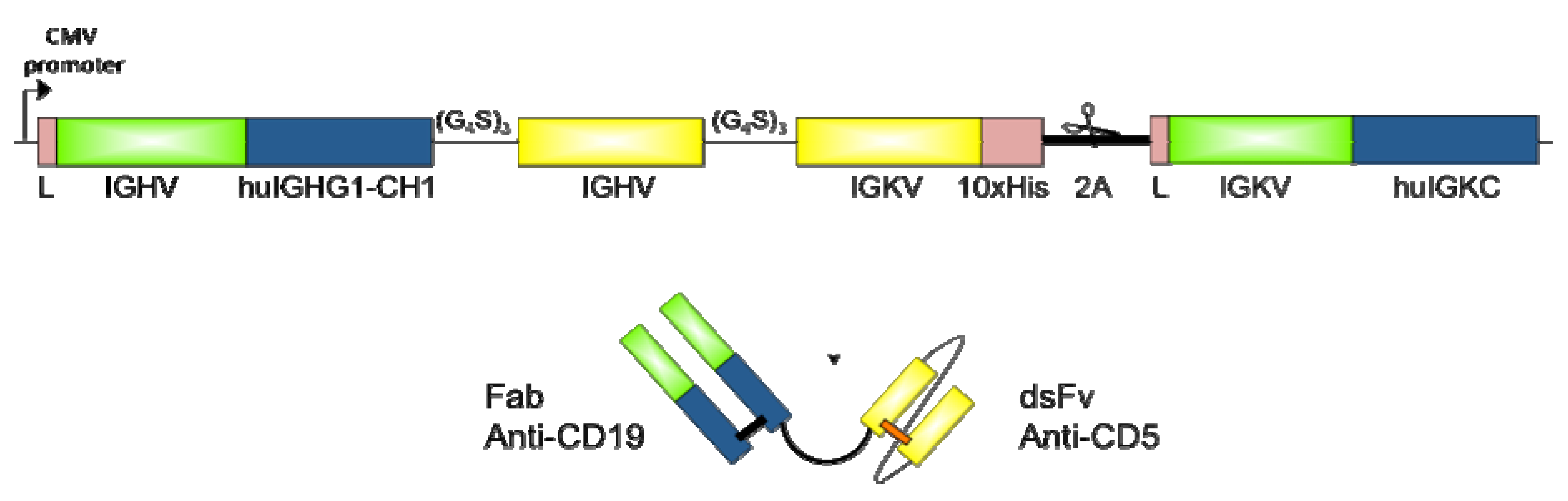
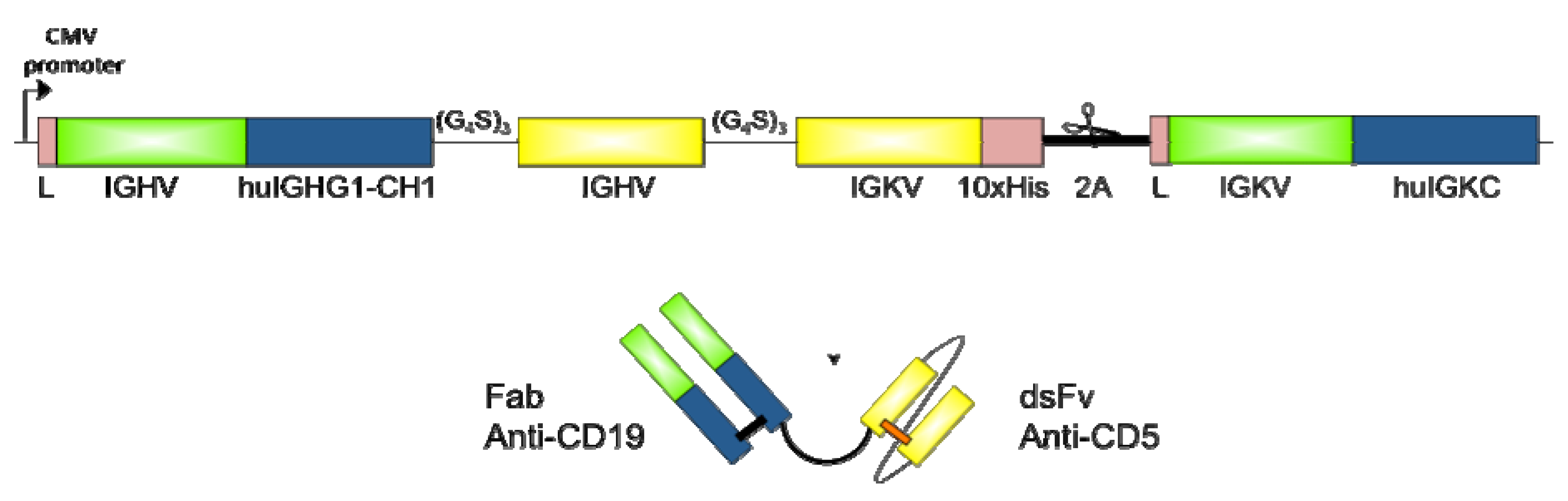
2.2. Design and Cloning of bsAb FabCD19xdsFvCD5
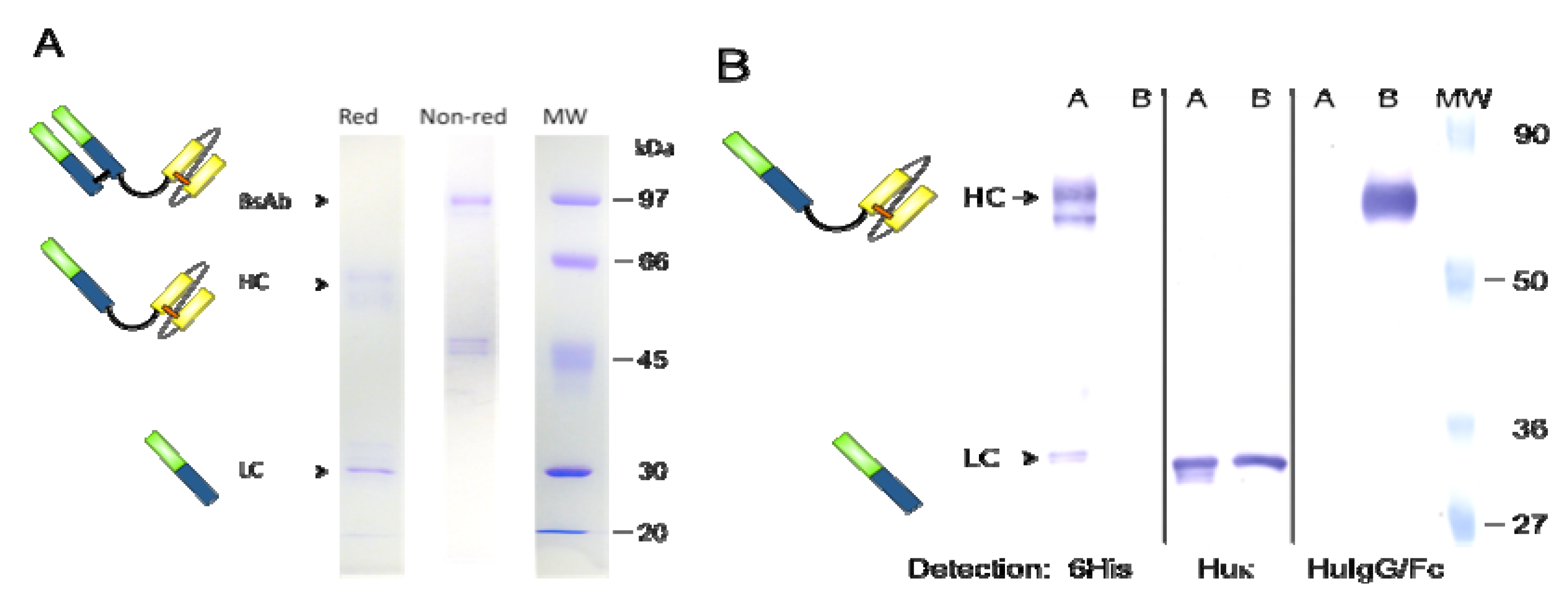
2.3. Expression and Purification of bsAb FabCD19xdsFvCD5
2.4. Western Blot Analysis
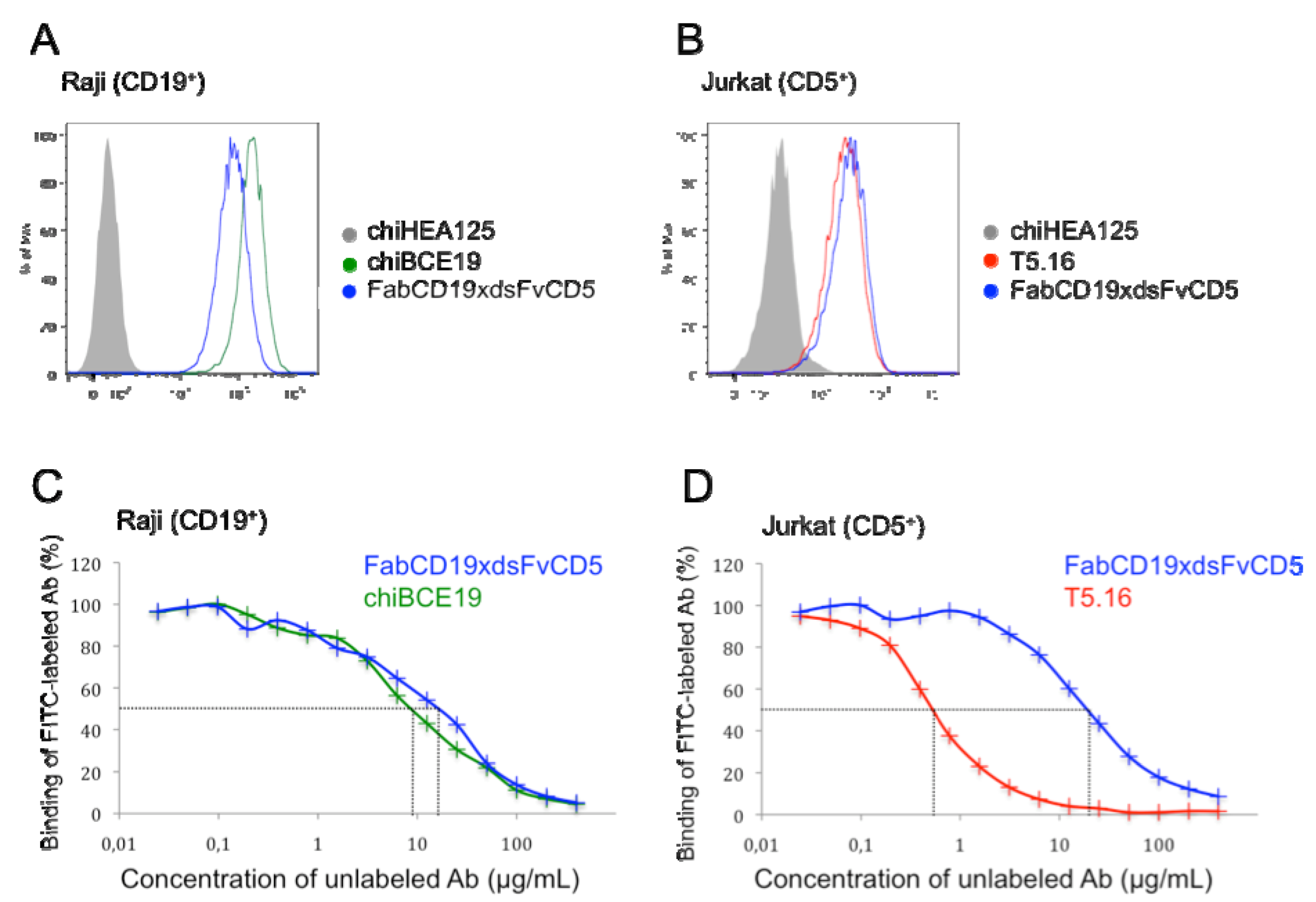
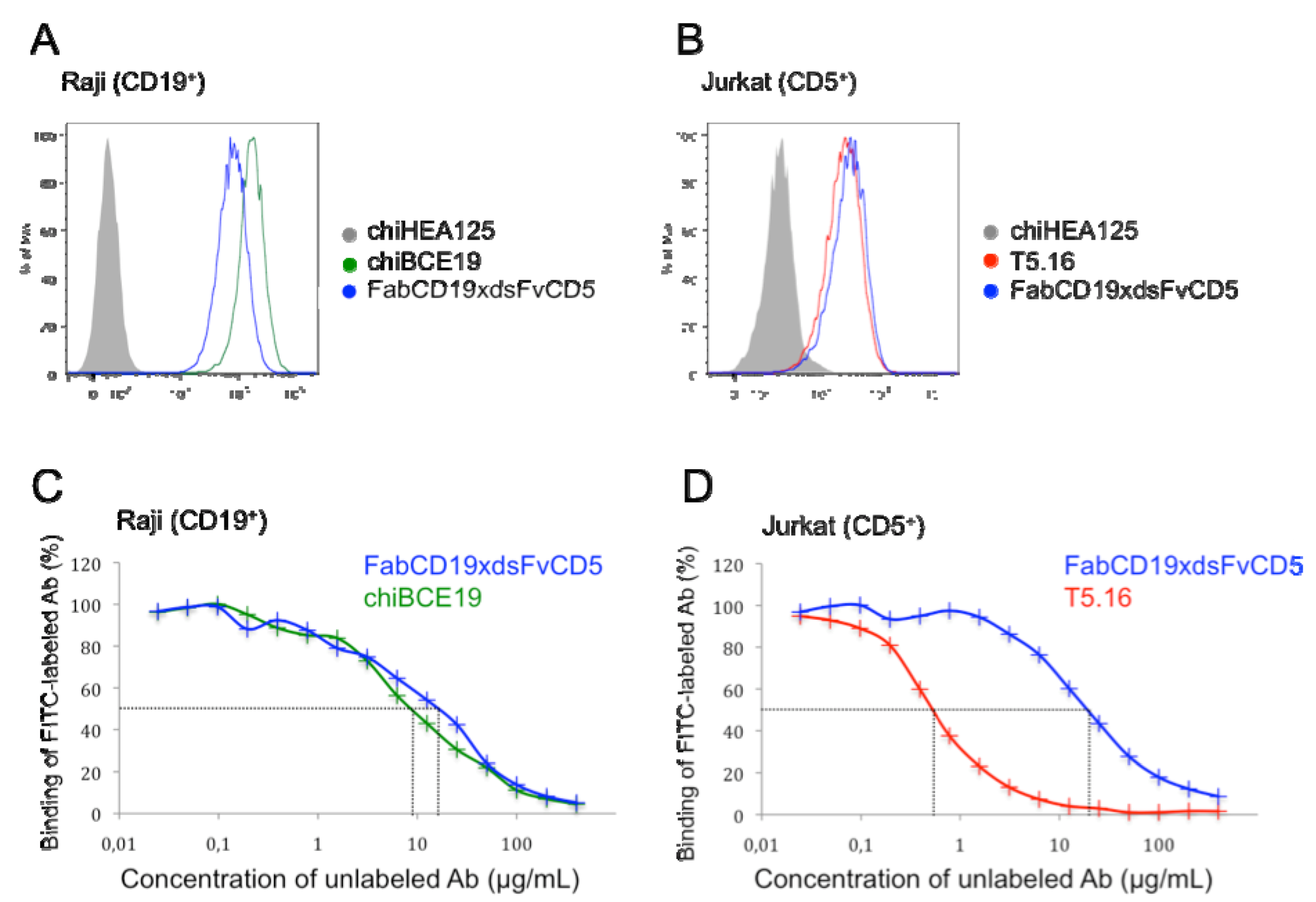
2.5. Flow Cytometry and Binding Competition Assay
2.6. Isolation and Activation of Peripheral Blood Mononuclear Cells
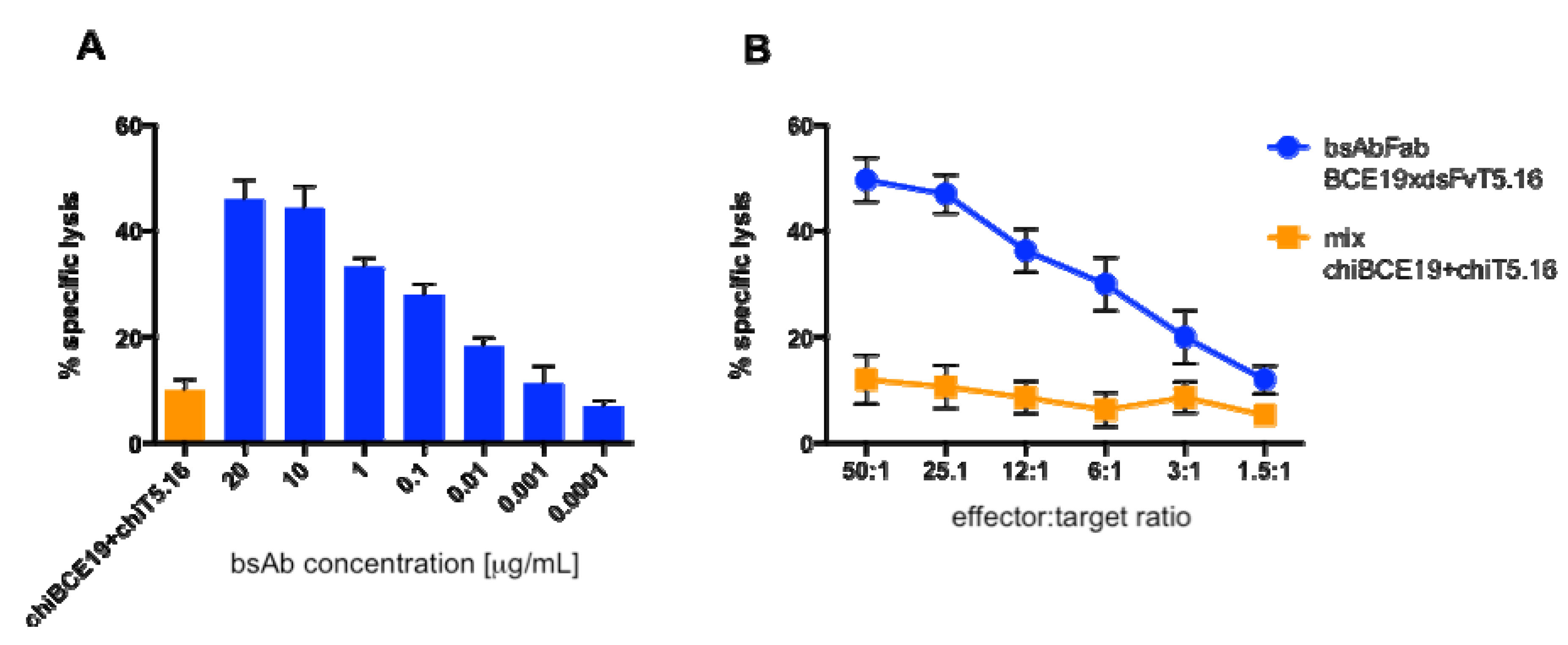
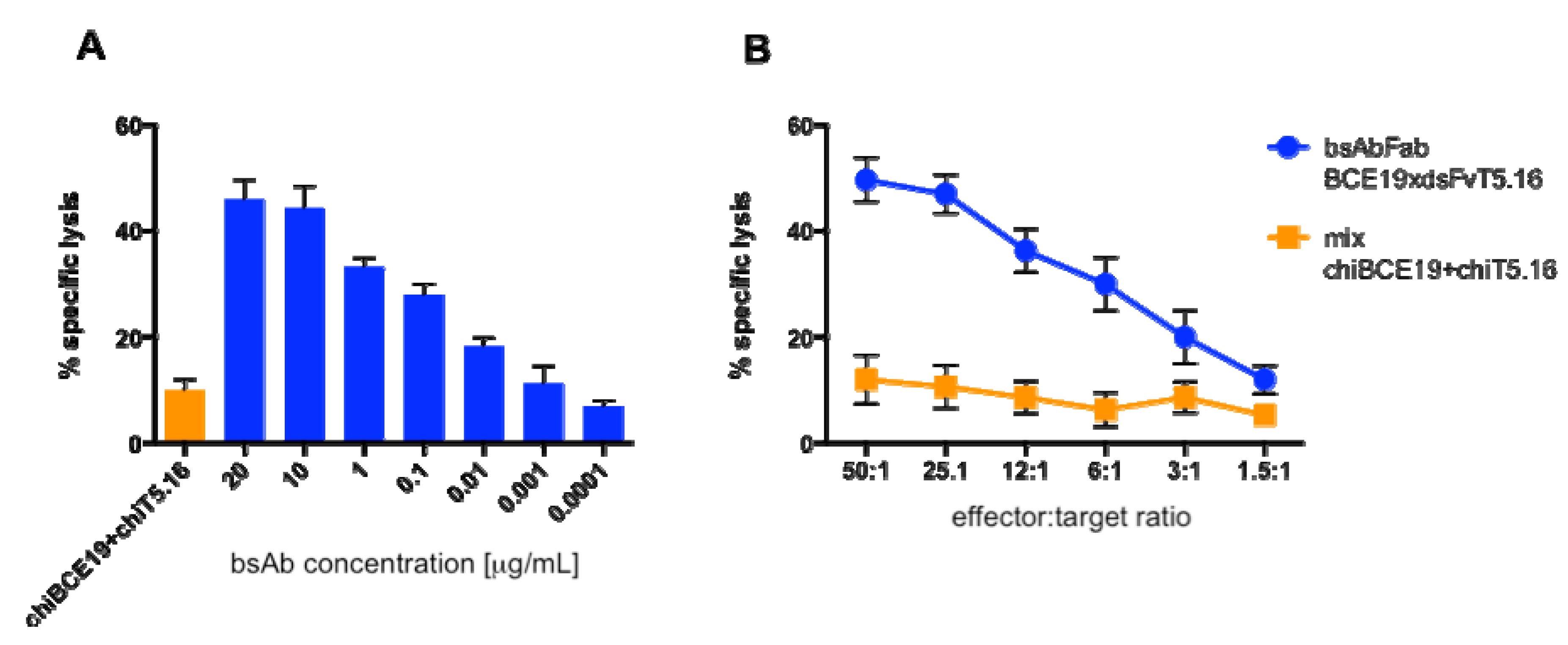
2.7. Cytotoxicity Assay
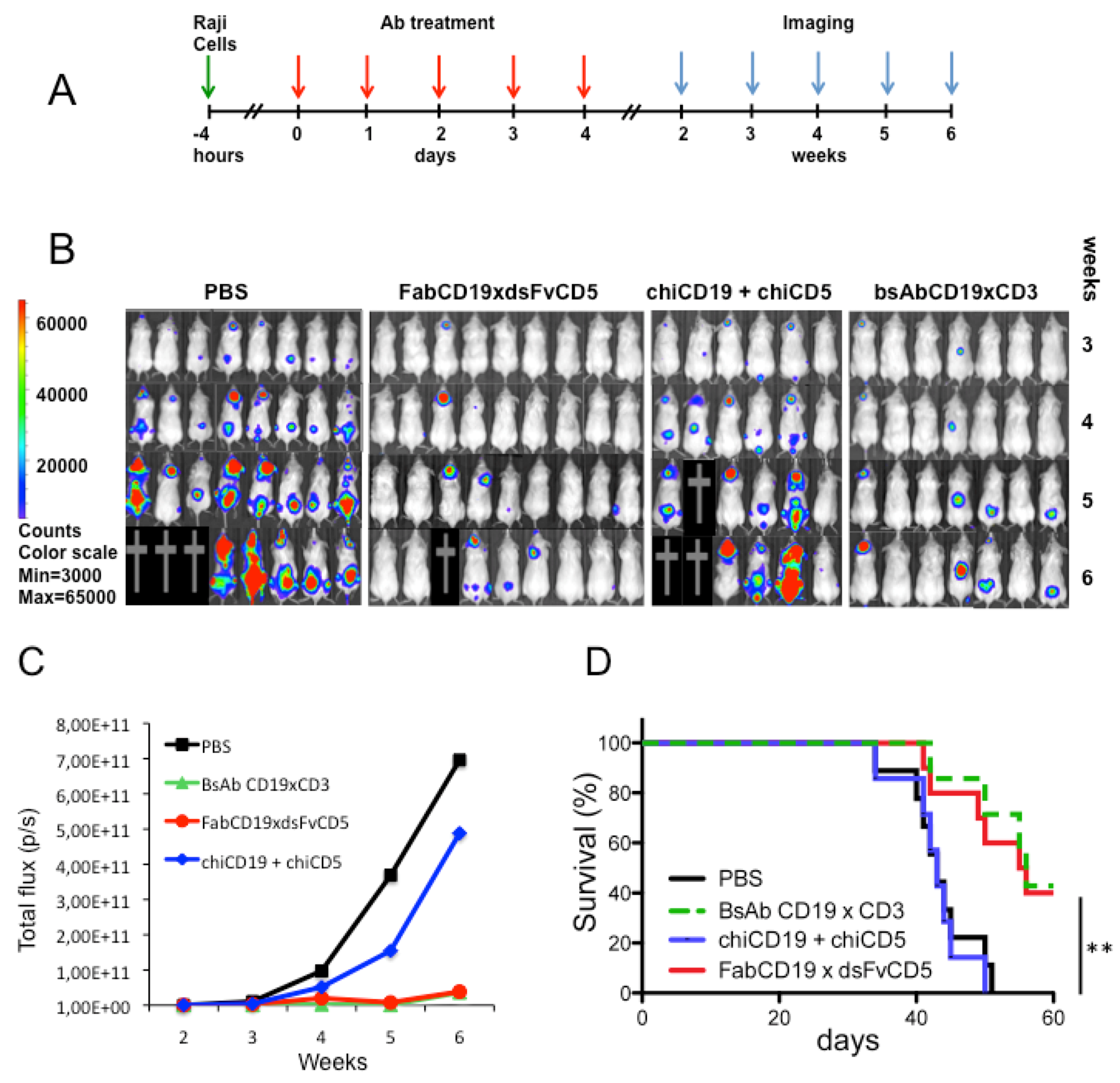
2.8. Raji Lymphoma Xenotransplantation Model and Antibody Treatment
2.9. Bioluminescent Imaging
2.10. Statistical Analysis
3. Results
3.1. Expression and Purification of bsAb FabCD19xdsFvCD5

3.2. Binding Characteristics of bsAb FabCD19xdsFvCD5
3.3. Cytotoxicity in Vitro
3.4. Antibody Treatment of Raji Lymphoma-Bearing Mice
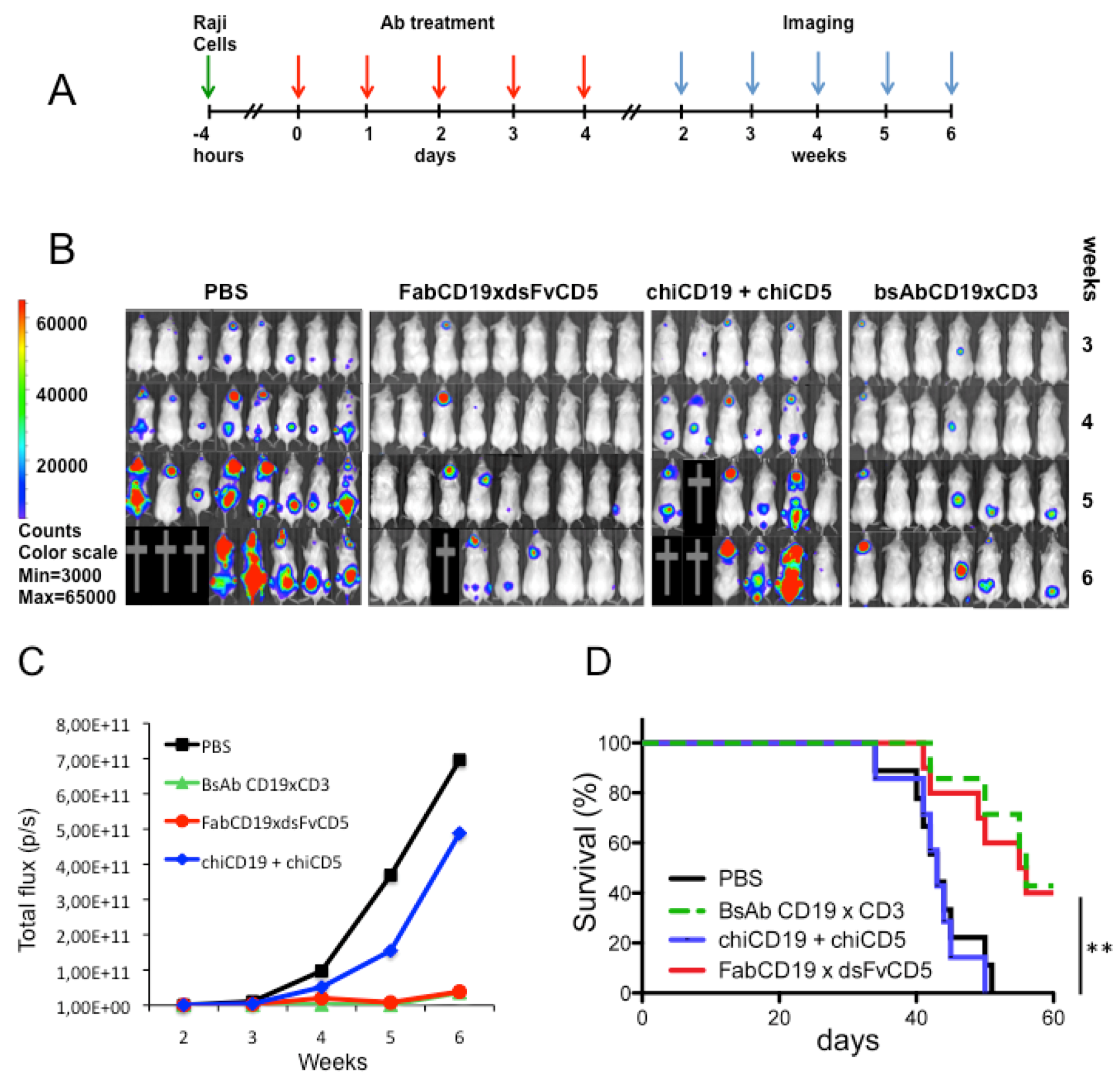
4. Discussion
5. Conclusions
Acknowledgements
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer Statistics, 2012. CA Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef]
- Lim, S.H.; Beers, S.A.; French, R.R.; Johnson, P.W.M.; Glennie, M.J.; Cragg, M.S. Anti-CD20 monoclonal antibodies: historical and future perspectives. Haematologica 2010, 95, 135–143. [Google Scholar] [CrossRef]
- Vacchelli, E.; Eggermont, A.; Galon, J.; Sautes-Fridman, C.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Monoclonal antibodies in cancer therapy. OncoImmunology 2013, 2, e22789. [Google Scholar] [CrossRef]
- Rezvani, A.R.; Maloney, D.G. Rituximab resistance. Best Pract. Res. Clin. Haematol. 2011, 24, 203–216. [Google Scholar] [CrossRef]
- Chames, P.; Baty, D. Bispecific antibodies for cancer therapy: The light at the end of the tunnel? MAbs 2009, 1, 539–547. [Google Scholar] [CrossRef]
- Pezzutto, A.; Dörken, B.; Rabinovitch, P.S.; Ledbetter, J.A.; Moldenhauer, G.; Clark, E.A. CD19 monoclonal antibody HD37 inhibits anti-immunoglobulin-induced B cell activation and proliferation. J. Immunol. 1987, 138, 2793–2799. [Google Scholar]
- Tedder, T.F. CD19: A promising B cell target for rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 572–577. [Google Scholar] [CrossRef]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar]
- Schneider, C.K.; Kalinke, U.; Löwer, J. TGN1412—A regulator’s perspective. Nat. Biotechnol. 2006, 24, 493–496. [Google Scholar] [CrossRef]
- Tabbekh, M.; Mokrani-Hammani, M.; Bismuth, G.; Mami-Chouaib, F. T-cell modulatory properties of CD5 and its role in antitumor immune responses. OncoImmunology 2013, 2, e22841. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Reinhardt, C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res. 2009, 69, 4941–4944. [Google Scholar] [CrossRef]
- Beck, A.; Wurch, T.; Bailly, C.; Corvaia, N. Strategies and challenges for the next generation of therapeutic antibodies. Nat. Rev. Immunol. 2010, 10, 345–352. [Google Scholar] [CrossRef]
- Chan, A.C.; Carter, P.J. Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. Immunol. 2010, 10, 301–316. [Google Scholar] [CrossRef]
- Müller, D.; Kontermann, R.E. Recombinant bispecific antibodies for cellular cancer immunotherapy. Curr. Opin. Mol. Ther. 2007, 9, 319–326. [Google Scholar]
- Müller, D.; Kontermann, R.E. Bispecific antibodies for cancer immunotherapy. Current perspectives. Biodrugs 2010, 24, 89–98. [Google Scholar] [CrossRef]
- Stamova, S.; Koristka, S.; Keil, J.; Arndt, C.; Feldmann, A.; Michalk, I.; Bartsch, H.; Bippes, C.C.; Schmitz, M.; Cartellieri, M.; et al. Cancer Immunotherapy by retargeting of immune effector cells via recombinant bispecific antibody constructs. Antibodies 2012, 1, 172–198. [Google Scholar] [CrossRef]
- Weiner, L.M. Building better magic bullets—Improving unconjugated monoclonal antibody therapy for cancer. Nat. Rev. Cancer 2007, 7, 701–706. [Google Scholar] [CrossRef]
- Oden, F. Max-Delbruck-Center for Molecular Medicine: Berlin, Germany, 2013; unpublished work.
- Zhang, H.; Moldenhauer, G.; Kipriyanov, S.M.; Braunagel, M.; Krauss, J.; Little, M. Construction, sequence and binding properties of an anti-CD5 single chain Fv antibody. Tumor Targeting 1996, 2, 314–321. [Google Scholar]
- Moldenhauer, G.; Momburg, F.; Möller, P.; Schwartz, R.; Hämmerling, G.J. Epithelium-specific surface glycoprotein of Mr 34,000 is a widely distributed human carcinoma marker. Br. J. Cancer 1987, 56, 714–721. [Google Scholar] [CrossRef]
- Momburg, F.; Moldenhauer, G.; Hämmerling, G.J.; Möller, P. Immunohistochemical study of the expression of a Mr 34,000 human epithelium-specific surface glycoprotein in normal and malignant tissues. Cancer Res. 1987, 47, 2883–2891. [Google Scholar]
- Lüttgau, S. German Cancer Research Center and National Center for Tumor Diseases: Heidelberg, Germany, 2013; unpublished work.
- Csoka, M.; Strauss, G.; Debatin, K.M.; Moldenhauer, G. Activation of T cell cytotoxicity against autologous common acute lymphoblastic leukemia (cALL) blasts by CD3xCD19 bispecific antibody. Leukemia 1996, 10, 1765–1772. [Google Scholar]
- Daniel, P.T.; Kroidl, A.; Kopp, J.; Sturm, I.; Moldenhauer, G.; Dörken, B.; Pezzutto, A. Immunotherapy of B-cell lymphoma with CD3x19 bispecific antibodies: costimulation via CD28 prevents "veto" apoptosis of antibody-targeted cytotoxic T cells. Blood 1998, 92, 4750–4757. [Google Scholar]
- Reiter, Y.; Brinkmann, U.; Webber, K.O.; Jung, S.-H.; Lee, B.; Pastan, I. Engineering interchain disulfide bonds into conserved framework regions of Fv fragments: Improved biochemical characteristics of recombinant immunotoxins containing disulfide-stabilized Fv. Protein Eng. 1994, 7, 697–704. [Google Scholar] [CrossRef]
- De Gast, G.C.; Van Houten, A.A.; Haagen, I.A.; Klein, S.; De Weger, R.A.; Van Dijk, A.; Philips, J.; Clark, M.; Bast, B.J.E.G. Clinical experience with CD3 x CD19 bispecific antibodies in patients with B cell malignancies. J. Hematother. 1995, 4, 433–437. [Google Scholar] [CrossRef]
- Manzke, O.; Tesch, H.; Borchmann, P.; Wolf, J.; Lackner, K.; Gossmann, A.; Diehl, V.; Bohlen, H. Locoregional treatment of low-grade B-cell lymphoma with CD3xCD19 bispecific antibodies and CD28 costimulation: I. clinical phase I evaluation. Int. J. Cancer 2001, 91, 508–515. [Google Scholar] [CrossRef]
- Kipriyanov, S.M.; Moldenhauer, G.; Strauss, G.; Little, M. Bispecific CD3xCD19 diabody for T cell-mediated lysis of malignant human B cells. Int. J. Cancer 1998, 77, 763–772. [Google Scholar] [CrossRef]
- Kipriyanov, S.M.; Moldenhauer, G; Schuhmacher, J.; Cochlovius, B.; Von der Lieth, C.-W.; Matys, E.R.; Little, M. Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics. J. Mol. Biol. 1999, 293, 41–56. [Google Scholar] [CrossRef]
- Cochlovius, B.; Kipriyanov, S.M.; Stassar, M.J.J.G.; Christ, O.; Schuhmacher, J.; Strauß, G.; Moldenhauer, G.; Little, M. Treatment of human B cell lymphoma xenografts with a CD3 x CD19 diabody and T cells. J. Immunol. 2000, 165, 888–895. [Google Scholar]
- Cochlovius, B.; Kipriyanov, S.M.; Stassar, M.J.J.G.; Schuhmacher, J.; Benner, A.; Moldenhauer, G.; Little, M. Cure of Burkitt’s lymphoma in severe combined immunodeficiency mice by T cells, tetravalent CD3 x CD19 tandem diabody, and CD28 costimulation. Cancer Res. 2000, 60, 4336–4341. [Google Scholar]
- Löffler, A.; Kufer, P.; Lutterbüse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmüller, G.; Dörken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19xCD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103. [Google Scholar]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008, 321, 974–977. [Google Scholar] [CrossRef]
- Topp, M.S.; Kufer, P.; Gökbuget, N.; Goebler, M.; Klinger, M.; Neumann, S.; Horst, H.-A.; Raff, T.; Viardot, A.; Schmid, M.; et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J. Clin. Oncol. 2011, 29, 2493–2498. [Google Scholar] [CrossRef]
- Wild, M.K.; Strittmatter, W.; Matzku, S.; Schraven, B.; Meuer, S.C. Tumor therapy with bispecific antibody: the targeting and triggering steps can be separated employing a CD2-based strategy. J. Immunol. 1999, 163, 2064–2072. [Google Scholar]
- Ferrini, S.; Prigione, I.; Mammoliti, S.; Colnaghi, M.I.; Ménard, S.; Moretta, A.; Moretta, L. Re-targeting of human lymphocytes expressing the T-cell receptor gamma/delta to ovarian carcinoma cells by the use of bispecific monoclonal antibodies. Int. J. Cancer 1989, 44, 245–250. [Google Scholar] [CrossRef]
- Tita-Nwa, F.; Moldenhauer, G.; Herbst, M.; Kleist, C.; Ho, A.D.; Kornacker, M. Cytokine-induced killer cells targeted by the novel bispecific antibody CD19xCD5 (HD37xT5.16) efficiently lyse B-lymphoma cells. Cancer Immunol. Immunother. 2007, 56, 1911–1920. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Antibodies, Fc receptors and cancer. Curr. Opin. Immunol. 2007, 19, 239–245. [Google Scholar] [CrossRef]
- Seimetz, D.; Lindhofer, H.; Bokemeyer, C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat. Rev. 2010, 36, 458–467. [Google Scholar] [CrossRef]
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: results of a prospective randomized phase II/III trial. Int. J. Cancer 2010, 127, 2209–2221. [Google Scholar] [CrossRef]
- Weatherill, E.E.; Cain, K.L.; Heywood, S.P.; Compson, J.E.; Heads, J.T.; Adams, R.; Humphreys, D.P. Towards a universal disulphide stabilized single chain Fv format: importance of interchain disulphide bond location and vL-vH orientation. Protein Eng. Des. Sel. 2012, 25, 321–329. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lüttgau, S.; Deppe, D.; Meyer, S.; Fertig, R.; Panjideh, H.; Lipp, M.; Schmetzer, O.; Pezzutto, A.; Breitling, F.; Moldenhauer, G. Immunotherapy of B-Cell Lymphoma with an Engineered Bispecific Antibody Targeting CD19 and CD5. Antibodies 2013, 2, 338-352. https://doi.org/10.3390/antib2020338
Lüttgau S, Deppe D, Meyer S, Fertig R, Panjideh H, Lipp M, Schmetzer O, Pezzutto A, Breitling F, Moldenhauer G. Immunotherapy of B-Cell Lymphoma with an Engineered Bispecific Antibody Targeting CD19 and CD5. Antibodies. 2013; 2(2):338-352. https://doi.org/10.3390/antib2020338
Chicago/Turabian StyleLüttgau, Sandra, Dorothée Deppe, Saskia Meyer, Regina Fertig, Hossein Panjideh, Martin Lipp, Oliver Schmetzer, Antonio Pezzutto, Frank Breitling, and Gerhard Moldenhauer. 2013. "Immunotherapy of B-Cell Lymphoma with an Engineered Bispecific Antibody Targeting CD19 and CD5" Antibodies 2, no. 2: 338-352. https://doi.org/10.3390/antib2020338