Comparison of Measurement-Based Methodologies to Apportion Secondary Organic Carbon (SOC) in PM2.5: A Review of Recent Studies


 ,
,
Abstract
:
1. Introduction and Objectives
2. Major Sources of SOA Precursors: Current Knowledge from Emission Inventories
3. Description of the Main Approaches to Apportion SOC Fraction
3.1. EC-Tracer Method
3.1.1. Principle
3.1.2. Limitations and Challenges
3.1.3. Review of Recent Studies Based on the EC-Tracer Method
3.1.3.1. America
3.1.3.2. Europe and the Middle East
3.1.3.3. Asia
3.2. Chemical Mass Balance (CMB)
3.2.1. Introduction
3.2.2. Limitations and Challenges
3.2.3. Review of Recent Studies Based on CMB Approach
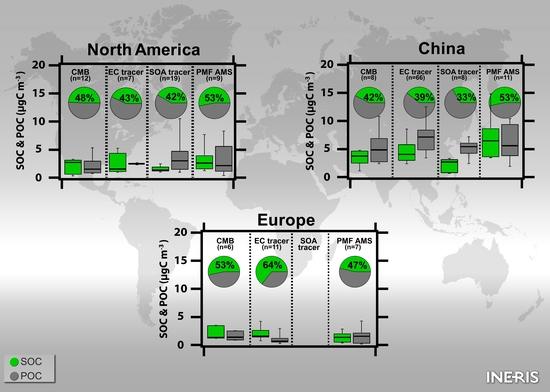
3.2.3.1. North America
3.2.3.2. Europe and the Middle East
3.2.3.3. Asia
3.3. SOA-Tracer Method
3.3.1. Introduction
3.3.2. Limitations and Challenges
3.3.3. Review of Recent Studies Based on the SOA-Tracer Method
3.3.3.1. North America
3.3.3.2. Europe
3.3.3.3. Asia
3.4. Positive Matrix Factorization (PMF) (Including AMS Data Analysis)
3.4.1. Introduction
3.4.2. Limitations and Challenges
3.4.3. Review of Recent Studies Based on the PMF Approach
3.4.3.1. Filter-Based PMF Studies
3.4.3.2. PMF-AMS/ACSM Based Studies
3.4.3.3. North America
3.4.3.4. Europe
3.4.3.5. Asia
3.5. 14C (Radiocarbon) Measurements
3.5.1. Introduction
3.5.2. Limitations and Challenges
3.5.3. Review of Recent Studies Based on 14C Measurements
3.5.3.1. North America
3.5.3.2. Europe
3.5.3.3. Asia
4. Review of the Studies Directly Comparing Different Methodologies
4.1. EC-Tracer vs. CMB
4.2. SOA-Tracer vs. Other Methodologies (EC-Tracer, CMB and PMF-Filter)
4.3. PMF vs. Other Methodologies (EC-Tracer, CMB)
5. Comparison Based on the Overall Picture Obtained from the Review of Recent Studies
5.1. Comparison of the Annual SOC Estimates Obtained Worldwide
5.2. Comparison of the Spring–Summer SOC Estimates Obtained Worldwide
6. Conclusions and Future Implications
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Docherty, K.S.; Stone, E.A.; Ulbrich, I.M.; DeCarlo, P.F.; Snyder, D.C.; Schauer, J.J.; Peltier, R.E.; Weber, R.J.; Murphy, S.M.; Seinfeld, J.H. Apportionment of primary and secondary organic aerosols in Southern California during the 2005 Study of Organic Aerosols in Riverside (SOAR-1). Environ. Sci. Technol. 2008, 42, 7655–7662. [Google Scholar] [CrossRef] [PubMed]
- Turpin, B.J.; Saxena, P.; Andrews, E. Measuring and simulating particulate organics in the atmosphere: Problems and prospects. Atmos. Environ. 2000, 34, 2983–3013. [Google Scholar] [CrossRef]
- Goldstein, A.H.; Galbally, I.E. Known and unexplored organic constituents in the earth’s atmosphere. Environ. Sci. Technol. 2007, 41, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Simoneit, B.R.; Mazurek, M.A. Organic matter of the troposphere—II. For Part I, see Simoneit et al. (1977). Natural background of biogenic lipid matter in aerosols over the rural western united states. Atmos. Environ. 1982, 16, 2139–2159. [Google Scholar] [CrossRef]
- Després, V.; Nowoisky, J.; Klose, M.; Conrad, R.; Andreae, M.; Pöschl, U. Characterization of primary biogenic aerosol particles in urban, rural, and high-alpine air by DNA sequence and restriction fragment analysis of ribosomal RNA genes. Biogeosciences 2007, 4, 1127–1141. [Google Scholar] [CrossRef] [Green Version]
- Jaenicke, R. Abundance of cellular material and proteins in the atmosphere. Science 2005, 308, 73. [Google Scholar] [CrossRef] [PubMed]
- Querol, X.; Viana, M.; Alastuey, A.; Amato, F.; Moreno, T.; Castillo, S.; Pey, J.; de la Rosa, J.; Sánchez de la Campa, A.; Artíñano, B.; et al. Source origin of trace elements in PM from regional background, urban and industrial sites of Spain. Atmos. Environ. 2007, 41, 7219–7231. [Google Scholar] [CrossRef]
- Seinfeld, J.; Pandis, S. Atmospheric Chemistry and Physics; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Volkamer, R.; Jimenez, J.L.; San Martini, F.; Dzepina, K.; Zhang, Q.; Salcedo, D.; Molina, L.T.; Worsnop, D.R.; Molina, M.J. Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected. Geophys. Res. Lett. 2006, 33. [Google Scholar] [CrossRef] [Green Version]
- Kanakidou, M.; Seinfeld, J.; Pandis, S.; Barnes, I.; Dentener, F.; Facchini, M.; Dingenen, R.V.; Ervens, B.; Nenes, A.; Nielsen, C. Organic aerosol and global climate modelling: A review. Atmos. Chem. Phys. 2005, 5, 1053–1123. [Google Scholar] [CrossRef]
- Ziemann, P.J.; Atkinson, R. Kinetics, products, and mechanisms of secondary organic aerosol formation. Chem. Soc. Rev. 2012, 41, 6582–6605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jimenez, J.L.; Canagaratna, M.R.; Allan, J.D.; Coe, H.; Ulbrich, I.; Alfarra, M.R.; Takami, A.; Middlebrook, A.M.; Sun, Y.L.; et al. Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes. Geophys. Res. Lett. 2007, 34. [Google Scholar] [CrossRef] [Green Version]
- Carlton, A.G.; Wiedinmyer, C.; Kroll, J.H. A review of Secondary Organic Aerosol (SOA) formation from isoprene. Atmos. Chem. Phys. 2009, 9, 4987–5005. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, M.K.; Subramanian, R.; Rogge, W.F.; Robinson, A.L. Sources of organic aerosol: Positive matrix factorization of molecular marker data and comparison of results from different source apportionment models. Atmos. Environ. 2007, 41, 9353–9369. [Google Scholar] [CrossRef]
- Zhao, Y.; Kreisberg, N.M.; Worton, D.R.; Isaacman, G.; Gentner, D.R.; Chan, A.W.H.; Weber, R.J.; Liu, S.; Day, D.A.; Russell, L.M.; et al. Sources of organic aerosol investigated using organic compounds as tracers measured during CalNex in Bakersfield. J. Geophys. Res.-Atmos. 2013, 118, 2012JD019248. [Google Scholar] [CrossRef]
- Griffin, R.J.; Cocker, D.R.; Flagan, R.C.; Seinfeld, J.H. Organic aerosol formation from the oxidation of biogenic hydrocarbons. J. Geophys. Res. 1999, 104, 3555. [Google Scholar] [CrossRef]
- Hallquist, M.; Wenger, J.; Baltensperger, U.; Rudich, Y.; Simpson, D.; Claeys, M.; Dommen, J.; Donahue, N.; George, C.; Goldstein, A. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155–5236. [Google Scholar] [CrossRef] [Green Version]
- Tsigaridis, K.; Daskalakis, N.; Kanakidou, M.; Adams, P.; Artaxo, P.; Bahadur, R.; Balkanski, Y.; Bauer, S.; Bellouin, N.; Benedetti, A. The AeroCom evaluation and intercomparison of organic aerosol in global models. Atmos. Chem. Phys. 2014, 14, 10845–10895. [Google Scholar] [CrossRef]
- Ciarelli, G.; Aksoyoglu, S.; Crippa, M.; Jimenez, J.L.; Nemitz, E.; Sellegri, K.; Äijälä, M.; Carbone, S.; Mohr, C.; O’Dowd, C.; et al. Evaluation of European air quality modelled by CAMx including the volatility basis set scheme. Atmos. Chem. Phys. 2016, 16, 10313–10332. [Google Scholar] [CrossRef]
- Grosjean, D. Particulate carbon in Los Angeles air. Sci. Total Environ. 1984, 32, 133–145. [Google Scholar] [CrossRef]
- Turpin, B.J.; Huntzicker, J.J. Identification of secondary organic aerosol episodes and quantitation of primary and secondary organic aerosol concentrations during SCAQS. Atmos. Environ. 1995, 29, 3527–3544. [Google Scholar] [CrossRef]
- Gray, H.A.; Cass, G.R.; Huntzicker, J.J.; Heyerdahl, E.K.; Rau, J.A. Characteristics of atmospheric organic and elemental carbon particle concentrations in Los Angeles. Environ. Sci. Technol. 1986, 20, 580–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, J.G.; Robinson, N.F.; Chow, J.C.; Henry, R.C.; Kim, B.; Pace, T.; Meyer, E.L.; Nguyen, Q. The USEPA/DRI chemical mass balance receptor model, CMB 7.0. Environ. Softw. 1990, 5, 38–49. [Google Scholar] [CrossRef]
- Paatero, P. Least squares formulation of robust non-negative factor analysis. Chemom. Intell. Lab. Syst. 1997, 37, 23–35. [Google Scholar] [CrossRef]
- Paatero, P.; Tapper, U. Positive matrix factorization: A non-negative factor model with optimal utilization of error estimates of data values. Environmetrics 1994, 5, 111–126. [Google Scholar] [CrossRef]
- Zhang, Y.; Sheesley, R.J.; Schauer, J.J.; Lewandowski, M.; Jaoui, M.; Offenberg, J.H.; Kleindienst, T.E.; Edney, E.O. Source apportionment of primary and secondary organic aerosols using positive matrix factorization (PMF) of molecular markers. Atmos. Environ. 2009, 43, 5567–5574. [Google Scholar] [CrossRef]
- Kleindienst, T.E.; Jaoui, M.; Lewandowski, M.; Offenberg, J.H.; Lewis, C.W.; Bhave, P.V.; Edney, E.O. Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location. Atmos. Environ. 2007, 41, 8288–8300. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Zhang, Y.; Liu, D.; Ding, P.; Shen, C.; Shen, K.; He, Q.; Ding, X.; Wang, X. Source apportionment using radiocarbon and organic tracers for PM2. 5 carbonaceous aerosols in guangzhou, south china: Contrasting local-and regional-scale haze events. Environ. Sci. Technol. 2014, 48, 12002–12011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szidat, S.; Ruff, M.; Perron, N.; Wacker, L.; Synal, H.-A.; Hallquist, M.; Shannigrahi, A.S.; Yttri, K.E.; Dye, C.; Simpson, D. Fossil and non-fossil sources of organic carbon (OC) and elemental carbon (EC) in Göteborg, Sweden. Atmos. Chem. Phys. 2009, 9, 1521–1535. [Google Scholar] [CrossRef] [Green Version]
- Gelencsér, A.; May, B.; Simpson, D.; Sánchez-Ochoa, A.; Kasper-Giebl, A.; Puxbaum, H.; Caseiro, A.; Pio, C.; Legrand, M. Source apportionment of PM2.5 organic aerosol over Europe: Primary/secondary, natural/anthropogenic, and fossil/biogenic origin. J. Geophys. Res. 2007, 112. [Google Scholar] [CrossRef]
- Weber, R.J.; Sullivan, A.P.; Peltier, R.E.; Russell, A.; Yan, B.; Zheng, M.; de Gouw, J.; Warneke, C.; Brock, C.; Holloway, J.S.; et al. A study of secondary organic aerosol formation in the anthropogenic-influenced southeastern United States. J. Geophys. Res.-Atmos. 2007, 112, D13302. [Google Scholar] [CrossRef]
- Blanchard, C.L.; Hidy, G.M.; Tanenbaum, S.; Edgerton, E.; Hartsell, B.; Jansen, J. Carbon in southeastern U.S. aerosol particles: Empirical estimates of secondary organic aerosol formation. Atmos. Environ. 2008, 42, 6710–6720. [Google Scholar] [CrossRef]
- DeCarlo, P.F.; Kimmel, J.R.; Trimborn, A.; Northway, M.J.; Jayne, J.T.; Aiken, A.C.; Gonin, M.; Fuhrer, K.; Horvath, T.; Docherty, K.S. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 2006, 78, 8281–8289. [Google Scholar] [CrossRef] [PubMed]
- Jayne, J.T.; Leard, D.C.; Zhang, X.; Davidovits, P.; Smith, K.A.; Kolb, C.E.; Worsnop, D.R. Development of an aerosol mass spectrometer for size and composition analysis of submicron particles. Aerosol Sci. Technol. 2000, 33, 49–70. [Google Scholar] [CrossRef]
- Sun, Y.L.; Zhang, Q.; Schwab, J.J.; Demerjian, K.L.; Chen, W.N.; Bae, M.S.; Hung, H.M.; Hogrefe, O.; Frank, B.; Rattigan, O.V.; et al. Characterization of the sources and processes of organic and inorganic aerosols in New York city with a high-resolution time-of-flight aerosol mass apectrometer. Atmos. Chem. Phys. 2011, 11, 1581–1602. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Q.; Chen, M.; Ge, X.; Ren, J.; Qin, D. Chemical composition, sources, and processes of urban aerosols during summertime in northwest China: Insights from high-resolution aerosol mass spectrometry. Atmos. Chem. Phys. 2014, 14, 12593–12611. [Google Scholar] [CrossRef]
- Zahardis, J.; Geddes, S.; Petrucci, G.A. Improved understanding of atmospheric organic aerosols via innovations in soft ionization aerosol mass spectrometry. Anal. Chem. 2011, 83, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Kleindienst, T.E.; Lewandowski, M.; Offenberg, J.H.; Edney, E.O.; Jaoui, M.; Zheng, M.; Ding, X.; Edgerton, E.S. Contribution of primary and secondary sources to organic aerosol and PM2.5 at SEARCH Network Sites. J. Air Waste Manag. Assoc. 2010, 60, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Kourtchev, I.; Warnke, J.; Maenhaut, W.; Hoffmann, T.; Claeys, M. Polar organic marker compounds in PM2.5 aerosol from a mixed forest site in western Germany. Chemosphere 2008, 73, 1308–1314. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Bian, Q.; Lau, A.K.H.; Yu, J.Z. Source apportioning of primary and secondary organic carbon in summer PM2.5 in Hong Kong using positive matrix factorization of secondary and primary organic tracer data. J. Geophys. Res.-Atmos. 2010, 115. [Google Scholar] [CrossRef]
- El Haddad, I.; Marchand, N.; Temime-Roussel, B.; Wortham, H.; Piot, C.; Besombes, J.L.; Baduel, C.; Voisin, D.; Armengaud, A.; Jaffrezo, J.L. Insights into the secondary fraction of the organic aerosol in a Mediterranean urban area: Marseille. Atmos. Chem. Phys. 2011, 11, 2059–2079. [Google Scholar] [CrossRef] [Green Version]
- Nozière, B.; Kalberer, M.; Claeys, M.; Allan, J.; D’Anna, B.; Decesari, S.; Finessi, E.; Glasius, M.; Grgić, I.; Hamilton, J.F.; et al. The molecular identification of organic compounds in the atmosphere: State of the art and challenges. Chem. Rev. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsigaridis, K.; Kanakidou, M. Secondary organic aerosol importance in the future atmosphere. Atmos. Environ. 2007, 41, 4682–4692. [Google Scholar] [CrossRef]
- Guenther, A.; Karl, T.; Harley, P.; Wiedinmyer, C.; Palmer, P.I.; Geron, C. Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature). Atmos. Chem. Phys. 2006, 6, 3181–3210. [Google Scholar] [CrossRef] [Green Version]
- Guenther, A.; Hewitt, C.N.; Erickson, D.; Fall, R.; Geron, C.; Graedel, T.; Harley, P.; Klinger, L.; Lerdau, M.; McKay, W.A.; et al. A global model of natural volatile organic compound emissions. J. Geophys. Res.-Atmos. 1995, 100, 8873–8892. [Google Scholar] [CrossRef]
- Acosta Navarro, J.C.; Smolander, S.; Struthers, H.; Zorita, E.; Ekman, A.M.; Kaplan, J.; Guenther, A.; Arneth, A.; Riipinen, I. Global emissions of terpenoid VOCs from terrestrial vegetation in the last millennium. J. Geophys. Res.-Atmos. 2014, 119, 6867–6885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettle, A.J.; Andreae, M.O. Flux of dimethylsulfide from the oceans: A comparison of updated data sets and flux models. J. Geophys. Res.-Atmos. 2000, 105, 26793–26808. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, K.; Sakaguchi, F. Molecular distributions of water soluble dicarboxylic acids in marine aerosols over the Pacific Ocean including tropics. J. Geophys. Res.-Atmos. 1999, 104, 3501–3509. [Google Scholar] [CrossRef] [Green Version]
- Facchini, M.C.; Decesari, S.; Rinaldi, M.; Carbone, C.; Finessi, E.; Mircea, M.; Fuzzi, S.; Moretti, F.; Tagliavini, E.; Ceburnis, D.; et al. Important source of marine secondary organic aerosol from biogenic amines. Environ. Sci. Technol. 2008, 42, 9116–9121. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.; Spracklen, D.; Williams, J.; Yassaa, N.; Sciare, J.; Bonsang, B.; Gros, V.; Peeken, I.; Lewis, A.; Alvain, S. Evaluation of the global oceanic isoprene source and its impacts on marine organic carbon aerosol. Atmos. Chem. Phys. 2009, 9, 1253–1262. [Google Scholar] [CrossRef] [Green Version]
- Odum, J.R.; Jungkamp, T.P.W.; Griffin, R.J.; Flagan, R.C.; Seinfeld, J.H. The atmospheric aerosol-forming potential of whole gasoline vapor. Science 1997, 276, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Utembe, S.; Jenkin, M.; Derwent, R.; Hayman, G.; Alfarra, M.; Coe, H.; McFiggans, G. Simulating regional scale secondary organic aerosol formation during the TORCH 2003 campaign in the southern UK. Atmos. Chem. Phys. 2006, 6, 403–418. [Google Scholar] [CrossRef] [Green Version]
- Kleeman, M.J.; Ying, Q.; Lu, J.; Mysliwiec, M.J.; Griffin, R.J.; Chen, J.; Clegg, S. Source apportionment of secondary organic aerosol during a severe photochemical smog episode. Atmos. Environ. 2007, 41, 576–591. [Google Scholar] [CrossRef]
- Chen, J.; Ying, Q.; Kleeman, M.J. Source apportionment of wintertime secondary organic aerosol during the California regional PM10/PM2.5 air quality study. Atmos. Environ. 2010, 44, 1331–1340. [Google Scholar] [CrossRef]
- Marta, G.V.; Manuel, S. Secondary organic aerosol formation from the oxidation of a mixture of organic gases in a chamber. Air Qual. 2010. [CrossRef]
- Tkacik, D.S.; Presto, A.A.; Donahue, N.M.; Robinson, A.L. Secondary organic aerosol formation from intermediate-volatility organic compounds: Cyclic, linear, and branched alkanes. Environ. Sci. Technol. 2012, 46, 8773–8781. [Google Scholar] [CrossRef] [PubMed]
- Boreddy, S.K.R.; Parvin, F.; Kawamura, K.; Zhu, C.; Lee, C.-T. Stable carbon and nitrogen isotopic compositions of fine aerosols (PM2.5) during an intensive biomass burning over Southeast Asia: Influence of SOA and aging. Atmos. Environ. 2018, 191, 478–489. [Google Scholar] [CrossRef]
- Boreddy, S.K.R.; Kawamura, K.; Tachibana, E. Long-term (2001–2013) observations of water-soluble dicarboxylic acids and related compounds over the western North Pacific: Trends, seasonality and source apportionment. Sci. Rep. 2017, 7, 8518. [Google Scholar] [CrossRef] [PubMed]
- Boreddy, S.K.R.; Kawamura, K.; Okuzawa, K.; Kanaya, Y.; Wang, Z. Temporal and diurnal variations of carbonaceous aerosols and major ions in biomass burning influenced aerosols over Mt. Tai in the North China Plain during MTX2006. Atmos. Environ. 2017, 154, 106–117. [Google Scholar] [CrossRef]
- Boreddy, S.K.R.; Haque, M.M.; Kawamura, K. Long-term (2001–2012) trends of carbonaceous aerosols from a remote island in the western North Pacific: An outflow region of Asian pollutants. Atmos. Chem. Phys. 2018, 18, 1291–1306. [Google Scholar] [CrossRef]
- Henze, D.; Seinfeld, J.; Ng, N.; Kroll, J.; Fu, T.-M.; Jacob, D.J.; Heald, C. Global modeling of secondary organic aerosol formation from aromatic hydrocarbons: High- vs. low-yield pathways. Atmos. Chem. Phys. 2008, 8, 2405–2420. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.L.; Donahue, N.M.; Shrivastava, M.K.; Weitkamp, E.A.; Sage, A.M.; Grieshop, A.P.; Lane, T.E.; Pierce, J.R.; Pandis, S.N. Rethinking organic aerosols: Semivolatile emissions and photochemical aging. Science 2007, 315, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hennigan, C.J.; May, A.A.; Tkacik, D.S.; de Gouw, J.A.; Gilman, J.B.; Kuster, W.C.; Borbon, A.; Robinson, A.L. Intermediate-volatility organic compounds: A large source of secondary organic aerosol. Environ. Sci. Technol. 2014, 48, 13743–13750. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.Q.; Ding, X.; He, Q.F.; Cong, Z.Y.; Yu, Q.Q.; Wang, X.M. Seasonal variation of secondary organic aerosol tracers in Central Tibetan Plateau. Atmos. Chem. Phys. 2015, 15, 8781–8793. [Google Scholar] [CrossRef]
- Donahue, N.M.; Robinson, A.L.; Pandis, S.N. Atmospheric organic particulate matter: From smoke to secondary organic aerosol. Atmos. Environ. 2009, 43, 94–106. [Google Scholar] [CrossRef]
- Schauer, J.J.; Kleeman, M.J.; Cass, G.R.; Simoneit, B.R.T. Measurement of Emissions from Air Pollution Sources. 4. C1−C27 Organic Compounds from Cooking with Seed Oils. Environ. Sci. Technol. 2001, 36, 567–575. [Google Scholar] [CrossRef]
- Schauer, J.J.; Kleeman, M.J.; Cass, G.R.; Simoneit, B.R.T. Measurement of Emissions from Air Pollution Sources. 2. C1 through C30 Organic Compounds from Medium Duty Diesel Trucks. Environ. Sci. Technol. 1999, 33, 1578–1587. [Google Scholar] [CrossRef]
- Sasaki, J.; Aschmann, S.M.; Kwok, E.S.C.; Atkinson, R.; Arey, J. Products of the gas-phase OH and NO3 radical-initiated reactions of naphthalene. Environ. Sci. Technol. 1997, 31, 3173–3179. [Google Scholar] [CrossRef]
- Wang, L.; Atkinson, R.; Arey, J. Dicarbonyl products of the OH radical-initiated reactions of naphthalene and the C1- and C2-alkylnaphthalenes. Environ. Sci. Technol. 2007, 41, 2803–2810. [Google Scholar] [CrossRef] [PubMed]
- Mihele, C.M.; Wiebe, H.A.; Lane, D.A. Particle formation and gas/particle partition measurements of the products of the naphthalene-OH radical reaction in a smog chamber. Polycycl. Aromat. Compd. 2002, 22, 729–736. [Google Scholar] [CrossRef]
- Zhang, H. Secondary organic aerosol from polycyclic aromatic hydrocarbons in Southeast Texas. Atmos. Environ. 2012, 55, 279–287. [Google Scholar] [CrossRef]
- Chan, A.W.H.; Kautzman, K.E.; Chhabra, P.S.; Surratt, J.D.; Chan, M.N.; Crounse, J.D.; Kürten, A.; Wennberg, P.O.; Flagan, R.C.; Seinfeld, J.H. Secondary organic aerosol formation from photooxidation of naphthalene and alkylnaphthalenes: Implications for oxidation of intermediate volatility organic compounds (IVOCs). Atmos. Chem. Phys. 2009, 9, 3049–3060. [Google Scholar] [CrossRef]
- Srivastava, D.; Tomaz, S.; Favez, O.; Lanzafame, G.M.; Golly, B.; Besombes, J.-L.; Alleman, L.Y.; Jaffrezo, J.-L.; Jacob, V.; Perraudin, E.; et al. Speciation of organic fraction does matter for source apportionment. Part 1: A one-year campaign in Grenoble (France). Sci. Total Environ. 2018, 624, 1598–1611. [Google Scholar] [CrossRef] [PubMed]
- Bruns, E.A.; El Haddad, I.; Slowik, J.G.; Kilic, D.; Klein, F.; Baltensperger, U.; Prévôt, A.S.H. Identification of significant precursor gases of secondary organic aerosols from residential wood combustion. Sci. Rep. 2016, 6, 27881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, L.; Kautzman, K.; Loza, C.; Schilling, K.; Coggon, M.; Chhabra, P.; Chan, M.; Chan, A.; Hersey, S.; Crounse, J. Secondary oranic aerosol formation from biomass burning intermediates: Phenol and methoxyphenols. Atmos. Chem. Phys. 2013, 13, 8019–8043. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics; John Wiley: Hoboken, NJ, USA, 1998; 1326p. [Google Scholar]
- Foster, T.L.; Caradonna, J.P. Fe2+-catalyzed heterolytic RO−OH bond cleavage and substrate oxidation: A functional synthetic non-heme iron monooxygenase system. J. Am. Chem. Soc. 2003, 125, 3678–3679. [Google Scholar] [CrossRef] [PubMed]
- Rutter, A.P.; Snyder, D.C.; Stone, E.A.; Shelton, B.; DeMinter, J.; Schauer, J.J. Preliminary assessment of the anthropogenic and biogenic contributions to secondary organic aerosols at two industrial cities in the upper Midwest. Atmos. Environ. 2014, 84, 307–313. [Google Scholar] [CrossRef]
- Mader, B.T.; Pankow, J.F. Gas/solid partitioning of semivolatile organic compounds (SOCs) to air filters. 3. An analysis of gas adsorption artifacts in measurements of atmospheric SOCs and organic carbon (OC) when using Teflon membrane filters and quartz fiber filters. Environ. Sci. Technol. 2001, 35, 3422–3432. [Google Scholar] [CrossRef] [PubMed]
- Albinet, A.; Papaiconomou, N.; Estager, J.; Suptil, J.; Besombes, J.-L. A new ozone denuder for aerosol sampling based on an ionic liquid coating. Anal. Bioanal. Chem. 2010, 396, 857–864. [Google Scholar] [CrossRef] [PubMed]
- McDow, S.R.; Huntzicker, J.J. Vapor adsorption artifact in the sampling of organic aerosol: Face velocity effects. Atmos. Environ. Part A 1990, 24, 2563–2571. [Google Scholar] [CrossRef]
- Turpin, B.J.; Huntzicker, J.J.; Hering, S.V. Investigation of organic aerosol sampling artifacts in the los angeles basin. Atmos. Environ. 1994, 28, 3061–3071. [Google Scholar] [CrossRef]
- Ding, Y.; Pang, Y.; Eatough, D.J. High-volume diffusion denuder sampler for the routine monitoring of fine particulate matter: I. Design and optimization of the PC-BOSS. Aerosol Sci. Technol. 2002, 36, 369–382. [Google Scholar] [CrossRef]
- Tsapakis, M.; Stephanou, E.G. Collection of gas and particle semi-volatile organic compounds: Use of an oxidant denuder to minimize polycyclic aromatic hydrocarbons degradation during high-volume air sampling. Atmos. Environ. 2003, 37, 4935–4944. [Google Scholar] [CrossRef]
- Subramanian, R.; Khlystov, A.Y.; Cabada, J.C.; Robinson, A.L. Positive and negative artifacts in particulate organic carbon measurements with denuded and undenuded sampler configurations special issue of aerosol science and technology on findings from the fine particulate matter supersites program. Aerosol Sci. Technol. 2004, 38, 27–48. [Google Scholar] [CrossRef]
- Goriaux, M.; Jourdain, B.; Temime, B.; Besombes, J.L.; Marchand, N.; Albinet, A.; Leoz-Garziandia, E.; Wortham, H. Field comparison of particulate PAH measurements using a low-flow denuder device and conventional sampling systems. Environ. Sci. Technol. 2006, 40, 6398–6404. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.M.; Pio, C.A.; Harrison, R.M.; Smith, D.J.T. Carbonaceous aerosol in urban and rural European atmospheres: Estimation of secondary organic carbon concentrations. Atmos. Environ. 1999, 33, 2771–2781. [Google Scholar] [CrossRef]
- Chu, S.-H. Stable estimate of primary OC/EC ratios in the EC tracer method. Atmos. Environ. 2005, 39, 1383–1392. [Google Scholar] [CrossRef]
- Saylor, R.D.; Edgerton, E.S.; Hartsell, B.E. Linear regression techniques for use in the EC tracer method of secondary organic aerosol estimation. Atmos. Environ. 2006, 40, 7546–7556. [Google Scholar] [CrossRef]
- Yu, S.; Dennis, R.L.; Bhave, P.V.; Eder, B.K. Primary and secondary organic aerosols over the United States: Estimates on the basis of observed organic carbon (OC) and elemental carbon (EC), and air quality modeled primary OC/EC ratios. Atmos. Environ. 2004, 38, 5257–5268. [Google Scholar] [CrossRef]
- Lim, H.-J.; Turpin, B.J. Origins of primary and secondary organic aerosol in Atlanta: Results of time-resolved measurements during the Atlanta supersite experiment. Environ. Sci. Technol. 2002, 36, 4489–4496. [Google Scholar] [CrossRef] [PubMed]
- Saffari, A.; Hasheminassab, S.; Shafer, M.M.; Schauer, J.J.; Chatila, T.A.; Sioutas, C. Nighttime aqueous-phase secondary organic aerosols in Los Angeles and its implication for fine particulate matter composition and oxidative potential. Atmos. Environ. 2016, 133, 112–122. [Google Scholar] [CrossRef]
- Pachon, J.E.; Balachandran, S.; Hu, Y.; Weber, R.J.; Mulholland, J.A.; Russell, A.G. Comparison of SOC estimates and uncertainties from aerosol chemical composition and gas phase data in Atlanta. Atmos. Environ. 2010, 44, 3907–3914. [Google Scholar] [CrossRef]
- Strader, R.; Lurmann, F.; Pandis, S.N. Evaluation of secondary organic aerosol formation in winter. Atmos. Environ. 1999, 33, 4849–4863. [Google Scholar] [CrossRef]
- Cabada, J.C.; Pandis, S.N.; Subramanian, R.; Robinson, A.L.; Polidori, A.; Turpin, B. Estimating the secondary organic aerosol contribution to PM2.5 using the EC Tracer method special issue of aerosol science and technology on findings from the fine particulate matter supersites program. Aerosol Sci. Technol. 2004, 38, 140–155. [Google Scholar] [CrossRef]
- Plaza, J.; Gómez-Moreno, F.J.; Núñez, L.; Pujadas, M.; Artíñano, B. Estimation of secondary organic aerosol formation from semi-continuous OC–EC measurements in a Madrid suburban area. Atmos. Environ. 2006, 40, 1134–1147. [Google Scholar] [CrossRef]
- Lonati, G.; Ozgen, S.; Giugliano, M. Primary and secondary carbonaceous species in PM2.5 samples in Milan (Italy). Atmos. Environ. 2007, 41, 4599–4610. [Google Scholar] [CrossRef]
- Yuan, Z.; Yu, J.; Lau, A.; Louie, P.; Fung, J.C.H. Application of positive matrix factorization in estimating aerosol secondary organic carbon in Hong Kong and its relationship with secondary sulfate. Atmos. Chem. Phys. 2006, 6, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yu, J.Z.; Ho, S.S.H.; Xu, J.; Wu, W.-S.; Wan, C.H.; Wang, X.; Wang, X.; Wang, L. The chemical composition of inorganic and carbonaceous materials in PM2.5 in Nanjing, China. Atmos. Environ. 2005, 39, 3735–3749. [Google Scholar] [CrossRef]
- Favez, O.; Cachier, H.; Sciare, J.; Alfaro, S.C.; El-Araby, T.M.; Harhash, M.A.; Abdelwahab, M.M. Seasonality of major aerosol species and their transformations in Cairo megacity. Atmos. Environ. 2008, 42, 1503–1516. [Google Scholar] [CrossRef]
- Na, K.; Sawant, A.A.; Song, C.; Cocker, D.R., III. Primary and secondary carbonaceous species in the atmosphere of Western Riverside County, California. Atmos. Environ. 2004, 38, 1345–1355. [Google Scholar] [CrossRef]
- Chow, J.C.; Watson, J.G.; Pritchett, L.C.; Pierson, W.R.; Frazier, C.A.; Purcell, R.G. The DRI thermal/optical reflectance carbon analysis system: Description, evaluation and applications in U.S. Air quality studies. Atmos. Environ. Part A 1993, 27, 1185–1201. [Google Scholar] [CrossRef]
- Birch, M.E.; Cary, R.A. Elemental carbon-based method for occupational monitoring of particulate diesel exhaust: Methodology and exposure issues. Analyst 1996, 121, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, F.; Viana, M.; Yttri, K.; Genberg, J.; Putaud, J.-P. Toward a standardised thermal-optical protocol for measuring atmospheric organic and elemental carbon: The EUSAAR protocol. Atmos. Meas. Tech. 2010, 3, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Chow, J.C.; Watson, J.G.; Chen, L.W.A.; Chang, M.C.O.; Robinson, N.F.; Trimble, D.; Kohl, S. The IMPROVE A Temperature Protocol for Thermal/Optical Carbon Analysis: Maintaining Consistency with a Long-Term Database. J. Air Waste Manag. Assoc. 2007, 57, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Karanasiou, A.; Minguillón, M.; Viana, M.; Alastuey, A.; Putaud, J.-P.; Maenhaut, W.; Panteliadis, P.; Močnik, G.; Favez, O.; Kuhlbusch, T. Thermal-optical analysis for the measurement of elemental carbon (EC) and organic carbon (OC) in ambient air a literature review. Atmos. Meas. Tech. Discuss. 2015, 8, 9649–9712. [Google Scholar] [CrossRef]
- Chiappini, L.; Verlhac, S.; Aujay, R.; Maenhaut, W.; Putaud, J.P.; Sciare, J.; Jaffrezo, J.L.; Liousse, C.; Galy-Lacaux, C.; Alleman, L.Y.; et al. Clues for a standardised thermal-optical protocol for the assessment of organic and elemental carbon within ambient air particulate matter. Atmos. Meas. Tech. 2014, 7, 1649–1661. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Huang, X.H.H.; Ng, W.M.; Griffith, S.M.; Yu, J.Z. Inter-comparison of NIOSH and IMPROVE protocols for OC and EC determination: Implications for inter-protocol data conversion. Atmos. Meas. Tech. 2016, 9, 4547–4560. [Google Scholar] [CrossRef]
- Mancilla, Y.; Herckes, P.; Fraser, M.P.; Mendoza, A. Secondary organic aerosol contributions to PM2.5 in Monterrey, Mexico: Temporal and seasonal variation. Atmos. Res. 2015, 153, 348–359. [Google Scholar] [CrossRef]
- Martinez, M.A.; Caballero, P.; Carrillo, O.; Mendoza, A.; Mejia, G.M. Chemical characterization and factor analysis of PM2.5 in two sites of Monterrey, Mexico. J. Air Waste Manag. Assoc. 2012, 62, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.A.; Snyder, D.C.; Sheesley, R.J.; Sullivan, A.; Weber, R.; Schauer, J. Source apportionment of fine organic aerosol in Mexico City during the MILAGRO experiment 2006. Atmos. Chem. Phys. 2008, 8, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Turpin, B.J.; Huntzicker, J.J.; Larson, S.M.; Cass, G.R. Los Angeles summer midday particulate carbon: Primary and secondary aerosol. Environ. Sci. Technol. 1991, 25, 1788–1793. [Google Scholar] [CrossRef]
- Polidori, A.; Turpin, B.J.; Lim, H.J.; Cabada, J.C.; Subramanian, R.; Pandis, S.N.; Robinson, A.L. Local and regional secondary organic aerosol: Insights from a year of semi-continuous carbon measurements at Pittsburgh. Aerosol Sci. Technol. 2006, 40, 861–872. [Google Scholar] [CrossRef]
- Yu, S.; Bhave, P.V.; Dennis, R.L.; Mathur, R. Seasonal and regional variations of primary and secondary organic aerosols over the Continental United States: Semi-empirical estimates and model evaluation. Environ. Sci. Technol. 2007, 41, 4690–4697. [Google Scholar] [CrossRef] [PubMed]
- Day, M.C.; Zhang, M.; Pandis, S.N. Evaluation of the ability of the EC tracer method to estimate secondary organic carbon. Atmos. Environ. 2015, 112, 317–325. [Google Scholar] [CrossRef]
- Sunder Raman, R.; Hopke, P.K.; Holsen, T.M. Carbonaceous aerosol at two rural locations in New York State: Characterization and behavior. J. Geophys. Res.-Atmos. 2008, 113, D12202. [Google Scholar] [CrossRef]
- Dreyfus, M.A.; Adou, K.; Zucker, S.M.; Johnston, M.V. Organic aerosol source apportionment from highly time-resolved molecular composition measurements. Atmos. Environ. 2009, 43, 2901–2910. [Google Scholar] [CrossRef]
- Vega, E.; Eidels, S.; Ruiz, H.; López-Veneroni, D.; Sosa, G.; Gonzalez, E.; Gasca, J.; Mora, V.; Reyes, E.; Sánchez-Reyna, G. Particulate air pollution in Mexico City: A detailed view. Aerosol Air Qual. Res. 2010, 10, 193–211. [Google Scholar] [CrossRef]
- Murillo, J.H.; Marin, J.F.R.; Roman, S.R.; Guerrero, V.H.B.; Arias, D.S.; Ramos, A.C.; Gonzalez, B.C.; Baumgardner, D.G. Temporal and spatial variations in organic and elemental carbon concentrations in PM10/PM2.5 in the metropolitan area of Costa Rica, Central America. Atmos. Pollut. Res. 2013, 4, 53–63. [Google Scholar] [CrossRef]
- Seguel, R. Estimations of primary and secondary organic carbon formation in PM2.5 aerosols of Santiago City, Chile. Atmos. Environ. 2009, 43, 2125–2131. [Google Scholar] [CrossRef]
- Toro Araya, R.; Flocchini, R.; Morales Segura, R.G.; Leiva Guzman, M.A. Carbonaceous aerosols in fine particulate matter of Santiago Metropolitan Area, Chile. Sci. World J. 2014, 2014, 794590. [Google Scholar] [CrossRef] [PubMed]
- Favez, O.; Cachier, H.; Sciare, J.; Le Moullec, Y. Characterization and contribution to PM2.5 of semi-volatile aerosols in Paris (France). Atmos. Environ. 2007, 41, 7969–7976. [Google Scholar] [CrossRef]
- Laongsri, B.; Harrison, R.M. Atmospheric behaviour of particulate oxalate at UK urban background and rural sites. Atmos. Environ. 2013, 71, 319–326. [Google Scholar] [CrossRef]
- Pant, P.; Yin, J.; Harrison, R.M. Sensitivity of a Chemical Mass Balance model to different molecular marker traffic source profiles. Atmos. Environ. 2014, 82, 238–249. [Google Scholar] [CrossRef]
- Mirante, F.; Salvador, P.; Pio, C.; Alves, C.; Artiñano, B.; Caseiro, A.; Revuelta, M.A. Size fractionated aerosol composition at roadside and background environments in the Madrid urban atmosphere. Atmos. Res. 2014, 138, 278–292. [Google Scholar] [CrossRef]
- Yubero, E.; Galindo, N.; Nicolás, J.; Crespo, J.; Calzolai, G.; Lucarelli, F. Temporal variations of PM1 major components in an urban street canyon. Environ. Sci. Pollut. Res. 2015, 22, 13328–13335. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, D.; Liakakou, E.; Gerasopoulos, E.; Theodosi, C.; Mihalopoulos, N. Long-term characterization of organic and elemental carbon in the PM2.5 fraction: The case of Athens, Greece. Atmos. Chem. Phys. 2014, 14, 13313–13325. [Google Scholar] [CrossRef]
- Samara, C.; Voutsa, D.; Kouras, A.; Eleftheriadis, K.; Maggos, T.; Saraga, D.; Petrakakis, M. Organic and elemental carbon associated to PM10 and PM2.5 at urban sites of northern Greece. Environ. Sci. Pollut. Res. 2014, 21, 1769–1785. [Google Scholar] [CrossRef] [PubMed]
- Błaszczak, B.; Rogula-Kozłowska, W.; Mathews, B.; Juda-Rezler, K.; Klejnowski, K.; Rogula-Kopiec, P. Chemical compositions of PM2.5 at two non-urban sites from the polluted region in Europe. Aerosol Air Qual. Res. 2016, 16, 2333–2348. [Google Scholar] [CrossRef]
- Grivas, G.; Cheristanidis, S.; Chaloulakou, A. Elemental and organic carbon in the urban environment of Athens. Seasonal and diurnal variations and estimates of secondary organic carbon. Sci. Total Environ. 2012, 414, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Wagener, S.; Langner, M.; Hansen, U.; Moriske, H.-J.; Endlicher, W. Assessing the influence of seasonal and spatial variations on the estimation of secondary organic carbon in urban particulate matter by applying the EC-tracer method. Atmosphere 2014, 5, 252–272. [Google Scholar] [CrossRef]
- Pietrogrande, M.C.; Bacco, D.; Ferrari, S.; Ricciardelli, I.; Scotto, F.; Trentini, A.; Visentin, M. Characteristics and major sources of carbonaceous aerosols in PM2.5 in Emilia Romagna Region (Northern Italy) from four-year observations. Sci. Total Environ. 2016, 553, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.M.; Yin, J. Sources and processes affecting carbonaceous aerosol in central England. Atmos. Environ. 2008, 42, 1413–1423. [Google Scholar] [CrossRef]
- Khan, M.B.; Masiol, M.; Formenton, G.; Di Gilio, A.; de Gennaro, G.; Agostinelli, C.; Pavoni, B. Carbonaceous PM2.5 and secondary organic aerosol across the Veneto region (NE Italy). Sci. Total Environ. 2016, 542 Pt A, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Pandis, S.N.; Harley, R.A.; Cass, G.R.; Seinfeld, J.H. Secondary organic aerosol formation and transport. Atmos. Environ. Part A 1992, 26, 2269–2282. [Google Scholar] [CrossRef]
- Odum, J.R.; Hoffmann, T.; Bowman, F.; Collins, D.; Flagan, R.C.; Seinfeld, J.H. Gas/particle partitioning and secondary organic aerosol yields. Environ. Sci. Technol. 1996, 30, 2580–2585. [Google Scholar] [CrossRef]
- Abdeen, Z.; Qasrawi, R.; Heo, J.; Wu, B.; Shpund, J.; Vanger, A.; Sharf, G.; Moise, T.; Brenner, S.; Nassar, K.; et al. Spatial and temporal variation in fine particulate matter mass and chemical composition: The Middle East consortium for aerosol research study. Sci. World J. 2014, 2014, 878704. [Google Scholar] [CrossRef] [PubMed]
- Park, S.S.; Cho, S.Y. Tracking sources and behaviors of water-soluble organic carbon in fine particulate matter measured at an urban site in Korea. Atmos. Environ. 2011, 45, 60–72. [Google Scholar] [CrossRef]
- Kim, W.; Lee, H.; Kim, J.; Jeong, U.; Kweon, J. Estimation of seasonal diurnal variations in primary and secondary organic carbon concentrations in the urban atmosphere: EC tracer and multiple regression approaches. Atmos. Environ. 2012, 56, 101–108. [Google Scholar] [CrossRef]
- Batmunkh, T.; Kim, Y.J.; Lee, K.Y.; Cayetano, M.G.; Jung, J.S.; Kim, S.Y.; Kim, K.C.; Lee, S.J.; Kim, J.S.; Chang, L.S.; et al. Time-resolved measurements of PM2.5 carbonaceous aerosols at Gosan, Korea. J. Air Waste Manag. Assoc. 2011, 61, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-K.; Heo, J.-B.; Ban, S.-J.; Yi, S.-M.; Zoh, K.-D. Chemical characteristics of PM2.5 aerosol in Incheon, Korea. Atmos. Environ. 2012, 60, 583–592. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Naito, S.; Oohashi, H. Seasonal variation of PM2.5 components observed in an industrial area of Chiba Prefecture, Japan. Asian J. Atmos. Environ. 2015, 9, 66–77. [Google Scholar] [CrossRef]
- Khan, M.F.; Shirasuna, Y.; Hirano, K.; Masunaga, S. Characterization of PM2.5, PM2.5–10 and PM>10 in ambient air, Yokohama, Japan. Atmos. Res. 2010, 96, 159–172. [Google Scholar] [CrossRef]
- Chou, C.-K.; Lee, C.; Cheng, M.; Yuan, C.; Chen, S.; Wu, Y.; Hsu, W.; Lung, S.; Hsu, S.; Lin, C. Seasonal variation and spatial distribution of carbonaceous aerosols in Taiwan. Atmos. Chem. Phys. 2010, 10, 9563–9578. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.-J.; Zhang, Y.; Bozzetti, C.; Ho, K.-F.; Cao, J.-J.; Han, Y.; Daellenbach, K.R.; Slowik, J.G.; Platt, S.M.; Canonaco, F.; et al. High secondary aerosol contribution to particulate pollution during haze events in China. Nature 2014, 514, 218–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, M.; Wang, H.; Chen, Y.; Yang, F.; Zhang, X.; Zou, Q.; Zhang, R.; Ma, Y.; He, K. Characteristics of aerosol pollution during heavy haze events in Suzhou, China. Atmos. Chem. Phys. 2016, 16, 7357–7371. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Hu, M.; Zamora, M.L.; Peng, J.; Shang, D.; Zheng, J.; Du, Z.; Wu, Z.; Shao, M.; Zeng, L.; et al. Elucidating severe urban haze formation in China. Proc. Natl. Acad. Sci. USA 2014, 111, 17373–17378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.K. Haze formation in China: Importance of secondary aerosol. J. Environ. Sci. 2015, 33, 261–262. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.J.; Duan, F.K.; Su, H.; Ma, Y.L.; Cheng, Y.; Zheng, B.; Zhang, Q.; Huang, T.; Kimoto, T.; Chang, D.; et al. Exploring the severe winter haze in Beijing: The impact of synoptic weather, regional transport and heterogeneous reactions. Atmos. Chem. Phys. 2015, 15, 2969–2983. [Google Scholar] [CrossRef]
- Han, T.; Liu, X.; Zhang, Y.; Qu, Y.; Zeng, L.; Hu, M.; Zhu, T. Role of secondary aerosols in haze formation in summer in the Megacity Beijing. J. Environ. Sci. 2015, 31, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Yang, L.-X.; Yi, H.; Yuan, Q.; Yan, C.; Dong, C.; Meng, C.-P.; Yao, L.; Yang, F.; Wang, W.-X. Influence of seasonal variation and long-range transport of carbonaceous aerosols on haze formation at a seaside background site, China. Aerosol Air Qual. Res. 2015, 15, 1251–1260. [Google Scholar] [CrossRef]
- Zhang, F.; Xu, L.; Chen, J.; Chen, X.; Niu, Z.; Lei, T.; Li, C.; Zhao, J. Chemical characteristics of PM2.5 during haze episodes in the urban of Fuzhou, China. Particuology 2013, 11, 264–272. [Google Scholar] [CrossRef]
- Ji, D.; Zhang, J.; He, J.; Wang, X.; Pang, B.; Liu, Z.; Wang, L.; Wang, Y. Characteristics of atmospheric organic and elemental carbon aerosols in urban Beijing, China. Atmos. Environ. 2016, 125 Pt A, 293–306. [Google Scholar] [CrossRef]
- Zhang, R.; Tao, J.; Ho, K.; Shen, Z.; Wang, G.; Cao, J.; Liu, S.; Zhang, L.; Lee, S. Characterization of atmospheric organic and elemental carbon of PM2. 5 in a typical semi-arid area of Northeastern China. Aerosol Air Qual. Res. 2012, 12, 792. [Google Scholar] [CrossRef]
- Niu, Z.; Zhang, F.; Chen, J.; Yin, L.; Wang, S.; Xu, L. Carbonaceous species in PM2.5 in the coastal urban agglomeration in the Western Taiwan Strait Region, China. Atmos. Res. 2013, 122, 102–110. [Google Scholar] [CrossRef]
- Li, W.; Bai, Z. Characteristics of organic and elemental carbon in atmospheric fine particles in Tianjin, China. Particuology 2009, 7, 432–437. [Google Scholar] [CrossRef]
- Lv, B.; Zhang, B.; Bai, Y. A systematic analysis of PM2.5 in Beijing and its sources from 2000 to 2012. Atmos. Environ. 2016, 124 Pt B, 98–108. [Google Scholar] [CrossRef]
- Zhou, J.; Xing, Z.; Deng, J.; Du, K. Characterizing and sourcing ambient PM2.5 over key emission regions in China I: Water-soluble ions and carbonaceous fractions. Atmos. Environ. 2016, 135, 20–30. [Google Scholar] [CrossRef]
- Gu, J.; Bai, Z.; Liu, A.; Wu, L.; Xie, Y.; Li, W.; Dong, H.; Zhang, X. Characterization of atmospheric organic carbon and element carbon of PM2.5 and PM10 at Tianjin, China. Aerosol Air Qual. Res 2010, 10, 167–176. [Google Scholar] [CrossRef]
- Cheng, Y.; He, K.-B.; Duan, F.-K.; Du, Z.-Y.; Zheng, M.; Ma, Y.-L. Characterization of carbonaceous aerosol by the stepwise-extraction thermal–optical-transmittance (SE-TOT) method. Atmos. Environ. 2012, 59, 551–558. [Google Scholar] [CrossRef]
- Yao, L.; Yang, L.; Chen, J.; Wang, X.; Xue, L.; Li, W.; Sui, X.; Wen, L.; Chi, J.; Zhu, Y.; et al. Characteristics of carbonaceous aerosols: Impact of biomass burning and secondary formation in summertime in a rural area of the North China Plain. Sci. Total Environ. 2016, 557–558, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chen, T.; Benjamin, G.; Helene, C.; Yu, T.; Liu, W.; Wang, X. Characteristics of carbonaceous particles in Beijing during winter and summer 2003. Adv. Atmos. Sci. 2006, 23, 468–473. [Google Scholar] [CrossRef]
- Lin, P.; Hu, M.; Deng, Z.; Slanina, J.; Han, S.; Kondo, Y.; Takegawa, N.; Miyazaki, Y.; Zhao, Y.; Sugimoto, N. Seasonal and diurnal variations of organic carbon in PM2.5 in Beijing and the estimation of secondary organic carbon. J. Geophys. Res.-Atmos. 2009, 114. [Google Scholar] [CrossRef]
- Cao, J.; Lee, S.; Chow, J.C.; Watson, J.G.; Ho, K.; Zhang, R.; Jin, Z.; Shen, Z.; Chen, G.; Kang, Y. Spatial and seasonal distributions of carbonaceous aerosols over China. J. Geophys. Res.-Atmos. 2007, 112. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Ho, K.F.; Lee, S.C.; Tsang, P.K.; Ho, S.S.H.; Zou, C.W.; Zou, S.C.; Cao, J.J.; Xu, H.M. Characteristics of carbonaceous aerosol in PM2.5: Pearl Delta River Region, China. Atmos. Res. 2012, 104–105, 227–236. [Google Scholar] [CrossRef]
- Feng, Y.; Chen, Y.; Guo, H.; Zhi, G.; Xiong, S.; Li, J.; Sheng, G.; Fu, J. Characteristics of organic and elemental carbon in PM2.5 samples in Shanghai, China. Atmos. Res. 2009, 92, 434–442. [Google Scholar] [CrossRef]
- Li, B.; Zhang, J.; Zhao, Y.; Yuan, S.; Zhao, Q.; Shen, G.; Wu, H. Seasonal variation of urban carbonaceous aerosols in a typical city Nanjing in Yangtze River Delta, China. Atmos. Environ. 2015, 106, 223–231. [Google Scholar] [CrossRef]
- Hu, W.; Hu, M.; Deng, Z.; Xiao, R.; Kondo, Y.; Takegawa, N.; Zhao, Y.; Guo, S.; Zhang, Y. The characteristics and origins of carbonaceous aerosol at a rural site of PRD in summer of 2006. Atmos. Chem. Phys. 2012, 12, 1811–1822. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Wang, T.; Wang, Z.; Li, W.; Xu, Z.; Wang, X.; Yuan, C.; Poon, C.N.; Louie, P.K.K.; Luk, C.W.Y.; et al. Photochemical evolution of organic aerosols observed in urban plumes from Hong Kong and the Pearl River Delta of China. Atmos. Environ. 2014, 88, 219–229. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, J.; Chen, J.; Xu, Y.; Xu, L. Pollution characteristics of organic and elemental carbon in PM2.5 in Xiamen, China. J. Environ. Sci. 2011, 23, 1342–1349. [Google Scholar] [CrossRef]
- Tao, J.; Ho, K.-F.; Chen, L.; Zhu, L.; Han, J.; Xu, Z. Effect of chemical composition of PM2.5 on visibility in Guangzhou, China, 2007 spring. Particuology 2009, 7, 68–75. [Google Scholar] [CrossRef]
- Fan, X.; Song, J.; Peng, P.A. Temporal variations of the abundance and optical properties of water soluble Humic-Like Substances (HULIS) in PM2.5 at Guangzhou, China. Atmos. Res. 2016, 172–173, 8–15. [Google Scholar] [CrossRef]
- Lai, S.; Zhao, Y.; Ding, A.; Zhang, Y.; Song, T.; Zheng, J.; Ho, K.F.; Lee, S.-C.; Zhong, L. Characterization of PM2.5 and the major chemical components during a 1-year campaign in rural Guangzhou, Southern China. Atmos. Res. 2016, 167, 208–215. [Google Scholar] [CrossRef]
- Qiao, T.; Zhao, M.; Xiu, G.; Yu, J. Simultaneous monitoring and compositions analysis of PM1 and PM2.5 in Shanghai: Implications for characterization of haze pollution and source apportionment. Sci. Total Environ. 2016, 557–558, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Guo, Z.; Lin, T.; Rose, N.L. Seasonal variation of carbonaceous pollutants in PM2.5 at an urban ‘supersite’ in Shanghai, China. Chemosphere 2016, 146, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Tan, J.; Cheng, D.; Bi, X.; Deng, W.; Sheng, G.; Fu, J.; Wong, M. Sources and characteristics of carbonaceous aerosol in two largest cities in Pearl River Delta Region, China. Atmos. Environ. 2007, 41, 2895–2903. [Google Scholar] [CrossRef]
- Wu, C.; Yu, J.Z. Determination of primary combustion source organic carbon-to-elemental carbon (OC/EC) ratio using ambient OC and EC measurements: Secondary OC-EC correlation minimization method. Atmos. Chem. Phys. 2016, 16, 5453–5465. [Google Scholar] [CrossRef]
- Ding, X.; Wang, X.-M.; Gao, B.; Fu, X.-X.; He, Q.-F.; Zhao, X.-Y.; Yu, J.-Z.; Zheng, M. Tracer-based estimation of secondary organic carbon in the Pearl River Delta, south China. J. Geophys. Res. 2012, 117. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Li, M.; Zhang, P.; Gong, S.; Zhong, M.; Wu, M.; Zheng, M.; Chen, C.; Wang, H.; Lou, S. Investigation of the sources and seasonal variations of secondary organic aerosols in PM2.5 in Shanghai with organic tracers. Atmos. Environ. 2013, 79, 614–622. [Google Scholar] [CrossRef]
- Chen, Y.; Xie, S.; Luo, B.; Zhai, C. Characteristics and origins of carbonaceous aerosol in the Sichuan Basin, China. Atmos. Environ. 2014, 94, 215–223. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, T.; Guo, J.; Gao, R.; Xue, L.; Zhang, J.; Zhou, Y.; Zhou, X.; Zhang, Q.; Wang, W. Formation of secondary organic carbon and cloud impact on carbonaceous aerosols at Mount Tai, North China. Atmos. Environ. 2012, 46, 516–527. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, Z.; Gao, R.; Xue, L.; Yuan, C.; Wang, T.; Gao, X.; Wang, X.; Nie, W.; Xu, Z.; et al. Formation of secondary organic carbon and long-range transport of carbonaceous aerosols at Mount Heng in South China. Atmos. Environ. 2012, 63, 203–212. [Google Scholar] [CrossRef]
- Kumar, A.; Ram, K.; Ojha, N. Variations in carbonaceous species at a high-altitude site in western India: Role of synoptic scale transport. Atmos. Environ. 2016, 125 Pt B, 371–382. [Google Scholar] [CrossRef]
- Sudheer, A.K.; Rengarajan, R.; Sheel, V. Secondary organic aerosol over an urban environment in a semi–arid region of western India. Atmos. Pollut. Res. 2015, 6, 11–20. [Google Scholar] [CrossRef]
- Pant, P.; Shukla, A.; Kohl, S.D.; Chow, J.C.; Watson, J.G.; Harrison, R.M. Characterization of ambient PM2.5 at a pollution hotspot in New Delhi, India and inference of sources. Atmos. Environ. 2015, 109, 178–189. [Google Scholar] [CrossRef]
- Pipal, A.S.; Gursumeeran Satsangi, P. Study of carbonaceous species, morphology and sources of fine (PM2.5) and coarse (PM10) particles along with their climatic nature in India. Atmos. Res. 2015, 154, 103–115. [Google Scholar] [CrossRef]
- Safai, P.D.; Raju, M.P.; Rao, P.S.P.; Pandithurai, G. Characterization of carbonaceous aerosols over the urban tropical location and a new approach to evaluate their climatic importance. Atmos. Environ. 2014, 92, 493–500. [Google Scholar] [CrossRef]
- Hooda, R.K.; Hyvärinen, A.P.; Vestenius, M.; Gilardoni, S.; Sharma, V.P.; Vignati, E.; Kulmala, M.; Lihavainen, H. Atmospheric aerosols local–regional discrimination for a semi-urban area in India. Atmos. Res. 2016, 168, 13–23. [Google Scholar] [CrossRef]
- Joseph, A.E.; Unnikrishnan, S.; Kumar, R. Chemical characterization and mass closure of fine aerosol for different land use patterns in Mumbai city. Aerosol Air Qual. Res. 2012, 12, 61–72. [Google Scholar] [CrossRef]
- Rengarajan, R.; Sudheer, A.; Sarin, M. Aerosol acidity and secondary organic aerosol formation during wintertime over urban environment in western India. Atmos. Environ. 2011, 45, 1940–1945. [Google Scholar] [CrossRef]
- Watson, J.G.; Cooper, J.A.; Huntzicker, J.J. The effective variance weighting for least squares calculations applied to the mass balance receptor model. Atmos. Environ. 1984, 18, 1347–1355. [Google Scholar] [CrossRef]
- Watson, J.G.; Zhu, T.; Chow, J.C.; Engelbrecht, J.; Fujita, E.M.; Wilson, W.E. Receptor modeling application framework for particle source apportionment. Chemosphere 2002, 49, 1093–1136. [Google Scholar] [CrossRef]
- Schauer, J.J.; Rogge, W.F.; Hildemann, L.M.; Mazurek, M.A.; Cass, G.R.; Simoneit, B.R. Source apportionment of airborne particulate matter using organic compounds as tracers. Atmos. Environ. 1996, 30, 3837–3855. [Google Scholar] [CrossRef]
- Li, A.; Jang, J.-K.; Scheff, P.A. Application of EPA CMB8.2 model for source apportionment of sediment PAHs in Lake Calumet, Chicago. Environ. Sci. Technol. 2003, 37, 2958–2965. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.G.; Chow, J.C.; Fujita, E.M. Review of volatile organic compound source apportionment by chemical mass balance. Atmos. Environ. 2001, 35, 1567–1584. [Google Scholar] [CrossRef]
- Bullock, K.R.; Duvall, R.M.; Norris, G.A.; McDow, S.R.; Hays, M.D. Evaluation of the CMB and PMF models using organic molecular markers in fine particulate matter collected during the Pittsburgh Air Quality Study. Atmos. Environ. 2008, 42, 6897–6904. [Google Scholar] [CrossRef]
- Stone, E.A.; Zhou, J.; Snyder, D.C.; Rutter, A.P.; Mieritz, M.; Schauer, J.J. A comparison of summertime secondary organic aerosol source contributions at contrasting urban locations. Environ. Sci. Technol. 2009, 43, 3448–3454. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Hu, M.; Guo, Q.; Zhang, X.; Zheng, M.; Zheng, J.; Chang, C.C.; Schauer, J.J.; Zhang, R. Primary sources and secondary formation of organic aerosols in Beijing, China. Environ. Sci. Technol. 2012, 46, 9846–9853. [Google Scholar] [CrossRef] [PubMed]
- Simoneit, B.R.; Schauer, J.J.; Nolte, C.; Oros, D.R.; Elias, V.O.; Fraser, M.; Rogge, W.; Cass, G.R. Levoglucosan, a tracer for cellulose in biomass burning and atmospheric particles. Atmos. Environ. 1999, 33, 173–182. [Google Scholar] [CrossRef]
- Hennigan, C.J.; Sullivan, A.P.; Collett, J.L.; Robinson, A.L. Levoglucosan stability in biomass burning particles exposed to hydroxyl radicals. Geophys. Res. Lett. 2010, 37. [Google Scholar] [CrossRef] [Green Version]
- Mochida, M.; Kawamura, K.; Fu, P.; Takemura, T. Seasonal variation of levoglucosan in aerosols over the western North Pacific and its assessment as a biomass-burning tracer. Atmos. Environ. 2010, 44, 3511–3518. [Google Scholar] [CrossRef] [Green Version]
- Kessler, S.H.; Smith, J.D.; Che, D.L.; Worsnop, D.R.; Wilson, K.R.; Kroll, J.H. Chemical sinks of organic aerosol: Kinetics and products of the heterogeneous oxidation of erythritol and levoglucosan. Environ. Sci. Technol. 2010, 44, 7005–7010. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Mungall, E.L.; Lee, A.K.Y.; Aljawhary, D.; Abbatt, J.P.D. Aqueous-phase photooxidation of levoglucosanndash; a mechanistic study using aerosol time-of-flight chemical ionization mass spectrometry (Aerosol ToF-CIMS). Atmos. Chem. Phys. 2014, 14, 9695–9706. [Google Scholar] [CrossRef]
- Chen, L.-W.A.; Watson, J.G.; Chow, J.C.; DuBois, D.W.; Herschberger, L. Chemical mass balance source apportionment for combined PM2.5 measurements from US non-urban and urban long-term networks. Atmos. Environ. 2010, 44, 4908–4918. [Google Scholar] [CrossRef]
- Lee, S.; Liu, W.; Wang, Y.; Russell, A.G.; Edgerton, E.S. Source apportionment of PM2.5: Comparing PMF and CMB results for four ambient monitoring sites in the southeastern United States. Atmos. Environ. 2008, 42, 4126–4137. [Google Scholar] [CrossRef]
- Zheng, M.; Zhao, X.; Cheng, Y.; Yan, C.; Shi, W.; Zhang, X.; Weber, R.J.; Schauer, J.J.; Wang, X.; Edgerton, E.S. Sources of primary and secondary organic aerosol and their diurnal variations. J. Hazard. Mater. 2014, 264, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, R.; Donahue, N.M.; Bernardo-Bricker, A.; Rogge, W.F.; Robinson, A.L. Insights into the primary–secondary and regional–local contributions to organic aerosol and PM2.5 mass in Pittsburgh, Pennsylvania. Atmos. Environ. 2007, 41, 7414–7433. [Google Scholar] [CrossRef]
- Ke, L.; Ding, X.; Tanner, R.L.; Schauer, J.J.; Zheng, M. Source contributions to carbonaceous aerosols in the Tennessee Valley Region. Atmos. Environ. 2007, 41, 8898–8923. [Google Scholar] [CrossRef]
- Sheesley, R.J.; Schauer, J.J.; Zheng, M.; Wang, B. Sensitivity of molecular marker-based CMB models to biomass burning source profiles. Atmos. Environ. 2007, 41, 9050–9063. [Google Scholar] [CrossRef]
- Zheng, M.; Cass, G.R.; Ke, L.; Wang, F.; Schauer, J.J.; Edgerton, E.S.; Russell, A.G. Source apportionment of daily fine particulate matter at Jefferson Street, Atlanta, GA, during summer and winter. J. Air Waste Manag. Assoc. 2007, 57, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Subramoney, P.; Karnae, S.; Farooqui, Z.; John, K.; Gupta, A.K. Identification of PM2.5 sources affecting a semi-arid coastal region using a chemical mass balance model. Aerosol Air Qual. Res. 2013, 13, 60–71. [Google Scholar] [CrossRef]
- Shirmohammadi, F.; Hasheminassab, S.; Saffari, A.; Schauer, J.J.; Delfino, R.J.; Sioutas, C. Fine and ultrafine particulate organic carbon in the Los Angeles basin: Trends in sources and composition. Sci. Total Environ. 2016, 541, 1083–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minguillón, M.C.; Arhami, M.; Schauer, J.J.; Sioutas, C. Seasonal and spatial variations of sources of fine and quasi-ultrafine particulate matter in neighborhoods near the Los Angeles–Long Beach harbor. Atmos. Environ. 2008, 42, 7317–7328. [Google Scholar] [CrossRef]
- Heo, J.; Dulger, M.; Olson, M.R.; McGinnis, J.E.; Shelton, B.R.; Matsunaga, A.; Sioutas, C.; Schauer, J.J. Source apportionments of PM2.5 organic carbon using molecular marker Positive Matrix Factorization and comparison of results from different receptor models. Atmos. Environ. 2013, 73, 51–61. [Google Scholar] [CrossRef]
- Hasheminassab, S.; Daher, N.; Schauer, J.J.; Sioutas, C. Source apportionment and organic compound characterization of ambient ultrafine particulate matter (PM) in the Los Angeles Basin. Atmos. Environ. 2013, 79, 529–539. [Google Scholar] [CrossRef]
- Ding, X.; Zheng, M.; Yu, L.; Zhang, X.; Weber, R.J.; Yan, B.; Russell, A.G.; Edgerton, E.S.; Wang, X. Spatial and seasonal trends in biogenic secondary organic aerosol tracers and water-soluble organic carbon in the Southeastern United States. Environ. Sci. Technol. 2008, 42, 5171–5176. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Pye, H.O.T.; He, J.; Chen, Y.; Murphy, B.N.; Ng, N.L. Large contributions from biogenic monoterpenes and sesquiterpenes to organic aerosol in the Southeastern United States. Atmos. Chem. Phys. Discuss. 2018, 2018, 1–47. [Google Scholar] [CrossRef]
- Sareen, N.; Waxman, E.M.; Turpin, B.J.; Volkamer, R.; Carlton, A.G. Potential of aerosol liquid water to facilitate organic aerosol formation: Assessing knowledge gaps about precursors and partitioning. Environ. Sci. Technol. 2017, 51, 3327–3335. [Google Scholar] [CrossRef] [PubMed]
- Carlton, A.; Jimenez, J.; Ambrose, J.; Brown, S.; Baker, K.; Brock, C.; Cohen, R.; Edgerton, S.; Farkas, C.; Farmer, D.; et al. The Southeast Atmosphere Studies (SAS): Coordinated investigation and discovery to answer critical questions about fundamental atmospheric processes. Bull. Am. Meteorol. Soc. 2018, 99, 547–567. [Google Scholar] [CrossRef]
- Zhang, H.; Yee, L.D.; Lee, B.H.; Curtis, M.P.; Worton, D.R.; Isaacman-VanWertz, G.; Offenberg, J.H.; Lewandowski, M.; Kleindienst, T.E.; Beaver, M.R.; et al. Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States. Proc. Natl. Acad. Sci. USA 2018, 115, 2038–2043. [Google Scholar] [CrossRef] [PubMed]
- El Haddad, I.; Marchand, N.; Dron, J.; Temime-Roussel, B.; Quivet, E.; Wortham, H.; Jaffrezo, J.L.; Baduel, C.; Voisin, D.; Besombes, J.L.; et al. Comprehensive primary particulate organic characterization of vehicular exhaust emissions in France. Atmos. Environ. 2009, 43, 6190–6198. [Google Scholar] [CrossRef]
- Favez, O.; El Haddad, I.; Piot, C.; Boréave, A.; Abidi, E.; Marchand, N.; Jaffrezo, J.L.; Besombes, J.L.; Personnaz, M.B.; Sciare, J.; et al. Inter-comparison of source apportionment models for the estimation of wood burning aerosols during wintertime in an Alpine city (Grenoble, France). Atmos. Chem. Phys. 2010, 10, 5295–5314. [Google Scholar] [CrossRef] [Green Version]
- Perrone, M.G.; Larsen, B.R.; Ferrero, L.; Sangiorgi, G.; De Gennaro, G.; Udisti, R.; Zangrando, R.; Gambaro, A.; Bolzacchini, E. Sources of high PM2.5 concentrations in Milan, Northern Italy: Molecular marker data and CMB modelling. Sci. Total Environ. 2012, 414, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Pirovano, G.; Colombi, C.; Balzarini, A.; Riva, G.; Gianelle, V.; Lonati, G. PM2.5 source apportionment in Lombardy (Italy): Comparison of receptor and chemistry-transport modelling results. Atmos. Environ. 2015, 106, 56–70. [Google Scholar] [CrossRef]
- Yin, J.; Harrison, R.M.; Chen, Q.; Rutter, A.; Schauer, J.J. Source apportionment of fine particles at urban background and rural sites in the UK atmosphere. Atmos. Environ. 2010, 44, 841–851. [Google Scholar] [CrossRef]
- Daher, N.; Ruprecht, A.; Invernizzi, G.; De Marco, C.; Miller-Schulze, J.; Heo, J.B.; Shafer, M.M.; Shelton, B.R.; Schauer, J.J.; Sioutas, C. Characterization, sources and redox activity of fine and coarse particulate matter in Milan, Italy. Atmos. Environ. 2012, 49, 130–141. [Google Scholar] [CrossRef]
- von Schneidemesser, E.; Schauer, J.J.; Hagler, G.S.W.; Bergin, M.H. Concentrations and sources of carbonaceous aerosol in the atmosphere of Summit, Greenland. Atmos. Environ. 2009, 43, 4155–4162. [Google Scholar] [CrossRef]
- von Schneidemesser, E.; Zhou, J.; Stone, E.A.; Schauer, J.J.; Qasrawi, R.; Abdeen, Z.; Shpund, J.; Vanger, A.; Sharf, G.; Moise, T.; et al. Seasonal and spatial trends in the sources of fine particle organic carbon in Israel, Jordan, and Palestine. Atmos. Environ. 2010, 44, 3669–3678. [Google Scholar] [CrossRef]
- von Schneidemesser, E.; Zhou, I.; Stone, E.A.; Schauer, J.I.; Shpund, J.; Brenner, S.; Qasrawi, R.; Abdeen, Z.; Sarnat, J.A. Spatial variability of carbonaceous aerosol concentrations in East and West Jerusalem. Environ. Sci. Technol. 2010, 44, 1911–1917. [Google Scholar] [CrossRef] [PubMed]
- Hamad, S.H.; Schauer, J.J.; Heo, J.; Kadhim, A.K.H. Source apportionment of PM2.5 carbonaceous aerosol in Baghdad, Iraq. Atmos. Res. 2015, 156, 80–90. [Google Scholar] [CrossRef]
- Shi, G.-L.; Tian, Y.-Z.; Zhang, Y.-F.; Ye, W.-Y.; Li, X.; Tie, X.-X.; Feng, Y.-C.; Zhu, T. Estimation of the concentrations of primary and secondary organic carbon in ambient particulate matter: Application of the CMB-Iteration method. Atmos. Environ. 2011, 45, 5692–5698. [Google Scholar] [CrossRef]
- Zheng, M.; Wang, F.; Hagler, G.S.W.; Hou, X.; Bergin, M.; Cheng, Y.; Salmon, L.G.; Schauer, J.J.; Louie, P.K.K.; Zeng, L.; et al. Sources of excess urban carbonaceous aerosol in the Pearl River Delta Region, China. Atmos. Environ. 2011, 45, 1175–1182. [Google Scholar] [CrossRef]
- Huang, L.; Wang, G. Chemical characteristics and source apportionment of atmospheric particles during heating period in Harbin, China. J. Environ. Sci. 2014, 26, 2475–2483. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y.-F.; Han, S.-Q.; Wu, J.-H.; Bi, X.-H.; Shi, G.-L.; Wang, J.; Yao, Q.; Cai, Z.-Y.; Liu, J.-L.; et al. Vertical characteristics of PM2.5 during the heating season in Tianjin, China. Sci. Total Environ. 2015, 523, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-C.; Yu, J.Z.; Ho, S.S.H.; Schauer, J.J.; Yuan, Z.; Lau, A.K.H.; Louie, P.K.K. Chemical characteristics and source apportionment of fine particulate organic carbon in Hong Kong during high particulate matter episodes in winter 2003. Atmos. Res. 2013, 120–121, 88–98. [Google Scholar] [CrossRef]
- Wang, J.; Ho, S.S.H.; Ma, S.; Cao, J.; Dai, W.; Liu, S.; Shen, Z.; Huang, R.; Wang, G.; Han, Y. Characterization of PM2.5 in Guangzhou, China: Uses of organic markers for supporting source apportionment. Sci. Total Environ. 2016, 550, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-C.; Yu, J.Z.; Ho, S.S.H.; Yuan, Z.; Lau, A.K.H.; Huang, X.-F. Chemical characteristics of PM2.5 and organic aerosol source analysis during cold front episodes in Hong Kong, China. Atmos. Res. 2012, 118, 41–51. [Google Scholar] [CrossRef]
- Stone, E.; Schauer, J.; Quraishi, T.A.; Mahmood, A. Chemical characterization and source apportionment of fine and coarse particulate matter in Lahore, Pakistan. Atmos. Environ. 2010, 44, 1062–1070. [Google Scholar] [CrossRef]
- Villalobos, A.M.; Amonov, M.O.; Shafer, M.M.; Devi, J.J.; Gupta, T.; Tripathi, S.N.; Rana, K.S.; McKenzie, M.; Bergin, M.H.; Schauer, J.J. Source apportionment of carbonaceous fine particulate matter (PM2.5) in two contrasting cities across the Indo–Gangetic Plain. Atmos. Pollut. Res. 2015, 6, 398–405. [Google Scholar] [CrossRef]
- Miller-Schulze, J.P.; Shafer, M.M.; Schauer, J.J.; Solomon, P.A.; Lantz, J.; Artamonova, M.; Chen, B.; Imashev, S.; Sverdlik, L.; Carmichael, G.R.; et al. Characteristics of fine particle carbonaceous aerosol at two remote sites in Central Asia. Atmos. Environ. 2011, 45, 6955–6964. [Google Scholar] [CrossRef]
- Lee, H.; Park, S.S.; Kim, K.W.; Kim, Y.J. Source identification of PM2.5 particles measured in Gwangju, Korea. Atmos. Res. 2008, 88, 199–211. [Google Scholar] [CrossRef]
- Kong, S.; Han, B.; Bai, Z.; Chen, L.; Shi, J.; Xu, Z. Receptor modeling of PM2.5, PM10 and TSP in different seasons and long-range transport analysis at a coastal site of Tianjin, China. Sci. Total Environ. 2010, 408, 4681–4694. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Hagler, G.S.; Ke, L.; Bergin, M.H.; Wang, F.; Louie, P.K.; Salmon, L.; Sin, D.W.; Yu, J.Z.; Schauer, J.J. Composition and sources of carbonaceous aerosols at three contrasting sites in Hong Kong. J. Geophys. Res.-Atmos. 2006, 111. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Ke, L.; Edgerton, E.S.; Schauer, J.J.; Dong, M.; Russell, A.G. Spatial distribution of carbonaceous aerosol in the southeastern United States using molecular markers and carbon isotope data. J. Geophys. Res.-Atmos. 2006, 111. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.; Liu, Y.; Ma, J.; Ma, Q.; Chu, B.; He, H. Heterogeneous kinetics of cis-pinonic acid with hydroxyl radical under different environmental conditions. J. Phys. Chem. A 2015, 119, 6583–6593. [Google Scholar] [CrossRef] [PubMed]
- Kostenidou, E.; Karnezi, E.; Kolodziejczyk, A.; Szmigielski, R.; Pandis, S. Physical and chemical properties of 3-methyl-1,2,3-butanetricarboxylic acid (MBTCA) aerosol. Environ. Sci. Technol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jaoui, M.; Kleindienst, T.E.; Lewandowski, M.; Offenberg, J.H.; Edney, E.O. Identification and quantification of aerosol polar oxygenated compounds bearing carboxylic or hydroxyl groups. 2. Organic tracer compounds from monoterpenes. Environ. Sci. Technol. 2005, 39, 5661–5673. [Google Scholar] [CrossRef] [PubMed]
- Martín-Reviejo, M.; Wirtz, K. Is benzene a precursor for secondary organic aerosol? Environ. Sci. Technol. 2005, 39, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Zhao, Y.; Chuang, W.K.; Robinson, A.L.; Donahue, N.M. Secondary organic aerosol production from pinanediol, a semi-volatile surrogate for first-generation oxidation products of monoterpenes. Atmos. Chem. Phys. Discuss. 2017, 2017, 1–46. [Google Scholar] [CrossRef]
- Lim, Y.B.; Ziemann, P.J. Products and mechanism of secondary organic aerosol formation from reactions of n-alkanes with OH radicals in the presence of NOx. Environ. Sci. Technol. 2005, 39, 9229–9236. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.B.; Ziemann, P.J. Effects of molecular structure on aerosol yields from OH radical-initiated reactions of linear, branched, and cyclic alkanes in the presence of NOx. Environ. Sci. Technol. 2009, 43, 2328–2334. [Google Scholar] [CrossRef] [PubMed]
- Shakya, K.M.; Griffin, R.J. Secondary organic aerosol from photooxidation of polycyclic aromatic hydrocarbons. Environ. Sci. Technol. 2010, 44, 8134–8139. [Google Scholar] [CrossRef] [PubMed]
- Kleindienst, T.E.; Jaoui, M.; Lewandowski, M.; Offenberg, J.H.; Docherty, K.S. The formation of SOA and chemical tracer compounds from the photooxidation of naphthalene and its methyl analogs in the presence and absence of nitrogen oxides. Atmos. Chem. Phys. 2012, 12, 8711–8726. [Google Scholar] [CrossRef] [Green Version]
- Riva, M.; Tomaz, S.; Cui, T.; Lin, Y.H.; Perraudin, E.; Gold, A.; Stone, E.A.; Villenave, E.; Surratt, J.D. Evidence for an unrecognized secondary anthropogenic source of organosulfates and sulfonates: Gas-phase oxidation of polycyclic aromatic hydrocarbons in the presence of sulfate aerosol. Environ. Sci. Technol. 2015, 49, 6654–6664. [Google Scholar] [CrossRef] [PubMed]
- Lamkaddam, H.; Gratien, A.; Pangui, E.; Cazaunau, M.; Picquet-Varrault, B.; Doussin, J.-F. High-NOx photooxidation of n-dodecane: Temperature dependence of SOA formation. Environ. Sci. Technol. 2017, 51, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Schilling Fahnestock, K.A.; Yee, L.D.; Loza, C.L.; Coggon, M.M.; Schwantes, R.; Zhang, X.; Dalleska, N.F.; Seinfeld, J.H. Secondary Organic Aerosol Composition from C12 Alkanes. J. Phys. Chem. A 2015, 119, 4281–4297. [Google Scholar] [CrossRef] [PubMed]
- Iinuma, Y.; Boge, O.; Grafe, R.; Herrmann, H. Methyl-nitrocatechols: Atmospheric tracer compounds for biomass burning secondary organic aerosols. Environ. Sci. Technol. 2010, 44, 8453–8459. [Google Scholar] [CrossRef] [PubMed]
- Surratt, J.D.; Lewandowski, M.; Offenberg, J.H.; Jaoui, M.; Kleindienst, T.E.; Edney, E.O.; Seinfeld, J.H. Effect of acidity on secondary organic aerosol formation from isoprene. Environ. Sci. Technol. 2007, 41, 5363–5369. [Google Scholar] [CrossRef] [PubMed]
- Iinuma, Y.; Müller, C.; Berndt, T.; Böge, O.; Claeys, M.; Herrmann, H. Evidence for the existence of organosulfates from β-pinene ozonolysis in ambient secondary organic aerosol. Environ. Sci. Technol. 2007, 41, 6678–6683. [Google Scholar] [CrossRef] [PubMed]
- Kiendler-Scharr, A.; Mensah, A.A.; Friese, E.; Topping, D.; Nemitz, E.; Prevot, A.S.H.; Äijälä, M.; Allan, J.; Canonaco, F.; Canagaratna, M.; et al. Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol. Geophys. Res. Lett. 2016, 43, 7735–7744. [Google Scholar] [CrossRef] [Green Version]
- Carlton, A.G.; Turpin, B.J.; Altieri, K.E.; Seitzinger, S.; Reff, A.; Lim, H.-J.; Ervens, B. Atmospheric oxalic acid and SOA production from glyoxal: Results of aqueous photooxidation experiments. Atmos. Environ. 2007, 41, 7588–7602. [Google Scholar] [CrossRef]
- Carlton, A.G.; Turpin, B.J.; Lim, H.-J.; Altieri, K.E.; Seitzinger, S. Link between isoprene and secondary organic aerosol (SOA): Pyruvic acid oxidation yields low volatility organic acids in clouds. Geophys. Res. Lett. 2006, 33. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.B.; Tan, Y.; Turpin, B.J. Chemical insights, explicit chemistry, and yields of secondary organic aerosol from OH radical oxidation of methylglyoxal and glyoxal in the aqueous phase. Atmos. Chem. Phys. 2013, 13, 8651–8667. [Google Scholar] [CrossRef]
- Liu, Y.; Siekmann, F.; Renard, P.; El Zein, A.; Salque, G.; El Haddad, I.; Temime-Roussel, B.; Voisin, D.; Thissen, R.; Monod, A. Oligomer and SOA formation through aqueous phase photooxidation of methacrolein and methyl vinyl ketone. Atmos. Environ. 2012, 49, 123–129. [Google Scholar] [CrossRef]
- Perri, M.J.; Seitzinger, S.; Turpin, B.J. Secondary organic aerosol production from aqueous photooxidation of glycolaldehyde: Laboratory experiments. Atmos. Environ. 2009, 43, 1487–1497. [Google Scholar] [CrossRef]
- Ervens, B.; Turpin, B.J.; Weber, R.J. Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): A review of laboratory, field and model studies. Atmos. Chem. Phys. 2011, 11, 11069–11102. [Google Scholar] [CrossRef]
- Brégonzio-Rozier, L.; Giorio, C.; Siekmann, F.; Pangui, E.; Morales, S.B.; Temime-Roussel, B.; Gratien, A.; Michoud, V.; Cazaunau, M.; DeWitt, H.L.; et al. Secondary organic aerosol formation from isoprene photooxidation during cloud condensation–evaporation cycles. Atmos. Chem. Phys. 2016, 16, 1747–1760. [Google Scholar] [CrossRef]
- Brégonzio-Rozier, L.; Siekmann, F.; Giorio, C.; Pangui, E.; Morales, S.B.; Temime-Roussel, B.; Gratien, A.; Michoud, V.; Ravier, S.; Cazaunau, M.; et al. Gaseous products and secondary organic aerosol formation during long term oxidation of isoprene and methacrolein. Atmos. Chem. Phys. 2015, 15, 2953–2968. [Google Scholar] [CrossRef]
- Giorio, C.; Monod, A.; Brégonzio-Rozier, L.; DeWitt, H.L.; Cazaunau, M.; Temime-Roussel, B.; Gratien, A.; Michoud, V.; Pangui, E.; Ravier, S.; et al. Cloud processing of secondary organic aerosol from isoprene and methacrolein photooxidation. J. Phys. Chem. A 2017, 121, 7641–7654. [Google Scholar] [CrossRef] [PubMed]
- El Haddad, I.; Yao, L.; Nieto-Gligorovski, L.; Michaud, V.; Temime-Roussel, B.; Quivet, E.; Marchand, N.; Sellegri, K.; Monod, A. In-cloud processes of methacrolein under simulated conditions—Part 2: Formation of secondary organic aerosol. Atmos. Chem. Phys. 2009, 9, 5107–5117. [Google Scholar] [CrossRef]
- Lim, H.-J.; Carlton, A.G.; Turpin, B.J. Isoprene forms secondary organic aerosol through cloud processing: model simulations. Environ. Sci. Technol. 2005, 39, 4441–4446. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Bian, Q.; Li, T.W.Y.; Lau, A.K.H.; Yu, J.Z. Contributions of isoprene, monoterpenes, β-caryophyllene, and toluene to secondary organic aerosols in Hong Kong during the summer of 2006. J. Geophys. Res.-Atmos. 2008, 113, D22206. [Google Scholar] [CrossRef]
- Srivastava, D.; Favez, O.; Bonnaire, N.; Lucarelli, F.; Perraudin, E.; Gros, V.; Villenave, E.; Albinet, A. Speciation of organic fractions does matter for aerosol source apportionment. Part 2: Intensive campaign in the Paris area (France). Sci. Total Environ. 2018, 634, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Jayarathne, T.; Rathnayake, C.M.; Stone, E.A. Local source impacts on primary and secondary aerosols in the Midwestern United States. Atmos. Environ. 2016, 130, 74–83. [Google Scholar] [CrossRef]
- Stone, E.A.; Hedman, C.J.; Zhou, J.; Mieritz, M.; Schauer, J.J. Insights into the nature of secondary organic aerosol in Mexico City during the MILAGRO experiment 2006. Atmos. Environ. 2010, 44, 312–319. [Google Scholar] [CrossRef]
- Offenberg, J.H.; Lewis, C.W.; Lewandowski, M.; Jaoui, M.; Kleindienst, T.E.; Edney, E.O. Contributions of toluene and α-pinene to SOA formed in an irradiated toluene/α-pinene/NOx/air mixture: Comparison of results using 14C content and SOA organic tracer methods. Environ. Sci. Technol. 2007, 41, 3972–3976. [Google Scholar] [CrossRef] [PubMed]
- Offenberg, J.H. Contributions of Biogenic and Anthropogenic Hydrocarbons to Secondary Organic Aerosol during 2006 in Research Triangle Park, NC. Aerosol Air Qual. Res. 2011. [Google Scholar] [CrossRef] [Green Version]
- Lewandowski, M.; Piletic, I.R.; Kleindienst, T.E.; Offenberg, J.H.; Beaver, M.R.; Jaoui, M.; Docherty, K.S.; Edney, E.O. Secondary organic aerosol characterisation at field sites across the United States during the spring–summer period. Int. J. Environ. Anal. Chem. 2013, 93, 1084–1103. [Google Scholar] [CrossRef]
- Lewandowski, M.; Jaoui, M.; Offenberg, J.H.; Kleindienst, T.E.; Edney, E.O.; Sheesley, R.J.; Schauer, J.J. Primary and secondary contributions to ambient PM in the midwestern United States. Environ. Sci. Technol. 2008, 42, 3303–3309. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Kawamura, K.; Chen, J.; Barrie, L.A. Isoprene, monoterpene, and sesquiterpene oxidation products in the high Arctic aerosols during late winter to early summer. Environ. Sci. Technol. 2009, 43, 4022–4028. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.-H.; Xie, Z.-Q.; Wang, X.-M.; Kang, H.; He, Q.-F.; Zhang, P. Secondary organic aerosols over oceans via oxidation of isoprene and monoterpenes from Arctic to Antarctic. Sci. Rep. 2013, 3, 2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, P.; Kawamura, K.; Chen, J.; Charrière, B.; Sempéré, R. Organic molecular composition of marine aerosols over the Arctic Ocean in summer: Contributions of primary emission and secondary aerosol formation. Biogeosciences 2013, 10, 653–667. [Google Scholar] [CrossRef]
- Kourtchev, I.; Hellebust, S.; Bell, J.M.; O’Connor, I.P.; Healy, R.M.; Allanic, A.; Healy, D.; Wenger, J.C.; Sodeau, J.R. The use of polar organic compounds to estimate the contribution of domestic solid fuel combustion and biogenic sources to ambient levels of organic carbon and PM2.5 in Cork Harbour, Ireland. Sci. Total Environ. 2011, 409, 2143–2155. [Google Scholar] [CrossRef] [PubMed]
- Kourtchev, I.; Copolovici, L.; Claeys, M.; Maenhaut, W. Characterization of atmospheric aerosols at a forested site in Central Europe. Environ. Sci. Technol. 2009, 43, 4665–4671. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.Q.; Kawamura, K.; Pochanart, P.; Tanimoto, H.; Kanaya, Y.; Wang, Z.F. Summertime contributions of isoprene, monoterpenes, and sesquiterpene oxidation to the formation of secondary organic aerosol in the troposphere over Mt. Tai, Central East China during MTX2006. Atmos. Chem. Phys. Discuss. 2009, 9, 16941–16972. [Google Scholar] [CrossRef]
- Fu, P.; Aggarwal, S.G.; Chen, J.; Li, J.; Sun, Y.; Wang, Z.; Chen, H.; Liao, H.; Ding, A.; Umarji, G.S.; et al. Molecular markers of secondary organic aerosol in Mumbai, India. Environ. Sci. Technol. 2016, 50, 4659–4667. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Kawamura, K.; Chen, J.; Ho, K.; Lee, S.; Gao, Y.; Cui, L.; Wang, T.; Fu, P. Anthropogenic and biogenic organic compounds in summertime fine aerosols (PM2.5) in Beijing, China. Atmos. Environ. 2016, 124 Pt B, 166–175. [Google Scholar] [CrossRef]
- Fu, P.; Kawamura, K. Diurnal variations of polar organic tracers in summer forest aerosols: A case study of a Quercus and Picea mixed forest in Hokkaido, Japan. Geochem. J. 2011, 45, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; He, Q.F.; Shen, R.Q.; Yu, Q.Q.; Wang, X.M. Spatial distributions of secondary organic aerosols from isoprene, monoterpenes, β-caryophyllene, and aromatics over China during summer. J. Geophys. Res.-Atmos. 2014, 119. [Google Scholar] [CrossRef]
- Fu, P.; Kawamura, K.; Kanaya, Y.; Wang, Z. Contributions of biogenic volatile organic compounds to the formation of secondary organic aerosols over Mt. Tai, Central East China. Atmos. Environ. 2010, 44, 4817–4826. [Google Scholar] [CrossRef] [Green Version]
- Fu, P.Q.; Kawamura, K.; Chen, J.; Li, J.; Sun, Y.L.; Liu, Y.; Tachibana, E.; Aggarwal, S.G.; Okuzawa, K.; Tanimoto, H.; et al. Diurnal variations of organic molecular tracers and stable carbon isotopic composition in atmospheric aerosols over Mt. Tai in the North China Plain: An influence of biomass burning. Atmos. Chem. Phys. 2012, 12, 8359–8375. [Google Scholar] [CrossRef]
- Ding, X.; He, Q.-F.; Shen, R.-Q.; Yu, Q.-Q.; Zhang, Y.-Q.; Xin, J.-Y.; Wen, T.-X.; Wang, X.-M. Spatial and seasonal variations of isoprene secondary organic aerosol in China: Significant impact of biomass burning during winter. Sci. Rep. 2016, 6, 20411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hantson, S.; Knorr, W.; Schurgers, G.; Pugh, T.A.; Arneth, A. Global isoprene and monoterpene emissions under changing climate, vegetation, CO2 and land use. Atmos. Environ. 2017, 155, 35–45. [Google Scholar] [CrossRef]
- Heald, C.L.; Henze, D.K.; Horowitz, L.W.; Feddema, J.; Lamarque, J.F.; Guenther, A.; Hess, P.G.; Vitt, F.; Seinfeld, J.H.; Goldstein, A.H.; et al. Predicted change in global secondary organic aerosol concentrations in response to future climate, emissions, and land use change. J. Geophys. Res.-Atmos. 2008, 113. [Google Scholar] [CrossRef] [Green Version]
- Lathiere, J.; Hauglustaine, D.; Friend, A.; Noblet-Ducoudré, N.D.; Viovy, N.; Folberth, G. Impact of climate variability and land use changes on global biogenic volatile organic compound emissions. Atmos. Chem. Phys. 2006, 6, 2129–2146. [Google Scholar] [CrossRef] [Green Version]
- Budisulistiorini, S.; Li, X.; Bairai, S.; Renfro, J.; Liu, Y.; Liu, Y.; McKinney, K.; Martin, S.; McNeill, V.; Pye, H. Examining the effects of anthropogenic emissions on isoprene-derived secondary organic aerosol formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee ground site. Atmos. Chem. Phys. 2015, 15, 8871–8888. [Google Scholar] [CrossRef]
- Steinbrecher, R.; Smiatek, G.; Köble, R.; Seufert, G.; Theloke, J.; Hauff, K.; Ciccioli, P.; Vautard, R.; Curci, G. Intra- and inter-annual variability of VOC emissions from natural and semi-natural vegetation in Europe and neighbouring countries. Atmos. Environ. 2009, 43, 1380–1391. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, L.; Sun, Y.; Favez, O.; Canonaco, F.; Albinet, A.; Couvidat, F.; Liu, D.; Jayne, J.T.; Wang, Z.; et al. Limited formation of isoprene epoxydiols-derived secondary organic aerosol under NOx-rich environments in Eastern China. Geophys. Res. Lett. 2017, 44. [Google Scholar] [CrossRef]
- Polissar, A.V.; Hopke, P.K.; Paatero, P.; Malm, W.C.; Sisler, J.F. Atmospheric aerosol over Alaska: 2. Elemental composition and sources. J. Geophys. Res.-Atmos. 1998, 103, 19045–19057. [Google Scholar] [CrossRef] [Green Version]
- Gianini, M.F.D.; Fischer, A.; Gehrig, R.; Ulrich, A.; Wichser, A.; Piot, C.; Besombes, J.L.; Hueglin, C. Comparative source apportionment of PM10 in Switzerland for 2008/2009 and 1998/1999 by Positive Matrix Factorisation. Atmos. Environ. 2012, 54, 149–158. [Google Scholar] [CrossRef]
- Sara, C.; Luisa, C.; Bernd, G. Positive Matrix Factorisation (PMF)—An introduction to the chemometric evaluation of environmental monitoring data using PMF. Inst. Environ. Sustain. 2009. [CrossRef]
- Yuan, Z.; Lau, A.K.H.; Zhang, H.; Yu, J.Z.; Louie, P.K.; Fung, J.C. Identification and spatiotemporal variations of dominant PM10 sources over Hong Kong. Atmos. Environ. 2006, 40, 1803–1815. [Google Scholar] [CrossRef]
- Ke, L.; Liu, W.; Wang, Y.; Russell, A.G.; Edgerton, E.S.; Zheng, M. Comparison of PM2.5 source apportionment using positive matrix factorization and molecular marker-based chemical mass balance. Sci. Total Environ. 2008, 394, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hopke, P.K.; Xia, X.; Rattigan, O.V.; Chalupa, D.C.; Utell, M.J. Source apportionment of airborne particulate matter using inorganic and organic species as tracers. Atmos. Environ. 2012, 55, 525–532. [Google Scholar] [CrossRef]
- Jaeckels, J.M.; Bae, M.-S.; Schauer, J.J. Positive matrix factorization (PMF) analysis of molecular marker measurements to quantify the sources of organic aerosols. Environ. Sci. Technol. 2007, 41, 5763–5769. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Fu, P.Q.; Kawamura, K.; Mizoguchi, Y.; Yamanoi, K. Seasonal variations of stable carbon isotopic composition and biogenic tracer compounds of water-soluble organic aerosols in a deciduous forest. Atmos. Chem. Phys. 2012, 12, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Worsnop, D.; Canagaratna, M.; Jimenez, J. Hydrocarbon-like and oxygenated organic aerosols in Pittsburgh: Insights into sources and processes of organic aerosols. Atmos. Chem. Phys. 2005, 5, 3289–3311. [Google Scholar] [CrossRef]
- Allan, J.D.; Delia, A.E.; Coe, H.; Bower, K.N.; Alfarra, M.R.; Jimenez, J.L.; Middlebrook, A.M.; Drewnick, F.; Onasch, T.B.; Canagaratna, M.R. A generalised method for the extraction of chemically resolved mass spectra from Aerodyne aerosol mass spectrometer data. J. Aerosol Sci. 2004, 35, 909–922. [Google Scholar] [CrossRef]
- Jimenez, J.L.; Jayne, J.T.; Shi, Q.; Kolb, C.E.; Worsnop, D.R.; Yourshaw, I.; Seinfeld, J.H.; Flagan, R.C.; Zhang, X.; Smith, K.A. Ambient aerosol sampling using the aerodyne aerosol mass spectrometer. J. Geophys. Res.-Atmos. 2003, 108. [Google Scholar] [CrossRef]
- Zhang, Q.; Jimenez, J.L.; Canagaratna, M.R.; Ulbrich, I.M.; Ng, N.L.; Worsnop, D.R.; Sun, Y. Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: A review. Anal. Bioanal. Chem. 2011, 401, 3045–3067. [Google Scholar] [CrossRef] [PubMed]
- Ng, N.L.; Canagaratna, M.R.; Zhang, Q.; Jimenez, J.L.; Tian, J.; Ulbrich, I.M.; Kroll, J.H.; Docherty, K.S.; Chhabra, P.S.; Bahreini, R.; et al. Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry. Atmos. Chem. Phys. 2010, 10, 4625–4641. [Google Scholar] [CrossRef]
- Ng, N.L.; Herndon, S.C.; Trimborn, A.; Canagaratna, M.R.; Croteau, P.L.; Onasch, T.B.; Sueper, D.; Worsnop, D.R.; Zhang, Q.; Sun, Y.L.; et al. An aerosol chemical speciation monitor (ACSM) for routine monitoring of the composition and mass concentrations of ambient aerosol. Aerosol Sci. Technol. 2011, 45, 780–794. [Google Scholar] [CrossRef]
- Petit, J.E.; Favez, O.; Sciare, J.; Crenn, V.; Sarda-Estève, R.; Bonnaire, N.; Močnik, G.; Dupont, J.C.; Haeffelin, M.; Leoz-Garziandia, E. Two years of near real-time chemical composition of submicron aerosols in the region of Paris using an Aerosol Chemical Speciation Monitor (ACSM) and a multi-wavelength Aethalometer. Atmos. Chem. Phys. 2015, 15, 2985–3005. [Google Scholar] [CrossRef]
- Petit, J.-E.; Amodeo, T.; Meleux, F.; Bessagnet, B.; Menut, L.; Grenier, D.; Pellan, Y.; Ockler, A.; Rocq, B.; Gros, V. Characterising an intense PM pollution episode in March 2015 in France from multi-site approach and near real time data: Climatology, variabilities, geographical origins and model evaluation. Atmos. Environ. 2017, 155, 68–84. [Google Scholar] [CrossRef]
- Fröhlich, R.; Cubison, M.J.; Slowik, J.G.; Bukowiecki, N.; Prévôt, A.S.H.; Baltensperger, U.; Schneider, J.; Kimmel, J.R.; Gonin, M.; Rohner, U.; et al. The ToF-ACSM: A portable aerosol chemical speciation monitor with TOFMS detection. Atmos. Meas. Tech. 2013, 6, 3225–3241. [Google Scholar] [CrossRef]
- Fröhlich, R.; Cubison, M.J.; Slowik, J.G.; Bukowiecki, N.; Canonaco, F.; Croteau, P.L.; Gysel, M.; Henne, S.; Herrmann, E.; Jayne, J.T.; et al. Fourteen months of on-line measurements of the non-refractory submicron aerosol at the Jungfraujoch (3580 m a.s.l.)—Chemical composition, origins and organic aerosol sources. Atmos. Chem. Phys. 2015, 15, 11373–11398. [Google Scholar] [CrossRef]
- Budisulistiorini, S.H.; Canagaratna, M.R.; Croteau, P.L.; Marth, W.J.; Baumann, K.; Edgerton, E.S.; Shaw, S.L.; Knipping, E.M.; Worsnop, D.R.; Jayne, J.T.; et al. Real-time continuous characterization of secondary organic aerosol derived from isoprene epoxydiols in Downtown Atlanta, Georgia, using the Aerodyne Aerosol Chemical Speciation Monitor. Environ. Sci. Technol. 2013, 47, 5686–5694. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.W.; Campuzano-Jost, P.; Palm, B.B.; Day, D.A.; Ortega, A.M.; Hayes, P.L.; Krechmer, J.E.; Chen, Q.; Kuwata, M.; Liu, Y.J.; et al. Characterization of a real-time tracer for isoprene epoxydiols-derived secondary organic aerosol (IEPOX-SOA) from aerosol mass spectrometer measurements. Atmos. Chem. Phys. 2015, 15, 11807–11833. [Google Scholar] [CrossRef]
- Chang, R.Y.W.; Leck, C.; Graus, M.; Müller, M.; Paatero, J.; Burkhart, J.F.; Stohl, A.; Orr, L.H.; Hayden, K.; Li, S.M.; et al. Aerosol composition and sources in the central Arctic Ocean during ASCOS. Atmos. Chem. Phys. 2011, 11, 10619–10636. [Google Scholar] [CrossRef] [Green Version]
- Crippa, M.; El Haddad, I.; Slowik, J.G.; DeCarlo, P.F.; Mohr, C.; Heringa, M.F.; Chirico, R.; Marchand, N.; Sciare, J.; Baltensperger, U.; et al. Identification of marine and continental aerosol sources in Paris using high resolution aerosol mass spectrometry. J. Geophys. Res.-Atmos. 2013, 118, 1950–1963. [Google Scholar] [CrossRef] [Green Version]
- Crenn, V.; Sciare, J.; Croteau, P.L.; Verlhac, S.; Fröhlich, R.; Belis, C.A.; Aas, W.; Äijälä, M.; Alastuey, A.; Artiñano, B.; et al. ACTRIS ACSM intercomparison—Part 1: Reproducibility of concentration and fragment results from 13 individual Quadrupole Aerosol Chemical Speciation Monitors (Q-ACSM) and consistency with co-located instruments. Atmos. Meas. Tech. 2015, 8, 5063–5087. [Google Scholar] [CrossRef] [Green Version]
- Fröhlich, R.; Crenn, V.; Setyan, A.; Belis, C.A.; Canonaco, F.; Favez, O.; Riffault, V.; Slowik, J.G.; Aas, W.; Aijälä, M.; et al. ACTRIS ACSM intercomparison—Part 2: Intercomparison of ME-2 organic source apportionment results from 15 individual, co-located aerosol mass spectrometers. Atmos. Meas. Tech. 2015, 8, 2555–2576. [Google Scholar] [CrossRef] [Green Version]
- Lanz, V.A.; Alfarra, M.R.; Baltensperger, U.; Buchmann, B.; Hueglin, C.; Prévôt, A.S.H. Source apportionment of submicron organic aerosols at an urban site by factor analytical modelling of aerosol mass spectra. Atmos. Chem. Phys. 2007, 7, 1503–1522. [Google Scholar] [CrossRef] [Green Version]
- Daellenbach, K.R.; Bozzetti, C.; Křepelová, A.; Canonaco, F.; Wolf, R.; Zotter, P.; Fermo, P.; Crippa, M.; Slowik, J.G.; Sosedova, Y.; et al. Characterization and source apportionment of organic aerosol using offline aerosol mass spectrometry. Atmos. Meas. Tech. 2016, 9, 23–39. [Google Scholar] [CrossRef] [Green Version]
- Daellenbach, K.R.; Stefenelli, G.; Bozzetti, C.; Vlachou, A.; Fermo, P.; Gonzalez, R.; Piazzalunga, A.; Colombi, C.; Canonaco, F.; Hueglin, C.; et al. Long-term chemical analysis and organic aerosol source apportionment at nine sites in central Europe: Source identification and uncertainty assessment. Atmos. Chem. Phys. 2017, 17, 13265–13282. [Google Scholar] [CrossRef]
- Bozzetti, C.; El Haddad, I.; Salameh, D.; Daellenbach, K.R.; Fermo, P.; Gonzalez, R.; Minguillón, M.C.; Iinuma, Y.; Poulain, L.; Elser, M.; et al. Organic aerosol source apportionment by offline-AMS over a full year in Marseille. Atmos. Chem. Phys. 2017, 17, 8247–8268. [Google Scholar] [CrossRef] [Green Version]
- Bozzetti, C.; Daellenbach, K.R.; Hueglin, C.; Fermo, P.; Sciare, J.; Kasper-Giebl, A.; Mazar, Y.; Abbaszade, G.; El Kazzi, M.; Gonzalez, R.; et al. Size-resolved identification, characterization, and quantification of primary biological organic aerosol at a European rural site. Environ. Sci. Technol. 2016, 50, 3425–3434. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sheesley, R.J.; Bae, M.-S.; Schauer, J.J. Sensitivity of a molecular marker based positive matrix factorization model to the number of receptor observations. Atmos. Environ. 2009, 43, 4951–4958. [Google Scholar] [CrossRef]
- Jaoui, M.; Lewandowski, M.; Kleindienst, T.E.; Offenberg, J.H.; Edney, E.O. β-caryophyllinic acid: An atmospheric tracer for β-caryophyllene secondary organic aerosol. Geophys. Res. Lett. 2007, 34, L05816. [Google Scholar] [CrossRef]
- Jimenez, J.; Canagaratna, M.; Donahue, N.; Prevot, A.; Zhang, Q.; Kroll, J.H.; DeCarlo, P.F.; Allan, J.D.; Coe, H.; Ng, N. Evolution of organic aerosols in the atmosphere. Science 2009, 326, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wen, T.; Li, X.; Wang, J.; Wang, Y. Characteristics of carbonaceous aerosols in Beijing based on two-year observation. Atmos. Pollut. Res. 2015, 6, 202–208. [Google Scholar] [CrossRef]
- Li, Y.J.; Lee, B.P.; Su, L.; Fung, J.C.H.; Chan, C.K. Seasonal characteristics of fine particulate matter (PM) based on high-resolution time-of-flight aerosol mass spectrometric (HR-ToF-AMS) measurements at the HKUST Supersite in Hong Kong. Atmos. Chem. Phys. 2015, 15, 37–53. [Google Scholar] [CrossRef]
- Crippa, M.; Canonaco, F.; Slowik, J.G.; El Haddad, I.; DeCarlo, P.F.; Mohr, C.; Heringa, M.F.; Chirico, R.; Marchand, N.; Temime-Roussel, B.; et al. Primary and secondary organic aerosol origin by combined gas-particle phase source apportionment. Atmos. Chem. Phys. 2013, 13, 8411–8426. [Google Scholar] [CrossRef]
- Huang, X.F.; He, L.Y.; Hu, M.; Canagaratna, M.R.; Sun, Y.; Zhang, Q.; Zhu, T.; Xue, L.; Zeng, L.W.; Liu, X.G.; et al. Highly time-resolved chemical characterization of atmospheric submicron particles during 2008 Beijing Olympic Games using an Aerodyne High-Resolution Aerosol Mass Spectrometer. Atmos. Chem. Phys. 2010, 10, 8933–8945. [Google Scholar] [CrossRef]
- Crippa, M.; Canonaco, F.; Lanz, V.A.; Äijälä, M.; Allan, J.D.; Carbone, S.; Capes, G.; Ceburnis, D.; Dall’Osto, M.; Day, D.A.; et al. Organic aerosol components derived from 25 AMS data sets across Europe using a consistent ME-2 based source apportionment approach. Atmos. Chem. Phys. 2014, 14, 6159–6176. [Google Scholar] [CrossRef] [Green Version]
- Hildemann, L.M.; Klinedinst, D.B.; Klouda, G.A.; Currie, L.A.; Cass, G.R. Sources of urban contemporary carbon aerosol. Environ. Sci. Technol. 1994, 28, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Stuiver, M.; Polach, H.A. Discussion reporting of 14 C data. Radiocarbon 1977, 19, 355–363. [Google Scholar] [CrossRef]
- Levin, I.; Naegler, T.; Kromer, B.; Diehl, M.; Francey, R.; Gomez-Pelaez, A.; Steele, P.; Wagenbach, D.; Weller, R.; Worthy, D. Observations and modelling of the global distribution and long-term trend of atmospheric 14CO2. Tellus B Chem. Phys. Meteorol. 2010, 62, 26–46. [Google Scholar] [CrossRef] [Green Version]
- Buchholz, B.A.; Fallon, S.J.; Zermeño, P.; Bench, G.; Schichtel, B.A. Anomalous elevated radiocarbon measurements of PM2.5. Nucl. Instrum. Methods Phys. Res. Sect. B 2013, 294, 631–635. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Liu, D.; Ding, P.; Shen, C.; Mo, Y.; Wang, X.; Luo, C.; Cheng, Z.; Szidat, S.; et al. Source apportionment and dynamic changes of carbonaceous aerosols during the haze bloom-decay process in China based on radiocarbon and organic molecular tracers. Atmos. Chem. Phys. 2016, 16, 2985–2996. [Google Scholar] [CrossRef] [Green Version]
- El Haddad, I.; D’Anna, B.; Temime-Roussel, B.; Nicolas, M.; Boreave, A.; Favez, O.; Voisin, D.; Sciare, J.; George, C.; Jaffrezo, J.L.; et al. Towards a better understanding of the origins, chemical composition and aging of oxygenated organic aerosols: Case study of a Mediterranean industrialized environment, Marseille. Atmos. Chem. Phys. 2013, 13, 7875–7894. [Google Scholar] [CrossRef]
- Schichtel, B.A.; Malm, W.C.; Bench, G.; Fallon, S.; McDade, C.E.; Chow, J.C.; Watson, J.G. Fossil and contemporary fine particulate carbon fractions at 12 rural and urban sites in the United States. J. Geophys. Res.-Atmos. 2008, 113. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Zheng, M.; Edgerton, E.S.; Jansen, J.J.; Wang, X. Contemporary or fossil origin: Split of estimated secondary organic carbon in the Southeastern United States. Environ. Sci. Technol. 2008, 42, 9122–9128. [Google Scholar] [CrossRef] [PubMed]
- Gilardoni, S.; Vignati, E.; Cavalli, F.; Putaud, J.; Larsen, B.; Karl, M.; Stenström, K.; Genberg, J.; Henne, S.; Dentener, F. Better constraints on sources of carbonaceous aerosols using a combined 14 C–macro tracer analysis in a European rural background site. Atmos. Chem. Phys. 2011, 11, 5685–5700. [Google Scholar] [CrossRef] [Green Version]
- Morino, Y.; Ohara, T.; Xu, J.; Hasegawa, S.; Zhao, B.; Fushimi, A.; Tanabe, K.; Kondo, M.; Uchida, M.; Yamaji, K. Diurnal variations of fossil and nonfossil carbonaceous aerosols in Beijing. Atmos. Environ. 2015, 122, 349–356. [Google Scholar] [CrossRef]
- Kaulfus, A.S.; Nair, U.; Jaffe, D.; Christopher, S.A.; Goodrick, S. Biomass burning smoke climatology of the United States: Implications for particulate matter air quality. Environ. Sci. Technol. 2017, 51, 11731–11741. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Choi, Y.; Souri, A.H.; Jeon, W.; Diao, L.; Pan, S.; Westenbarger, D. Effects of biomass burning emissions on air quality over the continental USA: A three-year comprehensive evaluation accounting for sensitivities due to boundary conditions and plume rise height. In Environmental Contaminants: Measurement, Modelling and Control; Gupta, T., Agarwal, A.K., Agarwal, R.A., Labhsetwar, N.K., Eds.; Springer: Singapore, 2018; pp. 245–278. [Google Scholar]
- Aiken, A.C.; Salcedo, D.; Cubison, M.J.; Huffman, J.A.; DeCarlo, P.F.; Ulbrich, I.M.; Docherty, K.S.; Sueper, D.; Kimmel, J.R.; Worsnop, D.R.; et al. Mexico City aerosol analysis during MILAGRO using high resolution aerosol mass spectrometry at the urban supersite (T0)—Part 1: Fine particle composition and organic source apportionment. Atmos. Chem. Phys. 2009, 9, 6633–6653. [Google Scholar] [CrossRef]
- Huang, X.-F.; Xue, L.; Tian, X.-D.; Shao, W.-W.; Sun, T.-L.; Gong, Z.-H.; Ju, W.-W.; Jiang, B.; Hu, M.; He, L.-Y. Highly time-resolved carbonaceous aerosol characterization in Yangtze River Delta of China: Composition, mixing state and secondary formation. Atmos. Environ. 2013, 64, 200–207. [Google Scholar] [CrossRef]
- Fountoukis, C.; Butler, T.; Lawrence, M.G.; Denier van der Gon, H.A.C.; Visschedijk, A.J.H.; Charalampidis, P.; Pilinis, C.; Pandis, S.N. Impacts of controlling biomass burning emissions on wintertime carbonaceous aerosol in Europe. Atmos. Environ. 2014, 87, 175–182. [Google Scholar] [CrossRef]
- Chen, J.; Li, C.; Ristovski, Z.; Milic, A.; Gu, Y.; Islam, M.S.; Wang, S.; Hao, J.; Zhang, H.; He, C.; et al. A review of biomass burning: Emissions and impacts on air quality, health and climate in China. Sci. Total Environ. 2017, 579, 1000–1034. [Google Scholar] [CrossRef] [PubMed]
- Denier van der Gon, H.A.C.; Bergström, R.; Fountoukis, C.; Johansson, C.; Pandis, S.N.; Simpson, D.; Visschedijk, A.J.H. Particulate emissions from residential wood combustion in Europe—Revised estimates and an evaluation. Atmos. Chem. Phys. 2015, 15, 6503–6519. [Google Scholar] [CrossRef] [Green Version]
- Viana, M.; Alastuey, A.; Querol, X.; Guerreiro, C.; Vogt, M.; Colette, A.; Collet, S.; Albinet, A.; Fraboulet, I.; Lacome, J.-M. Contribution of Residential Combustion to Ambient Air Pollution and Greenhouse Gas Emissions; ETC/ACM: Bilthoven, The Netherlands, 2015; Volume 1, ETC/ACM Technical Paper. [Google Scholar]
- Hodzic, A.; Kasibhatla, P.S.; Jo, D.S.; Cappa, C.D.; Jimenez, J.L.; Madronich, S.; Park, R.J. Rethinking the global secondary organic aerosol (SOA) budget: Stronger production, faster removal, shorter lifetime. Atmos. Chem. Phys. 2016, 16, 7917–7941. [Google Scholar] [CrossRef]
- Tie, X.; Li, G.; Ying, Z.; Guenther, A.; Madronich, S. Biogenic emissions of isoprenoids and NO in China and comparison to anthropogenic emissions. Sci. Total Environ. 2006, 371, 238–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Kong, L.; Cheng, T.; Chen, J.; Du, J.; Li, L.; Xia, X.; Leng, C.; Huang, G. Insights into summertime haze pollution events over Shanghai based on online water-soluble ionic composition of aerosols. Atmos. Environ. 2011, 45, 5131–5137. [Google Scholar] [CrossRef]
- Pernigotti, D.; Belis, C.A.; Spanò, L. SPECIEUROPE: The European data base for PM source profiles. Atmos. Pollut. Res. 2016, 7, 307–314. [Google Scholar] [CrossRef]
- Simon, H.; Beck, L.; Bhave, P.V.; Divita, F.; Hsu, Y.; Luecken, D.; Mobley, J.D.; Pouliot, G.A.; Reff, A.; Sarwar, G.; et al. The development and uses of EPA’s SPECIATE database. Atmos. Pollut. Res. 2010, 1, 196–206. [Google Scholar] [CrossRef]
- Zhang, Y.; Williams, B.J.; Goldstein, A.H.; Docherty, K.S.; Jimenez, J.L. A technique for rapid source apportionment applied to ambient organic aerosol measurements from a thermal desorption aerosol gas chromatograph (TAG). Atmos. Meas. Tech. 2016, 9, 5637–5653. [Google Scholar] [CrossRef] [Green Version]
- Williams, B.J.; Goldstein, A.H.; Kreisberg, N.M.; Hering, S.V.; Worsnop, D.R.; Ulbrich, I.M.; Docherty, K.S.; Jimenez, J.L. Major components of atmospheric organic aerosol in southern California as determined by hourly measurements of source marker compounds. Atmos. Chem. Phys. 2010, 10, 11577–11603. [Google Scholar] [CrossRef] [Green Version]
- Williams, B.J.; Jayne, J.T.; Lambe, A.T.; Hohaus, T.; Kimmel, J.R.; Sueper, D.; Brooks, W.; Williams, L.R.; Trimborn, A.M.; Martinez, R.E.; et al. The first combined thermal desorption aerosol gas chromatograph—Aerosol mass spectrometer (TAG-AMS). Aerosol Sci. Technol. 2014, 48, 358–370. [Google Scholar] [CrossRef]
- Gallimore, P.J.; Kalberer, M. Characterizing an extractive electrospray ionization (EESI) source for the online mass spectrometry analysis of organic aerosols. Environ. Sci. Technol. 2013, 47, 7324–7331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doezema, L.A.; Longin, T.; Cody, W.; Perraud, V.; Dawson, M.L.; Ezell, M.J.; Greaves, J.; Johnson, K.R.; Finlayson-Pitts, B.J. Analysis of secondary organic aerosols in air using extractive electrospray ionization mass spectrometry (EESI-MS). RSC Adv. 2012, 2, 2930–2938. [Google Scholar] [CrossRef]
- Gallimore, P.J.; Giorio, C.; Mahon, B.M.; Kalberer, M. Online molecular characterisation of organic aerosols in an atmospheric chamber using extractive electrospray ionisation mass spectrometry. Atmos. Chem. Phys. 2017, 17, 14485–14500. [Google Scholar] [CrossRef] [Green Version]
- Slowik, J.G.; Lopez-Hilfiker, F.; Pospisilova, V.; Qi, L.; Stefenelli, G.; Tong, Y.; Vogel, A.L.; Wang, L.; Yuan, B.; Xiao, M.; et al. Towards Quantitative Organic Aerosol Sampling with an Extractive Electrospray Ionization Time-Of-Flight Mass Spectrometer (EESI-TOF); AAAR: Raleigh, NC, USA, 2017. [Google Scholar]
- Stefenelli, G.; Lopez-Hilfiker, F.; Pospisilova, V.; Vogel, A.L.; Hueglin, C.; Rigler, M.; Baltensperger, U.; Prévôt, A.S.H.; Slowik, J.G. Investigation of Biogenic Influences and Day/Night Chemistry on Secondary Organic Aerosol by Extractive Electrospray Ionization Time-Of-Flight Mass Spectrometry (EESI-TOF); AAAR: Raleigh, NC, USA, 2017. [Google Scholar]
- Lopez-Hilfiker, F.D.; Mohr, C.; Ehn, M.; Rubach, F.; Kleist, E.; Wildt, J.; Mentel, T.F.; Lutz, A.; Hallquist, M.; Worsnop, D.; et al. A novel method for online analysis of gas and particle composition: Description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO). Atmos. Meas. Tech. 2014, 7, 983–1001. [Google Scholar] [CrossRef]
- Holzinger, R.; Williams, J.; Herrmann, F.; Lelieveld, J.; Donahue, N.M.; Röckmann, T. Aerosol analysis using a thermal-desorption proton-transfer-reaction mass spectrometer (TD-PTR-MS): A new approach to study processing of organic aerosols. Atmos. Chem. Phys. 2010, 10, 2257–2267. [Google Scholar] [CrossRef]
- Salvador, C.M.; Ho, T.T.; Chou, C.C.K.; Chen, M.J.; Huang, W.R.; Huang, S.H. Characterization of the organic matter in submicron urban aerosols using a Thermo-Desorption Proton-Transfer-Reaction Time-of-Flight Mass Spectrometer (TD-PTR-TOF-MS). Atmos. Environ. 2016, 140, 565–575. [Google Scholar] [CrossRef]
- Gkatzelis, G.I.; Tillmann, R.; Hohaus, T.; Müller, M.; Eichler, P.; Xu, K.M.; Schlag, P.; Schmitt, S.H.; Wegener, R.; Kaminski, M.; et al. Comparison of three aerosol chemical characterization techniques utilizing PTR-ToF-MS: A study on freshly formed and aged biogenic SOA. Atmos. Meas. Tech. Discuss. 2017, 2017, 1–31. [Google Scholar] [CrossRef]
- Eichler, P.; Müller, M.; D’Anna, B.; Wisthaler, A. A novel inlet system for online chemical analysis of semi-volatile submicron particulate matter. Atmos. Meas. Tech. 2015, 8, 1353–1360. [Google Scholar] [CrossRef]
- Slowik, J.G.; Vlasenko, A.; McGuire, M.; Evans, G.J.; Abbatt, J.P.D. Simultaneous factor analysis of organic particle and gas mass spectra: AMS and PTR-MS measurements at an urban site. Atmos. Chem. Phys. 2010, 10, 1969–1988. [Google Scholar] [CrossRef]
- Docherty, K.S.; Aiken, A.C.; Huffman, J.A.; Ulbrich, I.M.; DeCarlo, P.F.; Sueper, D.; Worsnop, D.R.; Snyder, D.C.; Peltier, R.E.; Weber, R.J.; et al. The 2005 Study of Organic Aerosols at Riverside (SOAR-1): Instrumental intercomparisons and fine particle composition. Atmos. Chem. Phys. 2011, 11, 12387–12420. [Google Scholar] [CrossRef] [Green Version]
- Healy, R.M.; Sciare, J.; Poulain, L.; Crippa, M.; Wiedensohler, A.; Prévôt, A.S.H.; Baltensperger, U.; Sarda-Estève, R.; McGuire, M.L.; Jeong, C.H.; et al. Quantitative determination of carbonaceous particle mixing state in Paris using single-particle mass spectrometer and aerosol mass spectrometer measurements. Atmos. Chem. Phys. 2013, 13, 9479–9496. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, Q.; Schwab, J.; Yang, T.; Ng, N.; Demerjian, K. Factor analysis of combined organic and inorganic aerosol mass spectra from high resolution aerosol mass spectrometer measurements. Atmos. Chem. Phys. 2012, 12, 8537–8551. [Google Scholar] [CrossRef]
- McGuire, M.; Chang, R.-W.; Slowik, J.; Jeong, C.-H.; Healy, R.; Lu, G.; Mihele, C.; Abbatt, J.; Brook, J.; Evans, G. Enhancing non-refractory aerosol apportionment from an urban industrial site through receptor modeling of complete high time-resolution aerosol mass spectra. Atmos. Chem. Phys. 2014, 14, 8017–8042. [Google Scholar] [CrossRef]
- Vlachou, A.; Daellenbach, K.R.; Bozzetti, C.; Chazeau, B.; Salazar, G.A.; Szidat, S.; Jaffrezo, J.L.; Hueglin, C.; Baltensperger, U.; El Haddad, I.; et al. Advanced source apportionment of carbonaceous aerosols by coupling offline AMS and radiocarbon size segregated measurements over a nearly two-year period. Atmos. Chem. Phys. Discuss. 2017, 2017, 1–25. [Google Scholar] [CrossRef]
- Petit, J.E.; Favez, O.; Sciare, J.; Canonaco, F.; Croteau, P.; Močnik, G.; Jayne, J.; Worsnop, D.; Leoz-Garziandia, E. Submicron aerosol source apportionment of wintertime pollution in Paris, France by double positive matrix factorization (PMF2) using an aerosol chemical speciation monitor (ACSM) and a multi-wavelength Aethalometer. Atmos. Chem. Phys. 2014, 14, 13773–13787. [Google Scholar] [CrossRef]
- Sofowote, U.M.; Healy, R.M.; Su, Y.; Debosz, J.; Noble, M.; Munoz, A.; Jeong, C.H.; Wang, J.M.; Hilker, N.; Evans, G.J.; et al. Understanding the PM2.5 imbalance between a far and near-road location: Results of high temporal frequency source apportionment and parameterization of black carbon. Atmos. Environ. 2018, 173, 277–288. [Google Scholar] [CrossRef]
- Ciarelli, G.; Aksoyoglu, S.; El Haddad, I.; Bruns, E.A.; Crippa, M.; Poulain, L.; Äijälä, M.; Carbone, S.; Freney, E.; O’Dowd, C.; et al. Modelling winter organic aerosol at the European scale with CAMx: Evaluation and source apportionment with a VBS parameterization based on novel wood burning smog chamber experiments. Atmos. Chem. Phys. 2017, 17, 7653–7669. [Google Scholar] [CrossRef]
- Aksoyoglu, S.; Keller, J.; Barmpadimos, I.; Oderbolz, D.; Lanz, V.; Prévôt, A.; Baltensperger, U. Aerosol modelling in Europe with a focus on Switzerland during summer and winter episodes. Atmos. Chem. Phys 2011, 11, 7355–7373. [Google Scholar] [CrossRef]
- Bessagnet, B.; Pirovano, G.; Mircea, M.; Cuvelier, C.; Aulinger, A.; Calori, G.; Ciarelli, G.; Manders, A.; Stern, R.; Tsyro, S.; et al. Presentation of the EURODELTA III intercomparison exercise—Evaluation of the chemistry transport models’ performance on criteria pollutants and joint analysis with meteorology. Atmos. Chem. Phys. 2016, 16, 12667–12701. [Google Scholar] [CrossRef]
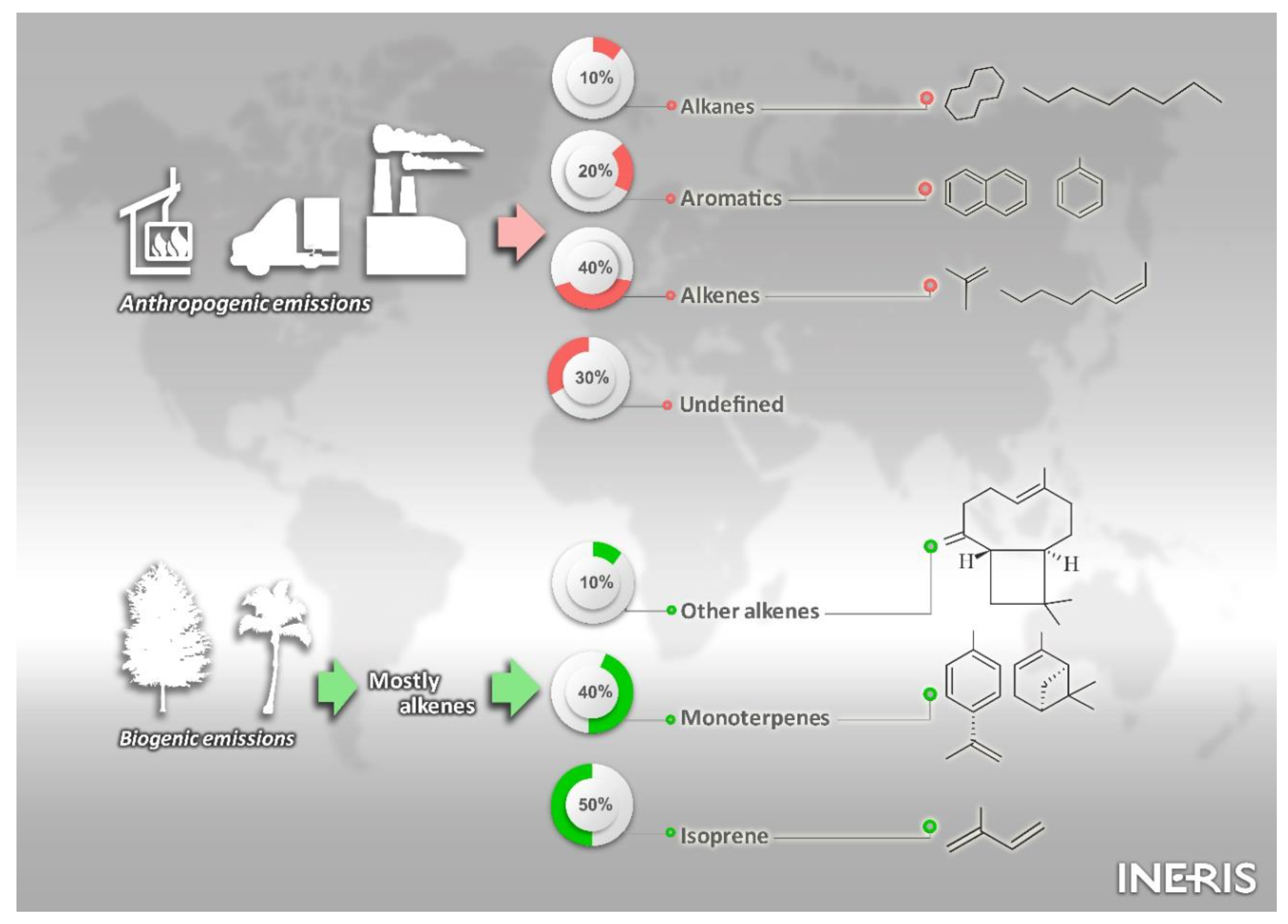
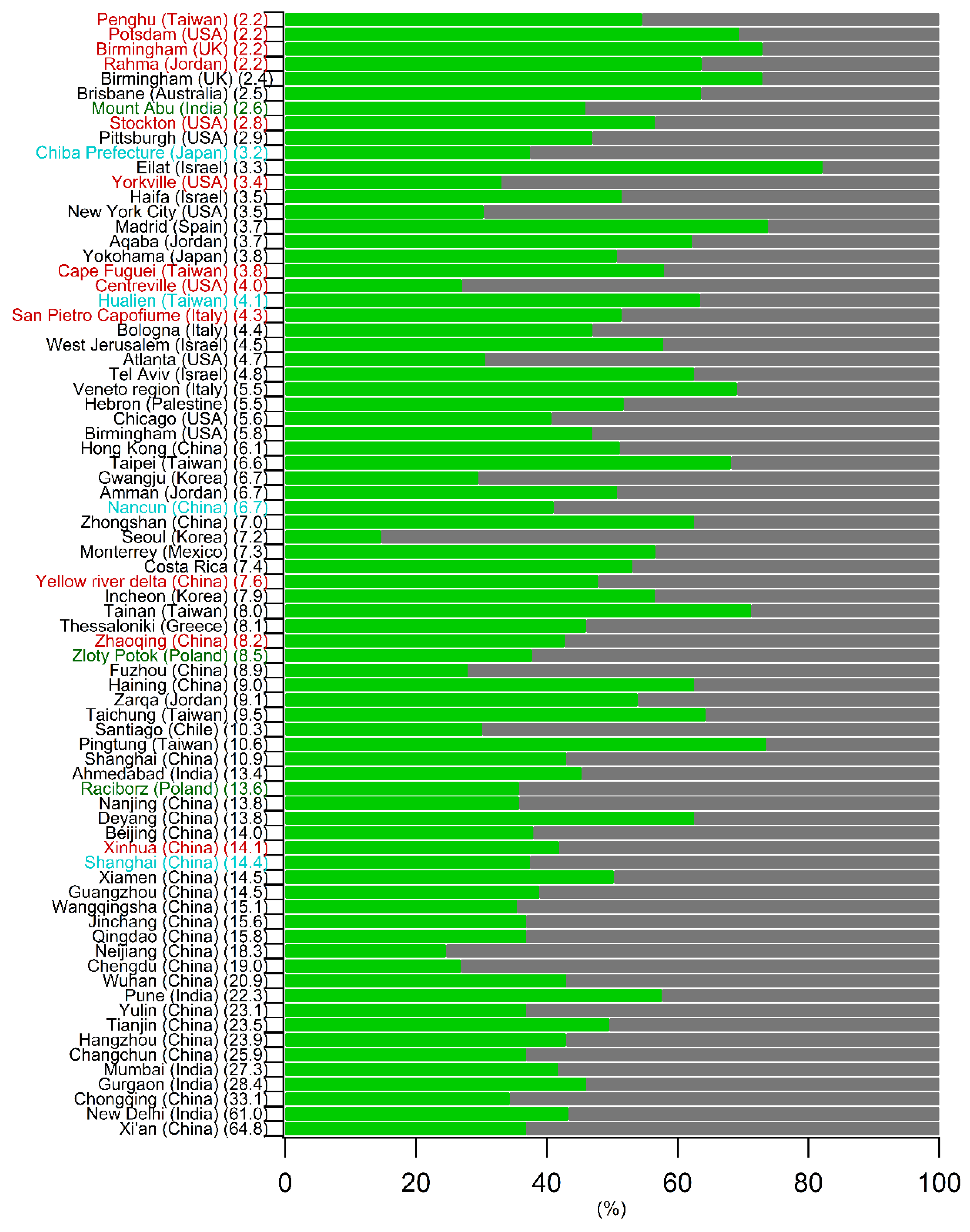

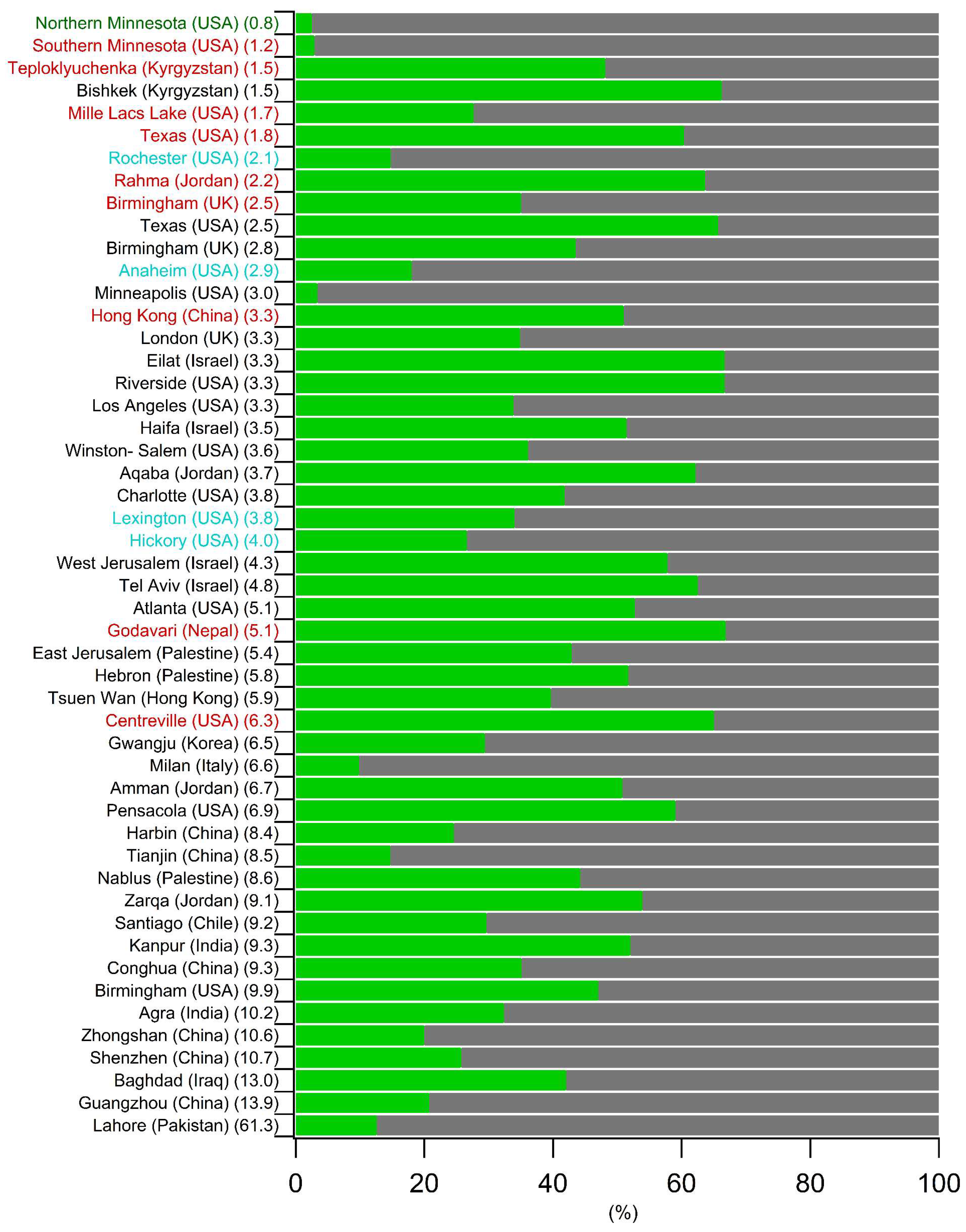

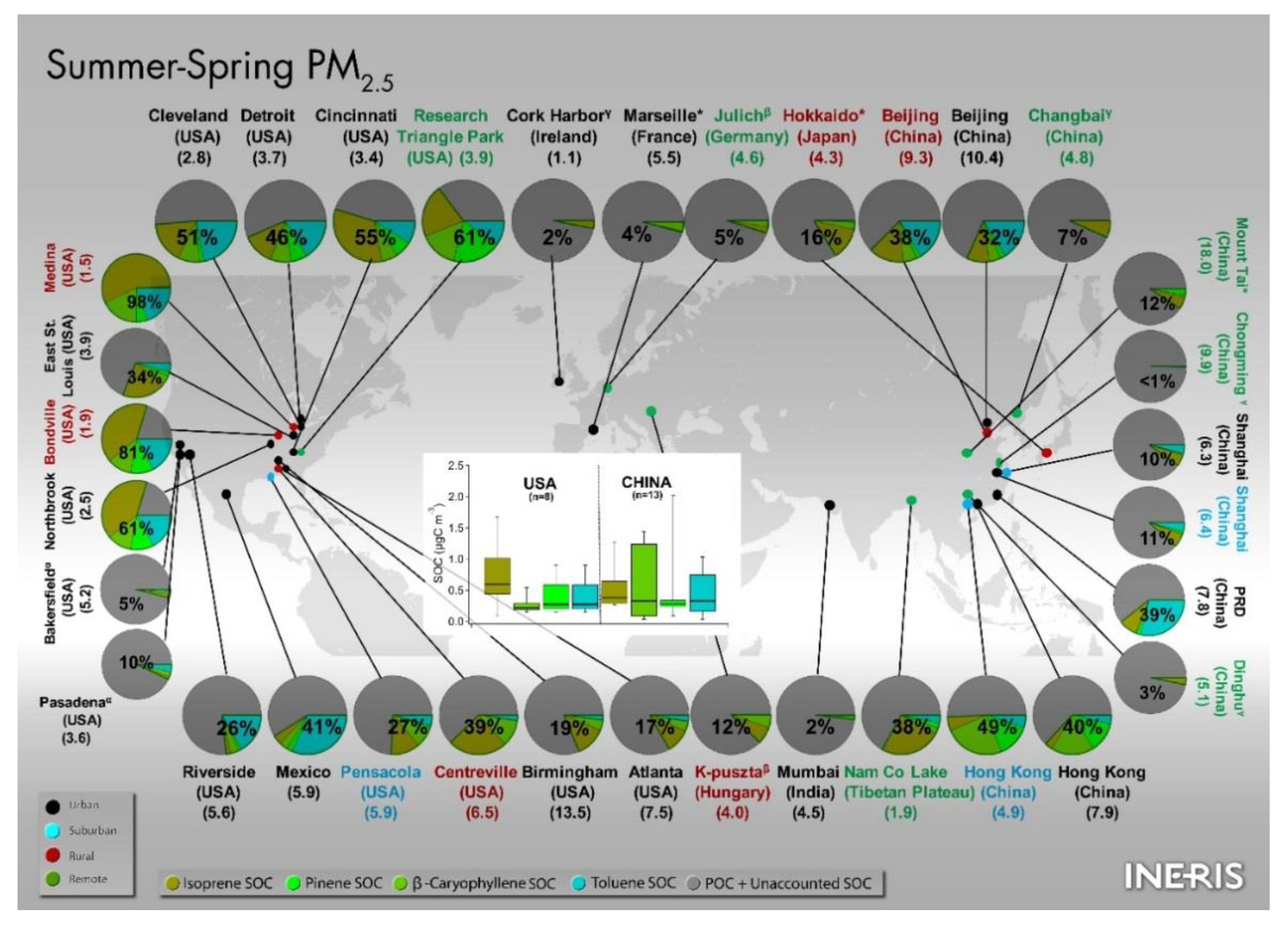
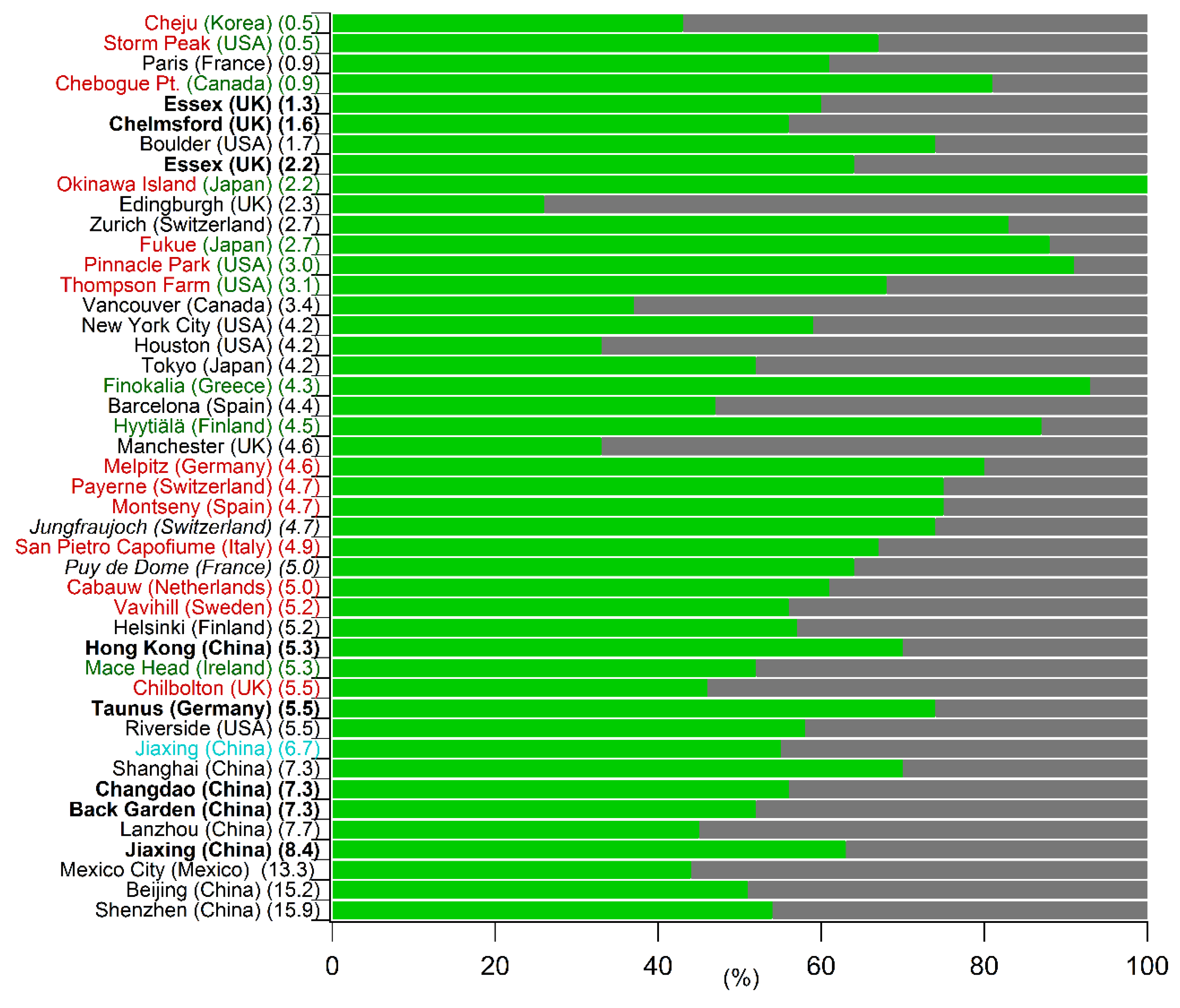

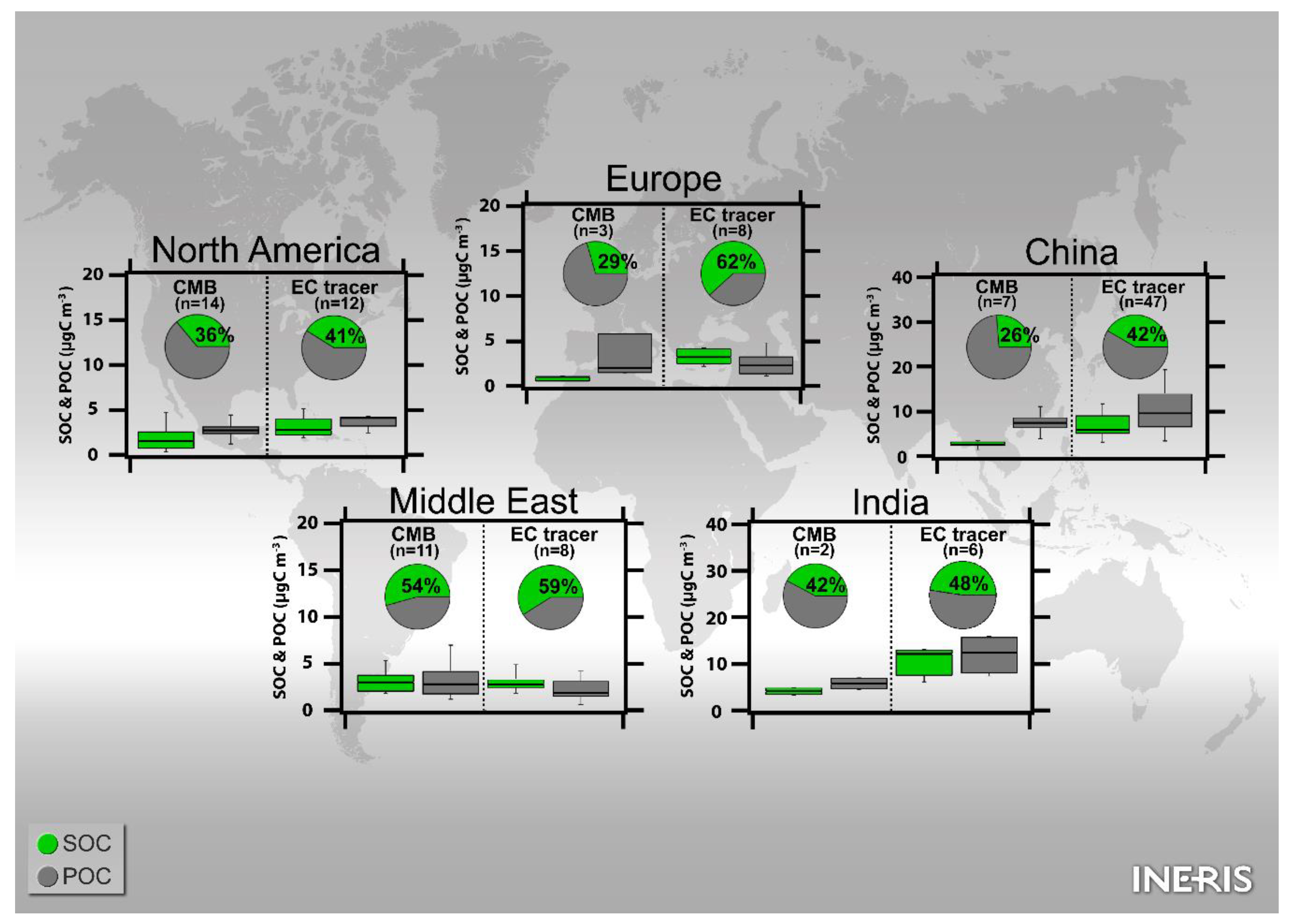


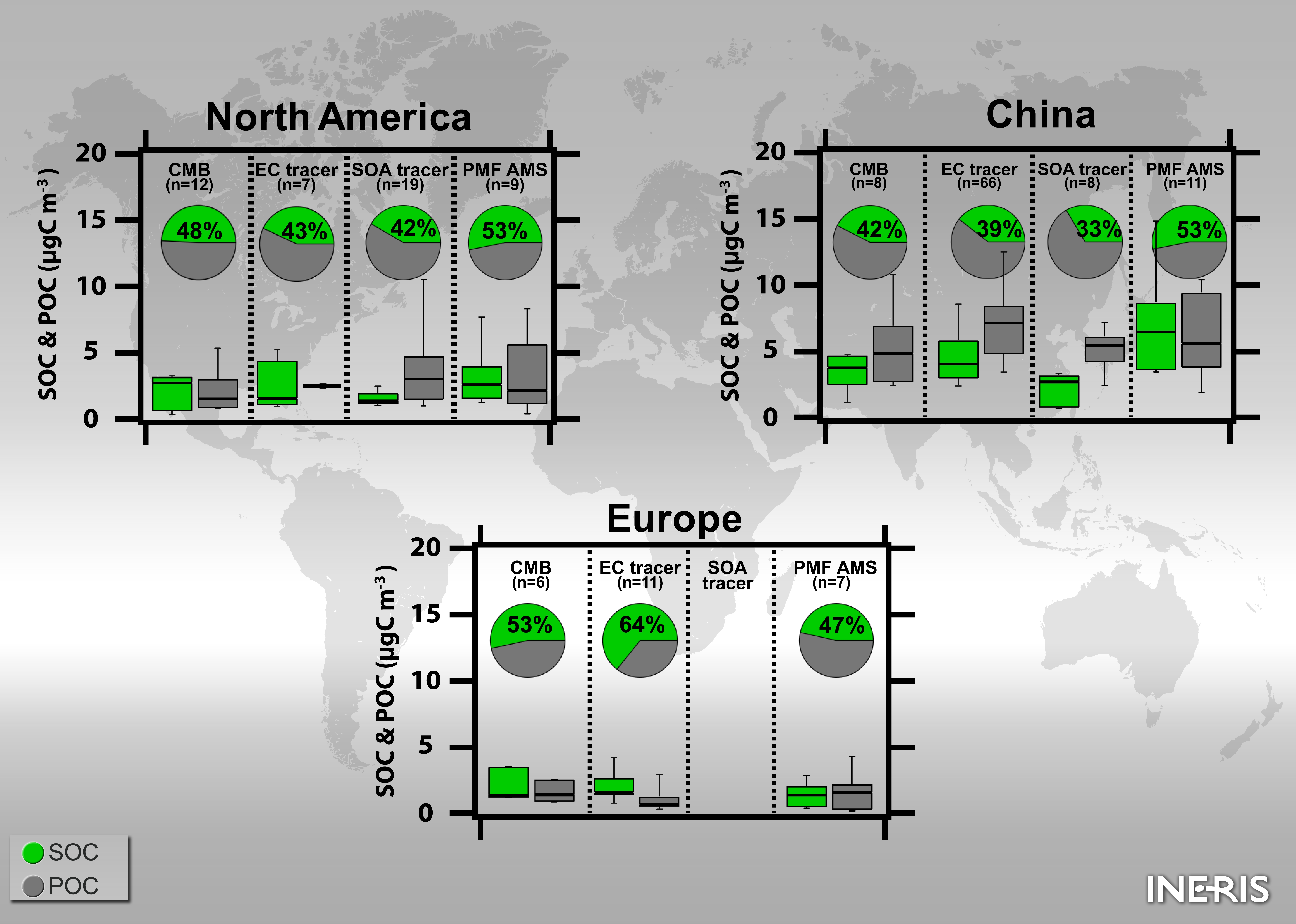{kind=link}
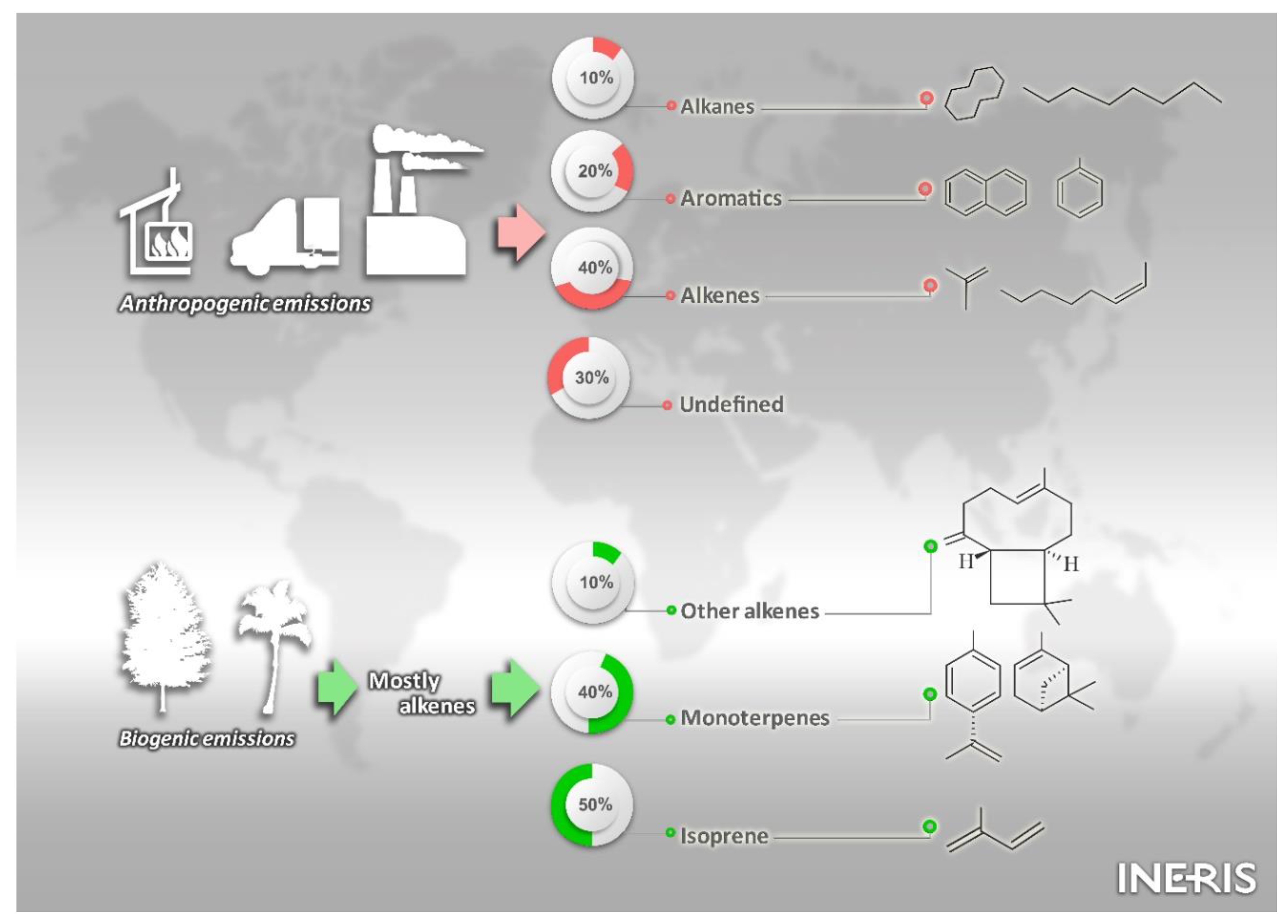{kind=link}
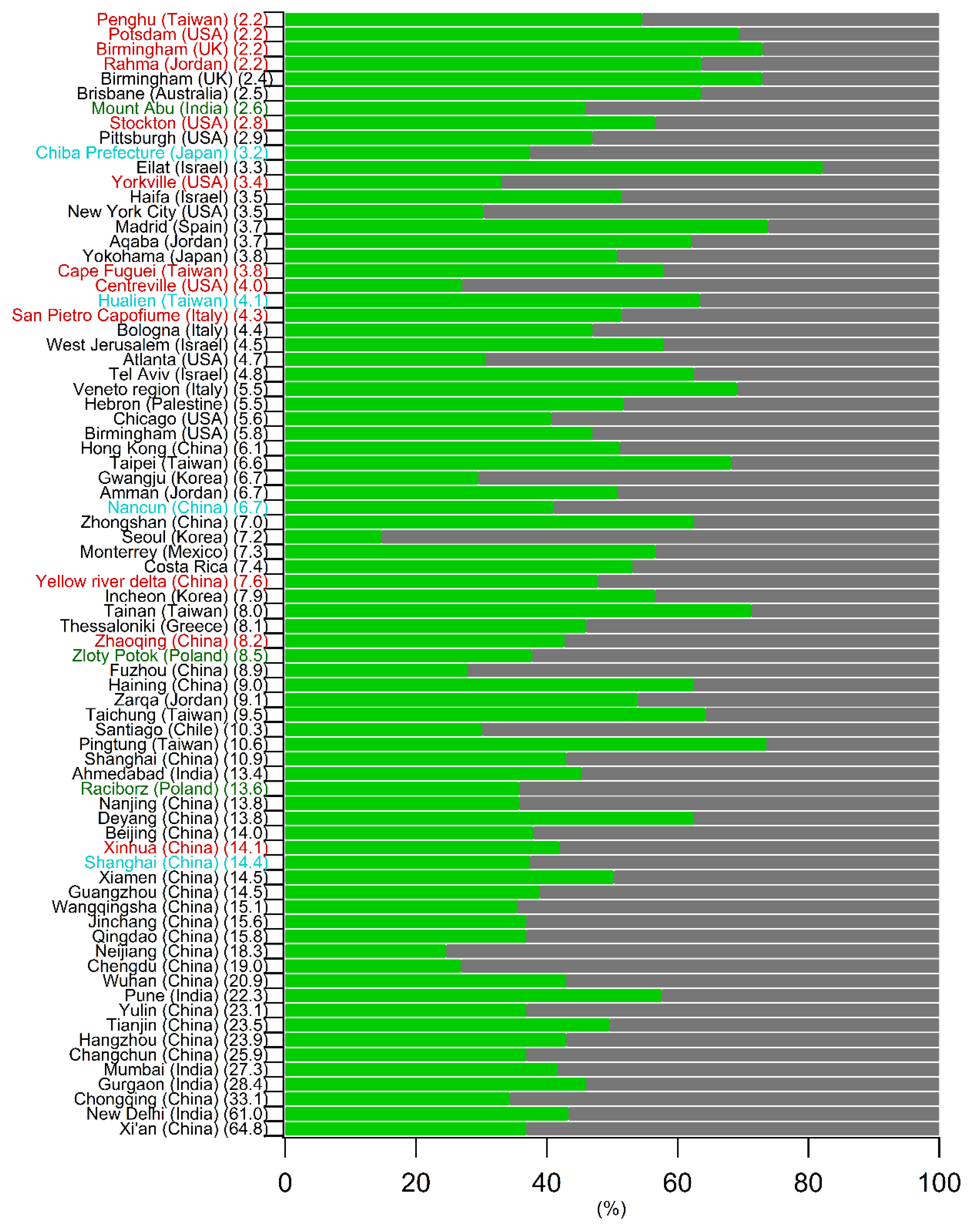{kind=link}
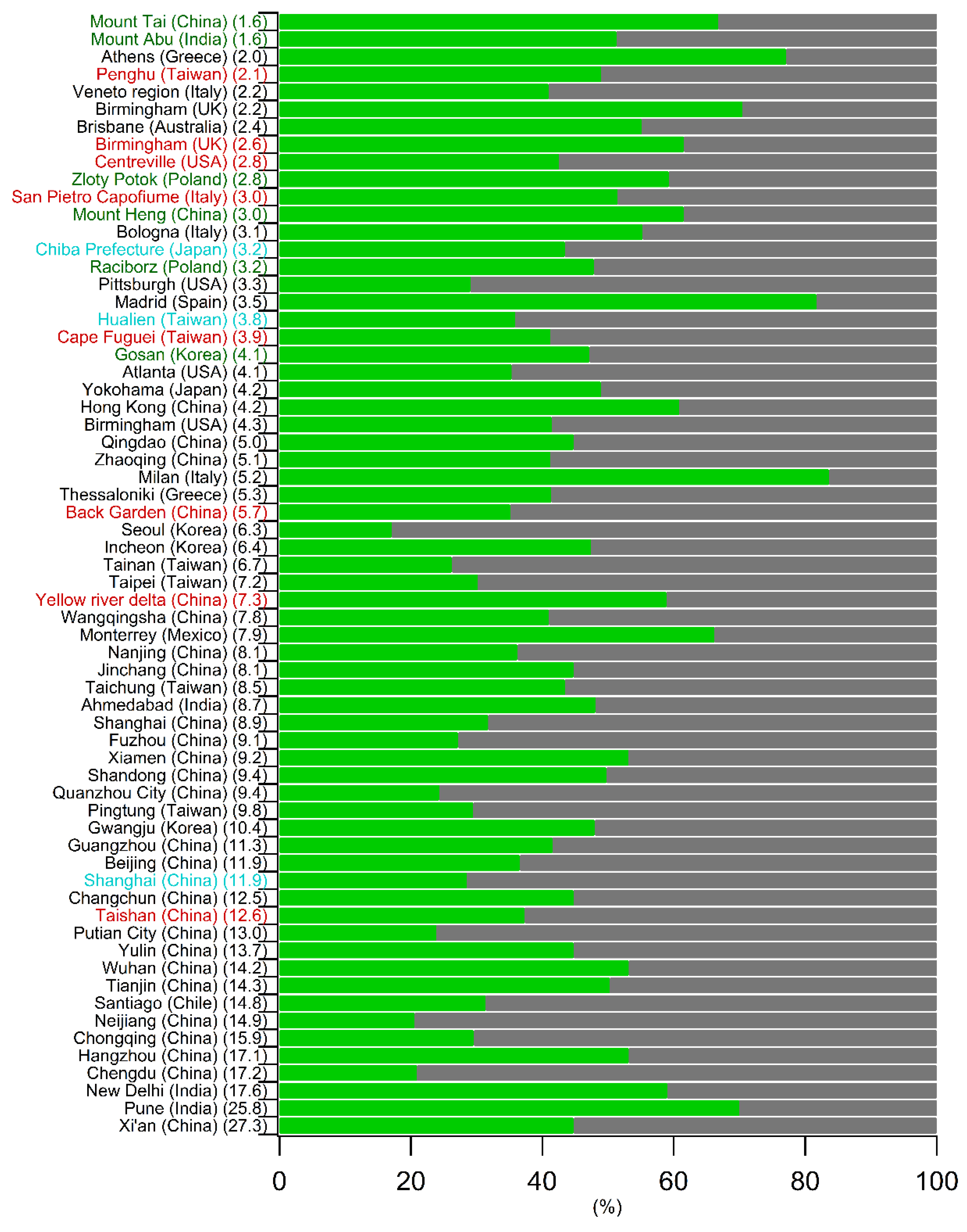{kind=link}
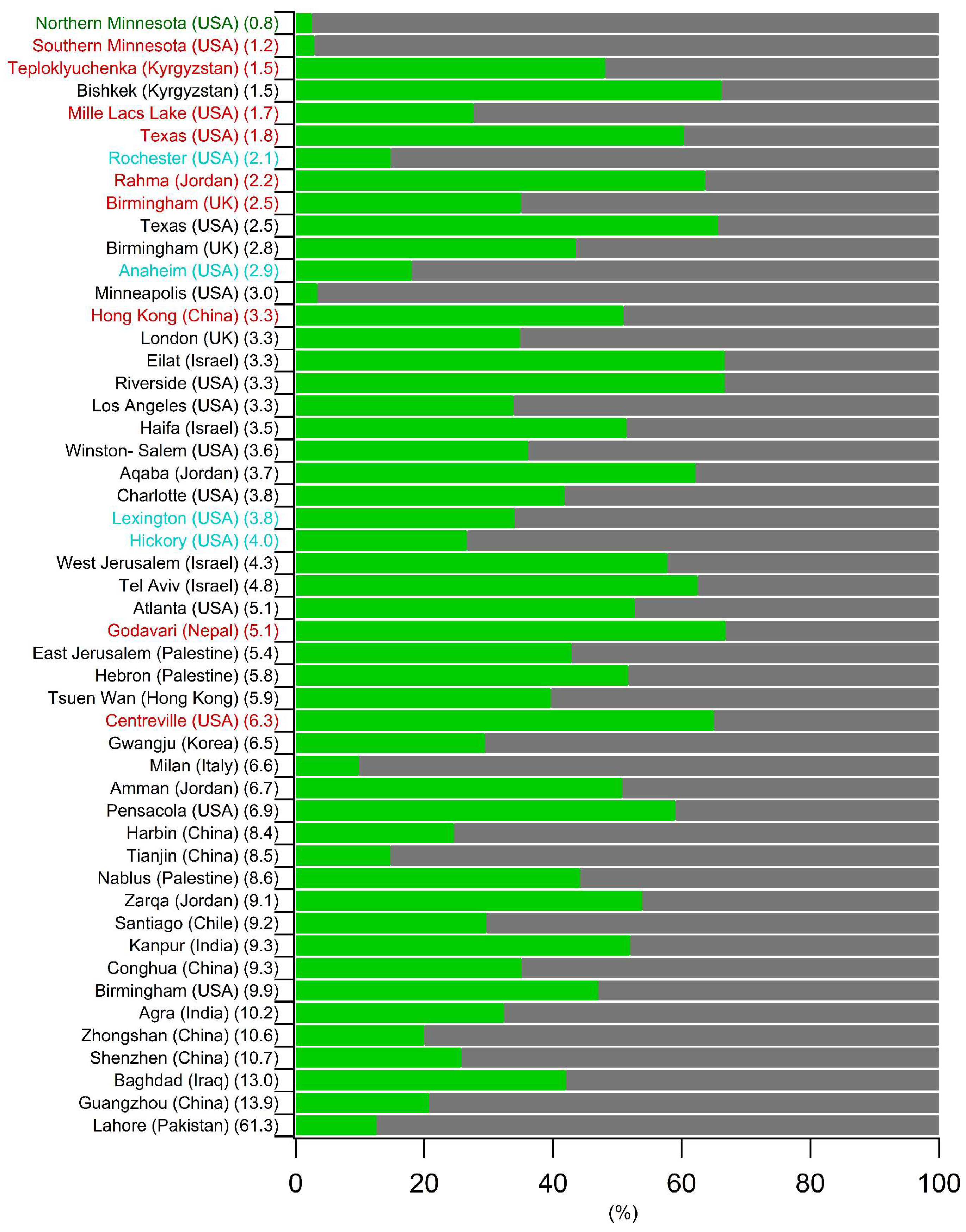{kind=link}
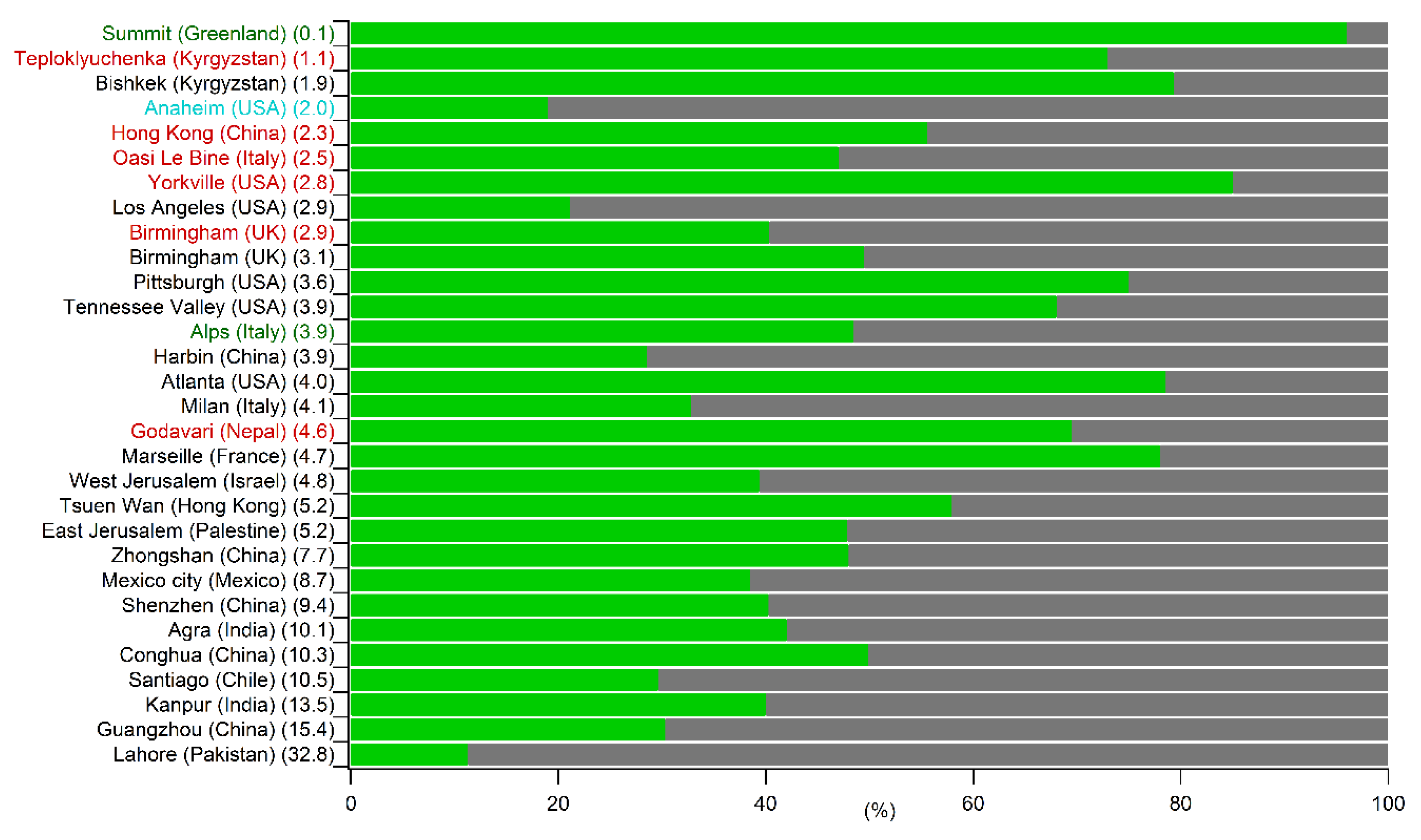{kind=link}
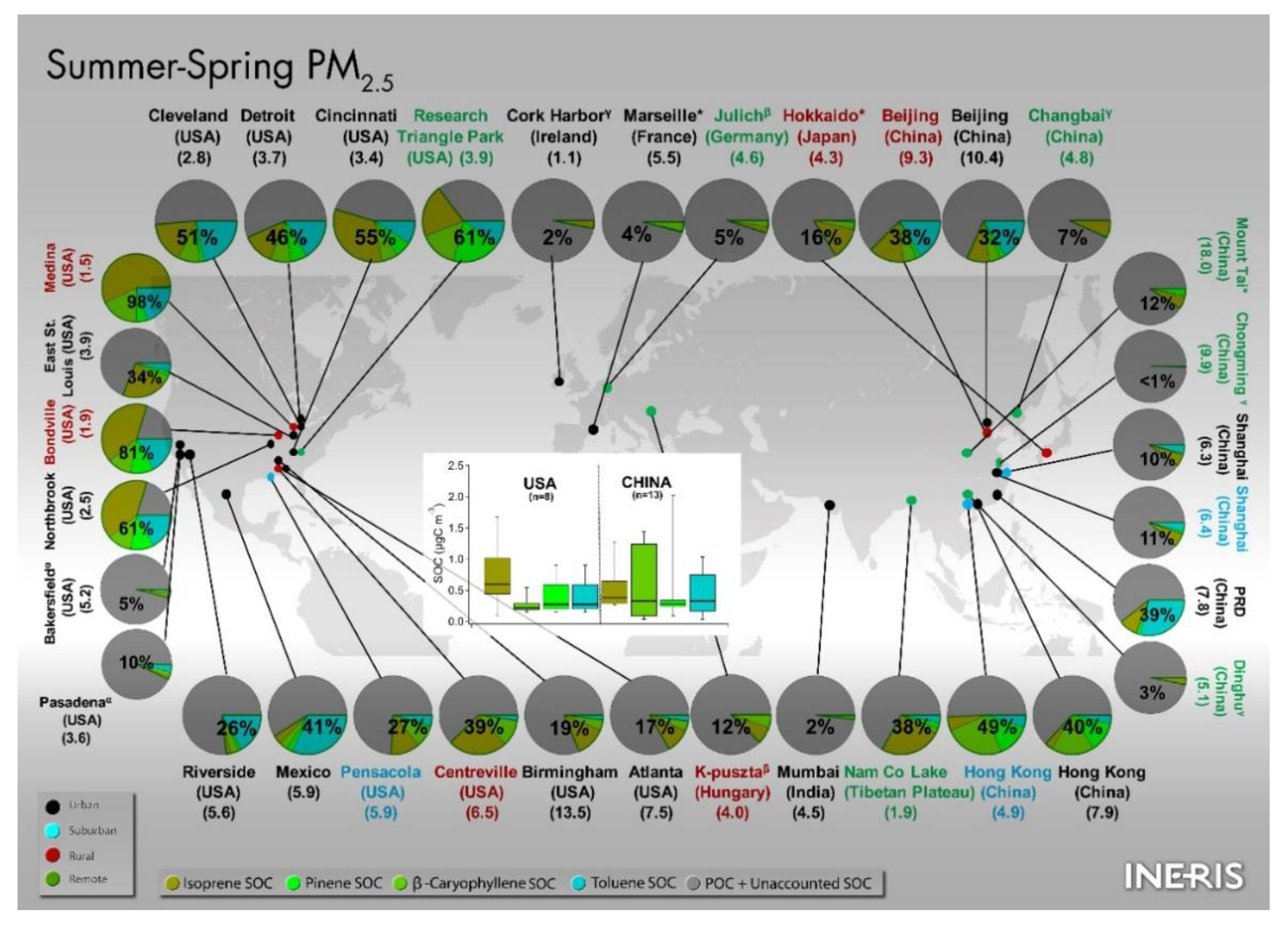{kind=link}
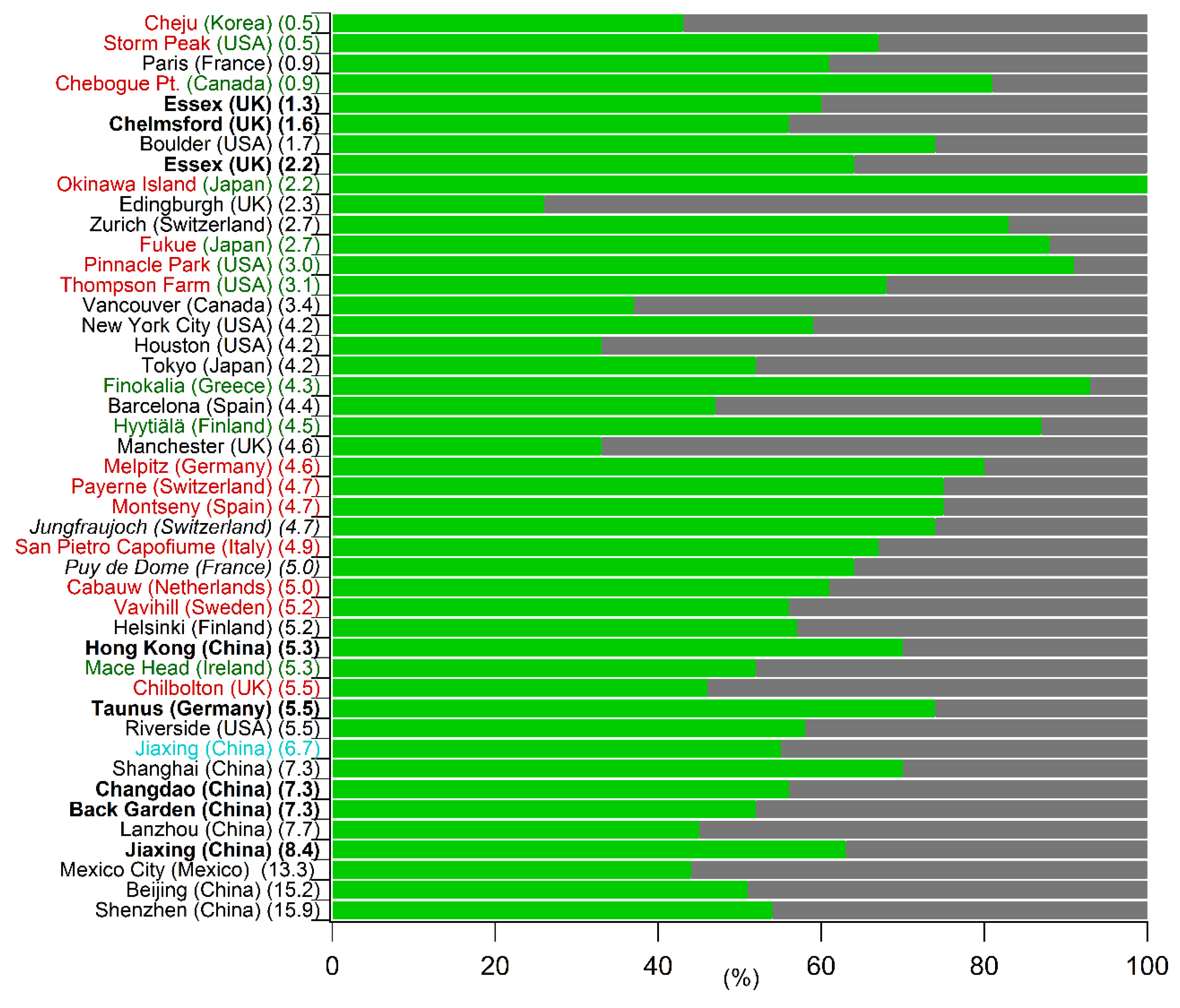{kind=link}
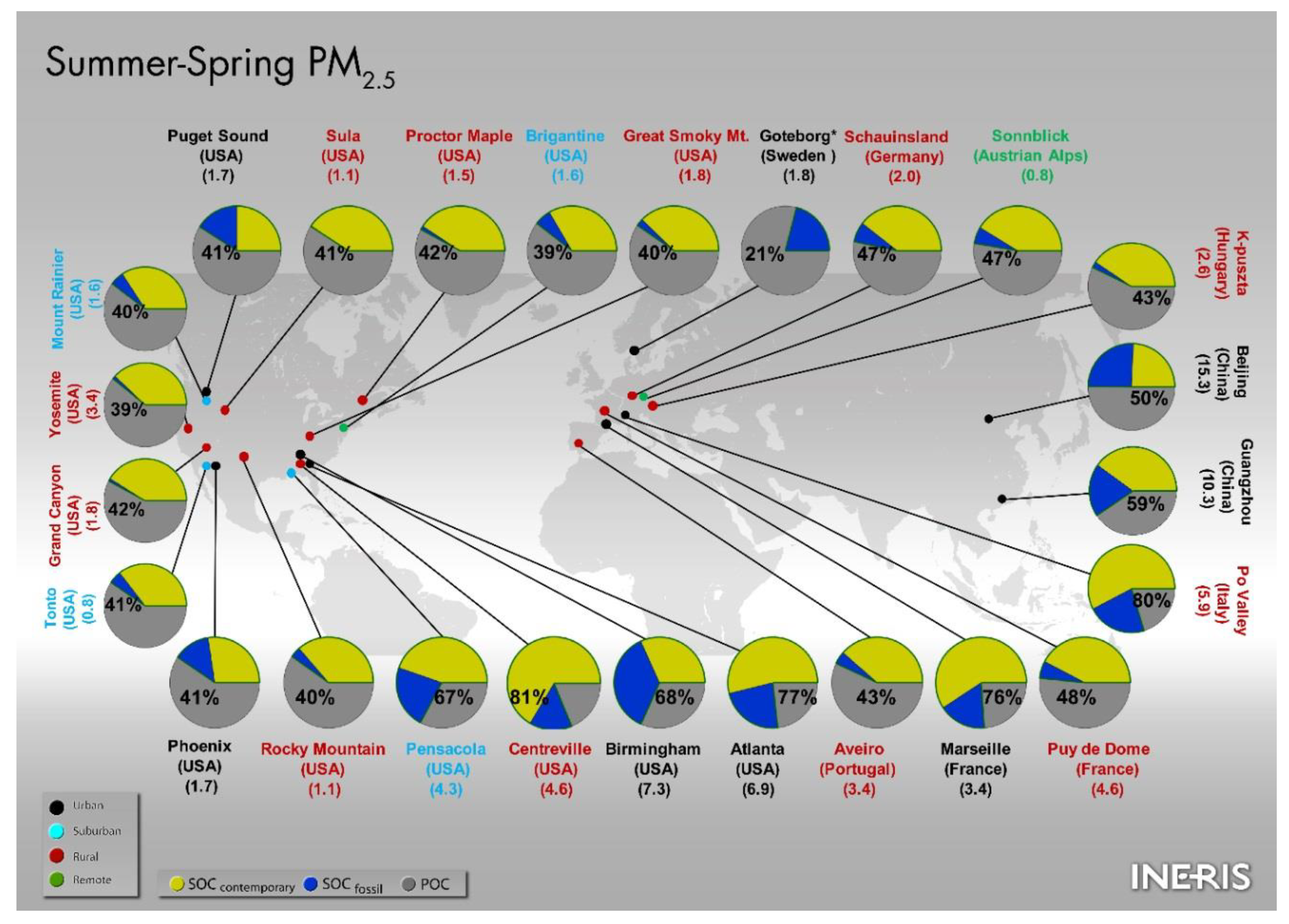{kind=link}
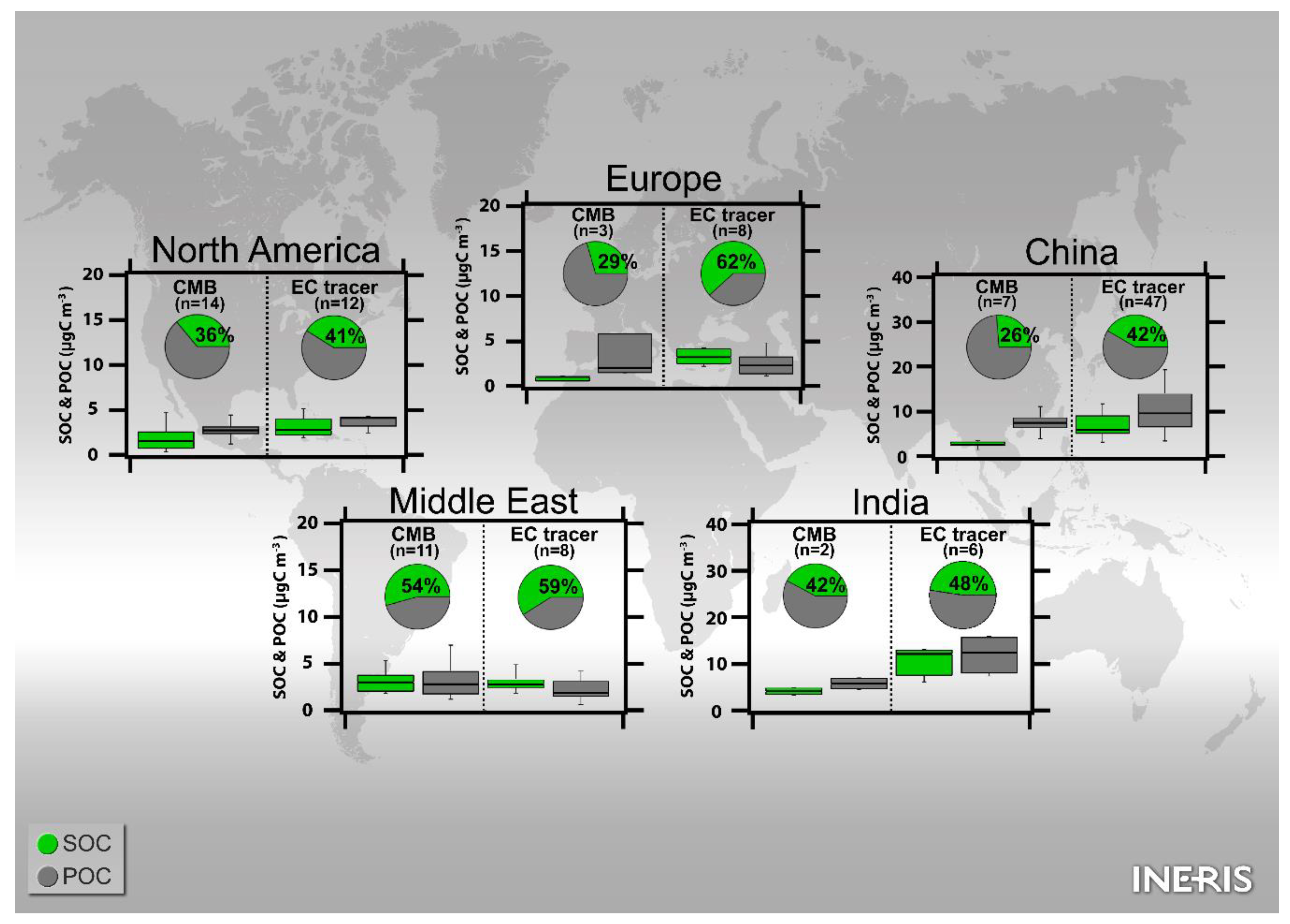{kind=link}
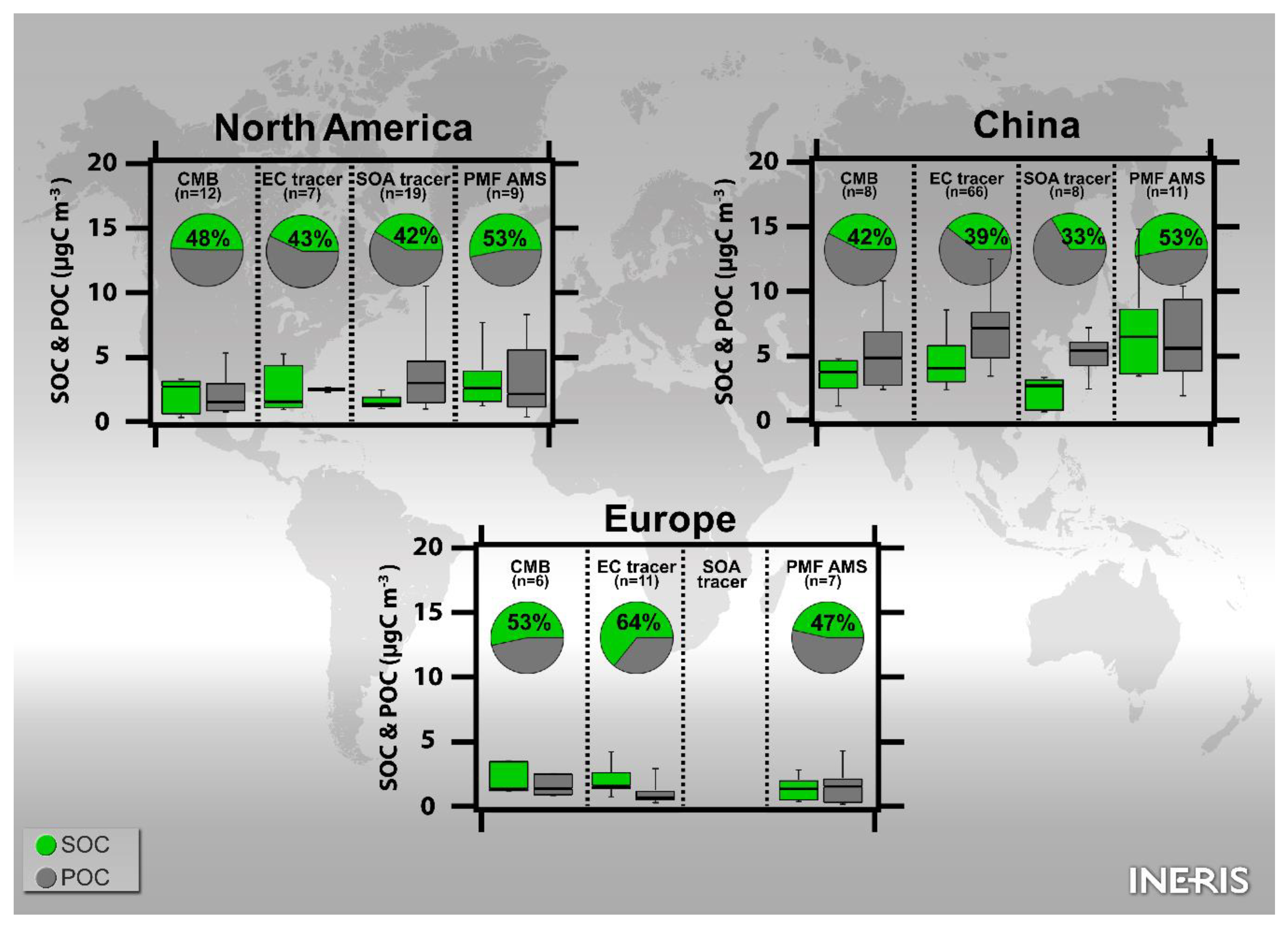{kind=link}
| Locations | Sampling Periods | Methodologies | Main Conclusions | References | |||
|---|---|---|---|---|---|---|---|
| EC-Tracer | CMB | SOA-Tracer | PMF | ||||
| Pittsburgh, Pennsylvania (USA) | Annual (July 2001–July 2002) | X | X | Good agreement (r2 = 0.71, slope = 0.75). Better in summer (r2 = 0.81, slope = 0.91) than in winter (r2 = 0.45, slope = 0.75). | [207] | ||
| London, Birmingham, Birmingham (UK) * | Summer (June 2010–August 2010) | X | X | SOC estimates were in broad agreement (urban sites, r2 = 0.70–0.92, slope = 0.80–0.92; rural site, r2 = 0.69–0.92, slope = 0.73–0.88). | [124] | ||
| Atlanta, Birmingham, Centreville, Yorkville (USA) | Annual (January 2000–December 2002) | X | X | √ 1 | Lower SOC estimates using PMF. Good correlation between SOC estimates using CMB and EC tracer method (CMB vs. EC, r = 0.67–0.84; CMB vs. PMF, r = 0.58–0.74; EC vs. PMF, r = 0.40–0.78). Comparable SOC levels (CMB vs. EC) at urban sites while at rural sites, larger SOC levels estimated using CMB. | [205] | |
| Atlanta (USA) | Summer/Winter (February 1999–December 2007) | X | X | √ 1 | Higher SOC estimates using CMB especially in winter. CMB vs. EC vs. PMF: summer: 2.0 ± 0.9 vs. 1.5 ± 1.4 vs. 1.4 ± 0.8 µg m−3; winter 1.8 ± 1.0 vs. 0.8 ± 2.0 vs. 0.9 ± 0.9 µg m−3. The highest uncertainty was obtained using the EC tracer method. The PMF uncertainties were significantly higher than the uncertainties in the CMB method. | [93] | |
| Pittsburgh (USA) | Annual (July 2001–August 2002) | X | X | √√√ 1,2 | All methods (EC tracer, CMB, PMF-filter and PMF-AMS) provided the same seasonal pattern with more SOA in summer than in winter. Summer, EC tracer vs. other approaches: 55–70% vs. 30–40% SOC in PM2.5 OC; winter, CMB vs. other approaches: 50 vs. 10% SOC in PM2.5 OC. PMF-filter vs. CMB: Non-winter: r2 = 0.55, slope = 0.72; in winter, poor correlation and low slope. | [14] | |
| Riverside (USA) | Summer (July 2005–August 2005) | X | X | √√ | SOA estimates were consistent for all the methods. Diurnal cycles of SOA/OA ratios were similar with maximum ratios observed during the early afternoon. However, the EC-tracer method apportioned SOA slightly differently throughout the evening/night. | [1] | |
| Birmingham, Centreville, Atlanta (USA) | Spring/Summer (August 2003–August 2005) | X | X | For Atlanta, SOC estimates were similar (EC vs. SOA tracer: spring: 1.4 vs. 1.3 µg m−3; summer: 1.2 vs. 1.4 µg m−3). For Birmingham and Centreville, the differences were significantly larger (on average, 1.8 vs. 2.8 µg m−3; 1.2 vs. 2.7 µg m−3, respectively). | [38] | ||
| Wangqingsha, Pearl River Delta (China) | Summer (August 2008–September 2008), Fall–Winter (November 2008–December 2008) | X | X | Good agreement in summer (r = 0.57, slope = 0.91, EC vs. SOA tracer SOC: 3.2 vs. 3.1 µg m−3) and better than in fall–winter (EC vs. SOA tracer SOC: 6.7 vs. 2.0 µg m−3). The minimum OC/EC ratio could be not representative of (OC/EC)p in winter season (biomass burning impacted). In fall–winter, other SOA precursors (“non-traditional” SOA) were probably significant but not considered in the SOA tracer method. | [178] | ||
| Marseille (France) | Summer (June 2008–July 2008) | X | X | Both methods followed different temporal trends (only biogenic SOC was considered for the SOA tracer method) (r2 = 0.18). CMB vs. SOA tracer: 2.1–8.5 vs. 0.0–0.6 µg m−3. | [41] | ||
| Hong Kong, (China) | Summer (July 2006–August 2006) | X | X | √ 1,2 | SOC estimates showed very similar time evolutions throughout the sampling period. The average SOC from CMB, PMF-filter and SOA tracer method were: 7.8, 6.8 and 5.0 µg m−3 during high pollution episodes (regional transport) and 1.2, 0.7 and 0.5 µg m−3 under the influence of local emissions (local days). | [40] | |
| Detroit, Cincinnati, East St. Louis, Northbrook, Bondville (USA) | Annual (March 2004–February 2005) | X | √ 2 | SOA estimates were highly consistent except for few months with high secondary contributions (r2 = 0.76, slope = 1.01). Underestimation by PMF when the secondary contributions were very low. | [26] | ||
| Shanghai (China) 2 sites: 1 urban + 1 suburban | Winter (January 2010, January 2011)/Spring (April 2010–May 2010)/Summer (July 2010)/Autumn (October 2010–November 2010) | X | X | √ 1,2 | SOA contributions might be underestimated with the SOA tracer method (only terpenes and aromatic compounds considered). EC vs. SOA: fall–winter: 2.8–8.8 vs. 0.1–0.4 µg m−3; spring–summer 1.5–2.2 vs. 0. 1–0.6 µg m−3. PMF (2.1–2.8 µg m−3) with no variation: a large part of the SOC was associated with nitrate and sulfate but not with the measured SOA tracers. SOA tracer and PMF-filter SOC estimates were significantly correlated (r2 = 0.68). As commercial standards for many of the tracers are not available, large uncertainty in the quantification of the SOA tracers. | [179] | |
| Atlanta (USA) | Summer (July 2001–August 2001)/Winter (January 2002) | X | √ 1 | Good correlation between SOC estimates (CMB vs. PM, r2 = 0.43–0.50, slope = 3.2–7.4). Larger SOC estimates using CMB may be due to the unresolved primary OC that would attribute to the CMB (high bias) and the SOA from the resolved primary sources that have not been included in the PMF SOA (low bias). | [303] | ||
| Hong Kong (China) 10 sites: 9 urbans + 1 traffic | 1998–2002 | X | √ 1 | The SOC estimates by the EC tracer method were consistently higher than PMF method. Overestimation by 70–212% for the summer samples and by 4–43% for the winter samples. The overestimation by the EC tracer method resulted from the inability of obtaining a single OC/EC ratio that represented a mixture of primary sources varying in time and space. | [98] | ||
| Mexico City (Mexico) | Spring (March 2006) | X | √√ | Better agreement using CMB estimates corrected from PMF LOA factor (local low nitrogen OA): 49% (PM2.5 OC) vs. 46% (PM1 OC); r2 = 0.40, slope = 1.01. | [348] | ||
| Guangzhou (China) | Summer (July 2006) | X | √√ | Good correlation between SOC and OOA from PMF (r2 = 0.60) but low regression slope (0.31) indicating that there was a substantial amount of noncarbon elements (e.g., O, N) in OOA. | [168] | ||
| Jiaxing, Yangtze River Delta (China) | Summer (July 2015)/Winter (December 2015) | X | √√ | Good agreement in summer with similar time trends (PMF vs. EC tracer: 7.2 vs. 6.8 µg m−3). In winter, strong biomass burning events led to overestimate SOA using the EC tracer method (3.9 vs. 7.0 µg m−3). | [349] | ||
| Beijing (China) | Fall (November 2013) | X | √√ | SOC estimates obtained using EC tracer method were consistent with those from PMF (r2 = 0.69) with very similar concentration levels. | [153] | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, D.; Favez, O.; Perraudin, E.; Villenave, E.; Albinet, A. Comparison of Measurement-Based Methodologies to Apportion Secondary Organic Carbon (SOC) in PM2.5: A Review of Recent Studies. Atmosphere 2018, 9, 452. https://doi.org/10.3390/atmos9110452
Srivastava D, Favez O, Perraudin E, Villenave E, Albinet A. Comparison of Measurement-Based Methodologies to Apportion Secondary Organic Carbon (SOC) in PM2.5: A Review of Recent Studies. Atmosphere. 2018; 9(11):452. https://doi.org/10.3390/atmos9110452
Chicago/Turabian StyleSrivastava, Deepchandra, Olivier Favez, Emilie Perraudin, Eric Villenave, and Alexandre Albinet. 2018. "Comparison of Measurement-Based Methodologies to Apportion Secondary Organic Carbon (SOC) in PM2.5: A Review of Recent Studies" Atmosphere 9, no. 11: 452. https://doi.org/10.3390/atmos9110452