
3D Models as a Tool to Assess the Anti-Tumor Efficacy of Therapeutic Antibodies: Advantages and Limitations

 ,
,
Abstract
:1. Introduction
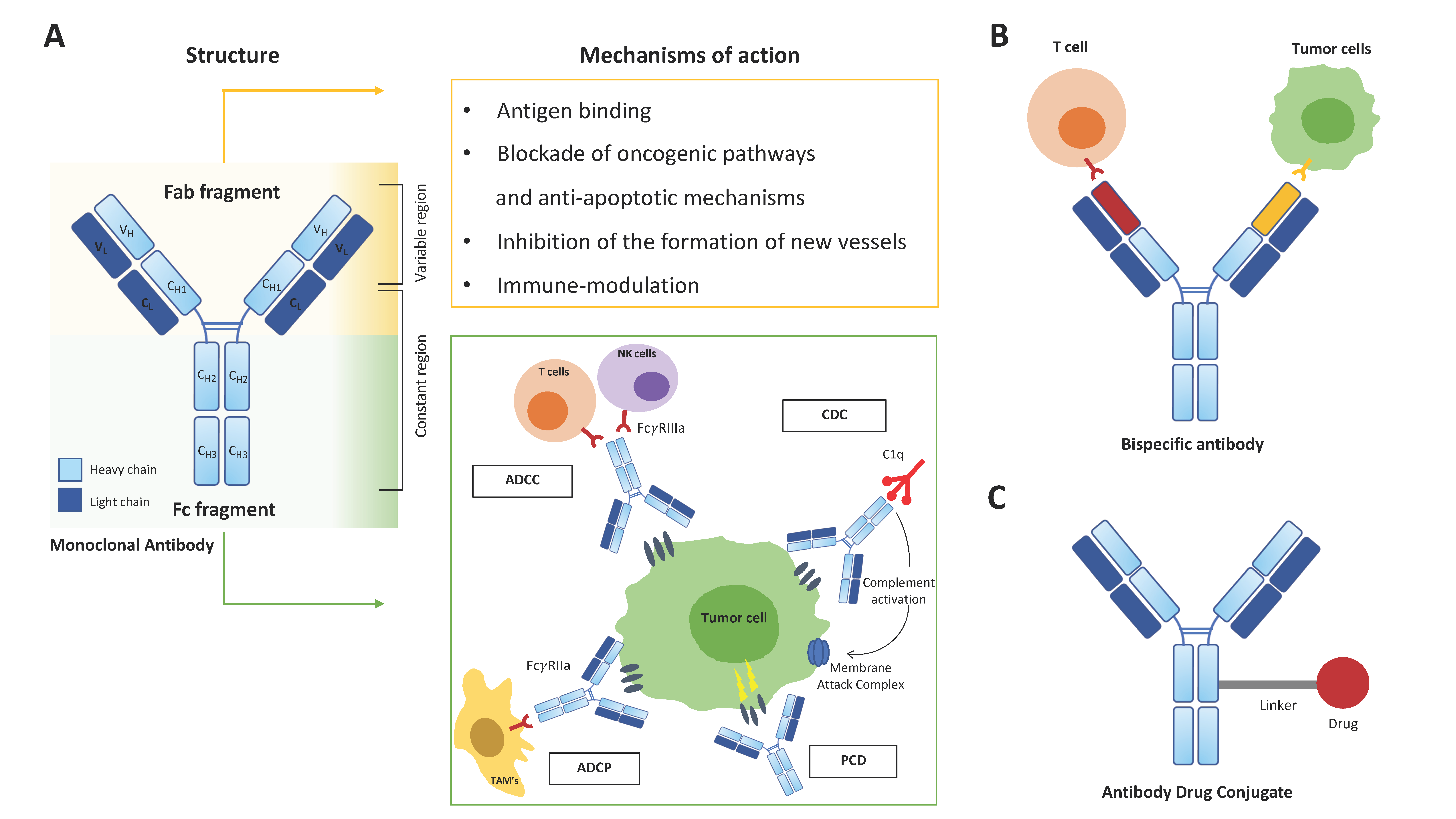
2. Therapeutic mAbs in Cancer Treatment
3. Moving from 2D to 3D Cultures to Model Tumor Microenvironment
3.1. Scaffold-Free Models
3.2. Scaffold-Based-Models
3.3. System-Based Models
4. Exploiting 3D Models for Therapeutic mAb Testing
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pento, J.T. Monoclonal Antibodies for the Treatment of Cancer. Anticancer Res. 2017, 37, 5935–5939. [Google Scholar] [PubMed] [Green Version]
- Modjtahedi, H.; Ali, S.; Essapen, S. Therapeutic application of monoclonal antibodies in cancer: Advances and challenges. Br. Med. Bull. 2012, 104, 41–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging new therapeutic antibody derivatives for cancer treatment. Sig. Transduct. Target Ther. 2022, 7, 39. [Google Scholar] [CrossRef]
- Smietana, K.; Siatkowski, M.; Møller, M. Trends in clinical success rates. Nat. Rev. Drug Discov. 2016, 15, 379–380. [Google Scholar] [CrossRef]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef]
- Brown, D.G.; Wobst, H.J.; Kapoor, A.; Kenna, L.A.; Southall, N. Clinical development times for innovative drugs. Nat. Rev. Drug Discov. 2021; Epub ahead of print. [Google Scholar] [CrossRef]
- Alemany-Ribes, M.; Semino, C.E. Bioengineering 3D environments for cancer models. Adv. Drug Deliv. Rev. 2014, 79–80, 40–49. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Taylor, P.C.; Adams, A.C.; Hufford, M.M.; de la Torre, I.; Winthrop, K.; Gottlieb, R.L. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol. 2021, 21, 382–393. [Google Scholar] [CrossRef]
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat. Rev. Drug Discov. 2021, 20, 491–495. [Google Scholar] [CrossRef]
- Kaplon, H.; Chenoweth, A.; Crescioli, S.; Reichert, J.M. Antibodies to watch in 2022. MAbs 2022, 14, 2014296. [Google Scholar] [CrossRef]
- Paci, A.; Desnoyer, A.; Delahousse, J.; Blondel, L.; Maritaz, C.; Chaput, N.; Mir, O.; Broutin, S. Pharmacokinetic/pharmacodynamic relationship of therapeutic monoclonal antibodies used in oncology: Part 1, monoclonal antibodies, antibody-drug conjugates and bispecific T-cell engagers. Eur. J. Cancer 2020, 128, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Freeman, C.L.; Sehn, L.H. A tale of two antibodies: Obinutuzumab versus rituximab. Br. J. Haematol. 2018, 182, 29–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salles, G.; Barrett, M.; Foà, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-cell hematologic malignancies: A review of 20 years of clinical experience. Adv. Ther. 2017, 34, 2232–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roschewski, M.; Staudt, L.M.; Wilson, W.H. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat. Rev. Clin. Oncol. 2014, 11, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Isenberg, D.A. Biologic therapies for systemic lupus erythematosus: Where are we now? Curr. Opin. Rheumatol. 2020, 32, 597–608. [Google Scholar] [CrossRef]
- Cencioni, M.T.; Mattoscio, M.; Magliozzi, R.; Bar-Or, A.; Muraro, P.A. B cells in multiple sclerosis—From targeted depletion to immune reconstitution therapies. Nat. Rev. Neurol. 2021, 17, 399–414. [Google Scholar] [CrossRef]
- McClure, M.; Gopaluni, S.; Jayne, D.; Jones, R. B cell therapy in ANCA-associated vasculitis: Current and emerging treatment options. Nat. Rev. Rheumatol. 2018, 14, 580–591. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 548–567. [Google Scholar] [CrossRef] [Green Version]
- Van de Donk, N.W.C.J.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef]
- Palladini, G.; Milani, P.; Malavasi, F.; Merlini, G. Daratumumab in the Treatment of Light-Chain (AL) Amyloidosis. Cells 2021, 10, 545. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; Introna, M. Mechanism of action of therapeutic monoclonal antibodies: Promises and pitfalls of in vitro and in vivo assays. Arch. Biochem. Biophys. 2012, 526, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Redman, J.M.; Hill, E.M.; AlDeghaither, D.; Weiner, L.M. Mechanisms of action of therapeutic antibodies for cancer. Mol. Immunol. 2015, 67, 28–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overdijk, M.B.; Jansen, J.H.; Nederend, M.; Lammerts van Bueren, J.J.; Groen, R.W.; Parren, P.W.; Leusen, J.H.; Boross, P. The Therapeutic CD38 Monoclonal Antibody Daratumumab Induces Programmed Cell Death via Fcγ Receptor-Mediated Cross-Linking. J. Immunol. 2016, 197, 807–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castel, P.; Rauen, K.A.; McCormick, F. The duality of human oncoproteins: Drivers of cancer and congenital disorders. Nat. Rev. Cancer 2020, 20, 383–397. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Li, Y.M.; Zhou, B.P.; Deng, J.; Pan, Y.; Hay, N.; Hung, M.C. A hypoxia-independent hypoxia-inducible factor-1 activation pathway induced by phosphatidylinositol-3 kinase/Akt in HER2 overexpressing cells. Cancer Res. 2005, 65, 3257–3263. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.Y.; Bang, Y.J. HER2-targeted therapies—A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef]
- Chen, J.S.; Lan, K.; Hung, M.C. Strategies to target HER2/neu overexpression for cancer therapy. Drug Resist. Updates 2003, 6, 129–136. [Google Scholar] [CrossRef]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [Green Version]
- Bejarano, L.; Jordāo, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Heath, V.L.; Bicknell, R. Anticancer strategies involving the vasculature. Nat. Rev. Clin. Oncol. 2009, 6, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular endothelial growth factor. Trends Cardiovasc. Med. 1993, 3, 244–250. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Ribatti, D.; Solimando, A.G.; Pezzella, F. The Anti-VEGF(R) Drug Discovery Legacy: Improving Attrition Rates by Breaking the Vicious Cycle of Angiogenesis in Cancer. Cancers 2021, 13, 3433. [Google Scholar] [CrossRef]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Tolaney, S.M.; Boucher, Y.; Duda, D.G.; Martin, J.D.; Seano, G.; Ancukiewicz, M.; Barry, W.T.; Goel, S.; Lahdenrata, J.; Isakoff, S.J.; et al. Role of vascular density and normalization in response to neoadjuvant bevacizumab and chemotherapy in breast cancer patients. Proc. Natl. Acad. Sci. USA 2015, 112, 14325–14330. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Kim, B.Y.S.; Chan, C.K.; Hahn, S.M.; Weissman, I.L.; Jiang, W. Improving immune-vascular crosstalk for cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Hlushchuk, R.; Barré, S.; Djonov, V. Morphological Aspects of Tumor Angiogenesis. Methods Mol. Biol. 2016, 1464, 13–24. [Google Scholar] [PubMed]
- Briukhovetska, D.; Dörr, J.; Endres, S.; Libby, P.; Dinarello, C.A.; Kobold, S. Interleukins in cancer: From biology to therapy. Nat. Rev. Cancer. 2021, 21, 481–499. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 70, 1120–1133.e17. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.A.; Bakouny, Z.; Hirsch, L.; Flippot, R.; Van Allen, E.M.; Wu, C.J.; Choueiri, T.K. Beyond conventional immune-checkpoint inhibition—Novel immunotherapies for renal cell carcinoma. Nat. Rev. Clin. Oncol. 2021, 18, 199–214. [Google Scholar] [CrossRef]
- Wolchok, J. Putting the Immunologic Brakes on Cancer. Cell 2018, 175, 1452–1454. [Google Scholar] [CrossRef] [Green Version]
- Korman, A.J.; Peggs, K.S.; Allison, J.P. Checkpoint blockade in cancer immunotherapy. Adv. Immunol. 2006, 90, 297–339. [Google Scholar]
- Kraehenbuehl, L.; Weng, C.H.; Eghbali, S.; Wolchok, J.D.; Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 2022, 19, 37–50. [Google Scholar] [CrossRef]
- Bottazzi, B.; Riboli, E.; Mantovani, A. Aging, inflammation and cancer. Semin. Immunol. 2018, 40, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garlanda, C.; Mantovani, A. Interleukin-1 in tumor progression, therapy, and prevention. Cancer Cell 2021, 39, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Gottschlich, A.; Endres, S.; Kobold, S. Therapeutic Strategies for Targeting IL-1 in Cancer. Cancers 2021, 13, 477. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef]
- Chen, A.Y.; Wolchok, J.D.; Bass, A.R. TNF in the era of immune checkpoint inhibitors: Friend or foe? Nat. Rev. Rheumatol. 2021, 17, 213–223. [Google Scholar] [CrossRef]
- Molfino, A.; Amabile, M.I.; Rossi Fanelli, F.; Muscaritoli, M. Novel therapeutic options for cachexia and sarcopenia. Expert Opin. Biol. Ther. 2016, 16, 1239–1244. [Google Scholar] [CrossRef]
- Kang, J.H.; Bluestone, J.A.; Young, A. Predicting and Preventing Immune Checkpoint Inhibitor Toxicity: Targeting Cytokines. Trends Immunol. 2021, 42, 293–311. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Zhao, H.; Luo, F.; Xue, J.; Li, S.; Xu, R.H. Emerging immunological strategies: Recent advances and future directions. Front. Med. 2021, 15, 805–828. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; Choblet, S.; Iwaszkiewicz, J.; Cérutti, P.; Ozil, A.; Loisel, S.; Pugnière, M.; Ubiali, G.; Zoete, V.; Michielin, O.; et al. Design and Validation of a Novel Generic Platform for the Production of Tetravalent IgG1-like Bispecific Antibodies. J. Immunol. 2016, 196, 3199–3211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713.e16. [Google Scholar] [CrossRef]
- García-Alonso, S.; Ocaña, A.; Pandiella, A. Resistance to Antibody-Drug Conjugates. Cancer Res. 2018, 78, 2159–2165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell. Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Zimmer, J.; Castriconi, R.; Scaglione, S. Editorial: Recent 3D Tumor Models for Testing Immune-Mediated Therapies. Front. Immunol. 2021, 12, 798493. [Google Scholar] [CrossRef]
- Graham, M.L.; Prescott, M.J. The multifactorial role of the 3Rs in shifting the harm-benefit analysis in animal models of disease. Eur. J. Pharmacol. 2015, 759, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Bédard, P.; Gauvin, S.; Ferland, K.; Caneparo, C.; Pellerin, È.; Chabaud, S.; Bolduc, S. Innovative Human Three-Dimensional Tissue-Engineered Models as an Alternative to Animal Testing. Bioengineering 2020, 7, 115. [Google Scholar] [CrossRef]
- Zitvogel, L.; Pitt, J.M.; Daillère, R.; Smyth, M.J.; Kroemer, G. Mouse models in oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Y.; Kenny, P.; Lee, E.H.; Bissell, M.J. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat. Methods 2007, 4, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.T.; Hughes-Fulford, M. Monolayer and spheroid culture of human liver hepatocellular carcinoma cell line cells demonstrate distinct global gene expression patterns and functional phenotypes. Tissue Eng. Part A. 2009, 15, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Riedl, A.; Schlederer, M.; Pudelko, K.; Stadler, M.; Walter, S.; Unterleuthner, D.; Unger, C.; Kramer, N.; Hengstschläger, M.; Kenner, L.; et al. Comparison of cancer cells in 2D vs. 3D culture reveals differences in AKT-mTOR-S6K signaling and drug responses. J. Cell Sci. 2017, 130, 203–218. [Google Scholar]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Bray, L.J.; Hutmacher, D.W.; Bock, N. Addressing Patient Specificity in the Engineering of Tumor Models. Front. Bioeng. Biotechnol. 2019, 7, 217. [Google Scholar] [CrossRef] [Green Version]
- Gunti, S.; Hoke, A.T.K.; Vu, K.P.; London, N.R., Jr. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers 2021, 13, 874. [Google Scholar] [CrossRef]
- Weiswald, L.B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Nunes, A.S.; Barros, A.S.; Costa, E.C.; Moreira, A.F.; Correia, I.J. 3D tumor spheroids as in vitro models to mimic in vivo human solid tumors resistance to therapeutic drugs. Biotechnol. Bioeng. 2019, 116, 206–226. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Kim, T.H. Recent Advances in Multicellular Tumor Spheroid Generation for Drug Screening. Biosensors 2021, 11, 445. [Google Scholar] [CrossRef]
- Chesnais, F.; Hue, J.; Roy, E.; Branco, M.; Stokes, R.; Pellon, A.; Le Caillec, J.; Elbahtety, E.; Battilocchi, M.; Danovi, D.; et al. High-content image analysis to study phenotypic heterogeneity in endothelial cell monolayers. J. Cell Sci. 2022, 135, jcs259104. [Google Scholar] [CrossRef] [PubMed]
- Carletti, E.; Motta, A.; Migliaresi, C. Scaffolds for tissue engineering and 3D cell culture. Methods Mol. Biol. 2011, 695, 17–39. [Google Scholar] [PubMed]
- Navran, S. The application of low shear modeled microgravity to 3-D cell biology and tissue engineering. Biotechnol. Annu. Rev. 2008, 14, 275–296. [Google Scholar] [PubMed]
- Grimm, D.; Wehland, M.; Pietsch, J.; Aleshcheva, G.; Wise, P.; van Loon, J.; Ulbrich, C.; Magnusson, N.E.; Infanger, M.; Bauer, J. Growing tissues in real and simulated microgravity: New methods for tissue engineering. Tissue Eng. Part B Rev. 2014, 20, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Ferrarini, M.; Steimberg, N.; Boniotti, J.; Berenzi, A.; Belloni, D.; Mazzoleni, G.; Ferrero, E. 3D-Dynamic Culture Models of Multiple Myeloma. Methods Mol. Biol. 2017, 1612, 177–190. [Google Scholar]
- Holton, A.B.; Sinatra, F.L.; Kreahling, J.; Conway, A.J.; Landis, D.A.; Altiok, S. Microfluidic Biopsy Trapping Device for the Real-Time Monitoring of Tumor Microenvironment. PLoS ONE 2017, 12, e0169797. [Google Scholar] [CrossRef] [Green Version]
- Datta, P.; Dey, M.; Ataie, Z.; Unutmaz, D.; Ozbolat, I.T. 3D bioprinting for reconstituting the cancer microenvironment. NPJ Precis. Oncol. 2020, 4, 18. [Google Scholar] [CrossRef]
- Augustine, R.; Kalva, S.N.; Ahmad, R.; Zahid, A.A.; Hasan, S.; Nayeem, A.; McClements, L.; Hasan, A. 3D Bioprinted cancer models: Revolutionizing personalized cancer therapy. Transl. Oncol. 2021, 14, 101015. [Google Scholar] [CrossRef]
- Sbrana, F.V.; Pinos, R.; Barbaglio, F.; Ribezzi, D.; Scagnoli, F.; Scarfò, L.; Redwan, I.N.; Martinez, H.; Farè, S.; Ghia, P.; et al. 3D Bioprinting Allows the Establishment of Long-Term 3D Culture Model for Chronic Lymphocytic Leukemia Cells. Front. Immunol. 2021, 12, 639572. [Google Scholar] [CrossRef]
- Marei, I.; Abu Samaan, T.; Al-Quradaghi, M.A.; Farah, A.A.; Mahmud, S.H.; Ding, H.; Triggle, C.R. 3D Tissue-Engineered Vascular Drug Screening Platforms: Promise and Considerations. Front. Cardiovasc. Med. 2022, 4, 847554. [Google Scholar] [CrossRef]
- Seano, G.; Chiaverina, G.; Gagliardi, P.A.; di Blasio, L.; Sessa, R.; Bussolino, F.; Primo, L. Modeling human tumor angiogenesis in a three-dimensional culture system. Blood 2013, 121, e129–e137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal-Crespo, A.; Matas-Céspedes, A.; Rodriguez, V.; Rossi, C.; Valero, J.G.; Serrat, N.; Sanjuan-Pla, A.; Menéndez, P.; Roué, G.; López-Guillermo, A.; et al. Daratumumab displays in vitro and in vivo anti-tumor activity in models of B-cell non-Hodgkin lymphoma and improves responses to standard chemo-immunotherapy regimens. Haematologica 2020, 105, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Foxall, R.; Narang, P.; Glaysher, B.; Hub, E.; Teal, E.; Coles, M.C.; Ashton-Key, M.; Beers, S.A.; Cragg, M.S. Developing a 3D B Cell Lymphoma Culture System to Model Antibody Therapy. Front. Immunol. 2021, 11, 605231. [Google Scholar] [CrossRef] [PubMed]
- Courau, T.; Bonnereau, J.; Chicoteau, J.; Bottois, H.; Remark, R.; Assante Miranda, L.; Toubert, A.; Blery, M.; Aparicio, T.; Allez, M.; et al. Cocultures of human colorectal tumor spheroids with immune cells reveal the therapeutic potential of MICA/B and NKG2A targeting for cancer treatment. J. Immunother. Cancer 2019, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Varesano, S.; Zocchi, M.R.; Poggi, A. Zoledronate Triggers Vδ2 T Cells to Destroy and Kill Spheroids of Colon Carcinoma: Quantitative Image Analysis of Three-Dimensional Cultures. Front. Immunol. 2018, 9, 998. [Google Scholar] [CrossRef]
- Pece, R.; Tavella, S.; Costa, D.; Varesano, S.; Camodeca, C.; Cuffaro, D.; Nuti, E.; Rossello, A.; Alfano, M.; D’Arrigo, C.; et al. Inhibitors of ADAM10 reduce Hodgkin lymphoma cell growth in 3D microenvironments and enhance brentuximab-vedotin effect. Haematologica 2022, 107, 909–920. [Google Scholar]
- Sargenti, A.; Musmeci, F.; Bacchi, F.; Delprete, C.; Cristaldi, D.A.; Cannas, F.; Bonetti, S.; Pasqua, S.; Gazzola, D.; Costa, D.; et al. Physical Characterization of Colorectal Cancer Spheroids and Evaluation of NK Cell Infiltration Through a Flow-Based Analysis. Front. Immunol. 2020, 11, 564887. [Google Scholar] [CrossRef]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, R.W.; Aref, A.R.; Lizotte, P.H.; Ivanova, E.; Stinson, S.; Zhou, C.W.; Bowden, M.; Deng, J.; Liu, H.; Miao, D.; et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018, 8, 196–215. [Google Scholar] [CrossRef] [Green Version]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [Green Version]
- van Diest, E.; Hernández López, P.; Meringa, A.D.; Vyborova, A.; Karaiskaki, F.; Heijhuurs, S.; Gumathi Bormin, J.; van Dooremalen, S.; Nicolasen, M.J.T.; Gatti, L.C.D.E.; et al. Gamma delta TCR anti-CD3 bispecific molecules (GABs) as novel immunotherapeutic compounds. J. Immunother. Cancer. 2021, 9, e003850. [Google Scholar] [CrossRef]
- Villa, A.; Belloni, D.; Vergani, B.; Cenci, S.; Cavalli, G.; Biavasco, R.; Rodolfo, M.; Cangi, M.G.; Doglioni, C.; Dagna, L.; et al. 3D culture of Erdheim-Chester disease tissues unveils histiocyte metabolism as a new therapeutic target. Ann. Rheum. Dis. 2019, 78, 862–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candini, O.; Grisendi, G.; Foppiani, E.M.; Brogli, M.; Aramini, B.; Masciale, V.; Spano, C.; Petrachi, T.; Veronesi, E.; Conte, P.; et al. A Novel 3D In Vitro Platform for Pre-Clinical Investigations in Drug Testing, Gene Therapy, and Immuno-oncology. Sci. Rep. 2019, 9, 7154. [Google Scholar] [CrossRef] [Green Version]
- Ferrarini, M.; Steimberg, N.; Ponzoni, M.; Belloni, D.; Berenzi, A.; Girlanda, S.; Caligaris-Cappio, F.; Mazzoleni, G.; Ferrero, E. Ex-vivo dynamic 3-D culture of human tissues in the RCCS™ bioreactor allows the study of Multiple Myeloma biology and response to therapy. PLoS ONE 2013, 8, e716. [Google Scholar] [CrossRef]
- Ferrero, E.; Villa, A.; Stefanoni, D.; Nemkov, T.; D’Alessandro, A.; Tengesdal, I.; Belloni, D.; Molteni, R.; Vergani, B.; De Luca, G.; et al. Immunometabolic activation of macrophages leads to cytokine production in the pathogenesis of KRAS-mutated histiocytosis. Rheumatology 2022, 61, e93–e96. [Google Scholar] [CrossRef] [PubMed]
- Haroche, J.; Cohen-Aubart, F.; Rollins, B.; Donadieu, J.; Charlotte, F.; Idbaih, A.; Vaglio, A.; Abdel-Wahab, O.; Emile, J.F.; Amoura, Z. Histiocytoses: Emerging neoplasia behind inflammation. Lancet Oncol. 2017, 18, e113–e125. [Google Scholar] [CrossRef]
- Cavalli, G.; Dagna, L.; Biavasco, R.; Villa, A.; Doglioni, C.; Ferrero, E.; Ferrarini, M. Erdheim-Chester disease: An in vivo human model of Mϕ activation at the crossroad between chronic inflammation and cancer. J. Leukoc. Biol. 2020, 108, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Belloni, D.; Heltai, S.; Ponzoni, M.; Villa, A.; Vergani, B.; Pecciarini, L.; Marcatti, M.; Girlanda, S.; Tonon, G.; Ciceri, F.; et al. Modeling multiple myeloma-bone marrow interactions and response to drugs in a 3D surrogate microenvironment. Haematologica 2018, 103, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Barbaglio, F.; Belloni, D.; Scarfò, L.; Sbrana, F.V.; Ponzoni, M.; Bongiovanni, L.; Pavesi, L.; Zambroni, D.; Stamatopoulos, K.; Caiolfa, V.R.; et al. Three-dimensional co-culture model of chronic lymphocytic leukemia bone marrow microenvironment predicts patient-specific response to mobilizing agents. Haematologica 2021, 106, 2334–2344. [Google Scholar] [CrossRef]
- Mark, C.; Czerwinski, T.; Roessner, S.; Mainka, A.; Hörsch, F.; Heublein, L.; Winterl, A.; Sanokowski, S.; Richter, S.; Bauer, N.; et al. Cryopreservation impairs 3-D migration and cytotoxicity of natural killer cells. Nat. Commun. 2020, 11, 5224. [Google Scholar] [CrossRef]
- Boucherit, N.; Gorvel, L.; Olive, D. 3D Tumor Models and Their Use for the Testing of Immunotherapies. Front. Immunol. 2020, 11, 603640. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.H.; Ren, L.; Kim, S.M.; Seo, S.H.; Jung, C.R.; Kim, D.S.; Noh, J.Y.; Lee, S.Y.; Lee, H.; Cho, M.Y.; et al. A three-dimensional hyaluronic acid-based niche enhances the therapeutic efficacy of human natural killer cell-based cancer immunotherapy. Biomaterials 2020, 247, 119960. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Duchamp, M.; Oklu, R.; Ellisen, L.W.; Langer, R.; Khademhosseini, A. Bioprinting the Cancer Microenvironment. ACS Biomater. Sci. Eng. 2016, 2, 1710–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotecki, N.; Kindt, N.; Krayem, M.; Awada, A. New horizons in early drugs development in solid cancers. Curr. Opin. Oncol. 2021, 33, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Hübner, J.; Raschke, M.; Rütschle, I.; Gräßle, S.; Hasenberg, T.; Schirrmann, K.; Lorenz, A.; Schnurre, S.; Lauster, R.; Maschmeyer, I.; et al. Simultaneous evaluation of anti-EGFR-induced tumour and adverse skin effects in a microfluidic human 3D co-culture model. Sci. Rep. 2018, 8, 15010. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| 3D models | Advantages | Limitations | Applications | Refs. | |
|---|---|---|---|---|---|
| Spheroids |  | Limited culture requirements Increased cell-cell and cell-matrix interactions Nutrient and oxygen gradients High through-put drug screening Low costs | Mostly monocultures (cell lines) Difficult experimental standardization Uneasy setting of functional assays Complex quantification of response | Flow cytometry Immunohistochemistry Live imaging Immunofluorescence ATP content assay Glucose dosages | [93,94,95,96,97,98] |
| Patient-derived Organoids |  | Native tumor heterogeneity Preservation of TME complexity, including TILs High through-put drug screening Biobanking | Variable success rate Time -consuming High costs Need of advanced tools for analysis | Flow cytometry Immunohistochemistry qRT-PCR LIVE/DEAD assay Cytokine detection Single Cell Gene Enrichment Analysis Immunofluorescence | [99,100,101] |
| Hydrogels |  | Easy to handle Minimal culture requirements Easy drug testing and experimental standardization Low costs | Lack of TME complexity Limited architectural organization | Flow cytometry Confocal microscopy Cytokine detection | [102] |
| 3D culture inbioreactor |  | Patient specificity Native tumor and TME Assessment of tumor/TME functions and metabolism Drug testing | High costs Specific expertise required No high-throughput Need of advanced tools for analysis Complex experimental standardization | Confocal microscopy Cytokine detection Immunohistochemistry Glucose/lactate dosages Metabolomics | [103] |
 | Easy to handle Patient specificity Drug testing | Lack of TME complexity Limited architectural organization | Cell Viability Assay LIVE/DEAD assay | [104] | |
 Tumor cell;
Tumor cell;  T cell;
T cell;  Dendritic cell;
Dendritic cell;  Stromal cell;
Stromal cell;  Tissue sample;
Tissue sample;  Vessel.
Vessel. Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzzeloni, V.; Veschini, L.; Pedica, F.; Ferrero, E.; Ferrarini, M. 3D Models as a Tool to Assess the Anti-Tumor Efficacy of Therapeutic Antibodies: Advantages and Limitations. Antibodies 2022, 11, 46. https://doi.org/10.3390/antib11030046
Guzzeloni V, Veschini L, Pedica F, Ferrero E, Ferrarini M. 3D Models as a Tool to Assess the Anti-Tumor Efficacy of Therapeutic Antibodies: Advantages and Limitations. Antibodies. 2022; 11(3):46. https://doi.org/10.3390/antib11030046
Chicago/Turabian StyleGuzzeloni, Virginia, Lorenzo Veschini, Federica Pedica, Elisabetta Ferrero, and Marina Ferrarini. 2022. "3D Models as a Tool to Assess the Anti-Tumor Efficacy of Therapeutic Antibodies: Advantages and Limitations" Antibodies 11, no. 3: 46. https://doi.org/10.3390/antib11030046