3.1. Methods and Models
Simulations were made to illustrate important chemical processes relevant to air quality in this study. They were performed using a box modeling system described previously with the RACM2 mechanism [
31,
33]. All simulations were made with gas-phase chemistry for clear sky and constant meteorological conditions. Physical loss processes, such as deposition, were not included. The first set included simulations of simple mixtures without emissions. A second set of simulations were made for a polluted urban case was simulated that included emissions.
The rate of photolysis of O3 and any other compound is the product of the compound’s concentration and the reaction’s photolysis frequency, J. The photolysis frequency is the integral of the product of the wavelength dependent, λ, spherically integrated actinic flux, I(λ), the absorption cross-section, σ(λ), and the quantum yield, φ(λ), (probability that molecule will react after absorbing a photon of wavelength λ), Equation 1.
The photolysis rate coefficients for the photochemical reactions were calculated using the delta-Eddington radiative transfer model [
34]. The RACM2 mechanism includes 33 photolysis reactions. The absorption cross-sections and quantum yields have been revised to be consistent with recent recommendations [
7,
8]. Aldehydes that have photolysis reactions included in RACM2 are formaldehyde, acetaldehyde, a higher aldehyde, unsaturated aldehyde (formed from aromatic oxidation) and benzaldehyde. Two photolysis reactions are included for peroxyacetyl nitrate. The ketones with photolysis reactions in RACM2 are acetone, methethylketone, methylvinylketone and a higher ketone. The product yields glyoxal, methyl glyoxal and higher dicarbonyl species have been revised for RACM2 based on the recent data.
All simulations discussed below were made for the surface level with the conditions given in
Table 1. Simulations were performed for an O
3 only system, with variations upon the addition of NO
x, and VOCs. Specific cases included adjusting the VOC to NO
x ratio by doubling and halving ethene concentrations,
Table 2. Simulations were made with the RACM2 mechanism with initial conditions and emissions for a polluted urban atmosphere,
Table 1 and
Table 3. Note that in
Table 3 for temperatures of 298 K and pressures of 1 atmosphere, “Slow Alkanes” in RACM2 are defined to have HO
• rate constants less than 3.4 × 10
−12 cm
3 s
−1, “Medium Alkanes” have HO
• rate constants between 3.4 × 10
−12 and 6.8 × 10
−12 cm
3 s
−1and “Fast Alkanes” have HO
• rate constants greater than 6.8 × 10
−12 cm
3 s
−1. “Internal Alkenes” have double bond in an internal position and “Terminal Alkenes” have a double bond in the terminal position.
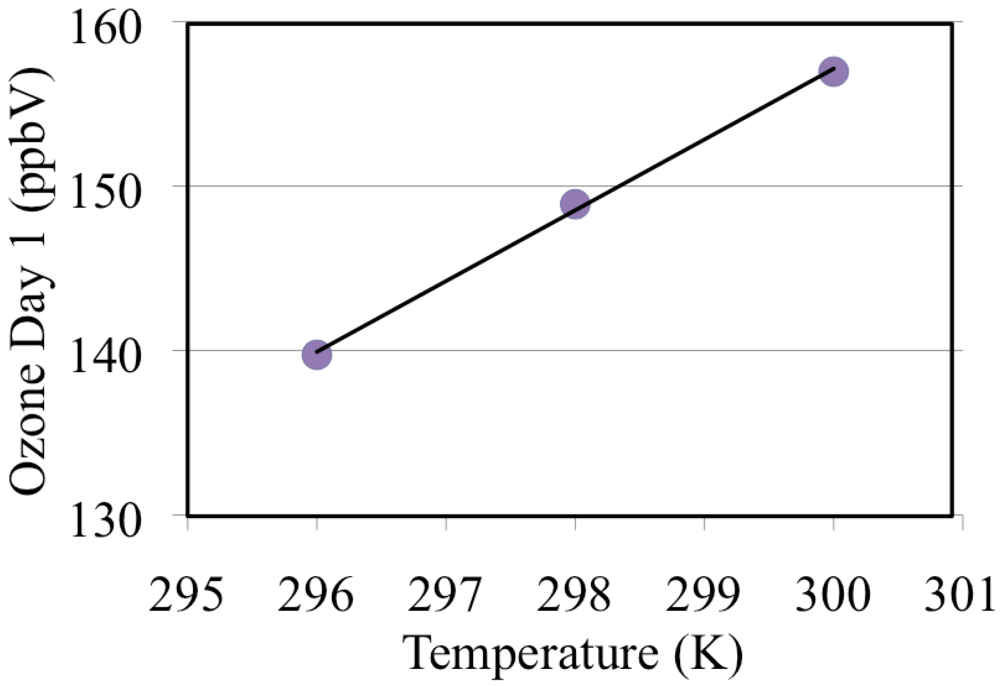
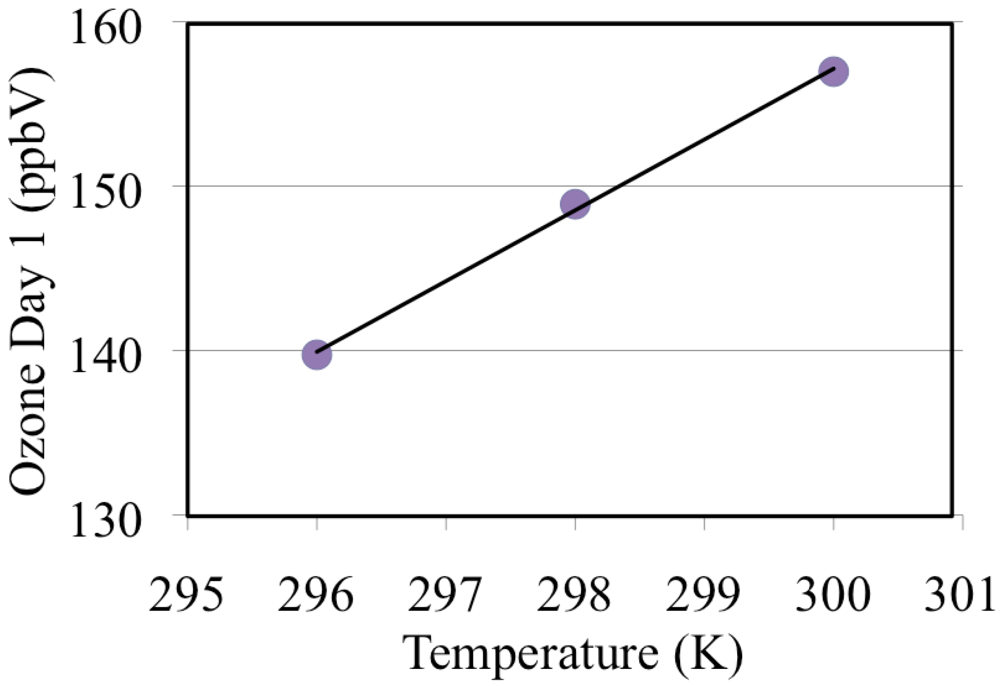
These simulations were repeated for a range of temperatures 296, 298 and 300 and these are discussed in section “3.8. The Effect of Temperature on Ozone Formation”.
Table 1.
Conditions used for all simulations.
Table 1.
Conditions used for all simulations.
| Initial Condition | Value |
|---|
| Start-Time | 6:00 |
| Duration | 48 h |
| Temperature | 298 K |
| Pressure | 1013.25 mbar |
| H2O Mixing Ratio | 15,500 ppm |
| H2 Mixing Ratio | 550 ppm |
| Date for photolysis calculation | 21 June |
| Latitude | 40° |
Table 2.
Simplified simulations made to illustrate atmospheric chemistry mechanisms.
Table 2.
Simplified simulations made to illustrate atmospheric chemistry mechanisms.
| Case | O3 (ppb) | NO2 (ppb) | Ethene (ppb) |
|---|
| O3 only | 30 | 0 | 0 |
| NO2 only | 0 | 30 | 0 |
| Ethene 4 * | 30 | 2.5 | 5 |
| Ethene 8 * | 30 | 2.5 | 10 |
| Ethene 16 * | 30 | 2.5 | 20 |
Table 3.
Initial concentrations and emissions used for the simulation of a polluted urban atmosphere. The initial VOC to NOx ratio is 19.
Table 3.
Initial concentrations and emissions used for the simulation of a polluted urban atmosphere. The initial VOC to NOx ratio is 19.
| Species | Initial Concentration (ppb) | Emission Rate (ppb min−1) |
|---|
| Inorganic | | |
| Ozone | 30 | --- |
| Nitric Oxide | 8. | 2.77 × 10−3 |
| Nitrogen Dioxide | 2 | 5.43 × 10−4 |
| Sulfur Dioxide | 30 | 2.94 × 10−3 |
| Carbon Monoxide | 1000 | 4.53 × 10−3 |
| Hydrogen | 550 | --- |
| Organic | | |
| Methane | 1800 | |
| Ethane | 3 | 8.03 × 10−5 |
| Slow Reacting Alkanes | 10 | 1.86 × 10−3 |
| Medium Reacting Alkanes | 2.5 | 6.81 × 10−4 |
| Fast Reacting Alkanes | 1.5 | 5.10 × 10−4 |
| Ethene | 2.0 | 6.78 × 10−4 |
| Internal Alkenes | 1.0 | 3.47 × 10−4 |
| Terminal Alkenes | 2.0 | 6.94 × 10−4 |
| Dienes | 0.5 | 1.74 × 10−4 |
| Benzene | 0.9 | 1.06 × 10−4 |
| Toluene | 2.0 | 6.05 × 10−4 |
| Xylene | 2.0 | 6.94 × 10−4 |
| o-Xylene | 1.0 | 3.44 × 10−4 |
| Methanol | 0.1 | --- |
| Ethanol | 0.1 | --- |
| Higher Alcohols | 0.1 | --- |
| Formaldehyde | 2.5 | 3.21 × 10−5 |
| Acetylene | 2.0 | 1.58 × 10−4 |
| Acetaldehyde | 1.0 | 3.15 × 10−5 |
| Higher Aldehyde | 0.5 | --- |
| Acetone | 0.3 | --- |
| Methyl Ethyl Ketone | 2.0 | --- |
| Higher Ketone | 2.0 | --- |
| Ethylene Glycol | 0.2 | --- |
| Methylglyoxal | 0.05 | --- |
| Methacrolein | 0.1 | --- |
| Methyl Vinyl Ketone | 0.1 | --- |
| Isoprene | 3.4 | 1.18 × 10−3 |
| α-Pinenes | 1.0 | 3.47 × 10−4 |
| d-Limonene | 1.0 | 3.47 × 10−4 |
3.2. The Inorganic Chemistry of Ozone Production
Ozone is a constituent of the natural troposphere due primarily to its production in the stratosphere [
35]. A fraction of stratospheric O
3 passes to the troposphere (transported by folding events, for example). Some of the O
3 is lost through photochemistry and other reactions and it is deposited to the Earth’s surface. Excited oxygen atoms, O(
1D), are produced by the photolysis of O
3.
The majority of the O(1D) are quenched to yield ground state oxygen atoms (O(3P)).
The O(3P) react with oxygen molecules to form O3, Reaction 5.
In Reaction 5, M represents molecular nitrogen, oxygen or other gaseous species that transfers excess energy from the transition state to stabilize the potential O3 molecule. Almost all of the O(3P) in the troposphere react with oxygen molecules (except in highly polluted emission plumes) because Reaction 5 is very fast in the troposphere due to its high oxygen concentrations and high pressure.
Although most of the O(1D) are quenched to reform O(3P) a fraction of the O(1D) react with water vapor to form HO•. The HO• radical is the most important oxidant found in the troposphere.
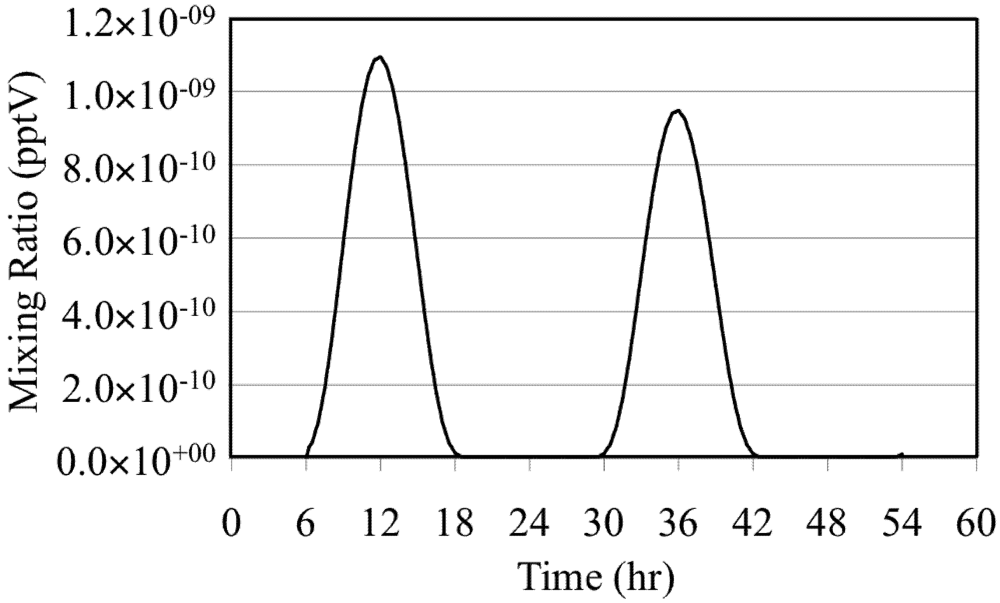
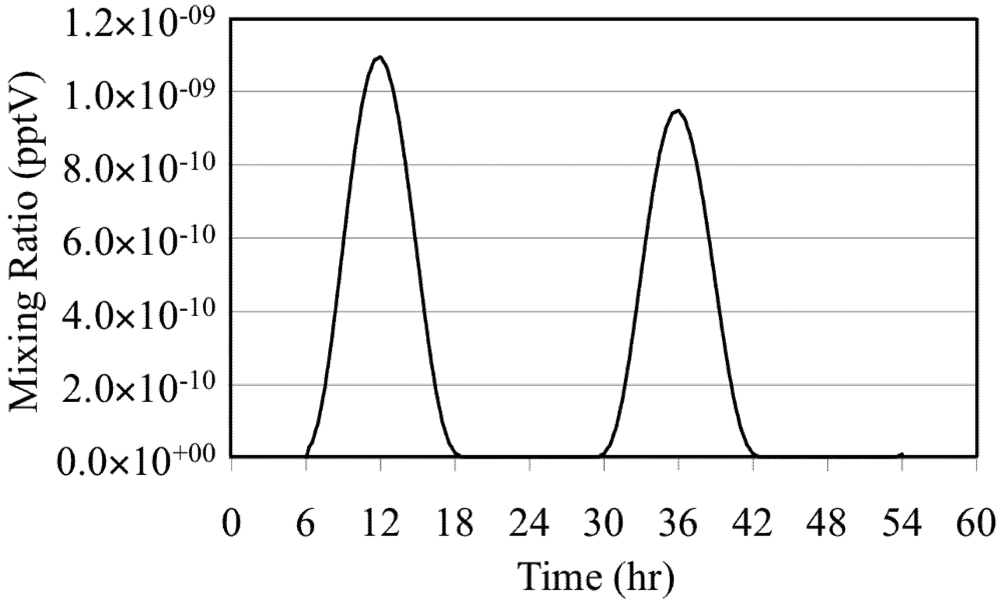
The effects of these reactions are illustrated by a simulation of a mixture of O
3 in background air without NO
x or reactive VOC. In the simulation O
3 is photolyzed during the daytime to produce O(
1D),
Figure 2. Note the extremely low mixing ratios of the O(
1D).
Figure 2.
Excited oxygen atom (O(1D)) for the O3 only case.
Figure 2.
Excited oxygen atom (O(1D)) for the O3 only case.
The mixing ratio of the O(
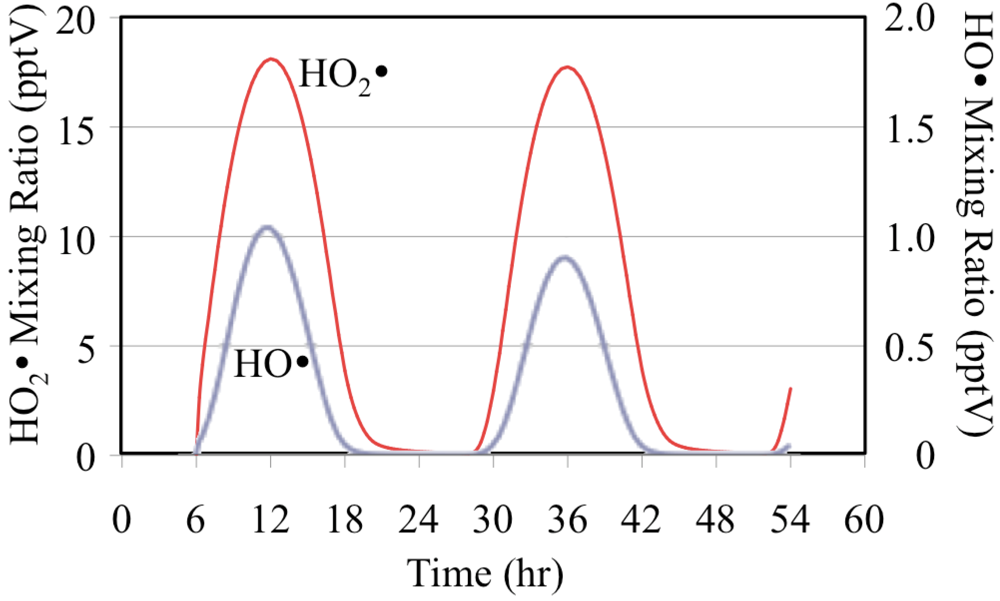
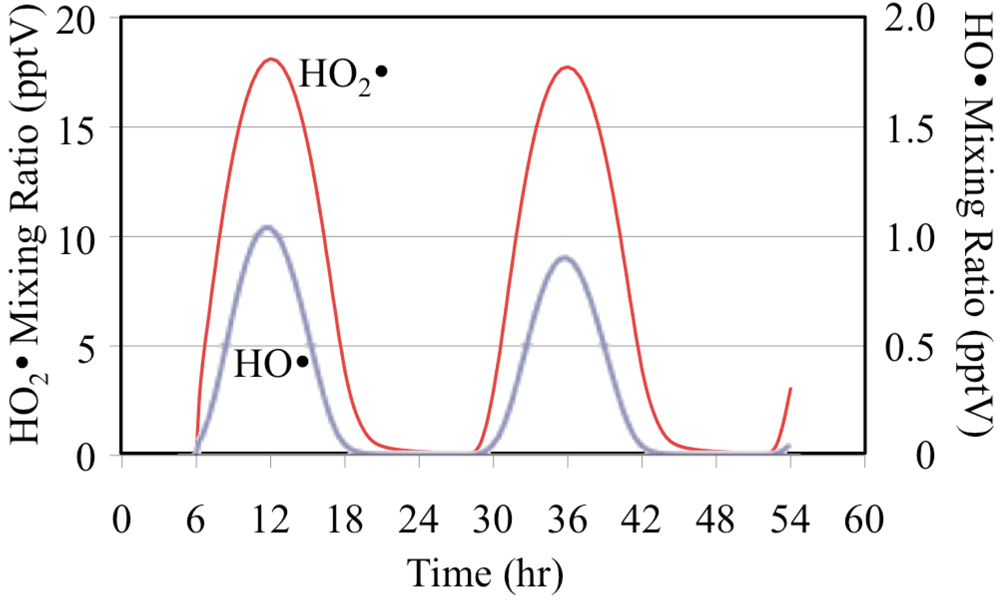
1D) follows the solar zenith angle with the peak mixing ratio occurring at the local solar noon due to its photochemical production. The HO
• reacts rapidly with O
3 to produce HO
2•,
Figure 3.
Figure 3.
HO• and HO2• radicals for the O3 only case.
Figure 3.
HO• and HO2• radicals for the O3 only case.
The net effect of the photolysis of O
3 and its reactions with HO
• and HO
2• is to reduce O
3 mixing ratios on the second day,
Figure 4. On the second day the lower mixing ratios of O
3 reduce the formation rate of O(
1D) from photolysis and therefore the O(
1D) mixing ratios are lower,
Figure 2. The lower second day O(
1D) mixing ratios reduce the mixing ratios of the HO
• radical,
Figure 3. The HO
2• mixing ratios are lower on the second day but the relative reduction is not as great for HO
2• as it is for O(
1D) and HO
•.
Figure 4.
Ozone mixing ratios for the O3 only case.
Figure 4.
Ozone mixing ratios for the O3 only case.
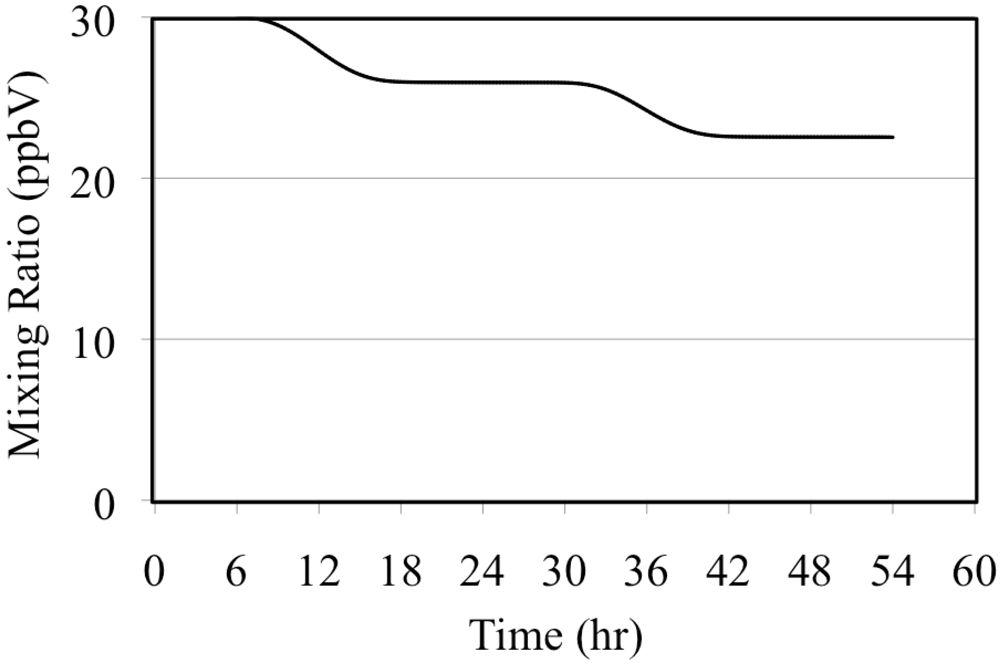
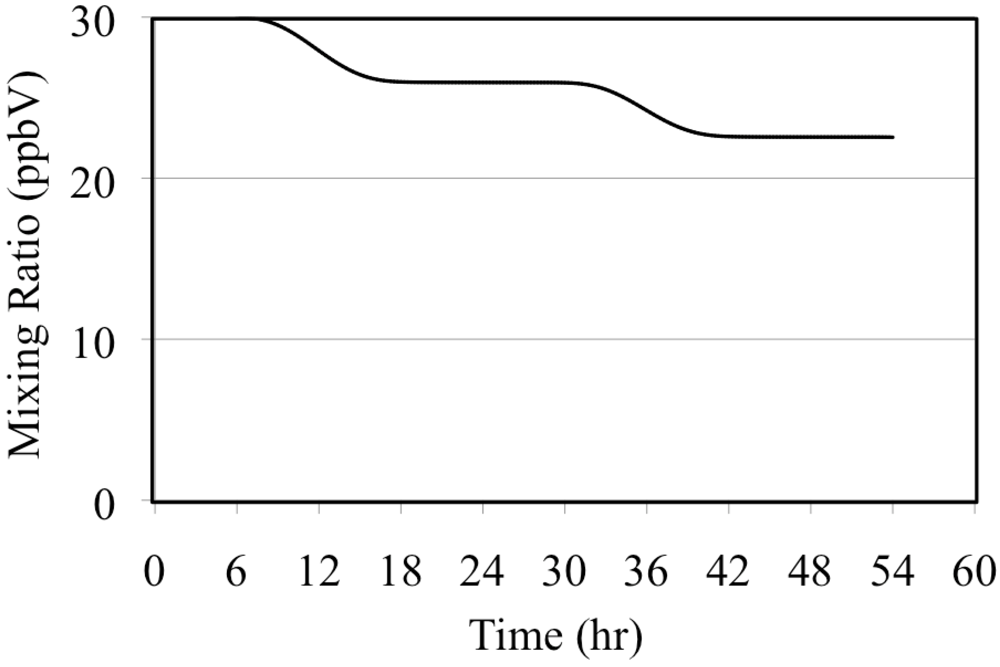
The photolysis of NO2 is the major source of O(3P). These react to produce tropospheric O3 through Reaction 5.
Reaction 9 is a major sink reaction for O3 and NO.
Reactions 8 and 9 play a major role in controlling tropospheric O
3 concentrations. As stated above, Reaction 5 is very fast in the troposphere so all of the O(
3P) produced by NO
2 photolysis can be assumed to react to produce O
3. If Reactions 8 and 9 are in equilibrium then the O
3 concentration is given by the O
3 photostationary state approximation (PSSA), Equation 10 [
36].
The brackets indicate chemical concentrations for the respective species,
JNO2 is the solar radiation dependant photolysis frequency of Reaction 8 and
k is the rate constant of Reaction 9. The value of
JNO2 depends on the intensity of solar radiation and for this reason
JNO2 follows the solar diurnal cycle. According to t34he PSSA the O
3 concentration follows the solar diurnal cycle if there is no carbon monoxide or reactive organic compounds present,
Figure 5.
Figure 5.
Ozone mixing ratios are shown for the NO2 only case.
Figure 5.
Ozone mixing ratios are shown for the NO2 only case.
3.3. The Effects of Hydrocarbons on Ozone Production
Real tropospheric O
3 production occurs when additional reactions involving CO, VOC, nitrogen oxides, HO
•, HO
2• and organic peroxy radicals (RO
2• and RCO
3•), increase the NO
2 to NO concentration ratio [
3,
36]. The reaction of HO
• radicals with CO and VOC produce peroxy radicals that convert NO to NO
2. Reaction 11 illustrates the formation of the hydroperoxy radical.
The formation of the hydroperoxy radical provides a key pathway to the “extra” conversions of NO to NO2 that are needed for O3 production.
There are many VOC compounds that are important for atmospheric chemistry [
3,
35]. For example, alkanes are hydrocarbons (containing only hydrogen and carbon atoms) and the atoms are bonded together with only single bonds. Methane (CH
4) is the simplest example of an alkane. Other examples include ethane (CH
3CH
3), propane (CH
3CH
2CH
3),
n-butane (CH
3CH
2CH
2CH
3) and tertiary-butane (CH
3)
3CH. The hydrogen atoms on the CH
3, CH
2 and CH groups are called primary, secondary and tertiary hydrogen atoms, respectively.
Alkanes react with HO
• through abstraction of a hydrogen atom, leading to the production of hydroperoxy radicals and organic peroxy radicals [
3]. Methane reacts with HO
• and it abstracts a hydrogen atom to form a methyl radical and water.
The methyl radical rapidly reacts with oxygen to produce a methyl peroxy radical.
The methyl peroxy radical reacts with NO to convert it to NO2.
The methoxy radical CH3O•, reacts with oxygen to form formaldehyde and a hydroperoxy radical.
The HO2• radical reacts with NO to make one more NO to NO2 conversion through Reaction 12. The net effect of the initial reaction of the HO• radical with an alkane is to produce several NO to NO2 conversions and these conversions increase the [NO2]/[NO] ratio by Equation 10, and so the concentration of O3 increases.
For alkanes with more complicated structures HO• may abstract any of an alkane’s hydrogen atoms but both reactivity differences between the different kinds of hydrogen atoms and the number of the kinds of hydrogen atoms affect the quantity of the different possible products. For alkanes, tertiary hydrogen atoms are more reactive than secondary and secondary hydrogen atoms are more reactive than primary hydrogen atoms. Reactions 17 and 18 illustrate the two possible channels for the reaction of n-butane with HO•.
Primary Hydrogen Abstraction
Secondary Hydrogen Abstraction
The two different radicals react with molecular oxygen to produce a primary organic peroxy radical, Reaction 19 and a secondary organic peroxy radical, Reaction 20.
The primary organic peroxy radical reacts with NO to produce a primary alkoxy radical that reacts with oxygen to yield n-butanal, CH3CH2CH2CHO and a HO2• radical.
The secondary organic peroxy radical reacts with NO to produce a secondary alkoxy radical that reacts with oxygen to yield methyl ethyl ketone, CH3(CO)CH2CH3 and a HO2• radical.
Alkenes are hydrocarbons with at least one double bond. Alkenes lead to greater O3 production than alkanes because of their higher rate constant for the reaction with HO•. Ethene, CH2=CH2, is the simplest example of an alkene while propene, CH3CH=CH2, is the next higher compound in the series. An alkene’s rate constant for its reactions with HO•, O3 and the resulting reaction products, depends very strongly on the location and number of double bonds in the alkene. For example, butene may have the double bond located at the end of the molecule, CH2=CHCH2CH3 (1-butene), or within the molecule, CH3CH=CHCH3 (2-butene). In this case the rate constant for the reaction of HO• with 2-butene is greater than its reaction with 1-butene.
In contrast to alkanes, the HO• reacts with alkenes through addition to either carbon atom of the double bond. For example, propene adds HO• to produce a radical that adds an oxygen molecule to form a peroxy radical (CH3(HCOH)CH2O2•), Reactions 25 and 26.
The CH3(HCOH)CH2O2• radical reacts with NO to produce NO2 and a hydroxy-carbonyl compound, Reactions 27 and 28.
The net effect is to convert NO to NO2 and this produces more O3 as discussed above.
The reaction of alkenes with O
3 forms many products including carbonyl compounds and HO
• [
4,
37,
38,
39]. The reactions of alkenes with O
3 are a small but dominant nighttime source of HO
•. For example, the O
3 molecule inserts itself across the double bond of propene, Reaction 29.
The CH3CH2OOOCH2product fragments through two different reactions to make acetaldehyde, formaldehyde, CH2O, and excited Criegee radicals, [•CH2OO•]≠ and [CH3•CHOO•]≠.
The excited Criegee radicals produce a wide variety of products, with one of the reactions producing HO
• radicals [
37].
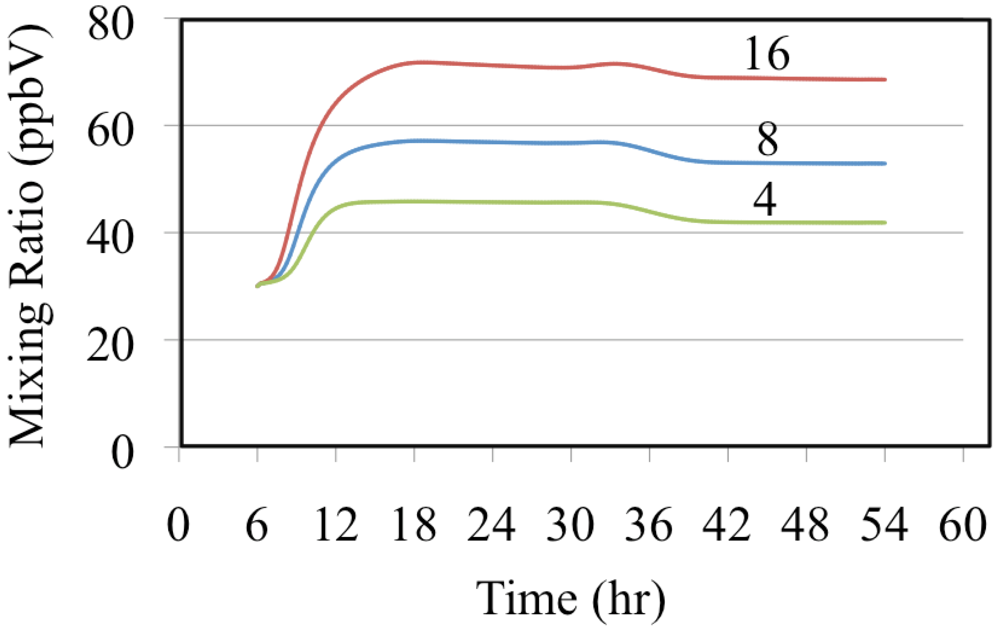
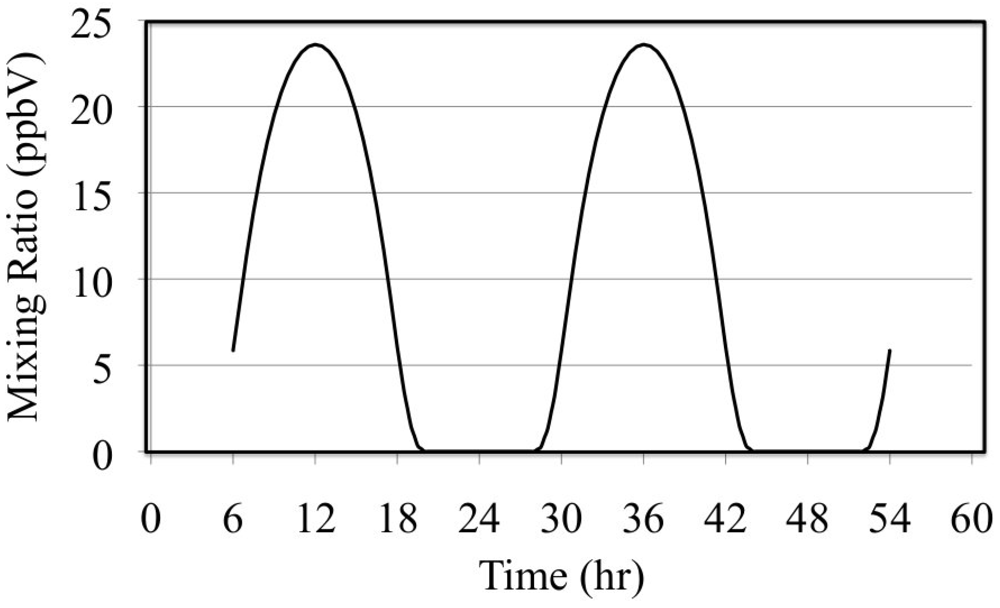
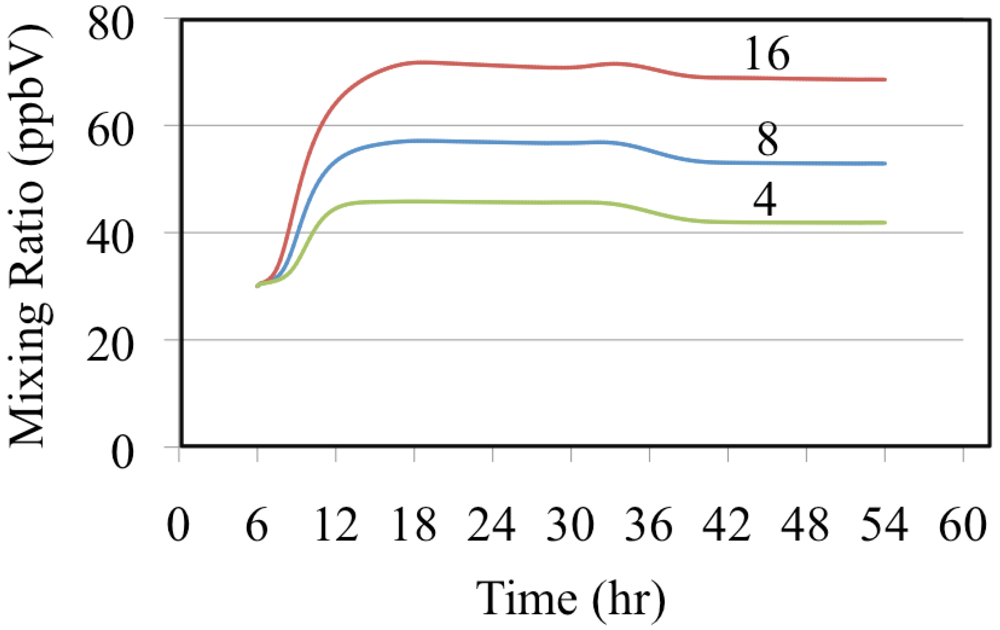
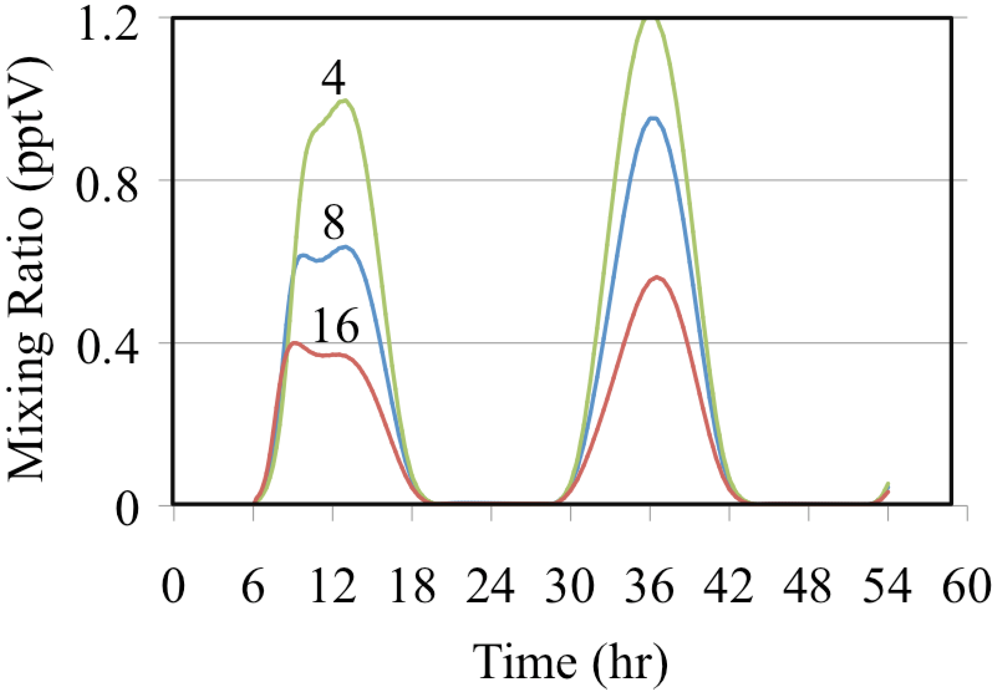
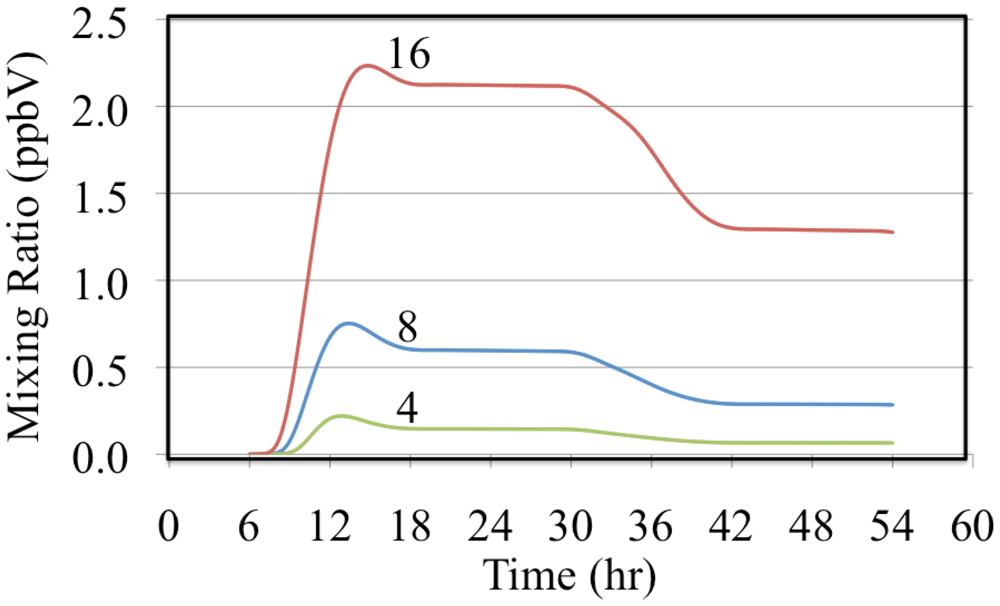
The effect of reactive organic compounds on O
3 formation is illustrated by three of simulations that initially contained O
3, ethene and NO
2. The VOC/NO
x ratios were 4, 8 and 16 ppbC/ppbN. The production of ozone for the chosen initial concentration of NO
2 was VOC limited because the ozone mixing ratio increased with increasing initial concentrations of ethene,
Figure 6.
Figure 6.
Ozone mixing ratios are shown for the ethene cases.
Figure 6.
Ozone mixing ratios are shown for the ethene cases.
Figure 6 shows that for these cases O
3 was formed rapidly on the first day but on the second day the air was aged and there was a small loss of O
3. Notice that the rate of O
3 formation during the morning of the first day depends on the VOC/NO
x ratio. The mixture with a VOC/NO
x ratio of 16 ppbC/ppbN forms O
3 between 8:00 and 10:00 at a rate of 9.2 ppb h
−1 while for VOC/NO
x ratios of 8 and 4 ppbC/PPbN the rates are 6.7 and 3.7 ppb h
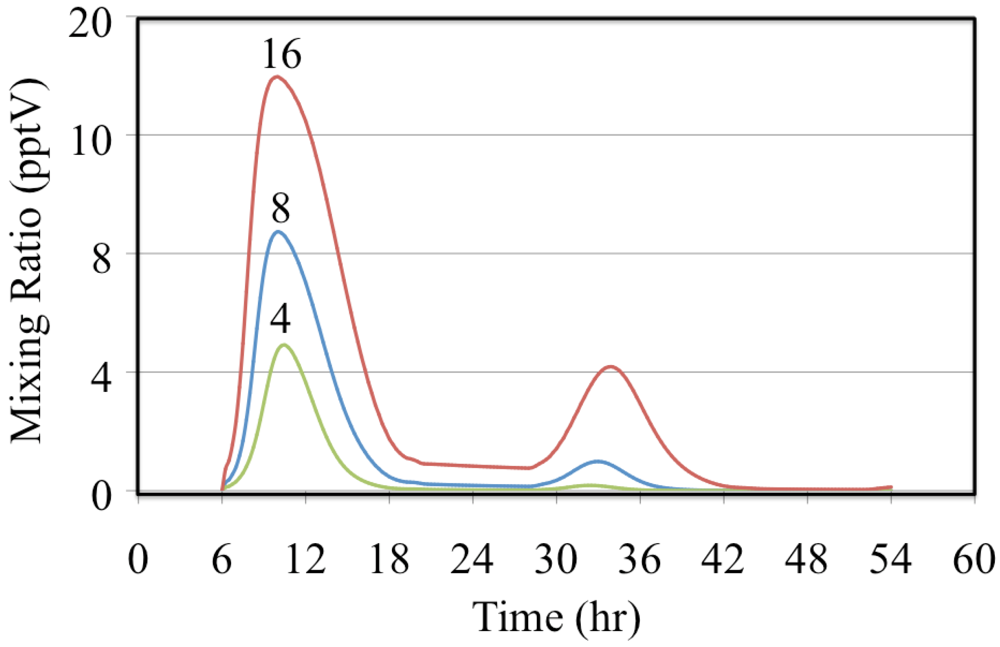
−1, respectively. The presence of ethene leads to the production of organic peroxy radicals (CH
2OH-CH
2O
2•, in this case),
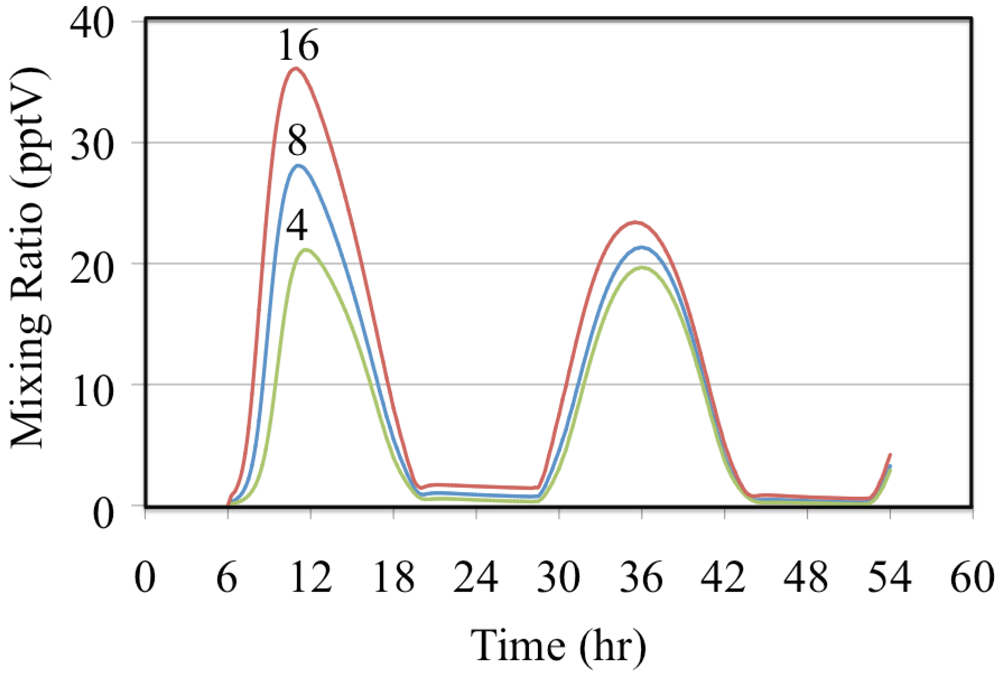
Figure 7 and HO
2• radicals,
Figure 8. Greater VOC/NO
x ratios lead to higher mixing ratios of peroxy radicals, CH
2OH-CH
2O
2 (in this case), and increased production of O
3 due to more rapid conversion of NO to NO
2.
Figure 7.
The CH2OH-CH2O2•, radical mixing ratios are shown for the ethene cases. The number indicates the initial VOC/NOx ratio in ppbC/ppbN.
Figure 7.
The CH2OH-CH2O2•, radical mixing ratios are shown for the ethene cases. The number indicates the initial VOC/NOx ratio in ppbC/ppbN.
Figure 8.
Hydroperoxy radical, HO2•, mixing ratios are shown for the ethene cases. The number indicates the initial VOC/NOx ratio in ppbC/ppbN.
Figure 8.
Hydroperoxy radical, HO2•, mixing ratios are shown for the ethene cases. The number indicates the initial VOC/NOx ratio in ppbC/ppbN.
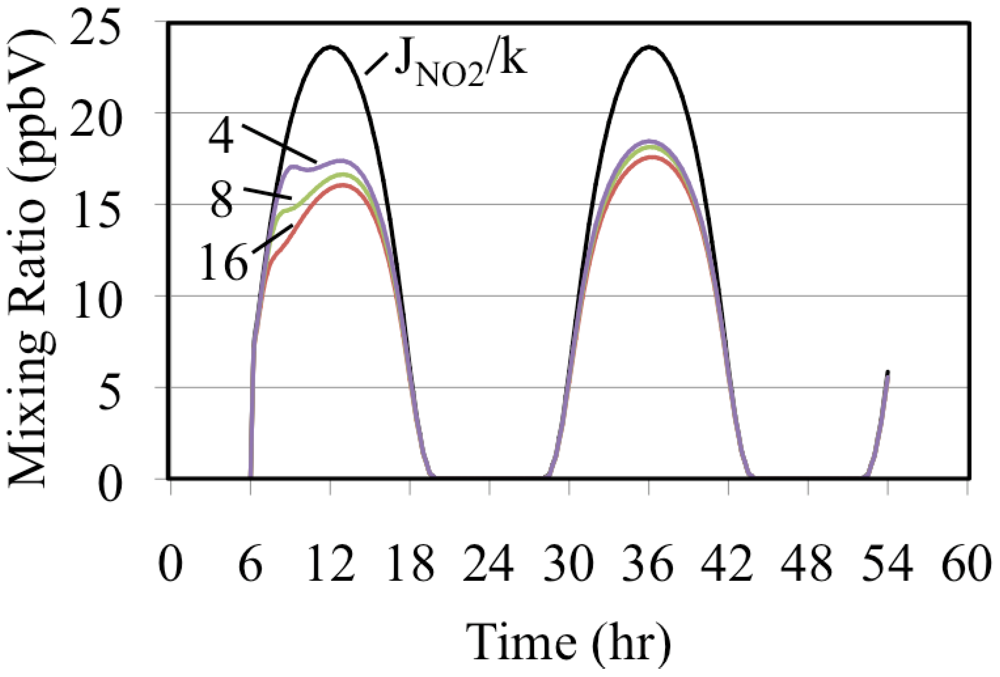
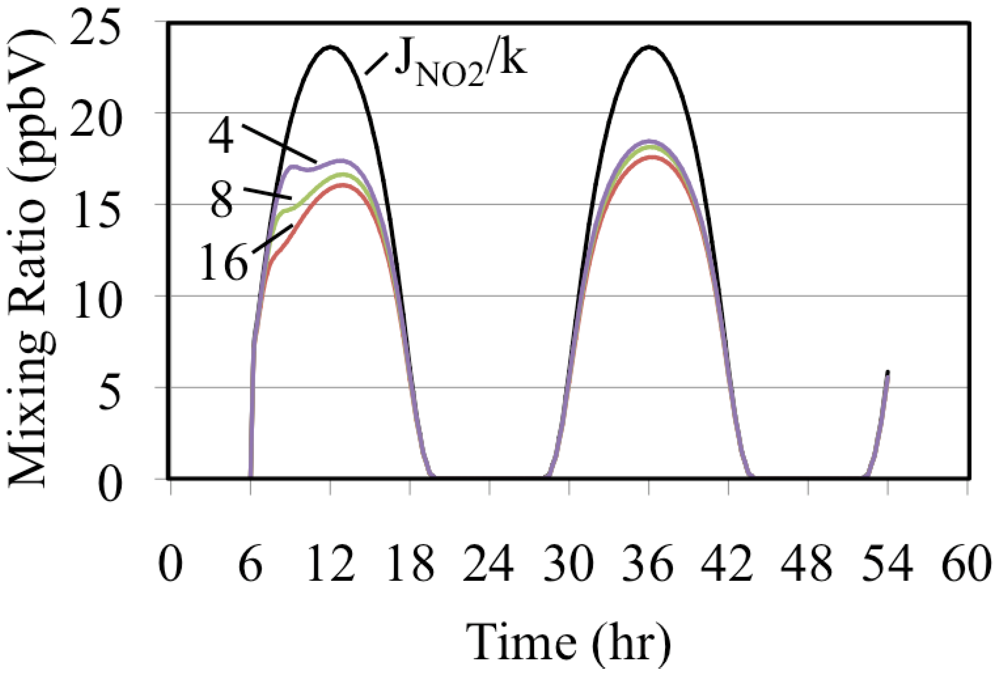
The formation of O3 occurs as a deviation from the PSSA, Equation 10. Equation 10 can be rearranged to give Equation 33.
If a simulated atmosphere obeyed the PSSA, the left hand and right hand sides of Equation 33 would be equal. Since
JNO2/
k, time dependent ratio of the photolysis frequency of NO
2 to the rate constant for the O
3 + NO reaction, is fixed for a given set of conditions it can be used as a basis for comparison with the ratio, [O
3] × [NO]/[NO
2].These are plotted on
Figure 9 for the three simulated ethene cases.
Figure 9.
The uppermost curve is the time dependent NO2 photolysis frequency divided by the rate constant for the O3 + NO reaction. The lower plots are the ratio, [O3] × [NO]/[NO2], for the three ethene cases. The number indicates the initial VOC/NOx ratio in ppbC/ppbN.
Figure 9.
The uppermost curve is the time dependent NO2 photolysis frequency divided by the rate constant for the O3 + NO reaction. The lower plots are the ratio, [O3] × [NO]/[NO2], for the three ethene cases. The number indicates the initial VOC/NOx ratio in ppbC/ppbN.
Figure 9 shows that the greater the initial mixing ratios of ethene the greater the deviation from the PSSA. Comparison of
Figure 6 and
Figure 9 show that the greater the deviation from the PSSA the greater the formation. This result is in accord with the higher mixing ratios of peroxy radicals that are associated with higher VOC/NO
x ratios. Increased peroxy radical concentrations provide faster conversion of NO to NO
2. This increases the production rate of O
3 by the increasing the NO
2 photolysis rate. The conversion of NO to NO
2 reduces the O
3 loss rate by reducing the rate of the O
3 reaction with NO. Increasing the O
3 production rate and decreasing its destruction rate at the same time has the net effect of increasing O
3 mixing ratios. The greater the O
3 production the greater the deviation from [O
3] × [NO]/[NO
2] for these reasons [
36].
HO
2• and HO
• radicals are in equilibrium and the partitioning between the concentrations of these two radicals depends on the NO concentration. In this case higher mixing ratios of HO
2• are associated with lower mixing ratios of HO
•,
Figure 8 and
Figure 10.
Figure 10.
Hydroxyl radical, HO•, mixing ratios are shown for the ethene cases. The number indicates the initial VOC/NOx ratio in ppbC/ppbN.
Figure 10.
Hydroxyl radical, HO•, mixing ratios are shown for the ethene cases. The number indicates the initial VOC/NOx ratio in ppbC/ppbN.
Aromatic compounds are the third type of hydrocarbon that is important for air pollution, and is the subject of ongoing research. The oxidation mechanism of aromatic compounds leads to the production of peroxy radicals and high molecular weight compounds that may condense to produce secondary organic aerosols. The chemistry of oxidation of aromatic compounds is very complicated and there are too many compounds and reactions to include in a mechanism to be routinely used for air quality modeling. There are at least several hundred reactions and products for a parent aromatic compound [
40]. The chemistry of a reaction of the HO
• radical with an organic substituent attached to an aromatic ring is the easiest to describe. However, only about 10% of the hydroxy radicals abstract hydrogen atoms from an alkyl group, attached to an aromatic ring. In this case the chemical mechanism will be that of the substituent group. For example, when HO
• reacts with the methyl group of toluene, the subsequent chemistry is similar to alkanes. A hydrogen atom is abstracted and an oxygen molecule adds to the CH
2• radical on the aromatic ring to make peroxy radical. The peroxy radical may react with NO to produce benzaldehyde.
The addition of HO
• to an aromatic ring is the dominant aromatic reaction [
40]. The subsequent reactions that follow this addition reaction may lead to either the breaking of the aromatic ring or ring-retaining products. In the case of the simplest aromatic compound, benzene, phenol is a ring-retaining product that is produced in high yield. Bloss
et al. [
41] supports a phenol yield of 0.52 while Berndt and Böge [
42] determined the yield of phenol to be 0.61 ± 0.07 in the presence and absence of NO
x.
A very large number of highly reactive compounds result when the ring breaks. The reactive compounds include a large number of dicarbonyl compounds that contain two carbonyl groups (C=O), are produced. These dicarbonyl compounds have a complicated and relatively unknown chemistry. Studies regarding the ring-opening products of the HO•-benzene reaction are sparse. The main aspects of their formation are uncertain due to lack of good experimental techniques for their quantification, a lack of commercially available standards, and their high reactivity.
For example, Gomez Alvarez
et al. [
43] confirmed the existence of dicarbonyl production and they found that the yields of dicarbonyls could be high. Fast ring-cleavage was observed, due to a peak in observed γ-dicarbonyls shortly after the chamber was opened to sunlight. Also, high yields of dicarbonyls (e.g., glyoxal) imply a high formation rate of HO
• into the system. They found that yields of glyoxal with values of 42 ± 3% and 36 ± 2% in two successive experiments. To be able to investigate the existence of higher molecular weight dicarbonyl compounds they had to synthesize
cis- and
trans-butenedial for calibration purposes. For one experiment they found total butenedial yields of 17 ± 9% with a breakdown of 8 ± 4%
cis-butenedial and 9 ± 5%
trans-butenedial. For a second experiment they found total butenedial yields of 15 ± 6%; the breakdown was 7 ± 3% and 7 ± 3% for the
cis and
trans isomers, respectively.
3.4. The Atmospheric Chemistry of Aldehydes, Ketones and Peroxyacetyl Nitrate
Aldehydes and ketones contain a carbonyl group, C=O. As presented above, aldehydes and ketones are oxidation products of VOC and many have biogenic and anthropogenic emission sources [
9,
35]. Formaldehyde is the simplest aldehyde. Higher molecular weight aldehydes follow the template RCHO; they have one hydrogen atom attached to the carbonyl and another organic functional group attached to it. Acetone, CH
3(CO)CH
3 is the simplest ketone. Higher molecular weight ketones follow the template R
1(CO)R
2; they have two organic functional groups attached to the carbonyl and these groups maybe the same or different.
Aldehydes and ketones react with HO• by abstraction of a hydrogen atom. The overall scheme has some similarity to the alkane oxidation scheme with some exceptions. Reaction 34 shows formaldehyde as an example of one exception.
The carbonyl radical, CHO•, does not add to oxygen to produce a peroxy radical but rather, it reacts with oxygen to produce the HO2• radical.
The carbonyl group is a strong chromophore for ultraviolet radiation. The absorption of ultraviolet radiation causes photo-dissociation of these compounds and some of the reaction channels lead to the production of radicals. For example, formaldehyde has two photolysis reactions that occur in the lower troposphere. Reaction 36 yields molecular products while Reaction 37 yields radical products.
The net effect of Reaction 37 is to produce two HO2• radicals because the CHO• reacts according to Reaction 35 and the hydrogen atom reacts with oxygen to produce another HO2•.
Under highly polluted urban conditions the photolysis of formaldehyde can be a source of HOx radicals that is as important as O3.
Acetaldehyde, CH
3CHO, is the next higher molecular weight aldehyde. It reacts in the polluted atmosphere to produce peroxyacetyl nitrate, CH
3(CO)O
2NO
2, (PAN) [
3]. PAN is an important compound because it serves as a reservoir of acetyl radicals and NO
2 and it is a strong lachrymator. The mechanism of PAN formation begins with HO
• abstracting the hydrogen atom that is attached to the carbonyl group in acetaldehyde. The addition of oxygen to the resulting CH
3CO
• radical produces the acetyl peroxy radical.
PAN decomposes thermally to reproduce acetyl peroxy radicals and NO2.
PAN is in equilibrium with NO
2 and the CH
3(CO)O
2 radical at temperatures near 25 °C. PAN is much more stable at lower temperatures and the lifetime of PAN becomes longer. PAN can be transported over long distances in the upper troposphere where the temperatures are colder and its photolysis becomes important. The photolysis of PAN proceeds by two pathways. The faster pathway forms the acetyl peroxy radical and NO
2. The slower photolysis route involves the destruction of PAN by the formation of the methyl peroxy radical, CO
2 and NO
3. Higher molecular weight aldehydes form acyl peroxy radicals that may react to produce higher homologs of PAN [
3,
44].
3.6. Radical Termination, the Production of Atmospheric Acids and Hydrogen Peroxides
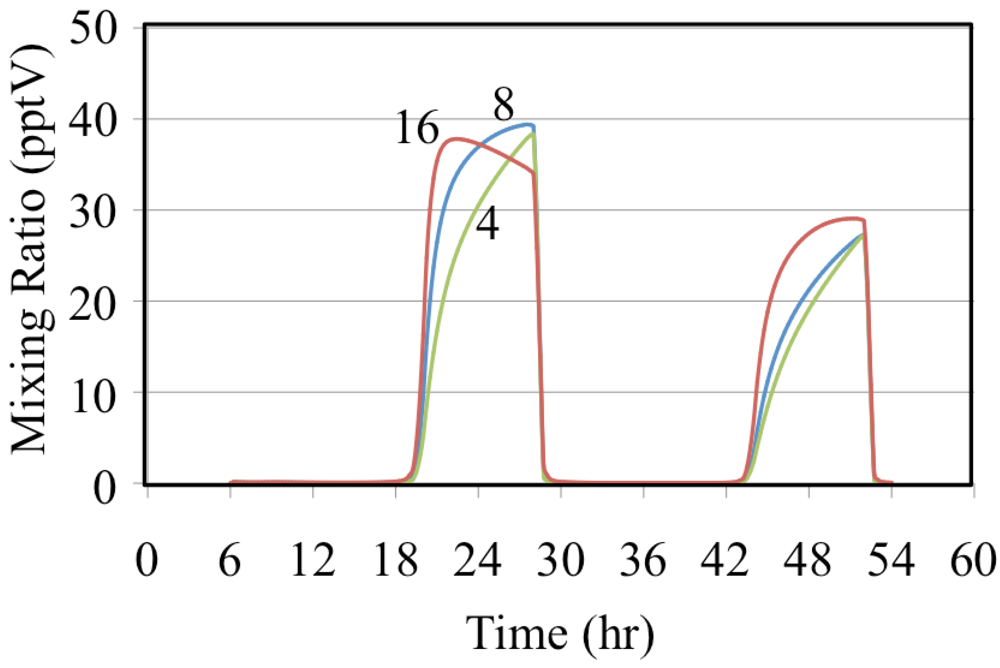
Tropospheric chemistry involves many chain reactions as discussed above. At some point the chain reactions must terminate. The reaction of HO• with NO2 to the form HNO3, Reaction 50, and the self-reaction of HO2• to form hydrogen peroxide, Reaction 51 are among the most important.
Note that the overall rate parameter for the self-reaction of HO
2• depends on atmospheric pressure and the water vapor concentration [
48].
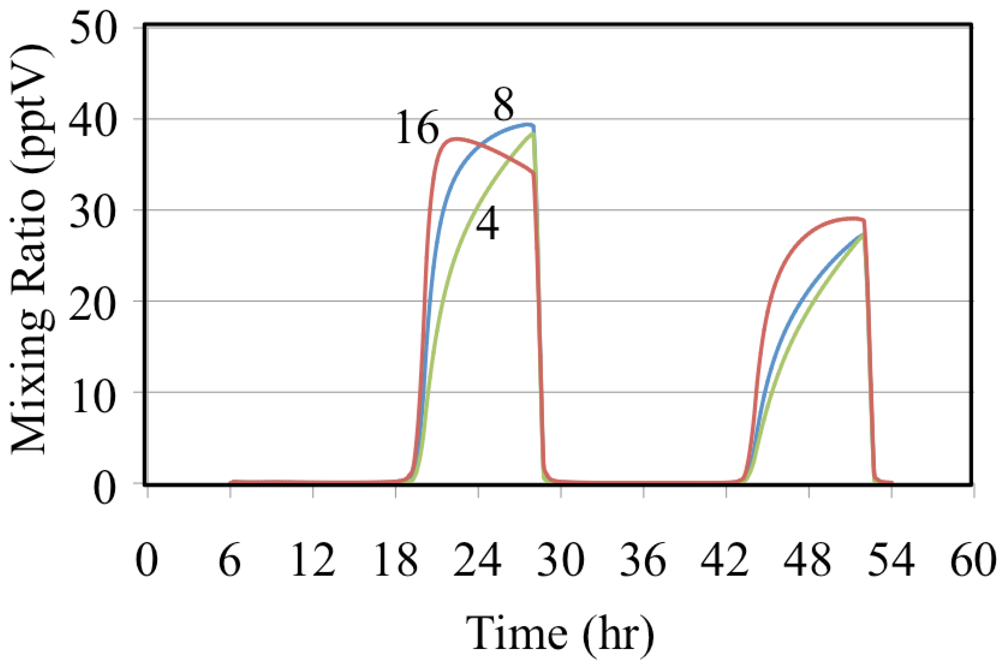
Figure 12 shows the formation of HNO
3 for the ethene cases where NO is relatively high.
Figure 12.
Nitric acid mixing ratios are shown for the ethene cases. The number indicates the initial VOC/NOx ratio in ppbC/ppbN.
Figure 12.
Nitric acid mixing ratios are shown for the ethene cases. The number indicates the initial VOC/NOx ratio in ppbC/ppbN.
Figure 12 shows that the mixing ratios of HNO
3 decrease as the VOC/NO
x ratio increases for the ethene simulations. This is consistent with the decrease in the mixing ratio of HO
• shown in
Figure 10. The reaction of HO
• with NO
2 is more important for urban conditions where NO concentrations are high while the self-reaction of HO
2• is more important for rural and remote conditions where NO concentrations are low.
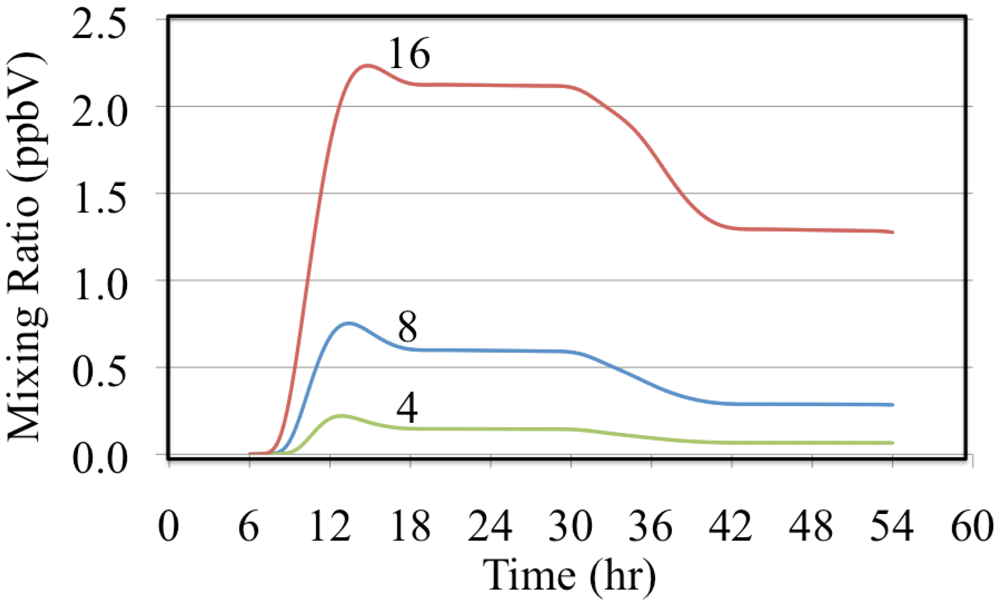
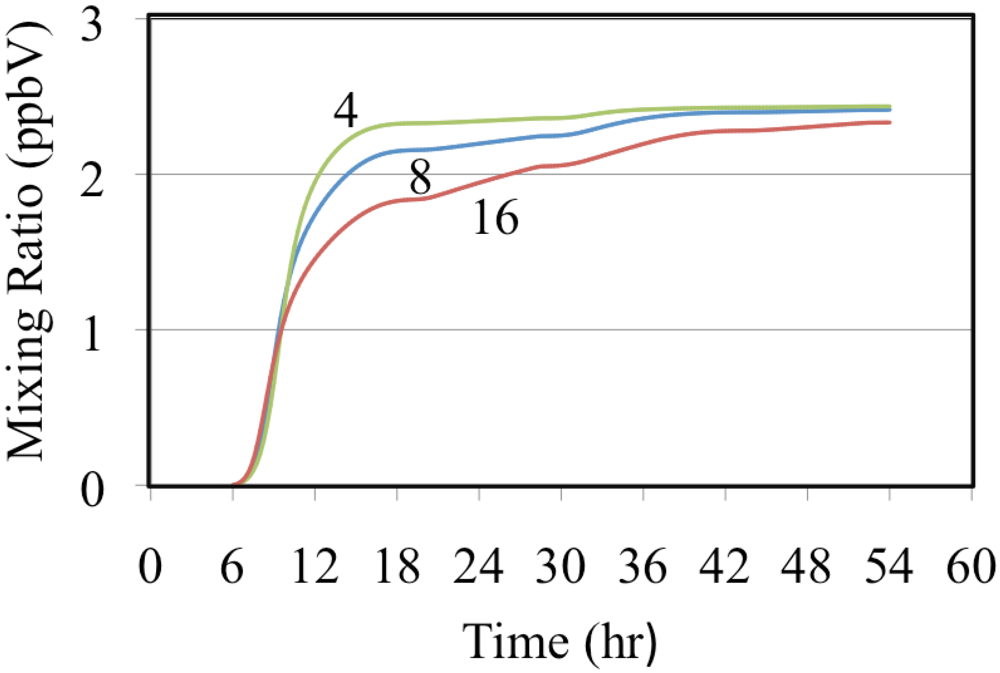
Figure 13 shows hydrogen peroxide formation for the ethene cases.
Figure 13 shows a typical trend with the mixing ratio of H
2O
2 depending on the VOC to NO
x ratio. Higher VOC concentrations relative to the NO concentration lead to the production of greater concentrations of HO
2•. Higher concentrations of HO
2• result in greater amounts of termination through the HO
2• self reaction, Reaction 51, producing more H
2O
2.
Some termination of the radical chains occurs through the reactions of HO2• with other peroxy radicals at low NO concentrations. These reactions typically lead to the production of organic hydrogen peroxides. For example the methylperoxy radical reacts with the HO2• radical to produce methyl hydrogen peroxide, CH3OOH.
Figure 13.
Hydrogen peroxide mixing ratios are shown for the ethene cases. The number indicates the initial VOC/NOx ratio in ppbC/ppbN.
Figure 13.
Hydrogen peroxide mixing ratios are shown for the ethene cases. The number indicates the initial VOC/NOx ratio in ppbC/ppbN.
The reactions of organic peroxy radicals are more complicated. For many organic peroxy radicals there are two reactions that occur. The first is the disproportion reaction that yields oxygen and alkoxy radicals.
The second type of organic peroxy radicals-radical reaction is the transfer of a hydrogen atom from a carbon atom that is adjacent to the peroxy oxygen atoms. The hydrogen atom is transferred to the oxygen atom that is adjacent to the other carbonyl group. For example, in the case of the self-reaction of the methylperoxy radical, formaldehyde and methanol are produced.
In the case of two different organic peroxy radicals there is the disproportion reaction and the transfer of the hydrogen atom that can go in both directions.
The transfer of a hydrogen atom can only go in one direction in the case of acyl organic peroxy radicals. For acyl organic peroxy radicals there is no adjacent hydrogen atom on the carbon atom adjacent to the peroxy oxygen atoms [
49]. For example the reaction of the methylperoxy radical with the acetyl peroxy radical there are two major reactions.
The CH3CO2• reacts with molecular oxygen to produce carbon dioxide and the methyl peroxy radical.
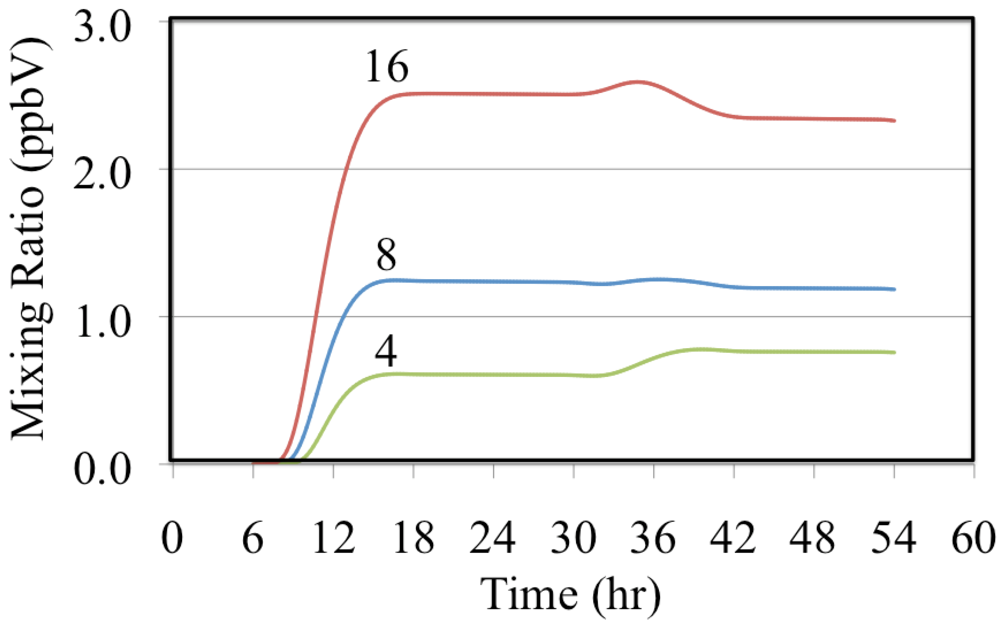
Figure 14 shows the formation of organic peroxides from the reaction of methyl peroxy radical with organic peroxy radicals to form peroxides of the form CH
3OOR.
Figure 14.
Organic peroxide mixing ratios are shown for the ethene cases. The number indicates the initial VOC/NOx ratio in ppbC/ppbN.
Figure 14.
Organic peroxide mixing ratios are shown for the ethene cases. The number indicates the initial VOC/NOx ratio in ppbC/ppbN.
The reactions of organic peroxy radicals with NO
3 radical may be highly important but there is relatively little data available as they are difficult to study in the laboratory. Organic peroxy radicals are expected to react with NO
3• as shown in Reaction 61 [
50].
But the reaction is not chain terminating because the RO will react to produce additional radicals.
Another reaction that is not chain terminating is the reaction of HO
• with SO
2, in contrast to its reaction with NO
2. The reaction of HO
• with SO
2 is an important source of sulfate and acid deposition but it does not greatly affect the atmospheric HO
x concentration. The reaction follows the following mechanism [
51].
3.7. The Behavior of a Complex Atmospheric Chemistry System
The purpose of this section is to illustrate the chemistry discussed above and to show how it applies to a more complex mixture that is closer to the real polluted atmosphere. There are important interactions between atmospheric inorganic chemistry and organic chemistry that affect the production of O
3, peroxy radicals, HNO
3 organic peroxides and many other species. These interactions are examined through the simulation of the complex mixture described in
Table 2 and
Table 3. The simulated case is a relatively realistic mixture of air pollutants with their emissions based on measurements made at Howard University’s atmospheric field site near Beltsville, Maryland.
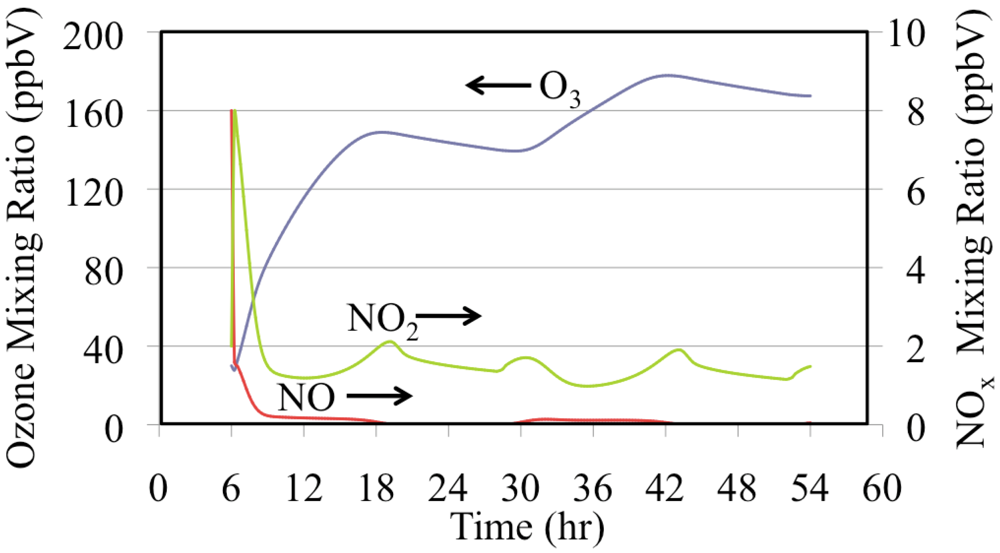
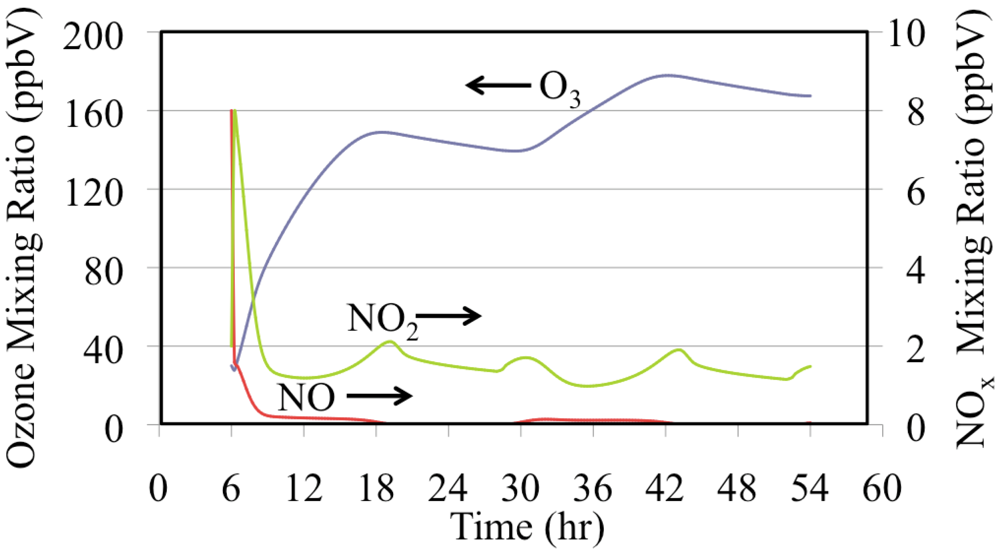
Figure 15 shows the production of O
3 from NO and NO
2. The mixing ratios of NO and O
3 were initialized to high values for this box model simulation. Under polluted urban conditions NO may accumulate near the surface during the nighttime and during the early morning rush hours. The onset of convection mixes O
3 from aloft to rapidly titrate the NO to produce NO
2. The photolysis of NO
2 produces O
3. This increase in O
3 drives down the NO mixing ratio further during the first few hours of the simulation. Emissions maintain a NO
2 mixing ratio of a few parts per billion for the entire episode. The mixing ratios of O
3 and NO
2 decrease during the nighttime due to the reaction of NO
2 with O
3 to produce NO
3 radical and N
2O
5. Nighttime chemistry converts NO
x to HNO
3, thereby removing reactive nitrogen from the atmosphere.
Figure 15.
Ozone, NO and NO2 mixing ratios are shown for the polluted urban atmosphere case.
Figure 15.
Ozone, NO and NO2 mixing ratios are shown for the polluted urban atmosphere case.
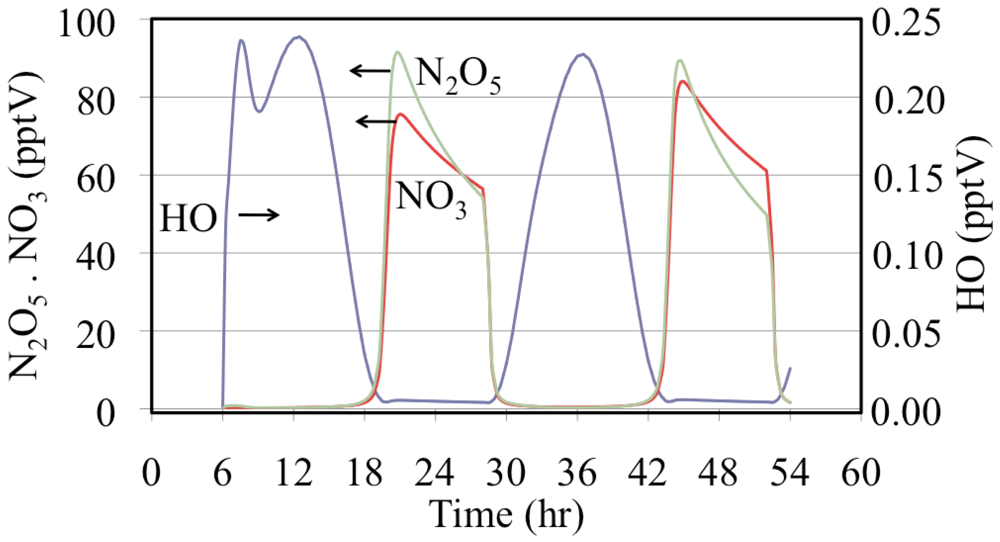
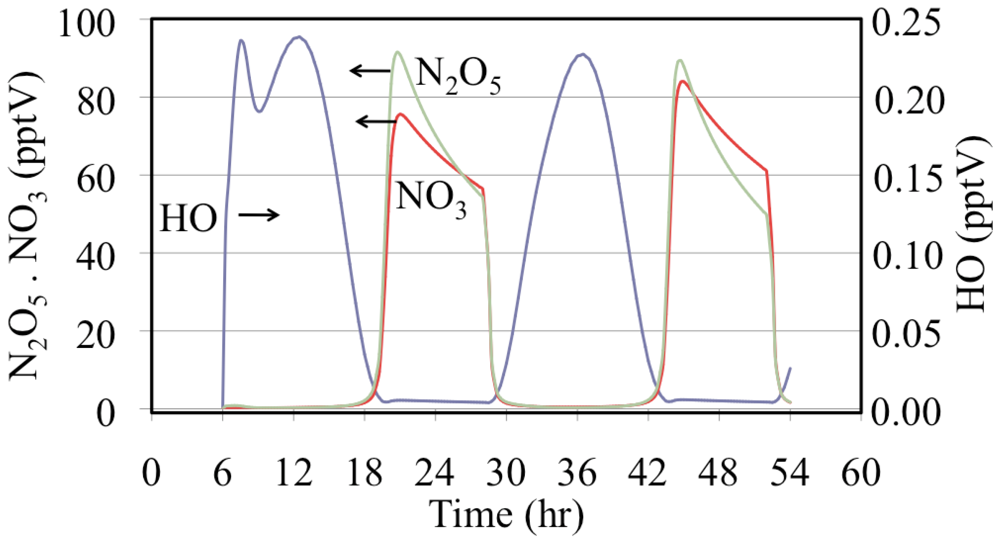
Figure 16.
Hydroxyl radical, nitrate radical and dinitrogen pentoxide mixing ratios are shown for the polluted urban atmosphere case.
Figure 16.
Hydroxyl radical, nitrate radical and dinitrogen pentoxide mixing ratios are shown for the polluted urban atmosphere case.
Figure 16 shows that after sunset the mixing ratios of N
2O
5 and NO
3 increase rapidly. Later during the night the NO
3 mixing ratios decrease due to the loss of NO
3 through titration by NO emissions and, to a lesser extent, through the reactions of NO
3 with aldehydes and alkenes. In addition N
2O
5 reacts with liquid water on aerosol and other surfaces although not included in this gas-phase simulation.
The time dependent profiles for the HO
• are more complicated than those in
Figure 10. The HO
• mixing ratios are much lower during the nighttime but they do not fall to zero due to their production from the reaction of O
3 with alkenes. Another feature is that on the first day there is a double peak due to changes between the major sources and sinks of HO
•. One change in the HO
• sink is due to a severe drop in the initial mixing ratios of biogenically emitted alkenes.
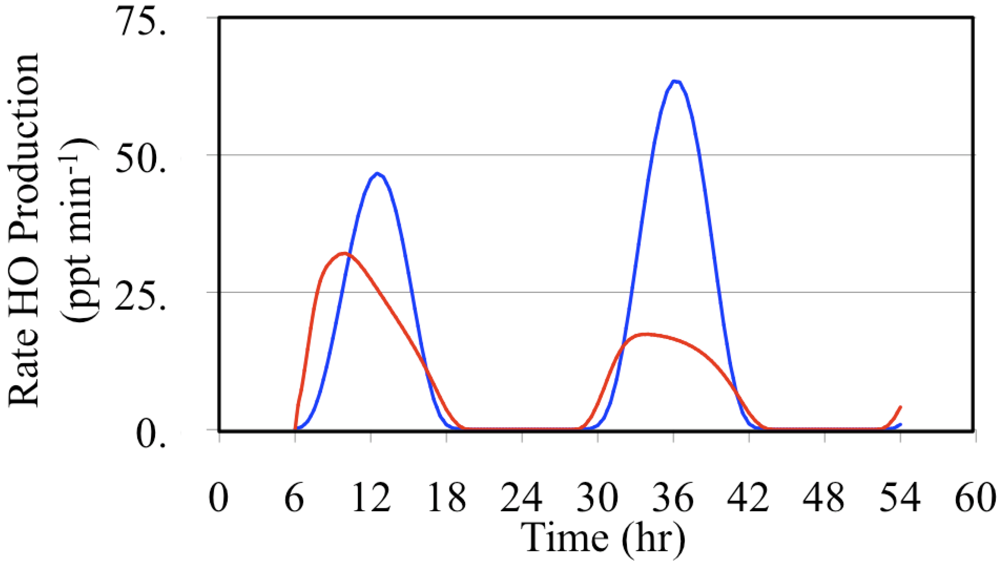
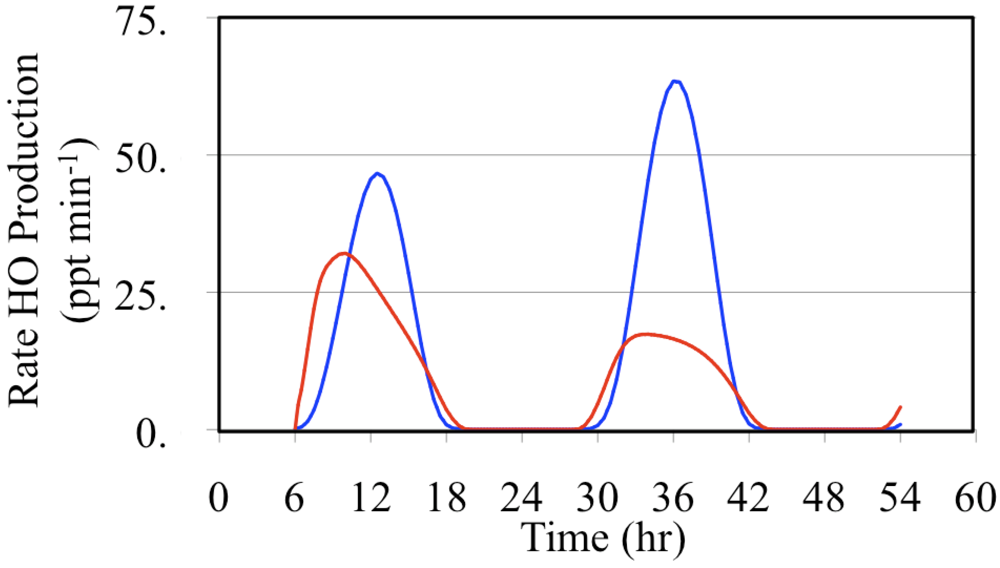
The major daytime sources of HO
• are the photolysis of formaldehyde and O
3,
Figure 17 and
Figure 18.
Figure 17 shows the production rates and
Figure 18 shows the relative production rates. In these figures the formaldehyde HO
• production rate was calculated by multiplying production rate of HO
2• from formaldehyde photolysis by the fraction of HO
2• radicals that react with NO.
Figure 17.
Production rates of HO• initiated through the photolysis of formaldehyde (red line) and the photolysis of O3 (blue line) are shown for the polluted urban atmosphere case.
Figure 17.
Production rates of HO• initiated through the photolysis of formaldehyde (red line) and the photolysis of O3 (blue line) are shown for the polluted urban atmosphere case.
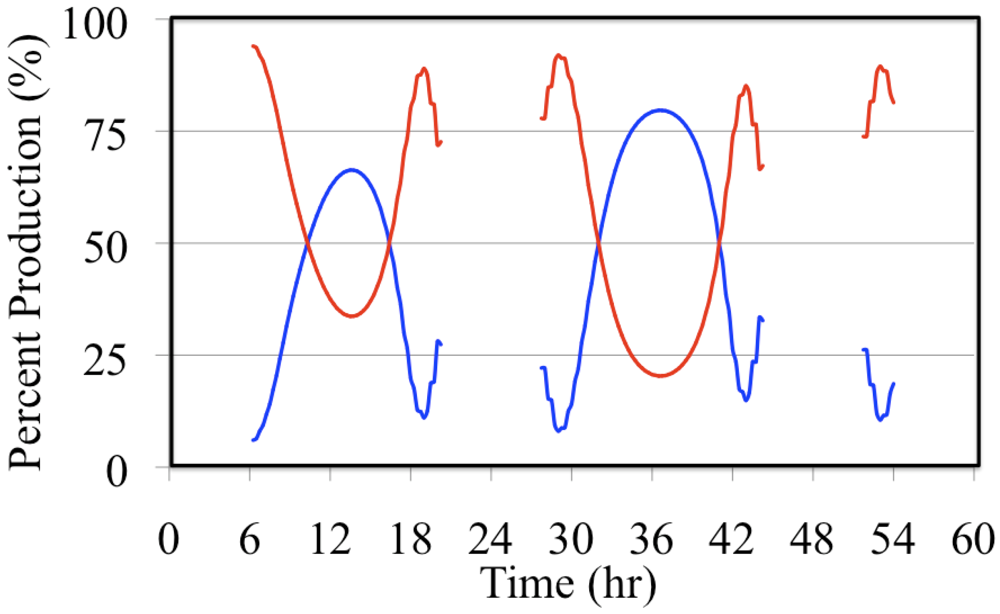
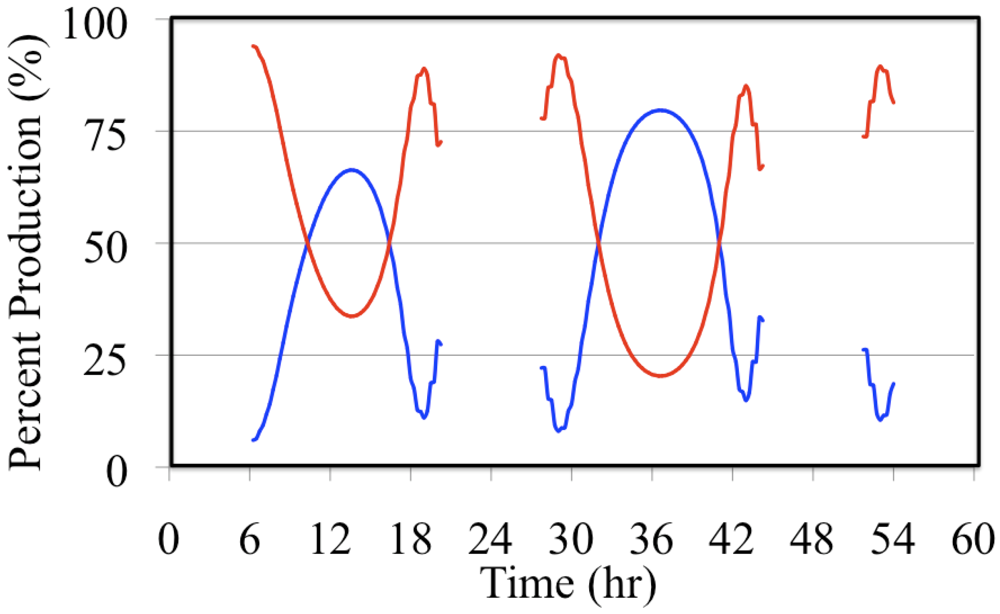
Figure 18.
Daytime relative production rates of HO• resulting from the photolysis of formaldehyde (red line) and the photolysis of O3 (blue line) are shown for the polluted urban atmosphere case.
Figure 18.
Daytime relative production rates of HO• resulting from the photolysis of formaldehyde (red line) and the photolysis of O3 (blue line) are shown for the polluted urban atmosphere case.
Formaldehyde photolysis is the more important source during the early morning and late afternoon hours. The time dependent profile of the formaldehyde photolysis source is skewed toward the morning hours while the time dependent profile of the O
3 photolysis source mirrors the O
3 photolysis rate constant. However, when these two HO
• sources are examined on a percentage basis the profiles appear to be much more symmetrical,
Figure 18.
Reaction of HO
• is almost equally divided between inorganic and organic species,
Table 4. A very large fraction of HO
• reacts with CO. For VOC, the reaction of HO
• with aldehydes and other products of hydrocarbon oxidation were the second most important class of hydroxyl radical reactions while the reaction of HO
• with the hydrocarbons was the third.
Table 4.
Relative percentages of HO• radicals reacting with chemical species over the entire simulated period.
Table 4.
Relative percentages of HO• radicals reacting with chemical species over the entire simulated period.
| Species | Percent (%) |
|---|
| Inorganic | |
| CO | 35.12 |
| SO2 | 4.33 |
| NOx | 3.05 |
| H2O2 | 2.17 |
| O3 | 1.53 |
| Radicals (HO2 + NO3) | 0.81 |
| H2 | 0.60 |
| HNOy | 0.15 |
| Total Inorganic | 47.76 |
| VOC | |
| CH4 | 1.85 |
| Hydrocarbons | 14.50 |
| Aldehydes, and other hydrocarbon products | 35.89 |
| Total Organic | 52.24 |
Different classes of VOC contribute differently to the loss of HO
•,
Figure 19.
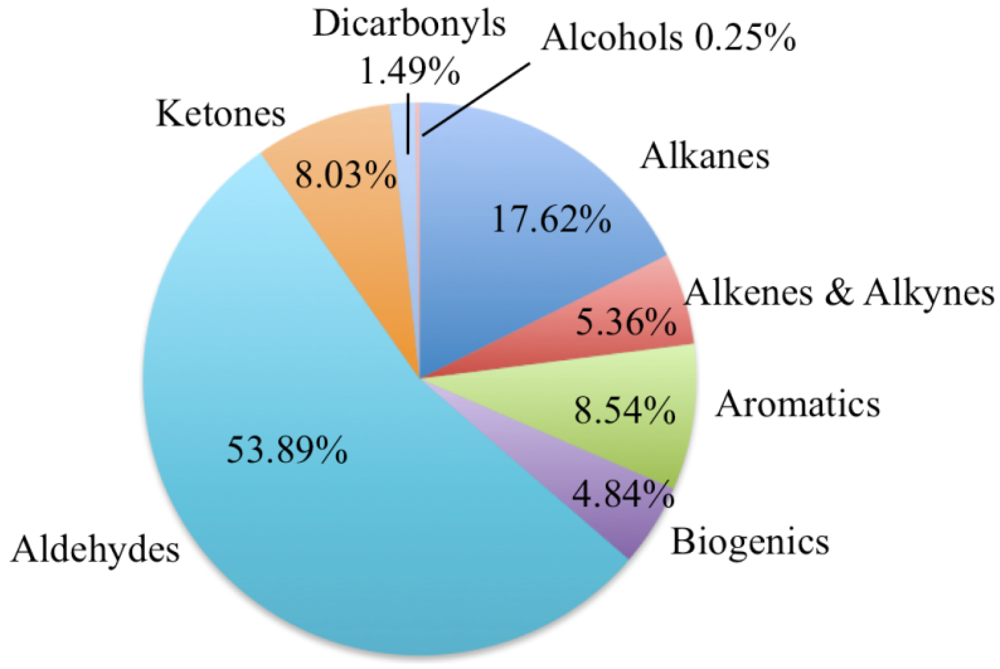
Figure 19 shows the relative fraction of HO
• that react with each class of VOC at noon on the second simulated day. By the second day aldehydes and ketones are the most important organic sink of HO
•. They are among the first generation of oxidation products and highly reactive with respect to HO
•.
Figure 19.
This figure shows the relative fraction of HO• that react with each class of VOC for the polluted urban atmosphere case. The time for the plot is noon on the second simulated day.
Figure 19.
This figure shows the relative fraction of HO• that react with each class of VOC for the polluted urban atmosphere case. The time for the plot is noon on the second simulated day.
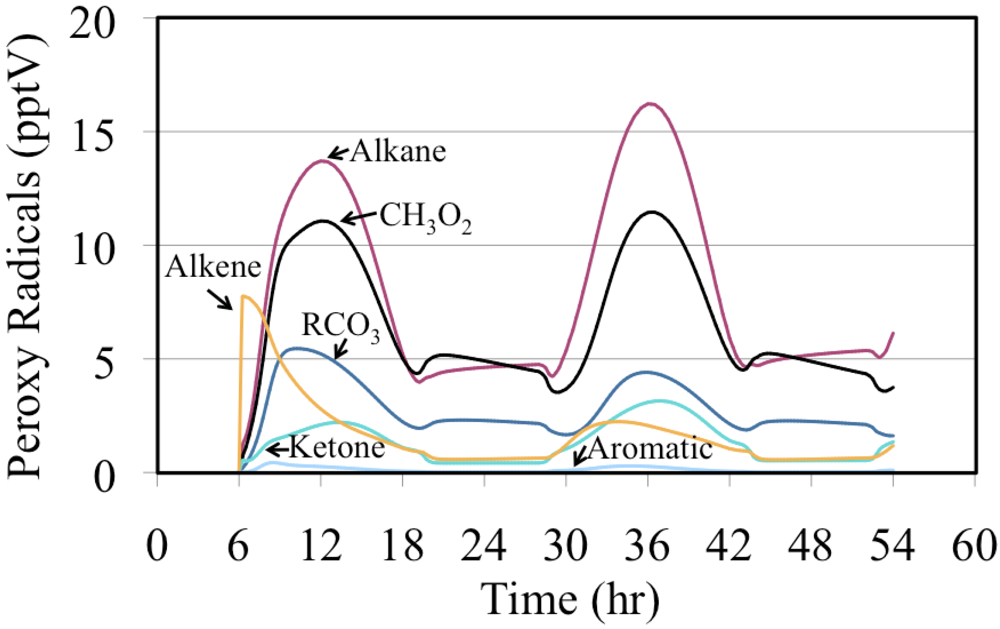
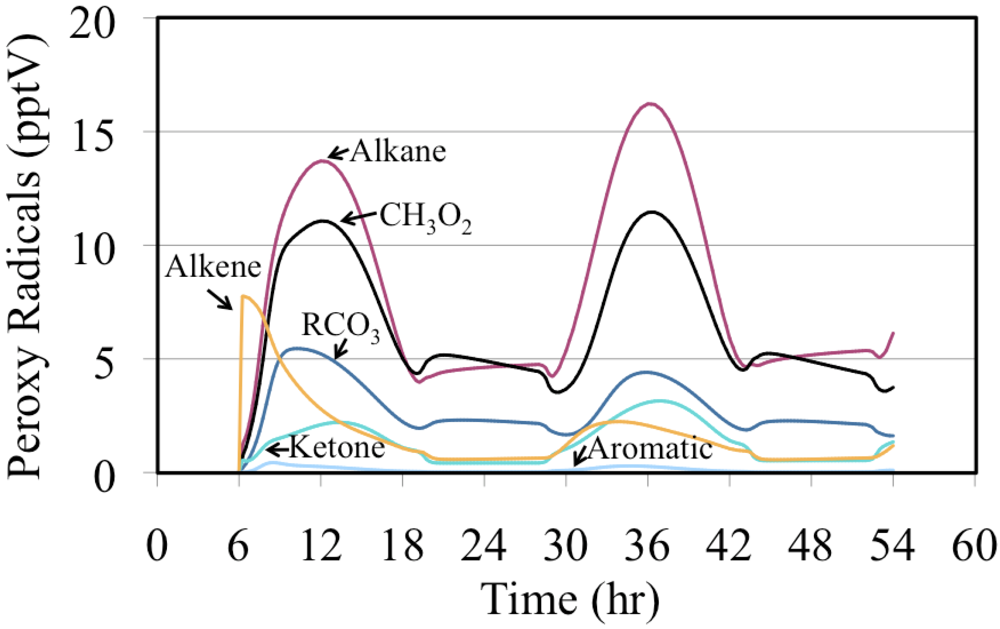
The mixing ratios for the organic peroxy radicals produced from the oxidation of VOC are shown in
Figure 20. In this case the peroxy radicals produced from alkenes was very high initially but their production dropped relatively quickly. For most of the simulation the peroxy radicals produced from alkanes, methyl peroxy radical (CH
3O
2) and acyl peroxy radicals (RCO
3) had the highest mixing ratios.
Figure 20.
The mixing ratios of organic peroxy radicals by organic class are shown for the polluted urban atmosphere case.
Figure 20.
The mixing ratios of organic peroxy radicals by organic class are shown for the polluted urban atmosphere case.
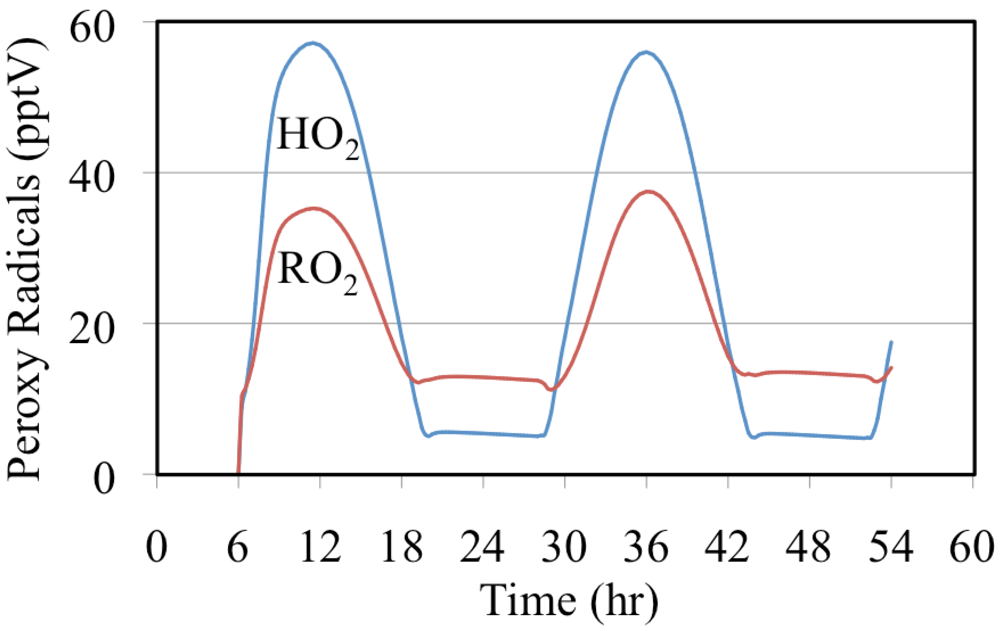
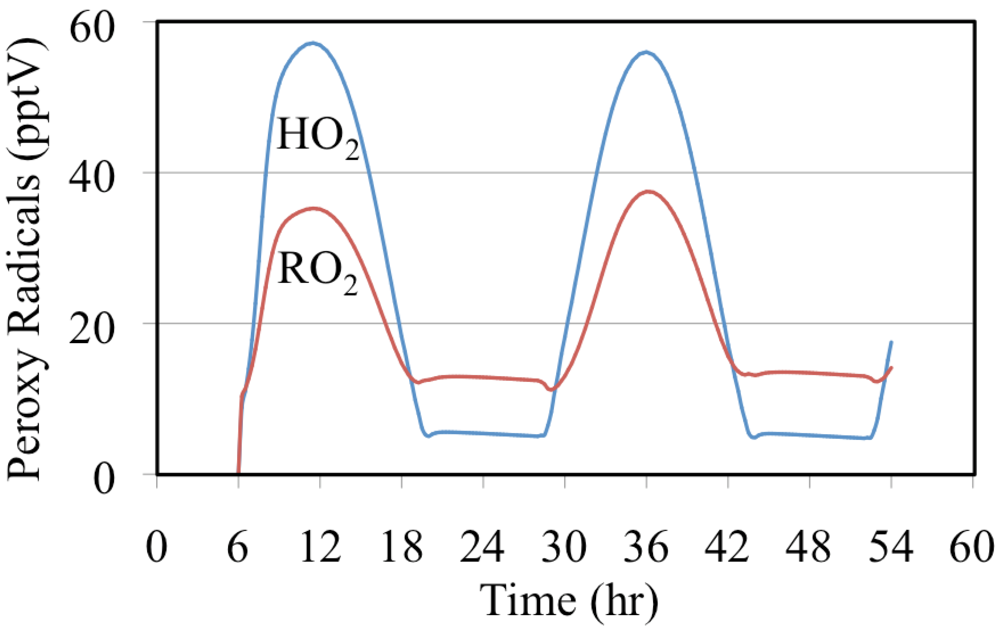
The mixing ratios of HO
2• radicals and the total mixing ratios of the organic peroxy radicals are shown in
Figure 21. Both profiles peak near noon with the HO
2• radicals having the highest daytime mixing ratios while the total mixing ratio of the organic peroxy radicals having the highest nighttime value. The high nighttime organic peroxy radical mixing ratios are produced through the reactions of O
3 and NO
3 with aldehydes and alkenes.
Figure 21.
The mixing ratio of HO2• and the total mixing ratio of organic peroxy radicals are shown for the polluted urban atmosphere case.
Figure 21.
The mixing ratio of HO2• and the total mixing ratio of organic peroxy radicals are shown for the polluted urban atmosphere case.
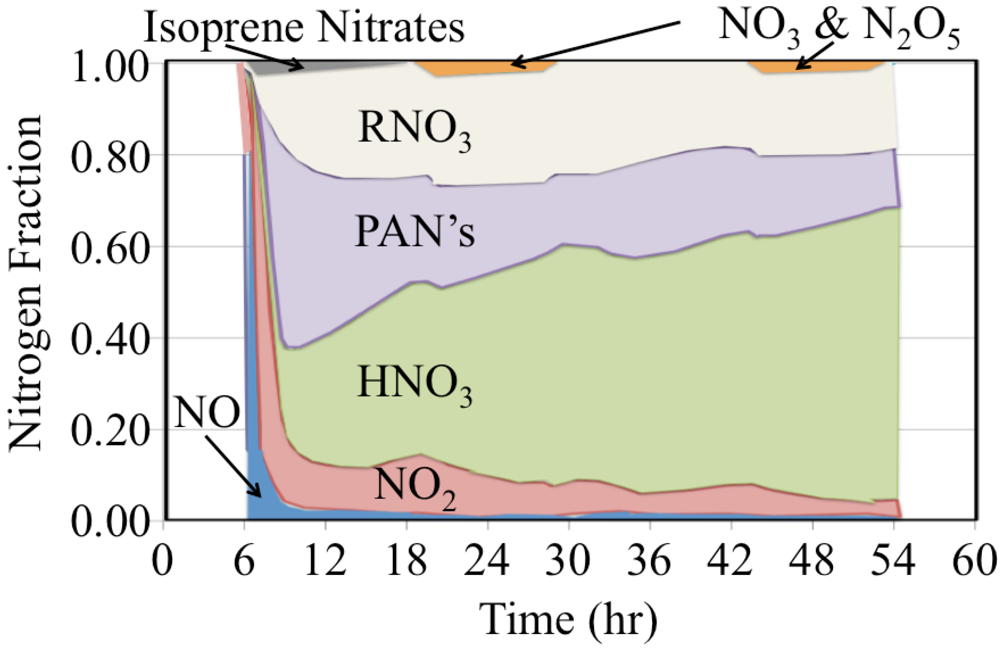
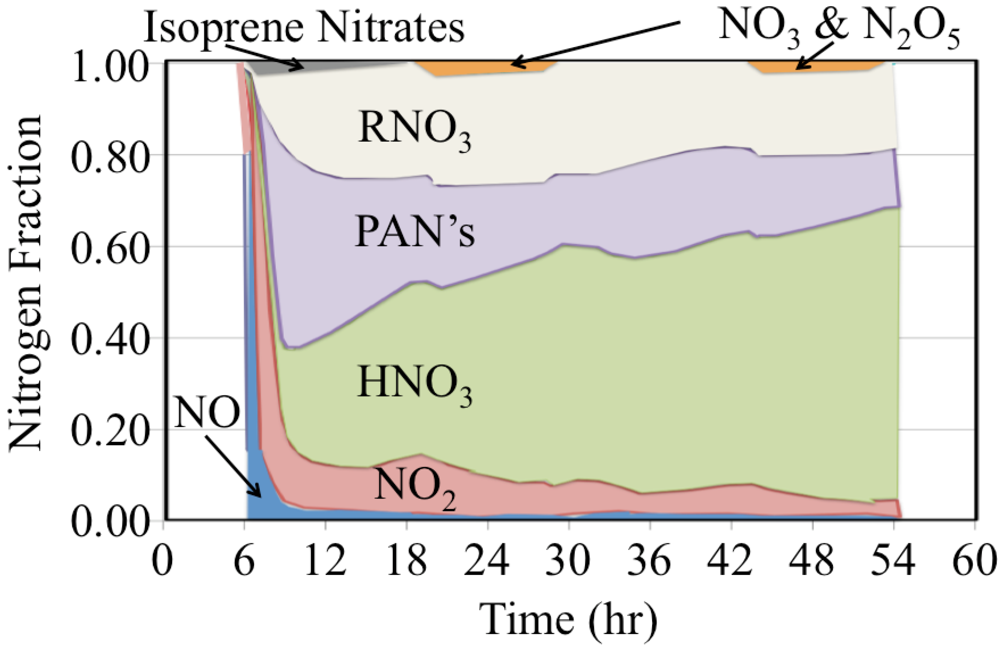
The initial atmosphere was assumed to be NO
2 and NO,
Figure 22. These were rapidly converted to a mixture that also included organic nitrates, peroxyactyl nitrates and HNO
3.
Figure 22.
The time dependent fate of the nitrogen containing species is shown as a stack plot for the polluted urban atmosphere case.
Figure 22.
The time dependent fate of the nitrogen containing species is shown as a stack plot for the polluted urban atmosphere case.
Organic nitrates are produced from the reaction of organic peroxy radicals with NO, Reaction 65.
The yield of organic nitrates tends to increase for higher molecular weight organic peroxy radicals [
45]. The peroxyactyl nitrates were produced from the reaction of acetyl peroxy radicals with NO
2. There is strong production of HNO
3 during the day and nighttime. However the HNO
3 production rate is somewhat slower at sunrise and sunset due to the lower concentrations of HO
• or NO
3 radicals during these times. The figure shows that over the long term of the simulation that nitrogen oxide emissions are converted to HNO
3 and organic nitrates.
In summary of this section, the polluted urban atmosphere simulation showed that there is a complex interplay between the daytime and nighttime chemistry induced by the HO• and NO3 radicals, respectively. Both of these radicals react to convert NOx to HNO3 while oxidizing organic compounds. The nighttime reactions of O3 and NO3 with aldehydes and alkenes produced HO•, HO2• and organic peroxy radical production during the night.
The loss of NOx reduces O3 formation because it is the photolysis of NO2 that leads to the production of O3. While the mixture began with NO and NO2 as the only reactive nitrogen containing species as the air aged these were converted to HNO3 and organic nitrates. The loss of NOx was somewhat underestimated in this gas-phase simulation because the heterogeneous conversion of N2O5 to HNO3 was not included. Dry and wet depositions of HNO3 are very significant sinks for atmospheric nitrogen but these were not included in our simulations. In addition, sulfate, hydrogen peroxide and organic peroxides are removed from the atmosphere through deposition.
The daytime formaldehyde photolysis was a more important source of HO• radicals than O3 photolysis during the early morning and late afternoon hours and as the air aged aldehydes and ketones became the most important organic sink of HO• radicals. The reaction of HO• with VOC produced organic peroxy radicals, while peroxy radicals with the highest mixing ratios were radicals produced from alkanes, the methyl peroxy radical (CH3O2•) and acyl peroxy radicals (RCO3•).



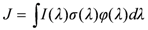

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}