Endothelial Protease Activated Receptor 1 (PAR1) Signalling Is Required for Lymphocyte Transmigration across Brain Microvascular Endothelial Cells





Abstract
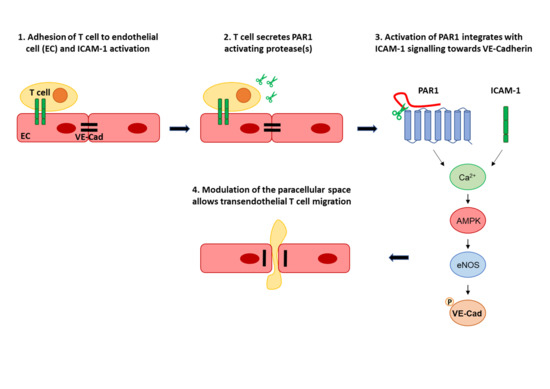
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Endothelial Cell Culture
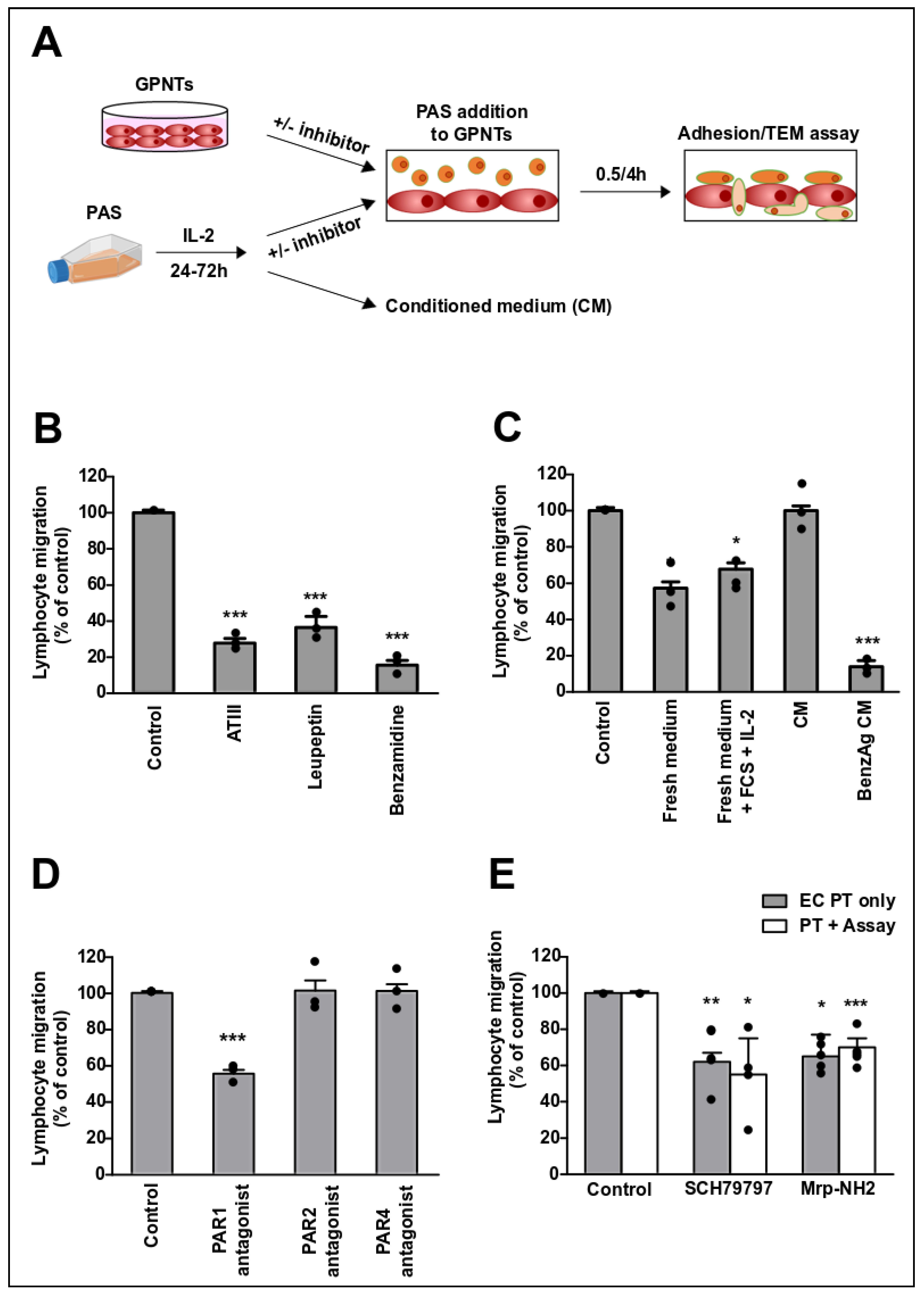
2.4. Lymphocyte Coculture, Adhesion and Migration Assays In Vitro
2.5. RT-PCR
2.6. siRNA Knockdown of PAR1
2.7. Immunoblotting
2.8. VE-Cadherin Plasmids
2.9. Data Analysis and Statistics
3. Results
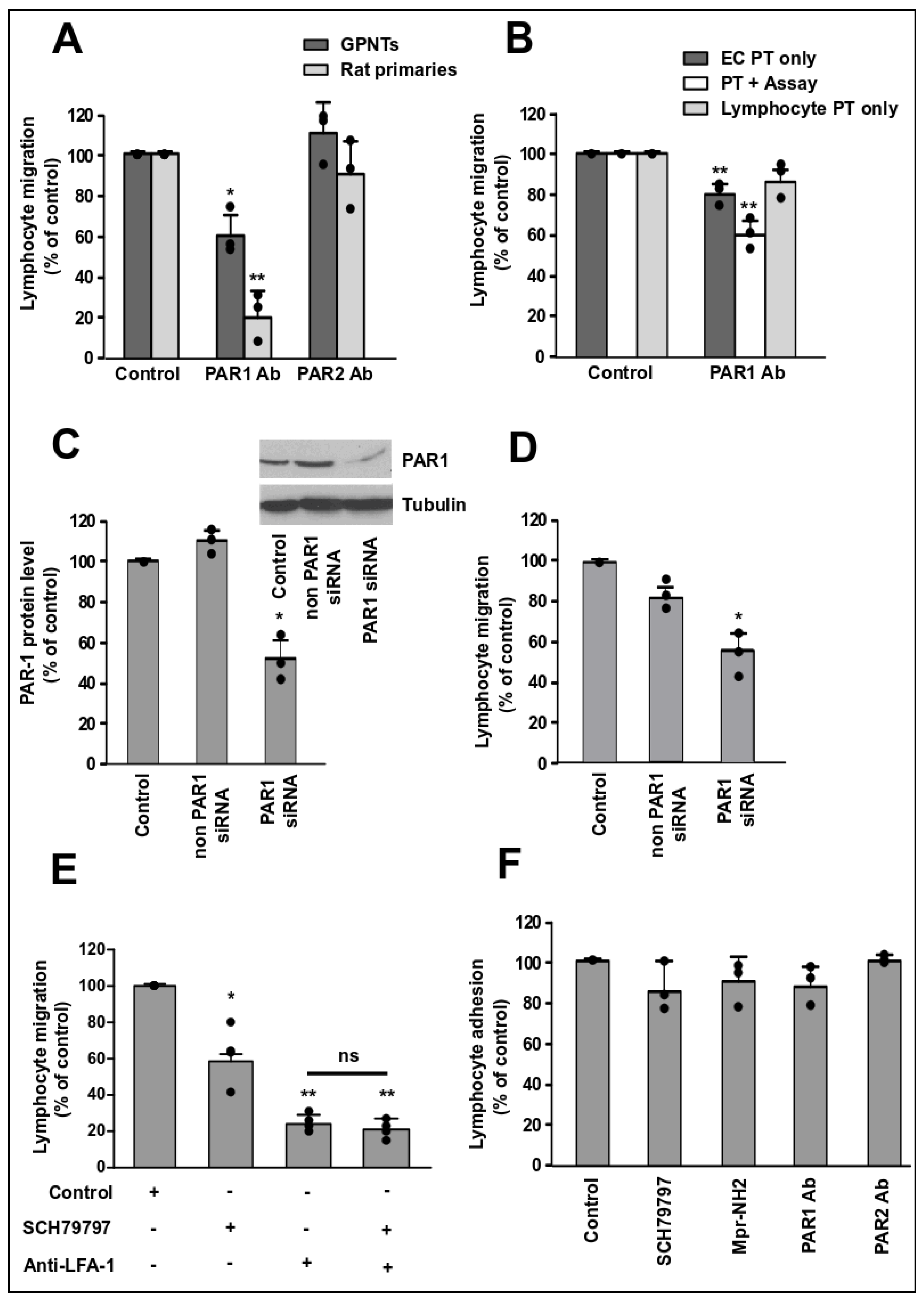
3.1. Endothelial PAR-1 Is Required for Lymphocyte Migration across Rat Brain Microvascular ECs
3.2. PAR1 Activation Leads to MAPK, AMPK and eNOS Activation in Brain Microvascular ECs
3.3. Transendothelial Migration Requires Ca2+, AMPK, eNOS and VEC
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| aPC | activated protein C |
| ATIII | anti-thrombin III |
| BBB | blood brain barrier |
| BMVEC | brain microvascular endothelial cell |
| EAE | experimental autoimmune encephalomyelitis |
| EC | endothelial cell |
| eNOS | endothelial nitric oxide synthase |
| GPCR | G protein-coupled receptor |
| LFA-1 | lymphocyte function-associated antigen 1 |
| Mpr-NH2 | Mercaptopropionyl-Phe-Cha-Arg-Lys-Pro-Asn-Asp-Lys-NH2 |
| tcY-NH2 | trans-Cinnamoyl-Tyr-Pro-Gly-Lys-Phe-NH2 |
| PAR | protease-activated receptor |
| PLN | peripheral lymph node |
| S1P | sphingosine 1-phosphate |
| TEER | transendothelial electrical resistance |
| TEM | transendothelial migration |
References
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.; Heasman, S.J.; Alvarez, J.I.; Prat, A.; Lyck, R.; Engelhardt, B. Review: Leucocyte-endothelial cell crosstalk at the blood-brain barrier: A prerequisite for successful immune cell entry to the brain. Neuropathol. Appl. Neurobiol. 2011, 37, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. The regulation of transendothelial migration: New knowledge and new questions. Cardiovasc. Res. 2015, 107, 310–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beraud, E.; Balzano, C.; Zamora, A.J.; Varriale, S.; Bernard, D.; Ben-Nun, A. Pathogenic and non-pathogenic T lymphocytes specific for the encephalitogenic epitope of myelin basic protein: Functional characteristics and vaccination properties. J. Neuroimmunol. 1993, 47, 41–53. [Google Scholar] [CrossRef]
- Pryce, G.; Male, D.; Campbell, I.; Greenwood, J. Factors controlling T-cell migration across rat cerebral endothelium in vitro. J. Neuroimmunol. 1997, 75, 84–94. [Google Scholar] [CrossRef]
- Oppenheimer-Marks, N.; Davis, L.S.; Bogue, D.T.; Ramberg, J.; Lipsky, P.E. Differential utilization of ICAM-1 and VCAM-1 during the adhesion and transendothelial migration of human T lymphocytes. J. Immunol. 1991, 147, 2913–2921. [Google Scholar]
- Etienne-Manneville, S.; Manneville, J.B.; Adamson, P.; Wilbourn, B.; Greenwood, J.; Couraud, P.O. ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J. Immunol. 2000, 165, 3375–3383. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, R.; Gegg, M.; Longbottom, R.; Adamson, P.; Turowski, P.; Greenwood, J. ICAM-1-mediated endothelial nitric oxide synthase activation via calcium and AMP-activated protein kinase is required for transendothelial lymphocyte migration. Mol. Biol. Cell 2009, 20, 995–1005. [Google Scholar] [CrossRef] [Green Version]
- Dragoni, S.; Hudson, N.; Kenny, B.A.; Burgoyne, T.; McKenzie, J.A.; Gill, Y.; Blaber, R.; Futter, C.E.; Adamson, P.; Greenwood, J.; et al. Endothelial MAPKs Direct ICAM-1 Signaling to Divergent Inflammatory Functions. J. Immunol. 2017, 198, 4074–4085. [Google Scholar] [CrossRef] [Green Version]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef]
- Adamson, P.; Wilbourn, B.; Etienne-Manneville, S.; Calder, V.; Beraud, E.; Milligan, G.; Couraud, P.-O.; Greenwood, J. Lymphocyte trafficking through the blood-brain barrier is dependent on endothelial cell heterotrimeric G-protein signaling. FASEB J. 2002, 16, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci. 2012, 33, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuberger, D.M.; Schuepbach, R.A. Protease-activated receptors (PARs): Mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb. J. 2019, 17, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Protease-activated receptors (PARs)--biology and role in cancer invasion and metastasis. Cancer Metastasis Rev. 2015, 34, 775–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Vanlandewijck, M.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. Single-cell RNA sequencing of mouse brain and lung vascular and vessel-associated cell types. Sci. Data 2018, 5, 180160. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Nieman, M.T. The domino effect triggered by the tethered ligand of the protease activated receptors. Thromb. Res. 2020, 196, 87–98. [Google Scholar] [CrossRef]
- Willis Fox, O.; Preston, R.J.S. Molecular basis of protease-activated receptor 1 signaling diversity. J. Thromb. Haemost. 2020, 18, 6–16. [Google Scholar] [CrossRef]
- Alabanza, L.M.; Bynoe, M.S. Thrombin induces an inflammatory phenotype in a human brain endothelial cell line. J. Neuroimmunol. 2012, 245, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Brailoiu, E.; Shipsky, M.M.; Yan, G.; Abood, M.E.; Brailoiu, G.C. Mechanisms of modulation of brain microvascular endothelial cells function by thrombin. Brain Res. 2017, 1657, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Ukropec, J.A.; Hollinger, M.K.; Salva, S.M.; Woolkalis, M.J. SHP2 association with VE-cadherin complexes in human endothelial cells is regulated by thrombin. J. Biol. Chem. 2000, 275, 5983–5986. [Google Scholar] [CrossRef] [Green Version]
- Niessen, F.; Furlan-Freguia, C.; Fernández, J.A.; Mosnier, L.O.; Castellino, F.J.; Weiler, H.; Rosen, H.; Griffin, J.H.; Ruf, W. Endogenous EPCR/aPC-PAR1 signaling prevents inflammation-induced vascular leakage and lethality. Blood 2009, 113, 2859–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feistritzer, C.; Riewald, M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood 2005, 105, 3178–3184. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; Kim, Y.R.; Ahn, S.M.; Lee, S.K.; Shin, H.K.; Choi, B.T. Protease activated receptor-1 antagonist ameliorates the clinical symptoms of experimental autoimmune encephalomyelitis via inhibiting breakdown of blood-brain barrier. J. Neurochem. 2015, 135, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.; Pryce, G.; Devine, L.; Male, D.K.; dos Santos, W.L.C.; Calder, V.L.; Adamson, P. SV40 large T immortalised cell lines of the rat blood-brain and blood-retinal barriers retain their phenotypic and immunological characteristics. J. Neuroimmunol. 1996, 71, 51–63. [Google Scholar] [CrossRef]
- Hudson, N.; Powner, M.B.; Sarker, M.H.; Burgoyne, T.; Campbell, M.; Ockrim, Z.K.; Martinelli, R.; Futter, C.E.; Grant, M.B.; Fraser, P.A.; et al. Differential apicobasal VEGF signaling at vascular blood-neural barriers. Dev. Cell 2014, 30, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Turowski, P.; Martinelli, R.; Crawford, R.; Wateridge, D.; Papageorgiou, A.P.; Lampugnani, M.G.; Gamp, A.C.; Vestweber, D.; Adamson, P.; Dejana, E.; et al. Phosphorylation of vascular endothelial cadherin controls lymphocyte emigration. J. Cell Sci. 2008, 121, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Metcalf, M.; Bunnett, N.W. Biased signaling of protease-activated receptors. Front. Endocrinol. (Lausanne) 2014, 5, 67. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; Soh, U.J.; Trejo, J. Proteases display biased agonism at protease-activated receptors: Location matters! Mol. Interv. 2009, 9, 87–96. [Google Scholar] [CrossRef]
- Wessel, F.; Winderlich, M.; Holm, M.; Frye, M.; Rivera-Galdos, R.; Vockel, M.; Linnepe, R.; Ipe, U.; Stadtmann, A.; Zarbock, A.; et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat. Immunol. 2014, 15, 223–230. [Google Scholar] [CrossRef]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [Green Version]
- Hailfinger, S.; Rebeaud, F.; Thome, M. Adapter and enzymatic functions of proteases in T-cell activation. Immunol. Rev. 2009, 232, 334–347. [Google Scholar] [CrossRef]
- van Daalen, K.R.; Reijneveld, J.F.; Bovenschen, N. Modulation of Inflammation by Extracellular Granzyme A. Front. Immunol. 2020, 11, 931. [Google Scholar] [CrossRef]
- Lee, P.R.; Johnson, T.P.; Gnanapavan, S.; Giovannoni, G.; Wang, T.; Steiner, J.P.; Medynets, M.; Vaal, M.J.; Gartner, V.; Nath, A. Protease-activated receptor-1 activation by granzyme B causes neurotoxicity that is augmented by interleukin-1beta. J. Neuroinflamm. 2017, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Haile, Y.; Carmine-Simmen, K.; Olechowski, C.; Kerr, B.; Bleackley, R.C.; Giuliani, F. Granzyme B-inhibitor serpina3n induces neuroprotection in vitro and in vivo. J. Neuroinflamm. 2015, 12, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeglinski, M.R.; Granville, D.J. Granzymes in cardiovascular injury and disease. Cell. Signal. 2020, 76, 109804. [Google Scholar] [CrossRef] [PubMed]
- Pejler, G. Novel Insight into the in vivo Function of Mast Cell Chymase: Lessons from Knockouts and Inhibitors. J. Innate Immun. 2020, 12, 357–372. [Google Scholar] [CrossRef]
- Göbel, K.; Asaridou, C.M.; Merker, M.; Eichler, S.; Herrmann, A.M.; Geuß, E.; Ruck, T.; Schüngel, L.; Groeneweg, L.; Narayanan, V.; et al. Plasma kallikrein modulates immune cell trafficking during neuroinflammation via PAR2 and bradykinin release. Proc. Natl. Acad. Sci. USA 2019, 116, 271–276. [Google Scholar] [CrossRef] [Green Version]
- Schulze-Topphoff, U.; Prat, A.; Prozorovski, T.; Siffrin, V.; Paterka, M.; Herz, J.; Bendix, I.; Ifergan, I.; Schadock, I.; Mori, M.A.; et al. Activation of kinin receptor B1 limits encephalitogenic T lymphocyte recruitment to the central nervous system. Nat. Med. 2009, 15, 788–793. [Google Scholar] [CrossRef] [Green Version]
- Alberelli, M.A.; De Candia, E. Functional role of protease activated receptors in vascular biology. Vascul. Pharmacol. 2014, 62, 72–81. [Google Scholar] [CrossRef]
- Mihara, K.; Ramachandran, R.; Renaux, B.; Saifeddine, M.; Hollenberg, M.D. Neutrophil elastase and proteinase-3 trigger G protein-biased signaling through proteinase-activated receptor-1 (PAR1). J. Biol. Chem. 2013, 288, 32979–32990. [Google Scholar] [CrossRef] [Green Version]
- Allingham, M.J.; van Buul, J.D.; Burridge, K. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J. Immunol. 2007, 179, 4053–4064. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activating Protease (gene) | Expression in T Cells | Inhibition by ATIII ‡ | ||
|---|---|---|---|---|
| hCD4+ (Th1) * | mCD4+ † | |||
| PAR1 | Thrombin13, 27, 28 (f2) | no | no | |
| Activated Protein C13, 27, 28 (proc) | very low | no | weak, but § | |
| Factor VIIa13 (f7) | no | no | ||
| Factor Xa13, 27, 28 (f10) | no | no | ||
| Trypsin13, 28 (prss1,2,3) | very low (prss1) | yes (prss2) | yes, but ¶ | |
| Plasmin13, 27, 28 (plg) | no | no | ||
| MMPs13, 27, 28 (mmp1,2,3,8,9,12,13) | yes (mmp9) | yes (mmp1,2,8,9) | no | |
| Neutrophil Elastase13, 27 (elane) | no | no | ||
| Proteinase-313, 27 (prtn3) | no | no | ||
| Granzymes13, 27, 28 (gzma,gzmb,gzmk) | high | yes | yes | |
| Chymase13 (cma1) | no | yes | yes # | |
| Cathepsin G13 (ctsg) | no | no | ||
| Kallikreins13 (klk4,5,6,14) | yes (klk14) | no | yes | |
| Calpain-113 (capn1) | high | yes | no ** | |
| PAR2 | Trypsin13, 27, 28 (prss1,2,3) | very low (prss1) | yes (prss2) | yes, but ¶ |
| Tryptase13, 27, 28 (tpsab1) | no | no | ||
| Factor VIIa27, 28 (f7) | no | no | ||
| Factor Xa27, 28 (f10) | no | no | ||
| Kallikreins13, 27, 28 (klk4,5,6,14) | yes (klk14) | no | yes | |
| Neutrophil Elastase13, 27 (elane) | no | no | ||
| Proteinase-313, 27 (prtn3) | no | no | ||
| Cathepsins13, 27 (ctss, ctsg) | high (ctss) | yes (ctss) | n/d | |
| Granzyme A28 (gzma) | high | yes | yes | |
| Matripase28 (st14) | no | yes | yes | |
| Thrombin13 (f2) | no | no | ||
| Activated Protein C13 (proc) | very low | no | weak, but § | |
| Chymase13 (cma1) | no | yes | yes | |
| Plasmin13 (plg) | no | no | ||
| Testisin13 (prss21) | yes | no | n/d | |
| Calpain-213 (capn2) | high | yes | no ** | |
| PAR3 | Thrombin13, 27, 28 (f2) | no | no | |
| Activated Protein C13, 27 (proc) | very low | no | weak, but § | |
| Factor Xa13 (f10) | no | no | ||
| Trypsin13 (prss1,2,3) | very low (prss1) | yes (prss2) | yes, but ¶ | |
| PAR4 | Thrombin13, 27, 28 (f2) | no | no | |
| Trypsin13, 27, 28 (prss1,2,3) | very low (prss1) | yes (prss2) | yes, but ¶ | |
| Factor Xa28 (f10) | no | no | ||
| Plasmin27, 28 (plg) | no | no | ||
| Cathepsin G13, 27, 28 (ctsg) | no | no | ||
| MASP127 (masp1) | no | no | ||
| Kallikrein1413, 28 (klk14) | yes | no | yes | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dragoni, S.; Papageorgiou, A.; Araiz, C.; Greenwood, J.; Turowski, P. Endothelial Protease Activated Receptor 1 (PAR1) Signalling Is Required for Lymphocyte Transmigration across Brain Microvascular Endothelial Cells. Cells 2020, 9, 2723. https://doi.org/10.3390/cells9122723
Dragoni S, Papageorgiou A, Araiz C, Greenwood J, Turowski P. Endothelial Protease Activated Receptor 1 (PAR1) Signalling Is Required for Lymphocyte Transmigration across Brain Microvascular Endothelial Cells. Cells. 2020; 9(12):2723. https://doi.org/10.3390/cells9122723
Chicago/Turabian StyleDragoni, Silvia, Anna Papageorgiou, Caroline Araiz, John Greenwood, and Patric Turowski. 2020. "Endothelial Protease Activated Receptor 1 (PAR1) Signalling Is Required for Lymphocyte Transmigration across Brain Microvascular Endothelial Cells" Cells 9, no. 12: 2723. https://doi.org/10.3390/cells9122723
APA StyleDragoni, S., Papageorgiou, A., Araiz, C., Greenwood, J., & Turowski, P. (2020). Endothelial Protease Activated Receptor 1 (PAR1) Signalling Is Required for Lymphocyte Transmigration across Brain Microvascular Endothelial Cells. Cells, 9(12), 2723. https://doi.org/10.3390/cells9122723