Iron: An Essential Element of Cancer Metabolism


1
Molecular Biotechnology Center, Department of Molecular Biotechnology and Health Sciences, University of Torino, 10126 Turin, Italy
2
Department of Clinical and Biological Science, University of Turin, AOU San Luigi Gonzaga, 10043 Orbassano, Italy
*
Authors to whom correspondence should be addressed.
Cells 2020, 9(12), 2591; https://doi.org/10.3390/cells9122591
Submission received: 6 November 2020
/
Revised: 24 November 2020
/
Accepted: 30 November 2020
/
Published: 3 December 2020
(This article belongs to the Special Issue Metabolic Rewiring in Cancer Cells: From Tumor Onset to Novel Therapies)
{kind=link}
{kind=link}
Abstract
:Cancer cells undergo considerable metabolic changes to foster uncontrolled proliferation in a hostile environment characterized by nutrient deprivation, poor vascularization and immune infiltration. While metabolic reprogramming has been recognized as a hallmark of cancer, the role of micronutrients in shaping these adaptations remains scarcely investigated. In particular, the broad electron-transferring abilities of iron make it a versatile cofactor that is involved in a myriad of biochemical reactions vital to cellular homeostasis, including cell respiration and DNA replication. In cancer patients, systemic iron metabolism is commonly altered. Moreover, cancer cells deploy diverse mechanisms to increase iron bioavailability to fuel tumor growth. Although iron itself can readily participate in redox reactions enabling vital processes, its reactivity also gives rise to reactive oxygen species (ROS). Hence, cancer cells further rely on antioxidant mechanisms to withstand such stress. The present review provides an overview of the common alterations of iron metabolism occurring in cancer and the mechanisms through which iron promotes tumor growth.
1. Introduction
Iron is an essential constituent of hemoproteins and iron-sulfur proteins that are responsible for a plethora of systemic functions, ranging from oxygen transport to cell metabolism and DNA synthesis [1]. Research in the field of iron metabolism has been focused on erythrocytes, while accumulating studies highlight the importance of an adequate iron supply in non-erythroid cells. Besides blood disorders, disrupted iron homeostasis has been linked to pathogenesis of metabolic diseases, including diabetes and cancer [2,3]. While a trace amount of iron is essential for cell survival, excessive iron promotes reactive oxygen species (ROS) generation [4,5]. In particular, cancer cells have to adapt to this double-edged sword as they require unphysiological amounts of iron to sustain cell proliferation which, meanwhile, contributes to increasing oxidative stress. The latter promotes deleterious protein, lipid, and DNA modifications and further metabolic changes, thereby selecting cells with most aggressive phenotype for tumor growth.
1.1. Epidemiology Linking Iron to Cancer
Results from large epidemiological studies have concluded that red meat intake is positively associated with liver, lung, pancreatic, breast, esophageal, and colorectal cancers [6,7,8,9,10,11]. The latter has been given particular attention in epidemiology, and high heme content in red meat was found to contribute to colorectal cancer [12,13]. Notably, in vitro data and human meta analyses collectively indicate that meat-derived heme catalyzes the formation of lipid peroxides [14] and nitroso-compounds (notably nitrosyl-heme and nitrosothiols, [15]), which can give rise to sequential formation of reactive species, eventually leading to stable carcinogenic DNA adducts [16]. Besides dietary iron intake, accumulating clinical studies have found a positive correlation of increased systemic iron levels with different types of cancer in humans (reviewed by Torti et al. [3]). Notably, high transferrin saturation (above 40%) was associated with increased risk of cancer in both genders [17]. Patients suffering from iron overload due to hemochromatosis or homozygous β-thalassemia were also reported to present a higher risk of cancer development, especially liver cancers [18,19,20]. Similarly, patients receiving blood transfusions (which increase iron levels) are also more susceptible to develop cancer [21]. In contrast, iron reduction by regular blood donations, or by phlebotomy in peripheral arterial disease patients, has been associated with a lower risk for cancer [22,23].
1.2. Systemic Iron Metabolism
Total body iron depends on age, gender, diet, and state of health and typically varies between 3 and 4 g in healthy adults, of which approximately two-thirds is bound in hemoglobin of red blood cells that is continuously recycled by the reticulo-endothelial system (particularly in the spleen and the bone marrow) [24,25]. Dietary iron absorption is relatively poor, as only 10% of ingested iron (from a balanced meal that contains both heme and non-heme iron) is absorbed in the duodenum and generally yields a negligible 1–2 mg of bioavailable iron [24]. After absorption, iron is exported from enterocytes to the blood system through the basolateral membrane transporter ferroportin (FPN), prior to its binding to transferrin, which allows its circulation in the bloodstream [24,26,27]. Of note, transferrin is the predominant iron-binding protein in circulation; other homologues include melanotransferrin, which is lowly expressed on cell membranes [28,29], and lactoferrin, a glycoprotein secreted by exocrine glands and immune cells with iron-sequestrating properties [30]. Nevertheless, both melanotransferrin and lactoferrin are unable to bind to transferrin receptors (TfRs) and have a minor role in iron metabolism [31,32,33]. Indeed, all cells require iron, but the vast majority of iron is used by the bone marrow for erythropoiesis [34]. Unused iron from peripheral tissues is then stored by the liver, which plays a major role in the maintenance of systemic iron homeostasis by secreting hepcidin. Hepcidin is a peptidic hormone which binds to FPN and induces its internalization and lysosomal degradation, leading to reduced circulating iron levels [35,36,37,38].
1.3. Cellular Iron Metabolism
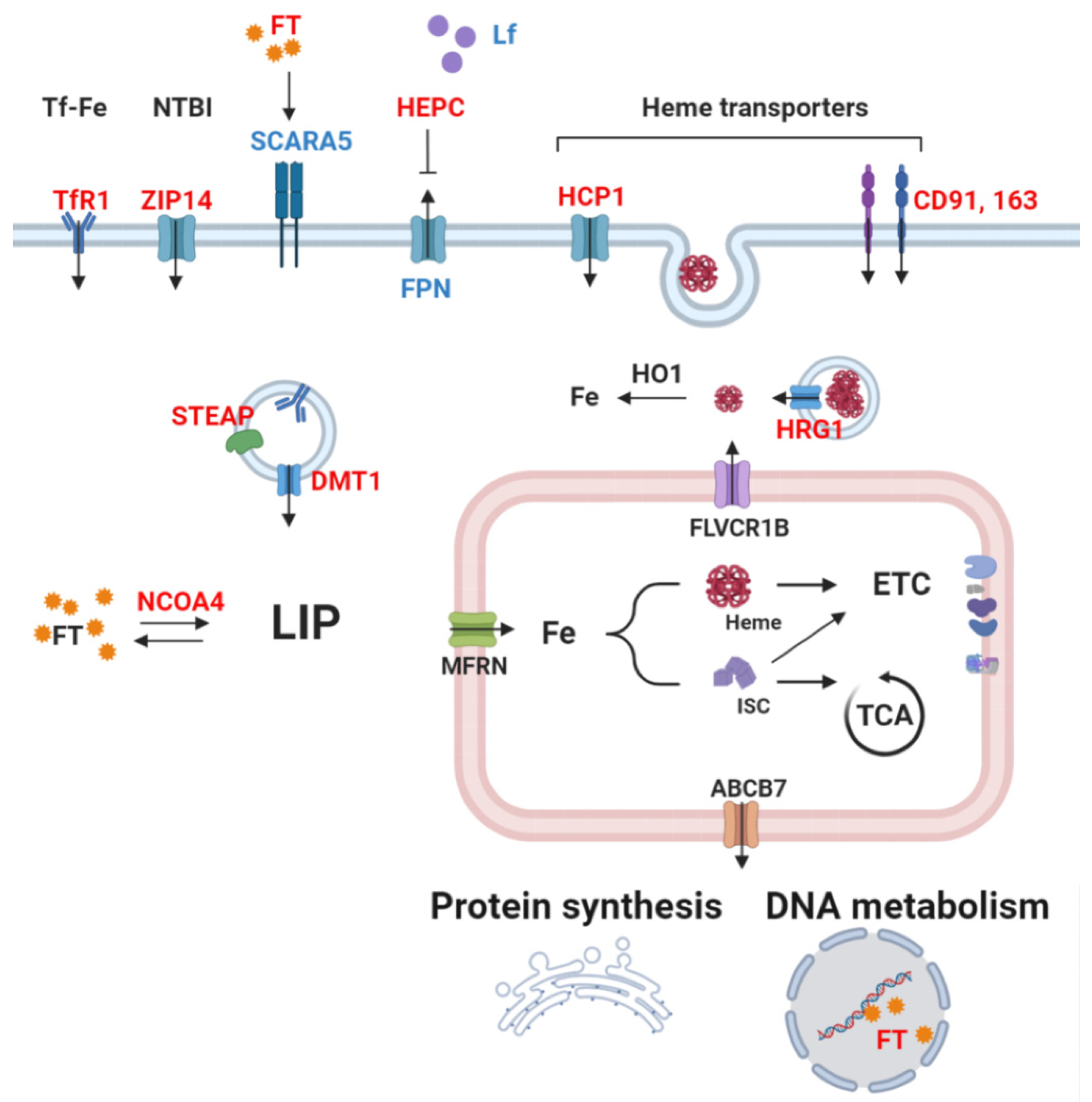
Since iron is both essential and toxic, iron uptake, trafficking, and storage are tightly controlled cellular processes that maintain homeostasis. In non-erythroid peripheral tissues, transferrin-bound iron (TBI) binds to its receptor (TfR1) and enters into the cells through endocytosis, is reduced to Fe (II) by six-transmembrane epithelial antigen of prostate (STEAP) reductases, and is released from the endosome by dimetal transporter 1 (DMT1) [39,40]. Unlike TfR1, TfR2 shows low affinity to Tf and is mainly expressed in the liver, where it acts as an iron-sensor, regulating hepcidin production [41]. For heme, several proteins (mostly non-selective) are involved in the uptake, including heme carrier protein 1 (HCP1) and clusters of differentiation 91 and 163 (CD91 and CD163), whose expression and role depend on the cell type. Circulating heme can also be imported via endocytosis, requiring heme-responsive gene 1 (HRG1) to release heme in the intracellular space, which can then be degraded by heme oxygenase 1 (HO1) to yield iron [42]. Iron can remain free in the cytosol, constituting the cytosolic labile iron pool (LIP) readily available for catalytic uses, but a major amount enters mitochondria through mitoferrins (MFRN) 1 and 2 in erythroid and non-erythroid cells, respectively [43,44]. Most of the imported iron is used for heme and iron sulfur-clusters synthesis that mainly takes place in the mitochondria. The biosynthesis pathways of both heme and iron-sulfur clusters have been extensively described previously [45,46,47]. The excess of iron in the cytosol is sequestrated by its storage protein ferritin (FT), composed of heavy and light subunits (FTH and FTL) [48]. During storage, iron is oxidized to the ferric state by FTH, while FTL stabilizes the complex, favoring mineralization [49]. Ferritin degradation by a process similar to autophagy (a process termed ferritinophagy), requiring a cargo receptor nuclear receptor coactivator 4 (NCOA4), releases bioavailable iron back to the cytosol [50]. How iron is reduced back to the ferrous state remains to be elucidated.
Intracellular iron homeostasis is mainly maintained by iron-regulatory proteins (IRP) 1 and 2, which regulate iron uptake, storage, and excretion by post-transcriptional modifications of major proteins involved in iron trafficking [51]. In iron deficiency, IRPs bind to iron-responsive elements (IREs) located in the 5′ or 3′ untranslated region (UTR) of target transcripts. Binding of IRPs to 5′UTR IRE results in translation repression of the mRNA (such as for ferritin and ferroportin), while binding to 3′UTR IRE stabilizes the mRNA and increases translation (such as for transferrin receptor 1) [51]. Although having the same role in the regulation of iron metabolism, the two IRPs function with distinct iron-sensing mechanisms. In iron-replete conditions, IRP1 is an iron-sulfur protein known as aconitase, which exerts enzymatic activity, converting citrate to isocitrate [52], whereas IRP2 is degraded by the ubiquitin-proteasome system after recognition by F-box and leucine-rich repeat protein 15 (FBXL5) [53]. In iron deficiency, the lack of iron-sulfur clusters triggers the functional switch of IRP1 and stabilizes IRP2.
2. Iron as a Catalyst of Cancer Metabolism
2.1. Hemoproteins and Cancer
Heme is a prosthetic group that consists of iron complexed with a protoporphyrin IX ring found in various proteins involved in oxygen binding, metabolism, detoxification, and even signaling [37]. Heme plays a major role in both anti-oxidant reactions. Most catalases (anti-oxidant enzymes that break down hydrogen peroxide to water and oxygen) are heme-containing enzymes that have a ferric protoporphyrin IX (or heme b) in their active sites [54]. It is noteworthy that hydrogen peroxide has toxic effects, as it participates directly in the Fenton reaction, producing hydroxyl radicals, but can also play the role of a messenger, activating inflammatory and apoptotic pathways [55]. Increased catalase expression has been reported in several cancer cell types [56], and recent meta-analyses revealed a correlation between a specific polymorphism of catalase and prostate cancer [57,58]. Besides antioxidant defense, heme also can participate in oxidative stress and inflammation. For instance, nitric oxide synthase (NOS) requires heme as a cofactor to produce NO from L-arginine. Inducible NOS (iNOS or -NOS2) isoform has been commonly found upregulated in tumors, where NO is viewed as an oncogenic molecule that promotes a wide range of pro-tumoral effects, including genome instability, cell invasion, and angiogenesis [59,60]. Importantly, several studies have reported that NO can inhibit M1 macrophage polarization and T-cell differentiation, resulting in immunosuppression [61]. On the contrary, it is widely known that NO generated by immune cells can exert direct anti-tumor effects in animal models [62]. The multifaceted controversial roles of NO in cancer, that are likely dependent on its concentration, have been increasingly acknowledged in recent years.
Similar to the effects exerted by NO, prostanoids (prostaglandins and thromboxane) are arachidonic acid-derived lipophilic signaling molecules that mediate inflammation and vasodilation. Their syntheses are catalyzed by cyclooxygenases COX1 and COX2, which require a heme prosthetic group as well [63]. Both isoforms, especially the second, have been extensively investigated for their role in cancer-related inflammation. COX2 is an inducible form that has been associated with excessive inflammation and carcinogenesis along with increased angiogenesis, tissue invasion, resistance to apoptosis, and immune defense [64,65]. Of note, patients under COX2 inhibitor use presented reduced risk for colon, breast, and lung cancers [65,66,67,68]. However, further investigation is necessary to understand whether iron contributes directly to the pro-tumoral effects of COX and NOS.
2.2. Iron-Sulfur Proteins in Cancer
Iron-sulfur clusters (ISCs) are inorganic complexes formed by two or more atoms of iron and sulfur from cysteine residues in proteins. As both iron and sulfur can donate or accept electrons, the resulting coordination presents a wide range of oxidation states [69,70]. Hence, ISCs represent versatile cofactors on which many reactions rely. Several components of its biosynthetic pathway, including cysteine disulfurase (NFS1), frataxin (FTX), and glutathione, have been shown to foster tumor progression and resistance to therapies in different models [71,72,73]. However, frataxin was also reported to induce mitochondrial oxidative metabolism in colon cancer cells, thereby suppressing tumor growth [74].
A recently characterized class of iron-sulfur proteins, named CDGSH iron-sulfur domain-containing protein (NEET), has also gained interest in the field of cancer. In non-malignant cells, NEET proteins were shown to be involved in remarkably broad processes, including autophagy, fatty acid metabolism, and insulin secretion [75,76,77]. MitoNEET and nutrient-deprivation autophagy factor-1(NAF1) have been reported to predict negative prognosis in breast, liver, pancreatic, and cervical cancers [78,79,80]. Coherently, mitoNEET and NAF1 promoted tumor growth by inducing mitochondrial oxidative metabolism and resistance to autophagy in breast cancer cells [81,82,83].
Interestingly, iron availability directly regulates mitochondrial biogenesis and oxidative metabolism [84]. Indeed, both heme and ISCs are essential to mitochondrial function as they enable several steps of the tricarboxylic acid (TCA) cycle and electron transport chain (ETC).
In humans, two reactions of the TCA cycle require ISCs: succinate dehydrogenase (SDH, also complex II of the ETC) and aconitase (ACO) [85]. Deficient expression or activity of these enzymes typically denotes mitochondrial dysfunction that has long been linked to tumorigenesis. Aconitase catalyzes the second step of the TCA cycle, which consists in the isomerization of citrate to iso-citrate via cis-aconitate formation. The conformation of aconitase changes according to the ISC and determines its enzymatic activity: liaison with a [3Fe-4S] cluster is found in the inactive form, while the acquirement of an additional iron atom forming [4Fe-4S] activates the enzyme [86,87]. Decreased mitochondrial aconitase ACO2 has been reported in gastric cancer, wherein it predicted poor prognosis, and in human breast tumor biopsies as well as breast cancer cell lines [88]. In addition, the induction of ACO2 enhanced mitochondrial oxidative metabolism and sustained ROS production, leading to reduced breast cancer cell proliferation [88]. A cytosolic isoform, more commonly termed iron-regulatory protein 1 (IRP1) or iron-responsive element binding protein (IRE-BP), exerts the same catalytic function but serves as a major iron-sensor and mRNA-binding protein, as discussed above [89].
Succinate dehydrogenase (SDH) catalyzes the oxidation of succinate to fumarate, which is then hydrated by fumarate hydratase into malate. Mutations inducing SDH-subunit B (SDHB) loss of function or deficiency have been shown to cause the development of paragangliomas, pheochromocytomas, and gastrointestinal and renal cancers [90,91,92,93,94]. Furthermore, SDHB-silencing was sufficient to induce ROS generation and metabolic switch, which increased cancer cell proliferation and tumor growth in vivo [95,96]. Indeed, excessive levels of succinate resulting from SDH stabilize hypoxia-inducible factor 1α (HIF1α) and initiate tumorigenesis; hence, it is considered as an oncometabolite [97].
In addition to the enzymes catalyzing the TCA cycle, iron is a step-limiting factor of the ETC, as all four complexes require at least one ISC, heme, or a combination of both to support the electron-transferring activity [98,99]. Consequently, a functional ETC is essential for efficient ATP generation in order to sustain cell proliferation. An impaired ETC is widely known to cause mitochondrial ROS that activate signaling pathways, fostering tumorigenesis [100,101]. Nevertheless, excessive ROS also trigger apoptosis; hence, inhibitors of ETCs have emerged as anti-cancer strategy [102].
Finally, many enzymes involved in DNA replication and repair carry at least one ISC that is essential for the enzymatic activity. The presence of ISCs is required for functional activity of DNA polymerases, glycosylases, and helicases, as well as exonucleases [103,104,105,106]. Importantly, recent findings show that eukaryotic DNA polymerases require an ISC that is essential for stable interactions between different subunits of the multimeric structure and its binding to double-stranded DNA, and lack of this ISC leads to poor fidelity of replication and genome defects [103,107,108]. Furthermore, iron chelation has been shown to inhibit ribonucleotide reductase (RNR), the enzyme that catalyzes the reduction of ribonucleotide to deoxynucleotides, as well as a class of histone lysine demethylases that enable gene transcription of several oncogenes [109,110,111,112]. Similarly, iron is also essential for nucleotide catabolism. Xanthine oxidoreductases (XORs) catalyze the oxidation of hypoxanthine to xanthine and, ultimately, to uric acid, along with the production of superoxide and hydrogen peroxide. The presence of ISCs is required for the catalytic reaction which contributes directly to oxidative stress by producing ROS and reactive nitrogen species (RNS), while uric acid can further trigger inflammation [113]. In fact, increased serum levels of XOR and uricemia are often associated to metabolic disorders, including cancer [114,115]. Inhibitors of XOR, such as febuxostat and allupurinol, have been reported to inhibit cell migration and exert cytotoxic effects on human breast cancer cells and hormone-refractory prostate cancer cells in vitro, respectively [116,117]. Hence, iron availability controls cell proliferation by regulating DNA synthesis, repair, transcription, and catabolism.
2.3. Free Iron as an Enzymatic Cofactor
Although it is evident that intracellular iron is mostly complexed within heme or ISCs, free iron can also directly act as a cofactor catalyzing enzymatic reactions. A notable example consists of the family of prolyl/asparagyl hydroxylases (PHDs) that catalyze post-translational hydroxylation of hypoxia-inducible factor 1α (HIF1α), resulting in its ubiquitination and subsequent proteasomal degradation. Besides oxygen, this modification is, in fact, dependent on the presence of 2 oxo-glutarate as the substrate and ferrous iron (Fe2+) as the cofactor. The requirement of iron in the degradation of HIF1α was first evidenced by the normoxic stabilization of HIF1α upon desferoxamine treatment in vitro, and chaperone proteins PCBP1 and PCBP2 were later identified to mediate the delivery of free iron to PHDs [118,119]. Once stabilized, HIF1α translocates into the nucleus, where it interacts with HIF1β (also known as ARNT) to bind hypoxia-response element (HRE) [120]. In cancer, HIF1α stabilization mediates many malignant properties, including defective angiogenesis and aerobic glycolysis, a phenotype termed the Warburg effect [121].
Similarly, deoxyhypusine hydroxylase (DOHH), the enzyme responsible for the last step of hypusine synthesis, requires iron for catalysis [122,123]. Hypusine is a highly conserved amino acid found solely in eukaryotic translational initiation factor 5A (eIF5A). Functional DOHH is, therefore, essential to cellular viability, as mature eIF5A regulates eukaryotic cell proliferation [124]. Interestingly, decreased levels of miR-331-3p and miR-642-5p were found in human prostate tumors overexpressing DDOH, and overexpression of these miRNAs reduced DOHH levels and hindered proliferation in vitro [125]. Likewise, pharmacological inhibition or RNA interference-mediated silencing of DOHH reduced cervical cancer cell proliferation [126].
3. Alterations of Iron Metabolism in Cancer
3.1. Systemic Iron Alterations in Cancer
Similar to what occurs in microbial infection, iron metabolism is altered at the systemic level in order to withhold iron stores within the host cells to limit uptake by pathogens. Anemia is commonly found in cancer patients and mostly arises from chronic inflammation characterized by iron restriction and decreased erythropoiesis, known as anemia of chronic disease or of inflammation [127]. For instance, the inflammatory cytokine interleukin 6 (IL-6) mediates hepcidin upregulation through the Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) pathway [128,129,130,131], while tumor necrosis factor-α (TNF-α) inhibits erythropoiesis [132]. In vitro studies have further shown that other cytokines, such as interleukin 1-β (IL-1β), also promote hepcidin synthesis [133,134]. Importantly, immune cells, including macrophages and neutrophils, also secrete hepcidin through TLR-4 induction as an innate immune response [135,136,137]. It is noteworthy that bone morphogenic proteins (BMPs), a class of transforming growth factor β ligands whose upregulation has been reported in various cancers, including breast, prostate, and bladder cancers, also induce hepcidin secretion by cancer cells [138]. Consistently, circulating plasma hepcidin is typically high and correlates with disease stage in breast, non-small lung, urothelial, and renal cancers [139,140,141,142,143]. Elevated serum ferritin has also been reported in many types of cancer, including neuroblastomas, lymphomas, colorectal, cervical, and breast cancers, and has been used as both a diagnostic tool and a prognostic factor [144,145,146,147,148,149]. Interestingly, excessive circulating ferritin, which modulates cancer cell metabolism and stimulates cancer cell proliferation, originates largely from tumor-associated macrophages (TAMs) [150].
3.2. Cellular Iron Dysregulation in Cancer
Cancer cells display peculiar alterations in iron metabolism that depict an overall increased iron turnover, with an enhanced affinity for iron that could be considered as a hallmark of cancer [151] (Figure 1). TfR1 overexpression has been commonly observed in both cancer cells in vitro and tumor tissues, including glioma, leukemia, and breast, ovarian, prostate, colorectal, and liver cancers [152]. Consequently, high TfR1 levels are typically associated with poor prognosis. In contrast, TfR1 silencing impeded oxidative phosphorylation and proliferation of pancreatic adenocarcinoma cells [153] and decreased breast tumor growth and lung metastases in mice [154]. Interestingly, p53 induction markedly downregulated TfR1 levels and led to cell cycle arrest in human lung cancer cells [155]. Similarly to TfR1, melanotransferrin is notably upregulated in melanoma tissues and, to a lesser degree, in liposarcoma and breast and lung cancers [156]. Moreover, melanotransferrin overexpression is associated with high tumor grade and metastases in human colorectal cancer [157]. Interestingly, despite a minor role in iron metabolism, gene silencing of melanotransferrin markedly reduced melanoma cell proliferation and tumor growth in mice [158]. A soluble form of melanotransferrin characterized to have low efficiency in delivering iron was found to foster melanoma cell migration, invasion, and endothelial cell angiogenesis [29,159,160]. In contrast to its homologues, lactoferrin (Lf) has been shown to possess anti-tumoral activity, with abnormally low levels found in several cancers. In particular, hypermethylation of Lf promoter was shown to cause its silencing in prostate cancer cells [161]. Mitogen-activated protein kinase (MAPK) pathway activation is observed in nasopharyngeal and gastric cancer tissues [162,163]. Contrarily, Lf supplementation reduced cancer cell proliferation in vitro and impeded tumor growth in vivo in many murine models [164]. Moreover, higher levels of Lf are correlated to better prognosis in breast carcinoma patients [165]. Lf supplementation decreased colon cancer progression in humans [166] and significantly improved clinical prognosis in colorectal cancer patients receiving chemotherapy and in breast carcinoma patients [167]. Although it is unclear whether such tumor suppressive effects are mediated by its iron-binding role, Lf has emerged as a promising prognostic factor and adjuvant therapy.
TfR2 is commonly upregulated in human cancer cell lines as well [168,169], and was shown to act as a signaling protein activating the mitogen-activated protein kinase (MAPK) pathway in human leukemia cells [170]. Studies in glioblastoma and leukemia have established a correlation between TfR2 expression and tumor grade, but TfR2 overexpression also increased sensitivity to chemotherapy and, thus, is associated to favorable prognosis [169,171]. Other proteins involved in the uptake of iron that show abnormally high expression in cancers include DMT1 as well as heme importers HCP1, HRG1, CD91, and CD163, which are associated to dismal prognosis [42,172,173,174,175,176,177,178,179]. For instance, DMT1-imported iron was shown to activate cyclin-dependent kinase 1 (CDK1) and JAK1/STAT1, contributing to colorectal tumorigenesis, and DMT1 inhibitor impeded tumor growth in vivo [172]. CD91 was shown to promote migration and invasion through matrix metalloproteinase (MMP) induction in human glioblastoma cells and via extracellular signal-regulated kinase (ERK) pathway activation in human thyroid carcinoma cells [180,181]. Finally, a scarcely investigated yet noteworthy iron importer is ZIP14, whose main substrate is zinc but that can also mediate the uptake of NTBI and intracellular release of TBI from endosomes [182]. In liver cancer cells, knockdown of tumor suppressor p53 induced iron uptake through ZIP14, highlighting its potential implication in tumorigenesis [183] (Figure 2).
Lipocalin 2 (LCN2 or NGAL) is a soluble iron-binding glycoprotein expressed mainly by neutrophils [188]. Data about its role in cancer remain controversial and vary depending on iron saturation and on the cancer type. Interestingly, LCN2 can either import or export iron and, consequently, promotes proliferation or apoptosis [189,190,191]. A clear correlation between LCN2 levels and tumor grade was found in in breast and thyroid cancers, whereas the opposite was suggested in ovarian and pancreatic cancers [192]. Similarly, both LCN2 overexpression and knockout reduced orthotopic pancreatic cancer tumor growth in mouse models [193,194,195]. However, these results collectively indicate that LCN2 is also involved in angiogenesis and inflammation within the tumor.
Since iron uptake is strongly enhanced, it is not surprising that cancer cells present aberrant intracellular iron storage and trafficking in order to cope with increased risk of iron-related oxidative stress. Reduction of ferric to bioactive ferrous iron by STEAP reductases is required to release endosomal TBI to the cytosol. Particularly, STEAP 1 and 2 are overexpressed in various human cancers and were shown to drive cancer cell proliferation and resistance to apoptosis [196,197,198]. In addition to the abnormally high levels of circulating ferritin, some cancers exhibit alterations in both ferritin expression and intracellular localization. Variable clinical outcomes have been reported accordingly. For example, glioblastoma and breast cancer cells exhibited nuclear expression of FTL and FTH, respectively [199,200]. In the latter, nuclear expression of FTH was shown to prevent iron-derived oxidative DNA damage [200]. Although both FTH and FTL are upregulated in head and neck cancers, only FTH predicted negative prognosis [201,202], whereas the opposite was found in glioblastoma [199]. In melanoma cells, FTL is required to withstand oxidative stress and prevent apoptosis [203]. Coherently, knockdown of FTH increased sensitivity to apoptosis triggered by ROS in mesothelioma cells [204]. Of note, FTH overexpression has been linked to chemotherapy (doxorubicin and cisplatin)-resistance in breast and ovarian cancers [200,205]. Interestingly, p53 activation induced both FTH and FTL expression in human lung cancer cells [155]. NCOA4, which is responsible for ferritin degradation and cytosolic iron increase, was found overexpressed in transformed endometriotic cells and pancreatic cancer cell lines and correlated to prostate cancer risk [206,207,208]. Furthermore, inactivation of p53 and activation of transforming protein p21 (HRAS) and proto-oncogene MYC led to overexpressed NCOA4 [206]. Noteworthily, ferritin heavy chains can be imported by endocytosis after binding to TfR1 [209]. Scavenger receptor class A member 5 (SCARA5) was also shown to mediate ferritin uptake [210,211].
SCARA5 downregulation was found in human breast cancer tissues and cell lines and correlated with tumor size and metastatic potential. In the same in vitro models, overexpression of SCARA5 inhibited ERK, AKT, and Signal Transducer and Activator of Transcription 3(STAT3) pathways, impeded cell proliferation, migration, colony formation, and induced apoptosis [212]. Moreover, promoter hypermethylation was found to mediate the downregulation of SCARA5 in hepatocarcinoma cells and in human breast tumors [213]. Therefore, SCARA5 has been considered to play a role as a tumor suppressor, although its contribution to physiological iron metabolism is not fully clear.
In leukemia and lung cancer cells, increased heme synthesis and availability also increased cell proliferation by enhancing mitochondrial oxidative metabolism [214,215,216]. Furthermore, recent studies have demonstrated that labile heme fosters cell proliferation by inhibiting p53 [187] and inducing MYC [186]. Interestingly, heme was shown to interfere directly with the p53 gene in vitro and in vivo while inducing proteosomal degradation of p53 protein [187]. Nevertheless, controversial studies exist when it comes to the effect of heme degradation by HO1 in cancer cells—some found that HO1 deficiency led to defective DNA, carcinogenesis, and resistance to ferroptosis (discussed hereafter); other data, however, indicated that HO1 induction promotes cancer cells antioxidant potential and cancer growth [217,218]. Notably, elevated HO1 activity induced matrix metalloproteinase 1 (MMP1) and stimulated migration and invasion in human breast cancer cells [219].
Despite sharing almost identical roles, IRPs show rather distinct patterns in tumors. Downregulation of IRP1 was found in hepatocellular carcinoma and predicted tumor stage and prognosis [172], and IRP1 overexpression in human non-small cell lung carcinoma cells suppressed tumor growth in mice [220]. In contrast, colorectal tumors showed IRP2 overexpression, which results from enhanced MAPK signaling and is associated with proto-oncogene B-Raf mutations [221]. In prostate cancer, IRP2 is predominantly upregulated, and decreased tumor growth due to apoptosis induction was observed after knockdown of IRP2, but not IRP1, in prostate cancer cell lines [222]. Similarly, although both IRPs show increased expression in breast cancer cells compared to non-malignant mammary epithelial cells, TfR1 and FT levels were altered only by IRP2 knockdown, which led to decreased tumor growth in vivo. Moreover, IRP2 expression correlated with histological grade and molecular subtype of human breast cancer. [223]. Thus, although both are potential prognostic factors, one IRP might prevail over the other without having the same expression trend, according to the cancer type. In cancer cells, other notable regulators of cellular iron metabolism include hypoxia-inducible factor 1α (HIF1α), the proto-oncogene MYC, and nuclear factor erythroid 2-related factor 2 (NRF2). In oxidative stress or iron overload conditions, NRF2 induces FTH, FTL, ferroportin, and heme oxygenase transcription to prevent excessive ROS formation promoted by iron availability [224]. Constitutive activation of NRF2 has been consistently found in various cancer types and linked to poor prognosis [225,226]. However, due to its pleiotropic effects, whether cancer progression promoted by NRF2 is iron-dependent or not has not been clear. In B cell lymphoma cells, MYC activates the transcription of TfR1 and DMT1 and represses that of FTH and FTL to increase the labile iron pool [185]. Similarly, hypoxia induces TfR1, DMT1, and hepcidin transcription through stabilization of HIF1α, leading to increased intracellular iron content [130,184,227].
In line with elevated hepcidin levels, decreased iron export is also a feature of several cancers [228,229,230,231]. In breast and pancreatic cancers, low FPN expression is associated with worse prognosis, while higher FPN expression in breast cancer patients corresponded to a cohort of patients presenting very high progression-free survival [231]. Coherently, overexpression of FPN in breast cancer cells showed decreased proliferation, colony formation, and tumor growth as well as liver metastases [230,232]. Likewise, FPN transcripts were downregulated in multiple myeloma cells isolated from patients compared to plasma cells from healthy donors and correlated with negative clinical outcomes. Furthermore, activation of STAT3 was found to mediate myeloma cell proliferation [233]. In several cell lines of prostate cancer, low FPN levels resulting from hepcidin upregulation were shown to promote proliferation, migration, and resistance to apoptosis [234], and FPN overexpression induced p53 and autophagy and reduced tumor growth in vivo [235]. Interestingly, human colorectal tumors were reported to upregulate FPN, but a histological analysis revealed aberrant cytoplasmic localization of the transporter—hence, non-functional FPN [236]. In summary, cancer cells commonly enhance iron import and suppress its export, while some uncertainty remains regarding how they cope with increased labile iron.
4. Iron-Induced Oxidative Stress and Ferroptosis
Ferroptosis is an iron-dependent form of regulated cell death that occurs as a consequence of excessive lipid peroxidation [237]. The occurrence of ferroptosis essentially depends on the availability of redox active iron. Free iron participates directly in radical-generating reactions, notably Fenton’s reaction, in which ferrous iron reacts with hydrogen peroxide, yielding the hydroxyl radical (Fe2+ + H2O2 → Fe3+ + OH + OH−). ROS then trigger lipid peroxidation in the presence of unsaturated fatty acids, giving rise to a chain of oxidative reactions which propagate through the polyunsaturated fatty acids (PUFAs) of phospholipids constituting the cell membrane. The build-up of oxidized lipids ultimately leads to cell death [238]. Indeed, cells are equipped with anti-oxidant enzymes that neutralize lipid ROS to prevent further oxidative damage, notably glutathione peroxidase 4 (GPX4), which reduces oxidized lipids [239]. Beside iron chelators, lipophilic antioxidants, including vitamin E [240], ferrostatin-1 [241], and coenzyme Q10 (CoQ10, also known as ubiquinone) [242], have also been shown to prevent ferroptosis. Likewise, ferroptosis suppressor protein 1 (FSP1), previously known as AIFM2, was shown to protect cells from ferroptosis by catalyzing NAD(P)H-dependent regeneration of CoQ10, representing a parallel and synergistic pathway alongside GPX4-mediated lipid-ROS detoxification [242]. In contrast, ferroptosis is inducible in vitro by inhibiting GPX4 or by raising intracellular iron levels and ROS. Consequently, the expression levels of iron-regulating proteins, such as TfR1, FPN, or FT, determine the sensitivity of cancer cells to ferroptosis [243,244]. Similarly, increased ROS generation resulting from artesunate treatment caused ferroptotic death in PDAC cell lines expressing constitutively active KRAS [245]. Moreover, recent studies have found that cancer cells in a mesenchymal or drug tolerant persister state are more sensitive to GPX4 inhibitors and, hence, prone to ferroptosis [246,247]. An increasing number of studies have recently validated the efficacy of ferroptosis induction in reducing cell proliferation and tumor growth in vivo, either alone or in association with standard chemotherapy [248,249]. Although the role of ferroptosis in physiological conditions remains largely unknown, these studies underscore the promising clinical prospect of ferroptotic inducers as anticancer therapies.
5. Iron in the Tumor Environment
A hallmark of malignant cells is the ability to reshape the environment where they reside, inducing angiogenesis, remodeling the extracellular matrix, and evading the host to sustain their growth [250]. Initially, immune cells recognize cancer cells as foreign and, therefore, they remodel their metabolism to compete for available iron with malignant cells [251]. Afterwards, chronic inflammation may result in evasion of immune surveillance by cancer cells, altering the immune cells toward an anti-inflammatory iron-releasing phenotype [252,253]. Polarization of tumor-associated macrophages (TAMs) determines their iron metabolism phenotype [253]: pro-inflammatory, M1-like TAMs exhibit upregulated FT levels coupled with decreased FPN, hence showing an iron-sequestering phenotype [254]. Interestingly, iron-loading in macrophages was associated with improved survival in lung cancer patients [255]. In contrast, anti-inflammatory, M2-like TAMs instead release iron and support tumor growth [256]. In particular, FPN is not the only route of iron secretion by M2-like TAMs, as its knockdown did not alter iron levels in breast cancer cells, but also, LCN2 expression mediates iron secretion by macrophages in the tumor microenvironment [257]. Iron-loading in TAMs can directly contribute to tumor growth, by feeding iron to cancer cells, or indirectly, by fostering angiogenesis. For instance, LCN2 expression by TAMs promotes lymphangiogenesis and metastasis progression in a murine breast cancer models [258]. Concerning angiogenesis, the effect of iron seems different according to the dose. While iron supplementation inhibited vascular endothelial growth factor (VEGF) signaling in endothelial cells in vitro and reduced lung carcinoma vascularization in vivo [259], LCN2 supplementation resulted in ROS accumulation and increased brain endothelial cell migration which was reversible upon iron chelation [260]. Hence, low dose of iron can inhibit angiogenesis, whereas high dose promotes oxidative stress that is widely recognized to promote angiogenesis [261].
Finally, cancer-associated fibroblasts (CAF) can also alter tumor iron metabolism, as fibroblast IL-6 secretion was shown to foster cancer cell hepcidin expression which supported iron retention in cancer cells in breast cancer spheroids [262].
Iron metabolism in the tumor microenvironment is an emerging field of research, and beside the complexity of its role in all the different cell populations involved, an in-depth understanding of how iron is handled in its niche will potentially provide novel anticancer strategies to interfere with iron metabolism at microenvironment level.
6. Effects of Iron Supplementation or Chelation on Tumorigenesis
Accumulating studies have identified iron as a relevant promoter of tumorigenesis in different aspects, including genetic and epigenetic alterations, tumor initiation, cell motility, and invasiveness; hence, a large pool of data supports iron deprivation as a therapeutic strategy in cancers.
Repeated administration of iron led to high incidence of renal cell carcinoma in rats, which was associated with large-scale genomic modifications, presumably due to increased oxidative stress [263]. Similarly, iron supplementation induced hypomethylation of oncogenes involved in PI3K/AKT, MAPK/ERK, and RAP1/RAS pathways in colonocytes [264]. In vitro, treatment with iron triggered increased migration and invasion via ROS production in human lung carcinoma and melanoma cells [265]. Treatment of colon cancer cells with ferric chloride induced an aggressive, mesenchymal phenotype with loss of intercellular adhesion by E-cadherin, which could be rescued by iron chelation with deferoxamine (DFO) [266]. Moreover, DFO reduced histone demethylases in human breast cancer cells and raised their sensitivity to chemotherapies in vitro [267]. In line with these findings, iron deprivation in vitro by iron chelators led to cell cycle inhibition and apoptosis of human colon, liver, and breast cancers and neuroblastoma, notably through activation of p53 and cyclin-dependent kinases [187,235,268]. High-throughput screening further identified iron chelators DFO, deferasirox, and ciclopirox as inhibitors of Wnt/β-catenin signaling, which is pivotal to cancer initiation and maintenance [269]. DFO was also shown to repress stem-like properties, including markers of stem-like cells and sphere-forming ability, in cholangiocarcinoma cells [270]. However, DFO-induced iron deficiency also led to normoxic stabilization of hypoxia-inducible factor-1α as well as epithelial-to-mesenchymal transition (EMT) in colorectal cancer cells, both of which have been linked to increased tumor growth [271]. In conclusion, in addition to the evident roles of iron in essential processes, such as cell respiration and genome replication, these findings highlight the multiple facets of iron as a peculiar metabolic cofactor and signaling element as well as initiator of oxidative stress and indicate, altogether, that modulating iron levels can impact different steps of tumorigenesis.
7. Clinical Application of Iron Chelation/Normalization Therapies
The phenotype of iron addiction in cancer cells represents, indeed, a therapeutic target of particular interest. Targeted therapies currently under commercial development are mainly aiming at TfR1 normalization or antagonism. Strategies including monoclonal antibodies, targeting peptides, and liposomal plasmid-based gene therapy have been proposed as treatments for various solid tumors [48,272]. Many have entered clinical trials, but limited data are available up to date. On the other hand, iron chelators, such as deferoxamine (DFO), deferasirox (DFX), and deferiprone (DFP), have been successively introduced to the market over recent decades, initially as treatments for iron overload conditions [48]. In recent years, their well characterized pharmacokinetic properties and promising in vitro anti-cancer effects have prompted the initiation of multiple preclinical studies and, eventually, clinical studies. In particular, DFO and DFX have shown efficacy alone or as adjuvants to chemotherapy in human xenografts of gastric, esophageal, pancreatic, liver, and breast cancers in mice [273,274,275,276,277]. However, in patients presenting advanced stages of hepatocarcinoma, DFX did not impact tumor progression and increased anorexia and serum creatinine [276]. Interestingly, DFP not only chelates LIP but also promotes intracellular ROS production and single-strand DNA breaks at lower doses in human hepatocarcinoma cells in vitro [278] and was recently found to reduce orthotopic prostate tumor growth in mice [279]. Other chelators that are less selective and specific are ciclopirox olamine, tachpyridine, and thiosemicarbazones, as well as epigallocatechin, curcumin, and silybin—three natural compounds that have remarkably broad activities. Ciclopirox olamine is a fungicide that initially showed promising effects in different mouse models [280,281,282,283] and in human hematological malignancies but showed a very poor pharmacokinetic profile (low solubility and rapid metabolism and clearance) [284]. A prodrug was consequently developed and was shown to be effective in urothelial cancers in rats and dog and is now under a phase I clinical trial for advanced solid tumors [285] (NCT03348514). Similar to DFP, tachpyridine also depletes intracellular iron while inducing oxidative stress [286]. Beside iron, it also binds zinc and copper; nevertheless, tachpyridine owes its cytotoxicity predominantly to its iron-chelating ability [287]. Thiosemicarbazones constitute another class of iron chelator that present ideal pharmacokinetic features, with the most prominent one being triapine. The latter showed efficacy in leukemia and ovarian and lung cancers and could even cross the blood–brain barrier; hence, it would be a potential candidate for the treatment of brain metastases [109]. Despite the observation of increased methemoglobin levels with high doses of triapine, results from a phase II trial proved its efficacy as an adjuvant therapy (with cisplatin) in advanced uterine cervix or vaginal cancers without significant toxicity and led to a phase III trial [288] (NCT02466971). Collectively, adjuvant therapy targeting iron metabolism will likely be available as an anticancer therapeutic option in the near future.
8. Concluding Remarks
Although iron is one of the most abundant elements on Earth, it constitutes a rate-limiting factor for physiological metabolism to sustain life. In particular, cancer cells rely heavily on the availability of iron and commonly exhibit striking alterations of iron metabolism, which have been shown to directly contribute to disease progression. Indeed, iron as a cofactor enables cell respiration, protein translation and even DNA replication and repair—required to sustain tumor growth. Furthermore, the tumor microenvironment can also reprogram iron metabolism and, accordingly, either cope with or fight against malignant cells. Accumulating clinical data indicate that such alterations in iron metabolism can serve as diagnostic tool and even determine prognosis. Importantly, the increased avidity of cancer cells for iron is a potential target for therapy, as it might be exploited as a trojan horse to burst oxidative stress and induce ferroptosis. Modulating iron levels or its metabolism has shown promising outcomes in vitro, and some strategies have even reached success in early phases of clinical trials as adjuvant in anticancer therapies. However, as iron is essential to all types of cells, targeting malignant cells with selectivity remains tricky, and chronic administration of iron chelators might induce systemic iron deficiency, which is also deleterious. Understanding the mechanisms to sensitize cancer cells to iron chelation or overload and targeting the tumor microenvironment will provide further progress to the development of iron-targeted anticancer strategies.
Author Contributions
Conceptualization, M.Y.H. and P.E.P.; Supervision A.R. and P.E.P.; Writing and Editing, M.Y.H., E.M., A.R., P.E.P.; Illustration, M.Y.H.; Funding acquisition, P.E.P.; All authors have read and agreed to the published version of the manuscript.
Funding
The laboratory work is supported by AIRC (P.E.P.; MFAG 21564).
Acknowledgments
We thank Valentina Rausch for her valuable comments on the manuscript. Figures were made with BioRender.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ABCB7 | ATP-Binding Cassette sub-family B member 7 |
| ACO | Aconitase |
| BMP | Bone-Morphogenetic Protein |
| CAF | Cancer-Associated Fibroblast |
| CAT | Catalase |
| CD | Cluster of Differentiation |
| CDK1 | Cyclin-Dependent Kinase 1 |
| CoQ10 | Coenzyme Q10 |
| COX | Cyclooxygenase |
| CP | Ceruloplasmin |
| DFO | Desferoxamine |
| DMT1 | Divalent Metal Transporter 1 |
| DOHH | Deoxyhypusine Hydroxylase |
| Dycb | Duodenal cytochrome b |
| EMT | Epithelial to Mesenchymal Transition |
| ETC | Electron Transport Chain |
| ERFE | Erythroferrone |
| ERK | Extracellular signal Regulated Kinase |
| FBXL15 | F-Box and Leucine Rich Repeat Protein 15 |
| FSP1 | Ferroptosis Suppressor Protein 1 |
| FTX | Frataxin |
| FT | Ferritin |
| FTH | Ferritin Heavy Chain |
| FTL | Ferritin Light Chain |
| FPN | Ferroportin |
| GPX4 | Glutathione Peroxidase 4 |
| HCP1 | Heme Carrier Protein 1 |
| HRG1 | Heme-Responsive Gene 1 |
| HEPC | Hepcidin |
| HEPH | Hephestin |
| HIF1 | hypoxia-inducible factor 1 |
| HO1 | Heme Oxygenase 1 |
| IL | Interleukin |
| IRE | Iron Responsive Element |
| IRP1/2 | Iron Regulatory Protein 1/2 |
| ISC | Iron-Sulfur Cluster |
| ISCU | ISC Assembly Enzyme |
| JAK | Janus Kinase |
| KRAS | Ki-ras2 Kirsten rat sarcoma viral oncogene homologue |
| LCN2 | Lipocalin2 |
| Lf | Lactoferrin |
| LIP | Labile Iron Pool |
| MAPK | Mitogen Activated Protein Kinase |
| MFRN | Mitoferrin |
| MMP | Matrix Metallo-Proteinase |
| NAF1 | Nutrient-deprivation Autophagy Factor-1 |
| NCOA4 | Nuclear Receptor Coactivator 4 |
| NEET | CDGSH iron-sulfur domain-containing protein |
| NFKB | Nuclear Factor Kappa-light-chain-enhancer of activated B cells |
| NFS1 | Cysteine Disulfurase |
| NO | Nitric Oxide |
| NOS | NO Synthase |
| NRF2 | Nuclear Factor Erythroid 2-Related Factor 2 |
| NTBI | Non-Transferrin Bound Iron |
| PI3K/Akt | Phosphatidylinositol 3-Kinase/protein kinase B |
| PHD | Prolyl Hydroxylase |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| RBC | Red Blood Cells |
| ROS | Reactive Oxygen Species |
| SCARA5 | Scavenger receptor class A member 5 |
| SDH | Succinate Dehydrogenase |
| STAT | Signal Transducer and Activator of Transcription |
| STEAP | Six-Transmembrane Epithelial Antigen of Prostate |
| TAM | Tumor-Associated Macrophages |
| Tf | Transferrin |
| TfR | Transferrin receptor |
| TBI | Transferrin-Bound Iron |
| TCA cycle | Tricarboxylic Acid cycle/ Krebs cycle |
| TNF-α | Tumor Necrosis Factor-α |
| VEGF | Vascular Endothelial Growth Factor |
| XOR | Xanthine Oxido-Reductase |
References
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, S.; Fonseca, V.A.; Alam, M.G.; Shah, S.V. The role of iron in diabetes and its complications. Diabetes Care 2007, 30, 1926–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torti, S.V.; Manz, D.H.; Paul, B.T.; Blanchette-Farra, N.; Torti, F.M. Iron and Cancer. Annu Rev. Nutr. 2018, 38, 97–125. [Google Scholar] [CrossRef] [PubMed]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Nakamura, T.; Naguro, I.; Ichijo, H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1398–1409. [Google Scholar] [CrossRef]
- Cross, A.J.; Leitzmann, M.F.; Gail, M.H.; Hollenbeck, A.R.; Schatzkin, A.; Sinha, R. A prospective study of red and processed meat intake in relation to cancer risk. PLoS Med. 2007, 4, e325. [Google Scholar] [CrossRef]
- Tasevska, N.; Sinha, R.; Kipnis, V.; Subar, A.F.; Leitzmann, M.F.; Hollenbeck, A.R.; Caporaso, N.E.; Schatzkin, A.; Cross, A.J. A prospective study of meat, cooking methods, meat mutagens, heme iron, and lung cancer risks. Am. J. Clin. Nutr. 2009, 89, 1884–1894. [Google Scholar] [CrossRef] [Green Version]
- Larsson, S.C.; Wolk, A. Red and processed meat consumption and risk of pancreatic cancer: Meta-analysis of prospective studies. Br. J. Cancer 2012, 106, 603–607. [Google Scholar] [CrossRef]
- Paluszkiewicz, P.; Smolinska, K.; Debinska, I.; Turski, W.A. Main dietary compounds and pancreatic cancer risk. The quantitative analysis of case-control and cohort studies. Cancer Epidemiol. 2012, 36, 60–67. [Google Scholar] [CrossRef]
- Guo, J.; Wei, W.; Zhan, L. Red and processed meat intake and risk of breast cancer: A meta-analysis of prospective studies. Breast Cancer Res. Treat. 2015, 151, 191–198. [Google Scholar] [CrossRef]
- Ferrucci, L.M.; Cross, A.J.; Graubard, B.I.; Brinton, L.A.; McCarty, C.A.; Ziegler, R.G.; Ma, X.; Mayne, S.T.; Sinha, R. Intake of meat, meat mutagens, and iron and the risk of breast cancer in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Br. J. Cancer 2009, 101, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, A.J.; Ferrucci, L.M.; Risch, A.; Graubard, B.I.; Ward, M.H.; Park, Y.; Hollenbeck, A.R.; Schatzkin, A.; Sinha, R. A large prospective study of meat consumption and colorectal cancer risk: An investigation of potential mechanisms underlying this association. Cancer Res. 2010, 70, 2406–2414. [Google Scholar] [CrossRef] [Green Version]
- Bastide, N.M.; Pierre, F.H.; Corpet, D.E. Heme iron from meat and risk of colorectal cancer: A meta-analysis and a review of the mechanisms involved. Cancer Prev. Res. 2011, 4, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Bastide, N.M.; Chenni, F.; Audebert, M.; Santarelli, R.L.; Tache, S.; Naud, N.; Baradat, M.; Jouanin, I.; Surya, R.; Hobbs, D.A.; et al. A central role for heme iron in colon carcinogenesis associated with red meat intake. Cancer Res. 2015, 75, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Kuhnle, G.G.; Story, G.W.; Reda, T.; Mani, A.R.; Moore, K.P.; Lunn, J.C.; Bingham, S.A. Diet-induced endogenous formation of nitroso compounds in the GI tract. Free Radic. Biol. Med. 2007, 43, 1040–1047. [Google Scholar] [CrossRef]
- Steinberg, P. Red Meat-Derived Nitroso Compounds, Lipid Peroxidation Products and Colorectal Cancer. Foods 2019, 8, 252. [Google Scholar] [CrossRef] [Green Version]
- Stevens, R.G.; Graubard, B.I.; Micozzi, M.S.; Neriishi, K.; Blumberg, B.S. Moderate elevation of body iron level and increased risk of cancer occurrence and death. Int. J. Cancer 1994, 56, 364–369. [Google Scholar] [CrossRef]
- Elmberg, M.; Hultcrantz, R.; Ekbom, A.; Brandt, L.; Olsson, S.; Olsson, R.; Lindgren, S.; Loof, L.; Stal, P.; Wallerstedt, S.; et al. Cancer risk in patients with hereditary hemochromatosis and in their first-degree relatives. Gastroenterology 2003, 125, 1733–1741. [Google Scholar] [CrossRef]
- Fargion, S.; Valenti, L.; Fracanzani, A.L. Hemochromatosis gene (HFE) mutations and cancer risk: Expanding the clinical manifestations of hereditary iron overload. Hepatology 2010, 51, 1119–1121. [Google Scholar] [CrossRef]
- Musallam, K.M.; Cappellini, M.D.; Wood, J.C.; Taher, A.T. Iron overload in non-transfusion-dependent thalassemia: A clinical perspective. Blood Rev. 2012, 26 (Suppl. 1), S16–S19. [Google Scholar] [CrossRef]
- Hjalgrim, H.; Edgren, G.; Rostgaard, K.; Reilly, M.; Tran, T.N.; Titlestad, K.E.; Shanwell, A.; Jersild, C.; Adami, J.; Wikman, A.; et al. Cancer incidence in blood transfusion recipients. J. Natl. Cancer Inst. 2007, 99, 1864–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merk, K.; Mattsson, B.; Mattsson, A.; Holm, G.; Gullbring, B.; Bjorkholm, M. The incidence of cancer among blood donors. Int. J. Epidemiol. 1990, 19, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Zacharski, L.R.; Chow, B.K.; Howes, P.S.; Shamayeva, G.; Baron, J.A.; Dalman, R.L.; Malenka, D.J.; Ozaki, C.K.; Lavori, P.W. Decreased cancer risk after iron reduction in patients with peripheral arterial disease: Results from a randomized trial. J. Natl. Cancer Inst. 2008, 100, 996–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohgo, Y.; Ikuta, K.; Ohtake, T.; Torimoto, Y.; Kato, J. Body iron metabolism and pathophysiology of iron overload. Int. J. Hematol. 2008, 88, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Yiannikourides, A.; Latunde-Dada, G.O. A Short Review of Iron Metabolism and Pathophysiology of Iron Disorders. Medicines 2019, 6, 85. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: Expression and regulation. Biochim. Biophys. Acta 2012, 1823, 1426–1433. [Google Scholar] [CrossRef] [Green Version]
- Fuqua, B.K.; Vulpe, C.D.; Anderson, G.J. Intestinal iron absorption. J. Trace Elem. Med. Biol. 2012, 26, 115–119. [Google Scholar] [CrossRef]
- Suryo Rahmanto, Y.; Bal, S.; Loh, K.H.; Yu, Y.; Richardson, D.R. Melanotransferrin: Search for a function. Biochim. Biophys. Acta 2012, 1820, 237–243. [Google Scholar] [CrossRef]
- Food, M.R.; Sekyere, E.O.; Richardson, D.R. The soluble form of the membrane-bound transferrin homologue, melanotransferrin, inefficiently donates iron to cells via nonspecific internalization and degradation of the protein. Eur. J. Biochem. 2002, 269, 4435–4445. [Google Scholar] [CrossRef]
- Rosa, L.; Cutone, A.; Lepanto, M.S.; Paesano, R.; Valenti, P. Lactoferrin: A Natural Glycoprotein Involved in Iron and Inflammatory Homeostasis. Int. J. Mol. Sci. 2017, 18, 1985. [Google Scholar] [CrossRef]
- Richardson, D.R.; Morgan, E.H. The transferrin homologue, melanotransferrin (p97), is rapidly catabolized by the liver of the rat and does not effectively donate iron to the brain. Biochim. Biophys. Acta 2004, 1690, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Lambert, L.A. Molecular evolution of the transferrin family and associated receptors. Biochim. Biophys. Acta 2012, 1820, 244–255. [Google Scholar] [CrossRef]
- Ward, P.P.; Mendoza-Meneses, M.; Cunningham, G.A.; Conneely, O.M. Iron status in mice carrying a targeted disruption of lactoferrin. Mol. Cell Biol. 2003, 23, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- De Domenico, I.; Ward, D.M.; Kaplan, J. Hepcidin and ferroportin: The new players in iron metabolism. Semin. Liver Dis. 2011, 31, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Rossi, E. Hepcidin--the iron regulatory hormone. Clin. Biochem. Rev. 2005, 26, 47–49. [Google Scholar]
- Pagani, A.; Nai, A.; Silvestri, L.; Camaschella, C. Hepcidin and Anemia: A Tight Relationship. Front. Physiol. 2019, 10, 1294. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [Green Version]
- Andrews, N.C. The iron transporter DMT1. Int. J. Biochem. Cell Biol. 1999, 31, 991–994. [Google Scholar] [CrossRef]
- Trinder, D.; Baker, E. Transferrin receptor 2: A new molecule in iron metabolism. Int. J. Biochem. Cell Biol. 2003, 35, 292–296. [Google Scholar] [CrossRef]
- Fiorito, V.; Chiabrando, D.; Petrillo, S.; Bertino, F.; Tolosano, E. The Multifaceted Role of Heme in Cancer. Front. Oncol. 2019, 9, 1540. [Google Scholar] [CrossRef] [Green Version]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharm. 2014, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Rouault, T.A. Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry 2010, 49, 4945–4956. [Google Scholar] [CrossRef]
- Lill, R. Function and biogenesis of iron-sulphur proteins. Nature 2009, 460, 831–838. [Google Scholar] [CrossRef]
- Crielaard, B.J.; Lammers, T.; Rivella, S. Targeting iron metabolism in drug discovery and delivery. Nat. Rev. Drug Discov. 2017, 16, 400–423. [Google Scholar] [CrossRef] [Green Version]
- Carmona, U.; Li, L.; Zhang, L.; Knez, M. Ferritin light-chain subunits: Key elements for the electron transfer across the protein cage. Chem. Commun. (Camb.) 2014, 50, 15358–15361. [Google Scholar] [CrossRef]
- Dowdle, W.E.; Nyfeler, B.; Nagel, J.; Elling, R.A.; Liu, S.; Triantafellow, E.; Menon, S.; Wang, Z.; Honda, A.; Pardee, G.; et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. [Google Scholar] [CrossRef]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 2012, 1823, 1468–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volz, K. The functional duality of iron regulatory protein 1. Curr. Opin. Struct. Biol. 2008, 18, 106–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi-Makise, N.; Ward, D.M.; Kaplan, J. On the mechanism of iron sensing by IRP2: New players, new paradigms. Nat. Chem. Biol. 2009, 5, 874–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Forsberg, L.; Lyrenas, L.; de Faire, U.; Morgenstern, R. A common functional C-T substitution polymorphism in the promoter region of the human catalase gene influences transcription factor binding, reporter gene transcription and is correlated to blood catalase levels. Free Radic. Biol. Med. 2001, 30, 500–505. [Google Scholar] [CrossRef]
- Liu, K.; Liu, X.; Wang, M.; Wang, X.; Kang, H.; Lin, S.; Yang, P.; Dai, C.; Xu, P.; Li, S.; et al. Two common functional catalase gene polymorphisms (rs1001179 and rs794316) and cancer susceptibility: Evidence from 14,942 cancer cases and 43,285 controls. Oncotarget 2016, 7, 62954–62965. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.D.; Sun, Y.; Chen, N.; Huang, L.; Huang, J.W.; Zhu, M.; Wang, T.; Ji, Y.L. The Role of Catalase C262T Gene Polymorphism in the Susceptibility and Survival of Cancers. Sci. Rep. 2016, 6, 26973. [Google Scholar] [CrossRef]
- Wink, D.A.; Ridnour, L.A.; Hussain, S.P.; Harris, C.C. The reemergence of nitric oxide and cancer. Nitric Oxide 2008, 19, 65–67. [Google Scholar] [CrossRef] [Green Version]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.; Yan, Y.; Zhang, R.; Xiong, H. Regulation of iNOS on Immune Cells and Its Role in Diseases. Int. J. Mol. Sci. 2018, 19, 3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouzer, C.A.; Marnett, L.J. Cyclooxygenases: Structural and functional insights. J. Lipid Res. 2009, 50, S29–S34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef] [Green Version]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in cancer: A review. J. Cell Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef]
- Harris, R.E.; Beebe-Donk, J.; Alshafie, G.A. Similar reductions in the risk of human colon cancer by selective and nonselective cyclooxygenase-2 (COX-2) inhibitors. BMC Cancer 2008, 8, 237. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.E.; Beebe-Donk, J.; Alshafie, G.A. Reduced risk of human lung cancer by selective cyclooxygenase 2 (COX-2) blockade: Results of a case control study. Int. J. Biol. Sci. 2007, 3, 328–334. [Google Scholar] [CrossRef]
- Harris, R.E.; Beebe-Donk, J.; Alshafie, G.A. Reduction in the risk of human breast cancer by selective cyclooxygenase-2 (COX-2) inhibitors. BMC Cancer 2006, 6, 27. [Google Scholar] [CrossRef]
- Cardenas-Rodriguez, M.; Chatzi, A.; Tokatlidis, K. Iron-sulfur clusters: From metals through mitochondria biogenesis to disease. J. Biol. Inorg. Chem. 2018, 23, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Rouault, T.A. Biogenesis of iron-sulfur clusters in mammalian cells: New insights and relevance to human disease. Dis. Model. Mech. 2012, 5, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Guccini, I.; Serio, D.; Condo, I.; Rufini, A.; Tomassini, B.; Mangiola, A.; Maira, G.; Anile, C.; Fina, D.; Pallone, F.; et al. Frataxin participates to the hypoxia-induced response in tumors. Cell Death Dis. 2011, 2, e123. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, T.J.; Thierbach, R.; Voigt, A.; Drewes, G.; Mietzner, B.; Steinberg, P.; Pfeiffer, A.F.; Ristow, M. Induction of oxidative metabolism by mitochondrial frataxin inhibits cancer growth: Otto Warburg revisited. J. Biol. Chem. 2006, 281, 977–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, N.C.; Nguyen, M.; Germain, M.; Shore, G.C. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2010, 29, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Kusminski, C.M.; Holland, W.L.; Sun, K.; Park, J.; Spurgin, S.B.; Lin, Y.; Askew, G.R.; Simcox, J.A.; McClain, D.A.; Li, C.; et al. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat. Med. 2012, 18, 1539–1549. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.E.; Crail, J.P.; Laffoon, M.M.; Fernandez, W.G.; Menze, M.A.; Konkle, M.E. Identification of disulfide bond formation between MitoNEET and glutamate dehydrogenase 1. Biochemistry 2013, 52, 8969–8971. [Google Scholar] [CrossRef] [Green Version]
- Mittler, R.; Darash-Yahana, M.; Sohn, Y.S.; Bai, F.; Song, L.; Cabantchik, I.Z.; Jennings, P.A.; Onuchic, J.N.; Nechushtai, R. NEET Proteins: A New Link Between Iron Metabolism, Reactive Oxygen Species, and Cancer. Antioxid. Redox Signal. 2019, 30, 1083–1095. [Google Scholar] [CrossRef]
- Chen, B.; Shen, S.; Wu, J.; Hua, Y.; Kuang, M.; Li, S.; Peng, B. CISD2 associated with proliferation indicates negative prognosis in patients with hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 13725–13738. [Google Scholar]
- Liu, L.; Xia, M.; Wang, J.; Zhang, W.; Zhang, Y.; He, M. CISD2 expression is a novel marker correlating with pelvic lymph node metastasis and prognosis in patients with early-stage cervical cancer. Med. Oncol. 2014, 31, 183. [Google Scholar] [CrossRef]
- Salem, A.F.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; Lisanti, M.P. Mitochondrial biogenesis in epithelial cancer cells promotes breast cancer tumor growth and confers autophagy resistance. Cell Cycle 2012, 11, 4174–4180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darash-Yahana, M.; Pozniak, Y.; Lu, M.; Sohn, Y.S.; Karmi, O.; Tamir, S.; Bai, F.; Song, L.; Jennings, P.A.; Pikarsky, E.; et al. Breast cancer tumorigenicity is dependent on high expression levels of NAF-1 and the lability of its Fe-S clusters. Proc. Natl. Acad. Sci. USA 2016, 113, 10890–10895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, Y.S.; Tamir, S.; Song, L.; Michaeli, D.; Matouk, I.; Conlan, A.R.; Harir, Y.; Holt, S.H.; Shulaev, V.; Paddock, M.L.; et al. NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 14676–14681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rensvold, J.W.; Ong, S.E.; Jeevananthan, A.; Carr, S.A.; Mootha, V.K.; Pagliarini, D.J. Complementary RNA and protein profiling identifies iron as a key regulator of mitochondrial biogenesis. Cell Rep. 2013, 3, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Stiban, J.; So, M.; Kaguni, L.S. Iron-Sulfur Clusters in Mitochondrial Metabolism: Multifaceted Roles of a Simple Cofactor. Biochemistry 2016, 81, 1066–1080. [Google Scholar] [CrossRef]
- Robbins, A.H.; Stout, C.D. The structure of aconitase. Proteins 1989, 5, 289–312. [Google Scholar] [CrossRef]
- Robbins, A.H.; Stout, C.D. Structure of activated aconitase: Formation of the [4Fe-4S] cluster in the crystal. Proc. Natl. Acad. Sci. USA 1989, 86, 3639–3643. [Google Scholar] [CrossRef] [Green Version]
- Ciccarone, F.; Di Leo, L.; Lazzarino, G.; Maulucci, G.; Di Giacinto, F.; Tavazzi, B.; Ciriolo, M.R. Aconitase 2 inhibits the proliferation of MCF-7 cells promoting mitochondrial oxidative metabolism and ROS/FoxO1-mediated autophagic response. Br. J. Cancer 2020, 122, 182–193. [Google Scholar] [CrossRef]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharm. 2014, 5, 176. [Google Scholar] [CrossRef] [Green Version]
- Bardella, C.; Pollard, P.J.; Tomlinson, I. SDH mutations in cancer. Biochim. Biophys. Acta 2011, 1807, 1432–1443. [Google Scholar] [CrossRef] [Green Version]
- Gill, A.J. Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology 2012, 44, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.; Woodward, E.R.; Killick, P.; Morris, M.R.; Astuti, D.; Latif, F.; Maher, E.R. Germline SDHB mutations and familial renal cell carcinoma. J. Natl. Cancer Inst. 2008, 100, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; Shuch, B.; Vocke, C.D.; Metwalli, A.R.; Bratslavsky, G.; Middelton, L.; Yang, Y.; Wei, M.H.; Pautler, S.E.; Peterson, J.; et al. Succinate dehydrogenase kidney cancer: An aggressive example of the Warburg effect in cancer. J. Urol. 2012, 188, 2063–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalla Pozza, E.; Dando, I.; Pacchiana, R.; Liboi, E.; Scupoli, M.T.; Donadelli, M.; Palmieri, M. Regulation of succinate dehydrogenase and role of succinate in cancer. Semin. Cell Dev. Biol. 2020, 98, 4–14. [Google Scholar] [CrossRef]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [Green Version]
- Tseng, P.L.; Wu, W.H.; Hu, T.H.; Chen, C.W.; Cheng, H.C.; Li, C.F.; Tsai, W.H.; Tsai, H.J.; Hsieh, M.C.; Chuang, J.H.; et al. Decreased succinate dehydrogenase B in human hepatocellular carcinoma accelerates tumor malignancy by inducing the Warburg effect. Sci. Rep. 2018, 8, 3081. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Vander Heiden, M.G. Altered metabolite levels in cancer: Implications for tumour biology and cancer therapy. Nat. Rev. Cancer 2016, 16, 680–693. [Google Scholar] [CrossRef]
- Winge, D.R. Sealing the mitochondrial respirasome. Mol. Cell Biol. 2012, 32, 2647–2652. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Barrientos, T.; Andrews, N.C. Iron and copper in mitochondrial diseases. Cell Metab. 2013, 17, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, L.; Neuzil, J. Targeting mitochondria as an anticancer strategy. Cancer Commun. 2019, 39, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netz, D.J.; Stith, C.M.; Stumpfig, M.; Kopf, G.; Vogel, D.; Genau, H.M.; Stodola, J.L.; Lill, R.; Burgers, P.M.; Pierik, A.J. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat. Chem. Biol. 2011, 8, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stehling, O.; Vashisht, A.A.; Mascarenhas, J.; Jonsson, Z.O.; Sharma, T.; Netz, D.J.; Pierik, A.J.; Wohlschlegel, J.A.; Lill, R. MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science 2012, 337, 195–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gari, K.; Leon Ortiz, A.M.; Borel, V.; Flynn, H.; Skehel, J.M.; Boulton, S.J. MMS19 links cytoplasmic iron-sulfur cluster assembly to DNA metabolism. Science 2012, 337, 243–245. [Google Scholar] [CrossRef]
- Paul, V.D.; Lill, R. Biogenesis of cytosolic and nuclear iron-sulfur proteins and their role in genome stability. Biochim. Biophys. Acta 2015, 1853, 1528–1539. [Google Scholar] [CrossRef] [Green Version]
- Baranovskiy, A.G.; Siebler, H.M.; Pavlov, Y.I.; Tahirov, T.H. Iron-Sulfur Clusters in DNA Polymerases and Primases of Eukaryotes. Methods Enzym. 2018, 599, 1–20. [Google Scholar] [CrossRef]
- Jozwiakowski, S.K.; Kummer, S.; Gari, K. Human DNA polymerase delta requires an iron-sulfur cluster for high-fidelity DNA synthesis. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Finch, R.A.; Liu, M.; Grill, S.P.; Rose, W.C.; Loomis, R.; Vasquez, K.M.; Cheng, Y.; Sartorelli, A.C. Triapine (3-aminopyridine-2-carboxaldehyde- thiosemicarbazone): A potent inhibitor of ribonucleotide reductase activity with broad spectrum antitumor activity. Biochem. Pharm. 2000, 59, 983–991. [Google Scholar] [CrossRef]
- Hoyes, K.P.; Hider, R.C.; Porter, J.B. Cell cycle synchronization and growth inhibition by 3-hydroxypyridin-4-one iron chelators in leukemia cell lines. Cancer Res. 1992, 52, 4591–4599. [Google Scholar]
- Cooper, C.E.; Lynagh, G.R.; Hoyes, K.P.; Hider, R.C.; Cammack, R.; Porter, J.B. The relationship of intracellular iron chelation to the inhibition and regeneration of human ribonucleotide reductase. J. Biol. Chem. 1996, 271, 20291–20299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodaverdian, V.; Tapadar, S.; MacDonald, I.A.; Xu, Y.; Ho, P.Y.; Bridges, A.; Rajpurohit, P.; Sanghani, B.A.; Fan, Y.; Thangaraju, M.; et al. Deferiprone: Pan-selective Histone Lysine Demethylase Inhibition Activity and Structure Activity Relationship Study. Sci. Rep. 2019, 9, 4802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, T.; Okamoto, K. The role of the [2Fe-2s] cluster centers in xanthine oxidoreductase. J. Inorg. Biochem. 2000, 82, 43–49. [Google Scholar] [CrossRef]
- Battelli, M.G.; Bortolotti, M.; Polito, L.; Bolognesi, A. Metabolic syndrome and cancer risk: The role of xanthine oxidoreductase. Redox Biol. 2019, 21, 101070. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine oxidoreductase in cancer: More than a differentiation marker. Cancer Med. 2016, 5, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Choi, S.Y.; Choi, H.J.; Ryu, H.M.; Kim, Y.J.; Jung, H.Y.; Cho, J.H.; Kim, C.D.; Park, S.H.; Kwon, T.H.; et al. The emerging role of xanthine oxidase inhibition for suppression of breast cancer cell migration and metastasis associated with hypercholesterolemia. FASEB J. 2019, 33, 7301–7314. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Yoshida, T.; Goda, A.E.; Horinaka, M.; Yano, K.; Shiraishi, T.; Wakada, M.; Mizutani, Y.; Miki, T.; Sakai, T. Anti-gout agent allopurinol exerts cytotoxicity to human hormone-refractory prostate cancer cells in combination with tumor necrosis factor-related apoptosis-inducing ligand. Mol. Cancer Res. 2008, 6, 1852–1860. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.L.; Semenza, G.L. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: Implications for models of hypoxia signal transduction. Blood 1993, 82, 3610–3615. [Google Scholar] [CrossRef] [Green Version]
- Nandal, A.; Ruiz, J.C.; Subramanian, P.; Ghimire-Rijal, S.; Sinnamon, R.A.; Stemmler, T.L.; Bruick, R.K.; Philpott, C.C. Activation of the HIF prolyl hydroxylase by the iron chaperones PCBP1 and PCBP2. Cell Metab. 2011, 14, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000, 88, 1474–1480. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Frey, A.G.; Nandal, A.; Park, J.H.; Smith, P.M.; Yabe, T.; Ryu, M.S.; Ghosh, M.C.; Lee, J.; Rouault, T.A.; Park, M.H.; et al. Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase. Proc. Natl. Acad. Sci. USA 2014, 111, 8031–8036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, V.V.; Emerson, J.P.; Martinho, M.; Kim, Y.S.; Munck, E.; Park, M.H.; Que, L., Jr. Human deoxyhypusine hydroxylase, an enzyme involved in regulating cell growth, activates O2 with a nonheme diiron center. Proc. Natl. Acad. Sci. USA 2009, 106, 14814–14819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.H. The post-translational synthesis of a polyamine-derived amino acid, hypusine, in the eukaryotic translation initiation factor 5A (eIF5A). J. Biochem. 2006, 139, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epis, M.R.; Giles, K.M.; Kalinowski, F.C.; Barker, A.; Cohen, R.J.; Leedman, P.J. Regulation of expression of deoxyhypusine hydroxylase (DOHH), the enzyme that catalyzes the activation of eIF5A, by miR-331-3p and miR-642-5p in prostate cancer cells. J. Biol. Chem. 2012, 287, 35251–35259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memin, E.; Hoque, M.; Jain, M.R.; Heller, D.S.; Li, H.; Cracchiolo, B.; Hanauske-Abel, H.M.; Pe’ery, T.; Mathews, M.B. Blocking eIF5A modification in cervical cancer cells alters the expression of cancer-related genes and suppresses cell proliferation. Cancer Res. 2014, 74, 552–562. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Ganz, T. Anemia of inflammation. Hematol. Oncol. Clin. N. Am. 2014, 28, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Pietrangelo, A.; Dierssen, U.; Valli, L.; Garuti, C.; Rump, A.; Corradini, E.; Ernst, M.; Klein, C.; Trautwein, C. STAT3 is required for IL-6-gp130-dependent activation of hepcidin in vivo. Gastroenterology 2007, 132, 294–300. [Google Scholar] [CrossRef]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Invest. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, G.; Chauvet, C.; Viatte, L.; Danan, J.L.; Bigard, X.; Devaux, I.; Beaumont, C.; Kahn, A.; Vaulont, S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Invest. 2002, 110, 1037–1044. [Google Scholar] [CrossRef]
- Nemeth, E.; Valore, E.V.; Territo, M.; Schiller, G.; Lichtenstein, A.; Ganz, T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003, 101, 2461–2463. [Google Scholar] [CrossRef] [PubMed]
- Buck, I.; Morceau, F.; Grigorakaki, C.; Dicato, M.; Diederich, M. Linking anemia to inflammation and cancer: The crucial role of TNFalpha. Biochem. Pharm. 2009, 77, 1572–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.; Peng, H.; Gelbart, T.; Wang, L.; Beutler, E. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc. Natl. Acad. Sci. USA 2005, 102, 1906–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matak, P.; Chaston, T.B.; Chung, B.; Srai, S.K.; McKie, A.T.; Sharp, P.A. Activated macrophages induce hepcidin expression in HuH7 hepatoma cells. Haematologica 2009, 94, 773–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurl, I.; Theurl, M.; Seifert, M.; Mair, S.; Nairz, M.; Rumpold, H.; Zoller, H.; Bellmann-Weiler, R.; Niederegger, H.; Talasz, H.; et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood 2008, 111, 2392–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sow, F.B.; Florence, W.C.; Satoskar, A.R.; Schlesinger, L.S.; Zwilling, B.S.; Lafuse, W.P. Expression and localization of hepcidin in macrophages: A role in host defense against tuberculosis. J. Leukoc. Biol. 2007, 82, 934–945. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Zinkernagel, A.S.; Datta, V.; Lauth, X.; Johnson, R.S.; Nizet, V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 2006, 107, 3727–3732. [Google Scholar] [CrossRef] [Green Version]
- Bach, D.H.; Park, H.J.; Lee, S.K. The Dual Role of Bone Morphogenetic Proteins in Cancer. Mol. Oncolytics 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Scimeca, M.; Bonanno, E. New highlight in breast cancer development: The key role of hepcidin and iron metabolism. Ann. Transl. Med. 2018, 6, S56. [Google Scholar] [CrossRef]
- Ciniselli, C.M.; De Bortoli, M.; Taverna, E.; Varinelli, L.; Pizzamiglio, S.; Veneroni, S.; Bonini, C.; Orlandi, R.; Verderio, P.; Bongarzone, I. Plasma hepcidin in early-stage breast cancer patients: No relationship with interleukin-6, erythropoietin and erythroferrone. Expert Rev. Proteom. 2015, 12, 695–701. [Google Scholar] [CrossRef]
- Orlandi, R.; De Bortoli, M.; Ciniselli, C.M.; Vaghi, E.; Caccia, D.; Garrisi, V.; Pizzamiglio, S.; Veneroni, S.; Bonini, C.; Agresti, R.; et al. Hepcidin and ferritin blood level as noninvasive tools for predicting breast cancer. Ann. Oncol. 2014, 25, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, L.; Ma, Y.; Wu, X.; Jin, L.; Yu, F. Increased hepcidin expression in non-small cell lung cancer tissue and serum is associated with clinical stage. Thorac. Cancer 2014, 5, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Shibata, Y.; Inoue, S.; Igarash, A.; Tokairin, Y.; Yamauchi, K.; Kimura, T.; Nemoto, T.; Sato, M.; Nakano, H.; et al. Serum hepcidin and iron are associated with non-small cell lung cancer stage. Eur. Respir. J. 2016. [Google Scholar] [CrossRef]
- Hann, H.W.; Evans, A.E.; Siegel, S.E.; Wong, K.Y.; Sather, H.; Dalton, A.; Hammond, D.; Seeger, R.C. Prognostic importance of serum ferritin in patients with Stages III and IV neuroblastoma: The Childrens Cancer Study Group experience. Cancer Res. 1985, 45, 2843–2848. [Google Scholar] [PubMed]
- Hann, H.W.; Lange, B.; Stahlhut, M.W.; McGlynn, K.A. Prognostic importance of serum transferrin and ferritin in childhood Hodgkin’s disease. Cancer 1990, 66, 313–316. [Google Scholar] [CrossRef]
- Koyama, S.; Fujisawa, S.; Watanabe, R.; Itabashi, M.; Ishibashi, D.; Ishii, Y.; Hattori, Y.; Nakajima, Y.; Motohashi, K.; Takasaki, H.; et al. Serum ferritin level is a prognostic marker in patients with peripheral T-cell lymphoma. Int. J. Lab. Hematol. 2017, 39, 112–117. [Google Scholar] [CrossRef]
- Lee, S.; Song, A.; Eo, W. Serum Ferritin as a Prognostic Biomarker for Survival in Relapsed or Refractory Metastatic Colorectal Cancer. J. Cancer 2016, 7, 957–964. [Google Scholar] [CrossRef]
- Ito, H.; Takagi, Y.; Ando, Y.; Kubo, A.; Hashimoto, S.; Tsutsui, F.; Kurihara, S. Serum ferritin levels in patients with cervical cancer. Obs. Gynecol. 1980, 55, 358–362. [Google Scholar] [CrossRef]
- Marcus, D.M.; Zinberg, N. Measurement of serum ferritin by radioimmunoassay: Results in normal individuals and patients with breast cancer. J. Natl. Cancer Inst. 1975, 55, 791–795. [Google Scholar] [CrossRef]
- Alkhateeb, A.A.; Connor, J.R. The significance of ferritin in cancer: Anti-oxidation, inflammation and tumorigenesis. Biochim. Biophys. Acta 2013, 1836, 245–254. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and cancer: 2020 Vision. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar] [PubMed]
- Jeong, S.M.; Hwang, S.; Seong, R.H. Transferrin receptor regulates pancreatic cancer growth by modulating mitochondrial respiration and ROS generation. Biochem. Biophys. Res. Commun. 2016, 471, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.P.; Elliott, R.L.; Head, J.F. Manipulation of iron transporter genes results in the suppression of human and mouse mammary adenocarcinomas. Anticancer Res. 2010, 30, 759–765. [Google Scholar]
- Zhang, F.; Wang, W.; Tsuji, Y.; Torti, S.V.; Torti, F.M. Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J. Biol. Chem. 2008, 283, 33911–33918. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.P.; Woodbury, R.G.; Hart, C.E.; Hellstrom, I.; Hellstrom, K.E. Quantitative analysis of melanoma-associated antigen p97 in normal and neoplastic tissues. Proc. Natl. Acad. Sci. USA 1981, 78, 539–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dus-Szachniewicz, K.; Ostasiewicz, P.; Wozniak, M.; Kolodziej, P.; Wisniewski, J.R.; Ziolkowski, P. Pattern of Melanotransferrin Expression in Human Colorectal Tissues: An Immunohistochemical Study on Potential Clinical Application. Anticancer Res. 2015, 35, 6551–6561. [Google Scholar]
- Dunn, L.L.; Sekyere, E.O.; Suryo Rahmanto, Y.; Richardson, D.R. The function of melanotransferrin: A role in melanoma cell proliferation and tumorigenesis. Carcinogenesis 2006, 27, 2157–2169. [Google Scholar] [CrossRef]
- Demeule, M.; Bertrand, Y.; Michaud-Levesque, J.; Jodoin, J.; Rolland, Y.; Gabathuler, R.; Beliveau, R. Regulation of plasminogen activation: A role for melanotransferrin (p97) in cell migration. Blood 2003, 102, 1723–1731. [Google Scholar] [CrossRef] [Green Version]
- Heybeli, N.; Yesildag, A.; Oyar, O.; Gulsoy, U.K.; Tekinsoy, M.A.; Mumcu, E.F. Diagnostic ultrasound treatment increases the bone fracture-healing rate in an internally fixed rat femoral osteotomy model. J. Ultrasound Med. 2002, 21, 1357–1363. [Google Scholar] [CrossRef]
- Shaheduzzaman, S.; Vishwanath, A.; Furusato, B.; Cullen, J.; Chen, Y.; Banez, L.; Nau, M.; Ravindranath, L.; Kim, K.H.; Mohammed, A.; et al. Silencing of Lactotransferrin expression by methylation in prostate cancer progression. Cancer Biol. 2007, 6, 1088–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, G.; Zhou, Y.; Yi, W.; Yi, H. Lactotransferrin expression is downregulated and affects the mitogen-activated protein kinase pathway in gastric cancer. Oncol. Lett. 2015, 9, 2409–2413. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zeng, Z.; Zhang, W.; Xiong, W.; Wu, M.; Tan, Y.; Yi, W.; Xiao, L.; Li, X.; Huang, C.; et al. Lactotransferrin: A candidate tumor suppressor-Deficient expression in human nasopharyngeal carcinoma and inhibition of NPC cell proliferation by modulating the mitogen-activated protein kinase pathway. Int. J. Cancer 2008, 123, 2065–2072. [Google Scholar] [CrossRef]
- Rodrigues, L.; Teixeira, J.; Schmitt, F.; Paulsson, M.; Mansson, H.L. Lactoferrin and cancer disease prevention. Crit. Rev. Food Sci. Nutr. 2009, 49, 203–217. [Google Scholar] [CrossRef] [Green Version]
- Ieni, A.; Barresi, V.; Licata, L.; Cardia, R.; Fazzari, C.; Nuciforo, G.; Caruso, F.; Caruso, M.; Adamo, V.; Tuccari, G. Immunoexpression of lactoferrin in triple-negative breast cancer patients: A proposal to select a less aggressive subgroup. Oncol. Lett. 2017, 13, 3205–3209. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, H.; Kozu, T.; Iinuma, G.; Ohashi, Y.; Saito, Y.; Saito, D.; Akasu, T.; Alexander, D.B.; Futakuchi, M.; Fukamachi, K.; et al. Cancer prevention by bovine lactoferrin: From animal studies to human trial. Biometals 2010, 23, 399–409. [Google Scholar] [CrossRef]
- Moastafa, T.M.; El-Sissy Ael, D.; El-Saeed, G.K.; Koura, M.S. Study on the Therapeutic Benefit on Lactoferrin in Patients with Colorectal Cancer Receiving Chemotherapy. Int. Sch. Res. Not. 2014, 2014, 184278. [Google Scholar] [CrossRef]
- Calzolari, A.; Oliviero, I.; Deaglio, S.; Mariani, G.; Biffoni, M.; Sposi, N.M.; Malavasi, F.; Peschle, C.; Testa, U. Transferrin receptor 2 is frequently expressed in human cancer cell lines. Blood Cells Mol. Dis. 2007, 39, 82–91. [Google Scholar] [CrossRef]
- Calzolari, A.; Larocca, L.M.; Deaglio, S.; Finisguerra, V.; Boe, A.; Raggi, C.; Ricci-Vitani, L.; Pierconti, F.; Malavasi, F.; De Maria, R.; et al. Transferrin receptor 2 is frequently and highly expressed in glioblastomas. Transl. Oncol. 2010, 3, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Calzolari, A.; Raggi, C.; Deaglio, S.; Sposi, N.M.; Stafsnes, M.; Fecchi, K.; Parolini, I.; Malavasi, F.; Peschle, C.; Sargiacomo, M.; et al. TfR2 localizes in lipid raft domains and is released in exosomes to activate signal transduction along the MAPK pathway. J. Cell Sci. 2006, 119, 4486–4498. [Google Scholar] [CrossRef] [Green Version]
- Nakamaki, T.; Kawabata, H.; Saito, B.; Matsunawa, M.; Suzuki, J.; Adachi, D.; Tomoyasu, S.; Phillip Koeffler, H. Elevated levels of transferrin receptor 2 mRNA, not transferrin receptor 1 mRNA, are associated with increased survival in acute myeloid leukaemia. Br. J. Haematol. 2004, 125, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Ramakrishnan, S.K.; Weisz, K.; Triner, D.; Xie, L.; Attili, D.; Pant, A.; Gyorffy, B.; Zhan, M.; Carter-Su, C.; et al. Iron Uptake via DMT1 Integrates Cell Cycle with JAK-STAT3 Signaling to Promote Colorectal Tumorigenesis. Cell Metab. 2016, 24, 447–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boult, J.; Roberts, K.; Brookes, M.J.; Hughes, S.; Bury, J.P.; Cross, S.S.; Anderson, G.J.; Spychal, R.; Iqbal, T.; Tselepis, C. Overexpression of cellular iron import proteins is associated with malignant progression of esophageal adenocarcinoma. Clin. Cancer Res. 2008, 14, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, P.; Liao, Z.; Ren, Z.; Zhao, J.; Song, F.; Wang, G.; Chen, K.; Yang, J. Roles of low-density lipoprotein receptor-related protein 1 in tumors. Chin. J. Cancer 2016, 35, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.; Horlad, H.; Ohnishi, K.; Nakagawa, T.; Yamada, S.; Kitada, S.; Motoshima, T.; Kamba, T.; Nakayama, T.; Fujimoto, N.; et al. CD163-positive cancer cells are potentially associated with high malignant potential in clear cell renal cell carcinoma. Med. Mol. Morphol. 2018, 51, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Zhang, D.; Gong, B.; Wang, P.; Liu, F. CD163 as a novel target gene of STAT3 is a potential therapeutic target for gastric cancer. Oncotarget 2017, 8, 87244–87262. [Google Scholar] [CrossRef] [Green Version]
- Shabo, I.; Stal, O.; Olsson, H.; Dore, S.; Svanvik, J. Breast cancer expression of CD163, a macrophage scavenger receptor, is related to early distant recurrence and reduced patient survival. Int. J. Cancer 2008, 123, 780–786. [Google Scholar] [CrossRef]
- Garvin, S.; Oda, H.; Arnesson, L.G.; Lindstrom, A.; Shabo, I. Tumor cell expression of CD163 is associated to postoperative radiotherapy and poor prognosis in patients with breast cancer treated with breast-conserving surgery. J. Cancer Res. Clin. Oncol. 2018, 144, 1253–1263. [Google Scholar] [CrossRef] [Green Version]
- Hiyama, K.; Matsui, H.; Tamura, M.; Shimokawa, O.; Hiyama, M.; Kaneko, T.; Nagano, Y.; Hyodo, I.; Tanaka, J.; Miwa, Y.; et al. Cancer cells uptake porphyrins via heme carrier protein 1. J. Porphyr. Phthalocyanines 2013, 17, 36–43. [Google Scholar] [CrossRef]
- Song, H.; Li, Y.; Lee, J.; Schwartz, A.L.; Bu, G. Low-density lipoprotein receptor-related protein 1 promotes cancer cell migration and invasion by inducing the expression of matrix metalloproteinases 2 and 9. Cancer Res. 2009, 69, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Langlois, B.; Perrot, G.; Schneider, C.; Henriet, P.; Emonard, H.; Martiny, L.; Dedieu, S. LRP-1 promotes cancer cell invasion by supporting ERK and inhibiting JNK signaling pathways. PLoS ONE 2010, 5, e11584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydemir, T.B.; Cousins, R.J. The Multiple Faces of the Metal Transporter ZIP14 (SLC39A14). J. Nutr. 2018, 148, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Zhang, A.S.; Wortham, A.M.; Jue, S.; Knutson, M.D.; Enns, C.A. The Tumor Suppressor, P53, Decreases the Metal Transporter, ZIP14. Nutrients 2017, 9, 1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lok, C.N.; Ponka, P. Identification of a hypoxia response element in the transferrin receptor gene. J. Biol. Chem. 1999, 274, 24147–24152. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, K.A.; Yu, D.; Zeller, K.I.; Kim, J.W.; Racke, F.; Thomas-Tikhonenko, A.; Dang, C.V. Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol. Cell Biol. 2006, 26, 2373–2386. [Google Scholar] [CrossRef] [Green Version]
- Canesin, G.; Di Ruscio, A.; Li, M.; Ummarino, S.; Hedblom, A.; Choudhury, R.; Krzyzanowska, A.; Csizmadia, E.; Palominos, M.; Stiehm, A.; et al. Scavenging of Labile Heme by Hemopexin Is a Key Checkpoint in Cancer Growth and Metastases. Cell Rep. 2020, 32, 108181. [Google Scholar] [CrossRef]
- Shen, J.; Sheng, X.; Chang, Z.; Wu, Q.; Wang, S.; Xuan, Z.; Li, D.; Wu, Y.; Shang, Y.; Kong, X.; et al. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 2014, 7, 180–193. [Google Scholar] [CrossRef] [Green Version]
- Cowland, J.B.; Borregaard, N. Molecular characterization and pattern of tissue expression of the gene for neutrophil gelatinase-associated lipocalin from humans. Genomics 1997, 45, 17–23. [Google Scholar] [CrossRef]
- Tong, Z.; Wu, X.; Ovcharenko, D.; Zhu, J.; Chen, C.S.; Kehrer, J.P. Neutrophil gelatinase-associated lipocalin as a survival factor. Biochem. J. 2005, 391, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, C.A.; Yan, L.; Louis, G.; Yang, J.; Kutok, J.L.; Moses, M.A. The matrix metalloproteinase-9/neutrophil gelatinase-associated lipocalin complex plays a role in breast tumor growth and is present in the urine of breast cancer patients. Clin. Cancer Res. 2005, 11, 5390–5395. [Google Scholar] [CrossRef] [Green Version]
- Devireddy, L.R.; Gazin, C.; Zhu, X.; Green, M.R. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 2005, 123, 1293–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.A.M.; Richardson, K.L.; Kabir, T.D.; Trinder, D.; Ganss, R.; Leedman, P.J. Altered Iron Metabolism and Impact in Cancer Biology, Metastasis, and Immunology. Front. Oncol. 2020, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Chou, S.B.; Swidnicka-Siergiejko, A.K.; Badi, N.; Chavez-Tomar, M.; Lesinski, G.B.; Bekaii-Saab, T.; Farren, M.R.; Mace, T.A.; Schmidt, C.; Liu, Y.; et al. Lipocalin-2 Promotes Pancreatic Ductal Adenocarcinoma by Regulating Inflammation in the Tumor Microenvironment. Cancer Res. 2017, 77, 2647–2660. [Google Scholar] [CrossRef] [Green Version]
- Tong, Z.; Kunnumakkara, A.B.; Wang, H.; Matsuo, Y.; Diagaradjane, P.; Harikumar, K.B.; Ramachandran, V.; Sung, B.; Chakraborty, A.; Bresalier, R.S.; et al. Neutrophil gelatinase-associated lipocalin: A novel suppressor of invasion and angiogenesis in pancreatic cancer. Cancer Res. 2008, 68, 6100–6108. [Google Scholar] [CrossRef] [Green Version]
- Feng, M.; Feng, J.; Chen, W.; Wang, W.; Wu, X.; Zhang, J.; Xu, F.; Lai, M. Lipocalin2 suppresses metastasis of colorectal cancer by attenuating NF-kappaB-dependent activation of snail and epithelial mesenchymal transition. Mol. Cancer 2016, 15, 77. [Google Scholar] [CrossRef] [Green Version]
- Moreaux, J.; Kassambara, A.; Hose, D.; Klein, B. STEAP1 is overexpressed in cancers: A promising therapeutic target. Biochem. Biophys. Res. Commun. 2012, 429, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, T.G.; Bach, H.; Cossarizza, A.; Matsumoto, I. The STEAP protein family: Versatile oxidoreductases and targets for cancer immunotherapy with overlapping and distinct cellular functions. Biol. Cell 2012, 104, 641–657. [Google Scholar] [CrossRef]
- Patel, A.J.; Som, R. eComment. The evidence for stress ulcer prophylaxis in patients undergoing cardiac surgery. Interact. Cardiovasc. Thorac. Surg. 2012, 14, 628. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Li, Y.; Liu, B.; Zhang, S.; Wu, L.; Zhu, X.; Chen, Q. Expression of Ferritin Light Chain (FTL) Is Elevated in Glioblastoma, and FTL Silencing Inhibits Glioblastoma Cell Proliferation via the GADD45/JNK Pathway. PLoS ONE 2016, 11, e0149361. [Google Scholar] [CrossRef] [Green Version]
- Shpyleva, S.I.; Tryndyak, V.P.; Kovalchuk, O.; Starlard-Davenport, A.; Chekhun, V.F.; Beland, F.A.; Pogribny, I.P. Role of ferritin alterations in human breast cancer cells. Breast Cancer Res. Treat. 2011, 126, 63–71. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, L.; Han, Y.; Li, F.; Zheng, A.; Xu, Y.; Wang, F.; Xiao, B.; Chen, C.; Tao, Z. Ferritin: A potential serum marker for lymph node metastasis in head and neck squamous cell carcinoma. Oncol. Lett. 2019, 17, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.Q.; De Marchi, T.; Timmermans, A.M.; Beekhof, R.; Trapman-Jansen, A.M.; Foekens, R.; Look, M.P.; van Deurzen, C.H.; Span, P.N.; Sweep, F.C.; et al. Ferritin heavy chain in triple negative breast cancer: A favorable prognostic marker that relates to a cluster of differentiation 8 positive (CD8+) effector T-cell response. Mol. Cell Proteom. 2014, 13, 1814–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldi, A.; Lombardi, D.; Russo, P.; Palescandolo, E.; De Luca, A.; Santini, D.; Baldi, F.; Rossiello, L.; Dell’Anna, M.L.; Mastrofrancesco, A.; et al. Ferritin contributes to melanoma progression by modulating cell growth and sensitivity to oxidative stress. Clin. Cancer Res. 2005, 11, 3175–3183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aung, W.; Hasegawa, S.; Furukawa, T.; Saga, T. Potential role of ferritin heavy chain in oxidative stress and apoptosis in human mesothelial and mesothelioma cells: Implications for asbestos-induced oncogenesis. Carcinogenesis 2007, 28, 2047–2052. [Google Scholar] [CrossRef]
- Salatino, A.; Aversa, I.; Battaglia, A.M.; Sacco, A.; Di Vito, A.; Santamaria, G.; Chirillo, R.; Veltri, P.; Tradigo, G.; Di Cello, A.; et al. H-Ferritin Affects Cisplatin-Induced Cytotoxicity in Ovarian Cancer Cells through the Modulation of ROS. Oxid. Med. Cell Longev. 2019, 2019, 3461251. [Google Scholar] [CrossRef]
- Rockfield, S.; Flores, I.; Nanjundan, M. Expression and function of nuclear receptor coactivator 4 isoforms in transformed endometriotic and malignant ovarian cells. Oncotarget 2018, 9, 5344–5367. [Google Scholar] [CrossRef] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Emami, N.C.; Kachuri, L.; Meyers, T.J.; Das, R.; Hoffman, J.D.; Hoffmann, T.J.; Hu, D.; Shan, J.; Feng, F.Y.; Ziv, E.; et al. Association of imputed prostate cancer transcriptome with disease risk reveals novel mechanisms. Nat. Commun. 2019, 10, 3107. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Fang, C.J.; Ryan, J.C.; Niemi, E.C.; Lebron, J.A.; Bjorkman, P.J.; Arase, H.; Torti, F.M.; Torti, S.V.; Nakamura, M.C.; et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl. Acad. Sci. USA 2010, 107, 3505–3510. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Y.; Paragas, N.; Ned, R.M.; Qiu, A.; Viltard, M.; Leete, T.; Drexler, I.R.; Chen, X.; Sanna-Cherchi, S.; Mohammed, F.; et al. Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev. Cell 2009, 16, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.; Cheng, C.; Wu, Y.; Guo, L.; Kong, D.; Zhang, Z.; Wang, Y.; Zheng, E.; Liu, Y.; He, Y. Interactions of ferritin with scavenger receptor class A members. J. Biol. Chem. 2020, 295, 15727–15741. [Google Scholar] [CrossRef] [PubMed]
- You, K.; Su, F.; Liu, L.; Lv, X.; Zhang, J.; Zhang, Y.; Liu, B. SCARA5 plays a critical role in the progression and metastasis of breast cancer by inactivating the ERK1/2, STAT3, and AKT signaling pathways. Mol. Cell Biochem. 2017, 435, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Ulker, D.; Ersoy, Y.E.; Gucin, Z.; Muslumanoglu, M.; Buyru, N. Downregulation of SCARA5 may contribute to breast cancer via promoter hypermethylation. Gene 2018, 673, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, Y.; Wang, Y.; Lian, S.; Lynch, J.; Nagai, S.; Fanshawe, B.; Kandilci, A.; Janke, L.J.; Neale, G.; Fan, Y.; et al. Upregulated heme biosynthesis, an exploitable vulnerability in MYCN-driven leukemogenesis. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Hooda, J.; Cadinu, D.; Alam, M.M.; Shah, A.; Cao, T.M.; Sullivan, L.A.; Brekken, R.; Zhang, L. Enhanced heme function and mitochondrial respiration promote the progression of lung cancer cells. PLoS ONE 2013, 8, e63402. [Google Scholar] [CrossRef] [Green Version]
- Sohoni, S.; Ghosh, P.; Wang, T.; Kalainayakan, S.P.; Vidal, C.; Dey, S.; Konduri, P.C.; Zhang, L. Elevated Heme Synthesis and Uptake Underpin Intensified Oxidative Metabolism and Tumorigenic Functions in Non-Small Cell Lung Cancer Cells. Cancer Res. 2019, 79, 2511–2525. [Google Scholar] [CrossRef] [Green Version]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [Green Version]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, J.H.; Kim, E.H.; Na, H.K.; Cha, Y.N.; Chung, J.H.; Surh, Y.J. 15-Deoxy-Delta12,14-prostaglandin J2 upregulates the expression of heme oxygenase-1 and subsequently matrix metalloproteinase-1 in human breast cancer cells: Possible roles of iron and ROS. Carcinogenesis 2009, 30, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Fillebeen, C.; Wang, J.; Pantopoulos, K. Overexpression of iron regulatory protein 1 suppresses growth of tumor xenografts. Carcinogenesis 2007, 28, 785–791. [Google Scholar] [CrossRef] [Green Version]
- Horniblow, R.D.; Bedford, M.; Hollingworth, R.; Evans, S.; Sutton, E.; Lal, N.; Beggs, A.; Iqbal, T.H.; Tselepis, C. BRAF mutations are associated with increased iron regulatory protein-2 expression in colorectal tumorigenesis. Cancer Sci. 2017, 108, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Manz, D.H.; Torti, S.V.; Torti, F.M. Iron-responsive element-binding protein 2 plays an essential role in regulating prostate cancer cell growth. Oncotarget 2017, 8, 82231–82243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Deng, Z.; Hatcher, H.; Miller, L.D.; Di, X.; Tesfay, L.; Sui, G.; D’Agostino, R.B., Jr.; Torti, F.M.; Torti, S.V. IRP2 regulates breast tumor growth. Cancer Res. 2014, 74, 497–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Rolfs, A.; Kvietikova, I.; Gassmann, M.; Wenger, R.H. Oxygen-regulated transferrin expression is mediated by hypoxia-inducible factor-1. J. Biol. Chem. 1997, 272, 20055–20062. [Google Scholar] [CrossRef] [Green Version]
- Xue, D.; Zhou, C.X.; Shi, Y.B.; Lu, H.; He, X.Z. Decreased expression of ferroportin in prostate cancer. Oncol. Lett. 2015, 10, 913–916. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.G.; Roberts, K.; Brookes, M.J.; Joy, H.; Martin, A.; Ismail, T.; Spychal, R.; Iqbal, T.; Tselepis, C. Increased hepcidin expression in colorectal carcinogenesis. World J. Gastroenterol. 2008, 14, 1339–1345. [Google Scholar] [CrossRef]
- Toshiyama, R.; Konno, M.; Eguchi, H.; Asai, A.; Noda, T.; Koseki, J.; Asukai, K.; Ohashi, T.; Matsushita, K.; Iwagami, Y.; et al. Association of iron metabolic enzyme hepcidin expression levels with the prognosis of patients with pancreatic cancer. Oncol. Lett. 2018, 15, 8125–8133. [Google Scholar] [CrossRef]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R., Jr.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2010, 2, 43ra56. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhang, S.; Chen, Y.; Zhang, D.; Yuan, L.; Cong, H.; Liu, S. An important role of the hepcidin-ferroportin signaling in affecting tumor growth and metastasis. Acta Biochim. Biophys. Sin. (Shanghai) 2015, 47, 703–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Wang, H.; Xia, J.; Yang, Y.; Jin, Z.; Xu, H.; Shi, J.; De Domenico, I.; Tricot, G.; Zhan, F. Decreased ferroportin promotes myeloma cell growth and osteoclast differentiation. Cancer Res. 2015, 75, 2211–2221. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Li, R.; Cheng, G.; Li, Z.; Zhang, Z.; Li, J.; Zhang, G.; Bi, C.; Hu, C.; Yang, L.; et al. Role of hepcidin and iron metabolism in the onset of prostate cancer. Oncol. Lett. 2018, 15, 9953–9958. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.; Manz, D.H.; Torti, S.V.; Torti, F.M. Effects of Ferroportin-Mediated Iron Depletion in Cells Representative of Different Histological Subtypes of Prostate Cancer. Antioxid. Redox Signal. 2019, 30, 1043–1061. [Google Scholar] [CrossRef]
- Brookes, M.J.; Hughes, S.; Turner, F.E.; Reynolds, G.; Sharma, N.; Ismail, T.; Berx, G.; McKie, A.T.; Hotchin, N.; Anderson, G.J.; et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut 2006, 55, 1449–1460. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Gregolin, C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim. Biophys. Acta 1985, 839, 62–70. [Google Scholar] [CrossRef]
- Hinman, A.; Holst, C.R.; Latham, J.C.; Bruegger, J.J.; Ulas, G.; McCusker, K.P.; Amagata, A.; Davis, D.; Hoff, K.G.; Kahn-Kirby, A.H.; et al. Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS ONE 2018, 13, e0201369. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016, 7, e2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Danhier, P.; Banski, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 556–572. [Google Scholar] [CrossRef]
- Nairz, M.; Haschka, D.; Demetz, E.; Weiss, G. Iron at the interface of immunity and infection. Front. Pharm. 2014, 5, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, O.; Porto, G.; Rema, A.; Faria, F.; Cruz Paula, A.; Gomez-Lazaro, M.; Silva, P.; Martins da Silva, B.; Lopes, C. Local iron homeostasis in the breast ductal carcinoma microenvironment. Bmc Cancer 2016, 16, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, M.; Columbano, A.; Ammarah, U.; Mazzone, M.; Menga, A. Understanding Metal Dynamics between Cancer Cells and Macrophages: Competition or Synergism? Front. Oncol. 2020, 10, 646. [Google Scholar] [CrossRef] [PubMed]
- Costa da Silva, M.; Breckwoldt, M.O.; Vinchi, F.; Correia, M.P.; Stojanovic, A.; Thielmann, C.M.; Meister, M.; Muley, T.; Warth, A.; Platten, M.; et al. Iron Induces Anti-tumor Activity in Tumor-Associated Macrophages. Front. Immunol. 2017, 8, 1479. [Google Scholar] [CrossRef] [Green Version]
- Thielmann, C.M.; Costa da Silva, M.; Muley, T.; Meister, M.; Herpel, E.; Muckenthaler, M.U. Iron accumulation in tumor-associated macrophages marks an improved overall survival in patients with lung adenocarcinoma. Sci. Rep. 2019, 9, 11326. [Google Scholar] [CrossRef] [Green Version]
- Cronin, S.J.F.; Woolf, C.J.; Weiss, G.; Penninger, J.M. The Role of Iron Regulation in Immunometabolism and Immune-Related Disease. Front. Mol. Biosci. 2019, 6, 116. [Google Scholar] [CrossRef] [Green Version]
- Mertens, C.; Mora, J.; Oren, B.; Grein, S.; Winslow, S.; Scholich, K.; Weigert, A.; Malmstrom, P.; Forsare, C.; Ferno, M.; et al. Macrophage-derived lipocalin-2 transports iron in the tumor microenvironment. Oncoimmunology 2018, 7, e1408751. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Oren, B.; Mora, J.; Mertens, C.; Dziumbla, S.; Popp, R.; Weigert, A.; Grossmann, N.; Fleming, I.; Brune, B. Lipocalin 2 from macrophages stimulated by tumor cell-derived sphingosine 1-phosphate promotes lymphangiogenesis and tumor metastasis. Sci. Signal. 2016, 9, ra64. [Google Scholar] [CrossRef]
- Kir, D.; Saluja, M.; Modi, S.; Venkatachalam, A.; Schnettler, E.; Roy, S.; Ramakrishnan, S. Cell-permeable iron inhibits vascular endothelial growth factor receptor-2 signaling and tumor angiogenesis. Oncotarget 2016, 7, 65348–65363. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Du, Y.; Lok, J.; Lo, E.H.; Xing, C. Lipocalin-2 enhances angiogenesis in rat brain endothelial cells via reactive oxygen species and iron-dependent mechanisms. J. Neurochem. 2015, 132, 622–628. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.W.; Byzova, T.V. Oxidative stress in angiogenesis and vascular disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchette-Farra, N.; Kita, D.; Konstorum, A.; Tesfay, L.; Lemler, D.; Hegde, P.; Claffey, K.P.; Torti, F.M.; Torti, S.V. Contribution of three-dimensional architecture and tumor-associated fibroblasts to hepcidin regulation in breast cancer. Oncogene 2018, 37, 4013–4032. [Google Scholar] [CrossRef] [PubMed]
- Akatsuka, S.; Yamashita, Y.; Ohara, H.; Liu, Y.T.; Izumiya, M.; Abe, K.; Ochiai, M.; Jiang, L.; Nagai, H.; Okazaki, Y.; et al. Fenton reaction induced cancer in wild type rats recapitulates genomic alterations observed in human cancer. PLoS ONE 2012, 7, e43403. [Google Scholar] [CrossRef] [PubMed]
- Horniblow, R.D.; Lai, S.; Beggs, A.; Iqbal, T.H.; Tselepis, C. Epigenetic dna methylation modifications following chronic iron exposure to colonocytes in vitro. Gut 2017, 66, A175. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Talbott, S.J.; Rojanasakul, Y.; Nimmannit, U.; Pongrakhananon, V.; Wang, L.; Chanvorachote, P. Regulation of lung cancer cell migration and invasion by reactive oxygen species and caveolin-1. J. Biol. Chem. 2010, 285, 38832–38840. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Hittelman, W.; Lu, T.; Ji, P.; Arlinghaus, R.; Shmulevich, I.; Hamilton, S.R.; Zhang, W. NGAL decreases E-cadherin-mediated cell-cell adhesion and increases cell motility and invasion through Rac1 in colon carcinoma cells. Lab. Invest. 2009, 89, 531–548. [Google Scholar] [CrossRef] [Green Version]
- Pogribny, I.P.; Tryndyak, V.P.; Pogribna, M.; Shpyleva, S.; Surratt, G.; Gamboa da Costa, G.; Beland, F.A. Modulation of intracellular iron metabolism by iron chelation affects chromatin remodeling proteins and corresponding epigenetic modifications in breast cancer cells and increases their sensitivity to chemotherapeutic agents. Int. J. Oncol. 2013, 42, 1822–1832. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.X.; Richardson, D.R. The effect of potent iron chelators on the regulation of p53: Examination of the expression, localization and DNA-binding activity of p53 and the transactivation of WAF1. Carcinogenesis 2003, 24, 1601–1614. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Christova, T.; Perusini, S.; Alizadeh, S.; Bao, R.Y.; Miller, B.W.; Hurren, R.; Jitkova, Y.; Gronda, M.; Isaac, M.; et al. Wnt inhibitor screen reveals iron dependence of beta-catenin signaling in cancers. Cancer Res. 2011, 71, 7628–7639. [Google Scholar] [CrossRef] [Green Version]
- Raggi, C.; Gammella, E.; Correnti, M.; Buratti, P.; Forti, E.; Andersen, J.B.; Alpini, G.; Glaser, S.; Alvaro, D.; Invernizzi, P.; et al. Dysregulation of Iron Metabolism in Cholangiocarcinoma Stem-like Cells. Sci. Rep. 2017, 7, 17667. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wu, Y.; Yan, Q.; Ma, F.; Shi, X.; Zhao, Y.; Peng, Y.; Wang, J.; Jiang, B. Deferoxamine enhances cell migration and invasion through promotion of HIF-1alpha expression and epithelial-mesenchymal transition in colorectal cancer. Oncol. Rep. 2014, 31, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.R.; Bernabeu, E.; Rodriguez, J.A.; Patel, S.; Kozman, M.; Chiappetta, D.A.; Holler, E.; Ljubimova, J.Y.; Helguera, G.; Penichet, M.L. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim. Biophys. Acta 2012, 1820, 291–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.L.; Lee, D.H.; Na, Y.J.; Kim, B.R.; Jeong, Y.A.; Lee, S.I.; Kang, S.; Joung, S.Y.; Lee, S.Y.; Oh, S.C.; et al. Iron chelator-induced apoptosis via the ER stress pathway in gastric cancer cells. Tumour Biol. 2016, 37, 9709–9719. [Google Scholar] [CrossRef] [PubMed]
- Ford, S.J.; Obeidy, P.; Lovejoy, D.B.; Bedford, M.; Nichols, L.; Chadwick, C.; Tucker, O.; Lui, G.Y.; Kalinowski, D.S.; Jansson, P.J.; et al. Deferasirox (ICL670A) effectively inhibits oesophageal cancer growth in vitro and in vivo. Br. J. Pharm. 2013, 168, 1316–1328. [Google Scholar] [CrossRef] [Green Version]
- Harima, H.; Kaino, S.; Takami, T.; Shinoda, S.; Matsumoto, T.; Fujisawa, K.; Yamamoto, N.; Yamasaki, T.; Sakaida, I. Deferasirox, a novel oral iron chelator, shows antiproliferative activity against pancreatic cancer in vitro and in vivo. BMC Cancer 2016, 16, 702. [Google Scholar] [CrossRef] [Green Version]
- Saeki, I.; Yamamoto, N.; Yamasaki, T.; Takami, T.; Maeda, M.; Fujisawa, K.; Iwamoto, T.; Matsumoto, T.; Hidaka, I.; Ishikawa, T.; et al. Effects of an oral iron chelator, deferasirox, on advanced hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 8967–8977. [Google Scholar] [CrossRef]
- Tury, S.; Assayag, F.; Bonin, F.; Chateau-Joubert, S.; Servely, J.L.; Vacher, S.; Becette, V.; Caly, M.; Rapinat, A.; Gentien, D.; et al. The iron chelator deferasirox synergises with chemotherapy to treat triple-negative breast cancers. J. Pathol. 2018, 246, 103–114. [Google Scholar] [CrossRef]
- Cragg, L.; Hebbel, R.P.; Miller, W.; Solovey, A.; Selby, S.; Enright, H. The iron chelator L1 potentiates oxidative DNA damage in iron-loaded liver cells. Blood 1998, 92, 632–638. [Google Scholar] [CrossRef]
- Leftin, A.; Zhao, H.; Turkekul, M.; de Stanchina, E.; Manova, K.; Koutcher, J.A. Iron deposition is associated with differential macrophage infiltration and therapeutic response to iron chelation in prostate cancer. Sci. Rep. 2017, 7, 11632. [Google Scholar] [CrossRef]
- Eberhard, Y.; McDermott, S.P.; Wang, X.; Gronda, M.; Venugopal, A.; Wood, T.E.; Hurren, R.; Datti, A.; Batey, R.A.; Wrana, J.; et al. Chelation of intracellular iron with the antifungal agent ciclopirox olamine induces cell death in leukemia and myeloma cells. Blood 2009, 114, 3064–3073. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Shen, T.; Luo, Y.; Liu, L.; Chen, W.; Xu, B.; Han, X.; Pang, J.; Rivera, C.A.; Huang, S. The antitumor activity of the fungicide ciclopirox. Int. J. Cancer 2010, 127, 2467–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Milasta, S.; Hu, D.; AlTahan, A.M.; Interiano, R.B.; Zhou, J.; Davidson, J.; Low, J.; Lin, W.; Bao, J.; et al. Targeting Histone Demethylases in MYC-Driven Neuroblastomas with Ciclopirox. Cancer Res. 2017, 77, 4626–4638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihailidou, C.; Papakotoulas, P.; Papavassiliou, A.G.; Karamouzis, M.V. Superior efficacy of the antifungal agent ciclopirox olamine over gemcitabine in pancreatic cancer models. Oncotarget 2018, 9, 10360–10374. [Google Scholar] [CrossRef] [PubMed]
- Minden, M.D.; Hogge, D.E.; Weir, S.; Kasper, J.; Patton, L.; Jitkova, Y.; Gronda, M.; Hurren, R.; Rajewski, L.; Schorno, K.; et al. Clinical proof of concept trial of oral ciclopirox olamine in patients with relapsed/refractory hematologic malignancy. Blood 2012, 120. [Google Scholar] [CrossRef]
- Weir, S.J.; Wood, R.; Schorno, K.; Brinker, A.E.; Ramamoorthy, P.; Heppert, K.; Rajewski, L.; Tanol, M.; Ham, T.; McKenna, M.J.; et al. Preclinical Pharmacokinetics of Fosciclopirox, a Novel Treatment of Urothelial Cancers, in Rats and Dogs. J. Pharm. Exp. 2019, 370, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Samuni, A.M.; Krishna, M.C.; DeGraff, W.; Russo, A.; Planalp, R.P.; Brechbiel, M.W.; Mitchell, J.B. Mechanisms underlying the cytotoxic effects of Tachpyr--a novel metal chelator. Biochim. Biophys. Acta 2002, 1571, 211–218. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M.; Whitman, S.P.; Brechbiel, M.W.; Park, G.; Planalp, R.P. Tumor cell cytotoxicity of a novel metal chelator. Blood 1998, 92, 1384–1389. [Google Scholar] [CrossRef]
- Kunos, C.A.; Andrews, S.J.; Moore, K.N.; Chon, H.S.; Ivy, S.P. Randomized Phase II Trial of Triapine-Cisplatin-Radiotherapy for Locally Advanced Stage Uterine Cervix or Vaginal Cancers. Front. Oncol. 2019, 9, 1067. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Simplified scheme of cellular iron metabolism in cancer cells. Red: upregulation reported in cancer. Blue: downregulation reported in cancer. Cancer cells commonly show increased uptake of iron by transferrin receptor 1 (TfR1). After binding of transferrin-bound iron (Tf-Fe or TBI) on its receptor, the complex is carried to the intracellular space by endocytosis. Coordination of six-transmembrane epithelial antigen of prostate (STEAP) and divalent metal transporter (DMT1) releases free iron in the cytosol. Circulating free, non-transferrin-bound iron (NTBI) can also be imported by ZIP14. Hepcidin (HEPC) secretion by the liver downregulates ferroportin (FPN) levels, consequently causing iron accumulation in the intracellular space. Cancer cells also deploy other iron-acquisition methods, e.g., via heme importers, including HCP1, CD91, and CD193. Heme-responsive gene 1 (HRG1) allows the cytosolic release of heme imported by endocytosis. Moreover, increased ferritinophagy, and hence the degradation of ferritin–iron (FT) complexes by NCOA4, further increases bioavailable iron. Altogether, these mechanisms lead to elevated labile iron pool (LIP). After incorporation in enzymes’ prosthetic moieties or alone as a cofactor by the mitochondria, iron can fuel the TCA cycle and the electron transport chain or be exported to the cytosol and participate in translational process as well as DNA replication or repair.
Figure 1.
Simplified scheme of cellular iron metabolism in cancer cells. Red: upregulation reported in cancer. Blue: downregulation reported in cancer. Cancer cells commonly show increased uptake of iron by transferrin receptor 1 (TfR1). After binding of transferrin-bound iron (Tf-Fe or TBI) on its receptor, the complex is carried to the intracellular space by endocytosis. Coordination of six-transmembrane epithelial antigen of prostate (STEAP) and divalent metal transporter (DMT1) releases free iron in the cytosol. Circulating free, non-transferrin-bound iron (NTBI) can also be imported by ZIP14. Hepcidin (HEPC) secretion by the liver downregulates ferroportin (FPN) levels, consequently causing iron accumulation in the intracellular space. Cancer cells also deploy other iron-acquisition methods, e.g., via heme importers, including HCP1, CD91, and CD193. Heme-responsive gene 1 (HRG1) allows the cytosolic release of heme imported by endocytosis. Moreover, increased ferritinophagy, and hence the degradation of ferritin–iron (FT) complexes by NCOA4, further increases bioavailable iron. Altogether, these mechanisms lead to elevated labile iron pool (LIP). After incorporation in enzymes’ prosthetic moieties or alone as a cofactor by the mitochondria, iron can fuel the TCA cycle and the electron transport chain or be exported to the cytosol and participate in translational process as well as DNA replication or repair.

Figure 2.
Crosstalk between oncogenes, signaling pathways, and iron regulators. Red: upregulation reported in cancer. Blue: downregulation reported in cancer. Several oncogenic transcription factors, signaling pathways, and proto-oncogenes directly regulate key players of iron metabolism. Notably, TfR1 upregulation can be caused by activation of HIF1 and MYC overexpression of IRP2, or loss of wild-type p53 [51,155,184,185]. Similarly, ferritin (FT) decrease can result from activation of MYC, IRP2, or p53 mutation [51,155,185]. Such a vicious cycle is sustained as increased LIP promotes CDK, JAK/STAT, PI3K, MAPK/ERK pathways [172], whereas heme can further enhance ERK and MYC and inhibits p53 [181,186,187].
Figure 2.
Crosstalk between oncogenes, signaling pathways, and iron regulators. Red: upregulation reported in cancer. Blue: downregulation reported in cancer. Several oncogenic transcription factors, signaling pathways, and proto-oncogenes directly regulate key players of iron metabolism. Notably, TfR1 upregulation can be caused by activation of HIF1 and MYC overexpression of IRP2, or loss of wild-type p53 [51,155,184,185]. Similarly, ferritin (FT) decrease can result from activation of MYC, IRP2, or p53 mutation [51,155,185]. Such a vicious cycle is sustained as increased LIP promotes CDK, JAK/STAT, PI3K, MAPK/ERK pathways [172], whereas heme can further enhance ERK and MYC and inhibits p53 [181,186,187].

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hsu, M.Y.; Mina, E.; Roetto, A.; Porporato, P.E. Iron: An Essential Element of Cancer Metabolism. Cells 2020, 9, 2591. https://doi.org/10.3390/cells9122591
AMA Style
Hsu MY, Mina E, Roetto A, Porporato PE. Iron: An Essential Element of Cancer Metabolism. Cells. 2020; 9(12):2591. https://doi.org/10.3390/cells9122591
Chicago/Turabian StyleHsu, Myriam Y., Erica Mina, Antonella Roetto, and Paolo E. Porporato. 2020. "Iron: An Essential Element of Cancer Metabolism" Cells 9, no. 12: 2591. https://doi.org/10.3390/cells9122591
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.