Deferiprone (DFP) Targets Cancer Stem Cell (CSC) Propagation by Inhibiting Mitochondrial Metabolism and Inducing ROS Production


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Models and Other Reagents
2.2. Cell Viability
2.3. Mammosphere Formation Efficiency (MFE)
2.4. ALDEFLUOR Assay
2.5. Seahorse XFe-96 Metabolic Flux Analysis
2.6. ROS Staining
2.7. Mitochondrial Superoxide Assessment
2.8. Statistical Analysis
3. Results
3.1. Evaluating the Effects of DFP on Cell Survival
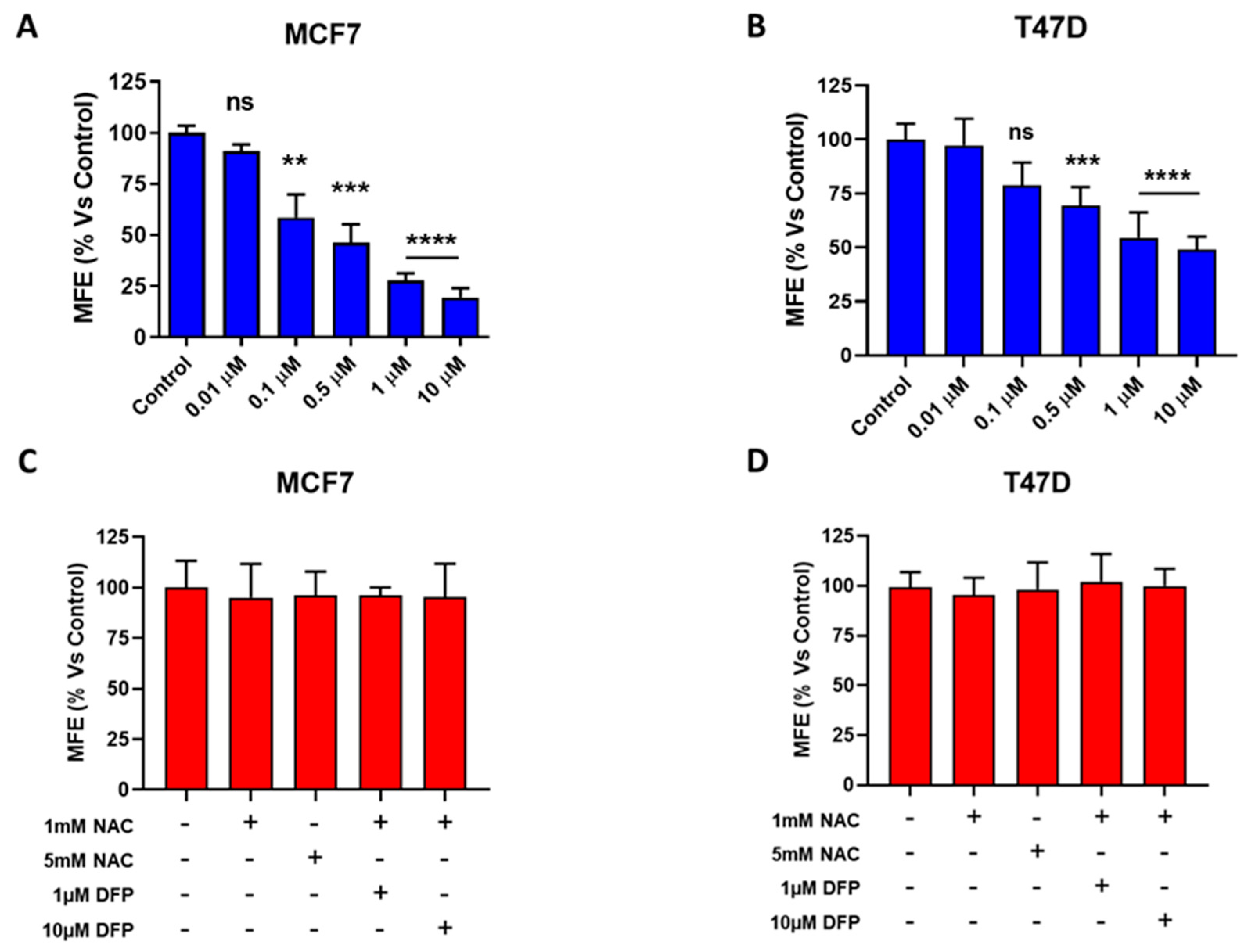
3.2. Effects of DFP on CSC Propagation and ALDH Activity
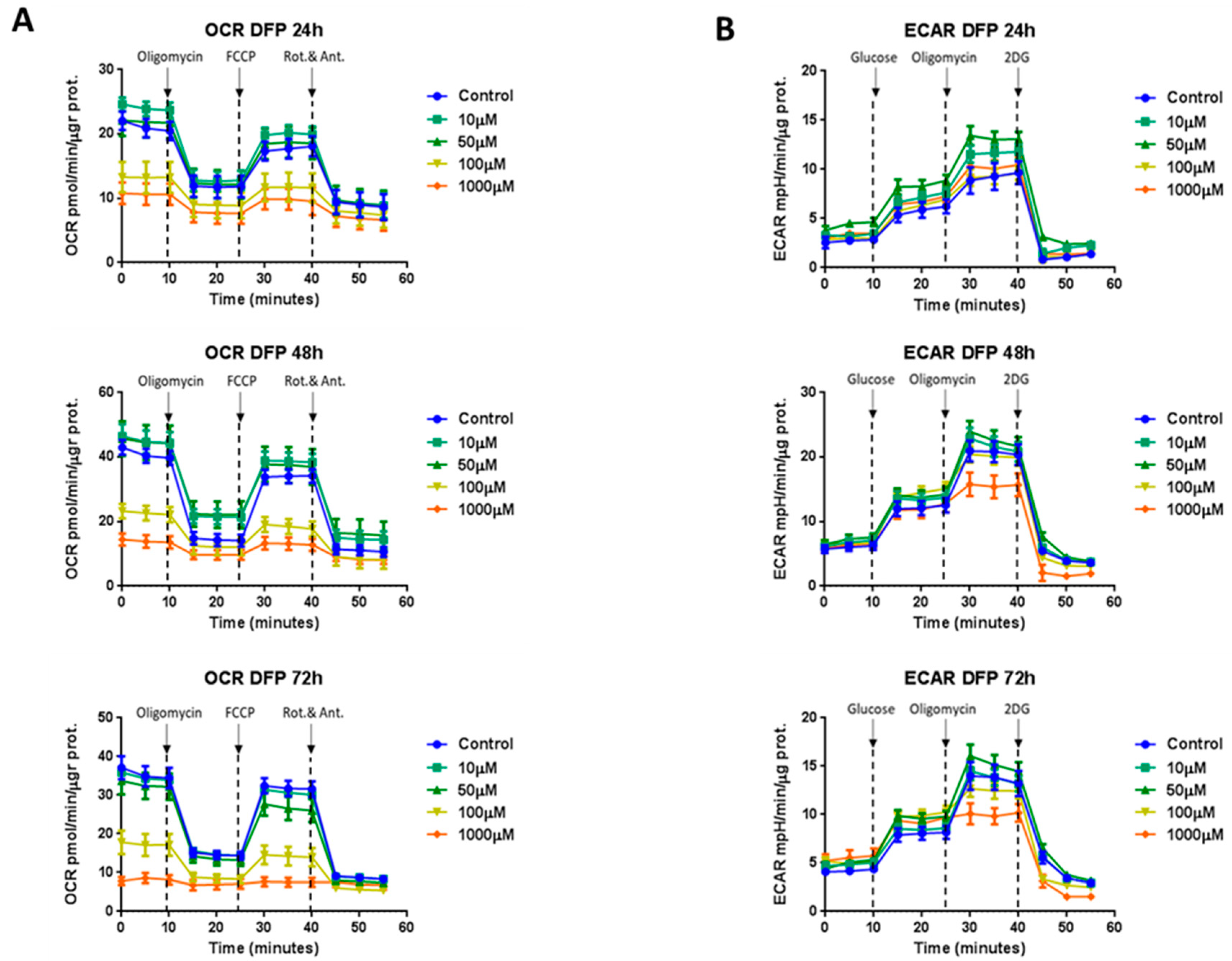
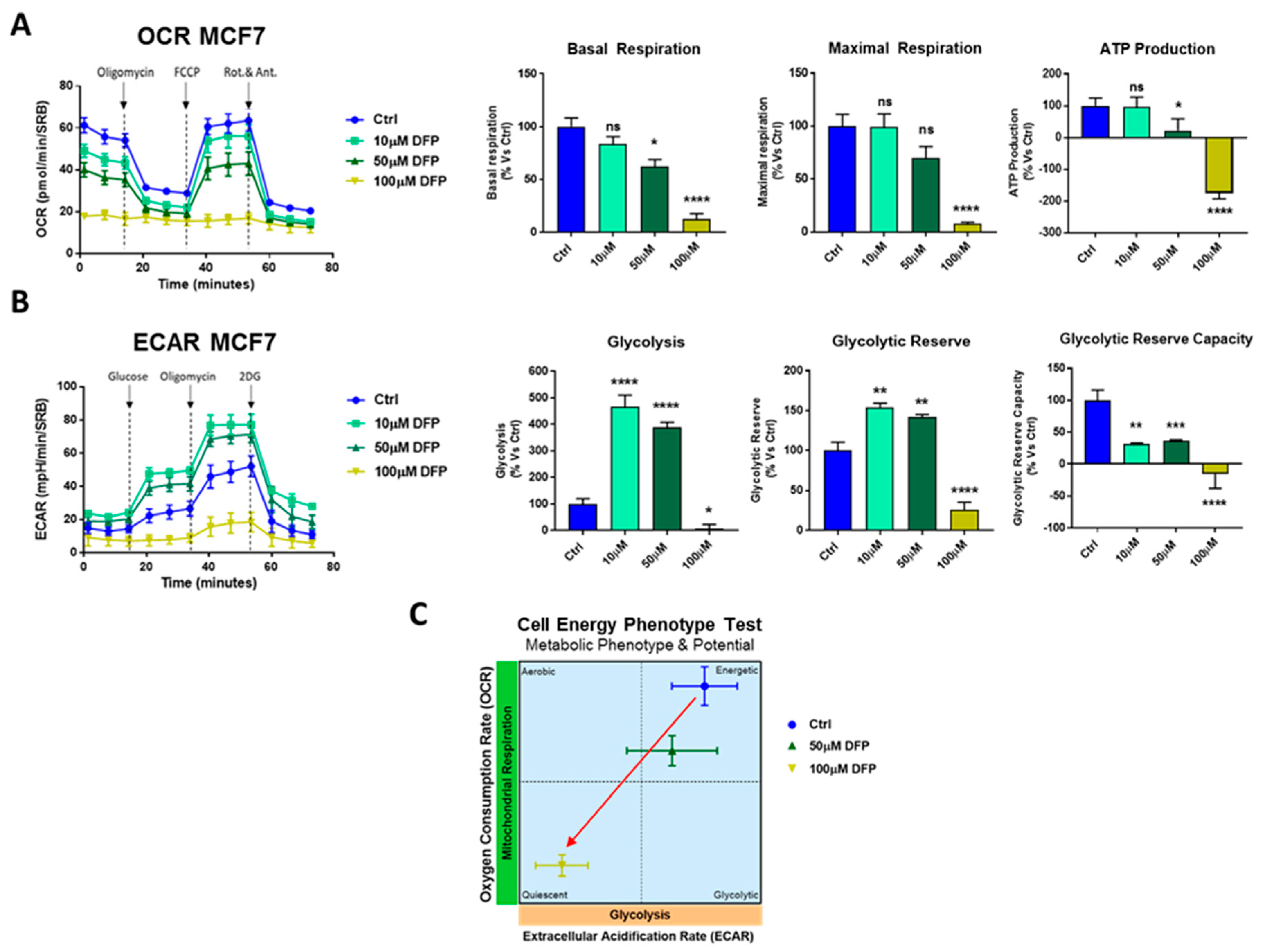
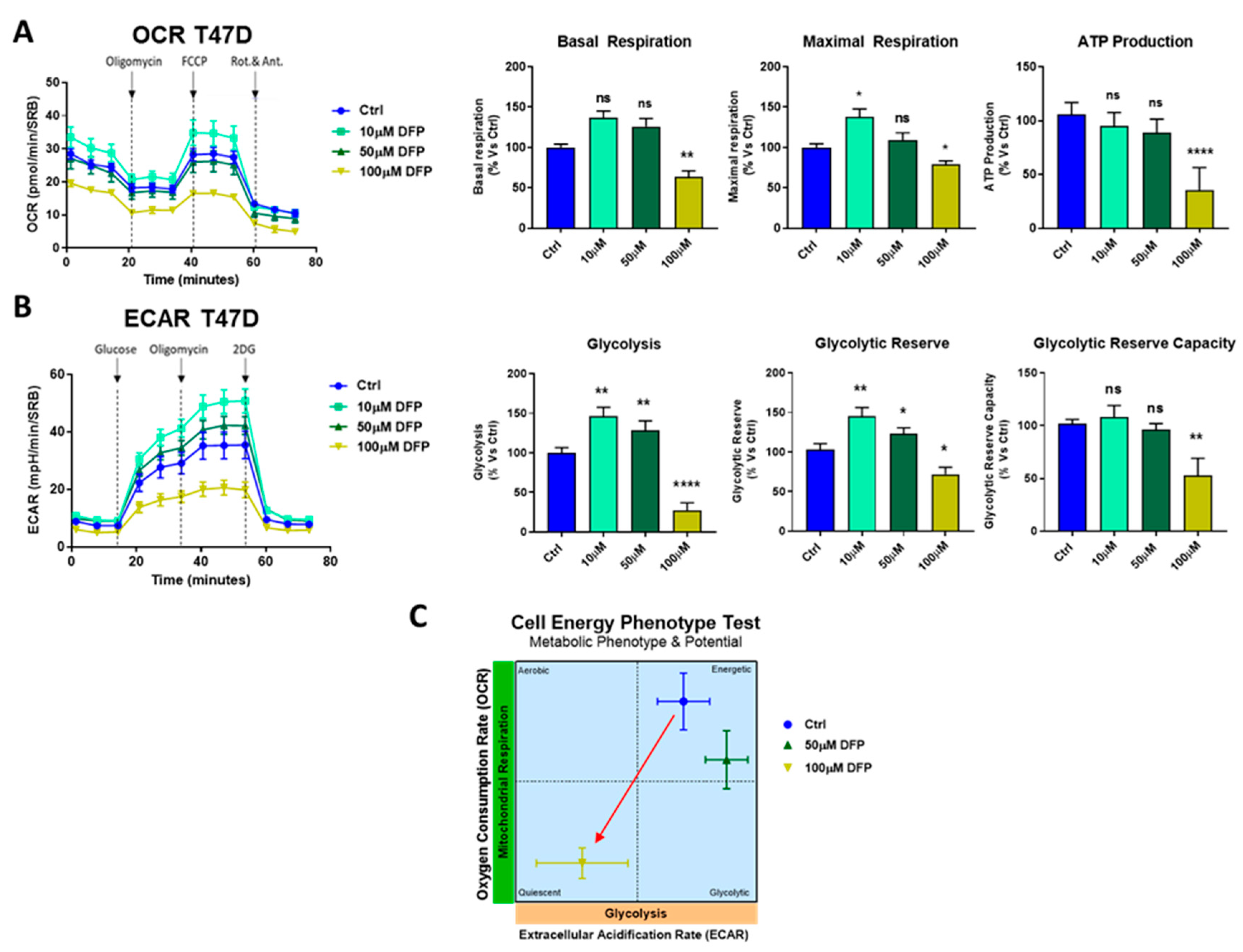
3.3. Effects of DFP on Mitochondrial Metabolism and Glycolysis
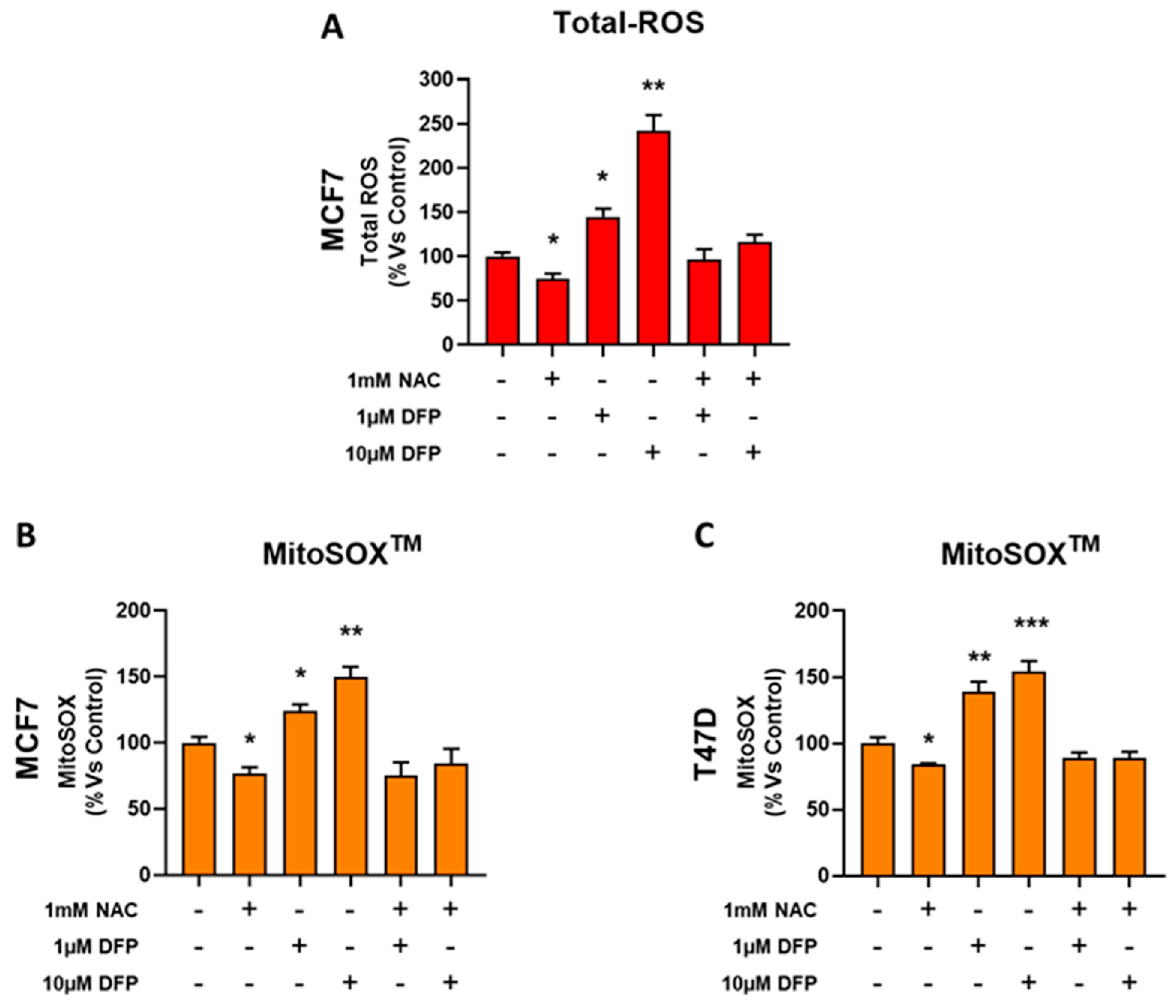
3.4. DFP Induces ROS and Mitochondrial Superoxide Production
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Luo, M.; Wicha, M.S. Targeting cancer stem cell redox metabolism to enhance therapy responses. Semin. Radiat. Oncol. 2019, 29, 42–54. [Google Scholar] [CrossRef]
- Luo, M.; Clouthier, S.G.; Deol, Y.; Liu, S.; Nagrath, S.; Azizi, E.; Wicha, M.S. Breast cancer stem cells: Current advances and clinical implications. Methods Mol. Biol. 2015, 1293, 1–49. [Google Scholar] [PubMed]
- Lamb, R.; Harrison, H.; Smith, D.L.; Townsend, P.A.; Jackson, T.; Ozsvari, B.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Howell, A.; Lisanti, M.P.; et al. Targeting tumor-initiating cells: Eliminating anabolic cancer stem cells with inhibitors of protein synthesis or by mimicking caloric restriction. Oncotarget 2015, 6, 4585–4601. [Google Scholar] [CrossRef] [Green Version]
- Peiris-Pages, M.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Metastasis and oxidative stress: Are antioxidants a metabolic driver of progression? Cell Metab. 2015, 22, 956–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorillo, M.; Sotgia, F.; Lisanti, M.P. “Energetic” Cancer Stem Cells (e-CSCs): A new hyper-metabolic and proliferative tumor cell phenotype, driven by mitochondrial energy. Front. Oncol. 2018, 8, 677. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Scatena, C.; Roncella, M.; di Paolo, A.; Aretini, P.; Menicagli, M.; Fanelli, G.; Marini, C.; Mazzanti, C.M.; Ghilli, M.; Sotgia, F.; et al. Doxycycline, an inhibitor of mitochondrial biogenesis, effectively reduces Cancer Stem Cells (CSCs) in early breast cancer patients: A clinical pilot study. Front. Oncol. 2018, 8, 452. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef] [Green Version]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and cancer. Mol. Cell. 2016, 61, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Alix-Panabieres, C.; Cayrefourcq, L.; Mazard, T.; Maudelonde, T.; Assenat, E.; Assou, S. Molecular portrait of metastasis-competent circulating tumor cells in colon cancer reveals the crucial role of genes regulating energy metabolism and DNA repair. Clin. Chem. 2017, 63, 700–713. [Google Scholar] [CrossRef]
- Sotgia, F.; Ozsvari, B.; Fiorillo, M.; de Francesco, E.M.; Bonuccelli, G.; Lisanti, M.P. A mitochondrial based oncology platform for targeting cancer stem cells (CSCs): MITO-ONC-RX. Cell Cycle 2018, 17, 2091–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacevic, Z.; Kalinowski, D.S.; Lovejoy, D.B.; Yu, Y.; Rahmanto, Y.S.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. The medicinal chemistry of novel iron chelators for the treatment of cancer. Curr. Top. Med. Chem. 2011, 11, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Ohara, T.; Tomono, Y.; Boyi, X.; Yingfu, S.; Omori, K.; Matsukawa, A. A novel, nontoxic iron chelator, super-polyphenol, effectively induces apoptosis in human cancer cell lines. Oncotarget 2018, 9, 32751–32760. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yu, L.; Ding, J.; Chen, Y. Iron metabolism in cancer. Int. J. Mol. Sci. 2018, 20, 95. [Google Scholar] [CrossRef] [Green Version]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer. 2013, 13, 342–355. [Google Scholar] [CrossRef] [Green Version]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. NY Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharm. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Luo, B.; Li, X.; Lu, W.; Yang, J.; Hu, Y.; Huang, P.; Wen, S. Inhibition of cancer growth in vitro and in vivo by a novel ROS-modulating agent with ability to eliminate stem-like cancer cells. Cell Death Dis. 2017, 8, e2887. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Qutub, A.A.; Popel, A.S. Reactive oxygen species regulate hypoxia-inducible factor 1alpha differentially in cancer and ischemia. Mol. Cell. Biol. 2008, 28, 5106–5119. [Google Scholar] [CrossRef] [Green Version]
- Bystrom, L.M.; Guzman, M.L.; Rivella, S. Iron and reactive oxygen species: Friends or foes of cancer cells? Antioxid Redox Signal. 2014, 20, 1917–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef] [PubMed]
- Piga, A.; Roggero, S.; Salussolia, I.; Massano, D.; Serra, M.; Longo, F. Deferiprone. Ann. NY Acad. Sci. 2010, 1202, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Simoes, R.V.; Veeraperumal, S.; Serganova, I.S.; Kruchevsky, N.; Varshavsky, J.; Blasberg, R.G.; Ackerstaff, E.; Koutcher, J.A. Inhibition of prostate cancer proliferation by Deferiprone. NMR Biomed. 2017, 30. [Google Scholar] [CrossRef] [PubMed]
- Ozsvari, B.; Fiorillo, M.; Bonuccelli, G.; Cappello, A.R.; Frattaruolo, L.; Sotgia, F.; Trowbridge, R.; Foster, R.; Lisanti, M.P. Mitoriboscins: Mitochondrial-based therapeutics targeting cancer stem cells (CSCs), bacteria and pathogenic yeast. Oncotarget 2017, 8, 67457–67472. [Google Scholar] [CrossRef] [Green Version]
- Shaw, F.L.; Harrison, H.; Spence, K.; Ablett, M.P.; Simoes, B.M.; Farnie, G.; Clarke, R.B. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J. Mammary Gland Biol. Neoplasia 2012, 17, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Bedaquiline, an FDA-approved antibiotic, inhibits mitochondrial function and potently blocks the proliferative expansion of stem-like cancer cells (CSCs). Aging 2016, 8, 1593–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorillo, M.; Toth, F.; Sotgia, F.; Lisanti, M.P. Doxycycline, Azithromycin and Vitamin C (DAV): A potent combination therapy for targeting mitochondria and eradicating cancer stem cells (CSCs). Aging 2019, 11, 2202–2216. [Google Scholar] [CrossRef]
- Fiorillo, M.; Peiris-Pages, M.; Sanchez-Alvarez, R.; Bartella, L.; di Donna, L.; Dolce, V.; Sindona, G.; Sotgia, F.; Cappello, A.R.; Lisanti, M.P. Bergamot natural products eradicate cancer stem cells (CSCs) by targeting mevalonate, Rho-GDI-signalling and mitochondrial metabolism. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 984–996. [Google Scholar] [CrossRef] [PubMed]
- Frattaruolo, L.; Fiorillo, M.; Brindisi, M.; Curcio, R.; Dolce, V.; Lacret, R.; Truman, A.W.; Sotgia, F.; Lisanti, M.P.; Cappello, A.R. Thioalbamide, a thioamidated peptide from amycolatopsis alba, affects tumor growth and stemness by inducing metabolic dysfunction and oxidative stress. Cells 2019, 8, 1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsura, Y.; Ohara, T.; Noma, K.; Ninomiya, T.; Kashima, H.; Kato, T.; Sato, H.; Komoto, S.; Narusaka, T.; Tomono, Y.; et al. A novel combination cancer therapy with iron chelator targeting cancer stem cells via suppressing stemness. Cancers 2019, 11, 177. [Google Scholar] [CrossRef] [Green Version]
- Ninomiya, T.; Ohara, T.; Noma, K.; Katsura, Y.; Katsube, R.; Kashima, H.; Kato, T.; Tomono, Y.; Tazawa, H.; Kagawa, S.; et al. Iron depletion is a novel therapeutic strategy to target cancer stem cells. Oncotarget 2017, 8, 98405–98416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, W.R.; Fedorka, S.R.; Gad, I.; Shah, R.; Alqahtani, H.D.; Koranne, R.; Kuganesan, N.; Dlamini, S.; Rogers, T.; Al-Hamashi, A.; et al. Small-molecule ferroptotic agents with potential to selectively target cancer stem cells. Sci. Rep. 2019, 9, 5926. [Google Scholar] [CrossRef] [Green Version]
- Piga, A.; Galanello, R.; Forni, G.L.; Cappellini, M.D.; Origa, R.; Zappu, A.; Donato, G.; Bordone, E.; Lavagetto, A.; Zanaboni, L.; et al. Randomized phase II trial of deferasirox (Exjade, ICL670), a once-daily, orally-administered iron chelator, in comparison to deferoxamine in thalassemia patients with transfusional iron overload. Haematologica 2006, 91, 873–880. [Google Scholar] [PubMed]
- Fisher, S.A.; Brunskill, S.J.; Doree, C.; Chowdhury, O.; Gooding, S.; Roberts, D.J. Oral deferiprone for iron chelation in people with thalassaemia. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.L.; Hatef, A.; ul-Haq, M.I.; Nair, N.; Unniappan, S.; Kizhakkedathu, J.N. Clinically approved iron chelators influence zebrafish mortality, hatching morphology and cardiac function. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorillo, M.; Tóth, F.; Brindisi, M.; Sotgia, F.; Lisanti, M.P. Deferiprone (DFP) Targets Cancer Stem Cell (CSC) Propagation by Inhibiting Mitochondrial Metabolism and Inducing ROS Production. Cells 2020, 9, 1529. https://doi.org/10.3390/cells9061529
Fiorillo M, Tóth F, Brindisi M, Sotgia F, Lisanti MP. Deferiprone (DFP) Targets Cancer Stem Cell (CSC) Propagation by Inhibiting Mitochondrial Metabolism and Inducing ROS Production. Cells. 2020; 9(6):1529. https://doi.org/10.3390/cells9061529
Chicago/Turabian StyleFiorillo, Marco, Fanni Tóth, Matteo Brindisi, Federica Sotgia, and Michael P. Lisanti. 2020. "Deferiprone (DFP) Targets Cancer Stem Cell (CSC) Propagation by Inhibiting Mitochondrial Metabolism and Inducing ROS Production" Cells 9, no. 6: 1529. https://doi.org/10.3390/cells9061529