Antibody-Directed Phototherapy (ADP)

Abstract
:1. Introduction
1.1. Antibody-Drug Conjugates (ADCs)
1.2. Photodynamic Therapy (PDT)

| A: Photosensitisers approved for clinical use | ||
|---|---|---|
| Drug | Details | Indication |
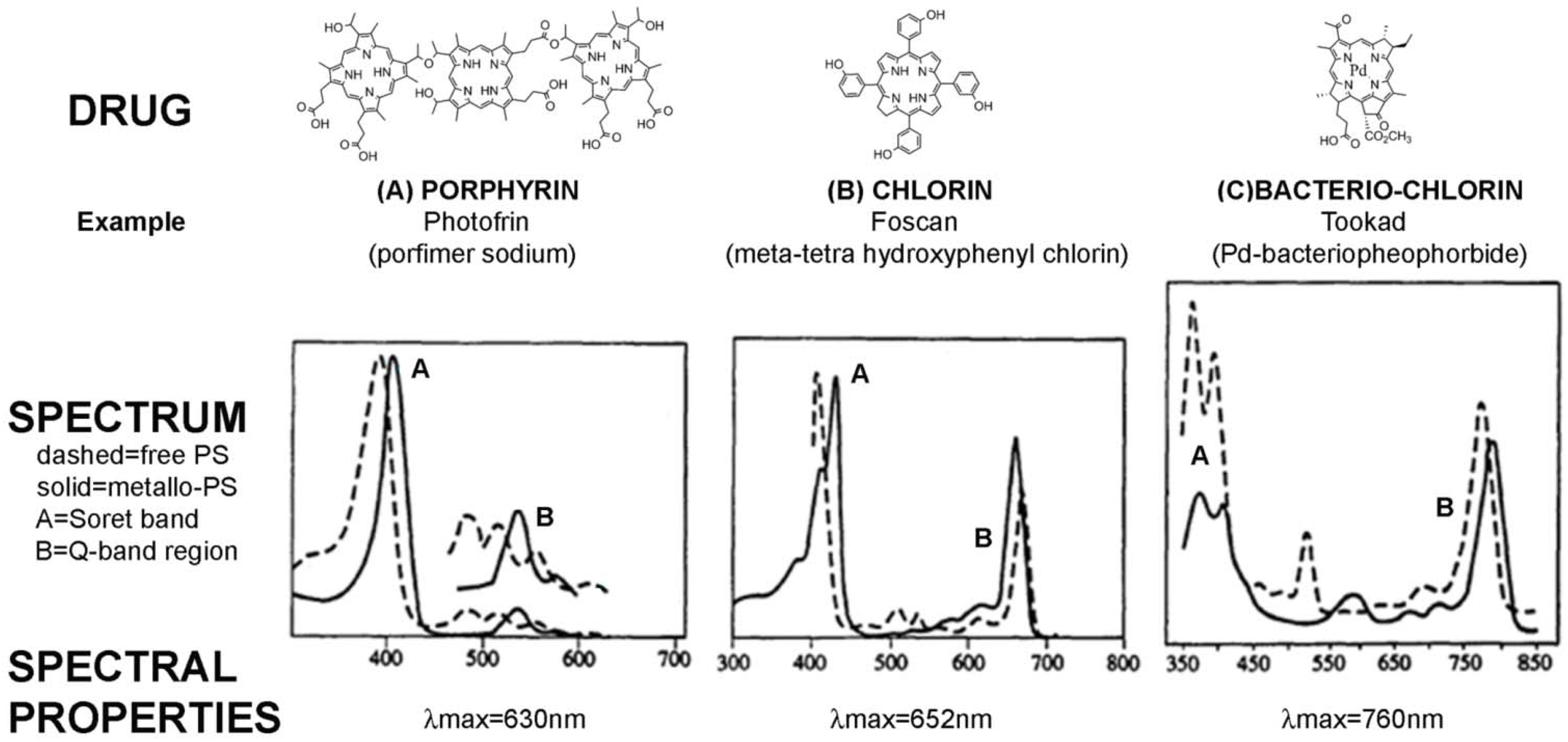
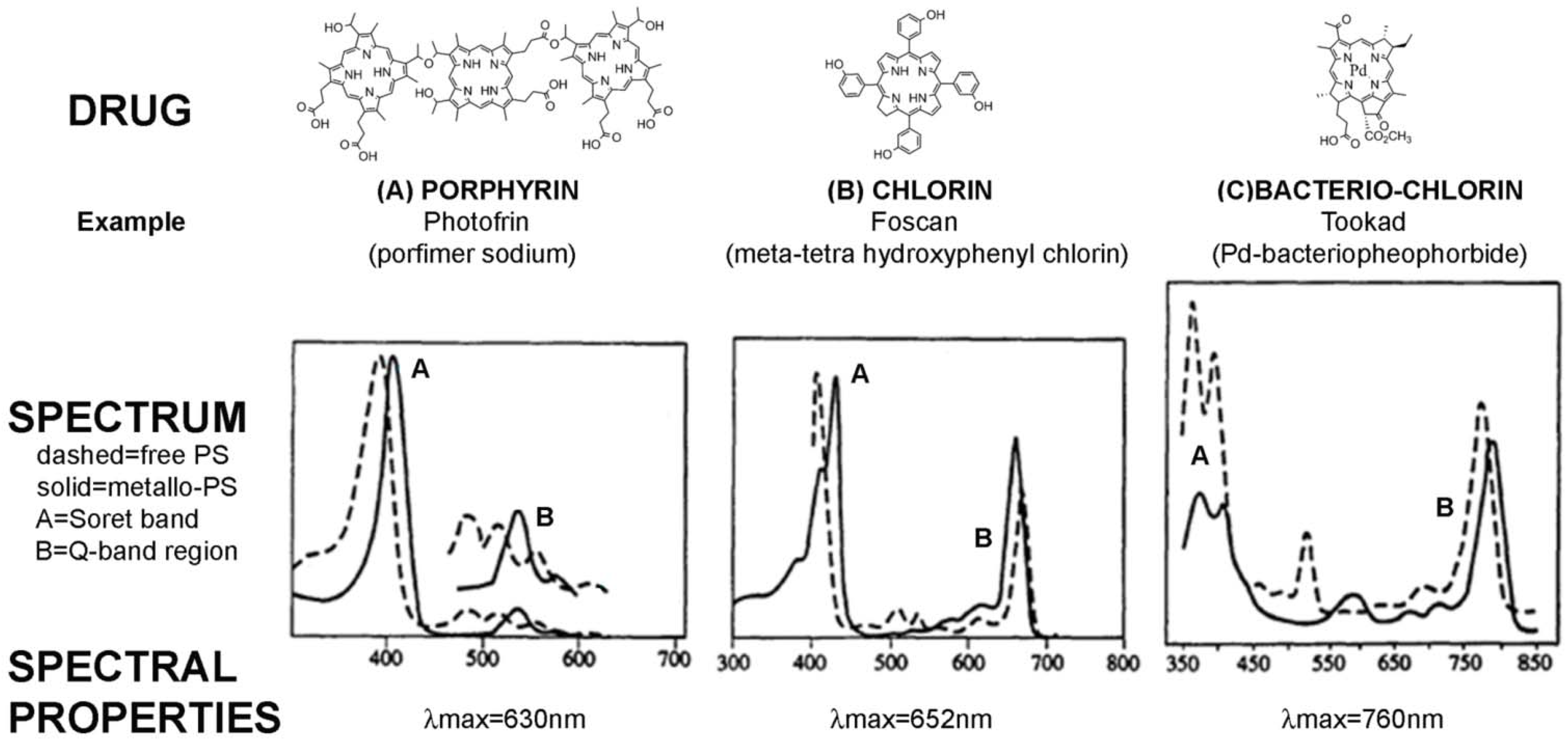
| Photofrin (Porfimer sodium) | A first generation PS-drug which is a mixture of hematoporphyrin derivatives. Absorbs at 630 nm, so limited tissue penetration (<5 mm) [24] | Oesophagus (dysplasia and cancer) |
| Stomach cancer | ||
| Lung cancer | ||
| Bladder cancer | ||
| Cervical cancer | ||
| Foscan (mTHPC) | One of the most potent PS-drugs known but associated with temporary pain and skin photosensitivity [20,21] | Head and neck cancer |
| Levulan (ALA) | A precursor compound (5-aminolevulinic acid) which is taken up by cells and converted to protoporphyrin IX; a potent PS-drug. Has good tissue selectivity (can be 10-fold) but the drug-light interval is up to 18 h to allow for PS conversion [25]. | Basal cell carcinoma |
| Bowens disease | ||
| Actinic Keratosis | ||
| Metvix (Methyl derivative of ALA) | Has reduced side effects and increased skin penetration compared to 5-ALA [25] | Basal cell carcinoma |
| Bowens disease | ||
| Actinic Keratosis | ||
| Photosense (aluminum phthalocyanine) | An aqueous solution of sodium salts of sulphonated aluminum phthalocyanine. A composition of aluminion phtalocyanines with different degrees of sulfonation. Photosense is an relatively hydrophilic PS. | Skin cancer |
| Radachlorin (Bremachlorin) | An aqueous composition of 3 chlorophyll a derivatives [26] | Skin cancer |
| Laserphyrin / Aptocine (Talaporfin sodium) | A water soluble photosensitizer consisting of chlorin e6 and L-aspartic acid | Lung cancer (Undergoing trials in US for range of solid tumours) |
| Visudyne (Verteporfin) | Almost a blockbuster drug with >$400 m sales at its peak in the early 2,000 s. Made up of the PS benzoporphyrin derivative (BPD), has quick clearance and thus a short drug-light interval (15 min). Absorbs at 690 nm. Ophthalmologist shine the laser into the eye destroying the blood vessels which are growing aberrantly over the macula improving sight [15]. | Age-related macular degeneration |
| B: Photosensitisers undergoing clinical trials | ||
| Drug | Details | Indication |
| Tookad (Palladium bacteriopheophorbide) | In phase II/III clinical trials for prostate cancer. | Prostate cancer |
| Amphinex (TPCS2a) | In Phase I/II clinical trial. First in class photosensitiser using photochemical internalisation of bleomycin [29] | Head and neck cancers |
| Lu-Tex (Motexafin lutetium) | A tripyrrolic porphyrin which absorbs at 732 nm [30] | Breast cancer |
| Photochlor (HPPH) | A lipophilic, chlorin derivative which absorbs at 665 nm [23] | Oesophageal cancer |
| Advantage | Reason |
|---|---|
| Low/non invasive | Intravenously or topically administered drug, followed by surface or endoscope/hollow needle illumination avoids the need for surgery. |
| Precise surgical tool / Low scarring | Dual specify of preferential tumour localisation and laser directionality. Lower energy radiation and precise generation of cytotoxin prevents collateral damage to structural and neighbouring tissues. |
| Low side effects | Low systemic toxicity of the drug in its inactive form. Light activation at the sight of disease and local biological effect prevents other tissues being damaged. |
| Compatible with other modalities | The PDT mechanism of action does not interfere with other established treatments including chemotherapy or immunotherapy. |
| Low risk of resistance/ Repeatable | The nature of ROS and its generation in multiple cellular compartments reduces the likelihood of tumour up regulation of alternative circumventive pathways. Hence multiple drug doses or illumination doses can be carried out. |
| Short treatment times | In the clinic, PDT requires short admittance times |
| Cost effective | PDT treatment can cost less than surgery and conventional chemotherapy financially and life-years saved |
| Not immunosuppressive | Much lower chance of any immunosuppression than many other comparable treatment modalities for cancer. Conversely in many cases immune-activation mechanisms post PDT can lead to enhanced tumour eradication, or the development of a tumour vaccine response. |
| Disadvantage | Reason |
| Poor selectivity / Skin photosensitivity | Although many PS exhibit significant tumour localization, PDT drugs do not have a high selectivity, in part due to their high hydrophobicity. Many PS-drugs remain in the body for weeks and accumulate in the skin; patients have to stay out of direct sunlight to prevent skin damage/ inflammation. This is being addressed by the development of specifically targeted PS, like in ADP. |
| Lack of approved drugs | Clinical development of new drugs is hindered by poor understanding and/or an unwillingness of pharmaceutical companies to explore the area. Complicated dosimetry and a requirement for equipped PDT centres has led to a lack of robust, randomized clinical trials. |
| Limited light penetration into tissues | Light propagation through tissues is heavily limited by refraction, reflection and scattering processes and the absorption patterns of tissue chromophores. This is being addressed by the continual improvement of laser and light delivery technology including interstitial PDT under image guidance, light diffusing fibres and use of red-absorbing drugs [40] |
| Not being as immediately applicable to systemic diseases | PDT is not as immediately applicable to systemic/disseminated disease This is being addressed by the development of the specific targeting of PS, like in ADP, and the development and further understanding of PDT-induced immune responses in particular antitumor-specific immunity [41] |
| A degree of oxygen dependence | Many of the effective mechanisms of PDT depend on the availability of molecular oxygen in the target tissue during light irradiation. This is being addressed as some PS can preferentially initiate oxygen independent pathways, and light-dose fractionation can allow time for tissue re-oxygenation [42] |
| Difficult patient dosimetry | Due to the range of different PS available, its multiple applications and multiple factors involved in its administration, there are difficulties in prescribing the correct patient dosimetry for each situation. Over-illumination of some drugs can lead to drug inactivation (photobleaching). |
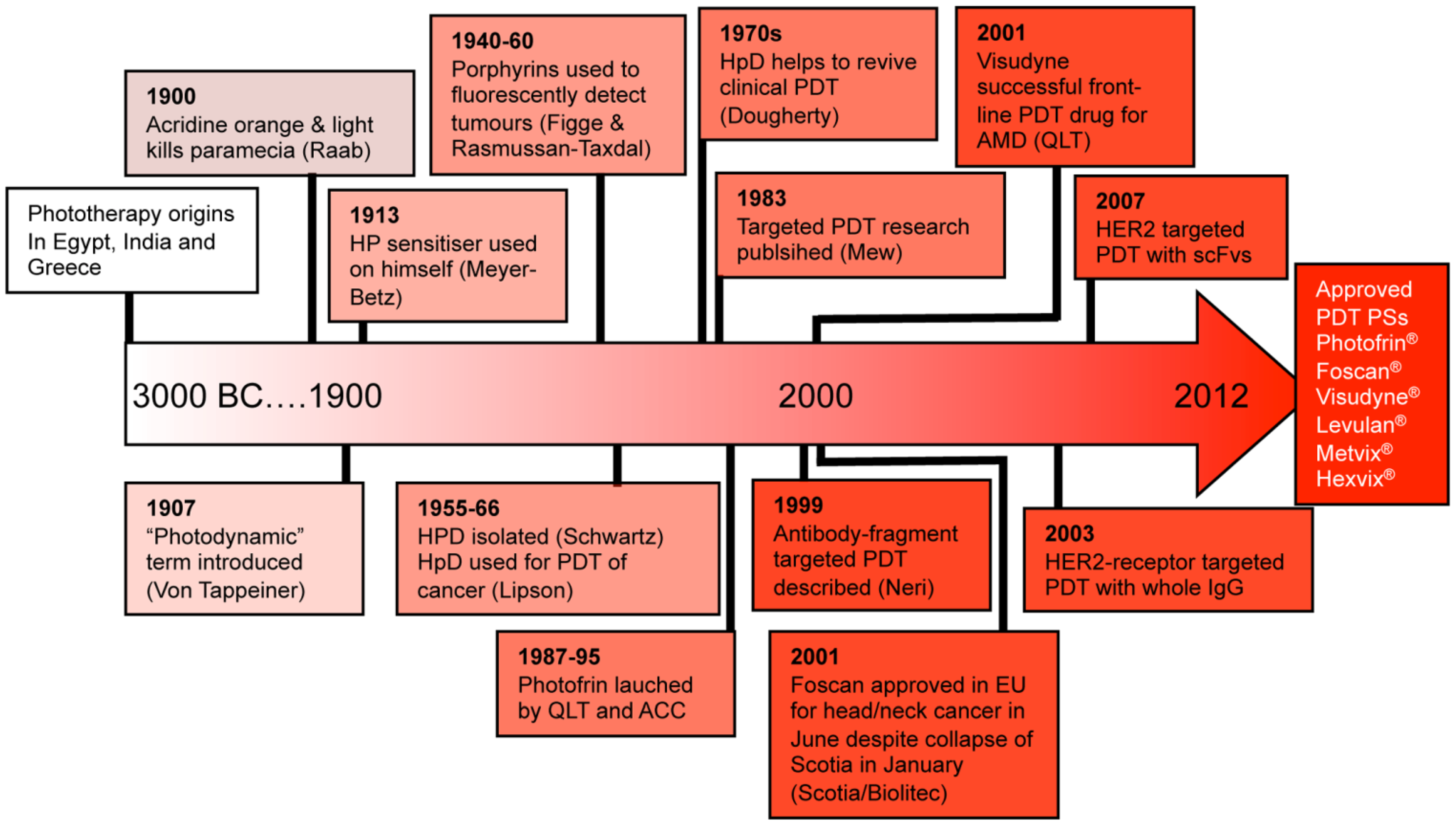
1.3. Antibody-Directed Phototherapy (ADP)
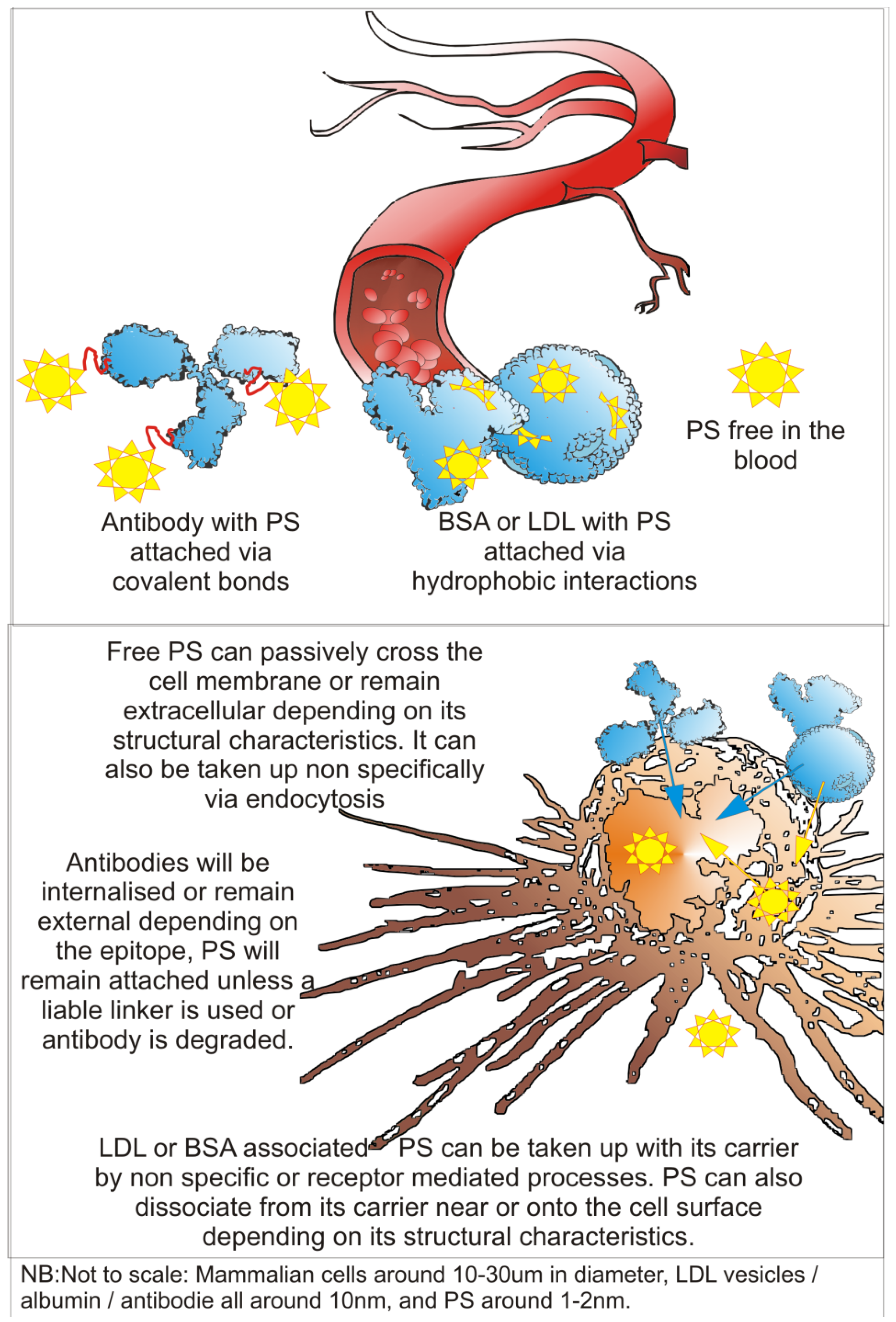
2. Photodynamic Therapy (PDT)
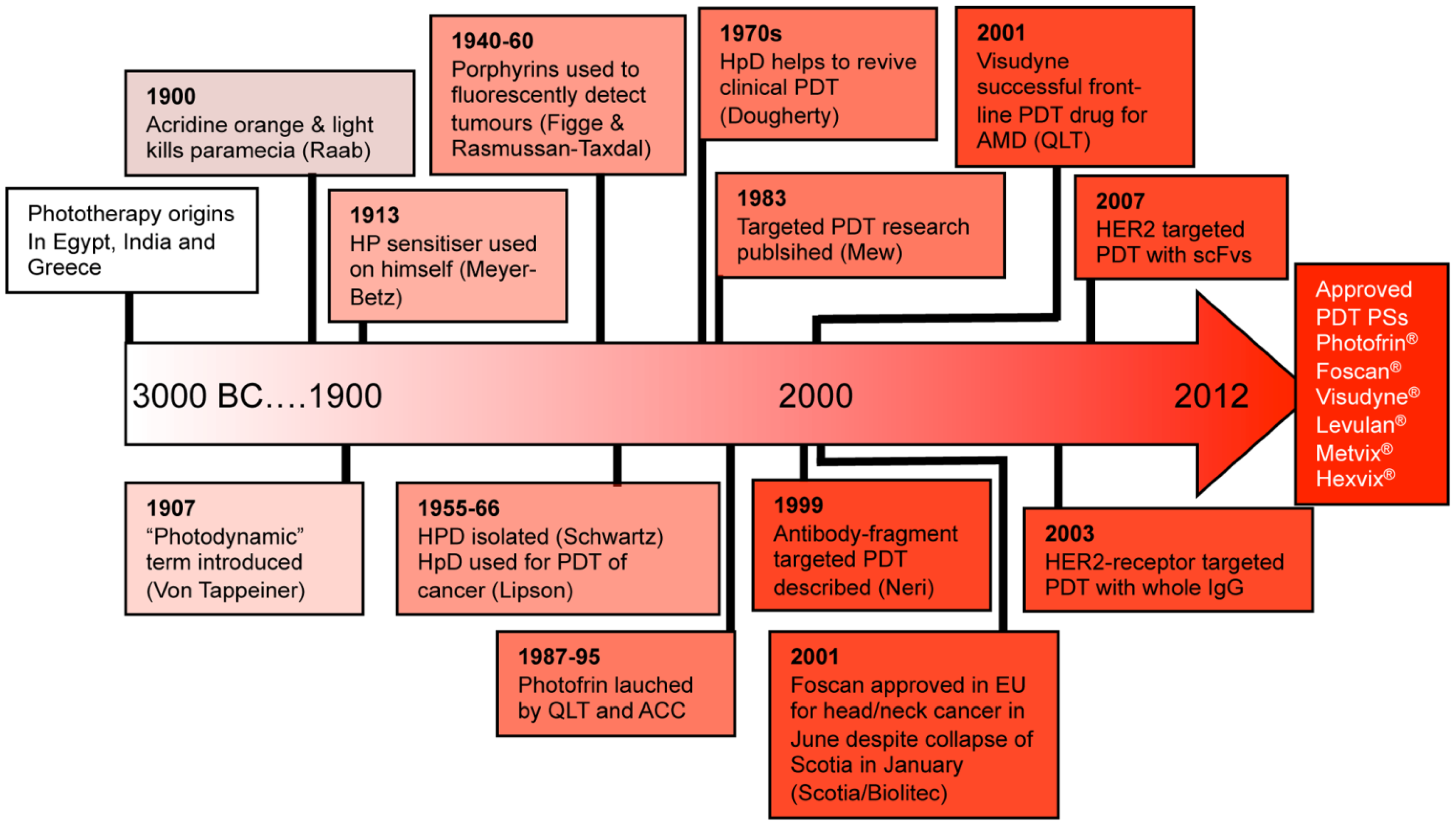
2.1. PS-Drug Development
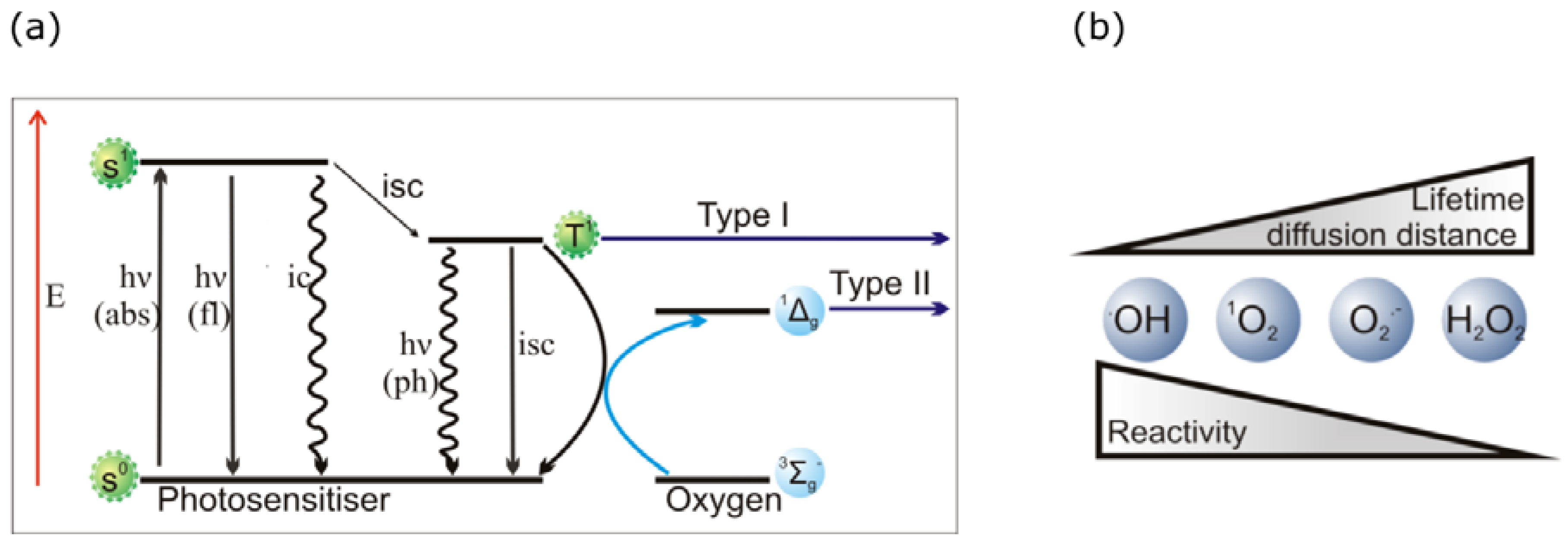
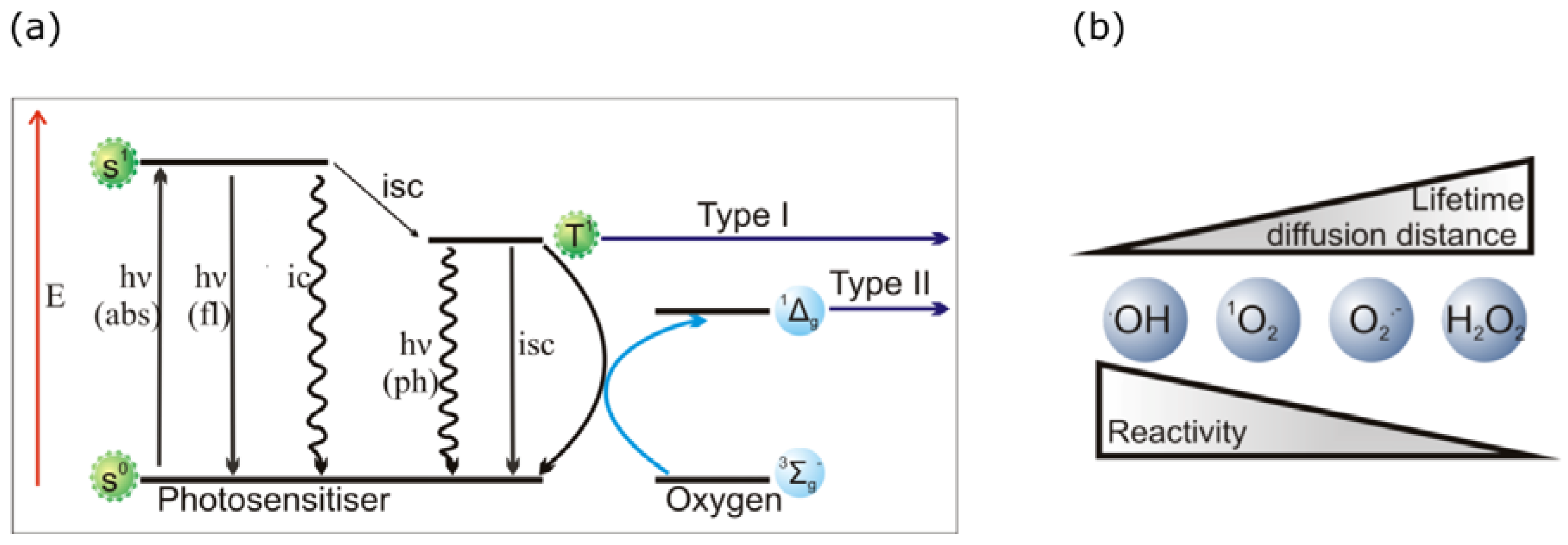
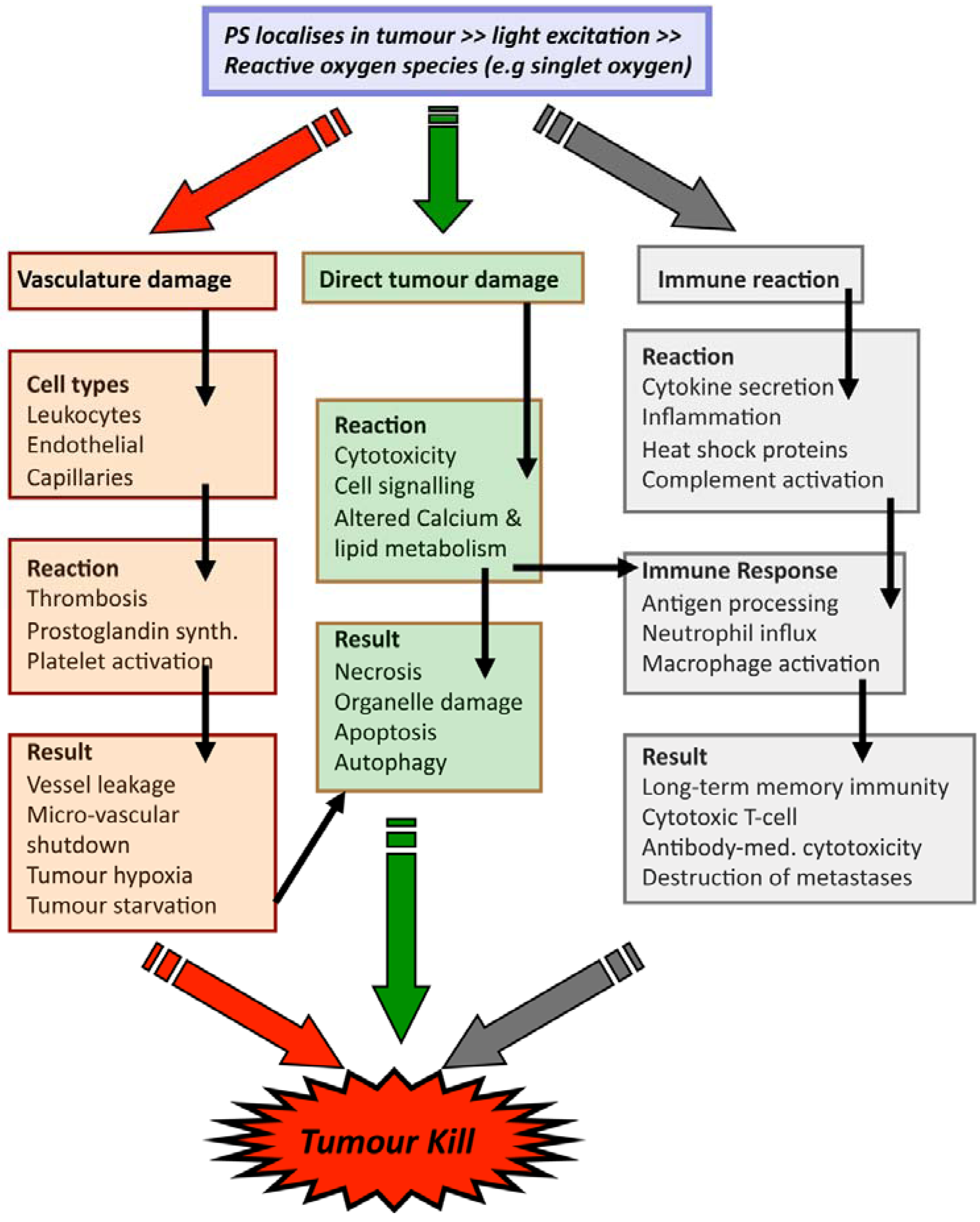
2.2. PDT Mechanisms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
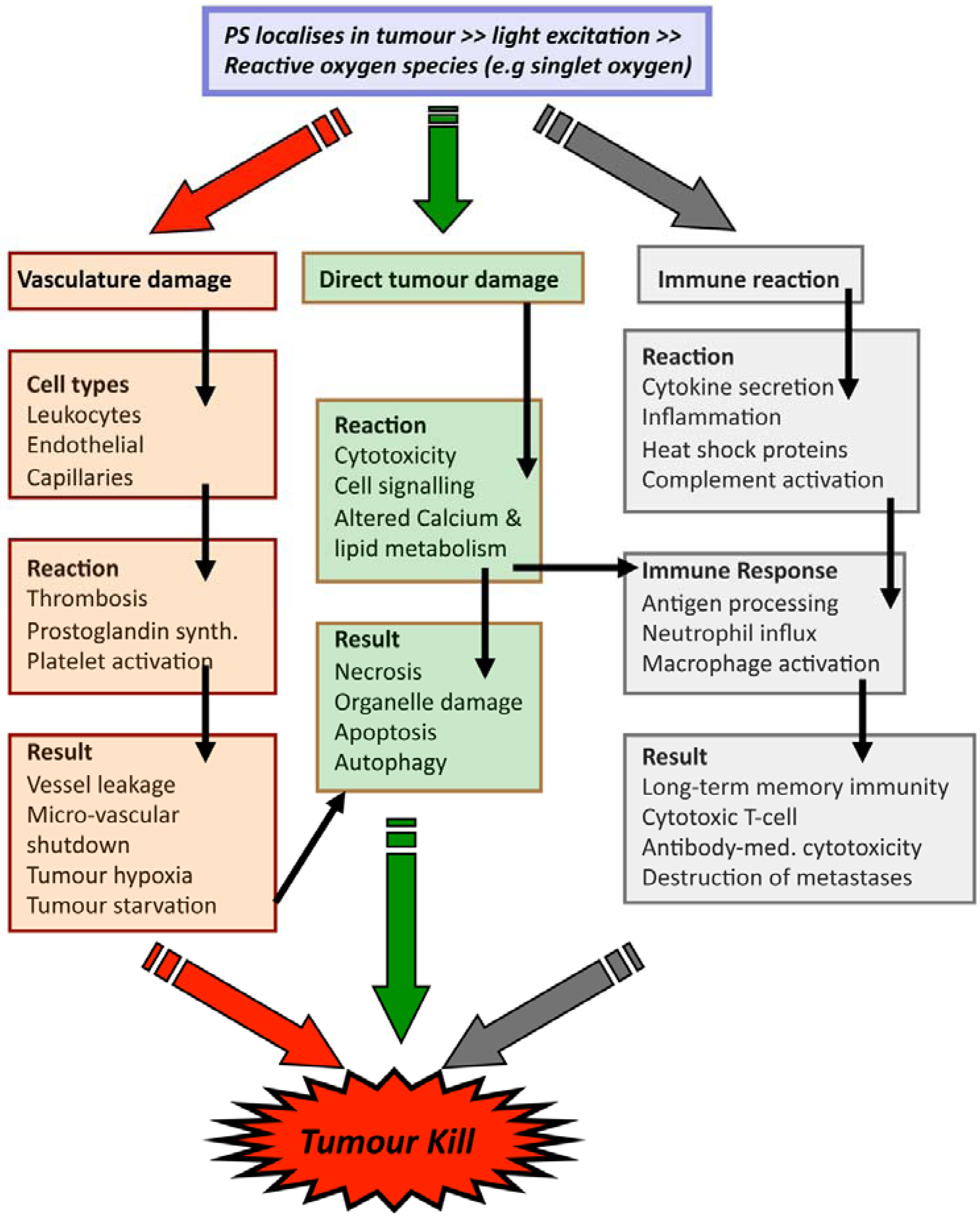
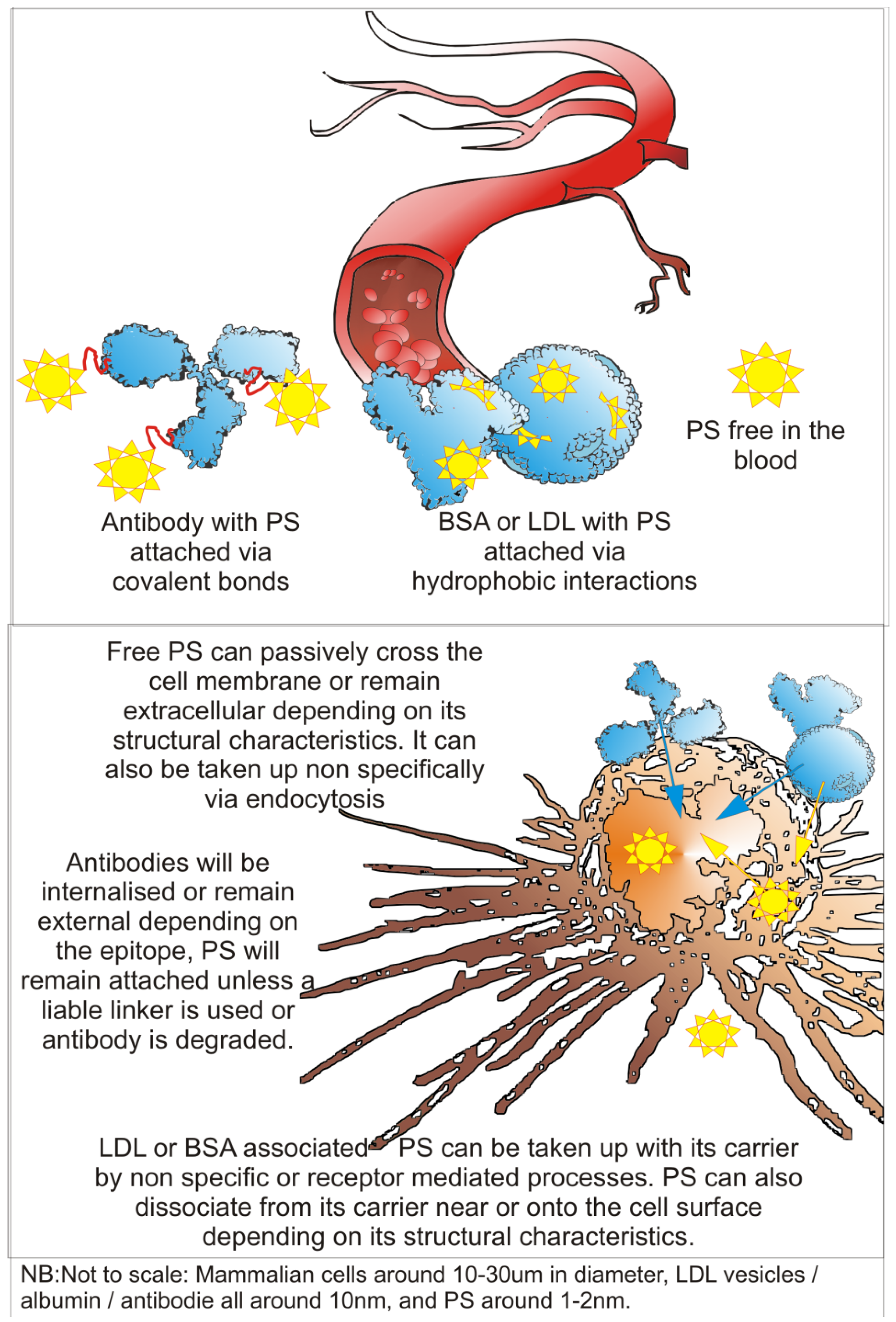
3. Technical Aspects of Antibody-Directed Phototherapy (ADP)
3.1. ADP Requirements
3.2. Antibody Component of an ADP-Drug
3.3. Drug and Linker Components of an ADP-Drug
| Reactive chemical group on antibody | Reactive chemical group / linking group on PS | Type of bond formed | Reference |
|---|---|---|---|
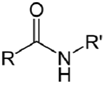
| NH2 (Amine) | COO- (Carboxylate) Activated via a carbodiimide to an O-acylisourea 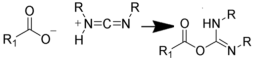 NB: Effciency can be increased by reaction via an NHS intermediate (see below) | Amide bond | [100,101,102,103,104] |
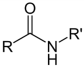 | |||
| NH2 (Amine) | N-Hydroxysuccinimide (NHS) Ester or Sulfo-NHS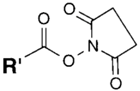 NB: NHS esters also may be formed in situ from a carboxylate by coupling the carbodiimide reaction above with the addition of NHS ester. NB: NHS esters also may be formed in situ from a carboxylate by coupling the carbodiimide reaction above with the addition of NHS ester. | Amide bond | [94,105,106,107,108,109,110] |
 | |||
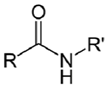
| NH2 (Amine) | Esterification of carboxyilic acid groups on PS to TFP esters (Tetrafluorophenyl esters)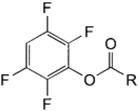 | Amide bond | [111,112,113,114] |
 | |||
| NH2 (Amine) | NCS (Isothiocyanate) | Isothiourea bond | [115,116,117,118,119] |
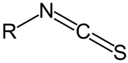 | 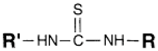 | ||
| SH (Sulfhydryl) | Maleimide | Thioether bond | [120] |
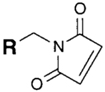 | 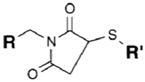 | ||
| SH (Sulfhydryl) | PS pre-loaded onto a Poly-Lysine Linker (NHS ester on PS to linker amine as above), heterobifunctional linking compound SPDP (N-succinimidyl 3-(2-pyridyldithio) propionate) used to conjugate amine group on the PS/linker to a sulfhydryl group on the antibody | Disulphide / Amide | [121,122,123,124,125,126,127] |
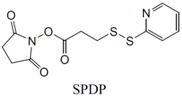 | 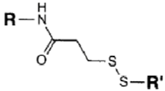 | ||
| NH2 (Amine) | PS pre-loaded onto a PVA Linker (carboxylate on PS to linker amine as above),Introduction of sulfhydryl groups to linker via 3-mercaptopropionic acid and use of heterobifunctional linking compound Sulfo-MBS (m-maleimidobenzoyl-N-hydroxysuccinimide ester) to conjugate amine group on the antibody to a sulfhydryl groups on the PS/linker | Thioether / Amide | [128,129,130] |
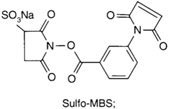 | 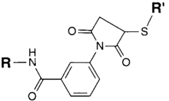 | ||
| CHO (Aldehyde) | NH2 (Amine) on the PS.MAb prepared for conjugation by carbohydrate oxidation with sodium periodate—opens sugar rings exposing free aldehyde groups which can react with the amine group. | Secondary amine linkage (once shchiff base linkage is reduced) | [131,132,133,134] |
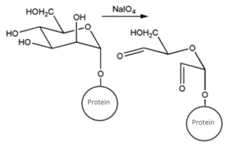 | 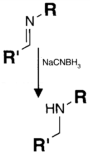 | ||
| CHO (Aldehyde) | PS pre-loaded onto a PGA Linker with the terminal carboxylate group protected (amine on PS to linker carboxylate as above), introduction of a hydrazide functional group to the terminal carboxylate of the linker by reaction of the activated ester (NHS) with hydrazine hydrate. | Hydrazone Linkage | [135,136,137,138] |
MAb prepared for conjugation by carbohydrate oxidation with sodium periodate (as above)
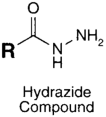 | 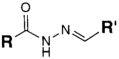 |
3.4. Issues in Making ADP-Drugs
4. ADP-Drug Development
4.1. First ADP-Drugs
4.2. ADP-Drug Development for Cancer with Whole Immunoglobulins
4.3. ADP-Drug Development for Cancer Using Antibody Fragments
5. The Outlook for ADP
6. Conclusions
Acknowledgements
References
- Deonarain, M.P. Recombinant antibodies for cancer therapy. Expert Opin. Biol. Ther. 2008, 8, 1123–1141. [Google Scholar] [CrossRef]
- Carter, P.J. Potent antibody therapeutics by design. Nat. Rev. Immunol. 2006, 6, 343–357. [Google Scholar] [CrossRef]
- Business Insights, L. The Cancer Market Outlook to 2016; Business Insights Ltd (Product Code: BI00042-009): London, UK, 2011. [Google Scholar]
- Aggarwal, S. What's fueling the biotech engine—2008. Nat. Biotechnol. 2009, 27, 987–993. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration Home Page. Available online: http://www.fda.gov/default.htm (accessed on 12 April 2013).
- The Antibody Society Home Page. Available online: http://www.antibodysociety.org/ (accessed on 12 April 2013).
- ClinicalTrials.gov Home Page. Available online: http://www.clinicaltrials.gov/ (accessed on 12 April 2013).
- Nahta, R.; Yu, D.; Hung, M.-C.; Hortobagyi, G.N.; Esteva, F.J. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat. Clin. Pract. Oncol. 2006, 3, 269–280. [Google Scholar] [CrossRef]
- Alley, S.C.; Okeley, N.M.; Senter, P.D. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537. [Google Scholar] [CrossRef]
- Hughes, B. Antibody-drug conjugates for cancer: Poised to deliver? Nat. Rev. Drug Discov. 2010, 9, 665–667. [Google Scholar] [CrossRef]
- Adair, J.R.; Howard, P.W.; Hartley, J.A.; Williams, D.G.; Chester, K.A. Antibody-drug conjugates—A perfect synergy. Expert Opin. Biol. Ther. 2012, 12, 1191–1206. [Google Scholar] [CrossRef]
- Brown, S.B.; Brown, E.A.; Walker, I. The present and future role of photodynamic therapy in cancer treatment. Lancet Oncol. 2004, 5, 497–508. [Google Scholar] [CrossRef]
- Dolmans, D.E.J.G.J.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef]
- Brown, S.B.; Mellish, K.J. Verteporfin: A milestone in opthalmology and photodynamic therapy. Expert Opin. Pharmacother. 2001, 2, 351–361. [Google Scholar] [CrossRef]
- Babilas, P.; Schreml, S.; Landthaler, M.; Szeimies, R.-M. Photodynamic therapy in dermatology: State-of-the-art. Photodermatol. Photoimmunol. Photomed. 2010, 26, 118–132. [Google Scholar] [CrossRef]
- Celli, J.P.; Spring, B.Q.; Rizvi, I.; Evans, C.L.; Samkoe, K.S.; Verma, S.; Pogue, B.W.; Hasan, T. Imaging and photodynamic therapy: mechanisms, monitoring, and optimization. Chem. Rev. 2010, 110, 2795–2838. [Google Scholar] [CrossRef]
- Bonnett, R. Chemical Aspects of Photodynamic Therapy; Gordon and Breach Science Publishers: Amsterdam, The Netherlands, 2000. [Google Scholar]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Lou, P.-J.; Jones, L.; Hopper, C. Clinical outcomes of photodynamic therapy for head-and-neck cancer. Technol. Cancer Res. Treat. 2003, 2, 311–317. [Google Scholar]
- Hopper, C.; Niziol, C.; Sidhu, M. The cost-effectiveness of Foscan mediated photodynamic therapy (Foscan-PDT) compared with extensive palliative surgery and palliative chemotherapy for patients with advanced head and neck cancer in the UK. Oral Oncol. 2004, 40, 372–382. [Google Scholar] [CrossRef]
- Nyst, H.J.; Tan, I.B.; Stewart, F.A.; Balm, A.J.M. Is photodynamic therapy a good alternative to surgery and radiotherapy in the treatment of head and neck cancer? Photodiagnosis Photodyn. Ther. 2009, 6, 3–11. [Google Scholar] [CrossRef]
- Allison, R.R.; Sibata, C.H. Oncologic photodynamic therapy photosensitizers: A clinical review. Photodiagnosis Photodyn. Ther. 2010, 7, 61–75. [Google Scholar] [CrossRef]
- Schweitzer, V.G.; Somers, M.L. PHOTOFRIN-mediated photodynamic therapy for treatment of early stage (Tis-T2N0M0) SqCCa of oral cavity and oropharynx. Lasers Surg. Med. 2010, 42, 1–8. [Google Scholar]
- Fien, S.M.; Oseroff, A.R. Photodynamic therapy for non-melanoma skin cancer. J. Natl. Compr. Canc. Netw. 2007, 5, 531–540. [Google Scholar]
- Kochneva, E.V.; Filonenko, E.V.; Vakulovskaya, E.G.; Scherbakova, E.G.; Seliverstov, O.V.; Markichev, N.A.; Reshetnickov, A.V. Photosensitizer Radachlorin(R): Skin cancer PDT phase II clinical trials. Photodiagnosis Photodyn. Ther. 2010, 7, 258–267. [Google Scholar] [CrossRef]
- Moore, C.M.; Pendse, D.; Emberton, M. Photodynamic therapy for prostate cancer—A review of current status and future promise. Nat. Clin. Pract. Urol. 2009, 6, 18–30. [Google Scholar] [CrossRef]
- Trachtenberg, J.; Weersink, R.A.; Davidson, S.R.H.; Haider, M.A.; Bogaards, A.; Gertner, M.R.; Evans, A.; Scherz, A.; Savard, J.; Chin, J.L.; et al. Vascular-targeted photodynamic therapy (padoporfin, WST09) for recurrent prostate cancer after failure of external beam radiotherapy: a study of escalating light doses. BJU Int. 2008, 102, 556–562. [Google Scholar] [CrossRef]
- Selbo, P.K.; Weyergang, A.; Bonsted, A.; Bown, S.G.; Berg, K. Photochemical internalization of therapeutic macromolecular agents: A novel strategy to kill multidrug-resistant cancer cells. J. Pharmacol. Exp. Ther. 2006, 319, 604–612. [Google Scholar] [CrossRef]
- Josefsen, L.B.; Boyle, R.W. Photodynamic therapy: Novel third-generation photosensitizers one step closer? Br. J. Pharmacol. 2008, 154, 1–3. [Google Scholar] [CrossRef]
- Maisch, T. Anti-microbial photodynamic therapy: Useful in the future? Lasers Med. Sci. 2007, 22, 83–91. [Google Scholar]
- Wormald, R.; Evans, J.; Smeeth, L.; Henshaw, K. Photodynamic therapy for neovascular age-related macular degeneration. Cochrane Database Syst Rev 2007, CD002030. [Google Scholar]
- Tandon, Y.K.; Yang, M.F.; Baron, E.D. Role of photodynamic therapy in psoriasis: A brief review. Photodermatol. Photoimmunol. Photomed. 2008, 24, 222–230. [Google Scholar] [CrossRef]
- van der Snoek, E.M.; Robinson, D.J.; van Hellemond, J.J.; Neumann, H.A. A review of photodynamic therapy in cutaneous leishmaniasis. J. Eur. Acad. Dermatol. Venereol. 2008, 22, 918–922. [Google Scholar] [CrossRef]
- Wilson, B.C.; Patterson, M.S. The physics, biophysics and technology of photodynamic therapy. Phys. Med. Biol. 2008, 53, R61–R109. [Google Scholar] [CrossRef]
- Choudhary, S.; Nouri, K.; Elsaie, M.L. Photodynamic therapy in dermatology: A review. Lasers Med. Sci. 2009, 24, 971–980. [Google Scholar] [CrossRef]
- Dai, T.; Huang, Y.Y.; Hamblin, M.R. Photodynamic therapy for localized infections—State of the art. Photodiagnosis Photodyn. Ther. 2009, 6, 170–188. [Google Scholar] [CrossRef]
- Huang, Z.; Xu, H.; Meyers, A.D.; Musani, A.I.; Wang, L.; Tagg, R.; Barqawi, A.B.; Chen, Y.K. Photodynamic therapy for treatment of solid tumors—Potential and technical challenges. Technol. Cancer Res. Treat. 2008, 7, 309–320. [Google Scholar]
- Allison, R.R.; Mota, H.C.; Bagnato, V.S.; Sibata, C.H. Bio-nanotechnology and photodynamic therapy—State of the art review. Photodiagnosis Photodyn. Ther. 2008, 5, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Mang, T.S. Lasers and light sources for PDT: Past, present and future. Photodiagnosis Photodyn. Ther. 2004, 1, 43–48. [Google Scholar] [CrossRef]
- Firczuk, M.; Nowis, D.; Gołąb, J. PDT-induced inflammatory and host responses. Photochem. Photobiol. Sci. 2011, 10, 653–663. [Google Scholar] [CrossRef]
- Freitas, I.; Baronzio, G.F. Tumor hypoxia, reoxygenation and oxygenation strategies: Possible role in photodynamic therapy. J. Photochem. Photobiol. B Biol. 1991, 11, 3–30. [Google Scholar] [CrossRef]
- Ferrari, M. Frontiers in cancer nanomedicine: Directing mass transport through biological barriers. Trends Biotechnol. 2010, 28, 181–188. [Google Scholar] [CrossRef]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part three—Photosensitizer pharmacokinetics, biodistribution, tumor localization and modes of tumor destruction. Photodiagnosis Photodyn. Ther. 2005, 2, 91–106. [Google Scholar] [CrossRef]
- Sternberg, E.D.; Dolphin, D.; Brückner, C. Porphyrin-based photosensitizers for use in photodynamic therapy. Tetrahedron 1998, 54, 4151–4202. [Google Scholar] [CrossRef]
- Konan, Y.N.; Gurny, R.; Allemann, E. State of the art in the delivery of photosensitizers for photodynamic therapy. J. Photochem. Photobiol. B 2002, 66, 89–106. [Google Scholar] [CrossRef]
- Sharman, W.M.; van Lier, J.E.; Allen, C.M. Targeted photodynamic therapy via receptor mediated delivery systems. Adv. Drug Deliv. Rev. 2004, 56, 53–76. [Google Scholar] [CrossRef]
- Bechet, D.; Couleaud, P.; Frochot, C.; Viriot, M.L.; Guillemin, F.; Barberi-Heyob, M. Nanoparticles as vehicles for delivery of photodynamic therapy agents. Trends Biotechnol. 2008, 26, 612–621. [Google Scholar] [CrossRef]
- Bugaj, A.M. Targeted photodynamic therapy—A promising strategy of tumor treatment. Photochem. Photobiol. Sci. 2011, 10, 1097–1109. [Google Scholar] [CrossRef]
- Macdonald, I.J.; Dougherty, T.J. Basic principles of photodynamic therapy. J. Porphyr. Phthalocyanines 2001, 05, 105–129. [Google Scholar] [CrossRef]
- Hamblin, M.R.; Mróz, P.; Mroz, P. Advances in Photodynamic Therapy: Basic, Translational and Clinical; Artech House: Norwood, MA, USA, 2008. [Google Scholar]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef]
- Redmond, R.W.; Kochevar, I.E. Spatially resolved cellular responses to singlet oxygen. Photochem. Photobiol. 2006, 82, 1178–1186. [Google Scholar] [CrossRef]
- Robertson, C.A.; Evans, D.H.; Abrahamse, H. Photodynamic therapy (PDT): A short review on cellular mechanisms and cancer research applications for PDT. J. Photochem. Photobiol. B Biol. 2009, 96, 1–8. [Google Scholar] [CrossRef]
- Oleinick, N.L.; Morris, R.L.; Belichenko, I. The role of apoptosis in response to photodynamic therapy: What, where, why, and how. Photochem. Photobiol. Sci. 2002, 1, 1–21. [Google Scholar] [CrossRef]
- Boyle, R.W.; Dolphin, D. Structure and biodistribution relationships of photodynamic sensitizers. Photochem. Photobiol. 1996, 64, 469–485. [Google Scholar] [CrossRef]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part one—Photosensitizers, photochemistry and cellular localization. Photodiagnosis Photodyn. Ther. 2004, 1, 279–293. [Google Scholar] [CrossRef]
- Mojzisova, H.; Bonneau, S.; Brault, D. Structural and physico-chemical determinants of the interactions of macrocyclic photosensitizers with cells. Eur. Biophys. J. 2007, 36, 943–953. [Google Scholar] [CrossRef]
- Almeida, R.D.; Manadas, B.J.; Carvalho, A.P.; Duarte, C.B. Intracellular signaling mechanisms in photodynamic therapy. Biochim. Biophys. Acta 2004, 1704, 59–86. [Google Scholar]
- Tsaytler, P.A.; C O'Flaherty, M.; Sakharov, D.V.; Krijgsveld, J.; Egmond, M.R. Immediate protein targets of photodynamic treatment in carcinoma cells. J. Proteome Res. 2008, 7, 3868–3878. [Google Scholar] [CrossRef]
- Baglo, Y.; Sousa, M.M.L.; Slupphaug, G.; Hagen, L.; Håvåg, S.; Helander, L.; Zub, K.A.; Krokan, H.E.; Gederaas, O.A. Photodynamic therapy with hexyl aminolevulinate induces carbonylation, posttranslational modifications and changed expression of proteins in cell survival and cell death pathways. Photochem. Photobiol. Sci. 2011, 10, 1137–1145. [Google Scholar] [CrossRef]
- Moor, A.C. Signaling pathways in cell death and survival after photodynamic therapy. J. Photochem. Photobiol. B Biol. 2000, 57, 1–13. [Google Scholar] [CrossRef]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part two—Cellular signaling, cell metabolism and modes of cell death. Photodiagnosis Photodyn. Ther. 2005, 2, 1–23. [Google Scholar] [CrossRef]
- Buytaert, E.; Dewaele, M.; Agostinis, P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim. Biophys. Acta 2007, 1776, 86–107. [Google Scholar]
- Pazos, M.d.C.; Nader, H.B. Effect of photodynamic therapy on the extracellular matrix and associated components. Braz. J. Med. Biol. Res. 2007, 40, 1025–1035. [Google Scholar] [CrossRef]
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 2006, 6, 535–545. [Google Scholar] [CrossRef]
- van Duijnhoven, F.H.; Aalbers, R.I.J.M.; Rovers, J.P.; Terpstra, O.T.; Kuppen, P.J.K. The immunological consequences of photodynamic treatment of cancer, a literature review. Immunobiology 2003, 207, 105–113. [Google Scholar] [CrossRef]
- Korbelik, M. Cancer vaccines generated by photodynamic therapy. Photochem. Photobiol. Sci. 2011, 10, 664–669. [Google Scholar] [CrossRef]
- Wilson, B.C.; Patterson, M.S.; Lilge, L. Implicit and explicit dosimetry in photodynamic therapy: A New paradigm. Lasers Med. Sci. 1997, 12, 182–199. [Google Scholar] [CrossRef]
- Caldwell, J.; Gardner, I.; Swales, N. An introduction to drug disposition: The basic principles of absorption, distribution, metabolism, and excretion. Toxicol. Pathol. 1995, 23, 102–114. [Google Scholar] [CrossRef]
- Ochsner, M. Photophysical and photobiological processes in the photodynamic therapy of tumours. J. Photochem. Photobiol. B 1997, 39, 1–18. [Google Scholar] [CrossRef]
- Henderson, B.W.; Gollnick, S.O.; Snyder, J.W.; Busch, T.M.; Kousis, P.C.; Cheney, R.T.; Morgan, J. Choice of oxygen-conserving treatment regimen determines the inflammatory response and outcome of photodynamic therapy of tumors. Cancer Res. 2004, 64, 2120–2126. [Google Scholar]
- Bachor, R.; Hautmann, R.; Hasan, T. Comparison of two routes of photosensitizer administration for photodynamic therapy of bladder cancer. Urol. Res. 1994, 22, 21–23. [Google Scholar] [CrossRef]
- Kwitniewski, M.; Juzeniene, A.; Glosnicka, R.; Moan, J. Immunotherapy: A way to improve the therapeutic outcome of photodynamic therapy? Photochem. Photobiol. Sci. 2008, 7, 1011–1017. [Google Scholar] [CrossRef]
- Bown, S.G.; Rogowska, A.Z.; Whitelaw, D.E.; Lees, W.R.; Lovat, L.B.; Ripley, P.; Jones, L.; Wyld, P.; Gillams, A.; Hatfield, A.W. Photodynamic therapy for cancer of the pancreas. Gut 2002, 50, 549–557. [Google Scholar] [CrossRef]
- Nelson, A.L.; Reichert, J.M. Development trends for therapeutic antibody fragments. Nat. Biotechnol. 2009, 27, 331–337. [Google Scholar] [CrossRef]
- Deyev, S.M.; Lebedenko, E.N. Multivalency: The hallmark of antibodies used for optimization of tumor targeting by design. Bioessays 2008, 30, 904–918. [Google Scholar] [CrossRef]
- Salfeld, J.G. Isotype selection in antibody engineering. Nat. Biotechnol. 2007, 25, 1369–1372. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Golay, J.; Introna, M. Mechanism of action of therapeutic monoclonal antibodies: Promises and pitfalls of in vitro and in vivo assays. Arch. Biochem. Biophys. 2012, 526, 146–153. [Google Scholar] [CrossRef]
- Arnold, J.N.; Wormald, M.R.; Sim, R.B.; Rudd, P.M.; Dwek, R.A. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu. Rev. Immunol. 2007, 25, 21–50. [Google Scholar] [CrossRef]
- Rothlisberger, D.; Honegger, A.; Pluckthun, A. Domain interactions in the Fab fragment: a comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J. Mol. Biol. 2005, 347, 773–789. [Google Scholar] [CrossRef]
- Quintero-Hernández, V.; Del Pozo-Yauner, L.; Pedraza-Escalona, M.; Juárez-González, V.R.; Alcántara-Recillas, I.; Possani, L.D.; Becerril, B. Evaluation of three different formats of a neutralizing single chain human antibody against toxin Cn2: Neutralization capacity versus thermodynamic stability. Immunol. Lett. 2012, 143, 152–160. [Google Scholar] [CrossRef]
- Carmichael, J.A.; Power, B.E.; Garrett, T.P.J.; Yazaki, P.J.; Shively, J.E.; Raubischek, A.A.; Wu, A.M.; Hudson, P.J. The crystal structure of an anti-CEA scFv diabody assembled from T84.66 scFvs in V(L)-to-V(H) orientation: implications for diabody flexibility. J. Mol. Biol. 2003, 326, 341–351. [Google Scholar]
- Kipriyanov, S.M.; Moldenhauer, G.; Braunagel, M.; Reusch, U.; Cochlovius, B.; Le Gall, F.; Kouprianova, O.A.; Von der Lieth, C.W.; Little, M. Effect of domain order on the activity of bacterially produced bispecific single-chain Fv antibodies. J. Mol. Biol. 2003, 330, 99–111. [Google Scholar] [CrossRef]
- Cuesta, A.M.; Sainz-Pastor, N.; Bonet, J.; Oliva, B.; Alvarez-Vallina, L. Multivalent antibodies: when design surpasses evolution. Trends Biotechnol. 2010, 28, 355–362. [Google Scholar] [CrossRef]
- Todorovska, A.; Roovers, R.C.; Dolezal, O.; Kortt, A.A.; Hoogenboom, H.R.; Hudson, P.J. Design and application of diabodies, triabodies and tetrabodies for cancer targeting. J. Immunol. Methods 2001, 248, 47–66. [Google Scholar] [CrossRef]
- Kenanova, V.; Wu, A.M. Tailoring antibodies for radionuclide delivery. Expert Opin. Drug Deliv. 2006, 3, 53–70. [Google Scholar] [CrossRef]
- Yuan, F.; Dellian, M.; Fukumura, D.; Leunig, M.; Berk, D.A.; Torchilin, V.P.; Jain, R.K. Vascular permeability in a human tumor xenograft: Molecular size dependence and cutoff size. Cancer Res. 1995, 55, 3752–3756. [Google Scholar]
- Batra, S.K.; Jain, M.; Wittel, U.A.; Chauhan, S.C.; Colcher, D. Pharmacokinetics and biodistribution of genetically engineered antibodies. Curr. Opin. Biotechnol. 2002, 13, 603–608. [Google Scholar] [CrossRef]
- Rudnick, S.I.; Adams, G.P. Affinity and avidity in antibody-based tumor targeting. Cancer Biother. Radiopharm. 2009, 24, 155–161. [Google Scholar] [CrossRef]
- Dahle, J.; Bagdonas, S.; Kaalhus, O.; Olsen, G.; Steen, H.B.; Moan, J. The bystander effect in photodynamic inactivation of cells. Biochim. Biophys. Acta 2000, 1475, 273–280. [Google Scholar] [CrossRef]
- Rubio, N.; Fleury, S.P.; Redmond, R.W. Spatial and temporal dynamics of in vitro photodynamic cell killing: Extracellular hydrogen peroxide mediates neighbouring cell death. Photochem. Photobiol. Sci. 2009, 8, 457–464. [Google Scholar] [CrossRef]
- Birchler, M.; Viti, F.; Zardi, L.; Spiess, B.; Neri, D. Selective targeting and photocoagulation of ocular angiogenesis mediated by a phage-derived human antibody fragment. Nat. Biotechnol. 1999, 17, 984–988. [Google Scholar] [CrossRef]
- Conlon, K.A.; Berrios, M. Light-induced proteolysis of myosin heavy chain by Rose Bengal-conjugated antibody complexes. J. Photochem. Photobiol. B 2001, 65, 22–28. [Google Scholar] [CrossRef]
- Conlon, K.A.; Rosenquist, T.; Berrios, M. Site-directed photochemical disruption of the actin cytoskeleton by actin-binding Rose Bengal-conjugates. J. Photochem. Photobiol. B 2002, 68, 140–146. [Google Scholar] [CrossRef]
- Hermanson, G.T. Bioconjugate Techniques; Academic Press: London, UK, 2008. [Google Scholar]
- Bullous, A.J.; Alonso, C.M.A.; Boyle, R.W. Photosensitiser-antibody conjugates for photodynamic therapy. Photochem. Photobiol. Sci. 2011, 10, 721–750. [Google Scholar] [CrossRef]
- Giuntini, F.; Alonso, C.M.A.; Boyle, R.W. Synthetic approaches for the conjugation of porphyrins and related macrocycles to peptides and proteins. Photochem. Photobiol. Sci. 2011, 10, 759–791. [Google Scholar] [CrossRef]
- Mew, D.; Lum, V.; Wat, C.K.; Towers, G.H.; Sun, C.H.; Walter, R.J.; Wright, W.; Berns, M.W.; Levy, J.G. Ability of specific monoclonal antibodies and conventional antisera conjugated to hematoporphyrin to label and kill selected cell lines subsequent to light activation. Cancer Res. 1985, 45, 4380–4386. [Google Scholar]
- Mew, D.; Wat, C.K.; Towers, G.H.; Levy, J.G. Photoimmunotherapy: treatment of animal tumors with tumor-specific monoclonal antibody-hematoporphyrin conjugates. J. Immunol. 1983, 130, 1473–1477. [Google Scholar]
- Steele, J.K.; Liu, D.; Stammers, A.T.; Whitney, S.; Levy, J.G. Suppressor deletion therapy: selective elimination of T suppressor cells in vivo using a hematoporphyrin conjugated monoclonal antibody permits animals to reject syngeneic tumor cells. Cancer Immunol. Immunother. 1988, 26, 125–131. [Google Scholar]
- Carcenac, M.; Dorvillius, M.; Garambois, V.; Glaussel, F.; Larroque, C.; Langlois, R.; Hynes, N.E.; van Lier, J.E.; Pèlegrin, A. Internalisation enhances photo-induced cytotoxicity of monoclonal antibody-phthalocyanine conjugates. Br. J. Cancer 2001, 85, 1787–1793. [Google Scholar] [CrossRef]
- Carcenac, M.; Larroque, C.; Langlois, R.; van Lier, J.E.; Artus, J.C.; Pèlegrin, A. Preparation, phototoxicity and biodistribution studies of anti-carcinoembryonic antigen monoclonal antibody-phthalocyanine conjugates. Photochem. Photobiol. 1999, 70, 930–936. [Google Scholar]
- Savellano, M.D.; Hasan, T. Targeting cells that overexpress the epidermal growth factor receptor with polyethylene glycolated BPD verteporfin photosensitizer immunoconjugates. Photochem. Photobiol. 2003, 77, 431–439. [Google Scholar] [CrossRef]
- Savellano, M.D.; Pogue, B.W.; Hoopes, P.J.; Vitetta, E.S.; Paulsen, K.D. Multiepitope HER2 targeting enhances photoimmunotherapy of HER2-overexpressing cancer cells with pyropheophorbide-a immunoconjugates. Cancer Res. 2005, 65, 6371–6379. [Google Scholar] [CrossRef]
- Bhatti, M.; Yahioglu, G.; Milgrom, L.R.; Garcia-Maya, M.; Chester, K.A.; Deonarain, M.P. Targeted photodynamic therapy with multiply-loaded recombinant antibody fragments. Int. J. Cancer 2008, 122, 1155–1163. [Google Scholar]
- Soukos, N.S.; Hamblin, M.R.; Keel, S.; Fabian, R.L.; Deutsch, T.F.; Hasan, T. Epidermal growth factor receptor-targeted immunophotodiagnosis and photoimmunotherapy of oral precancer in vivo. Cancer Res. 2001, 61, 4490–4496. [Google Scholar]
- Fabbrini, M.; Trachsel, E.; Soldani, P.; Bindi, S.; Alessi, P.; Bracci, L.; Kosmehl, H.; Zardi, L.; Neri, D.; Neri, P. Selective occlusion of tumor blood vessels by targeted delivery of an antibody-photosensitizer conjugate. Int. J. Cancer 2006, 118, 1805–1813. [Google Scholar] [CrossRef]
- Palumbo, A.; Hauler, F.; Dziunycz, P.; Schwager, K.; Soltermann, A.; Pretto, F.; Alonso, C.; Hofbauer, G.F.; Boyle, R.W.; Neri, D. A chemically modified antibody mediates complete eradication of tumours by selective disruption of tumour blood vessels. Br. J. Cancer 2011, 104, 1106–1115. [Google Scholar] [CrossRef]
- Vrouenraets, M.B.; Visser, G.W.M.; Stigter, M.; Oppelaar, H.; Snow, G.B.; van Dongen, G.A.M.S. Comparison of aluminium (III) phthalocyanine tetrasulfonate- and meta-tetrahydroxyphenylchlorin-monoclonal antibody conjugates for their efficacy in photodynamic therapy in vitro. Int. J. Cancer 2002, 98, 793–798. [Google Scholar]
- Vrouenraets, M.B.; Visser, G.W.; Stewart, F.A.; Stigter, M.; Oppelaar, H.; Postmus, P.E.; Snow, G.B.; van Dongen, G.A. Development of meta-tetrahydroxyphenylchlorin-monoclonal antibody conjugates for photoimmunotherapy. Cancer Res. 1999, 59, 1505–1513. [Google Scholar]
- Vrouenraets, M.B.; Visser, G.W.; Loup, C.; Meunier, B.; Stigter, M.; Oppelaar, H.; Stewart, F.A.; Snow, G.B.; van Dongen, G.A. Targeting of a hydrophilic photosensitizer by use of internalizing monoclonal antibodies: A new possibility for use in photodynamic therapy. Int. J. Cancer 2000, 88, 108–114. [Google Scholar] [CrossRef]
- Vrouenraets, M.B.; Visser, G.W.; Stigter, M.; Oppelaar, H.; Snow, G.B.; van Dongen, G.A. Targeting of aluminum (III) phthalocyanine tetrasulfonate by use of internalizing monoclonal antibodies: improved efficacy in photodynamic therapy. Cancer Res. 2001, 61, 1970–1975. [Google Scholar]
- Sutton, J.M.; Clarke, O.J.; Fernandez, N.; Boyle, R.W. Porphyrin, chlorin, and bacteriochlorin isothiocyanates: Useful reagents for the synthesis of photoactive bioconjugates. Bioconjug. Chem. 2002, 13, 249–263. [Google Scholar] [CrossRef]
- Hudson, R.; Carcenac, M.; Smith, K.; Madden, L.; Clarke, O.J.; Pèlegrin, A.; Greenman, J.; Boyle, R.W. The development and characterisation of porphyrin isothiocyanate-monoclonal antibody conjugates for photoimmunotherapy. Br. J. Cancer 2005, 92, 1442–1449. [Google Scholar] [CrossRef]
- Staneloudi, C.; Smith, K.A.; Hudson, R.; Malatesti, N.; Savoie, H.; Boyle, R.W.; Greenman, J. Development and characterization of novel photosensitizer: scFv conjugates for use in photodynamic therapy of cancer. Immunology 2007, 120, 512–517. [Google Scholar] [CrossRef]
- Smith, K.; Malatesti, N.; Cauchon, N.; Hunting, D.; Lecomte, R.; van Lier, J.E.; Greenman, J.; Boyle, R.W. Mono- and tri-cationic porphyrin-monoclonal antibody conjugates: Photodynamic activity and mechanism of action. Immunology 2011, 132, 256–265. [Google Scholar] [CrossRef]
- Malatesti, N.; Smith, K.; Savoie, H.; Greenman, J.; Boyle, R.W. Synthesis and in vitro investigation of cationic 5,15-diphenyl porphyrin-monoclonal antibody conjugates as targeted photodynamic sensitisers. Int. J. Oncol. 2006, 28, 1561–1569. [Google Scholar]
- Alonso, C.M.A.; Palumbo, A.; Bullous, A.J.; Pretto, F.; Neri, D.; Boyle, R.W. Site-specific and stoichiometric conjugation of cationic porphyrins to antiangiogenic monoclonal antibodies. Bioconjug. Chem. 2010, 21, 302–313. [Google Scholar] [CrossRef]
- Hamblin, M.R.; Del Governatore, M.; Rizvi, I.; Hasan, T. Biodistribution of charged 17.1A photoimmunoconjugates in a murine model of hepatic metastasis of colorectal cancer. Br. J. Cancer 2000, 83, 1544–1551. [Google Scholar] [CrossRef]
- Duska, L.R.; Hamblin, M.R.; Bamberg, M.P.; Hasan, T. Biodistribution of charged F(ab')2 photoimmunoconjugates in a xenograft model of ovarian cancer. Br. J. Cancer 1997, 75, 837–844. [Google Scholar] [CrossRef]
- Duska, L.R.; Hamblin, M.R.; Miller, J.L.; Hasan, T. Combination photoimmunotherapy and cisplatin: effects on human ovarian cancer ex vivo. J. Natl. Cancer Inst. 1999, 91, 1557–1563. [Google Scholar] [CrossRef]
- Hamblin, M.R.; Miller, J.L.; Hasan, T. Effect of charge on the interaction of site-specific photoimmunoconjugates with human ovarian cancer cells. Cancer Res. 1996, 56, 5205–5210. [Google Scholar]
- Del Governatore, M.; Hamblin, M.R.; Shea, C.R.; Rizvi, I.; Molpus, K.G.; Tanabe, K.K.; Hasan, T. Experimental photoimmunotherapy of hepatic metastases of colorectal cancer with a 17.1A chlorin(e6) immunoconjugate. Cancer Res. 2000, 60, 4200–4205. [Google Scholar]
- Molpus, K.L.; Hamblin, M.R.; Rizvi, I.; Hasan, T. Intraperitoneal photoimmunotherapy of ovarian carcinoma xenografts in nude mice using charged photoimmunoconjugates. Gynecol. Oncol. 2000, 76, 397–404. [Google Scholar] [CrossRef]
- Del Governatore, M.; Hamblin, M.R.; Piccinini, E.E.; Ugolini, G.; Hasan, T. Targeted photodestruction of human colon cancer cells using charged 17.1A chlorin e6 immunoconjugates. Br. J. Cancer 2000, 82, 56–64. [Google Scholar]
- Jiang, F.N.; Jiang, S.; Liu, D.; Richter, A.; Levy, J.G. Development of technology for linking photosensitizers to a model monoclonal antibody. J. Immunol. Methods 1990, 134, 139–149. [Google Scholar] [CrossRef]
- Jiang, F.N.; Allison, B.; Liu, D.; Levy, J.G. Enhanced photodynamic killing of target cells by either monoclonal antibody or low density lipoprotein mediated delivery systems. J. Control Release 1992, 19, 41–58. [Google Scholar] [CrossRef]
- Jiang, F.N.; Liu, D.J.; Neyndorff, H.; Chester, M.; Jiang, S.Y.; Levy, J.G. Photodynamic killing of human squamous cell carcinoma cells using a monoclonal antibody-photosensitizer conjugate. J. Natl. Cancer Inst. 1991, 83, 1218–1225. [Google Scholar] [CrossRef]
- Berthiaume, F.; Reiken, S.R.; Toner, M.; Tompkins, R.G.; Yarmush, M.L. Antibody-targeted photolysis of bacteria in vivo. Biotechnology (N.Y.) 1994, 12, 703–706. [Google Scholar]
- Strong, L.H.; Berthiaume, F.; Yarmush, M.L. Control of fibroblast populated collagen lattice contraction by antibody targeted photolysis of fibroblasts. Lasers Surg. Med. 1997, 21, 235–247. [Google Scholar] [CrossRef]
- Thorpe, W.P.; Toner, M.; Ezzell, R.M.; Tompkins, R.G.; Yarmush, M.L. Dynamics of photoinduced cell plasma membrane injury. Biophys. J. 1995, 68, 2198–2206. [Google Scholar] [CrossRef]
- Lu, X.M.; Fischman, A.J.; Stevens, E.; Lee, T.T.; Strong, L.; Tompkins, R.G.; Yarmush, M.L. Sn-chlorin e6 antibacterial immunoconjugates. An in vitro and in vivo analysis. J. Immunol. Methods 1992, 156, 85–99. [Google Scholar] [CrossRef]
- Hasan, T.; Lin, A.; Yarmush, D.; Oseroff, A.; Yarmush, M. Monoclonal antibody-chromophore conjugates as selective phototoxins. J. Control Release 1989, 10, 107–117. [Google Scholar] [CrossRef]
- Goff, B.A.; Hermanto, U.; Rumbaugh, J.; Blake, J.; Bamberg, M.; Hasan, T. Photoimmunotherapy and biodistribution with an OC125-chlorin immunoconjugate in an in vivo murine ovarian cancer model. Br. J. Cancer 1994, 70, 474–480. [Google Scholar] [CrossRef]
- Goff, B.A.; Bamberg, M.; Hasan, T. Photoimmunotherapy of human ovarian carcinoma cells ex vivo. Cancer Res. 1991, 51, 4762–4767. [Google Scholar]
- Goff, B.A.; Blake, J.; Bamberg, M.P.; Hasan, T. Treatment of ovarian cancer with photodynamic therapy and immunoconjugates in a murine ovarian cancer model. Br. J. Cancer 1996, 74, 1194–1198. [Google Scholar] [CrossRef]
- Savellano, M.D.; Owusu-Brackett, N.; Son, J.; Ganga, T.; Leung, N.L.; Savellano, D.H. Photodynamic Tumor Eradication With a Novel Targetable Photosensitizer: Strong Vascular Effects and Dependence on Treatment Repetition Versus Potentiation. Photochem. Photobiol. 2012. [Google Scholar] [CrossRef]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef]
- Pèlegrin, A.; Folli, S.; Buchegger, F.; Mach, J.P.; Wagnières, G.; van den Bergh, H. Antibody-fluorescein conjugates for photoimmunodiagnosis of human colon carcinoma in nude mice. Cancer 1991, 67, 2529–2537. [Google Scholar] [CrossRef]
- Kuimova, M.K.; Bhatti, M.; Deonarain, M.; Yahioglu, G.; Levitt, J.A.; Stamati, I.; Suhling, K.; Phillips, D. Fluorescence characterisation of multiply-loaded anti-HER2 single chain Fv-photosensitizer conjugates suitable for photodynamic therapy. Photochem. Photobiol. Sci. 2007, 6, 933–939. [Google Scholar] [CrossRef]
- Bhatti, M.; MacRobert, A.; Henderson, B.; Shepherd, P.; Cridland, J.; Wilson, M. Antibody-targeted lethal photosensitization of Porphyromonas gingivalis. Antimicrob. Agents Chemother. 2000, 44, 2615–2618. [Google Scholar] [CrossRef]
- Stuchinskaya, T.; Moreno, M.; Cook, M.J.; Edwards, D.R.; Russell, D.A. Targeted photodynamic therapy of breast cancer cells using antibody-phthalocyanine-gold nanoparticle conjugates. Photochem. Photobiol. Sci. 2011, 10, 822–831. [Google Scholar] [CrossRef] [Green Version]
- Bulina, M.E.; Chudakov, D.M.; Britanova, O.V.; Yanushevich, Y.G.; Staroverov, D.B.; Chepurnykh, T.V.; Merzlyak, E.M.; Shkrob, M.A.; Lukyanov, S.; Lukyanov, K.A. A genetically encoded photosensitizer. Nat. Biotechnol. 2005, 24, 95–99. [Google Scholar]
- Vegh, R.B.; Solntsev, K.M.; Kuimova, M.K.; Cho, S.; Liang, Y.; Loo, B.L.W.; Tolbert, L.M.; Bommarius, A.S. Reactive oxygen species in photochemistry of the red fluorescent protein "Killer Red". Chem. Commun. (Camb.) 2011, 47, 4887–4889. [Google Scholar]
- Serebrovskaya, E.; Stremovsky, O.; Chudakov, D.; Lukyanov, K.; Deyev, S. Genetically encoded immunophotosensitizer. Russ. J. Bioorg. Chem. 2011, 37, 123–129. [Google Scholar] [CrossRef]
- Serebrovskaya, E.O.; Edelweiss, E.F.; Stremovskiy, O.A.; Lukyanov, K.A.; Chudakov, D.M.; Deyev, S.M. Targeting cancer cells by using an antireceptor antibody-photosensitizer fusion protein. Proc. Natl. Acad. Sci. USA 2009, 106, 9221–9225. [Google Scholar]
- Oseroff, A.R.; Ohuoha, D.; Hasan, T.; Bommer, J.C.; Yarmush, M.L. Antibody-targeted photolysis: Selective photodestruction of human T-cell leukemia cells using monoclonal antibody-chlorin e6 conjugates. Proc. Natl. Acad. Sci. USA 1986, 83, 8744–8748. [Google Scholar]
- Yarmush, M.L.; Thorpe, W.P.; Strong, L.; Rakestraw, S.L.; Toner, M.; Tompkins, R.G. Antibody Targeted Photolysis. Crit. Rev. Ther. Drug Carrier Syst. 1993, 10, 197–252. [Google Scholar]
- Savellano, M.D.; Hasan, T. Photochemical targeting of epidermal growth factor receptor: A mechanistic study. Clin. Cancer Res. 2005, 11, 1658–1668. [Google Scholar] [CrossRef]
- Kaspar, M.; Zardi, L.; Neri, D. Fibronectin as target for tumor therapy. Int. J. Cancer 2006, 118, 1331–1339. [Google Scholar] [CrossRef]
- Pini, A.; Viti, F.; Santucci, A.; Carnemolla, B.; Zardi, L.; Neri, P.; Neri, D. Design and use of a phage display library. Human antibodies with subnanomolar affinity against a marker of angiogenesis eluted from a two-dimensional gel. J. Biol. Chem. 1998, 273, 21769–21776. [Google Scholar]
- Stamati, I.; Kuimova, M.K.; Lion, M.; Yahioglu, G.; Phillips, D.; Deonarain, M.P. Novel photosensitisers derived from pyropheophorbide-a: uptake by cells and photodynamic efficiency in vitro. Photochem. Photobiol. Sci. 2010, 9, 1033–1041. [Google Scholar] [CrossRef]
- Folli, S.; Wagnières, G.; Pèlegrin, A.; Calmes, J.M.; Braichotte, D.; Buchegger, F.; Chalandon, Y.; Hardman, N.; Heusser, C.; Givel, J.C. Immunophotodiagnosis of colon carcinomas in patients injected with fluoresceinated chimeric antibodies against carcinoembryonic antigen. Proc. Natl. Acad. Sci. USA 1992, 89, 7973–7977. [Google Scholar] [CrossRef]
- Collins, H.A.; Khurana, M.; Moriyama, E.H.; Mariampillai, A.; Dahlstedt, E.; Balaz, M.; Kuimova, M.K.; Drobizhev, M.; Yang, V.X.D.; Phillips, D.; et al. Blood-vessel closure using photosensitizers engineered for two-photon excitation. Nat. Photonics. 2008, 2, 420–424. [Google Scholar] [CrossRef]
- Khurana, M.; Collins, H.A.; Karotki, A.; Anderson, H.L.; Cramb, D.T.; Wilson, B.C. Quantitative in vitro demonstration of two-photon photodynamic therapy using photofrin and visudyne. Photochem. Photobiol. 2007, 83, 1441–1448. [Google Scholar] [CrossRef]
- Pawlicki, M.; Collins, H.A.; Denning, R.G.; Anderson, H.L. Two-photon absorption and the design of two-photon dyes. Angew. Chem. Int. Ed. Engl. 2009, 48, 3244–3266. [Google Scholar] [CrossRef]
- Kuroki, M.; Hachimine, K.; Abe, H.; Shibaguchi, H.; Kuroki, M.; Maekawa, S.-I.; Yanagisawa, J.; Kinugasa, T.; Tanaka, T.; Yamashita, Y. Sonodynamic therapy of cancer using novel sonosensitizers. Anticancer Res. 2007, 27, 3673–3677. [Google Scholar]
- Ma, X.; Pan, H.; Yi, J. Combination sonodynamic therapy with immunoadjuvant may be a promising new modality for cancer treatment. Med. Hypotheses 2009, 72, 418–420. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Kobayashi, H.; Narita, T.; Kanehira, K.; Sonezaki, S.; Kudo, N.; Kubota, Y.; Terasaka, S.; Houkin, K. Sonodynamic therapy using water-dispersed TiO2-polyethylene glycol compound on glioma cells: Comparison of cytotoxic mechanism with photodynamic therapy. Ultrason. Sonochem. 2011, 18, 1197–1204. [Google Scholar] [CrossRef]
- Huang, X.; Jain, P.K.; El-Sayed, I.H.; El-Sayed, M.A. Plasmonic photothermal therapy (PPTT) using gold nanoparticles. Lasers Med. Sci. 2008, 23, 217–228. [Google Scholar] [CrossRef]
- Huang, X.; Qian, W.; El-Sayed, I.H.; El-Sayed, M.A. The potential use of the enhanced nonlinear properties of gold nanospheres in photothermal cancer therapy. Lasers Surg. Med. 2007, 39, 747–753. [Google Scholar] [CrossRef]
- El-Sayed, I.H.; Huang, X.; El-Sayed, M.A. Selective laser photo-thermal therapy of epithelial carcinoma using anti-EGFR antibody conjugated gold nanoparticles. Cancer Lett. 2006, 239, 129–135. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, W.; Wang, S.; Joly, A.G. Investigation of water-soluble X-ray luminescence nanoparticles for photodynamic activation. Appl. Phys. Lett. 2008, 92, 043901. [Google Scholar] [CrossRef]
- Samia, A.C.S.; Dayal, S.; Burda, C. Quantum dot-based energy transfer: perspectives and potential for applications in photodynamic therapy. Photochem. Photobiol. 2006, 82, 617–625. [Google Scholar] [CrossRef]
- Juzenas, P.; Chen, W.; Sun, Y.-P.; Coelho, M.A.N.; Generalov, R.; Generalova, N.; Christensen, I.L. Quantum dots and nanoparticles for photodynamic and radiation therapies of cancer. Adv. Drug Deliv. Rev. 2008, 60, 1600–1614. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, J. Using nanoparticles to enable simultaneous radiation and photodynamic therapies for cancer treatment. J. Nanosci. Nanotechnol. 2006, 6, 1159–1166. [Google Scholar] [CrossRef]
- Vaidya, A.; Sun, Y.; Feng, Y.; Emerson, L.; Jeong, E.-K.; Lu, Z.-R. Contrast-enhanced MRI-guided photodynamic cancer therapy with a pegylated bifunctional polymer conjugate. Pharm. Res. 2008, 25, 2002–2011. [Google Scholar] [CrossRef]
- Huang, P.; Li, Z.; Lin, J.; Yang, D.; Gao, G.; Xu, C.; Bao, L.; Zhang, C.; Wang, K.; Song, H.; Hu, H.; Cui, D. Photosensitizer-conjugated magnetic nanoparticles for in vivo simultaneous magnetofluorescent imaging and targeting therapy. Biomaterials 2011, 32, 3447–3458. [Google Scholar] [CrossRef]
- Detty, M.R. Direct 1270 nm irradiation as an alternative to photosensitized generation of singlet oxygen to induce cell death. Photochem. Photobiol. 2012, 88, 2–4. [Google Scholar] [CrossRef]
- Laptev, R.; Nisnevitch, M.; Siboni, G.; Malik, Z.; Firer, M.A. Intracellular chemiluminescence activates targeted photodynamic destruction of leukaemic cells. Br. J. Cancer 2006, 95, 189–196. [Google Scholar]
- Embleton, M.L.; Nair, S.P.; Cookson, B.D.; Wilson, M. Antibody-directed photodynamic therapy of methicillin resistant Staphylococcus aureus. Microb. Drug Resist. 2004, 10, 92–97. [Google Scholar] [CrossRef]
- Berki, T.; Németh, P. Novel method for in vitro depletion of T cells by monoclonal antibody-targeted photosensitization. J. Immunol. Methods 1998, 211, 139–146. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pye, H.; Stamati, I.; Yahioglu, G.; Butt, M.A.; Deonarain, M. Antibody-Directed Phototherapy (ADP). Antibodies 2013, 2, 270-305. https://doi.org/10.3390/antib2020270
Pye H, Stamati I, Yahioglu G, Butt MA, Deonarain M. Antibody-Directed Phototherapy (ADP). Antibodies. 2013; 2(2):270-305. https://doi.org/10.3390/antib2020270
Chicago/Turabian StylePye, Hayley, Ioanna Stamati, Gokhan Yahioglu, M. Adil Butt, and Mahendra Deonarain. 2013. "Antibody-Directed Phototherapy (ADP)" Antibodies 2, no. 2: 270-305. https://doi.org/10.3390/antib2020270