The Role of MYC and PP2A in the Initiation and Progression of Myeloid Leukemias


1
Medical Oncology Department, Sidney Kimmel Cancer Center, Thomas Jefferson University, Philadelphia, PA 19107, USA
2
Centro de Investigación Médica Aplicada (CIMA), University of Navarra, 31008 Pamplona, Spain
3
Biochemistry and Genetics Department, University of Navarra, 31008 Pamplona, Spain
4
IdiSNA, Instituto de Investigación Sanitaria de Navarra, 31008 Pamplona, Spain
*
Authors to whom correspondence should be addressed.
Cells 2020, 9(3), 544; https://doi.org/10.3390/cells9030544
Submission received: 30 January 2020
/
Revised: 23 February 2020
/
Accepted: 24 February 2020
/
Published: 26 February 2020
(This article belongs to the Special Issue Regulation and Function of the Myc Oncogene)
Abstract
:The MYC transcription factor is one of the best characterized PP2A substrates. Deregulation of the MYC oncogene, along with inactivation of PP2A, are two frequent events in cancer. Both proteins are essential regulators of cell proliferation, apoptosis, and differentiation, and they, directly and indirectly, regulate each other’s activity. Studies in cancer suggest that targeting the MYC/PP2A network is an achievable strategy for the clinic. Here, we focus on and discuss the role of MYC and PP2A in myeloid leukemias.
1. Introduction
Myeloid cells, including erythrocytes, monocytes/macrophages, granulocytes, and megakaryocyte/platelets and dendritic cells, are key effector cells of the innate immune defense against invading micro-organisms [1]. These cells are continuously generated from hematopoietic stem cells in the bone marrow through a well-timed and regulated process named myelopoiesis [2]. Deregulation of proliferation and lineage differentiation due to the genetic and epigenetic aberrations is linked to blood cell disorders such as acute myeloid leukemia (AML) and chronic myeloid leukemia (CML) [3].
Research in hematology has fundamentally improved our understanding of the biology of cancer and resulted in many innovative discoveries [4]. Many genetic aberrations have prognostic impact on myeloid leukemias, including the overexpression of the MYC, CIP2A, and SET oncogenes. MYC is a master transcription factor that regulates a wide spectrum of target genes, and is found altered in several types of cancer [5,6]. CIP2A and SET are endogenous inhibitors of the phosphatase 2A (PP2A), a tumor suppressor protein that is recurrently inactivated in acute and chronic myeloid leukemias [7]. PP2A is a key regulator of many oncoprotein signaling pathways involved in the regulation of cell growth and survival, including MYC. Mainly, PP2A activity leads to MYC degradation [8]; therefore, its role in regulating MYC biology is fundamental to maintain a correct balance between normal and aberrant phenotype. In this review, we focus on the role of MYC and PP2A in the myeloid leukemia initiation and progression.
1.1. Acute Myeloid Leukemia (AML)
AML is a clonal malignancy that results from genetic and epigenetic abnormalities which leads to uncontrolled proliferation and impaired differentiation of hematopoietic stem and progenitor cells [9,10,11]. Cytogenetic analysis for the detection of chromosomal abnormalities was the first prognostication tool in AML and still remains the backbone of current AML genomic classification [12]. With the advancement of genetic and next-generation sequencing approaches, recurrent mutations in AML have also been characterized [13,14]. The genetic profile of AML is notably heterogeneous, and only a few mutations (i.e., FLT3, NPM1, and DNMT3A) are present in more than a quarter of AML patients [15]. Transcription factors are a class of genes that are frequently altered in AML, such as PML, EVI1, GATA2, RUNX1, ETV6, and C/EBPα, which establish and maintain genetic networks governing the genesis and function of blood stem and progenitor cells [16]. Furthermore, a recent genomic analysis in over 1500 AML patients identified novel hotspot mutations in another critical transcription factor, such as MYC [17]. MYC high expression is also common in AML.
Scientific understanding of the AML biology has led to the development of targeted treatment approaches [4], i.e., the all-trans retinoic acid (ATRA) treatment for acute promyelocytic leukemia (APL) that carries the PML-RARα translocation [18] or the use of FLT3 kinase inhibitors in FLT3-mutated cases [19,20]. Nevertheless, the treatment of AML has been mostly unmodified for the past decades. Although many patients with AML respond to induction chemotherapy, relapse is common and represents the primary cause of treatment failure [9]. Interestingly, bone marrow environment has been suggested to play a role in this unresponsive phenotype, by providing shelter to leukemic stem cells, whose persistence eventually leads to therapy resistance [21].
1.2. Chronic Myeloid Leukemia (CML)
CML is a type of leukemia with an initial chronic phase (CP) characterized by a massive expansion of myeloid precursors and mature cells, followed by a late acute phase termed “blast crisis” (BC), which resembles AML [22]. CML biology is characterized by the presence of the reciprocal translocation t(9;22)(q34;q11) that generates a BCR-ABL fusion protein with aberrant and uncontrolled activity [23]. The development of a BCR/ABL kinase inhibitor, imatinib, was a significant major improvement in CML treatment [24]. However, imatinib, and second and third-generation tyrosine kinase inhibitors’ effectiveness is often inadequate as resistant CML stem cells and residual disease persist in many patients [25,26,27]. Genomic sequencing of CML patients has identified additional genetic changes, like mutations of the tumor suppressor genes RB1, TP53, and CDKN2A [28], and highlighted the fact that BCR-ABL kinase mutations are only identified in approximately 50% of patients with poor responses and disease progression [29]. Therefore, progress in our understanding of CML biology is still required.
1.3. MYC
MYC is a master transcription factor of the helix-loop-helix leucine zipper (HLHZ) family [30]. MYC forms an heterodimer with MAX and binds to E-box sequences in the regulatory regions of numerous target genes [31] involved in the control of proliferation and differentiation, metabolism, cell growth, among other processes [32,33]. The expression of MYC is tightly controlled in normal cells. However, the aberrant activity of MYC is one of the most common events in solid and hematopoietic neoplasias [31].
The MYC gene was first discovered as the oncogene carried by a retrovirus that induced a myeloid neoplasm in chicken, i.e., myelocytomatosis [34] and a few years later identified in human Burkitt lymphoma and in mouse plasmacytomas [35,36]. MYC is amplified or deregulated in both AML and CML; however, the mechanisms that lead to MYC overexpression in AML are still not well-defined. It has been suggested that the aberrant activity of rearranged transcription factors (i.e., RUNX1-ETO, PML-RARα, and PLZF-RARα) might increase MYC expression. Trisomy 8, where MYC is localized, could represent another potential mechanism. MYC upregulation is usually moderate in myeloid leukemias, yet, enough to generate radical changes in the myeloid precursor differentiation [37].
MYC is a short half-live protein; thus, its post-transcriptional regulation plays an essential role in its stability and function. Two interdependent phosphorylation sites that are critical for the regulation of MYC have been identified. Activation of the ERK cascade or cyclin-dependent kinases (CDKs) leads to the phosphorylation of MYC at Serine 62 (S62) [38]. This modification enhances MYC DNA binding and gene regulation. Phosphorylation of S62 also primes MYC for glycogen synthase kinase 3 (GSK3)-mediated phosphorylation at Threonine 58 (T58) [39], which initiates MYC turnover [8]. Dual phosphorylated MYC (S62 and T58) becomes target of the PP2A, which dephosphorylates the stabilizing S62 residue and marks MYC for ubiquitin-mediated proteosomal degradation [40] (Figure 1).
1.4. MYC in Myeloid Leukemias
The mechanisms behind MYC-driven oncogenesis in AML are still underexplored. Different murine models, either using engraftment of hematopoietic precursors over-expressing MYC or transgenic mice, demonstrated the myeloid leukemogenic activity of MYC [37] and highlighted the significance of MYC-driven abrogation of apoptosis as key molecular mechanism [41,42]. In APL, MYC cooperates with PML-RARα to accelerate the development of leukemia [43], while in AML, MYC protein expression is a poor prognostic factor, particularly in patients with high risk of relapse [44,45]. It has been reported that the inhibition of MYC in AML cell lines drastically induces cell differentiation [46], and studies in mice have demonstrated that even transient inactivation of MYC yields tumor regression [47], supporting the hypothesis that targeting MYC regulation could be a highly pursued goal to treat cancer patients.
In CML, it has been widely reported that the BCR-ABL aberrant kinase activity induces MYC transcription [48] and translation, and that the progress into blast crisis depends on this BCR-ABL-induced MYC expression [49]. The generation of a mouse model with an inducible BCR-ABL along with sequentially and spontaneously activated mutations, helped to recapitulate the human CML biology. Interestingly, Giotopoulos et al. identified potentially targetable pathways in CML such as ERG, MEK, RAF, JAK1/2, and confirmed that MYC is crucial for CML-progression to BC [50]. Additionally, MYC and its partner MAX bind to the BCR promoter, enhancing BCR-ABL mRNA and protein content, creating a feedback loop that is key for CML transition to BC [51]. Nevertheless, Reavie et al. recently showed that MYC is essential but not sufficient for CML induction and progression. Additional mutations, such as p53 loss, are needed to sustain CML pathobiology [52].
Yet, high levels of MYC correlate with poor response to imatinib and, at molecular level, MYC induces genomic instability [53]. Also, MYC’s negative regulation of p27kip1 protein stability seems to be critical. By inducing SKP2, a component of the ubiquitin ligase complex that targets p27kip1 for degradation, MYC prevents cell differentiation [54].
1.5. PP2A
Reversible phosphorylation is one of the mechanisms that cells use to maintain normal homeostasis [7], yet the role of phosphatases in cancer has just started to be fully considered. PP2A is a serine-threonine phosphatase that dephosphorylates serines or threonines residues of key factors involved in the regulation of cell proliferation, survival, and differentiation and that together with PP1, contributes >90% of the phosphatase activity of the cell [55]. Such wide-ranging substrate specificity is achieved by the formation of at least 100 distinct heterotrimeric holoenzymes, each containing a common core enzyme formed by the catalytic (C) and scaffold (A) subunits and a third variable regulatory subunit (B) that determine substrate specificity and subcellular localization [56]. In humans, the C and A subunits are each encoded by two different genes, giving rise to two isoforms of the C subunit (PPP2CA and PPP2CB), and A subunit (PPP2R1A and PPP2R1B). For the B-type subunits, 15 human genes have been described, giving rise to more than 23 isoforms belonging to four different families, PR55 ⁄ B (or B55, encoded by PPP2R2A to PPP2R2D), PR61 ⁄ B’ (or B56, encoded by PPP2R5A to PPP2R5E), PR72 ⁄ B’’ (encoded by PPP2R3A to PPP2R3C), and PR93 ⁄ PR110 ⁄ B’’’ (or the striatins, encoded by STRN, STRN3 and STRN4) (Table 1). This diversity in composition creates specificity and forms the basis for PP2A’s multiple physiological functions [57,58]. Particularly, complexes containing PP2A B56α, B56γ, B55α, and PR72/PR130 have been reported to specifically activate MYC, WNT, ERK MAPK, and PI3K/AKT pathways and, thus, considered actionable targets [56,59,60,61].
PP2A is frequently inactivated in both AML and CML [64,68], and several studies now recognize PP2A inactivation as an essential requirement for transforming human cells [56]. Mutations, mostly in the A subunit that impair the phosphatase function or assembly, have been reported in solid tumors. However, their incidence in myeloid leukemia is low, except for the PP2A-C subunit haploinsufficiency in AML with deletions of chromosome 5, a region that includes other tumor suppressor genes [65]. Altered expression of PP2A subunits is a recurrent event in AML. Mainly, PP2A-B55α is under-expressed in primary blast cells and correlates with increased AKT phosphorylation and shorter complete remission [7,69]. At protein level, B55α content also associates with increased MYC activity [70]. Nevertheless, current evidence suggests that the inhibition of PP2A is mostly achieved through non-genomic mechanisms, such as post-translational modifications of its subunits and/or overexpression of PP2A endogenous inhibitors [7].
The activity and localization of PP2A can be regulated by post-translational modifications [73]. Methylation and phosphorylation of residues from PP2A-C subunits modulate the formation of the complex. For instance, methylation of leucine 309 (L309) in the catalytic PP2A-C by the leucine carboxyl methyltransferase 1 (LCMT1) [74] is required for binding of the PR55/B55 subunit [75]. Phosphorylation of tyrosine 307 (Y307) considerably impairs PP2A phosphatase activity by inhibiting the interaction of PP2A-C with the PTPA-activating protein, which enables the assembly of the holoenzyme [58,76], and the regulatory subunits PR55/B55 and PR61/B56 [77]. Actually, increased phosphorylation of Y307 of PP2A-C inversely correlates with disease prognosis in AML and CML [68,78]. In contrast, threonine 304 (T304) phosphorylation prevents the assembly of PR55/B55 to the core enzyme [75].
In AML, our group and others showed that the hyperphosphorylation of PP2A-C and consequent inactivation is observed in 78% of AML patients, along with high levels of PP2A endogenous inhibitors SET and CIP2A [68,79,80,81]. In c-KIT-mutated AML, oncogenic receptor tyrosine kinase c-KIT, which activates proliferation, differentiation, and survival signaling pathways, requires PP2A inactivation for leukemogenesis. Roberts et al. have shown that myeloid precursor cells expressing oncogenic mutant c-KIT receptors display significantly reduced PP2A activity compared to c-KIT negative or wild type c-KIT. Furthermore, inhibition of PP2A by mutant c-KIT is associated with reduced protein expression of PP2A subunits, together with altered expression of PP2A endogenous inhibitor SET oncogene [63]. PP2A inhibitors are described in more detail in the next paragraph.
In CML, functional inactivation of PP2A by BCR-ABL is essential for the progression into blast crisis. One suggested mechanism of inactivation is the increased expression of the PP2A-Aα subunit observed in BCR-ABL+ cells, as the Aα subunit could sequester the catalytic or regulatory subunits and act as a PP2A inhibitor [82]. However, it is generally believed that BCR-ABL enhances the expression of the endogenous PP2A inhibitor SET with subsequent loss of PP2A activity [64,83]. Imatinib-derived inhibition of BCR-ABL dramatically reduces SET expression and restores PP2A activity, resulting in the dephosphorylation of STAT5, ERK1/2, AKT, BAD, and JAK2, substrates of both BCR-ABL and PP2A [78,84].
1.6. PP2A Endogenous Inhibitors
Recent reports have highlighted the role of the PP2A endogenous inhibitors ANP32A, SET, CIP2A, and ARPP19 (Figure 2), and their role in cancer progression [85,86]. Particularly, their alterations in myeloid leukemia are reported in Table 2.
ANP32A, (acidic nuclear phosphoprotein 32a), also known as I1PP2A (Inhibitor 1 of PP2A), is a potent inhibitor of PP2A catalytic subunit [87,88]. It is overexpressed in primary AML cells [89] and is a critical factor that contributes to acute megakaryoblastic leukemia progression [101].
SET, also known as Inhibitor 2 of PP2A (I2PP2A) or TAF-1 or PHAPII, is a multitask protein [102] that along with being a histone chaperone [103] and inhibitor of histone acetylation [104] binds directly to the catalytic subunit C of PP2A, impairing its activity [90,91]. This oncoprotein is overexpressed in both solid and hematological tumors [64,79]. In AML, SET is highly dependent on MYC-transcriptional activity and recruitment of RUNX1 and GATA2 on its promoter [102]. By impairing SET binding to PP2A, with molecules like FTY720 [82,105], CM-1231 [106], or OP449 [107,108] it is possible to re-establish PP2A activity, inhibiting tumor growth [91,109] and overcoming therapeutic resistance in preclinical models [92,108]. SET is known to interact with SETBP1 (SET-binding protein 1), which protects SET from protease cleavage. SETBP1 is also overexpressed in over 27% AML patients, and its high expression significantly correlates with shorter overall survival, mostly in patients over 60 years [79]. Mutations in the SETBP1 gene have been found, and the coexistence of SETBP1 and ASXL1 mutations is known to promote leukemogenesis by repressing TGFβ pathway genes [110]. Genetic mutations in the SETBP1 gene have also been found in CML and other myeloproliferative disorders with no BCR-ABL translocation [111,112]. Along with its SET stabilizing function, SETBP1 additionally promotes self-renewal of myeloid progenitors in vivo, wherein BCR-ABL positive conditions help generating aggressive leukemias in recipient mice [113]. In addition, our group has recently shown that p38β potentiates the inactivation of PP2A mediated by SET by two mechanisms: facilitating cytoplasmic translocation of SET through phosphorylation of CK2, and directly binding and stabilizing the SET protein [114].
CIP2A, also named KIAA1524, is a scaffold protein that stabilizes MYC by inhibiting the PP2A-regulated de-phosphorylation on the residue S62 [93]. Structure analysis of the CIP2A-PP2A interaction revealed that CIP2A forms homodimers. The dimer is stabilized by its interaction with the PP2A-B56α and B56γ subunits [94]. CIP2A specifically interacts with PP2A-B56α holoenzyme, which is the most ubiquitously expressed isoform of the B56 family [115] and the one that associates with MYC to negatively regulate its protein levels and activity [38]. Thus, in the absence of PP2A-B56α-mediated control, MYC can be aberrantly expressed. By preventing PP2A-B56α interaction with MYC, CIP2A gained scientific attention, and it is now considered a promising target for tumor treatment, particularly in AML [81,96,116], where it is overexpressed and promotes cell growth and neoplastic transformation [81]. Additionally, CIP2A has been reported to be a translocation partner with the mixed-lineage leukemia (MLL) gene [117]. In CML, CIP2A is also biologically and clinically important [96,118], as high levels correlate not only with increased levels of MYC, but with upregulation of the antiapoptotic protein BCL-XL [97], another important target of PP2A [119]. Additionally, CIP2A mRNA and protein content is regulated by MYC, creating a positive feedback loop between the two oncogenes in cancer [95]. By inhibiting MYC, by treating the cells with an inhibitor of MYC interaction with its partner MAX through the basic-helix-loop-helix-leucine zipper domain, 10058-F4 [120], Lucas et al. demonstrated that the levels of CIP2A and SET could be dramatically reduced, confirming once again that studying the inhibition of MYC is worth it [121].
Among the other PP2A negative regulators, ARPP19, together with ENSA and BOD1, associates with and inhibits PP2A to promote mitotic entry [98,122,123]. While its role in tumor progression is still underexplored, a recent study revealed that ARPP19 mRNA expression is an independent predictor for relapse in AML, and it might promote cell survival by regulating CIP2A and MYC expression [100].
1.7. Targeting the MYC/PP2A Axis in Leukemia
MYC is a desirable therapeutic target for several types of cancer. Yet, because of the absence of a clear ligand-binding domain and its unstructured nature, direct targeting of MYC is still a challenge [124,125].
Several molecules have been described as MYC inhibitors, although none of them are used in the clinic. The 10058-F4, which prevents the interaction of MYC with MAX, used in several preclinical studies, was able to reduce BCR-ABL kinase activity and CIP2A expression in CML patients [121]. In AML cells, it was reported that 10058-F4 inhibits growth, induces cell cycle arrest, and differentiation [126]. Besides, our group described that 10058-F4 treatment also reduces SET mRNA transcription, leading to PP2A re-establishment in AML cells [102].
Another indirect inhibitor of MYC is JQ1, a powerful inhibitor of BET bromodomain protein BRD4 which regulates MYC transcription [127]. The treatment with JQ1 or other BET inhibitors triggers MYC downregulation with consequent cell cycle arrest and apoptosis in mouse and human leukemia cells [128,129,130,131]. Some BET inhibitors are currently in early phase clinical trials for treating hematopoietic malignancies and solid tumors with exciting results [132]. However, toxic side effects [133] and cases of resistance have been reported [134].
Therefore, pointing at PP2A activation to circumvent MYC undruggability and target MYC protein stability is considered today a suitable and attractive approach. Even though the levels of MYC after PP2A activation have not been clearly defined in myeloid leukemias, it is known that the levels of PP2A activity indirectly correlate with the ones of MYC.
PP2A-activating drugs are known to be highly effective in reducing MYC activity in several types of cancers [135,136,137]. Notably, small molecules that prevent SET-PP2A interaction such as FTY720, OP449, and CM-1231 re-activates PP2A, inhibiting cell proliferation and promoting apoptosis in AML and CML cell lines and primary patient samples [62,63,68,83,92,106,108,138,139,140]. Combination of FTY720 or OP449 along with Idarubicin and Ara-C, drugs used in standard induction therapy in AML, or FLT3 inhibitors, as well as with tyrosine kinase inhibitors in CML, have a synergic effect and significantly reduced growth of leukemic cells [92,108,138,141].
2. Conclusions
Deregulation of the MYC oncogene, along with the inactivation of PP2A, is persistently found in myeloid leukemias, inducing tumor progression and conferring poor prognosis. Both proteins play pivotal roles in the initiation and development of the disease and constitute attractive therapeutic targets. As for other transcription factors, the development of molecules inhibiting MYC activity continues to be challenging. Thus, indirect approaches have been suggested. The discovery of the MYC/PP2A axis has set the foundation for the use of PP2A activating molecules to impair MYC stability. Ultimately, more knowledge of the MYC/PP2A biology is needed. Nonetheless, studies in leukemia cells and preclinical models hint that therapies targeting MYC/PP2A inhibitory network are a clinically feasible strategy for leukemia treatment.
Author Contributions
Conceptualization, writing, review and editing R.P. and M.D.O. All authors have read and agree to the published version of the manuscript.
Funding
This work was supported by grants from the ISCIII and Spanish Ministry of Economy and Competiveness (PI17/02272) and by CIBERONC (CB16/12/00489) (Co-financed with FEDER funds), Departamento de Salud of the Government of Navarra (29/2015) and Departamento de Desarrollo Económico of the Government of Navarra (PI020 PDX-AML) (M.D.O.) and from the Prostate Cancer Foundation (R.P.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nauseef, W.M.; Borregaard, N. Neutrophils at work. Nat. Immunol. 2014, 15, 602–611. [Google Scholar] [CrossRef]
- Engert, A.; Balduini, C.; Brand, A.; Coiffier, B.; Cordonnier, C.; Döhner, H.; de Wit, T.D.; Eichinger, S.; Fibbe, W.; Green, T.; et al. The European Hematology Association Roadmap for European Hematology Research: A consensus document. Haematologica 2016, 101, 115–208. [Google Scholar] [CrossRef]
- Vicente, C.; Conchillo, A.; García-Sánchez, M.A.; Odero, M.D. The role of the GATA2 transcription factor in normal and malignant hematopoiesis. Crit. Rev. Oncol. Hematol. 2012, 82, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Richard-Carpentier, G.; DiNardo, C.D. Single-agent and combination biologics in acute myeloid leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2019, 1, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Dunn, S.; Cowling, V.H. Myc and mRNA capping. Biochim. Biophys. Acta 2015, 1849, 501–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, C.J.; van Riggelen, J. MYC-Master Regulator of the Cancer Epigenome and Transcriptome. Genes 2017, 8, 142. [Google Scholar] [CrossRef] [PubMed]
- Arriazu, E.; Pippa, R.; Odero, M.D. Protein Phosphatase 2A as a Therapeutic Target in Acute Myeloid Leukemia. Front. Oncol. 2016, 6, 78. [Google Scholar] [CrossRef] [Green Version]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T.; et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell. Biol. 2004, 6, 308–318. [Google Scholar] [CrossRef]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chen, B.R.; Deshpande, A. Epigenetic Regulators in the Development, Maintenance, and Therapeutic Targeting of Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Almosailleakh, M.; Schwaller, J. Murine Models of Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pippa, R.; Odero, M.D. Importance of genetics in acute myeloid leukemia. An. Sist. Sanit. Navar. 2014, 37, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.D.; Hourigan, C.S. MRD evaluation of AML in clinical practice: Are we there yet? Hematol. Am. Soc. Hematol. Educ. Program 2019, 1, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churpek, J.E.; Bresnick, E.H. Transcription factor mutations as a cause of familial myeloid neoplasms. J. Clin. Investig. 2019, 129, 476–488. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Cortes, J.E. Mutations in AML: Prognostic and therapeutic implications. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 348–355. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Li, C.; Zhu, X. FLT3 inhibitors in acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 133. [Google Scholar] [CrossRef]
- Smith, C.C. The growing landscape of FLT3 inhibition in AML. Hematol. Am. Soc. Hematol. Educ. Program 2019, 1, 539–547. [Google Scholar] [CrossRef]
- Zhou, H.S.; Carter, B.Z.; Andreeff, M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabretta, B.; Perrotti, D. The biology of CML blast crisis. Blood 2004, 103, 4010–4022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowley, J.D. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Bhamidipati, P.K.; Kantarjian, H.; Cortes, J.; Cornelison, A.M.; Jabbour, E. Management of imatinib-resistant patients with chronic myeloid leukemia. Ther. Adv. Hematol. 2013, 4, 103–117. [Google Scholar] [CrossRef] [PubMed]
- García-Gutiérrez, V.; Hernández-Boluda, J.C. Tyrosine Kinase Inhibitors Available for Chronic Myeloid Leukemia: Efficacy and Safety. Front. Oncol. 2019, 9, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrotti, D.; Silvestri, G.; Stramucci, L.; Yu, J.; Trotta, R. Cellular and Molecular Networks in Chronic Myeloid Leukemia: The Leukemic Stem, Progenitor and Stromal Cell Interplay. Curr. Drug Targets 2017, 18, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.; Fioretos, T.; Mitelman, F. Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol. 2002, 107, 76–94. [Google Scholar] [CrossRef]
- Shanmuganathan, N.; Branford, S. The Hidden Pathogenesis of CML: Is BCR-ABL1 the First Event? Curr. Hematol. Malig. Rep. 2019, 14, 501–506. [Google Scholar] [CrossRef]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kress, T.R.; Sabò, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheiness, D.; Bishop, J.M. DNA and RNA from uninfected vertebrate cells contain nucleotide sequences related to the putative transforming gene of avian myelocytomatosis virus. J. Virol. 1979, 31, 514–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalla-Favera, R.; Gelmann, E.P.; Martinotti, S.; Franchini, G.; Papas, T.S.; Gallo, R.C.; Wong-Stall, F. Cloning and characterization of different human sequences related to the onc gene (v-myc) of avian myelocytomatosis virus (MC29). Proc. Natl. Acad. Sci. USA 1982, 21, 6497–6501. [Google Scholar] [CrossRef] [Green Version]
- Crews, S.; Barth, R.; Hood, L.; Prehn, J.; Calame, K. Mouse c-myc oncogene is located on chromosome 15 and translocated to chromosome 12 in plasmacytomas. Science 1982, 4579, 1319–1321. [Google Scholar] [CrossRef]
- Delgado, M.D.; Albajar, M.; Gomez-Casares, M.T.; Batlle, A.; León, J. MYC oncogene in myeloid neoplasias. Clin. Transl. Oncol. 2013, 15, 87–94. [Google Scholar] [CrossRef]
- Farrell, A.S.; Sears, R.C. MYC degradation. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef]
- Liu, L.; Eisenman, R.N. Regulation of c-Myc Protein Abundance by a Protein Phosphatase 2A-Glycogen Synthase Kinase 3β-Negative Feedback Pathway. Genes Cancer 2012, 3, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Arnold, H.K.; Sears, R.C. Protein phosphatase 2A regulatory subunit B56alpha associates with c-myc and negatively regulates c-myc accumulation. Mol. Cell. Biol. 2006, 26, 2832–2844. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Li, Q.; O’Neal, J.; Kreisel, F.; Le Beau, M.M.; Tomasson, M.H. c-Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood 2005, 106, 2452–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Z.; Luo, H.; Payton, J.E.; Cain, J.; Ley, T.J.; Opferman, J.T.; Tomasson, M.H. Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J. Clin. Investig. 2010, 120, 2109–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, L.; Wei, G.; Sevcikova, S.; Phan, V.; Jain, S.; Shieh, A.; Wong, J.C.; Li, M.; Dubansky, J.; Maunakea, M.L.; et al. Gain of MYC underlies recurrent trisomy of the MYC chromosome in acute promyelocytic leukemia. J. Exp. Med. 2010, 207, 2581–2594. [Google Scholar] [CrossRef] [PubMed]
- Ohanian, M.; Rozovski, U.; Kanagal-Shamanna, R.; Abruzzo, L.V.; Loghavi, S.; Kadia, T.; Futreal, A.; Bhalla, K.; Zuo, Z.; Huh, Y.O.; et al. MYC protein expression is an important prognostic factor in acute myeloid leukemia. Leuk. Lymphoma 2019, 60, 37–48. [Google Scholar] [CrossRef]
- Yun, S.; Sharma, R.; Chan, O.; Vincelette, N.D.; Sallman, D.A.; Sweet, K.; Padron, E.; Komrokji, R.; Lancet, J.E.; Abraham, I.; et al. Prognostic significance of MYC oncoprotein expression on survival outcome in patients with acute myeloid leukemia with myelodysplasia related changes (AML-MRC). Leuk. Res. 2019, 84, 106194. [Google Scholar] [CrossRef]
- Delgado, M.D.; León, J. Myc roles in hematopoiesis and leukemia. Genes Cancer 2010, 1, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Brondfield, S.; Umesh, S.; Corella, A.; Zuber, J.; Rappaport, A.R.; Gaillard, C.; Lowe, S.W.; Goga, A.; Kogan, S.C. Direct and indirect targeting of MYC to treat acute myeloid leukemia. Cancer Chemother. Pharmacol. 2015, 76, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Sawyers, C.L.; Callahan, W.; Witte, O.N. Dominant negative MYC blocks transformation by ABL oncogenes. Cell 1992, 70, 901–910. [Google Scholar] [CrossRef]
- Notari, M.; Neviani, P.; Santhanam, R.; Blaser, B.W.; Chang, J.S.; Galietta, A.; Willis, A.E.; Roy, D.C.; Caligiuri, M.A.; Marcucci, G.; et al. A MAPK/HNRPK pathway controls BCR/ABL oncogenic potential by regulating MYC mRNA translation. Blood 2006, 107, 2507–2516. [Google Scholar] [CrossRef]
- Giotopoulos, G.; van der Weyden, L.; Osaki, H.; Rust, A.G.; Gallipoli, P.; Meduri, E.; Horton, S.J.; Chan, W.I.; Foster, D.; Prinjha, R.K.; et al. A novel mouse model identifies cooperating mutations and therapeutic targets critical for chronic myeloid leukemia progression. J. Exp. Med. 2015, 212, 1551–1569. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Magistroni, V.; Piazza, R.; Citterio, S.; Mezzatesta, C.; Khandelwal, P.; Pirola, A.; Gambacorti-Passerini, C. BCR/ABL1 and BCR are under the transcriptional control of the MYC oncogene. Mol. Cancer 2015, 14, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reavie, L.; Buckley, S.M.; Loizou, E.; Takeishi, S.; Aranda-Orgilles, B.; Ndiaye-Lobry, D.; Abdel-Wahab, O.; Ibrahim, S.; Nakayama, K.I.; Aifantis, I. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer Cell 2013, 23, 362–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albajar, M.; Gómez-Casares, M.T.; Llorca, J.; Mauleon, I.; Vaqué, J.P.; Acosta, J.C.; Bermúdez, A.; Donato, N.; Delgado, M.D.; León, J. MYC in chronic myeloid leukemia: Induction of aberrant DNA synthesis and association with poor response to imatinib. Mol. Cancer Res. 2011, 5, 564–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Casares, M.T.; García-Alegria, E.; López-Jorge, C.E.; Ferrándiz, N.; Blanco, R.; Alvarez, S.; Vaqué, J.P.; Bretones, G.; Caraballo, J.M.; Sánchez-Bailón, P.; et al. MYC antagonizes the differentiation induced by imatinib in chronic myeloid leukemia cells through downregulation of p27(KIP1.). Oncogene 2013, 32, 2239–2246. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.G.; Zheng, A.; Jiang, L.; Rowse, M.; Stanevich, V.; Chen, H.; Li, Y.; Satyshur, K.A.; Johnson, B.; Gu, T.J.; et al. Methylation-regulated decommissioning of multimeric PP2A complexes. Nat. Commun. 2017, 8, 2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sablina, A.A.; Hector, M.; Colpaert, N.; Hahn, W.C. Identification of PP2A complexes and pathways involved in cell transformation. Cancer Res. 2010, 70, 10474–10484. [Google Scholar] [CrossRef] [Green Version]
- Ruvolo, P.P. The broken “Off” switch in cancer signaling: PP2A as a regulator of tumorigenesis, drug resistance, and immune surveillance. BBA Clin. 2016, 6, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Sents, W.; Ivanova, E.; Lambrecht, C.; Haesen, D.; Janssens, V. The biogenesis of active protein phosphatase 2A holoenzymes: A tightly regulated process creating phosphatase specificity. FEBS J. 2013, 280, 644–661. [Google Scholar] [CrossRef]
- Letourneux, C.; Rocher, G.; Porteu, F. B56-containing PP2A dephosphorylate ERK and their activity is controlled by the early gene IEX-1 and ERK. EMBO J. 2006, 4, 727–1738. [Google Scholar] [CrossRef] [Green Version]
- Ory, S.; Zhou, M.; Conrads, T.P.; Veenstra, T.D.; Morrison, D.K. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr. Biol. 2003, 16, 1356–1364. [Google Scholar] [CrossRef] [Green Version]
- Silverstein, A.M.; Barrow, C.A.; Davis, A.J.; Mumby, M.C. Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc. Natl. Acad. Sci. USA 2002, 7, 4221–4226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Huang, Q.; Lu, Y.; Li, X.; Huang, S. Reactivating PP2A by FTY720 as a novel therapy for AML with C-KIT tyrosine kinase domain mutation. J. Cell. Biochem. 2012, 113, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Smith, A.M.; McDougall, F.; Carpenter, H.; Horan, M.; Neviani, P.; Powell, J.A.; Thomas, D.; Guthridge, M.A.; Perrotti, D.; et al. Essential requirement for PP2A inhibition by the oncogenic receptor c-KIT suggests PP2A reactivation as a strategy to treat c-KIT+ cancers. Cancer Res. 2010, 70, 5438–5447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neviani, P.; Santhanam, R.; Trotta, R.; Notari, M.; Blaser, B.W.; Liu, S.; Mao, H.; Chang, J.S.; Galietta, A.; Uttam, A.; et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell 2005, 8, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, K.; Spitzer, B.; Kentsis, A. Therapeutic Re-Activation of Protein Phosphatase 2A in Acute Myeloid Leukemia. Front. Oncol. 2015, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Chen, X.; Rocha, K.; Epling-Burnette, P.K.; Djeu, J.Y.; Liu, Q.; Byrd, J.; Sokol, L.; Lawrence, N.; Pireddu, R.; et al. A critical role for phosphatase haplodeficiency in the selective suppression of deletion 5q MDS by lenalidomide. Proc. Natl. Acad. Sci. USA 2009, 106, 12974–12979. [Google Scholar] [CrossRef] [Green Version]
- Farrar, J.E.; Schuback, H.L.; Ries, R.E.; Wai, D.; Hampton, O.A.; Trevino, L.R.; Alonzo, T.A.; Guidry Auvil, J.M.; Davidsen, T.M.; Gesuwan, P.; et al. Genomic Profiling of Pediatric Acute Myeloid Leukemia Reveals a Changing Mutational Landscape from Disease Diagnosis to Relapse. Cancer Res. 2016, 76, 2197–2205. [Google Scholar] [CrossRef] [Green Version]
- Cristobal, I.; Garcia-Orti, L.; Cirauqui, C.; Alonso, M.M.; Calasanz, M.J.; Odero, M.D. PP2A impaired activity is a common event in acute myeloid leukemia and its activation by forskolin has a potent anti-leukemic effect. Leukemia 2011, 25, 606–614. [Google Scholar] [CrossRef]
- Ruvolo, P.P.; Qui, Y.H.; Coombes, K.R.; Zhang, N.; Ruvolo, V.R.; Borthakur, G.; Konopleva, M.; Andreeff, M.; Kornblau, S.M. Low expression of PP2A regulatory subunit B55α is associated with T308 phosphorylation of AKT and shorter complete remission duration in acute myeloid leukemia patients. Leukemia 2011, 25, 1711–1717. [Google Scholar] [CrossRef]
- Ruvolo, P.P.; Ruvolo, V.R.; Jacamo, R.; Burks, J.K.; Zeng, Z.; Duvvuri, S.R.; Zhou, L.; Qiu, Y.; Coombes, K.R.; Zhang, N.; et al. The protein phosphatase 2A regulatory subunit B55α is a modulator of signaling and microRNA expression in acute myeloid leukemia cells. Biochim. Biophys. Acta 2014, 1843, 1969–1977. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Chen, Y.; Chen, S.; Niu, Y.; Yang, L.; Li, B.; Lu, Y.; Geng, S.; Du, X.; Li, Y. Expression and distribution of PPP2R5C gene in leukemia. J. Hematol. Oncol. 2011, 4, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristóbal, I.; Cirauqui, C.; Castello-Cros, R.; Garcia-Orti, L.; Calasanz, M.J.; Odero, M.D. Downregulation of PPP2R5E is a common event in acute myeloid leukemia that affects the oncogenic potential of leukemic cells. Haematologica 2013, 98, e103–e104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bononi, A.; Agnoletto, C.; De Marchi, E.; Marchi, S.; Patergnani, S.; Bonora, M.; Giorgi, C.; Missiroli, S.; Poletti, F.; Rimessi, A.; et al. Protein kinases and phosphatases in the control of cell fate. Enzym. Res. 2011, 2011, 329098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanevich, V.; Jiang, L.; Satyshur, K.A.; Li, Y.; Jeffrey, P.D.; Li, Z.; Menden, P.; Semmelhack, M.F.; Xing, Y. The structural basis for tight control of PP2A methylation and function by LCMT-1. Mol. Cell 2011, 41, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssens, V.; Longin, S.; Goris, J. PP2A holoenzyme assembly: In cauda venenum (the sting is in the tail). Trends Biochem. Sci. 2008, 33, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Löw, C.; Quistgaard, E.M.; Kovermann, M.; Anandapadamanaban, M.; Balbach, J.; Nordlund, P. Structural basis for PTPA interaction with the invariant C-terminal tail of PP2A. Biol. Chem. 2014, 395, 881–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangodkar, J.; Farrington, C.C.; McClinch, K.; Galsky, M.D.; Kastrinsky, D.B.; Narla, G. All roads lead to PP2A: Exploiting the therapeutic potential of this phosphatase. FEBS J. 2016, 283, 1004–1024. [Google Scholar] [CrossRef] [Green Version]
- Perrotti, D.; Neviani, P. Protein phosphatase 2A: A target for anticancer therapy. Lancet Oncol. 2013, 14, e229–e238. [Google Scholar] [CrossRef] [Green Version]
- Cristobal, I.; Blanco, F.J.; Garcia-Orti, L.; Marcotegui, N.; Vicente, C.; Rifon, J.; Novo, F.J.; Bandres, E.; Calasanz, M.J.; Bernabeu, C.; et al. SETBP1 overexpression is a novel leukemogenic mechanism that predicts adverse outcome in elderly patients with acute myeloid leukemia. Blood 2010, 115, 615–625. [Google Scholar] [CrossRef]
- Cristobal, I.; Garcia-Orti, L.; Cirauqui, C.; Cortes-Lavaud, X.; Garcia-Sanchez, M.A.; Calasanz, M.J.; Odero, M.D. Overexpression of SET is a recurrent event associated with poor outcome and contributes to protein phosphatase 2A inhibition in acute myeloid leukemia. Haematologica 2012, 97, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Barragán, E.; Chillón, M.C.; Castelló-Cros, R.; Marcotegui, N.; Prieto, M.I.; Hoyos, M.; Pippa, R.; Llop, M.; Etxabe, A.; Cervera, J.; et al. CIP2A high expression is a poor prognostic factor in normal karyotype acute myeloid leukemia. Haematologica 2015, 100, e183–e185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrotti, D.; Neviani, P. ReSETting PP2A tumour suppressor activity in blast crisis and imatinib-resistant chronic myelogenous leukaemia. Br. J. Cancer 2006, 95, 775–781. [Google Scholar] [CrossRef] [Green Version]
- Neviani, P.; Santhanam, R.; Oaks, J.J.; Eiring, A.M.; Notari, M.; Blaser, B.W.; Liu, S.; Trotta, R.; Muthusamy, N.; Gambacorti-Passerini, C.; et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J. Clin. Investig. 2007, 117, 2408–2421. [Google Scholar] [CrossRef] [Green Version]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef] [PubMed]
- Pippa, R.; Boffo, S.; Odero, M.D.; Giordano, A. Data mining analysis of the PP2A cell cycle axis in mesothelioma patients. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kauko, O.; Westermarck, J. Non-genomic mechanisms of protein phosphatase 2A (PP2A) regulation in cancer. Int. J. Biochem. Cell Biol. 2018, 96, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Guo, H.; Damuni, Z. Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry 1995, 6, 1988–1996. [Google Scholar] [CrossRef]
- Li, M.; Makkinje, A.; Damuni, Z. Molecular identification of I1PP2A, a novel potent heat-stable inhibitor protein of protein phosphatase 2A. Biochemistry 1996, 22, 6988–7002. [Google Scholar] [CrossRef]
- Yang, X.; Lu, B.; Sun, X.; Han, C.; Fu, C.; Xu, K.; Wang, M.; Li, D.; Chen, Z.; Opal, P.; et al. ANP32A regulates histone H3 acetylation and promotes leukemogenesis. Leukemia 2018, 7, 1587–1597. [Google Scholar] [CrossRef]
- Li, M.; Makkinje, A.; Damuni, Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J. Biol. Chem. 1996, 271, 11059–11062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pippa, R.; Dominguez, A.; Christensen, D.J.; Moreno-Miralles, I.; Blanco-Prieto, M.J.; Vitek, M.P.; Odero, M.D. Effect of FTY720 on the SET-PP2A complex in acute myeloid leukemia; SET binding drugs have antagonistic activity. Leukemia 2014, 28, 1915–1918. [Google Scholar] [CrossRef] [PubMed]
- Neviani, P.; Harb, J.G.; Oaks, J.J.; Santhanam, R.; Walker, C.J.; Ellis, J.J.; Ferenchak, G.; Dorrance, A.M.; Paisie, C.A.; Eiring, A.M.; et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J. Clin. Investig. 2013. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; Puustinen, P.; Niemelä, M.; Ahola, R.; Arnold, H.; Böttzauw, T.; Ala-aho, R.; Nielsen, C.; Ivaska, J.; Taya, Y.; et al. CIP2A inhibits PP2A in human malignancies. Cell 2007, 130, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Okkeri, J.; Pavic, K.; Wang, Z.; Kauko, O.; Halonen, T.; Sarek, G.; Ojala, P.M.; Rao, Z.; Xu, W.; et al. Oncoprotein CIP2A is stabilized via interaction with tumor suppressor PP2A/B56. EMBO Rep. 2017, 18, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A.; Bockelman, C.; Hemmes, A.; Junttila, M.R.; Wiksten, J.P.; Lundin, M.; Junnila, S.; Murphy, D.J.; Evan, G.I.; Haglund, C.; et al. MYC-dependent regulation and prognostic role of CIP2A in gastric cancer. J. Natl. Cancer Inst. 2009, 101, 793–805. [Google Scholar] [CrossRef] [Green Version]
- Lucas, C.M.; Scott, L.J.; Carmell, N.; Holcroft, A.K.; Hills, R.K.; Burnett, A.K.; Clark, R.E. CIP2A- and SETBP1-mediated PP2A inhibition reveals AKT S473 phosphorylation to be a new biomarker in AML. Blood Adv. 2018, 2, 964–968. [Google Scholar] [CrossRef]
- Lucas, C.M.; Milani, M.; Butterworth, M.; Carmell, N.; Scott, L.J.; Clark, R.E.; Cohen, G.M.; Varadarajan, S. High CIP2A levels correlate with an antiapoptotic phenotype that can be overcome by targeting BCL-XL in chronic myeloid leukemia. Leukemia 2016, 30, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Gharbi-Ayachi, A.; Labbé, J.C.; Burgess, A.; Vigneron, S.; Strub, J.M.; Brioudes, E.; Van-Dorsselaer, A.; Castro, A.; Lorca, T. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 2010, 330, 1673–1677. [Google Scholar] [CrossRef]
- Andrade, E.C.; Musante, V.; Horiuchi, A.; Matsuzaki, H.; Brody, A.H.; Wu, T.; Greengard, P.; Taylor, J.R.; Nairn, A.C. ARPP-16 Is a Striatal-Enriched Inhibitor of Protein Phosphatase 2A Regulated by Microtubule-Associated Serine/Threonine Kinase 3 (Mast 3 Kinase). J. Neurosci. 2017, 37, 2709–2722. [Google Scholar] [CrossRef]
- Mäkelä, E.; Löyttyniemi, E.; Salmenniemi, U.; Kauko, O.; Varila, T.; Kairisto, V.; Itälä-Remes, M.; Westermarck, J. Arpp19 Promotes Myc and Cip2a Expression and Associates with Patient Relapse in Acute Myeloid Leukemia. Cancers 2019, 11, 1774. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Lu, B.; Han, C.; Qiu, W.; Jin, Q.; Li, D.; Li, Q.; Yang, Q.; Wen, Q.; Opal, P.; et al. ANP32A dysregulation contributes to abnormal megakaryopoiesis in acute megakaryoblastic leukemia. Blood Cancer J. 2017, 12, 661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pippa, R.; Dominguez, A.; Malumbres, R.; Endo, A.; Arriazu, E.; Marcotegui, N.; Guruceaga, E.; Odero, M.D. MYC-dependent recruitment of RUNX1 and GATA2 on the SET oncogene promoter enhances PP2A inactivation in acute myeloid leukemia. Oncotarget 2017, 8, 53989–54003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karetsou, Z.; Emmanouilidou, A.; Sanidas, I.; Liokatis, S.; Nikolakaki, E.; Politou, A.S.; Papamarcaki, T. Identification of distinct SET/TAF-Ibeta domains required for core histone binding and quantitative characterisation of the interaction. BMC Biochem. J. 2009, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.B.; McNamara, P.; Heo, S.; Turner, A.; Lane, W.S.; Chakravarti, D. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell 2001, 1, 119–130. [Google Scholar] [CrossRef] [Green Version]
- De Palma, R.M.; Parnham, S.R.; Li, Y.; Oaks, J.J.; Peterson, Y.K.; Szulc, Z.M.; Roth, B.M.; Xing, Y.; Ogretmen, B. The NMR-based characterization of the FTY720-SET complex reveals an alternative mechanism for the attenuation of the inhibitory SET-PP2A interaction. FASEB J. 2019, 33, 7647–7666. [Google Scholar] [CrossRef]
- Vicente, C.; Arriazu, E.; Martínez-Balsalobre, E.; Peris, I.; Marcotegui, N.; García-Ramírez, P.; Pippa, R.; Rabal, O.; Oyarzábal, J.; Guruceaga, E.; et al. A novel FTY720 analogue targets SET-PP2A interaction and inhibits growth of acute myeloid leukemia cells without inducing cardiac toxicity. Cancer Lett. 2020, 468, 1–13. [Google Scholar] [CrossRef]
- Switzer, C.H.; Cheng, R.Y.; Vitek, T.M.; Christensen, D.J.; Wink, D.A.; Vitek, M.P. Targeting SET/I(2)PP2A oncoprotein functions as a multi-pathway strategy for cancer therapy. Oncogene 2011, 30, 2504–2513. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; MacKenzie, R.J.; Pippa, R.; Eide, C.A.; Oddo, J.; Tyner, J.W.; Sears, R.; Vitek, M.P.; Odero, M.D.; Christensen, D.J.; et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin. Cancer Res. 2014, 20, 2092–2103. [Google Scholar] [CrossRef] [Green Version]
- Richard, N.P.; Pippa, R.; Cleary, M.M.; Puri, A.; Tibbitts, D.; Mahmood, S.; Christensen, D.J.; Jeng, S.; McWeeney, S.; Look, A.T.; et al. Combined targeting of SET and tyrosine kinases provides an effective therapeutic approach in human T-cell acute lymphoblastic leukemia. Oncotarget 2016, 7, 84214–84227. [Google Scholar] [CrossRef] [Green Version]
- Saika, M.; Inoue, D.; Nagase, R.; Sato, N.; Tsuchiya, A.; Yabushita, T.; Kitamura, T.; Goyama, S. ASXL1 and SETBP1 mutations promote leukaemogenesis by repressing TGFβ pathway genes through histone deacetylation. Sci. Rep. 2018, 8, 15873. [Google Scholar] [CrossRef]
- Fu, S.; Hu, Y.; Fu, Y.; Chen, F.; Liu, X.; Zhang, M.; Wang, X.; Tu, S.; Zhang, J. Novel BCR-ABL1 fusion and leukemic mutations of SETBP1, PAX5, and TP53 detected by next generation sequencing in chronic myeloid leukemia. Cancer Biol. Ther. 2016, 17, 1003–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piazza, R.; Valletta, S.; Winkelmann, N.; Redaelli, S.; Spinelli, R.; Pirola, A.; Antolini, L.; Mologni, L.; Donadoni, C.; Papaemmanuil, E.; et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat. Gen. 2012, 45, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Oakley, K.; Han, Y.; Vishwakarma, B.A.; Chu, S.; Bhatia, R.; Gudmundsson, K.O.; Keller, J.; Chen, X.; Vasko, V.; Jenkins, N.A.; et al. Setbp1 promotes the self-renewal of murine myeloid progenitors via activation of Hoxa9 and Hoxa10. Blood 2012, 119, 6099–6108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arriazu, E.; Vicente, C.; Pippa, R.; Peris, I.; Martínez-Balsalobre, E.; García-Ramírez, P.; Marcotegui, N.; Igea, A.; Alignani, D.; Rifón, J.; et al. A new regulatory mechanism of protein phosphatase 2A activity via SET in acute myeloid leukemia. Blood Cancer J. 2020, 1, 3. [Google Scholar] [CrossRef]
- Martens, E.; Stevens, I.; Janssens, V.; Vermeesch, J.; Götz, J.; Goris, J.; Van Hoof, C. Genomic organisation, chromosomal localisation tissue distribution and developmental regulation of the PR61/B’ regulatory subunits of protein phosphatase 2A in mice. J. Mol. Biol. 2004, 336, 971–986. [Google Scholar] [CrossRef]
- Wang, L.; Gu, F.; Ma, N.; Zhang, L.; Bian, J.M.; Cao, H.Y. CIP2A expression is associated with altered expression of epithelial-mesenchymal transition markers and predictive of poor prognosis in pancreatic ductal adenocarcinoma. Tumour Biol. 2013, 34, 2309–2313. [Google Scholar] [CrossRef]
- Coenen, E.A.; Zwaan, C.M.; Meyer, C.; Marschalek, R.; Pieters, R.; van der Veken, L.T.; Beverloo, H.B.; van den Heuvel-Eibrink, M.M. KIAA1524: A novel MLL translocation partner in acute myeloid leukemia. Leuk. Res. 2011, 35, 133–135. [Google Scholar] [CrossRef]
- Lucas, C.M.; Harris, R.J.; Giannoudis, A.; Copland, M.; Slupsky, J.R.; Clark, R.E. Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood 2011, 117, 6660–6668. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.S.; Bassik, M.C.; Suh, H.; Nishino, M.; Arroyo, J.D.; Hahn, W.C.; Korsmeyer, S.J.; Roberts, T.M. PP2A regulates BCL-2 phosphorylation and proteasome-mediated degradation at the endoplasmic reticulum. J. Biol. Chem. 2006, 281, 23003–23012. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Giap, C.; Lazo, J.S.; Prochownik, E.V. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003, 40, 6151–6159. [Google Scholar] [CrossRef] [Green Version]
- Lucas, C.M.; Harris, R.J.; Giannoudis, A.; Clark, R.E. c-Myc inhibition decreases CIP2A and reduces BCR-ABL1 tyrosine kinase activity in chronic myeloid leukemia. Haematologica 2015, 100, e179–e182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochida, S.; Maslen, S.L.; Skehel, M.; Hunt, T. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 2010, 330, 1670–1673. [Google Scholar] [CrossRef] [PubMed]
- Porter, I.M.; Schleicher, K.; Porter, M.; Swedlow, J.R. Bod1 regulates protein phosphatase 2A at mitotic kinetochores. Nat. Commun. 2013, 4, 2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darnell, J.E. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 10, 740–749. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.J.; Cheng, Y.C.; Liu, C.R.; Lin, S.; Liu, H.E. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp. Hematol. 2006, 34, 1480–1489. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 7327, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi Screen Identifies Brd4 as a Therapeutic Target in Acute Myeloid Leukaemia. Nature 2011, 7370, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Picaud, S.; Da Costa, D.; Thanasopoulou, A.; Filippakopoulos, P.; Fish, P.V.; Philpott, M.; Fedorov, O.; Brennan, P.; Bunnage, M.E.; Owen, D.R.; et al. PFI-1, a highly selective protein interaction inhibitor, targeting BET Bromodomains. Cancer Res. 2013, 11, 3336–3346. [Google Scholar] [CrossRef] [Green Version]
- Bui, M.H.; Lin, X.; Albert, D.H.; Li, L.; Lam, L.T.; Faivre, E.J.; Warder, S.E.; Huang, X.; Wilcox, D.; Donawho, C.K.; et al. Preclinical Characterization of BET Family Bromodomain Inhibitor ABBV-075 Suggests Combination Therapeutic Strategies. Cancer Res. 2017, 11, 2976–2989. [Google Scholar] [CrossRef] [Green Version]
- Coudé, M.M.; Braun, T.; Berrou, J.; Dupont, M.; Bertrand, S.; Masse, A.; Rafflou, E.; Itzykson, R.; Delord, M.; Riveiro, M.E.; et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 2015, 19, 17698–17712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Vakoc, C.R. Targeting Cancer Cells with BET Bromodomain Inhibitors. Cold Spring Harb. Perspect. Med. 2017, 7, a026674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pervaiz, M.; Mishra, P.; Gunther, S. Bromodomain Drug Discovery—The Past, the Present, and the Future. Chem. Rec. 2018, 12, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 7570, 538–542. [Google Scholar] [CrossRef]
- Farrell, A.S.; Allen-Petersen, B.; Daniel, C.J.; Wang, X.; Wang, Z.; Rodriguez, S.; Impey, S.; Oddo, J.; Vitek, M.P.; Lopez, C.; et al. Targeting Inhibitors of the Tumor Suppressor PP2A for the Treatment of Pancreatic Cancer. Mol. Cancer Res. 2014, 12, 924–939. [Google Scholar] [CrossRef] [Green Version]
- Janghorban, M.; Farrell, A.S.; Allen-Petersen, B.L.; Pelz, C.; Daniel, C.J.; Oddo, J.; Langer, E.M.; Christensen, D.J.; Sears, R.C. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 9157–9162. [Google Scholar] [CrossRef] [Green Version]
- Farrington, C.C.; Yuan, E.; Mazhar, S.; Izadmehr, S.; Hurst, L.; Allen-Petersen, B.L.; Janghorban, M.; Chung, E.; Wolczanski, G.; Galsky, M.; et al. Protein phosphatase 2A activation as a therapeutic strategy for managing MYC-driven cancers. J. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Smith, A.M.; Dun, M.D.; Lee, E.M.; Harrison, C.; Kahl, R.; Flanagan, H.; Panicker, N.; Mashkani, B.; Don, A.S.; Morris, J.; et al. Activation of protein phosphatase 2A in FLT3+ acute myeloid leukemia cells enhances the cytotoxicity of FLT3 tyrosine kinase inhibitors. Oncotarget 2016, 7, 47465–47478. [Google Scholar] [CrossRef] [Green Version]
- Kiyota, M.; Kuroda, J.; Yamamoto-Sugitani, M.; Shimura, Y.; Nakayama, R.; Nagoshi, H.; Mizutani, S.; Chinen, Y.; Sasaki, N.; Sakamoto, N.; et al. FTY720 induces apoptosis of chronic myelogenous leukemia cells via dual activation of BIM and BID and overcomes various types of resistance to tyrosine kinase inhibitors. Apoptosis 2013, 18, 1437–1446. [Google Scholar] [CrossRef]
- Young, M.M.; Bui, V.; Chen, C.; Wang, H.G. FTY720 induces non-canonical phosphatidylserine externalization and cell death in acute myeloid leukemia. Cell Death Dis. 2019, 10, 847. [Google Scholar] [CrossRef] [Green Version]
- Estella-Hermoso de Mendoza, A.; Castello-Cros, R.; Imbuluzqueta, E.; Cirauqui, C.; Pippa, R.; Odero, M.D.; Blanco-Prieto, M.J. Lipid Nanosystems Enhance the Bioavailability and the Therapeutic Efficacy of FTY720 in Acute Myeloid Leukemia. J. Biomed. Nanotechnol. 2015, 11, 691–701. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
MYC regulation by PP2A. MYC undergoes two critical phospho-modifications on S62 and T58 residues. The phosphorylation on T58 is regulated by GSK3β. To be active, GSK3β has to be dephosphorylated. The PP2A complex, which includes the B56α subunit, dephosphorylates and activates GSK3β. Interestingly, the B56α subunit is transcriptionally activated by MYC. Moreover, the presence of a phosphorylated T58 residue also recruits the PP2A-B56α complex, which dephosphorylated the S62 phospho-residue. Dephosphorylation of S62 residue eventually targets MYC for ubiquitin-mediated proteosomal degradation. See text for more details.
Figure 1.
MYC regulation by PP2A. MYC undergoes two critical phospho-modifications on S62 and T58 residues. The phosphorylation on T58 is regulated by GSK3β. To be active, GSK3β has to be dephosphorylated. The PP2A complex, which includes the B56α subunit, dephosphorylates and activates GSK3β. Interestingly, the B56α subunit is transcriptionally activated by MYC. Moreover, the presence of a phosphorylated T58 residue also recruits the PP2A-B56α complex, which dephosphorylated the S62 phospho-residue. Dephosphorylation of S62 residue eventually targets MYC for ubiquitin-mediated proteosomal degradation. See text for more details.

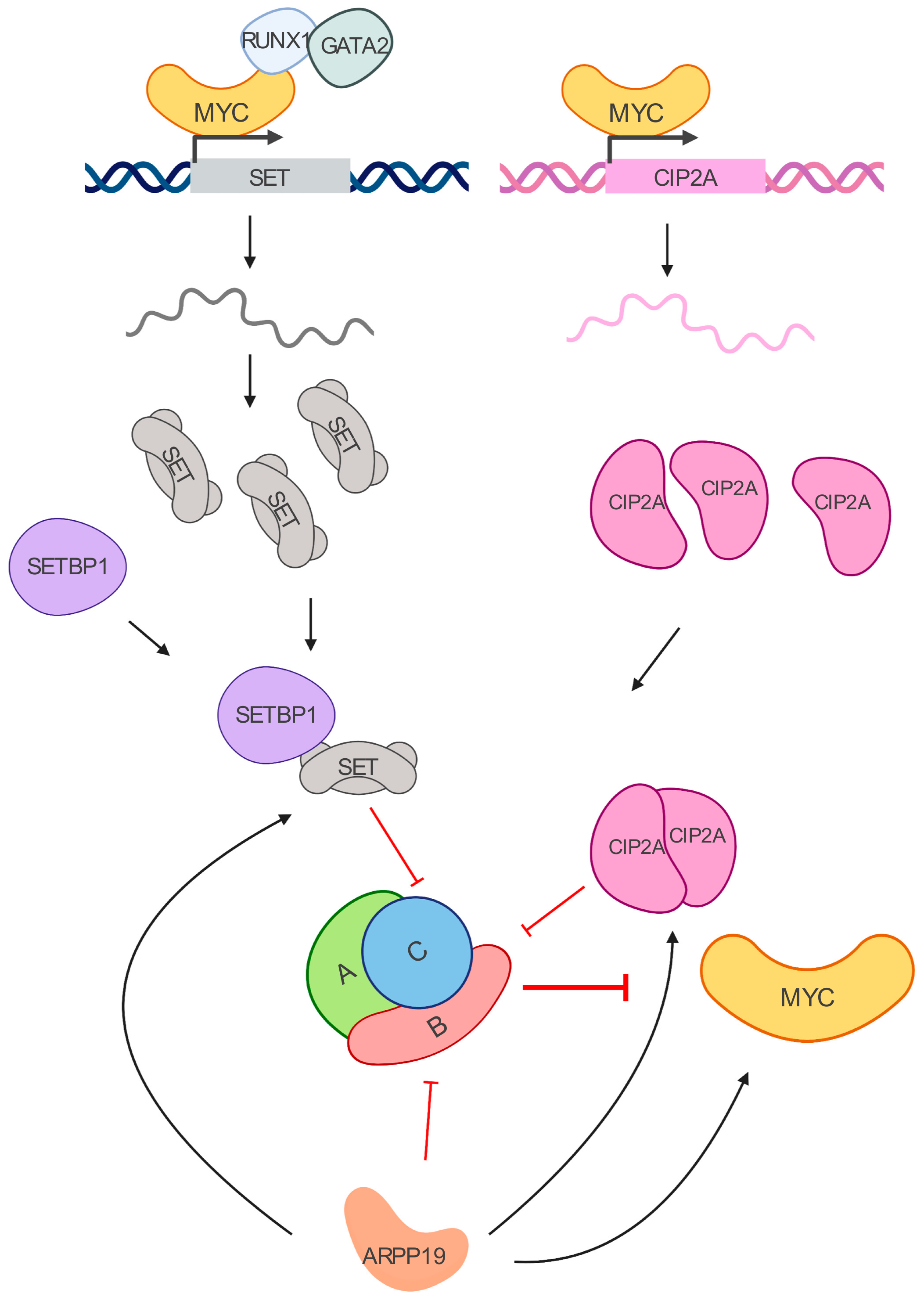
Figure 2.
MYC/PP2A inhibitory network. MYC transcriptionally regulates SET and CIP2A. SET binds to the PP2A-C subunit, while CIP2A interacts with PP2A-B56𝛼 subunit. Their increased expressions lead to PP2A inhibition in leukemia cells. SETBP1 is a SET binding protein overexpressed in myeloid leukemias that prevents SET cleavage. ARPP19 is another inhibitor of PP2A that has been recently reported to enhance the levels of CIP2A, SET, and MYC in AML.
Figure 2.
MYC/PP2A inhibitory network. MYC transcriptionally regulates SET and CIP2A. SET binds to the PP2A-C subunit, while CIP2A interacts with PP2A-B56𝛼 subunit. Their increased expressions lead to PP2A inhibition in leukemia cells. SETBP1 is a SET binding protein overexpressed in myeloid leukemias that prevents SET cleavage. ARPP19 is another inhibitor of PP2A that has been recently reported to enhance the levels of CIP2A, SET, and MYC in AML.

{kind=link}
{kind=link}
Table 1.
PP2A subunits and reported alterations in acute myeloid leukemia (AML) and chronic myeloid leukemia (CML).
Table 1.
PP2A subunits and reported alterations in acute myeloid leukemia (AML) and chronic myeloid leukemia (CML).
| Subunit. | Gene | Locus | Protein | Alteration in AML | Alteration in CML |
|---|---|---|---|---|---|
| A Structural | PPP2R1A | 19q13.41 | PR65/A𝛼 | Downregulation [62] .Oncogenic c-KIT mutations decrease protein levels [63]. | Increased levels in BCR/ABL cells [64]. |
| PPP2R1B | 11q23.1 | PR65/A𝛽 | Downregulation [65] | ||
| C Catalytic | PPP2CA | 5q31.1 | PP2A-C/C 𝛼 | Downregulation in TP53 mutant AML cases [65]. Decreased expression in del5q AML [66]. Gene deletion in pediatric AML [67]. Hyperphosphorylation of Y307 [68]. | Hyperphosphorylation of Y307 and T304 [64]. |
| PPP2CB | 8p12 | PP2A-C/C 𝛽 | |||
| B Regulatory | PPP2R2A | 8p21.2 | PR55/B55 𝛼 | Oncogenic c-KIT mutations decrease protein levels [63]. Downregulation at mRNA and protein level [62,69,70]. | |
| PPP2R2B | 5q32 | PR55/B55 𝛽 | High expression [65]. | ||
| PPP2R2C | 4p16.1 | PR55/B55 𝛾 | Downregulation [65]. | ||
| PPP2R2D | 10q26.3 | PR55/B55 𝛿 | |||
| B’ Regulatory | PPP2R5A | 1q32.3 | PR56/B56 𝛼 | Oncogenic c-KIT mutations decrease protein levels [63]. Gene amplification in pediatric AML [67]. | |
| PPP2R5B | 11q13.1 | PR56/B56 𝛽 | Downregulation [68]. High expression [65]. Gene deletion in pediatric AML [67]. | ||
| PPP2R5C | 14q32.31 | PR56/B56 𝛾 | Downregulation [68]. Oncogenic c-KIT mutations decrease protein levels [63]. Gene deletion in pediatric AML [67]. | Decreased expression in CML-BC compared to de novo CML [71]. | |
| PPP2R5D | 6p21.1 | PR55/B55 𝛿 | Oncogenic c-KIT mutations decrease protein levels [63] | ||
| PPP2R5E | 14q32.2 | PR55/B55 𝜀 | Downregulation [72]. | ||
| B” Regulatory | PPP2R3A | 3q22.2 | PR72/PR130 or B” 𝛼 | ||
| PPP2R3B | Yp11.32; Xp22.33 | PR48/PR70 or B” 𝛽 | Downregulation [65]. | ||
| PPP2R3C | 14q13.2 | B” 𝛾or G5PR | |||
| B’’’ Regulatory | STRN | 2p22.2 | Striatin | ||
| STRN3 | 14q13-q12 | Striatin3 | |||
| STRN4 | 19q13.2 | Striatin4 |
Table 2.
PP2A endogenous inhibitors and reported alterations in AML and CML.
| Inhibitor | Mechanism | MYC Association | Alteration in AML | Alteration in CML |
|---|---|---|---|---|
| ANP32A | Binds to and inactivates PP2A-C [87,88]. | High expression in primary AML cells [89]. | ||
| SET | Binds to and inactivates PP2A-C [90]. | MYC regulates SET transcriptional expression [91]. | High expression [68,80]. | High expression [64]. Associate with monosomy 7 and predict worse overall survival and progression-free survival [92]. |
| CIP2A | Binds to PP2A A, B56 subunits, preventing the dephosphorylation of MYC [93,94] | CIP2A prevents PP2A-dependent dephosphorylation of MYC on S62 [93]. MYC regulates CIP2A expression [95]. | High expression [81,96]. | High expression [97]. |
| ARPP19 | Binds and inhibits B55αδ in mitosis [98,99] | ARPP19 promotes MYC expression [100] | High expression [100] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pippa, R.; Odero, M.D. The Role of MYC and PP2A in the Initiation and Progression of Myeloid Leukemias. Cells 2020, 9, 544. https://doi.org/10.3390/cells9030544
AMA Style
Pippa R, Odero MD. The Role of MYC and PP2A in the Initiation and Progression of Myeloid Leukemias. Cells. 2020; 9(3):544. https://doi.org/10.3390/cells9030544
Chicago/Turabian StylePippa, Raffaella, and Maria D. Odero. 2020. "The Role of MYC and PP2A in the Initiation and Progression of Myeloid Leukemias" Cells 9, no. 3: 544. https://doi.org/10.3390/cells9030544
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.