Human Pluripotent Stem Cells-Based Therapies for Neurodegenerative Diseases: Current Status and Challenges


Abstract
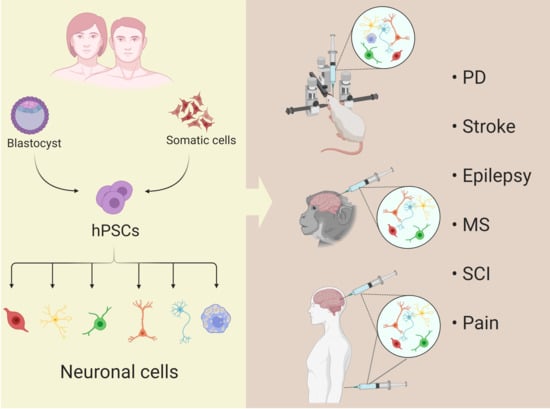
:
1. Introduction
2. Main Text
2.1. Parkinson’s Disease
2.2. Stroke
2.3. Epilepsy
2.4. Learning & Memory Disorders/Dementia (Alzheimer’s Disease)
2.5. Multiple Sclerosis
2.6. Spinal Cord Injury
2.7. Neuropathic Pain
3. Current Challenges
3.1. Transplantation of Stem Cells
3.1.1. Pluripotency and Cancer
3.1.2. Methods to Prevent Tumor Formation
3.1.3. Oncogenic Risks Associated with Reprogramming
3.1.4. The Epigenetic Landscape of Induced Pluripotent Stem Cells
3.1.5. Immune Rejection
3.2. Regeneration of the Central Nervous System
3.2.1. CNS Microenvironment
3.2.2. Glial Scarring
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schwartz, S.D.; Hubschman, J.-P.; Heilwell, G.; Franco-Cardenas, V.; Pan, C.K.; Ostrick, R.M.; Mickunas, E.; Gay, R.; Klimanskaya, I.; Lanza, R. Embryonic stem cell trials for macular degeneration: A preliminary report. Lancet 2012, 379, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.D.; Regillo, C.D.; Lam, B.L.; Eliott, D.; Rosenfeld, P.J.; Gregori, N.Z.; Hubschman, J.-P.; Davis, J.L.; Heilwell, G.; Spirn, M.; et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet 2015, 385, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Song, W.K.; Park, K.-M.; Kim, H.-J.; Lee, J.H.; Choi, J.; Chong, S.Y.; Shim, S.H.; Del Priore, L.V.; Lanza, R. Treatment of Macular Degeneration Using Embryonic Stem Cell-Derived Retinal Pigment Epithelium: Preliminary Results in Asian Patients. Stem Cell Rep. 2015, 4, 860–872. [Google Scholar] [CrossRef]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell–Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Boldt, H.C.; Bressler, S.B.; Fine, S.L.; Bressler, N.M. Age-related macular degeneration. Curr. Opin. Ophthalmol. 1990, 1, 247–257. [Google Scholar] [CrossRef]
- Lindvall, O.; Brundin, P.; Widner, H.; Rehncrona, S.; Gustavii, B.; Frackowiak, R.S.; Leenders, K.L.; Sawle, G.V.; Rothwell, J.C.; Marsden, C.D.; et al. Grafts of fetal dopamine neurons survive and improve motor function in Parkinson’s disease. Science 1990, 247, 574–577. [Google Scholar] [CrossRef]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.-Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef]
- Tsuji, O.; Sugai, K.; Yamaguchi, R.; Tashiro, S.; Nagoshi, N.; Kohyama, J.; Iida, T.; Ohkubo, T.; Itakura, G.; Isoda, M.; et al. Concise Review: Laying the Groundwork for a First-In-Human Study of an Induced Pluripotent Stem Cell-Based Intervention for Spinal Cord Injury. Stem Cells 2019, 37, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Merkle, F.T.; Ghosh, S.; Kamitaki, N.; Mitchell, J.; Avior, Y.; Mello, C.; Kashin, S.; Mekhoubad, S.; Ilic, D.; Charlton, M.; et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nat. Cell Biol. 2017, 545, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- Poewe, W. Treatments for Parkinson disease--past achievements and current clinical needs. Neurology 2009, 72, 65–73. [Google Scholar] [CrossRef]
- Figge, D.A.; Jaunarajs, K.L.E.; Standaert, D.G. Dynamic DNA Methylation Regulates Levodopa-Induced Dyskinesia. J. Neurosci. 2016, 36, 6514–6524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, N.S.; Cleren, C.; Singh, S.K.; Yang, L.; Beal, M.F.; Goldman, S.A. Functional engraftment of human ES cell–derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat. Med. 2006, 12, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Doi, D.; Morizane, A.; Kikuchi, T.; Onoe, H.; Hayashi, T.; Kawasaki, T.; Motono, M.; Sasai, Y.; Saiki, H.; Gomi, M.; et al. Prolonged Maturation Culture Favors a Reduction in the Tumorigenicity and the Dopaminergic Function of Human ESC-Derived Neural Cells in a Primate Model of Parkinson’s Disease. Stem Cells 2012, 30, 935–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriks, S.; Shim, J.-W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nat. Cell Biol. 2011, 480, 547–551. [Google Scholar] [CrossRef]
- Kirkeby, A.; Grealish, S.; Wolf, D.A.; Nelander, J.; Wood, J.; Lundblad, M.; Lindvall, O.; Parmar, M. Generation of Regionally Specified Neural Progenitors and Functional Neurons from Human Embryonic Stem Cells under Defined Conditions. Cell Rep. 2012, 1, 703–714. [Google Scholar] [CrossRef] [Green Version]
- Grealish, S.; Diguet, E.; Kirkeby, A.; Mattsson, B.; Heuer, A.; Bramoulle, Y.; Van Camp, N.; Perrier, A.L.; Hantraye, P.; Björklund, A.; et al. Human ESC-Derived Dopamine Neurons Show Similar Preclinical Efficacy and Potency to Fetal Neurons when Grafted in a Rat Model of Parkinson’s Disease. Cell Stem Cell 2014, 15, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Gantner, C.W.; De Luzy, I.R.; Kauhausen, J.A.; Moriarty, N.; Niclis, J.C.; Bye, C.R.; Penna, V.; Hunt, C.P.; Ermine, C.M.; Pouton, C.W.; et al. Viral Delivery of GDNF Promotes Functional Integration of Human Stem Cell Grafts in Parkinson’s Disease. Cell Stem Cell 2020, 26, 511–526.e5. [Google Scholar] [CrossRef]
- Hargus, G.; Cooper, O.; Deleidi, M.; Levy, A.; Lee, K.N.; Marlow, E.; Yow, A.; Soldner, F.; Hockemeyer, D.; Hallett, P.J.; et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc. Natl. Acad. Sci. USA 2010, 107, 15921–15926. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, T.; Morizane, A.; Doi, D.; Okita, K.; Nakagawa, M.; Yamakado, H.; Inoue, H.; Takahashi, R.; Takahashi, J. Idiopathic Parkinson’s disease patient-derived induced pluripotent stem cells function as midbrain dopaminergic neurons in rodent brains. J. Neurosci. Res. 2017, 95, 1829–1837. [Google Scholar] [CrossRef]
- Rhee, Y.-H.; Ko, J.-Y.; Chang, M.-Y.; Yi, S.-H.; Kim, D.; Kim, C.-H.; Shim, J.-W.; Jo, A.-Y.; Kim, B.-W.; Lee, H.; et al. Protein-based human iPS cells efficiently generate functional dopamine neurons and can treat a rat model of Parkinson disease. J. Clin. Investig. 2011, 121, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Wang, W.; Chen, B.; Chen, C.; Li, S.; Lu, X.; Duan, J.; Zhang, Y.; Zhang, Y.A.; Guo, W.; et al. Human induced pluripotent stem cell–derived neurons improve motor asymmetry in a 6-hydroxydopamine–induced rat model of Parkinson’s disease. Cytotherapy 2015, 17, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Morizane, A.; Doi, D.; Onoe, H.; Hayashi, T.; Kawasaki, T.; Saiki, H.; Miyamoto, S.; Takahashi, J. Survival of Human Induced Pluripotent Stem Cell–Derived Midbrain Dopaminergic Neurons in the Brain of a Primate Model of Parkinson’s Disease. J. Park. Dis. 2011, 1, 395–412. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, T.; Morizane, A.; Doi, D.; Magotani, H.; Onoe, H.; Hayashi, T.; Mizuma, H.; Takara, S.; Takahashi, R.; Inoue, H.; et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nat. Cell Biol. 2017, 548, 592–596. [Google Scholar] [CrossRef]
- Oki, K.; Tatarishvili, J.; Woods, J.; Koch, P.; Wattananit, S.; Mine, Y.; Monni, E.; Tornero, D.; Ahlenius, H.; Ladewig, J.; et al. Human-Induced Pluripotent Stem Cells form Functional Neurons and Improve Recovery After Grafting in Stroke-Damaged Brain. Stem Cells 2012, 30, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Tornero, D.; Tsupykov, O.; Granmo, M.; Rodriguez, C.; Grønning-Hansen, M.; Thelin, J.; Smozhanik, E.; Laterza, C.; Wattananit, S.; Ge, R.; et al. Synaptic inputs from stroke-injured brain to grafted human stem cell-derived neurons activated by sensory stimuli. Brain 2017, 140, 692–706. [Google Scholar] [CrossRef] [Green Version]
- Tornero, D.; Wattananit, S.; Madsen, M.G.; Koch, P.; Wood, J.; Tatarishvili, J.; Mine, Y.; Ge, R.; Monni, E.; Devaraju, K.; et al. Human induced pluripotent stem cell-derived cortical neurons integrate in stroke-injured cortex and improve functional recovery. Brain 2013, 136, 3561–3577. [Google Scholar] [CrossRef] [Green Version]
- Palma-Tortosa, S.; Tornero, D.; Hansen, M.G.; Monni, E.; Hajy, M.; Kartsivadze, S.; Aktay, S.; Tsupykov, O.; Parmar, M.; Deisseroth, K.; et al. Activity in grafted human iPS cell–derived cortical neurons integrated in stroke-injured rat brain regulates motor behavior. Proc. Natl. Acad. Sci. USA 2020, 117, 9094–9100. [Google Scholar] [CrossRef]
- Gomi, M.; Takagi, Y.; Morizane, A.; Doi, D.; Nishimura, M.; Miyamoto, S.; Takahashi, J. Functional recovery of the murine brain ischemia model using human induced pluripotent stem cell-derived telencephalic progenitors. Brain Res. 2012, 1459, 52–60. [Google Scholar] [CrossRef]
- Mohamad, O.; Drury-Stewart, D.; Song, M.; Faulkner, B.; Chen, D.; Yu, S.P.; Wei, L. Vector-Free and Transgene-Free Human iPS Cells Differentiate into Functional Neurons and Enhance Functional Recovery after Ischemic Stroke in Mice. PLoS ONE 2013, 8, e64160. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.-J.; Lee, N.; Park, I.-H.; Choi, C.; Jeon, I.; Kwon, J.; Oh, S.-H.; Shin, D.A.; Tae, J.; Lee, N.R.; et al. Therapeutic Potential of Human Induced Pluripotent Stem Cells in Experimental Stroke. Cell Transplant. 2013, 22, 1427–1440. [Google Scholar] [CrossRef] [PubMed]
- Hermanto, Y.; Sunohara, T.; Faried, A.; Takagi, Y.; Takahashi, J.; Maki, T.; Miyamoto, S. Transplantation of feeder-free human induced pluripotent stem cell-derived cortical neuron progenitors in adult male Wistar rats with focal brain ischemia. J. Neurosci. Res. 2017, 96, 863–874. [Google Scholar] [CrossRef]
- Baker, E.W.; Platt, S.R.; Lau, V.W.; Grace, H.E.; Holmes, S.P.; Wang, L.; Duberstein, K.J.; Howerth, E.W.; Kinder, H.A.; Stice, S.L.; et al. Induced Pluripotent Stem Cell-Derived Neural Stem Cell Therapy Enhances Recovery in an Ischemic Stroke Pig Model. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, M.; Cho, J.-H.; Leung, A.; Savvidis, G.; Ahn, S.; Moon, M.; Lee, P.K.; Han, J.J.; Azimi, N.; Kim, K.-S.; et al. hPSC-Derived Maturing GABAergic Interneurons Ameliorate Seizures and Abnormal Behavior in Epileptic Mice. Cell Stem Cell 2014, 15, 559–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, N.C.; Van Zandt, M.A.; Shrestha, S.; Lawrence, D.B.; Gupta, J.; Chen, C.Y.; Harrsch, F.A.; Boyi, T.; Dundes, C.E.; Aaron, G.; et al. Pluripotent stem cell-derived interneuron progenitors mature and restore memory deficits but do not suppress seizures in the epileptic mouse brain. Stem Cell Res. 2018, 33, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Shimizu, J.; Takai, K.; Arimitsu, N.; Saito, A.; Kono, T.; Umehara, T.; Ueda, Y.; Wakisaka, S.; Suzuki, T.; et al. Restoration of spatial memory dysfunction of human APP transgenic mice by transplantation of neuronal precursors derived from human iPS cells. Neurosci. Lett. 2013, 557, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Li, Y.; Zhang, T.; Jiang, M.; Qian, Y.; Zhang, M.; Sheng, N.; Feng, S.; Tang, K.; Yu, X.; et al. ESC-Derived Basal Forebrain Cholinergic Neurons Ameliorate the Cognitive Symptoms Associated with Alzheimer’s Disease in Mouse Models. Stem Cell Rep. 2015, 5, 776–790. [Google Scholar] [CrossRef] [Green Version]
- Takamatsu, K.; Ikeda, T.; Haruta, M.; Matsumura, K.; Ogi, Y.; Nakagata, N.; Uchino, M.; Ando, Y.; Nishimura, Y.; Senju, S. Degradation of amyloid beta by human induced pluripotent stem cell-derived macrophages expressing Neprilysin-2. Stem Cell Res. 2014, 13, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Bates, J.; Li, X.; Schanz, S.; Chandler-Militello, D.; Levine, C.; Maherali, N.; Studer, L.; Hochedlinger, K.; Windrem, M.; et al. Human iPSC-Derived Oligodendrocyte Progenitor Cells Can Myelinate and Rescue a Mouse Model of Congenital Hypomyelination. Cell Stem Cell 2013, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Douvaras, P.; Wang, J.; Zimmer, M.; Hanchuk, S.; O’Bara, M.A.; Sadiq, S.; Sim, F.J.; Goldman, J.; Fossati, V. Efficient Generation of Myelinating Oligodendrocytes from Primary Progressive Multiple Sclerosis Patients by Induced Pluripotent Stem Cells. Stem Cell Rep. 2014, 3, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Nori, S.; Okada, Y.; Yasuda, A.; Tsuji, O.; Takahashi, Y.; Kobayashi, Y.; Fujiyoshi, K.; Koike, M.; Uchiyama, Y.; Ikeda, E.; et al. Grafted human-induced pluripotent stem-cell-derived neurospheres promote motor functional recovery after spinal cord injury in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 16825–16830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, Y.; Abematsu, M.; Falk, A.; Tsujimura, K.; Sanosaka, T.; Juliandi, B.; Semi, K.; Namihira, M.; Komiya, S.; Smith, A.; et al. Treatment of a Mouse Model of Spinal Cord Injury by Transplantation of Human Induced Pluripotent Stem Cell-Derived Long-Term Self-Renewing Neuroepithelial-Like Stem Cells. Stem Cells 2012, 30, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Pomeshchik, Y.; Puttonen, K.A.; Kidin, I.; Ruponen, M.; Lehtonen, S.; Malm, T.; Åkesson, E.; Hovatta, O.; Koistinaho, J. Transplanted Human Induced Pluripotent Stem Cell-Derived Neural Progenitor Cells Do Not Promote Functional Recovery of Pharmacologically Immunosuppressed Mice with Contusion Spinal Cord Injury. Cell Transplant. 2015, 24, 1799–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.; Lee, K.-I.; Kim, H.-T.; You, Y.; Yoon, D.H.; Song, K.Y.; Cheong, E.; Ha, Y.; Hwang, D.-Y. Human-induced pluripotent stem cells generated from intervertebral disc cells improve neurologic functions in spinal cord injury. Stem Cell Res. Ther. 2015, 6, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Woodruff, G.; Wang, Y.; Graham, L.; Hunt, M.; Wu, D.; Boehle, E.; Ahmad, R.; Poplawski, G.; Brock, J.; et al. Long-Distance Axonal Growth from Human Induced Pluripotent Stem Cells after Spinal Cord Injury. Neuron 2014, 83, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Romanyuk, N.; Amemori, T.; Turnovcova, K.; Prochazka, P.; Onteniente, B.; Sykova, E.; Jendelova, P. Beneficial Effect of Human Induced Pluripotent Stem Cell-Derived Neural Precursors in Spinal Cord Injury Repair. Cell Transplant. 2015, 24, 1781–1797. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Okada, Y.; Itakura, G.; Iwai, H.; Nishimura, S.; Yasuda, A.; Nori, S.; Hikishima, K.; Konomi, T.; Fujiyoshi, K.; et al. Pre-Evaluated Safe Human iPSC-Derived Neural Stem Cells Promote Functional Recovery after Spinal Cord Injury in Common Marmoset without Tumorigenicity. PLoS ONE 2012, 7, e52787. [Google Scholar] [CrossRef] [Green Version]
- All, A.H.; Bazley, F.A.; Gupta, S.; Pashai, N.; Hu, C.; Pourmorteza, A.; Kerr, C.L. Human Embryonic Stem Cell-Derived Oligodendrocyte Progenitors Aid in Functional Recovery of Sensory Pathways following Contusive Spinal Cord Injury. PLoS ONE 2012, 7, e47645. [Google Scholar] [CrossRef] [Green Version]
- All, A.H.; Mohammad-Gharibani, P.; Gupta, S.; Bazley, F.A.; Pashai, N.; Chou, B.-K.; Shah, S.; Resar, L.M.; Cheng, L.; Gearhart, J.D.; et al. Early Intervention for Spinal Cord Injury with Human Induced Pluripotent Stem Cells Oligodendrocyte Progenitors. PLoS ONE 2015, 10, e0116933. [Google Scholar] [CrossRef] [Green Version]
- Fandel, T.M.; Trivedi, A.; Nicholas, C.R.; Zhang, H.; Chen, J.; Martinez, A.F.; Noble-Haeusslein, L.J.; Kriegstein, A.R. Transplanted Human Stem Cell-Derived Interneuron Precursors Mitigate Mouse Bladder Dysfunction and Central Neuropathic Pain after Spinal Cord Injury. Cell Stem Cell 2016, 19, 544–557. [Google Scholar] [CrossRef] [Green Version]
- Manion, J.; Khuong, T.; Harney, D.; Littleboy, J.B.; Ruan, T.; Loo, L.; Costigan, M.; Larance, M.; Caron, L.; Neely, G.G. Human induced pluripotent stem cell-derived GABAergic interneuron transplants attenuate neuropathic pain. Pain 2020, 161, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Tiklová, K.; Nolbrant, S.; Fiorenzano, A.; Björklund, Å.; Sharma, Y.; Heuer, A.; Gillberg, L.; Hoban, D.B.; Cardoso, T.; Adler, A.F.; et al. Single cell transcriptomics identifies stem cell-derived graft composition in a model of Parkinson’s disease. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Yamakado, H.; Moriwaki, Y.; Yamasaki, N.; Miyakawa, T.; Kurisu, J.; Uemura, K.; Inoue, H.; Takahashi, M.; Takahashi, R. α-Synuclein BAC transgenic mice as a model for Parkinson’s disease manifested decreased anxiety-like behavior and hyperlocomotion. Neurosci. Res. 2012, 73, 173–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, R.A.; Parmar, M.; Studer, L.; Takahashi, J. Human Trials of Stem Cell-Derived Dopamine Neurons for Parkinson’s Disease: Dawn of a New Era. Cell Stem Cell 2017, 21, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Parmar, M.; Grealish, S.; Henchcliffe, C. The future of stem cell therapies for Parkinson disease. Nat. Rev. Neurosci. 2020, 21, 103–115. [Google Scholar] [CrossRef]
- George, P.M.; Steinberg, G.K. Novel Stroke Therapeutics: Unraveling Stroke Pathophysiology and Its Impact on Clinical Treatments. Neuron 2015, 87, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Marei, H.E.-S.; Hasan, A.; Rizzi, R.; Althani, A.; Afifi, N.; Cenciarelli, C.; Caceci, T.; Shuaib, A. Potential of Stem Cell-Based Therapy for Ischemic Stroke. Front. Neurol. 2018, 9, 34. [Google Scholar] [CrossRef]
- Koch, P.; Opitz, T.; Steinbeck, J.A.; Ladewig, J.; Brüstle, O. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc. Natl. Acad. Sci. USA 2009, 106, 3225–3230. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Paudel, Y.N.; Shaikh, M.F.; Shah, S.; Kumari, Y.; Othman, I. Role of inflammation in epilepsy and neurobehavioral comorbidities: Implication for therapy. Eur. J. Pharmacol. 2018, 837, 145–155. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; De Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Prim. 2018, 4, 18025. [Google Scholar] [CrossRef]
- Hamlett, E.D.; Hjorth, E.; Ledreux, A.; Gilmore, A.; Schultzberg, M.; Granholm, A.C. RvE1 treatment prevents memory loss and neuroinflammation in the Ts65Dn mouse model of Down syndrome. Glia 2020, 68, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, J.; Jiang, W.; Cao, Z.; Zhao, F.; Cai, T.; Aschner, M.; Luo, W. The role of NLRP3-CASP1 in inflammasome-mediated neuroinflammation and autophagy dysfunction in manganese-induced, hippocampal-dependent impairment of learning and memory ability. Autophagy 2017, 13, 914–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, J.K.; Nation, D.A.; Initiative, F.T.A.D.N. Neuropsychological Profiles and Trajectories in Preclinical Alzheimer’s Disease. J. Int. Neuropsychol. Soc. 2018, 24, 693–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blurton-Jones, M.; Kitazawa, M.; Martinez-Coria, H.; Castello, N.A.; Müller, F.-J.; Loring, J.F.; Yamasaki, T.R.; Poon, W.W.; Green, K.N.; LaFerla, F.M. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc. Natl. Acad. Sci. USA 2009, 106, 13594–13599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouad, G.I. Stem cells as a promising therapeutic approach for Alzheimer’s disease: A review. Bull. Natl. Res. Cent. 2019, 43, 52. [Google Scholar] [CrossRef]
- Moghadam, F.H.; Alaie, H.; Karbalaie, K.; Tanhaei, S.; Esfahani, M.H.N.; Baharvand, H. Transplantation of primed or unprimed mouse embryonic stem cell-derived neural precursor cells improves cognitive function in Alzheimerian rats. Differentiation 2009, 78, 59–68. [Google Scholar] [CrossRef]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- Yue, C.; Jing, N. The promise of stem cells in the therapy of Alzheimer’s disease. Transl. Neurodegener. 2015, 4, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Gao, T.; Zhang, B.; Pu, J. Recent Advances: Decoding Alzheimer’s Disease With Stem Cells. Front. Aging Neurosci. 2018, 10, 77. [Google Scholar] [CrossRef]
- Al-Gharaibeh, A.; Culver, R.; Stewart, A.N.; Srinageshwar, B.; Spelde, K.; Frollo, L.; Kolli, N.; Story, D.; Paladugu, L.; Anwar, S.; et al. Induced Pluripotent Stem Cell-Derived Neural Stem Cell Transplantations Reduced Behavioral Deficits and Ameliorated Neuropathological Changes in YAC128 Mouse Model of Huntington’s Disease. Front. Neurosci. 2017, 11, 628. [Google Scholar] [CrossRef] [PubMed]
- Jeon, I.; Choi, C.; Lee, N.; Im, W.; Kim, M.; Oh, S.-H.; Park, I.-H.; Kim, H.S.; Song, J. In Vivo Roles of a Patient-Derived Induced Pluripotent Stem Cell Line (HD72-iPSC) in the YAC128 Model of Huntington’s Disease. Int. J. Stem Cells 2014, 7, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.-Y.; Du, Z.-W.; Zhang, S.-C. Differentiation of human oligodendrocytes from pluripotent stem cells. Nat. Protoc. 2009, 4, 1614–1622. [Google Scholar] [CrossRef] [Green Version]
- Izrael, M.; Zhang, P.; Kaufman, R.; Shinder, V.; Ella, R.; Amit, M.; Itskovitz-Eldor, J.; Chebath, J.; Revel, M. Human oligodendrocytes derived from embryonic stem cells: Effect of noggin on phenotypic differentiation in vitro and on myelination in vivo. Mol. Cell. Neurosci. 2007, 34, 310–323. [Google Scholar] [CrossRef]
- Molineaux, S.M.; Engh, H.; De Ferra, F.; Hudson, L.; Lazzarini, R.A. Recombination within the myelin basic protein gene created the dysmyelinating shiverer mouse mutation. Proc. Natl. Acad. Sci. USA 1986, 83, 7542–7546. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Yang, Y.; He, L.; Pang, M.; Luo, C.; Liu, B.; Rong, L. High-dose methylprednisolone for acute traumatic spinal cord injury. Neurology 2019, 93, e841–e850. [Google Scholar] [CrossRef]
- Alizadeh, A.; Dyck, S.M.; Karimi-Abdolrezaee, S. Traumatic Spinal Cord Injury: An Overview of Pathophysiology, Models and Acute Injury Mechanisms. Front. Neurol. 2019, 10, 282. [Google Scholar] [CrossRef] [Green Version]
- Khazaei, M.; Ahuja, C.S.; Nakashima, H.; Nagoshi, N.; Li, L.; Wang, J.; Chio, J.; Badner, A.; Seligman, D.; Ichise, A.; et al. GDNF rescues the fate of neural progenitor grafts by attenuating Notch signals in the injured spinal cord in rodents. Sci. Transl. Med. 2020, 12, eaau3538. [Google Scholar] [CrossRef]
- Salewski, R.P.; Mitchell, R.A.; Li, L.; Shen, C.; Milekovskaia, M.; Nagy, A.; Fehlings, M.G. Transplantation of Induced Pluripotent Stem Cell-Derived Neural Stem Cells Mediate Functional Recovery Following Thoracic Spinal Cord Injury Through Remyelination of Axons. Stem Cells Transl. Med. 2015, 4, 743–754. [Google Scholar] [CrossRef]
- Griffiths, D. Functional imaging of structures involved in neural control of the lower urinary tract. Handb. Clin. Neurol. 2015, 130, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.S.; Finnerup, N.B. Allodynia and hyperalgesia in neuropathic pain: Clinical manifestations and mechanisms. Lancet Neurol. 2014, 13, 924–935. [Google Scholar] [CrossRef] [PubMed]
- Calvo, M.; Davies, A.J.; Hébert, H.L.; Weir, G.A.; Chesler, E.J.; Finnerup, N.B.; Levitt, R.C.; Smith, B.H.; Neely, G.G.; Costigan, M.; et al. The Genetics of Neuropathic Pain from Model Organisms to Clinical Application. Neuron 2019, 104, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Finnerup, N.B.; Attal, N.; Aziz, Q.; Baron, R.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Cruccu, G.; Davis, K.D.; et al. The IASP classification of chronic pain for ICD-11: Chronic neuropathic pain. Pain 2019, 160, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.A.; Kohno, T.; Karchewski, L.A.; Scholz, J.; Baba, H.; Woolf, C.J. Partial Peripheral Nerve Injury Promotes a Selective Loss of GABAergic Inhibition in the Superficial Dorsal Horn of the Spinal Cord. J. Neurosci. 2002, 22, 6724–6731. [Google Scholar] [CrossRef]
- Manion, J.; Waller, M.A.; Clark, T.; Massingham, J.N.; Neely, G.G. Developing Modern Pain Therapies. Front. Neurosci. 2019, 13, 1370. [Google Scholar] [CrossRef]
- Khuong, T.M.; Neely, G.G.; Manion, J.; Oyston, L.J.; Lau, M.-T.; Towler, H.; Lin, Y.Q.; Neely, G.G. Nerve injury drives a heightened state of vigilance and neuropathic sensitization in Drosophila. Sci. Adv. 2019, 5, eaaw4099. [Google Scholar] [CrossRef] [Green Version]
- Paulozzi, L.J. Opioid Analgesic Involvement in Drug Abuse Deaths in American Metropolitan Areas. Am. J. Public Heal. 2006, 96, 1755–1757. [Google Scholar] [CrossRef]
- Walia, K.S.; Khan, E.A.; Ko, D.H.; Raza, S.S.; Khan, Y. Side Effects of Antiepileptics- A Review. Pain Pr. 2004, 4, 194–203. [Google Scholar] [CrossRef]
- Bráz, J.; Sharif-Naeini, R.; Vogt, D.; Kriegstein, A.; Alvarez-Buylla, A.; Rubenstein, J.L.; Basbaum, A.I. Forebrain GABAergic Neuron Precursors Integrate into Adult Spinal Cord and Reduce Injury-Induced Neuropathic Pain. Neuron 2012, 74, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Bráz, J.M.; Wang, X.; Guan, Z.; Basbaum, A.I. Transplant-mediated enhancement of spinal cord GABAergic inhibition reverses paclitaxel-induced mechanical and heat hypersensitivity. Pain 2015, 156, 1084–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savatier, P.; Lapillonne, H.; Jirmanova, L.; Vitelli, L.; Samarut, J. Analysis of the cell cycle in mouse embryonic stem cells. Methods Mol. Boil. (Cliftonn.J.) 2002, 185, 27–33. [Google Scholar]
- Kapinas, K.; Grandy, R.; Ghule, P.; Medina, R.; Becker, K.; Pardee, A.; Zaidi, S.K.; Lian, J.; Stein, J.; Van Wijnen, A.; et al. The abbreviated pluripotent cell cycle. J. Cell. Physiol. 2013, 228, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somay, C.; Grunt, T.; Mannhalter, C.; Dittrich, C. Relationship of myc protein expression to the phenotype and to the growth potential of HOC-7 ovarian cancer cells. Br. J. Cancer 1992, 66, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, B.; Lu, S.; Zhao, Y.; Liu, P. HN1 contributes to migration, invasion, and tumorigenesis of breast cancer by enhancing MYC activity. Mol. Cancer 2017, 16, 90. [Google Scholar] [CrossRef] [Green Version]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Yeo, J.-C.; Ng, H.-H. The transcriptional regulation of pluripotency. Cell Res. 2012, 23, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Park, I.-H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nat. Cell Biol. 2008, 451, 141–146. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-H.; Chu, T.-Y.; Ding, D.-C. WNT/β-Catenin signaling pathway regulates non-tumorigenesis of human embryonic stem cells co-cultured with human umbilical cord mesenchymal stem cells. Sci. Rep. 2017, 7, srep41913. [Google Scholar] [CrossRef] [Green Version]
- Samardzija, C.; Quinn, M.A.; Findlay, J.K.; Eahmed, N. Attributes of Oct4 in stem cell biology: Perspectives on cancer stem cells of the ovary. J. Ovarian Res. 2012, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comisso, E.; Scarola, M.; Rosso, M.; Piazza, S.; Marzinotto, S.; Ciani, Y.; Orsaria, M.; Mariuzzi, L.; Schneider, C.; Schoeftner, S.; et al. OCT4 controls mitotic stability and inactivates the RB tumor suppressor pathway to enhance ovarian cancer aggressiveness. Oncogene 2017, 36, 4253–4266. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-W.; Wu, X.-L.; Dong, C.-L.; Xie, X.-Y.; Wu, J.-F.; Zhang, X. The Differential Expression of OCT4 Isoforms in Cervical Carcinoma. PLoS ONE 2015, 10, e0118033. [Google Scholar] [CrossRef] [Green Version]
- Fujino, S.; Myoshi, N. Oct4 Gene Expression in Primary Colorectal Cancer Promotes Liver Metastasis. Stem Cells Int. 2019, 2019, 7896524-10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Huan, Z.; Wang, W.; Mao, Y.; Zhu, J.; Zhou, B.; Sun, J.; Zhang, X. Expression of Oct-4 is significantly associated with the development and prognosis of colorectal cancer. Oncol. Lett. 2015, 10, 691–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Zhou, H.; Gu, Z.; Gao, Q.; Shen, G. Oct4 promotes cancer cell proliferation and migration and leads to poor prognosis associated with the survivin/STAT3 pathway in hepatocellular carcinoma. Oncol. Rep. 2018, 40, 979–987. [Google Scholar] [CrossRef]
- Fu, T.-Y.; Hsieh, I.-C.; Cheng, J.-T.; Tsai, M.-H.; Hou, Y.-Y.; Lee, J.-H.; Liou, H.-H.; Huang, S.-F.; Chen, H.-C.; Yen, L.-M.; et al. Association of OCT4, SOX2, and NANOG expression with oral squamous cell carcinoma progression. J. Oral Pathol. Med. 2016, 45, 89–95. [Google Scholar] [CrossRef]
- Villodre, E.S.; Kipper, F.C.; Pereira, M.B.; Lenz, G. Roles of OCT4 in tumorigenesis, cancer therapy resistance and prognosis. Cancer Treat. Rev. 2016, 51, 1–9. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, L.; Huang, H.; Huang, Y.; Huang, L.; Wang, J.; Huang, S.; He, L.; Zhou, Y.; Jia, W.; et al. MiR-26b/KPNA2 axis inhibits epithelial ovarian carcinoma proliferation and metastasis through downregulating OCT. Oncotarget 2015, 6, 23793–23806. [Google Scholar] [CrossRef]
- Lee, T.K.W.; Castilho, A.; Cheung, V.C.H.; Tang, K.H.; Ma, S.; Ng, I.O.L. CD24+ Liver Tumor-Initiating Cells Drive Self-Renewal and Tumor Initiation through STAT3-Mediated NANOG Regulation. Cell Stem Cell 2011, 9, 50–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palla, A.; Piazzolla, D.; Alcazar, N.; Cañamero, M.; Graña, O.; Gómez-Lopez, G.; Dominguez, O.; Dueñas, M.; Paramio, J.M.; Serrano, M. The pluripotency factor NANOG promotes the formation of squamous cell carcinomas. Sci. Rep. 2015, 5, 10205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santaliz-Ruiz, L.E.I.; Xie, X.; Old, M.; Teknos, T.N.; Pan, Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int. J. Cancer 2014, 135, 2741–2748. [Google Scholar] [CrossRef]
- Wuebben, E.L.; Rizzino, A. The dark side of SOX2: Cancer - a comprehensive overview. Oncotarget 2017, 8, 44917–44943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.S.; Tang, C.; Rao, M.S.; Weissman, I.L.; Wu, J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013, 19, 998–1004. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.K. The Transcription Regulator Kruppel-Like Factor 4 and Its Dual Roles of Oncogene in Glioblastoma and Tumor Suppressor in Neuroblastoma. Forum Immunopathol. Dis. Ther. 2016, 7, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Blum, B.; Benvenisty, N. The Tumorigenicity of Human Embryonic Stem Cells. Adv. Cancer Res. 2008, 100, 133–158. [Google Scholar] [CrossRef]
- Edel, M.J.; Menchon, C.; Ménendez, S.; Consiglio, A.; Raya, A.; Belmonte, J.C.I. Rem2 GTPase maintains survival of human embryonic stem cells as well as enhancing reprogramming by regulating p53 and cyclin D1. Genes Dev. 2010, 24, 561–573. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-N.; Chung, S.-K.; Xu, Z.; Xu, Y. Oct4 Maintains the Pluripotency of Human Embryonic Stem Cells by Inactivating p53 Through Sirt1-Mediated Deacetylation. Stem Cells 2014, 32, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Kohno, S.; Kitajima, S.; Sasaki, N.; Takahashi, C. Retinoblastoma tumor suppressor functions shared by stem cell and cancer cell strategies. World J. Stem Cells 2016, 8, 170–184. [Google Scholar] [CrossRef]
- Närvä, E.; Autio, R.; Rahkonen, N.; Kong, L.; Harrison, N.J.; Kitsberg, D.; Borghese, L.; Itskovitz-Eldor, J.; Rasool, O.; Dvorak, P.; et al. High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nat. Biotechnol. 2010, 28, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Nori, S.; Okada, Y.; Nishimura, S.; Sasaki, T.; Itakura, G.; Kobayashi, Y.; Renault-Mihara, F.; Shimizu, A.; Koya, I.; Yoshida, R.; et al. Long-Term Safety Issues of iPSC-Based Cell Therapy in a Spinal Cord Injury Model: Oncogenic Transformation with Epithelial-Mesenchymal Transition. Stem Cell Rep. 2015, 4, 360–373. [Google Scholar] [CrossRef] [Green Version]
- Doi, D.; Samata, B.; Katsukawa, M.; Kikuchi, T.; Morizane, A.; Ono, Y.; Sekiguchi, K.; Nakagawa, M.; Parmar, M.; Takahashi, J. Isolation of Human Induced Pluripotent Stem Cell-Derived Dopaminergic Progenitors by Cell Sorting for Successful Transplantation. Stem Cell Rep. 2014, 2, 337–350. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-Y.; Moon, S.H.; Jeong, H.-C.; Yi, J.-Y.; Lee, T.-H.; Shim, S.H.; Rhee, Y.-H.; Lee, S.-H.; Oh, S.-J.; Han, M.-J.; et al. Inhibition of pluripotent stem cell-derived teratoma formation by small molecules. Proc. Natl. Acad. Sci. USA 2013, 110, E3281–E3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okubo, T.; Iwanami, A.; Kohyama, J.; Itakura, G.; Kawabata, S.; Nishiyama, Y.; Sugai, K.; Ozaki, M.; Iida, T.; Matsubayashi, K.; et al. Pretreatment with a γ-Secretase Inhibitor Prevents Tumor-like Overgrowth in Human iPSC-Derived Transplants for Spinal Cord Injury. Stem Cell Rep. 2016, 7, 649–663. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Ke, Q.; Chen, F.; Cai, B.; Gao, Y.; Ye, C.; Wang, D.; Zhang, L.; Lahn, B.T.; Li, W.; et al. Protecting against wayward human induced pluripotent stem cells with a suicide gene. Biomaterials 2012, 33, 3195–3204. [Google Scholar] [CrossRef]
- Schuldiner, M.; Itskovitz-Eldor, J.; Benvenisty, N. Selective Ablation of Human Embryonic Stem Cells Expressing a “Suicide” Gene. Stem Cells 2003, 21, 257–265. [Google Scholar] [CrossRef]
- Zhao, Q.; Lu, B.; George, S.K.; Yoo, J.J.; Atala, A. Safeguarding pluripotent stem cells for cell therapy with a non-viral, non-integrating episomal suicide construct. Biomaterials 2012, 33, 7261–7271. [Google Scholar] [CrossRef]
- Liang, Q.; Monetti, C.; Shutova, M.V.; Neely, E.J.; Hacibekiroglu, S.; Yang, H.; Kim, C.; Zhang, P.; Li, C.; Nagy, K.; et al. Linking a cell-division gene and a suicide gene to define and improve cell therapy safety. Nat. Cell Biol. 2018, 563, 701–704. [Google Scholar] [CrossRef]
- Qadir, M.M.F.; Álvarez-Cubela, S.; Belle, K.; Sapir, T.; Messaggio, F.; Johnson, K.B.; Umland, O.; Hardin, D.; Klein, D.; Pérez-Álvarez, I.; et al. A Double Fail-Safe Approach to Prevent Tumorigenesis and Select Pancreatic β Cells from Human Embryonic Stem Cells. Stem Cell Rep. 2019, 12, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Nau, R.; Sörgel, F.; Eiffert, H. Penetration of Drugs through the Blood-Cerebrospinal Fluid/Blood-Brain Barrier for Treatment of Central Nervous System Infections. Clin. Microbiol. Rev. 2010, 23, 858–883. [Google Scholar] [CrossRef] [Green Version]
- Ando, M.; Nishimura, T.; Yamazaki, S.; Yamaguchi, T.; Kawana-Tachikawa, A.; Hayama, T.; Nakauchi, Y.; Ando, J.; Ota, Y.; Takahashi, S.; et al. A Safeguard System for Induced Pluripotent Stem Cell-Derived Rejuvenated T Cell Therapy. Stem Cell Rep. 2015, 5, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Yagyu, S.; Hoyos, V.; Del Bufalo, F.; Brenner, M.K. An Inducible Caspase-9 Suicide Gene to Improve the Safety of Therapy Using Human Induced Pluripotent Stem Cells. Mol. Ther. 2015, 23, 1475–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.; Tchao, J.; Wu, L.; Carman, A.J. Precision installation of a highly efficient suicide gene safety switch in human induced pluripotent stem cells. Stem Cells Transl. Med. 2020, 9, 1378–1388. [Google Scholar] [CrossRef]
- Itakura, G.; Kawabata, S.; Ando, M.; Nishiyama, Y.; Sugai, K.; Ozaki, M.; Iida, T.; Ookubo, T.; Kojima, K.; Kashiwagi, R.; et al. Fail-Safe System against Potential Tumorigenicity after Transplantation of iPSC Derivatives. Stem Cell Rep. 2017, 8, 673–684. [Google Scholar] [CrossRef]
- Parr, C.J.C.; Katayama, S.; Miki, K.; Kuang, Y.; Yoshida, Y.; Morizane, A.; Takahashi, J.; Yamanaka, S.; Saito, H. MicroRNA-302 switch to identify and eliminate undifferentiated human pluripotent stem cells. Sci. Rep. 2016, 6, 32532. [Google Scholar] [CrossRef] [Green Version]
- Benabdallah, B.; Désaulniers-Langevin, C.; Colas, C.; Li, Y.; Rousseau, G.; Guimond, J.V.; Haddad, E.; Beauséjour, C. Natural Killer Cells Prevent the Formation of Teratomas Derived From Human Induced Pluripotent Stem Cells. Front. Immunol. 2019, 10, 2580. [Google Scholar] [CrossRef] [Green Version]
- Gore, A.; Li, Z.; Fung, H.-L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nat. Cell Biol. 2011, 471, 63–67. [Google Scholar] [CrossRef]
- Hussein, S.M.; Batada, N.N.; Vuoristo, S.; Ching, R.W.; Autio, R.; Närvä, E.; Ng, S.; Sourour, M.; Hamalainen, R.H.; Olsson, C.; et al. Copy number variation and selection during reprogramming to pluripotency. Nat. Cell Biol. 2011, 471, 58–62. [Google Scholar] [CrossRef]
- Perrera, V.; Martello, G. How Does Reprogramming to Pluripotency Affect Genomic Imprinting? Front. Cell Dev. Biol. 2019, 7, 76. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Hansen, N.F.; Zhao, L.; Du, Y.; Zou, C.; Donovan, F.X.; Chou, B.-K.; Zhou, G.; Li, S.; Dowey, S.N.; et al. Low Incidence of DNA Sequence Variation in Human Induced Pluripotent Stem Cells Generated by Nonintegrating Plasmid Expression. Cell Stem Cell 2012, 10, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, M.; Kasama, Y.; Araki, R.; Hoki, Y.; Sunayama, M.; Uda, M.; Nakamura, M.; Ando, S.; Abe, M. Induced Pluripotent Stem Cell Generation-Associated Point Mutations Arise during the Initial Stages of the Conversion of These Cells. Stem Cell Rep. 2014, 2, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; Devine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B 2009, 85, 348–362. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human Induced Pluripotent Stem Cells Free of Vector and Transgene Sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [Green Version]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nat. Cell Biol. 2009, 458, 766–770. [Google Scholar] [CrossRef]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.-H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly Efficient Reprogramming to Pluripotency and Directed Differentiation of Human Cells with Synthetic Modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Kim, C.-H.; Moon, J.-I.; Chung, Y.-G.; Chang, M.-Y.; Han, B.-S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of Human Induced Pluripotent Stem Cells by Direct Delivery of Reprogramming Proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, N.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of Induced Pluripotent Stem Cells Using Recombinant Proteins. Cell Stem Cell 2009, 4, 581. [Google Scholar] [CrossRef] [Green Version]
- Ohnuki, M.; Tanabe, K.; Sutou, K.; Teramoto, I.; Sawamura, Y.; Narita, M.; Nakamura, M.; Tokunaga, Y.; Watanabe, A.; Yamanaka, S.; et al. Dynamic regulation of human endogenous retroviruses mediates factor-induced reprogramming and differentiation potential. Proc. Natl. Acad. Sci. USA 2014, 111, 12426–12431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nat. Cell Biol. 2010, 467, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nat. Cell Biol. 2011, 471, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Ohi, Y.; Qin, H.; Hong, C.; Blouin, L.; Polo, J.M.; Guo, T.; Qi, Z.; Downey, S.L.; Manos, P.D.; Rossi, D.J.; et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat. Cell Biol. 2011, 13, 541–549. [Google Scholar] [CrossRef]
- Polo, J.M.; Liu, S.; Figueroa, M.E.; Kulalert, W.; Eminli, S.; Tan, K.Y.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 2010, 28, 848–855. [Google Scholar] [CrossRef] [Green Version]
- Nishino, K.; Toyoda, M.; Yamazaki-Inoue, M.; Fukawatase, Y.; Chikazawa, E.; Sakaguchi, H.; Akutsu, H.; Umezawa, A. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 2011, 7, e1002085. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-Y.; Hysolli, E.; Tanaka, Y.; Wang, B.; Jung, Y.W.; Pan, X.; Weissman, S.M.; Park, I.-H. X Chromosome of Female Cells Shows Dynamic Changes in Status during Human Somatic Cell Reprogramming. Stem Cell Rep. 2014, 2, 896–909. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.S.; Rowntree, R.K.; Mekhoubad, S.; Lee, J.T. X-chromosome inactivation and epigenetic fluidity in human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 4820–4825. [Google Scholar] [CrossRef] [Green Version]
- Anguera, M.C.; Sadreyev, R.; Zhang, Z.; Szanto, A.; Payer, B.; Sheridan, S.D.; Kwok, S.; Haggarty, S.J.; Sur, I.; Alvarez, J.; et al. Molecular Signatures of Human Induced Pluripotent Stem Cells Highlight Sex Differences and Cancer Genes. Cell Stem Cell 2012, 11, 75–90. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Zhang, Z.-N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent stem cells. Nat. Cell Biol. 2011, 474, 212–215. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Zhang, Z.-N.; Westenskow, P.D.; Todorova, D.; Hu, Z.; Lin, T.; Rong, Z.; Kim, J.; He, J.; Wang, M.; et al. Humanized Mice Reveal Differential Immunogenicity of Cells Derived from Autologous Induced Pluripotent Stem Cells. Cell Stem Cell 2015, 17, 353–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guha, P.; Morgan, J.W.; Mostoslavsky, G.; Rodrigues, N.P.; Boyd, A.S. Lack of Immune Response to Differentiated Cells Derived from Syngeneic Induced Pluripotent Stem Cells. Cell Stem Cell 2013, 12, 407–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, R.; Uda, M.; Hoki, Y.; Sunayama, M.; Nakamura, M.; Ando, S.; Sugiura, M.; Ideno, H.; Shimada, A.; Nifuji, A.; et al. Negligible immunogenicity of terminally differentiated cells derived from induced pluripotent or embryonic stem cells. Nat. Cell Biol. 2013, 494, 100–104. [Google Scholar] [CrossRef]
- Strnadel, J.; Carromeu, C.; Bardy, C.; Navarro, M.; Platoshyn, O.; Glud, A.N.; Marsala, S.; Kafka, J.; Miyanohara, A.; Kato, T.; et al. Survival of syngeneic and allogeneic iPSC–derived neural precursors after spinal grafting in minipigs. Sci. Transl. Med. 2018, 10, eaam6651. [Google Scholar] [CrossRef] [Green Version]
- López, M.M.; Valenzuela, J.E.; Álvarez, F.C.; López-Álvarez, M.R.; Cecilia, G.S.; Paricio, P.P. Long-term problems related to immunosuppression. Transpl. Immunol. 2006, 17, 31–35. [Google Scholar] [CrossRef]
- Li, L.; Baroja, M.L.; Majumdar, A.; Chadwick, K.; Rouleau, A.; Gallacher, L.; Ferber, I.; Lebkowski, J.; Martin, T.; Madrenas, J.; et al. Human Embryonic Stem Cells Possess Immune-Privileged Properties. Stem Cells 2004, 22, 448–456. [Google Scholar] [CrossRef]
- Drukker, M.; Katchman, H.; Katz, G.; Friedman, S.E.-T.; Shezen, E.; Hornstein, E.; Mandelboim, O.; Reisner, Y.; Benvenisty, N. Human Embryonic Stem Cells and Their Differentiated Derivatives Are Less Susceptible to Immune Rejection Than Adult Cells. Stem Cells 2006, 24, 221–229. [Google Scholar] [CrossRef]
- Perez-Cunningham, J.; Ames, E.; Smith, R.C.; Peter, A.K.; Naidu, R.; Nolta, J.A.; Murphy, W.J. Natural Killer Cell Subsets Differentially Reject Embryonic Stem Cells Based on Licensing. Transplantation 2014, 97, 992–998. [Google Scholar] [CrossRef]
- Sheldon, S.; Poulton, K.; Philip, H.; Marlene, R. HLA Typing and Its Influence on Organ Transplantation. Transplant. Immunol. 2006, 333, 157–174. [Google Scholar] [CrossRef]
- Lee, S.; Huh, J.Y.; Turner, D.M.; Lee, S.; Robinson, J.; Stein, J.E.; Shim, S.H.; Hong, C.P.; Kang, M.S.; Nakagawa, M.; et al. Repurposing the Cord Blood Bank for Haplobanking of HLA-Homozygous iPSCs and Their Usefulness to Multiple Populations. Stem Cells 2018, 36, 1552–1566. [Google Scholar] [CrossRef] [Green Version]
- Umekage, M.; Sato, Y.; Takasu, N. Overview: An iPS cell stock at CiRA. Inflamm. Regen. 2019, 39, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Gourraud, P.-A.; Gilson, L.; Girard, M.; Peschanski, M. The Role of Human Leukocyte Antigen Matching in the Development of Multiethnic “Haplobank” of Induced Pluripotent Stem Cell Lines. Stem Cells 2012, 30, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, B.; Ono, M.; Kagita, A.; Fujii, K.; Sasakawa, N.; Ueda, T.; Gee, P.; Nishikawa, M.; Nomura, M.; et al. Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell 2019, 24, 566–578.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morizane, A.; Kikuchi, T.; Hayashi, T.; Mizuma, H.; Takara, S.; Doi, H.; Mawatari, A.; Glasser, M.F.; Shiina, T.; Ishigaki, H.; et al. MHC matching improves engraftment of iPSC-derived neurons in non-human primates. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Badin, R.A.; Bugi, A.; Williams, S.; Vadori, M.; Michael, M.; Jan, C.; Nassi, A.; Lecourtois, S.; Blancher, A.; Cozzi, E.; et al. MHC matching fails to prevent long-term rejection of iPSC-derived neurons in non-human primates. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, T.; Miyagawa, S.; Fukushima, S.; Maeda, A.; Kashiyama, N.; Kawamura, A.; Miki, K.; Okita, K.; Yoshida, Y.; Shiina, T.; et al. Cardiomyocytes Derived from MHC-Homozygous Induced Pluripotent Stem Cells Exhibit Reduced Allogeneic Immunogenicity in MHC-Matched Non-human Primates. Stem Cell Rep. 2016, 6, 312–320. [Google Scholar] [CrossRef] [Green Version]
- Deuse, T.; Hu, X.; Gravina, A.; Wang, D.; Tediashvili, G.; De, C.; Thayer, W.O.; Wahl, A.; Garcia, J.V.; Reichenspurner, H.; et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019, 37, 252–258. [Google Scholar] [CrossRef]
- Jang, Y.; Choi, J.; Park, N.; Kang, J.; Kim, M.; Kim, Y.; Ju, J.H. Development of immunocompatible pluripotent stem cells via CRISPR-based human leukocyte antigen engineering. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, M.; Duan, S.; Franco, P.J.; Kenty, J.H.-R.; Hedrick, P.; Xia, Y.; Allen, A.; Ferreira, L.M.R.; Strominger, J.L.; et al. Generation of hypoimmunogenic human pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2019, 116, 10441–10446. [Google Scholar] [CrossRef] [Green Version]
- Gornalusse, G.G.; Hirata, R.K.; Funk, S.E.; Riolobos, L.; Lopes, V.S.; Manske, G.; Prunkard, D.; Colunga, A.G.; Hanafi, L.-A.; O Clegg, V.S.L.D.; et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 2017, 35, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Sorrells, S.F.; Paredes, M.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nat. Cell Biol. 2018, 555, 377–381. [Google Scholar] [CrossRef]
- Tobin, M.K.; Musaraca, K.; Disouky, A.; Shetti, A.; Bheri, A.; Honer, W.G.; Kim, N.; Dawe, R.J.; Bennett, D.A.; Arfanakis, K.; et al. Human Hippocampal Neurogenesis Persists in Aged Adults and Alzheimer’s Disease Patients. Cell Stem Cell 2019, 24, 974–982.e3. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.; Schwab, M.E.; Popovich, P.G. Central Nervous System Regenerative Failure: Role of Oligodendrocytes, Astrocytes, and Microglia. Cold Spring Harb. Perspect. Biol. 2014, 7, a020602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempf, A.; Schwab, M.E. Nogo-A Represses Anatomical and Synaptic Plasticity in the Central Nervous System. Physiology 2013, 28, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Gonzenbach, R.R.; Zoerner, B.; Schnell, L.; Weinmann, O.; Mir, A.K.; Schwab, M.E. Delayed Anti-Nogo-A Antibody Application after Spinal Cord Injury Shows Progressive Loss of Responsiveness. J. Neurotrauma 2012, 29, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-F.; Wang, J.-W.; Yang, J.-F.; Ma, Y.; Hua, Z.; Guo, Y.; Gu, X.-L. Nogo-A expression dynamically varies after spinal cord injury. Neural Regen. Res. 2015, 10, 225–229. [Google Scholar] [CrossRef]
- Liebscher, T.; Schnell, L.; Schnell, D.; Scholl, J.; Schneider, R.; Gullo, M.; Fouad, K.; Mir, A.; Rausch, M.; Kindler, D.; et al. Nogo-A antibody improves regeneration and locomotion of spinal cord-injured rats. Ann. Neurol. 2005, 58, 706–719. [Google Scholar] [CrossRef]
- Popovich, P.G.; Guan, Z.; Wei, P.; Huitinga, I.; Van Rooijen, N.; Stokes, B.T. Depletion of Hematogenous Macrophages Promotes Partial Hindlimb Recovery and Neuroanatomical Repair after Experimental Spinal Cord Injury. Exp. Neurol. 1999, 158, 351–365. [Google Scholar] [CrossRef]
- Stirling, D.P.; Khodarahmi, K.; Liu, J.; McPhail, L.T.; McBride, C.B.; Steeves, J.D.; Ramer, M.S.; Tetzlaff, W. Minocycline Treatment Reduces Delayed Oligodendrocyte Death, Attenuates Axonal Dieback, and Improves Functional Outcome after Spinal Cord Injury. J. Neurosci. 2004, 24, 2182–2190. [Google Scholar] [CrossRef] [Green Version]
- Gensel, J.C.; Nakamura, S.; Guan, Z.; Van Rooijen, N.; Ankeny, D.P.; Popovich, P.G. Macrophages promote axon regeneration with concurrent neurotoxicity. J. Neurosci. 2009, 29, 3956–3968. [Google Scholar] [CrossRef] [PubMed]
- Stirling, D.P.; Cummins, K.; Mishra, M.; Teo, W.; Yong, V.W.; Stys, P. Toll-like receptor 2-mediated alternative activation of microglia is protective after spinal cord injury. Brain 2014, 137, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.J.; Kim, H.W.; Gonzalez-Cano, R.; Choi, J.; Back, S.K.; Roh, S.E.; Johnson, E.; Gabriac, M.; Kim, M.-S.; Lee, J.; et al. Natural Killer Cells Degenerate Intact Sensory Afferents following Nerve Injury. Cell 2019, 176, 716–728.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padi, S.S.; Kulkarni, S.K. Minocycline prevents the development of neuropathic pain, but not acute pain: Possible anti-inflammatory and antioxidant mechanisms. Eur. J. Pharmacol. 2008, 601, 79–87. [Google Scholar] [CrossRef]
- Ohtake, Y.; Li, S. Molecular mechanisms of scar-sourced axon growth inhibitors. Brain Res. 2015, 1619, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monnier, P.P.; Sierra, A.; Schwab, J.M.; Henke-Fahle, S.; Mueller, B.K. The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol. Cell. Neurosci. 2003, 22, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, E.J.; Moon, L.D.F.; Popat, R.J.; King, V.R.; Bennett, G.S.; Patel, P.N.; Fawcett, J.W.; McMahon, S.B. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nat. Cell Biol. 2002, 416, 636–640. [Google Scholar] [CrossRef]
- Barritt, A.W.; Davies, M.; Marchand, F.; Hartley, R.; Grist, J.; Yip, P.; McMahon, S.B.; Bradbury, E.J. Chondroitinase ABC Promotes Sprouting of Intact and Injured Spinal Systems after Spinal Cord Injury. J. Neurosci. 2006, 26, 10856–10867. [Google Scholar] [CrossRef]
- Alilain, W.J.; Horn, K.P.; Hu, H.; Dick, T.E.; Silver, J. Functional regeneration of respiratory pathways after spinal cord injury. Nat. Cell Biol. 2011, 475, 196–200. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Jin, J.; Tilve, S.; Huang, Z.; Zhou, L.; Geller, H.M. Effect of chondroitin sulfate proteoglycans on neuronal cell adhesion, spreading and neurite growth in culture. Neural Regen. Res. 2018, 13, 289–297. [Google Scholar] [CrossRef]
- Tang, H.-B.; Jiang, X.-J.; Wang, C.; Liu, S.-C. S1P/S1PR3 signaling mediated proliferation of pericytes via Ras/pERK pathway and CAY10444 had beneficial effects on spinal cord injury. Biochem. Biophys. Res. Commun. 2018, 498, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Sabelström, H.; Stenudd, M.; Réu, P.; Dias, D.O.; Elfineh, M.; Zdunek, S.; Damberg, P.; Göritz, C.; Frisén, J. Resident Neural Stem Cells Restrict Tissue Damage and Neuronal Loss After Spinal Cord Injury in Mice. Science 2013, 342, 637–640. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, R.K.G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nat. Cell Biol. 2016, 532, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Haindl, M.T.; Köck, U.; Zeitelhofer-Adzemovic, M.; Fazekas, F.; Hochmeister, S. The formation of a glial scar does not prohibit remyelination in an animal model of multiple sclerosis. Glia 2018, 67, 467–481. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Cell Type Transplanted | Transplant Site | Animal Model | Readout | Reference |
|---|---|---|---|---|---|
| PD | hESC-DANs | Striatum | Rat, 6-OHDA | apomorphine-induced rotations, adjusting step test, cylinder test. | [13] |
| hESC-DANs | Striatum in mice, putamen in monkeys | Mouse Monkeys, MPTP | MRI, Neurological rating scale, video-based analysis of spontaneous movements | [14] | |
| hESC-midbrain DANs | Striatum in mice and rats, Putamen in monkeys | Mouse, 6-OHDA Rats, 6-OHDA Monkeys, MPTP | Amphetamine-induced rotations (Rats and mice), stepping test (rats), cylinder test (rats). | [15] | |
| hESC-midbrain DANs | Striatum | Rat, 6-OHDA | Amphetamine-induced rotation Cylinder test | [16] | |
| hESC-midbrain DANs | Striatum | Rat, 6-OHDA | MRI MR spectroscopy PET-Scan Amphetamine-induced rotations | [17] | |
| hESC-DA progenitor cells | Striatum | Rat, 6-OHDA Mouse, 6-OHDA | Amphetamine-induced rotations Cylinder test | [18] | |
| hiPSC-DANs | Striatum | Rat, 6-OHDA | Amphetamine- and apomorphine-induced rotations | [19] | |
| hiPSC-DANs | Striatum | Rat, 6-OHDA Mouse, α-Synuclein Tg | Amphetamine-induced rotations | [20] | |
| hiPSC-NPCs and hiPSC-DANs | Striatum | Rat, 6-OHDA | Amphetamine-induced rotations | [21] | |
| hiPSC-NSCs | Striatum | Rat, 6-OHDA | Turning-over test Rotation-rod test | [22] | |
| hiPSC-NPCs | Putamen | Monkey, MPTP | Raisin pick up test Neurological rating scale | [23] | |
| hiPSC-DA progenitor cells | Putamen | Monkey, MPTP | Neurological rating scale, Video-based analysis of spontaneous movements | [24] | |
| Stroke | hiPSC-lt-NES cells | Striatum, Cortex | Rat & Mouse, MCAo | Staircase and corridor tests | [25] |
| hiPSC-cortical fated lt-NES | Cortex | Rat, MCAo | Immunoelectron microscopy Rabies virus retrograde synaptic tracing electrophysiology | [26] | |
| hiPSC-cortical fated lt-NES | Cortex | Rat, MCAo | Cylinder and stepping test | [27] | |
| hiPSC-cortical fated lt-NES | Cortex | Rat, MCAo | Rabies virus retrograde synaptic tracing Immunoelectron microscopy Optogenetics Electrophysiology Cylinder test | [28] | |
| hiPSC-NPCs | Striatum | Mouse, MCAo | Modified neurological severity score (mNSS) | [29] | |
| hiPSC-NPCs | Penumbra region of the cortex | Mouse, MCAo | Adhesive removal test – latency and removal time | [30] | |
| hiPSC-NPCs | Striatum | Rat, MCAo | Rotarod test Stepping test mNSS Apomorphine-induced rotation tests | [31] | |
| hiPS-NPCs | Right cortex | Rat, Incision in common carotid artery | Vibrissae-elicited forelimb placing test Cylinder test | [32] | |
| hiPSC-NSC | Cortex surrounding lesion | Pig, MCAo | MRI and histology (no functional measurement) | [33] | |
| Epilepsy | hESC-MGE progenitors | Hippocampus | Mouse, Pilocarpine-induced TLE | EEG recording Novel object recognition test Locomotion test Handling test | [34] |
| hESC-MGE progenitors | Hippocampus | Mouse, Pilocarpine-induced TLE | Morris Water Maze test, EEG recording | [35] | |
| Learning and Memory/AD | hiPSC-NPCs (with cholinergic neuronal phenotype) | Bilateral hippocampus | Mouse, Tg PDAPP | Morris Water Maze test | [36] |
| hESC-BFCN Progenitors | Bilateral Hippocampus | Mouse, Tg 5XFAD and | Morris Water Maze test Electrophysiology (Whole-Cell patch-clamp) | [37] | |
| hiPSC-ML/NEP2 | Hippocampus | Mouse, Tg 5XFAD | Immuno-histochemistry (no functional assay) | [38] | |
| Multiple Sclerosis | hiPSC-OPCs | Corpus Callosum | Mouse, Shiverer/rag2 | Survival time | [39] |
| hiPSC-OPCs | Forebrain | Mouse, Shiverer/rag2 | Immuno-histochemistry (no functional assay) | [40] | |
| Spinal Cord Injury | hiPSC-NSCs | Lesion epicentre | Mouse, moderate contusive SCI at T10 level | Rotarod test BMS score DigiGait system | [41] |
| hiPSC-lt-NES | Lesion epicentre | Mouse, contusive SCI at T9 level | BMS locomotor scale | [42] | |
| hiPSC-NPCs | Lesion epicentre | Mouse, moderate contusive SCI at T11 level | BMS scale CatWalk-automated gait analysis | [43] | |
| hiPSC-NPCs | Lesion epicentre at T11 | Mouse, and compression injury T11 | Open-field, footprint analysis | [44] | |
| hiPSC-NSCs | Lesion epicenter at C5 | Rat, C5 lateral hemisection lesions | Grid-walking Grooming Vertical exploration (no functional improvement) | [45] | |
| hiPSC-NPCs | Lesion epicenter at T8 | Rat, balloon-induced compression lesion at T8 level | BBB test Beam walking test Rotarod test Plantar test | [46] | |
| hiPSC-NSCs | Lesion epicenter | Marmoset, moderate contusive SCI by weight-drop at C5 level | Open field rating scale Bar grip test Cage climbing test | [47] | |
| hESC-OPCs | Lesion epicenter at T8 of spinal cord. | Rat, contusive injury by weight-drop at T8 level | SSEP (Somatosensory Evoked Potentials) evaluation | [48] | |
| hiPSC-OPCs | T8 of spinal cord. | Rat, moderate contusive SCI by weight-drop at T8 level | BBB locomotor rating scale | [49] | |
| hESC-MGE (GABAergic progenitors) | Lumbar enlargement level L3–L5. | Mouse, moderate contusive SCI at T13 level | BMS scale, Open field, Von Frey, Over-grooming, Assessment of bladder function by analysis of voluntary Voiding Pattern and Cystometry | [50] | |
| Neuropathic Pain | hESC-MGE progenitors | Spinal Cord, Lumbar enlargement (L3–L5) | Mouse, moderate contusive SCI at T13 level | BMS scale, Open Field, Von Frey, Over-grooming. | [50] |
| hiPSC-GABAergic neurons | Spinal Cord, Lumbar enlargement L1. | Mouse, SNI | BMS scale Von Frey, Acetone Open Field, | [51] |
| Disease | Treatment Type | Phase | Clinical Trial Identifier | Country |
|---|---|---|---|---|
| PD | parthenogenetic hESC-NSC (ISC-hpNSC) | Phase I | NCT02452723 | Australia |
| HLA-matched hESC-NPC | Phase I/II | NCT03119636 | China | |
| hiPSC-DA Progenitors | Phase I/II | JMA-IIA00384 UMIN000033564 | Japan | |
| Amyotrophic Lateral Sclerosis (ALS) | hESC-Astrocystes (AstroRx) | Phase I/II | NCT03482050 | Israel |
| SCI | hESC-OPC (AST-OPC1) | Phase I/II | NCT02302157 | USA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ford, E.; Pearlman, J.; Ruan, T.; Manion, J.; Waller, M.; Neely, G.G.; Caron, L. Human Pluripotent Stem Cells-Based Therapies for Neurodegenerative Diseases: Current Status and Challenges. Cells 2020, 9, 2517. https://doi.org/10.3390/cells9112517
Ford E, Pearlman J, Ruan T, Manion J, Waller M, Neely GG, Caron L. Human Pluripotent Stem Cells-Based Therapies for Neurodegenerative Diseases: Current Status and Challenges. Cells. 2020; 9(11):2517. https://doi.org/10.3390/cells9112517
Chicago/Turabian StyleFord, Elizabeth, Jodie Pearlman, Travis Ruan, John Manion, Matthew Waller, Gregory G. Neely, and Leslie Caron. 2020. "Human Pluripotent Stem Cells-Based Therapies for Neurodegenerative Diseases: Current Status and Challenges" Cells 9, no. 11: 2517. https://doi.org/10.3390/cells9112517