Cathepsins in the Pathophysiology of Mucopolysaccharidoses: New Perspectives for Therapy




1
Department of Molecular Medicine and Medical Biotechnology, School of Medicine, University of Naples Federico II, 80131 Naples, Italy
2
Institute of Biomedical Research of Barcelona, Spanish Research Council, 08036 Barcelona, Spain
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cells 2020, 9(4), 979; https://doi.org/10.3390/cells9040979
Submission received: 25 March 2020
/
Revised: 13 April 2020
/
Accepted: 14 April 2020
/
Published: 15 April 2020
(This article belongs to the Special Issue Lysosomal Storage Disorders)
Abstract
:Cathepsins (CTSs) are ubiquitously expressed proteases normally found in the endolysosomal compartment where they mediate protein degradation and turnover. However, CTSs are also found in the cytoplasm, nucleus, and extracellular matrix where they actively participate in cell signaling, protein processing, and trafficking through the plasma and nuclear membranes and between intracellular organelles. Dysregulation in CTS expression and/or activity disrupts cellular homeostasis, thus contributing to many human diseases, including inflammatory and cardiovascular diseases, neurodegenerative disorders, diabetes, obesity, cancer, kidney dysfunction, and others. This review aimed to highlight the involvement of CTSs in inherited lysosomal storage disorders, with a primary focus to the emerging evidence on the role of CTSs in the pathophysiology of Mucopolysaccharidoses (MPSs). These latter diseases are characterized by severe neurological, skeletal and cardiovascular phenotypes, and no effective cure exists to date. The advance in the knowledge of the molecular mechanisms underlying the activity of CTSs in MPSs may open a new challenge for the development of novel therapeutic approaches for the cure of such intractable diseases.
1. Introduction
Cathepsins (CTSs) are a family of proteases expressed in all living organisms. In humans, CTSs comprise 15 proteolytic enzymes that are classified in three distinct groups based on the key amino acid within their active site, namely serine (CTS A and G), cysteine (CTS B, C, H, F, L, K, O, S, V, X, W), and aspartate (CTS D and E) [1]. These proteases, which mostly require mild acidic conditions for their optimal activity, are all synthesized as proenzymes. Although CTSs are mainly localized in the lysosomes where the acidic environment facilitates their proteolytic activity, they are also found in the cytoplasm, nucleus, and extracellular space where they participate in extracellular matrix (ECM) protein degradation, cell signaling, protein processing, and trafficking through the plasma and nuclear membranes and between intracellular organelles (Figure 1) [2,3,4,5,6,7]. While some CTSs are ubiquitously expressed in the whole body, some are expressed in a more restricted pattern, suggesting specific cellular functions for distinct CTSs.
CTSs have been shown to play essential roles in coagulation, digestion, hormone liberation, adipogenesis, peptide synthesis, immune response, and many other vital processes [1,8]. Abnormal expression and/or activity of CTSs have been associated with a variety of human diseases, including inflammatory and cardiovascular diseases, neurodegenerative disorders, diabetes, obesity, cancer, kidney dysfunction, and many others (Table 1).
The activity and stability of CTSs are tightly regulated by glycosaminoglycans (GAGs), a class of linear, negatively charged polysaccharides that comprise the non-sulfated hyaluronic acid (HA) and the sulfated chondroitin sulfate (CS), dermatan sulfate (DS), keratan sulfate (KS), heparin and heparan sulfate (HS). The protease–GAG interactions may enable autocatalytic activation of CTSs, promote conformational changes in the CTS structures that may increase their affinity for the substrate, thus enhancing their biological activity, and finally, protect proteases from alkaline pH-induced inactivation [101,102,103].
Most of the GAGs are covalently attached to a core protein, forming proteoglycans that are abundantly found at the cell surface and ECM [104,105]. Accumulation of undigested GAGs occurs in the lysosomes as well as on the cell surface and ECM in patients affected by Mucopolysaccharidoses (MPSs). These are a group of lysosomal storage diseases (LSDs) caused by mutations in genes encoding for lysosomal enzymes involved in GAG degradation [106]. Seven types of MPSs (I, II, III, IV, VI, VII and IX) are known to differ in the type of the accumulated GAG, their prevalence and the severity of the clinical manifestations [106,107,108]. The patients exhibit neurological disorders, skeletal and joint defects, hearing and vision impairment as well as cardiovascular and respiratory disease, and premature death [108]. Current therapeutic options available for MPSs include enzyme replacement therapy (ERT), hematopoietic stem cell transplantation (HSCT), substrate reduction therapy (SRT), chaperone therapy, and gene therapy [109,110]. Despite a definite improvement for some clinical manifestations, most of these therapies are not curative, but they only ameliorate some symptoms of the disease. Indeed, the treatment of neurological disorders, avascular cartilage lesions, and cardiac dysfunctions in MPS patients still represents an unmet clinical need [110].
In this review, we summarized the current knowledge about four LSDs due to CTS gene mutations, namely galactosialidosis, neuronal ceroid lipofuscinoses (NCLs) type 10 and type 13, and pycnodysostosis. More importantly, we highlighted the involvement of CTSs in the physiopathology of MPSs that has been scarcely considered thus far, and finally, we reviewed the various types of CTS inhibitors currently available for therapeutic applications. A deeper understanding of the molecular mechanisms underlying the role of CTSs in the onset and progression of MPSs may provide a new basis for the development of novel approaches for the treatment of such diseases.
2. Cathepsin Deficiency Causing Lysosomal Storage Diseases
Lysosomal storage diseases (LSDs) are a family of about 70 disorders caused by disruption of lysosomal homeostasis due to inherited gene mutations. A common feature for all LSDs is the abnormal storage of macromolecular substrates or monomeric compounds inside the endosomal/lysosomal compartment. LSDs are caused by both deficiency of lysosomal enzymes and defects in non-enzymatic soluble lysosomal proteins; however, the former accounts for most LSDs [111]. LSD combined incidence is estimated at 1:5000 live births [112]. The degree of protein function, the biochemistry of the stored material, and the affected cell type determine the clinical onset and symptoms exhibited by LSD patients [113].
To date, four LSDs are known to be caused by inactivating mutations in CTS genes. Defects in CTSA, CTSD, CTSF, and CTSK result in galactosialidosis, neuronal ceroid lipofuscinoses (NCLs) type 10 and type 13, and pycnodysostosis, respectively [13,43,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149] (Table 2).
Galactosialidosis (GSL, OMIM ID: 256540) is an autosomal recessive LSD caused by mutations in the gene encoding CTSA [13,114,115,116,117,118]. The protease CTSA, also known as protective protein/cathepsin A or PPCA, is a serine carboxypeptidase essential in beta-galactosidase (β-GAL) and neuraminidase-1 (NEU1) protein complex stabilization. Indeed, CTSA plays a protective role by preventing β-GAL and NEU-1 lysosomal degradation, although its catalytic activity is distinct from its protective function towards β-GAL and NEU1. Defects in CTSA expression and/or activity result in a complete or partial deficiency of NEU-1 and β-GAL enzyme activities, leading to the accumulation of sialylated oligosaccharides and glycoproteins in lysosomes, and excretion of the formers in body fluids. Moreover, CTSA participates in processing vasoactive peptides and the formation of elastic fibers [119].
The multiple biological functions of CTSA translate into a broad spectrum of clinical manifestations in patients affected by GSL, which are currently classified in three different forms: early infantile, late infantile, and juvenile/adult form [43]. Recurrent clinical features in the three GSL types include coarse facies, hepatosplenomegaly, dysostosis multiplex, growth retardation associated with muscular atrophy, heart involvement with cardiomegaly and thickening of the mitral and aortic valves, hearing loss, and neurological disorders [43,120,121]. To date, 28 different CTSA gene mutations have been linked to GSL, including deletions, splicing, and missense mutations [114,115,116,117,118]. Only one mutation (p.Val150Met) has been predicted to affect CTSA catalytic function, while the others are thought to most likely affect protein stability and folding [114]. Further investigation needs to clarify the link between the different mutations and the effect over CTSA, whether functional or structural. There are no effective treatments currently available for GSL patients other than supportive care.
Neuronal ceroid lipofuscinoses (NCLs), also known as Batten disease, are a clinically genetically heterogeneous group of neurodegenerative LSDs [43,121]. All NCL phenotypes exhibit early impairment of the vision, progressive decline in cognitive and motor functions, dementia, epilepsy, seizures, and, ultimately, premature death. At the cellular level, NCLs show intracellular accumulation of ceroid lipofuscins in the neurons of the central nervous system (CNS), resulting in different degrees of neurodegeneration [122]. Various types of NCLs are known due to over 430 mutations in 14 different genes (called CLNs), and they are classified into four groups based on the protein that the gene encodes such as soluble and transmembrane proteins localizing to the endoplasmic reticulum or the endosomal/lysosomal compartment [123]. CLN10 and CLN13 are included into the NCL-related group due to mutations in the genes that encode lysosomal soluble proteins/enzymes [43].
In particular, CLN10 (OMIM ID: 610127) is caused by mutations in the CTSD gene due to autosomal recessive inheritance [42]. According to the ClinVar Database, 21 mutations have been identified related to CLN10 and affecting the CTSD gene; they include 19 single nucleotide variants, one insertion, and one duplication. Among the 21 mutations, only nine mutations have been confirmed to be pathogenic and linked to the development of CLN10: six missense mutations (p.Phe229Ile, p.Trp383Cys [42], p.Gly149Val, p.Arg399His [124], p.Ser100Phe [125], p.Glu69Lys [126]), a nonsense mutation (c.764dup, p.Tyr255Ter [127]), an insertion (c.268_269insC, p.Gln90fs [128]), and one deletion (p.Phe229del [129]). All different mutations result in neuropathogenesis whose extension is determined by the degree of CTSD gene function loss. Therefore, while complete loss of CTSD activity translates in an early infantile form of CLN10 with patients dying within hours to weeks after birth, patients with residual CTSD activity develop late infantile, juvenile, or adult CLN10 with milder phenotypes [43,121,124]. It is not yet clear how CTSD deficiency causes neuropathies; however, experimental evidence suggests defective autophagy might be in part responsible, as CTSD is essential in degrading cellular components during macroautophagy [130]. Several papers have demonstrated a contribution of glial dysfunction and the involvement of various brain regions in the pathogenesis of NCLs [131]. A recent study has revealed a previously unrecognized role for CTSD in selectively modulating inhibitory synaptic vesicle trafficking and synaptic transmission, showing mechanistic evidence that GABAergic presynaptic endosomal dysfunction might account for the synaptic pathology observed in CTSD deficiency-related NCL diseases [132]. Enzyme replacement therapy (ERT) with recombinant pro-CTSD corrects defective proteolysis and autophagy in cellular and murine models of CNL10 [133]. However, to date, no therapy exists for the disease.
Mutations in the CTSF gene result in NCL type 13 (CLN13, OMIM ID: 615362), an adult-onset form of NCL, also known as type B Kufs disease [43,134,135,136,137]. Patients with CLN13 exhibit mental and motor deterioration in late adulthood [51]. To date, nine mutations with recessive inheritance are known to cause CLN13: six missense mutations (p.Gln321Arg, p.Gly458Ala, p.Ser480Leu, p.Tyr231Cys, p.Ile404Thr, and p.Cys326Phe), a nonsense mutation c.416C > A (p.S139*), a frameshift mutation (p.Ser319Leufs*27), and a mutation preventing the correct splicing of CTSF mRNA (c.213 + 1G>C) [134,135,136,137]. It has been shown that disease-causing CTSF mutants fail to cleave the lysosomal integral membrane protein type-2 (LIMP-2/SCARB2) required for normal biogenesis and maintenance of lysosomes and endosomes [138,139]; however, the exact mechanism by which CTSF deficiency translates in the clinical onset of CLN13 remains elusive. The biochemical and molecular mechanisms underlying NCLs have not been addressed yet. However, several cellular and animal models [140] of the diseases provided useful tools to study the pathogenesis of such devastating neurological disorders and to test novel therapeutic approaches as well [51].
Pycnodysostosis (PKND, OMIM ID: 265800) is an autosomal recessive LSD mainly affecting skeletal structures caused by mutations in the gene encoding CTSK [43,72,141,142]. To date, 48 different CTSK mutations have been reported including missense, nonsense, frameshift, splice-site mutations, and small insertions and deletions [43,72,142]. All the genetic modifications result either in the complete loss of the protein, defective folding and impaired enzyme activity, or faulty intracellular trafficking of the enzyme to the endo/lysosomal compartment. PKND is a specific form of osteopetrosis (increased bone density), and patients exhibit decreased bone resorption resulting in osteosclerosis, without affecting bone formation [141]. Thus, in patients with PKND, markers of bone formation such as type I collagen carboxy-terminal propeptide and osteocalcin are normal, whereas markers of bone resorption (cross-linked N- and C-telopeptides of type I collagen) are significantly decreased [143]. In vitro studies showed that mutant CTSK proteins do not degrade type I collagen, which constitutes 95% of the organic bone matrix [144]. Moreover, osteoclasts and fibroblasts from PKND specimens showed accumulation of undigested collagen fibrils in their endosomal/lysosomal compartments, reflecting the defective bone reabsorption [145]. Almost half of PKND patients have growth hormone deficiency with pituitary hypoplasia and low serum insulin-like growth factor-1 (IGF-1) levels [146,147]. However, opposite findings have been reported in in vitro cell studies using CTSK inhibitors in osteoclasts, demonstrating an increase in IGF-1 due to an impairment in the degradation of the bone matrix-secreted IGF-1 [148]. This paradox could be partially explained because PKND patients present defective osteoclastic resorption, which is responsible for the release of bone matrix embedded IGF-1. Pycnodysostosis does not correlate with increased mortality; however, it can cause significant morbidity such as recurrent fractures, osteolysis of the distal phalanges, craniosynostosis, respiratory sleep disorders, short stature, and dental problems [43]. To date, no therapy is effective for the cure of PKND, although growth hormone treatment has been shown to improve growth rates and final heights in patients with PKND [149]. Targeted enzyme or gene replacement therapies are being investigated for the cure of PKND [43].
3. Cathepsin Involvement in the Pathophysiology of Mucopolysaccharidoses
In MPSs, the lysosomal accumulation of undigested GAGs is considered the “primum movens” of the subsequent functional cell impairment; however, evidence demonstrates that the accumulation of storage material does not occur only in the lysosomes, but also on the cell surface and ECM where GAGs form proteoglycans through their covalent binding to a core protein [105,106,109]. The accumulation of storage material in non-lysosomal compartments accounts for impaired cell signaling and trafficking, protein unfolding, abnormal autophagy, alterations of intracellular calcium homeostasis, lysolipid accumulation, and modifications in other cellular processes that ultimately lead to the MPS phenotypes [150,151,152,153,154,155,156,157,158,159].
A variety of evidence demonstrate that abnormal expression and/or activity of both lysosomal and extra-lysosomal CTSs correlate with MPS major clinical manifestations such as neuropathology, bone and joint defects, and cardiovascular disorders (Table 3).
In particular, the cysteine CTSB, involved in the degradation of collagen [176] and responsible for heart dilatation [19], displayed a marked increase of its activity in MPS I mouse model, suggesting that the progressive heart failure and valve disease observed in these mice may be dependent on CTSB overexpression [160]. The in vivo treatment of MPS I mice with a CTSB inhibitor reduced aortic dilatation and heart valve thickening, and led to an improvement of cardiac function, suggesting that CTSB inhibition may have a potential benefit in the disease [161]. Elevated activity of CTSB was detected in the MPS VII dog model showing abnormalities in the collagen structure of mitral valve [162]. When affected dogs received an intravenous injection of a retroviral vector expressing canine β-glucuronidase (the deficient enzyme in MPS VII), a reduced CTSB activity was observed, which correlated with an improved signal for structurally intact-collagen. Furthermore, in both mouse and dog models of MPS I and MPS VII, aortic dilatation resulted in being associated with an up-regulation of the elastase CTSS as well [171,172,173]. Indeed, neonatal intravenous injection of a retroviral vector expressing α-L-iduronidase normalized CTSS mRNA levels and prevented aortic disease in MPS I mice [171]. Moreover, intravenous injection of a retroviral vector expressing β-glucuronidase to MPS VII dogs reduced RNA levels of CTSS and delayed the development of aortic dilatation [173].
Cardiac involvement, although firstly reported for MPS I, II, and VI affected patients, has been reported in all MPS patients [177]. Indeed, cardiac disease has been described in MPS III patients as well as in patients affected by the other MPS subtypes [178,179]. Multiple evidence demonstrated the involvement of CTSs in many cardiovascular diseases, including atherosclerosis, cardiac hypertrophy, cardiomyopathy, myocardial infarction, and hypertension, some of which are common clinical manifestations in MPSs [8,10,17,18,19,36,56,61,66,76,80,81]. In particular, CTSB results in being up-regulated in cardiomyocytes in response to hypertrophic stimuli both in vivo and in vitro [180]. Furthermore, CTSB was associated with an increased risk of cardiovascular events in patients with stable coronary heart disease [18]. A specific CTSB inhibitor, namely CA-074Me, reduced cardiac dysfunction, remodeling, and fibrosis in a rat model of myocardial infarction [181]. Studies aimed to explore the potential of CTS inhibitors for the treatment of cardiovascular diseases are ongoing [19], and targeting CTSs–based therapy might provide new avenues for the treatment of MPSs as well.
Beside cardiac disease, some MPS subtypes are characterized by central nervous system (CNS) degeneration for which there are currently no resolutive treatments [106,109]. The involvement of CNS in the disease manifests as mental retardation, intellectual disabilities, behavioral disorders, sleep disturbances, progressive neurodegeneration, and early death. Neurodegeneration occurs in the severe forms of MPS I, and it is prominent in MPS III affected patients [106,108,182]. Elevated transcripts of CTSD, CTSS, and CTSZ were detected in the cortex of MPS I and MPS IIIB mouse models [166]. Abnormal activity of CTSD in the cerebral cortex correlated with locomotion disorders and neuropathology in MPS I mice [167]. Up-regulation of CTSB was observed in the brain of MPS IIIA mice [163]. Overexpression of CTSB and CTSD was also found in the proteomic profile of MPS I mouse brain tissues [164]. Interestingly, enhanced expression and activity of CTSB resulted in being associated with increased deposition of amyloid plaques in the MPS I mouse brain, and the existence of a novel CTSB-associated amyloidogenic pathway leading to neurodegeneration was highlighted [165]. Since CTSB is a crucial regulator of the NLRP3 inflammasome, it likely contributes to the inflammasome-dependent pathway involved in MPS neuroinflammation [183]. On the other hand, the cysteine CTSB and the aspartate CTSD result to be up-regulated in a variety of neurological disorders [184,185,186]. The protease CTSS is preferentially expressed in cells of the macrophage/monocyte lineage, and inflammation stimulates its secretion from the microglia and macrophages [185,187]. The involvement of microglial CTSB, CTSD, and CTSS in neurodegenerative diseases supports the view that microglia-driven neuroinflammation contributes to the progression of neurodegeneration in MPS I and IIIB [183,188,189]. Indeed, molecular evidence of microgliosis has been well established in mouse and dog models of MPS I and MPS III A, B, and C subtypes [166,182,188,190]. Inhibition of CTSB has been shown to prevent neuronal death and behavioral disorders in a patient affected by the Niemann-Pick disease type A and in a mouse model of the disease [191]. Although further studies are needed to fully elucidate the pathophysiological role of CTSs in the CNS, the above findings strongly suggest that the specific inhibition of microglial CTSs might lead to neuroprotective outcomes in MPS phenotypes characterized by activated pro-inflammatory microglia.
Neurological phenotypes are also common in other MPS types than MPS I and III. Indeed, patients affected by the severe forms of Hunter syndrome (MPS II), which account for about 75% of the cases, exhibit impairment of cognitive skills, mental retardation, intense neurobehavioral symptoms, and death in the second decade of life [192]. In agreement with the previous findings in MPS I and III subtypes, a transcriptome analysis of the brain from the MPS II mouse model showed CTSD up-regulation in the cerebral cortex of affected mice [170]. The same study also highlighted dysregulation of CTSA, CTSC, CTSH, CTSL, and CTSS gene expression in MPS II mouse brain, although with different trends (up/down-regulation) in the cerebral cortex and the midbrain/diencephalon/hippocampus areas. Variation in CTS gene expression between different brain regions was also observed in the MPS VII mouse model, thus suggesting that different neuropathologic mechanisms may predominate in the different areas of brains [174]. Furthermore, a transcriptome analysis of MPS VII mouse brain showed that CTSA, CTSB, CTSC, CTSD, CTSH, CTSS, and CTSZ were highly up-regulated in all brain regions, while CTSK was only changed in the brain stem and was down-regulated. Integrated analysis of proteome and transcriptome changes confirmed CTSS and CTSZ dysregulation in the MPS VII mouse hippocampus [175]. MPS VII affected patients present a broad clinical spectrum of symptoms from severe to milder phenotypes; however, most of them display intellectual disabilities together with delayed speech development, hearing impairment, and behavioral disturbances [193].
The accumulation of undigested GAGs in the lysosomes of connective tissue cells and chondrocytes is responsible for musculoskeletal abnormalities commonly observed in almost all MPS subtypes [100,101,102,103,104,105,106,194,195,196]. However, in MPSs, the pathogenesis of the skeletal and joint disease, including growth impairment, may involve complex molecular mechanisms underlying alterations of cartilage and bone metabolism, as well as inflammatory pathways [197]. Indeed, metabolic inflammation is a significant cause of osteoarticular symptoms in MPS disorders [183,198]. On the other hand, CTSs have long been involved in skeletal and bone health and disease [199]. Recently, up-regulation of CTSA, CTSH, and CTSZ has been detected through transcriptomic and proteomic analyses in a rat model of spinal cord injury [200]. In VCP (valosin containing protein) knock out mice, up-regulation of CTSB and CTSD in skeletal muscle correlated with activation of the transcription factor EB [201]. The cysteine protease CTSK, which has long been known as a molecular marker of differentiated osteoclasts and is directly involved in the degradation of bone matrix proteins [201,202,203], plays a crucial role in skeletal pathologies frequently observed in MPSs and other LSDs as well [168]. In the murine model of MPS I, the accumulation of GAGs in bones had an inhibitory effect on CTSK activity, resulting in impaired osteoclast activity and decreased cartilage resorption, thus contributing to the bone pathology seen in the disease [169]. This finding makes CTSK a candidate therapeutic target for MPS types where current therapies have a limited effect on skeletal conditions.
4. Cathepsin Inhibitors and Their Therapeutic Applications
The inhibition of CTSs has been widely explored over the last decades in the field of chronic inflammatory diseases [27,37,204,205], cardiovascular diseases [10,19,181], osteoporosis [70,71,72,73], arthritis [28,206], kidney diseases [30,31,32,84], pancreatitis [207], obesity [208,209,210], cancer [25,34,48,74,82,211], neurodegenerative diseases [39,41,184,185,212,213], and many other pathological states. Multiple inhibitors are currently available, ranging from reversible covalent inhibitors to irreversible inhibitors [214,215,216,217,218] (Table 4).
The class of reversible inhibitors acts by engaging the target protein through non-covalent interactions, whereas irreversible inhibitors bind the target protein with stable covalent bonds [219]. Although both types of CTS inhibitors have shown to work efficiently, reversible inhibitors have been proved to exhibit higher selectivity [216,218,219,220]. Moreover, off-target effects with broad-spectrum inhibition of proteases, leading to unpredictable side effects in clinical trials, has represented the main concern for the therapeutic use of CTS inhibitors in humans. The recent focus on target- and ligand-binding drug design to selectively inhibit specific CTSs has provided excellent results in overcoming such issue [19,72,204,205,206,214,215,216,217,218,219,220,221,222]. In this context, several new strategies have been reported for successfully CTS targeting, including designed ankyrin repeat proteins (DARPins) with high CTSB blocking activity [222], non-peptide synthetic molecules with anti-CTSK [71] and anti-CTSD [223] activity, and naturally occurring asperphenamate [224]. Natural depsipeptides inhibitor of CTSD, namely izenamides A, B, and C, have been recently successfully tested [225], and quantum mechanics/molecular modeling studies of the mechanism of cysteine protease inhibition by dipeptidyl nitroalkenes provided promising results [226].
Selective CTSG inhibitors have been recently designed with the potential to improve chronic inflammatory diseases [205]. A variety of CTSC inhibitors have been developed and evaluated in preclinical/clinical trials to regulate serine protease activity in inflammatory and immunologic conditions [27,28]. Pepstatin A is a potent inhibitor for CTSD [227]. Although Pepstatin A can target other aspartyl proteases than CTSD, it is 26,000 times more specific for CTSD (Ki = 0.5 µmol/L) than for its next target renin (Ki = 13000 µmol/L). Pepstatin A has been proved effective in slowing down chronic kidney disease progression [30,32] and fatty liver disease [209] in experimental animal models. Due to the low bioavailability of this peptidic inhibitor, efforts have been made to design Pepstatin A analogues more suitable for the treatment of human diseases [228]. Highly specific and potent small-molecule inhibitors of CTSD other than Pepstatin A have been developed for the treatment of non-alcoholic fatty liver disease [229], as well as CTSD targeting by natural products has shown to be beneficial in cancer chemoprevention [216].
The aspartic protease CTSE plays an essential role in antigen processing within the class II MHC pathway [48], therefore broadly inhibiting CTSE can lead to undesirable side effects. Selective inhibitors of CTSE and CTSB, both involved in the polarization of microglia/macrophages in neurotoxic phenotypes leading to hypoxia/ischemia, are being tested as pharmacological agents for the treatment of ischemic brain injury [212]. Moreover, CTSS inhibitors have shown neuroprotective and anti-inflammatory effects in preclinical studies for the treatment of neurodegenerative diseases [184], although CTSS essential role in CNS homeostasis might limit its therapeutic applications [204]. However, molecular modeling-assisted design of CTSS inhibitors has provided novel scaffolds for improved CTSS inhibition [217]. Experimental evidences suggest that inhibition of CTSS attenuates the progression of atherosclerosis during chronic kidney disease [84], improves sugar levels during type2 diabetes [208], and prevents autoantigen presentation and autoimmunity [229]. CTSK inhibitors have been proved successful improving osteoporosis [72,73,230]; however, concerns emerged over off-target effects of the inhibitors against other CTSs and CTSK inhibition at nonbone sites (i.e., skin, and cardiovascular and cerebrovascular sites). Recently, novel selective inhibitors for CTSK have been developed, showing beneficial effects on bone and cartilage in preclinical osteoarthritis models with a safety profile [71,206].
The use of CTS inhibitors in cellular and animal models have contributed to deepening our understanding of the mechanisms of action and biological functions of these proteolytic enzymes.
5. Conclusions
Neuropathology, skeletal and joint defects, and cardiac disorders are among the most prominent clinical manifestations of MPSs, which are refractory to the current therapies [106,108,109]. Although no studies are available in humans, investigations in animal models have shown a beneficial effect of CTS inhibition, especially in ameliorating cardiac disease in MPSs and other LSDs [161,162,171,173]. Since CTSs have also been shown to play a role in the onset and progression of neuropathology and skeletal disorders in MPSs, affected patients might gain benefits from treatments with CTS targeting-based drugs. Therefore, it would be of great interest to test the effectiveness of the new generation of highly selective CTS inhibitors in MPSs. They could be used alone or in combination with the current therapeutic approaches to improve the quality and duration of life of these patients. However, there is a need for further investigations on the effective role of distinct CTSs in the pathophysiology of MPSs to recognize them as key players in the fight against such incurable diseases.
Author Contributions
Conceptualization, V.D.P., A.M. and L.M.P.; writing—original draft preparation, V.D.P., A.M. and L.M.P.; writing—review and editing, V.D.P., A.M. and L.M.P.; supervision, A.M. and L.M.P.; funding acquisition, A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by “Research Challenges” 2018 R + D+i Project from the Spanish Government to A.M.; A.M.’s salary is funded through a Ramon y Cajal Contract by the Spanish Government.
Acknowledgments
We apologize to all the authors whose work could not be cited due to space limitations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Patel, S.; Homaei, A.; El-Seedi, H.R.; Akhtar, N. Cathepsins: Proteases that are vital for survival but can also be fatal. Biomed. Pharmacother. 2018, 105, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Vidak, E.; Javoršek, U.; Vizovišek, M.; Turk, B. Cysteine cathepsins and their extracellular roles: Shaping the microenvironment. Cells 2019, 8, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallo-Medved, D.; Rudy, D.; Blum, G.; Bogyo, M.; Caglic, D.; Sloane, B.F. Live-cell imaging demonstrates extracellular matrix degradation in association with active cathepsin B in caveolae of endothelial cells during tube formation. Exp. Cell Res. 2009, 315, 1234–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bright, N.A.; Davis, L.J.; Luzio, J.P. Endolysosomes are the principal intracellular sites of acid hydrolase activity. Curr. Biol. 2016, 26, 2233–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, S.M.; Kanneganti, T.D. Regulation of lysosomal dynamics and autophagy by CTSB/cathepsin B. Autophagy 2016, 12, 2504–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soond, S.M.; Kozhevnikova, M.V.; Frolova, A.S.; Savvateeva, L.V.; Plotnikov, E.Y.; Townsend, P.A.; Han, Y.P.; Zamyatnin, A.A., Jr. Lost or Forgotten: The nuclear cathepsin protein isoforms in cancer. Cancer Lett. 2019, 462, 43–50. [Google Scholar] [CrossRef]
- Droga-Mazovec, G.; Bojic, L.; Petelin, A.; Ivanova, S.; Romih, R.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008, 283, 19140–19150. [Google Scholar] [CrossRef] [Green Version]
- Reiser, J.; Adair, B.; Reinheckel, T. Specialized roles for cysteine cathepsins in health and disease. J. Clin. Investig. 2010, 120, 3421–3431. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Cowling, R.T. Cathepsin A inhibitors to treat heart disease: Much potential, many questions. JACC Basic Transl. Sci. 2019, 4, 345–347. [Google Scholar] [CrossRef]
- Seyrantepe, V.; Hinek, A.; Peng, J.; Fedjaev, M.; Ernest, S.; Kadota, Y.; Canuel, M.; Itoh, K.; Morales, C.R.; Lavoie, J.; et al. Enzymatic activity of lysosomal carboxypeptidase (cathepsin) A is required for proper elastic fiber formation and inactivation of endothelin-1. Circulation 2008, 117, 1973–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fledrich, R.; Mannil, M.; Leha, A.; Ehbrecht, C.; Solari, A.; Pelayo-Negro, A.L.; Berciano, J.; Schlotter-Weigel, B.; Schnizer, T.J.; Prukop, T.; et al. Biomarkers predict outcome in Charcot-Marie-Tooth disease 1A. J. Neurol. Neurosurg. Psychiatry 2017, 88, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, I.; d’Azzo, A. Galactosialidosis: Historic aspects and overview of investigated and emerging treatment options. Expert Opin. Orphan Drugs 2017, 5, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Iodice, A.A.; Weinstock, I.M. Cathepsin A in nutritional and hereditary muscular dystrophy. Nature 1965, 207, 1102. [Google Scholar] [CrossRef]
- Bernstein, H.G.; Keilhoff, G. Putative roles of cathepsin B in Alzheimer’s disease pathology: The good, the bad, and the ugly in one? Neural Regen. Res. 2018, 13, 2100–2101. [Google Scholar] [CrossRef]
- Aggarwal, N.; Sloane, B.F. Cathepsin B: Multiple roles in cancer. Proteomics Clin. Appl. 2014, 8, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.Q.; Xu, M.; Yuan, Y.; Li, F.F.; Yang, Z.; Liu, Y.; Zhou, M.Q.; Bian, Z.Y.; Deng, W.; Gao, L.; et al. Cathepsin B deficiency attenuates cardiac remodeling in response to pressure overload via TNF-α/ASK1/JNK pathway. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1143–H1154. [Google Scholar] [CrossRef]
- Wuopio, J.; Hilden, J.; Bring, C.; Kastrup, J.; Sajadieh, A.; Jensen, G.B.; Kjøller, E.; Kolmos, H.J.; Larsson, A.; Jakobsen, J.C.; et al. Cathepsin B and S as markers for cardiovascular risk and all-cause mortality in patients with stable coronary heart disease during 10 years: A CLARICOR trial sub-study. Atherosclerosis 2018, 278, 97–102. [Google Scholar] [CrossRef]
- Liu, C.L.; Guo, J.; Zhang, X.; Sukhova, G.K.; Libby, P.; Shi, G.P. Cysteine protease cathepsins in cardiovascular disease: From basic research to clinical trials. Nat. Rev. Cardiol. 2018, 15, 351–370. [Google Scholar] [CrossRef]
- Canbay, A.; Guicciardi, M.E.; Higuchi, H.; Feldstein, A.; Bronk, S.F.; Rydzewski, R.; Taniai, M.; Gores, G.J. Cathepsin B inactivation attenuates hepatic injury and fibrosis during cholestasis. J. Clin. Investig. 2003, 112, 152–159. [Google Scholar] [CrossRef] [Green Version]
- Moles, A.; Tarrats, N.; Fernández-Checa, J.C.; Marí, M. Cathepsins B and D drive hepatic stellate cell proliferation and promote their fibrogenic potential. Hepatology 2009, 49, 1297–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerch, M.M.; Halangk, W. Human pancreatitis and the role of cathepsin B. Gut 2006, 55, 1228–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghdassi, A.A.; John, D.S.; Sendler, M.; Weiss, F.U.; Reinheckel, T.; Mayerle, J.; Lerch, M.M. Cathepsin D regulates cathepsin B activation and disease severity predominantly in inflammatory cells during experimental pancreatitis. J. Biol. Chem. 2018, 293, 1018–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, B.; Affara, N.I.; Cottone, L.; Junankar, S.; Johansson, M.; DeNardo, D.G.; Korets, L.; Reinheckel, T.; Sloane, B.F.; Bogyo, M.; et al. Cathepsin C is a tissue-specific regulator of squamous carcinogenesis. Genes Dev. 2013, 27, 2086–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaket, T.P.; Singh, M.P.; Khan, I.; Bhardwaj, M.; Kang, S.C. Targeting of cathepsin C induces autophagic dysregulation that directs ER stress mediated cellular cytotoxicity in colorectal cancer cells. Cell. Signal. 2018, 46, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Hamon, Y.; Legowska, M.; Hervé, V.; Dallet-Choisy, S.; Marchand-Adam, S.; Vanderlynden, L.; Demonte, M.; Williams, R.; Scott, C.J.; Si-Tahar, M.; et al. Neutrophilic cathepsin C is maturated by a multistep proteolytic process and secreted by activated cells during inflammatory lung diseases. J. Biol. Chem. 2016, 291, 8486–8499. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, B.; Caughey, G.H.; Chapple, I.; Gauthier, F.; Hirschfeld, J.; Jenne, D.E.; Kettritz, R.; Lalmanach, G.; Lamort, A.S.; Lauritzen, C.; et al. Therapeutic targeting of cathepsin C: From pathophysiology to treatment. Pharmacol. Ther. 2018, 190, 202–236. [Google Scholar] [CrossRef]
- Korkmaz, B.; Lesner, A.; Wysocka, M.; Gieldon, A.; Håkansson, M.; Gauthier, F.; Logan, D.T.; Jenne, D.E.; Lauritzen, C.; Pedersen, J. Structure-based design and in vivo anti-arthritic activity evaluation of a potent dipeptidyl cyclopropyl nitrile inhibitor of cathepsin C. Biochem. Pharmacol. 2019, 164, 349–367. [Google Scholar] [CrossRef]
- Hewitt, C.; McCormick, D.; Linden, G.; Turk, D.; Stern, I.; Wallace, I.; Southern, L.; Zhang, L.; Howard, R.; Bullon, P.; et al. The role of cathepsin C in Papillon-Lefèvre syndrome, prepubertal periodontitis, and aggressive periodontitis. Hum. Mutat. 2004, 23, 222–228. [Google Scholar] [CrossRef]
- Cocchiaro, P.; Fox, C.; Tregidgo, N.W.; Howarth, R.; Wood, K.M.; Situmorang, G.R.; Pavone, L.M.; Sheerin, N.S.; Moles, A. Lysosomal protease cathepsin D; a new driver of apoptosis during acute kidney injury. Sci. Rep. 2016, 6, 27112. [Google Scholar] [CrossRef] [Green Version]
- Cocchiaro, P.; De Pasquale, V.; Della Morte, R.; Tafuri, S.; Avallone, L.; Pizard, A.; Moles, A.; Pavone, L.M. The multifaceted role of the lysosomal protease cathepsins in kidney disease. Front. Cell Dev. Biol. 2017, 5, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, C.; Cocchiaro, P.; Oakley, F.; Howarth, R.; Callaghan, K.; Leslie, J.; Luli, S.; Wood, K.M.; Genovese, F.; Sheerin, N.S.; et al. Inhibition of lysosomal protease cathepsin D reduces renal fibrosis in murine chronic kidney disease. Sci. Rep. 2016, 6, 20101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Lin, Y.; Hu, X.; Wu, Z.; Guo, W. VPS52 induces apoptosis via cathepsin D in gastric cancer. J. Mol. Med. (Berl) 2017, 95, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Dubey, V.; Luqman, S. Cathepsin D as a promising target for the discovery of novel anticancer agents. Curr. Cancer Drug Targets 2017, 17, 404–422. [Google Scholar] [CrossRef]
- Basu, S.; Cheriyamundath, S.; Gavert, N.; Brabletz, T.; Haase, G.; Ben-Ze’ev, A. Increased expression of cathepsin D is required for L1-mediated colon cancer progression. Oncotarget 2019, 10, 5217–5228. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Yuan, X.; Li, F.; Zhang, J.; Zhu, W.; Wei, M.; Li, J.; Wang, X. Myocardial upregulation of Cathepsin D by ischemic heart disease promotes autophagic flux and protects against cardiac remodeling and heart failure. Circ. Heart Fail. 2017, 10, e004044. [Google Scholar] [CrossRef]
- Dey, T.; Kalita, J.; Weldon, S.; Taggart, C.C. Proteases and their inhibitors in chronic obstructive pulmonary disease. J. Clin. Med. 2018, 7, 244. [Google Scholar] [CrossRef] [Green Version]
- Afinogenova, Y.; Ruan, J.; Yang, R.; Kleytman, N.; Pastores, G.; Lischuk, A.; Mistry, P.K. Aberrant progranulin, YKL-40, cathepsin D and cathepsin S in Gaucher disease. Mol. Genet. Metab. 2019, 128, 62–67. [Google Scholar] [CrossRef]
- Chai, Y.L.; Chong, J.R.; Weng, J.; Howlett, D.; Halsey, A.; Lee, J.H.; Attems, J.; Aarsland, D.; Francis, P.T.; Chen, C.P.; et al. Lysosomal cathepsin D is upregulated in Alzheimer’s disease neocortex and may be a marker for neurofibrillary degeneration. Brain Pathol. 2019, 29, 63–74. [Google Scholar] [CrossRef]
- Xicoy, H.; Peñuelas, N.; Vila, M.; Laguna, A. Autophagic- and Lysosomal-related biomarkers for Parkinson’s disease: Lights and shadows. Cells 2019, 8, 1317. [Google Scholar] [CrossRef] [Green Version]
- Pal, P.; Sadhukhan, T.; Chakraborty, S.; Sadhukhan, S.; Biswas, A.; Das, S.K.; Ray, K.; Ray, J. Role of apolipoprotein E, cathepsin D, and brain-derived neurotrophic factor in Parkinson’s disease: A study from Eastern India. Neuromol. Med. 2019, 21, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Steinfeld, R.; Reinhardt, K.; Schreiber, K.; Hillebrand, M.; Kraetzner, R.; Bruck, W.; Saftig, P.; Gartner, J. Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am. J. Hum. Genet. 2006, 78, 988–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketterer, S.; Gomez-Auli, A.; Hillebrand, L.E.; Petrera, A.; Ketscher, A.; Reinheckel, T. Inherited diseases caused by mutations in cathepsin protease genes. FEBS J. 2017, 284, 1437–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macías-Vidal, J.; Guerrero-Hernández, M.; Estanyol, J.M.; Aguado, C.; Knecht, E.; Coll, M.J.; Bachs, O. Identification of lysosomal Npc1-binding proteins: Cathepsin D activity is regulated by NPC1. Proteomics 2016, 16, 150–158. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Homer, R.; Zhang, Y.; Petrache, I.; Mannam, P.; Lee, P.J. Cathepsin E promotes pulmonary emphysema via mitochondrial fission. Am. J. Pathol. 2014, 184, 2730–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, Y.; Zhang, J.; Imari, K.; Yamasaki, R.; Ni, J.; Wu, Z.; Yamamoto, K.; Kira, J.; Nakanishi, H.; Hayashi, Y. Cathepsin E in neutrophils contributes to the generation of neuropathic pain in experimental autoimmune encephalomyelitis. Pain 2019, 160, 2050–2062. [Google Scholar] [CrossRef]
- Gonçalves, N.P.; Moreira, J.; Martins, D.; Vieira, P.; Obici, L.; Merlini, G.; Saraiva, M.; Saraiva, M.J. Differential expression of Cathepsin E in transthyretin amyloidosis: From neuropathology to the immune system. J. Neuroinflammation 2017, 14, 115. [Google Scholar] [CrossRef]
- Pontious, C.; Kaul, S.; Hong, M.; Hart, P.A.; Krishna, S.G.; Lara, L.F.; Conwell, D.L.; Cruz-Monserrate, Z. Cathepsin E expression and activity: Role in the detection and treatment of pancreatic cancer. Pancreatology 2019, 19, 951–956. [Google Scholar] [CrossRef]
- Bras, J.; Djaldetti, R.; Alves, A.M.; Mead, S.; Darwent, L.; Lleo, A.; Molinuevo, J.L.; Blesa, R.; Singleton, A.; Hardy, J.; et al. Exome sequencing in a consanguineous family clinically diagnosed with early-onset Alzheimer’s disease identifies a homozygous CTSF mutation. Neurobiol. Aging 2016, 46, e1–e6. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Ortiz, G.; Pina-Sanchez, P.; Vazquez, K.; Duenas, A.; Taja, L.; Mendoza, P.; Garcia, J.A.; Salcedo, M. Overexpression of cathepsin F, matrix metalloproteinases 11 and 12 in cervical cancer. BMC Cancer 2005, 5, 68. [Google Scholar]
- Mole, S.E.; Anderson, G.; Band, H.A.; Berkovic, S.F.; Cooper, J.D.; Kleine Holthaus, S.M.; McKay, T.R.; Medina, D.L.; Rahim, A.A.; Schulz, A.; et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol. 2019, 18, 107–116. [Google Scholar] [CrossRef]
- Gao, S.; Zhu, H.; Zuo, X.; Luo, H. Cathepsin G and its role in inflammation and autoimmune diseases. Arch. Rheumatol. 2018, 33, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Zou, F.; Lai, X.; Li, J.; Lei, S.; Hu, L. Downregulation of cathepsin G reduces the activation of CD4+ T cells in murine autoimmune diabetes. Am. J. Transl. Res. 2017, 9, 5127–5137. [Google Scholar] [PubMed]
- Twigg, M.S.; Brockbank, S.; Lowry, P.; FitzGerald, S.P.; Taggart, C.; Weldon, S. The role of serine proteases and antiproteases in the cystic fibrosis lung. Mediat. Inflamm. 2015, 2015, 293053. [Google Scholar] [CrossRef] [Green Version]
- Gudmann, N.S.; Manon-Jensen, T.; Sand, J.M.B.; Diefenbach, C.; Sun, S.; Danielsen, A.; Karsdal, M.A.; Leeming, D.J. Lung tissue destruction by proteinase 3 and cathepsin G mediated elastin degradation is elevated in chronic obstructive pulmonary disease. Biochem. Biophys. Res. Commun. 2018, 503, 1284–1290. [Google Scholar] [CrossRef]
- Liu, R.; Chen, L.; Wu, W.; Chen, H.; Zhang, S. Neutrophil serine proteases and their endogenous inhibitors in coronary artery ectasia patients. Anatol. J. Cardiol. 2016, 16, 23–28. [Google Scholar] [CrossRef]
- Denadai-Souza, A.; Bonnart, C.; Tapias, N.S.; Marcellin, M.; Gilmore, B.; Alric, L.; Bonnet, D.; Burlet-Schiltz, O.; Hollenberg, M.D.; Vergnolle, N.; et al. Functional proteomic profiling of secreted serine proteases in health and inflammatory bowel disease. Sci. Rep. 2018, 8, 7834. [Google Scholar] [CrossRef]
- Aghdassi, A.A.; John, D.S.; Sendler, M.; Storck, C.; van den Brandt, C.; Krüger, B.; Weiss, F.U.; Mayerle, J.; Lerch, M.M. Absence of the neutrophil serine protease cathepsin G decreases neutrophil granulocyte infiltration but does not change the severity of acute pancreatitis. Sci. Rep. 2019, 9, 16774. [Google Scholar] [CrossRef]
- Krasavin, M.Y.; Gureev, M.A.; Garabadzhiu, A.V.; Pashkin, A.Y.; Zhukov, A.S.; Khairutdinov, V.R.; Samtsov, A.V.; Shvets, V.I. Inhibition of neutrophil elastase and cathepsin G as a new approach to the treatment of psoriasis: From fundamental biology to development of new target-specific drugs. Dokl. Biochem. Biophys. 2019, 487, 272–276. [Google Scholar] [CrossRef]
- Szekanecz, Z.; Koch, A.E. Macrophages and their products in rheumatoid arthritis. Curr. Opin. Rheumatol. 2007, 19, 289–295. [Google Scholar] [CrossRef]
- Sukhova, G.K.; Shi, G.P. Do cathepsins play a role in abdominal aortic aneurysm pathogenesis? Ann. N. Y. Acad. Sci. 2006, 1085, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Abisi, S.; Burnand, K.G.; Waltham, M.; Humphries, J.; Taylor, P.R.; Smith, A. Cysteine protease activity in the wall of abdominal aortic aneurysms. J. Vasc. Surg. 2007, 46, 1260–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldahmesh, M.A.; Khan, A.O.; Alkuraya, H.; Adly, N.; Anazi, S.; Al-Saleh, A.A.; Mohamed, J.Y.; Hijazi, H.; Prabakaran, S.; Tacke, M.; et al. Mutations in LRPAP1 are associated with severe myopia in humans. Am. J. Hum. Genet. 2013, 93, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahlios, J.; De la Herrán-Arita, A.K.; Mignot, E. The autoimmune basis of narcolepsy. Curr. Opin. Neurobiol. 2013, 23, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Fløyel, T.; Brorsson, C.; Nielsen, L.B.; Miani, M.; Bang-Berthelsen, C.H.; Friedrichsen, M.; Overgaard, A.J.; Berchtold, L.A.; Wiberg, A.; Poulsen, P.; et al. CTSH regulates β-cell function and disease progression in newly diagnosed type 1 diabetes patients. Proc. Natl. Acad. Sci. USA 2014, 111, 10305–10310. [Google Scholar]
- Zhao, H.; Qin, X.; Wang, S.; Sun, X.; Dong, B. Decreased cathepsin K levels in human atherosclerotic plaques are associated with plaque instability. Exp. Ther. Med. 2017, 14, 3471–3476. [Google Scholar] [CrossRef] [Green Version]
- Verbovšek, U.; Van Noorden, C.J.; Lah, T.T. Complexity of cancer protease biology: Cathepsin K expression and function in cancer progression. Semin. Cancer Biol. 2015, 35, 71–84. [Google Scholar] [CrossRef]
- Bühling, F.; Röcken, C.; Brasch, F.; Hartig, R.; Yasuda, Y.; Saftig, P.; Brömme, D.; Welte, T. Pivotal role of cathepsin K in lung fibrosis. Am. J. Pathol. 2004, 164, 2203–2216. [Google Scholar] [CrossRef] [Green Version]
- Salminen-Mankonen, H.J.; Morko, J.; Vuorio, E. Role of cathepsin K in normal joints and in the development of arthritis. Curr. Drug Targets 2007, 8, 315–323. [Google Scholar] [CrossRef]
- Lindström, E.; Rizoska, B.; Henderson, I.; Terelius, Y.; Jerling, M.; Edenius, C.; Grabowska, U. Nonclinical and clinical pharmacological characterization of the potent and selective cathepsin K inhibitor MIV-711. J. Transl. Med. 2018, 16, 125. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Wang, M.; Wang, Z.; Fu, Z.; Lu, A.; Zhang, G. Advances in the discovery of cathepsin K inhibitors on bone resorption. J. Enzyme Inhib. Med. Chem. 2018, 33, 890–904. [Google Scholar] [CrossRef] [PubMed]
- Drake, M.T.; Clarke, B.L.; Oursler, M.J.; Khosla, S. Cathepsin K inhibitors for osteoporosis: Biology, potential clinical utility, and lessons learned. Endocr. Rev. 2017, 38, 325–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brömme, D.; Panwar, P.; Turan, S. Cathepsin K osteoporosis trials, pycnodysostosis and mouse deficiency models: Commonalities and differences. Expert Opin. Drug Discov. 2016, 11, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Sudhan, D.R.; Siemann, D.W. Cathepsin L targeting in cancer treatment. Pharmacol. Ther. 2015, 155, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Xiang, Z.; Zhu, T.; Chen, J.; Zhong, M.Z.; Huang, J.; Wang, K.S.; Li, L.; Sun, L.Q.; Zhou, W.B. Cathepsin L interacts with CDK2-AP1 as a potential predictor of prognosis in patients with breast cancer. Oncol. Lett. 2020, 19, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Sukhova, G.K.; Yang, J.T.; Sun, J.; Ma, L.; Ren, A.; Xu, W.H.; Fu, H.; Dolganov, G.M.; Hu, C.; et al. Cathepsin L expression and regulation in human abdominal aortic aneurysm, atherosclerosis, and vascular cells. Atherosclerosis 2006, 184, 302–311. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, X.; Li, Y.; Lu, Y.; Zhong, H.; Jiang, W.; Chen, A.F.; Billiar, T.R.; Yuan, H.; Cai, J. Cathepsin L activity correlates with proteinuria in chronic kidney disease in humans. Int. Urol. Nephrol. 2017, 49, 1409–1417. [Google Scholar] [CrossRef]
- Garsen, M.; Rops, A.L.; Dijkman, H.; Willemsen, B.; van Kuppevelt, T.H.; Russel, F.G.; Rabelink, T.J.; Berden, J.H.; Reinheckel, T.; van der Vlag, J. Cathepsin L is crucial for the development of early experimental diabetic nephropathy. Kidney Int. 2016, 90, 1012–1022. [Google Scholar] [CrossRef]
- Schechter, I.; Ziv, E. Cathepsins S, B and L with aminopeptidases display β-secretase activity associated with the pathogenesis of Alzheimer’s disease. Biol. Chem. 2011, 392, 555–569. [Google Scholar] [CrossRef]
- Huang, S.; Cao, Y. Correlation of cathepsin S with coronary stenosis degree, carotid thickness, blood pressure, glucose and lipid metabolism and vascular endothelial function in atherosclerosis. Exp. Ther. Med. 2020, 19, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Andrault, P.M.; Panwar, P.; Mackenzie, N.C.W.; Brömme, D. Elastolytic activity of cysteine cathepsins K, S, and V promotes vascular calcification. Sci. Rep. 2019, 9, 9682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Wang, H.; Xu, J. Cathepsin S as a cancer target. Neoplasma 2015, 62, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Steubl, D.; Kumar, S.V.; Tato, M.; Mulay, S.R.; Larsson, A.; Lind, L.; Risérus, U.; Renders, L.; Heemann, U.; Carlsson, A.C.; et al. Circulating cathepsin-S levels correlate with GFR decline and sTNFR1 and sTNFR2 levels in mice and humans. Sci. Rep. 2017, 7, 43538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, B.F.; Figueiredo, J.L.; Aikawa, E. Cathepsin S as an inhibitor of cardiovascular inflammation and calcification in chronic kidney disease. Front. Cardiovasc. Med. 2018, 4, 88. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Lu, B.; Yang, Y.; Zhang, W.; Wang, X.; Zhou, H.; Wen, J.; Yang, Z.; Hu, R. Elevated circulating cathepsin S levels are associated with metabolic syndrome in overweight and obese individuals. Diabetes Metab. Res. Rev. 2019, 35, e3117. [Google Scholar] [CrossRef]
- Lv, B.J.; Lindholt, J.S.; Wang, J.; Cheng, X.; Shi, G.P. Plasma levels of cathepsins L, K, and V and risks of abdominal aortic aneurysms: A randomized population-based study. Atherosclerosis 2013, 230, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Toss, M.; Miligy, I.; Gorringe, K.; Mittal, K.; Aneja, R.; Ellis, I.; Green, A.; Rakha, E. Prognostic significance of cathepsin V (CTSV/CTSL2) in breast ductal carcinoma in situ. J. Clin. Pathol. 2020, 73, 76–82. [Google Scholar] [CrossRef]
- Rath, B.; Klameth, L.; Plangger, A.; Hochmair, M.; Ulsperger, E.; Huk, I.; Zeillinger, R.; Hamilton, G. Expression of proteolytic enzymes by small cell lung cancer circulating tumor cell lines. Cancers 2019, 11, 114. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Li, Q.; Chen, J.; Yi, P.; Xu, X.; Fan, Y.; Cui, B.; Yu, Y.; Li, X.; Du, Y.; et al. Salivary protease spectrum biomarkers of oral cancer. Int. J. Oral Sci. 2019, 11, 7. [Google Scholar] [CrossRef]
- Leng, Y.P.; Ma, Y.S.; Li, X.G.; Chen, R.F.; Zeng, P.Y.; Li, X.H.; Qiu, C.F.; Li, Y.P.; Zhang, Z.; Chen, A.F. l-Homocysteine-induced cathepsin V mediates the vascular endothelial inflammation in hyperhomocysteinaemia. Br. J. Pharmacol. 2018, 175, 1157–1172. [Google Scholar] [CrossRef]
- Tolosa, E.; Li, W.; Yasuda, Y.; Wienhold, W.; Denzin, L.K.; Lautwein, A.; Driessen, C.; Schnorrer, P.; Weber, E.; Stevanovic, S.; et al. Cathepsin V is involved in the degradation of invariant chain in human thymus and is overexpressed in myasthenia gravis. J. Clin. Investig. 2003, 112, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keegan, P.M.; Surapaneni, S.; Platt, M.O. Sickle cell disease activates peripheral blood mononuclear cells to induce cathepsins K and V activity in endothelial cells. Anemia 2012, 2012, 201781. [Google Scholar] [CrossRef] [PubMed]
- Kuester, D.; Vieth, M.; Peitz, U.; Kahl, S.; Stolte, M.; Roessner, A.; Weber, E.; Malfertheiner, P.; Wex, T. Upregulation of cathepsin W-expressing T cells is specific for autoimmune atrophic gastritis compared to other types of chronic gastritis. World J. Gastroenterol. 2005, 11, 5951–5957. [Google Scholar] [CrossRef] [PubMed]
- Bühling, F.; Peitz, U.; Krüger, S.; Küster, D.; Vieth, M.; Gebert, I.; Roessner, A.; Weber, E.; Malfertheiner, P.; Wex, T. Cathepsins K, L, B, X and W are differentially expressed in normal and chronically inflamed gastric mucosa. Biol. Chem. 2004, 385, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Kothapalli, R.; Bailey, R.D.; Kusmartseva, I.; Mane, S.; Epling-Burnette, P.K.; Loughran, T.P., Jr. Constitutive expression of cytotoxic proteases and down-regulation of protease inhibitors in LGL leukemia. Int. J. Oncol. 2003, 22, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Pišlar, A.; Tratnjek, L.; Glavan, G.; Živin, M.; Kos, J. Upregulation of cysteine protease cathepsin X in the 6-hydroxydopamine model of Parkinson’s disease. Front. Mol. Neurosci. 2018, 11, 412. [Google Scholar] [CrossRef]
- Pečar Fonović, U.; Kos, J. Cathepsin X cleaves profilin 1 C-terminal Tyr139 and influences clathrin-mediated endocytosis. PLoS ONE 2015, 10, e0137217. [Google Scholar] [CrossRef] [Green Version]
- Teller, A.; Jechorek, D.; Hartig, R.; Adolf, D.; Reißig, K.; Roessner, A.; Franke, S. Dysregulation of apoptotic signaling pathways by interaction of RPLP0 and cathepsin X/Z in gastric cancer. Pathol. Res. Pract. 2015, 211, 62–70. [Google Scholar] [CrossRef]
- Breznik, B.; Limbaeck Stokin, C.; Kos, J.; Khurshed, M.; Hira, V.V.V.; Bošnjak, R.; Lah, T.T.; Van Noorden, C.J.F. Cysteine cathepsins B, X and K expression in peri-arteriolar glioblastoma stem cell niches. J. Mol. Histol. 2018, 49, 481–497. [Google Scholar] [CrossRef] [Green Version]
- Krueger, S.; Kalinski, T.; Hundertmark, T.; Wex, T.; Küster, D.; Peitz, U.; Ebert, M.; Nägler, D.K.; Kellner, U.; Malfertheiner, P.; et al. Up-regulation of cathepsin X in Helicobacter pylori gastritis and gastric cancer. J. Pathol. 2005, 207, 32–42. [Google Scholar] [CrossRef]
- Almeida, P.C.; Nantes, I.L.; Rizzi, C.C.; Júdice, W.A.; Chagas, J.R.; Juliano, L.; Nader, H.B.; Tersariol, I.L. Cysteine proteinase activity regulation. A possible role of heparin and heparin-like glycosaminoglycans. J. Biol. Chem. 1999, 274, 30433–30438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caglic, D.; Pungercar, J.R.; Pejler, G.; Turk, V.; Turk, B. Glycosaminoglycans facilitate procathepsin B activation through disruption of propeptide-mature enzyme interactions. J. Biol. Chem. 2007, 282, 33076–33085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novinec, M.; Lenarčič, B.; Turk, B. Cysteine cathepsin activity regulation by glycosaminoglycans. Biomed. Res. Int. 2014, 2014, 309718. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef] [PubMed]
- De Pasquale, V.; Pavone, L.M. Heparan sulfate proteoglycans: The sweet side of development turns sour in mucopolysaccharidoses. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165539. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.F.; Muenzer, J. The Mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3421–3452. [Google Scholar]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef]
- Stapleton, M.; Arunkumar, N.; Kubaski, F.; Mason, R.W.; Tadao, O.; Tomatsu, S. Clinical presentation and diagnosis of mucopolysaccharidoses. Mol. Genet. Metab. 2018, 125, 4–17. [Google Scholar] [CrossRef]
- Fecarotta, S.; Gasperini, S.; Parenti, G. New treatments for the mucopolysaccharidoses: From pathophysiology to therapy. Ital. J. Pediatr. 2018, 44, 124. [Google Scholar] [CrossRef]
- Sawamoto, K.; Stapleton, M.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J.; Losada, J.C.; Suarez, D.A.; Tomatsu, S. Therapeutic options for mucopolysaccharidoses: Current and emerging treatments. Drugs 2019, 79, 1103–1134. [Google Scholar] [CrossRef]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.A.; Saftig, P. Lysosomal storage disorders - challenges, concepts and avenues for therapy: Beyond rare diseases. J. Cell Sci. 2019, 132, jcs221739. [Google Scholar] [CrossRef] [PubMed]
- Caciotti, A.; Catarzi, S.; Tonin, R.; Lugli, L.; Perez, C.R.; Michelakakis, H.; Mavridou, I.; Donati, M.A.; Guerrini, R.; d’Azzo, A.; et al. Galactosialidosis: Review and analysis of CTSA gene mutations. Orphanet J. Rare Dis. 2013, 8, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostadinov, S.; Shah, B.A.; Alroy, J.; Phornphutkul, C. A case of galactosialidosis with novel mutations of the protective protein/cathepsin a gene: Diagnosis prompted by trophoblast vacuolization on placental examination. Pediatr. Dev. Pathol. 2014, 17, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Kartal, A.; Aydın, K. A Turkish case of galactosialidosis with a new homozygous mutation in CTSA gene. Metab. Brain Dis. 2017, 32, 973–975. [Google Scholar] [CrossRef]
- Aldámiz-Echevarría, L.; Couce, M.L.; Villate, O.; Fernández-Marmiesse, A.; Piñán, M.Á. New CTSA mutation in early infantile galactosialidosis. Pediatr. Int. 2018, 60, 761–762. [Google Scholar] [CrossRef]
- Nakajima, H.; Ueno, M.; Adachi, K.; Nanba, E.; Narita, A.; Tsukimoto, J.; Itoh, K.; Kawakami, A. A new heterozygous compound mutation in the CTSA gene in galactosialidosis. Hum. Genome Var. 2019, 6, 22. [Google Scholar] [CrossRef]
- Timur, Z.K.; Akyildiz Demir, S.; Seyrantepe, V. Lysosomal cathepsin A plays a significant role in the processing of endogenous bioactive peptides. Front. Mol. Biosci. 2016, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Calhan, O.Y.; Seyrantepe, V. Mice with catalytically inactive cathepsin A display neurobehavioral alterations. Behav. Neurol. 2017, 2017, 4261873. [Google Scholar] [CrossRef]
- Radke, J.; Stenzel, W.; Goebel, H.H. Human NCL neuropathology. Biochim. Biophys. Acta 2015, 1852, 2262–2266. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.B.; Appu, A.P.; Sadhukhan, T.; Casey, S.; Mondal, A.; Zhang, Z.; Bagh, M.B. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol. Neurodegener. 2019, 14, 4. [Google Scholar] [CrossRef] [Green Version]
- Kollmann, K.; Uusi-Rauva, K.; Scifo, E.; Tyynelä, J.; Jalanko, A.; Braulke, T. Cell biology and function of neuronal ceroid lipofuscinosis-related proteins. Biochim. Biophys. Acta 2013, 1832, 1866–1881. [Google Scholar] [CrossRef] [PubMed]
- Hersheson, J.; Burke, D.; Clayton, R.; Anderson, G.; Jacques, T.S.; Mills, P.; Wood, N.W.; Gissen, P.; Clayton, P.; Fearnley, J.; et al. Cathepsin D deficiency causes juvenile-onset ataxia and distinctive muscle pathology. Neurology 2014, 83, 1873–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritchie, K.; Siintola, E.; Armao, D.; Lehesjoki, A.E.; Marino, T.; Powell, C.; Tennison, M.; Booker, J.M.; Koch, S.; Partanen, S.; et al. Novel mutation and the first prenatal screening of cathepsin D deficiency (CLN10). Acta Neuropathol. 2009, 117, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Doccini, S.; Sartori, S.; Maeser, S.; Pezzini, F.; Rossato, S.; Moro, F.; Toldo, I.; Przybylski, M.; Santorelli, F.M.; Simonati, A. Early infantile neuronal ceroid lipofuscinosis (CLN10 disease) associated with a novel mutationin CTSD. J. Neurol. 2016, 263, 1029–1032. [Google Scholar] [CrossRef]
- Siintola, E.; Partanen, S.; Strömme, P.; Haapanen, A.; Haltia, M.; Maehlen, J.; Lehesjoki, A.E.; Tyynelä, J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain 2006, 129, 1438–1445. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.; Yilmaz, U.; Kim, Y.J.; Steinfeld, R.; Meyberg-Solomayer, G.; Oehl-Jaschkowitz, B.; Tzschach, A.; Gortner, L.; Igel, J.; Schofer, O. Congenital CLN disease in two siblings. Wien. Med. Wochenschr. 2015, 165, 210–213. [Google Scholar] [CrossRef]
- Varvagiannis, K.; Hanquinet, S.; Billieux, M.H.; De Luca, R.; Rimensberger, P.; Lidgren, M.; Guipponi, M.; Makrythanasis, P.; Blouin, J.L.; Antonarakis, S.E.; et al. Congenital neuronal ceroid lipofuscinosis with a novel CTSD gene mutation: A rare cause of neonatal-onset neurodegenerative disorder. Neuropediatrics 2018, 49, 150–153. [Google Scholar] [CrossRef]
- Tatti, M.; Motta, M.; Di Bartolomeo, S.; Cianfanelli, V.; Salvioli, R. Cathepsin-mediated regulation of autophagy in saposin C deficiency. Autophagy 2013, 9, 241–243. [Google Scholar] [CrossRef] [Green Version]
- Nelvagal, H.R.; Lange, J.; Takahashi, K.; Tarczyluk-Wells, M.A.; Cooper, J.D. Pathomechanisms in the neuronal ceroid lipofuscinoses. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 165570. [Google Scholar] [CrossRef]
- Li, X.; Qin, L.; Li, Y.; Yu, H.; Zhang, Z.; Tao, C.; Liu, Y.; Xue, Y.; Zhang, X.; Xu, Z.; et al. Presynaptic endosomal Cathepsin D regulates the biogenesis of GABAergic synaptic vesicles. Cell Rep. 2019, 28, 1015–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, A.R.A.; Di Spiezio, A.; Thießen, N.; Schmidt, L.; Grötzinger, J.; Lüllmann-Rauch, R.; Damme, M.; Storck, S.E.; Pietrzik, C.U.; Fogh, J.; et al. Enzyme replacement therapy with recombinant pro-CTSD (cathepsin D) corrects defective proteolysis and autophagy in neuronal ceroid lipofuscinosis. Autophagy 2019, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.R.; Dahl, H.H.; Canafoglia, L.; Andermann, E.; Damiano, J.; Morbin, M.; Bruni, A.C.; Giaccone, G.; Cossette, P.; Saftig, P.; et al. Cathepsin F mutations cause Type B Kufs disease, an adult-onset neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 2013, 22, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Di Fabio, R.; Moro, F.; Pestillo, L.; Meschini, M.C.; Pezzini, F.; Doccini, S.; Casali, C.; Pierelli, F.; Simonati, A.; Santorelli, F.M. Pseudo-dominant inheritance of a novel CTSF mutation associated with type B Kufs disease. Neurology 2014, 83, 1769–1770. [Google Scholar] [CrossRef]
- van der Zee, J.; Mariën, P.; Crols, R.; Van Mossevelde, S.; Dillen, L.; Perrone, F.; Engelborghs, S.; Verhoeven, J.; D’aes, T.; Ceuterick-De Groote, C.; et al. Mutated CTSF in adult-onset neuronal ceroid lipofuscinosis and FTD. Neurol. Genet. 2016, 2, e102. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xu, H.; Yuan, Y.; Lian, Y.; Xie, N.; Ming, L. Novel compound heterozygous mutations causing Kufs disease type B. Int. J. Neurosci. 2018, 128, 573–576. [Google Scholar] [CrossRef]
- Peters, J.; Rittger, A.; Weisner, R.; Knabbe, J.; Zunke, F.; Rothaug, M.; Damme, M.; Berkovic, S.F.; Blanz, J.; Saftig, P.; et al. Lysosomal integral membrane protein type-2 (LIMP-2/SCARB2) is a substrate of cathepsin-F, a cysteine protease mutated in type-B-Kufs-disease. Biochem. Biophys. Res. Commun. 2015, 457, 334–340. [Google Scholar] [CrossRef]
- Jerič, B.; Dolenc, I.; Mihelič, M.; Klarić, M.; Zavašnik-Bergant, T.; Gunčar, G.; Turk, B.; Turk, V.; Stoka, V. N-terminally truncated forms of human cathepsin F accumulate in aggresome-like inclusions. Biochim. Biophys. Acta. 2013, 1833, 2254–2266. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.J.; Hughes, S.M.; Liu, W.; Morgan, A.; Tuxworth, R.I.; Russell, C. The contribution of multicellular model organisms to neuronal ceroid lipofuscinosis research. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 165614. [Google Scholar] [CrossRef]
- Markatos, K.; Mavrogenis, A.F.; Karamanou, M.; Androutsos, G. Pycnodysostosis: The disease of Henri de Toulouse-Lautrec. Eur. J. Orthop. Surg. Traumatol. 2018, 28, 1569–1572. [Google Scholar] [CrossRef]
- Turan, S. Current research on pycnodysostosis. Intractable Rare Dis. Res. 2014, 3, 91–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi, Y.; Atley, L.; Eyre, D.E.; Edelson, J.G.; Superti-Furga, A.; Yasuda, T.; Desnick, R.J.; Gelb, B.D. Determination of bone markers in pycnodysostosis: Effects of cathepsin K deficiency on bone matrix degradation. J. Bone Miner. Res. 1999, 14, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- Aguda, A.H.; Panwar, P.; Du, X.; Nguyen, N.T.; Brayer, G.D.; Brömme, D. Structural basis of collagen fiber degradation by cathepsin K. Proc. Natl. Acad. Sci. USA 2014, 111, 17474–17479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everts, V.; Hou, W.S.; Rialland, X.; Tigchelaar, W.; Saftig, P.; Brömme, D.; Gelb, B.D.; Beertsen, W. Cathepsin K deficiency in pycnodysostosis results in accumulation of non-digested phagocytosed collagen in fibroblasts. Calcif. Tissue Int. 2003, 73, 380–386. [Google Scholar] [CrossRef]
- Soliman, A.T.; Ramadan, M.A.; Sherif, A.; Aziz Bedair, E.S.; Rizk, M.M. Pycnodysostosis: Clinical, radiologic, and endocrine evaluation and linear growth after growth hormone therapy. Metabolism 2001, 50, 905–911. [Google Scholar] [CrossRef]
- Rothenbühler, A.; Piquard, C.; Gueorguieva, I.; Lahlou, N.; Linglart, A.; Bougnères, P. Near normalization of adult height and body proportions by growth hormone in pycnodysostosis. J. Clin. Endocrinol. Metab. 2010, 95, 2827–2831. [Google Scholar] [CrossRef] [Green Version]
- Fuller, K.; Lawrence, K.M.; Ross, J.L.; Grabowska, U.B.; Shiroo, M.; Samuelsson, B.; Chambers, T.J. Cathepsin K inhibitors prevent matrix-derived growth factor degradation by human osteoclasts. Bone 2008, 42, 200–211. [Google Scholar] [CrossRef]
- Bizaoui, V.; Michot, C.; Baujat, G.; Amouroux, C.; Baron, S.; Capri, Y.; Cohen-Solal, M.; Collet, C.; Dieux, A.; Geneviève, D.; et al. Pycnodysostosis: Natural history and management guidelines from 27 French cases and a literature review. Clin. Genet. 2019, 96, 309–316. [Google Scholar] [CrossRef]
- Pan, C.; Nelson, M.S.; Reyes, M.; Koodie, L.; Brazil, J.J.; Stephenson, E.J.; Zhao, R.C.; Peters, C.; Selleck, S.B.; Stringer, S.E.; et al. Functional abnormalities of heparan sulfate in mucopolysaccharidosis-I are associated with defective biologic activity of FGF-2 on human multipotent progenitor cells. Blood 2005, 106, 1956–1964. [Google Scholar] [CrossRef] [Green Version]
- McCarty, D.M.; DiRosario, J.; Gulaid, K.; Killedar, S.; Oosterhof, A.; van Kuppevelt, T.H.; Martin, P.T.; Fu, H. Differential distribution of heparan sulfate glycoforms and elevated expression of heparan sulfate biosynthetic enzyme genes in the brain of mucopolysaccharidosis IIIB mice. Metab. Brain Dis. 2011, 26, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Batzios, S.P.; Zafeiriou, D.I.; Papakonstantinou, E. Extracellular matrix components: An intricate network of possible biomarkers for lysosomal storage disorders? FEBS Lett. 2013, 587, 1258–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiattarella, G.G.; Cerulo, G.; De Pasquale, V.; Cocchiaro, P.; Paciello, O.; Avallone, L.; Belfiore, M.P.; Iacobellis, F.; Di Napoli, D.; Magliulo, F.; et al. The murine model of Mucopolysaccharidosis IIIB develops cardiopathies over time leading to heart failure. PLoS ONE 2015, 10, e0131662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingma, S.D.K.; Wagemans, T.; IJlst, L.; Bronckers, A.L.J.J.; van Kuppevelt, T.H.; Everts, V.; Wijburg, F.A.; van Vlies, N. Altered interaction and distribution of glycosaminoglycans and growth factors in mucopolysaccharidosis type I bone disease. Bone 2016, 88, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Urbani, A.; Salvalaio, M.; Bellesso, S.; Cieri, D.; Zancan, I.; Filocamo, M.; Bonaldo, P.; Szabò, I.; Tomanin, R.; et al. Perturbations in cell signaling elicit early cardiac defects in mucopolysaccharidosis type II. Hum. Mol. Genet. 2017, 26, 1643–1655. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, C.A.; Scudder, S.L.; Lin, Y.; Dozier, L.E.; Phan, D.; Allen, N.J.; Patrick, N.; Esko, J.D. Neurodevelopmental changes in excitatory synaptic structure and function in the cerebral cortex of Sanfilippo syndrome IIIA mice. Sci. Rep. 2017, 7, 46576. [Google Scholar] [CrossRef]
- Bigger, B.W.; Begley, D.J.; Virgintino, D.; Pshezhetsky, A.V. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2018, 125, 322–331. [Google Scholar] [CrossRef]
- De Pasquale, V.; Sarogni, P.; Pistorio, V.; Cerulo, G.; Paladino, S.; Pavone, L.M. Targeting heparan sulfate proteoglycans as a novel therapeutic strategy for Mucopolysaccharidoses. Mol. Ther. Methods Clin. Dev. 2018, 10, 8–16. [Google Scholar] [CrossRef]
- De Pasquale, V.; Pezone, A.; Sarogni, P.; Tramontano, A.; Schiattarella, G.G.; Avvedimento, V.E.; Paladino, S.; Pavone, L.M. EGFR activation triggers cellular hypertrophy and lysosomal disease in NAGLU-depleted cardiomyoblasts, mimicking the hallmarks of mucopolysaccharidosis IIIB. Cell Death Dis. 2018, 9, 40. [Google Scholar] [CrossRef]
- Baldo, G.; Tavares, A.M.; Gonzalez, E.; Poletto, E.; Mayer, F.Q.; Matte, U.D.; Giugliani, R. Progressive heart disease in mucopolysaccharidosis type I mice may be mediated by increased cathepsin B activity. Cardiovasc. Pathol. 2017, 27, 45–50. [Google Scholar] [CrossRef]
- Gonzalez, E.A.; Martins, G.R.; Tavares, A.M.V.; Viegas, M.; Poletto, E.; Giugliani, R.; Matte, U.; Baldo, G. Cathepsin B inhibition attenuates cardiovascular pathology in mucopolysaccharidosis I mice. Life Sci. 2018, 196, 102–109. [Google Scholar] [CrossRef]
- Bigg, P.W.; Baldo, G.; Sleeper, M.M.; O’Donnell, P.A.; Bai, H.; Rokkam, V.R.; Liu, Y.; Wu, S.; Giugliani, R.; Casal, M.L.; et al. Pathogenesis of mitral valve disease in mucopolysaccharidosis VII dogs. Mol. Genet. Metab. 2013, 110, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arfi, A.; Richard, M.; Gandolphe, C.; Bonnefont-Rousselot, D.; Therond, P.; Scherman, D. Neuroinflammatory and oxidative stress phenomena in MPS IIIA mouse model: The positive effect of long-term aspirin treatment. Mol. Genet. Metab. 2011, 103, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Baldo, G.; Lorenzini, D.M.; Santos, D.S.; Mayer, F.Q.; Vitry, S.; Bigou, S.; Heard, J.M.; Matte, U.; Giugliani, R. Shotgun proteomics reveals possible mechanisms for cognitive impairment in Mucopolysaccharidosis I mice. Mol. Genet. Metab. 2015, 114, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Viana, G.M.; Gonzalez, E.A.; Alvarez, M.M.P.; Cavalheiro, R.P.; do Nascimento, C.C.; Baldo, G.; D’Almeida, V.; de Lima, M.A.; Pshezhetsky, A.V.; Nader, H.B. Cathepsin B-associated activation of amyloidogenic pathway in murine Mucopolysaccharidosis type I brain cortex. Int. J. Mol. Sci. 2020, 21, 1459. [Google Scholar] [CrossRef] [Green Version]
- Ohmi, K.; Greenberg, D.S.; Rajavel, K.S.; Ryazantsev, S.; Li, H.H.; Neufeld, E.F. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl. Acad. Sci. USA 2003, 100, 1902–1907. [Google Scholar] [CrossRef] [Green Version]
- Pasqualim, G.; Baldo, G.; de Carvalho, T.G.; Tavares, A.M.; Giugliani, R.; Matte, U. Effects of enzyme replacement therapy started late in a murine model of mucopolysaccharidosis type I. PLoS ONE 2015, 10, e0117271. [Google Scholar] [CrossRef]
- Wilson, S.; Brömme, D. Potential role of cathepsin K in the pathophysiology of mucopolysaccharidoses. J. Pediatr. Rehabil. Med. 2010, 3, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.; Hashamiyan, S.; Clarke, L.; Saftig, P.; Mort, J.; Dejica, V.M.; Brömme, D. Glycosaminoglycan-mediated loss of cathepsin K collagenolytic activity in MPS I contributes to osteoclast and growth plate abnormalities. Am. J. Pathol. 2009, 175, 2053–2062. [Google Scholar] [CrossRef] [Green Version]
- Salvalaio, M.; D’Avanzo, F.; Rigon, L.; Zanetti, A.; D’Angelo, M.; Valle, G.; Scarpa, M.; Tomanin, R. Brain RNA-Seq profiling of the Mucopolysaccharidosis type II mouse model. Int. J. Mol. Sci. 2017, 18, 1072. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Tittiger, M.; Knutsen, R.H.; Kovacs, A.; Schaller, L.; Mecham, R.P.; Ponder, K.P. Upregulation of elastase proteins results in aortic dilatation in mucopolysaccharidosis I mice. Mol. Genet. Metab. 2008, 94, 298–304. [Google Scholar] [CrossRef] [Green Version]
- Baldo, G.; Wu, S.; Howe, R.A.; Ramamoothy, M.; Knutsen, R.H.; Fang, J.; Mecham, R.P.; Liu, Y.; Wu, X.; Atkinson, J.P.; et al. Pathogenesis of aortic dilatation in mucopolysaccharidosis VII mice may involve complement activation. Mol. Genet. Metab. 2011, 104, 608–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalf, J.A.; Linders, B.; Wu, S.; Bigg, P.; O’Donnell, P.; Sleeper, M.M.; Whyte, M.P.; Haskins, M.; Ponder, K.P. Upregulation of elastase activity in aorta in mucopolysaccharidosis I and VII dogs may be due to increased cytokine expression. Mol. Genet. Metab. 2010, 99, 396–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parente, M.K.; Rozen, R.; Seeholzer, S.H.; Wolfe, J.H. Integrated analysis of proteome and transcriptome changes in the mucopolysaccharidosis type VII mouse hippocampus. Mol. Genet. Metab. 2016, 118, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Parente, M.K.; Rozen, R.; Cearley, C.N.; Wolfe, J.H. Dysregulation of gene expression in a lysosomal storage disease varies between brain regions implicating unexpected mechanisms of neuropathology. PLoS ONE 2012, 7, e32419. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.D.; Manolson, M.F.; Downey, G.P.; Sodek, J.; McCulloch, C.A. A novel model system for characterization of phagosomal maturation, acidification, and intracellular collagen degradation in fibroblasts. J. Biol. Chem. 2000, 275, 35432–35441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunlin, E.A.; Harmatz, P.R.; Scarpa, M.; Furlanetto, B.; Kampmann, C.; Loehr, J.P.; Ponder, K.P.; Roberts, W.C.; Rosenfeld, H.M.; Giugliani, R. Cardiac disease in patients with mucopolysaccharidosis: Presentation, diagnosis and management. J. Inherit. Metab. Dis. 2011, 34, 1183–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolourchi, M.; Renella, P.; Wang, R.Y. Aortic root dilatation in Mucopolysaccharidosis I-VII. Int. J. Mol. Sci. 2016, 17, 2004. [Google Scholar] [CrossRef] [Green Version]
- Galimberti, C.; Madeo, A.; Di Rocco, M.; Fiumara, A. Mucopolysaccharidoses: Early diagnostic signs in infants and children. It. J. Pediatr. 2018, 44, 133. [Google Scholar] [CrossRef]
- Zhang, X.; Luo, S.; Wang, M.; Shi, G.P. Cysteinyl cathepsins in cardiovascular diseases. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140360. [Google Scholar] [CrossRef]
- Liu, A.; Gao, X.; Zhang, Q.; Cui, L. Cathepsin inhibition attenuates cardiac dysfunction and remodeling following myocardial infarction by inhibiting the NLRP3 pathway. Mol. Med. Rep. 2013, 8, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, F.L.; Holley, R.J.; Langford-Smith, K.J.; Badrinath, S.; Liao, A.; Langford-Smith, A.; Cooper, J.D.; Jones, S.A.; Wraith, J.E.; Wynn, R.F.; et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS ONE 2012, 7, e35787. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.; Bigger, B.W. The role of innate immunity in mucopolysaccharide diseases. J. Neurochem. 2019, 148, 631–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, A.; Banik, N.L.; Ray, S.K. New insights into the roles of endolysosomal cathepsins in the pathogenesis of Alzheimer’s disease: Cathepsin inhibitors as potential therapeutics. CNS Neurol. Disord. Drug Targets 2008, 7, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Lowry, J.R.; Klegeris, A. Emerging roles of microglial cathepsins in neurodegenerative disease. Brain Res. Bull. 2018, 139, 144–156. [Google Scholar] [CrossRef]
- Nakanishi, H. Microglial cathepsin B as a key driver of inflammatory brain diseases and brain aging. Neural Regen. Res. 2020, 15, 25–29. [Google Scholar] [CrossRef]
- Petanceska, S.; Canoll, P.; Devi, L.A. Expression of rat cathepsin S in phagocytic cells. J. Biol. Chem. 1996, 271, 4403–4409. [Google Scholar] [CrossRef] [Green Version]
- Martins, C.; Hůlková, H.; Dridi, L.; Dormoy-Raclet, V.; Grigoryeva, L.; Choi, Y.; Langford-Smith, A.; Wilkinson, F.L.; Ohmi, K.; DiCristo, G.; et al. Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 2015, 138, 336–355. [Google Scholar] [CrossRef] [Green Version]
- Archer, L.D.; Langford-Smith, K.J.; Bigger, B.W.; Fildes, J.E. Mucopolysaccharide diseases: A complex interplay between neuroinflammation, microglial activation and adaptive immunity. J. Inherit. Metab. Dis. 2014, 37, 1–12. [Google Scholar] [CrossRef]
- Winner, L.K.; Marshall, N.R.; Jolly, R.D.; Trim, P.J.; Duplock, S.K.; Snel, M.F.; Hemsley, K.M. Evaluation of disease lesions in the developing canine MPS IIIA brain. JIMD Rep. 2019, 43, 91–101. [Google Scholar]
- Gabandé-Rodríguez, E.; Pérez-Cañamás, A.; Soto-Huelin, B.; Mitroi, D.N.; Sánchez-Redondo, S.; Martínez-Sáez, E.; Venero, C.; Peinado, H.; Ledesma, M.D. Lipid-induced lysosomal damage after demyelination corrupts microglia protective function in lysosomal storage disorders. EMBO J. 2019, 38, e99553. [Google Scholar] [CrossRef]
- Eisengart, J.B.; King, K.E.; Shapiro, E.G.; Whitley, C.B.; Muenzer, J. The nature and impact of neurobehavioral symptoms in neuronopathic Hunter syndrome. Mol. Genet. Metab. Rep. 2019, 22, 100549. [Google Scholar] [CrossRef] [PubMed]
- Barone, R.; Pellico, A.; Pittalà, A.; Gasperini, S. Neurobehavioral phenotypes of neuronopathic mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oussoren, E.; Brands, M.M.; Ruijter, G.J.; der Ploeg, A.T.; Reuser, A.J. Bone, joint and tooth development in mucopolysaccharidoses: Relevance to therapeutic options. Biochim. Biophys. Acta 2011, 1812, 1542–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, P.G.; Baldo, G.; Mayer, F.Q.; Martinelli, B.; Meurer, L.; Giugliani, R.; Matte, U.; Xavier, R.M. Characterization of joint disease in mucopolysaccharidosis type I mice. Int. J. Exp. Pathol. 2013, 94, 305–311. [Google Scholar] [CrossRef]
- Spina, V.; Barbuti, D.; Gaeta, A.; Palmucci, S.; Soscia, E.; Grimaldi, M.; Leone, A.; Manara, R.; Polonara, G. The role of imaging in the skeletal involvement of mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 118. [Google Scholar] [CrossRef]
- Clarke, L.A.; Hollak, C.E. The clinical spectrum and pathophysiology of skeletal complications in lysosomal storage disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 219–235. [Google Scholar] [CrossRef]
- Opoka-Winiarska, V.; Jurecka, A.; Emeryk, A.; Tylki-Szymańska, A. Osteoimmunology in mucopolysaccharidoses type I, II, VI and VII. Immunological regulation of the osteoarticular system in the course of metabolic inflammation. Osteoarthr. Cartil. 2013, 21, 1813–1823. [Google Scholar] [CrossRef] [Green Version]
- Novack, D.V.; Mbalaviele, G. Osteoclasts-key players in skeletal health and disease. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Tica, J.; Bradbury, E.J.; Didangelos, A. Combined transcriptomics, proteomics and bioinformatics identify drug targets in spinal cord injury. Int. J. Mol. Sci. 2018, 19, 1461. [Google Scholar] [CrossRef] [Green Version]
- Arhzaouy, K.; Papadopoulos, C.; Schulze, N.; Pittman, S.K.; Meyer, H.; Weihl, C.C. VCP maintains lysosomal homeostasis and TFEB activity in differentiated skeletal muscle. Autophagy 2019, 15, 1082–1099. [Google Scholar] [CrossRef]
- Garnero, P.; Borel, O.; Byrjalsen, I.; Ferreras, M.; Drake, F.H.; McQueney, M.S.; Foged, N.T.; Delmas, P.D.; Delaissé, J.M. The collagenolytic activity of cathepsin K is unique among mammalian proteinases. J. Biol. Chem. 1998, 273, 32347–32352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecaille, F.; Bromme, D.; Lalmanach, G. Biochemical properties and regulation of cathepsin K activity. Biochimie 2008, 90, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Lee-Dutra, A.; Wiener, D.K.; Sun, S. Cathepsin S inhibitors: 2004-2010. Expert Opin. Ther. Pat. 2011, 21, 311–337. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, J.E.; Li, C.Y.; de Veer, S.J.; Wang, C.K.; Craik, D.J. Design of potent and selective Cathepsin G inhibitors based on the sunflower trypsin inhibitor-1 scaffold. J. Med. Chem. 2017, 60, 658–667. [Google Scholar] [CrossRef]
- Conaghan, P.G.; Bowes, M.A.; Kingsbury, S.R.; Brett, A.; Guillard, G.; Rizoska, B.; Sjögren, N.; Graham, P.; Jansson, Å.; Wadell, C.; et al. Disease-modifying effects of a novel Cathepsin K inhibitor in osteoarthritis: A randomized, placebo-controlled study. Ann. Intern. Med. 2019. [Google Scholar] [CrossRef]
- Van Acker, G.J.; Saluja, A.K.; Bhagat, L.; Singh, V.P.; Song, A.M.; Steer, M.L. Cathepsin B inhibition prevents trypsinogen activation and reduces pancreatitis severity. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G794–G800. [Google Scholar] [CrossRef] [Green Version]
- Lafarge, J.C.; Pini, M.; Pelloux, V.; Orasanu, G.; Hartmann, G.; Venteclef, N.; Sulpice, T.; Shi, G.P.; Clément, K.; Guerre-Millo, M. Cathepsin S inhibition lowers blood glucose levels in mice. Diabetologia 2014, 57, 1674–1683. [Google Scholar] [CrossRef]
- Houben, T.; Oligschlaeger, Y.; Hendrikx, T.; Bitorina, A.V.; Walenbergh, S.M.A.; van Gorp, P.J.; Gijbels, M.J.J.; Friedrichs, S.; Plat, J.; Schaap, F.G.; et al. Cathepsin D regulates lipid metabolism in murine steatohepatitis. Sci. Rep. 2017, 7, 3494. [Google Scholar] [CrossRef] [Green Version]
- Mizunoe, Y.; Kobayashi, M.; Tagawa, R.; Nakagawa, Y.; Shimano, H.; Higami, Y. Association between lysosomal dysfunction and obesity-related pathology: A key knowledge to prevent metabolic syndrome. Int. J. Mol. Sci. 2019, 20, E3688. [Google Scholar] [CrossRef] [Green Version]
- Rudzińska, M.; Parodi, A.; Soond, S.M.; Vinarov, A.Z.; Korolev, D.O.; Morozov, A.O.; Daglioglu, C.; Tutar, Y.; Zamyatnin, A.A., Jr. The role of cysteine cathepsins in cancer progression and drug resistance. Int. J. Mol. Sci. 2019, 20, 3602. [Google Scholar]
- Hook, G.; Jacobsen, J.S.; Grabstein, K.; Kindy, M.; Hook, V. Cathepsin B is a new drug target for traumatic brain injury therapeutics: Evidence for E64d as a promising lead drug candidate. Front. Neurol. 2015, 6, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romine, H.; Rentschler, K.M.; Smith, K.; Edwards, A.; Colvin, C.; Farizatto, K.; Pait, M.C.; Butler, D.; Bahr, B.A. Potential Alzheimer’s disease therapeutics among weak cysteine protease inhibitors exhibit mechanistic differences regarding extent of Cathepsin B up-regulation and ability to block calpain. Eur. Sci. J. 2017, 13, 38–59. [Google Scholar] [CrossRef] [PubMed]
- Sartori, G.R.; Leitão, A.; Montanari, C.A.; Laughton, C.A. Ligand-induced conformational selection predicts the selectivity of cysteine protease inhibitors. PLoS ONE 2019, 14, e0222055. [Google Scholar] [CrossRef] [PubMed]
- Dana, D.; Pathak, S.K. A review of small molecule inhibitors and functional probes of human Cathepsin L. Molecules 2020, 25, 698. [Google Scholar] [CrossRef] [Green Version]
- Houštecká, R.; Hadzima, M.; Fanfrlík, J.; Brynda, J.; Pallová, L.; Hánová, I.; Mertlíková-Kaiserová, H.; Lepšík, M.; Horn, M.; Smrčina, M.; et al. Biomimetic macrocyclic inhibitors of human Cathepsin D: Structure-activity relationship and binding mode analysis. J. Med. Chem. 2020, 63, 1576–1596. [Google Scholar] [CrossRef]
- Ahmad, S.; Bhagwati, S.; Kumar, S.; Banerjee, D.; Siddiqi, M.I. Molecular modeling assisted identification and biological evaluation of potent cathepsin S inhibitors. J. Mol. Graph. Model. 2020, 96, 107512. [Google Scholar] [CrossRef]
- Johnson, D.S.; Weerapana, E.; Cravatt, B.F. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med. Chem. 2010, 2, 949–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siklos, M.; BenAissa, M.; Thatcher, G.R. Cysteine proteases as therapeutic targets: Does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B 2015, 5, 506–519. [Google Scholar] [CrossRef] [Green Version]
- McConnell, R.M.; Inapudi, K.; Kadasala, N.; Yarlagadda, K.; Velusamy, P.; McConnell, M.S.; Green, A.; Trana, C.; Sayyar, K.; McConnell, J.S. New cathepsin D inhibitor library utilizing hydroxyethyl isosteres with cyclic tertiary amines. Med. Chem. 2012, 8, 1146–1154. [Google Scholar]
- Li, Y.Y.; Fang, J.; Ao, G.Z. Cathepsin B and L inhibitors: A patent review (2010 - present). Expert Opin. Ther. Pat. 2017, 27, 643–656. [Google Scholar] [CrossRef]
- Reinheckel, T. New Kid on the Block. Theranostics 2017, 7, 2965–2967. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Rani, M.; Khan, I.A.; Iqbal, A.; Khan, K.M.; Haleem, M.A.; Azim, M.K. Characterisation of hydrazides and hydrazine derivatives as novel aspartic protease inhibitors. J. Enzyme Inhib. Med. Chem. 2010, 25, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Sheng, L.; He, W.; Zou, C.; Hu, B.; Liu, J.; Ge, W.; Liu, Y.; Wang, J.; Ma, E. Discovery of novel cathepsin inhibitors with potent anti-metastatic effects in breast cancer cells. Bioorg. Chem. 2018, 81, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, Y.; Iwasaki, A.; Sumimoto, S.; Matsubara, T.; Sato, T.; Suenaga, K. Izenamides A and B, statine-containing depsipeptides, and an analogue from a marine Cyanobacterium. J. Nat. Prod. 2018, 81, 1673–1681. [Google Scholar] [CrossRef]
- Arafet, K.; González, F.V.; Moliner, V. Quantum mechanics/molecular mechanics studies of the mechanism of cysteine proteases inhibition by dipeptidyl nitroalkenes. Chemistry 2020, 26, 2002–2012. [Google Scholar] [CrossRef]
- Gacko, M.; Minarowska, A.; Karwowska, A.; Minarowski, Ł. Cathepsin D inhibitors. Folia Histochem. Cytobiol. 2007, 45, 291–313. [Google Scholar]
- Arodola, O.A.; Soliman, M.E. Hybrid 2D/3D-quantitative structure-activity relationship and modeling studies perspectives of pepstatin A analogs as cathepsin D inhibitors. Future Med. Chem. 2018, 10, 5–26. [Google Scholar] [CrossRef]
- Khurana, P.; Yadati, T.; Goyal, S.; Dolas, A.; Houben, T.; Oligschlaeger, Y.; Agarwal, A.K.; Kulkarni, A.; Shiri-Sverdlov, R. Inhibiting extracellular Cathepsin D reduces hepatic steatosis in Sprague-Dawley rats. Biomolecules 2019, 9, 171. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.C.; Dong, X.L.; Dai, Y.; Xiao, G.K.; Wang, X.L.; Wong, K.C.; Wong, M.S.; Yao, X.S. Discovery of a new class of Cathepsin K inhibitors in Rhizoma Drynariae as potential candidates for the treatment of osteoporosis. Int. J. Mol. Sci. 2016, 17, 2116. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Cellular localization of cathepsins (CTSs). Here, we provide some examples of CTS functions in the different locations. CTSs in the extracellular space can cleave extracellular matrix proteins, cell receptors, or cytokines [2]. CTSs can also be found in caveolae triggering cell-surface proteolytic events associated with cell migration [3], or in the endolysosomal [4] and autolysosomal compartments where they process the compartment’s cargo [5]. CTSs in the nuclei play an important role in cell cycle regulation [6], while in the cytosol mediate mitochondrial permeabilization and apoptosis through cleavage of Bid and release of Bax [7]. Some of the cellular components displayed in the figure have been adapted from Smart Servier Medical Art under Creative Commons Attribution 3.0 Unported License.
Figure 1.
Cellular localization of cathepsins (CTSs). Here, we provide some examples of CTS functions in the different locations. CTSs in the extracellular space can cleave extracellular matrix proteins, cell receptors, or cytokines [2]. CTSs can also be found in caveolae triggering cell-surface proteolytic events associated with cell migration [3], or in the endolysosomal [4] and autolysosomal compartments where they process the compartment’s cargo [5]. CTSs in the nuclei play an important role in cell cycle regulation [6], while in the cytosol mediate mitochondrial permeabilization and apoptosis through cleavage of Bid and release of Bax [7]. Some of the cellular components displayed in the figure have been adapted from Smart Servier Medical Art under Creative Commons Attribution 3.0 Unported License.


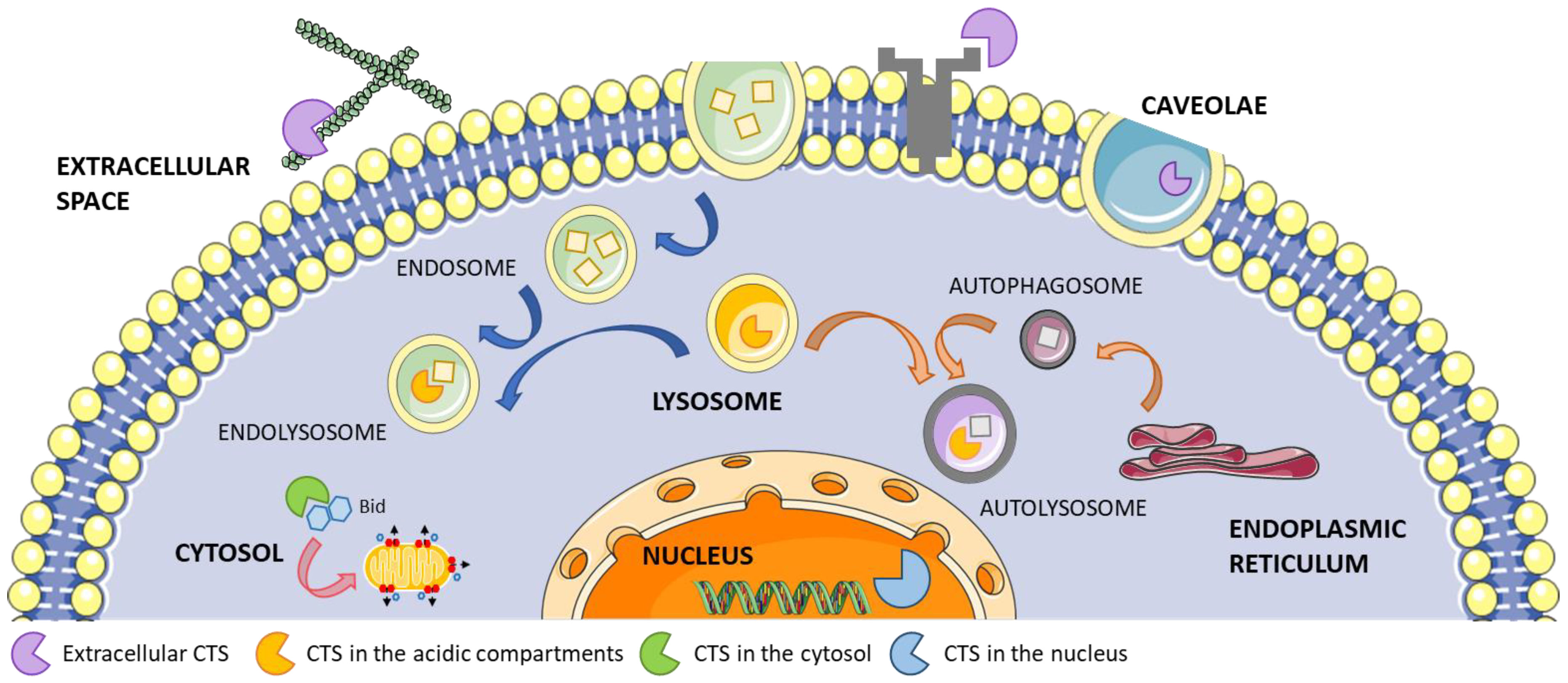{kind=link}
Table 1.
Cathepsin-related diseases.
| Cathepsin (CTS) | Type | MEROPS ID [9] | Disease | References |
|---|---|---|---|---|
| CTSA | Serine | S10.002 | Cardiovascular disease | [10,11] |
| Charcot-Marie Tooth diseases | [12] | |||
| Galactosialidosis | [13] | |||
| Muscular dystrophy | [14] | |||
| CTSB | Cysteine | C01.060 | Alzheimer’s disease | [15] |
| Cancer | [16] | |||
| Cardiovascular disease | [17,18,19] | |||
| Liver fibrosis | [20,21] | |||
| Pancreatitis | [22,23] | |||
| CTSC | Cysteine | C01.070 | Cancer | [24,25] |
| Inflammatory/autoimmune diseases | [26,27,28] | |||
| Papillon–Lefèvre syndrome | [29] | |||
| CTSD | Aspartate | A01.009 | Acute and chronic renal disease | [30,31,32] |
| Cancer | [33,34,35] | |||
| Cardiac disease | [36] | |||
| Chronic obstructive pulmonary disease | [37] | |||
| Gaucher disease | [38] | |||
| Liver fibrosis | [21] | |||
| Neurodegenerative disorders | [39,40,41] | |||
| Neuronal ceroid lipofuscinosis 10 | [42,43] | |||
| Niemann-Pick type C | [44] | |||
| Pancreatitis | [23] | |||
| CTSE | Aspartate | A01.010 | Chronic obstructive pulmonary disease | [37,45] |
| Neuropathies | [46,47] | |||
| Pancreatic cancer | [48] | |||
| CTSF | Cysteine | C01.018 | Alzheimer’s disease | [49] |
| Cancer | [50] | |||
| Neuronal ceroid lipofuscinosis 13 | [43,51] | |||
| CTSG | Serine | S01.133 | Autoimmune diseases | [52,53] |
| Cystic fibrosis | [54] | |||
| Chronic obstructive pulmonary disease | [37,55] | |||
| Coronary artery disease | [56] | |||
| Inflammatory bowel disease | [57] | |||
| Pancreatitis | [58] | |||
| Psoriasis | [59] | |||
| Rheumatoid arthritis | [60] | |||
| CTSH | Cysteine | C01.040 | Aortic aneurism | [61,62] |
| Myopia | [43,63] | |||
| Narcolepsy | [64] | |||
| Type I diabetes | [65] | |||
| CTSK | Cysteine | C01.036 | Aortic aneurism | [61,62] |
| Atherosclerosis | [66] | |||
| Cancer | [67] | |||
| Chronic obstructive pulmonary disease | [37] | |||
| Lung fibrosis | [68] | |||
| Osteoarthritis; Osteoporosis | [69,70,71,72,73] | |||
| Pycnodysostosis | [43,73] | |||
| CTSL | Cysteine | C01.032 | Cancer | [74,75] |
| Cardiovascular disease | [19,61,62,76] | |||
| Chronic kidney disease; Diabetic nephropathy | [77,78] | |||
| CTSO | Cysteine | C01.035 | Unknown | |
| CTSS | Cysteine | C01.034 | Alzheimer’s disease | [79] |
| Atherosclerosis | [18,19,80,81] | |||
| Cancer | [82] | |||
| Chronic obstructive pulmonary disease | [37] | |||
| Chronic kidney disease | [83,84] | |||
| Gaucher disease | [38] | |||
| Metabolic syndromes | [85] | |||
| CTSV | Cysteine | C01.009 | Atherosclerosis | [81,86] |
| Cancer | [87,88,89] | |||
| Hyperhomocysteinemia | [90] | |||
| Myasthenia gravis | [91] | |||
| Sickle cell disease | [92] | |||
| CTSW | Cysteine | C01.037 | Gastritis | [93,94] |
| Leukaemia | [95] | |||
| CTSX/Z | Cysteine | C01.013 | Ageing and neurodegeneration | [96] |
| Cancer | [88,97,98,99] | |||
| Helicobacter pylori | [14,100] |
Table 2.
Lysosomal storage diseases caused by cathepsin mutations.
| LSD Type | OMIM ID | Gene Deficiency | Biological Effect | References |
|---|---|---|---|---|
| Galactosialidosis | #256540 | CTSA | Accumulation of sialylated oligosaccharides and glycoproteins in lysosomes due to complete deficiency in neuraminidase-1 and partial deficiency in beta-galactosidase. | [13,43,119,121,122] |
| Neuronal ceroid lipofuscinosis 10 | #610127 | CTSD | Impaired lysosomal degradation and aberrant autophagy. | [42,43,130,131] |
| Neuronal ceroid lipofuscinosis 13 | #615362 | CTSF | Impaired lysosomal degradation and aberrant autophagy. | [43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140] |
| Pycnodysostosis | #265800 | CTSK | Impaired osteoclast-mediated bone resorption. | [43,141,142,145] |
Table 3.
Cathepsin involvement in mucopolysaccharidoses (MPS).
| MPS Type | OMIM | Cathepsin | Pathological Effect | References |
|---|---|---|---|---|
| MPS-I (Hurler Syndrome) | 607014 607015 607016 | CTSB | Cardiac disease | [160,161,162] |
| CTSB | Neurological disorders | [163,164,165] | ||
| CTSD | Neurological disorders | [164,166,167] | ||
| CTSS, CTSZ | Neurological disorders | [166] | ||
| CTSK | Skeletal disorders | [168,169] | ||
| MPS-II (Hunter Syndrome) | 309900 | CTSA, CTSC, CTSD, CTSH, CTSL, CTSS | Neurological disorders | [170] |
| MPS-III (Sanfilippo Syndrome) | 252900 252920 252930 252940 | CTSD, CTSS, CTSZ | Neurological disorders | [166] |
| CTSK | Skeletal disorders | [168] | ||
| MPS-IV (Morquio Syndrome) | 253000 | CTSK | Skeletal disorders | [168] |
| MPS-VI (Maroteaux-Lamy Syndrome) | 253200 | CTSK | Skeletal disorders | [168] |
| MPS-VII (Sly Syndrome) | 253220 | CTSS | Cardiac disease | [171,172,173] |
| CTSA, CTSB, CTSC, CTSD, CTSH, CTSK, CTSS, CTSZ | Neurological disorders | [174,175] | ||
| CTSK | Skeletal disorders | [168] |
Table 4.
Inhibitors of cathepsins.
| Irreversible Covalent Inhibitors | Reversible Non-Covalent Inhibitors |
|---|---|
| Epoxysuccinates | Aldehydes and ketones |
| Oxiranes and strained ring electrophiles | Cyclopropenones |
| Michael acceptors | α-Keto derivatives |
| Halomethyl ketones | Nitriles |
| Diazomethyl ketones | |
| Acyloxymethyl and other activated ketones |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
De Pasquale, V.; Moles, A.; Pavone, L.M. Cathepsins in the Pathophysiology of Mucopolysaccharidoses: New Perspectives for Therapy. Cells 2020, 9, 979. https://doi.org/10.3390/cells9040979
AMA Style
De Pasquale V, Moles A, Pavone LM. Cathepsins in the Pathophysiology of Mucopolysaccharidoses: New Perspectives for Therapy. Cells. 2020; 9(4):979. https://doi.org/10.3390/cells9040979
Chicago/Turabian StyleDe Pasquale, Valeria, Anna Moles, and Luigi Michele Pavone. 2020. "Cathepsins in the Pathophysiology of Mucopolysaccharidoses: New Perspectives for Therapy" Cells 9, no. 4: 979. https://doi.org/10.3390/cells9040979
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.