Acid Sphingomyelinase Impacts Canonical Transient Receptor Potential Channels 6 (TRPC6) Activity in Primary Neuronal Systems

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Rat Neuronal Cell Culture
2.2. ASM Mouse Strain and Murine Cell Culture
2.3. Genotyping of Mice
2.4. Enzymatic Activity Assay
2.5. Lipid Quantification by Liquid Chromatography Tandem-Mass Spectrometry (LC-MS/MS)
2.6. Mouse Neuronal Ca2+ Measurements
2.7. Mouse Synaptosomal Preparations and Synaptosomal Ca2+ Measurements
2.8. Rat Cortical Phospho-CREB Immunocytochemistry
2.9. Mouse Cortical Phospho-CREB Immunocytochemistry
2.10. Animal Welfare Declaration
2.11. Statistical Analyses
3. Results
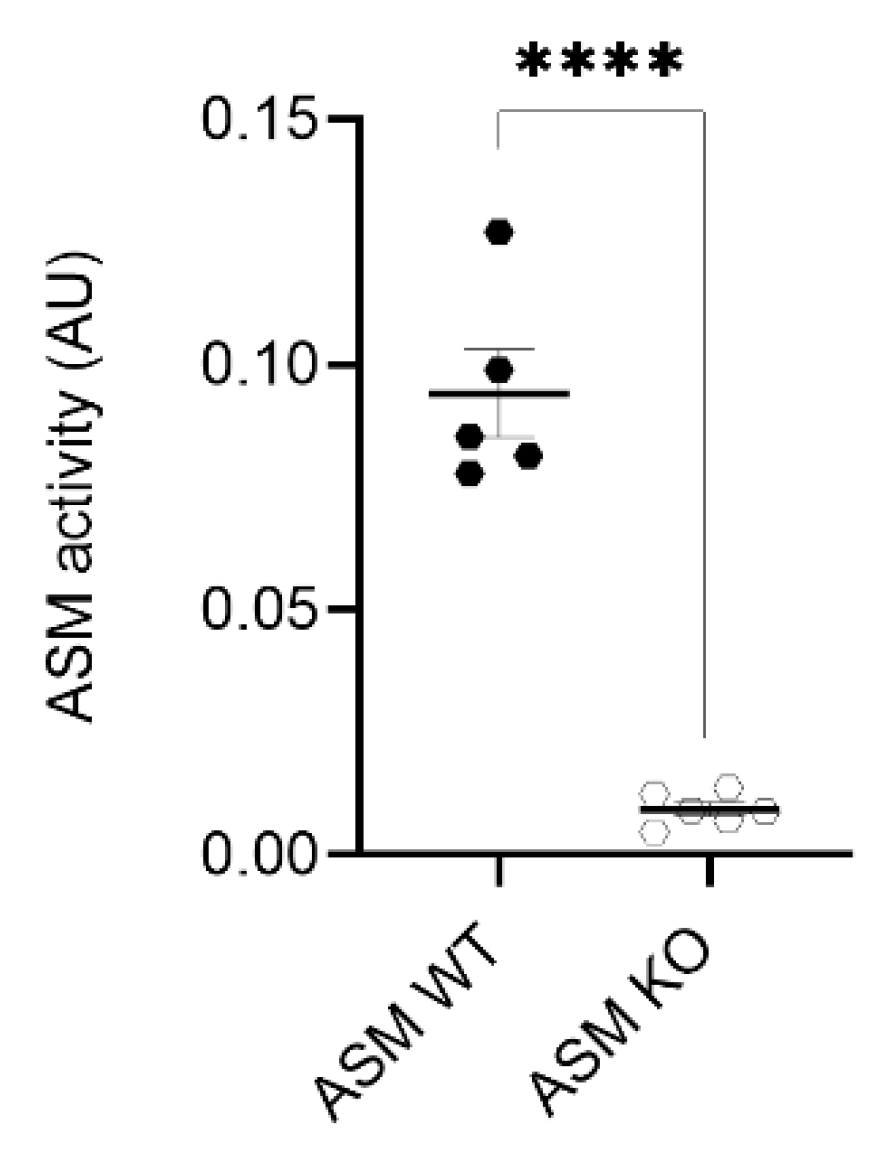
3.1. ASM Activity Is Decreased in Cortices of ASM KO Mice
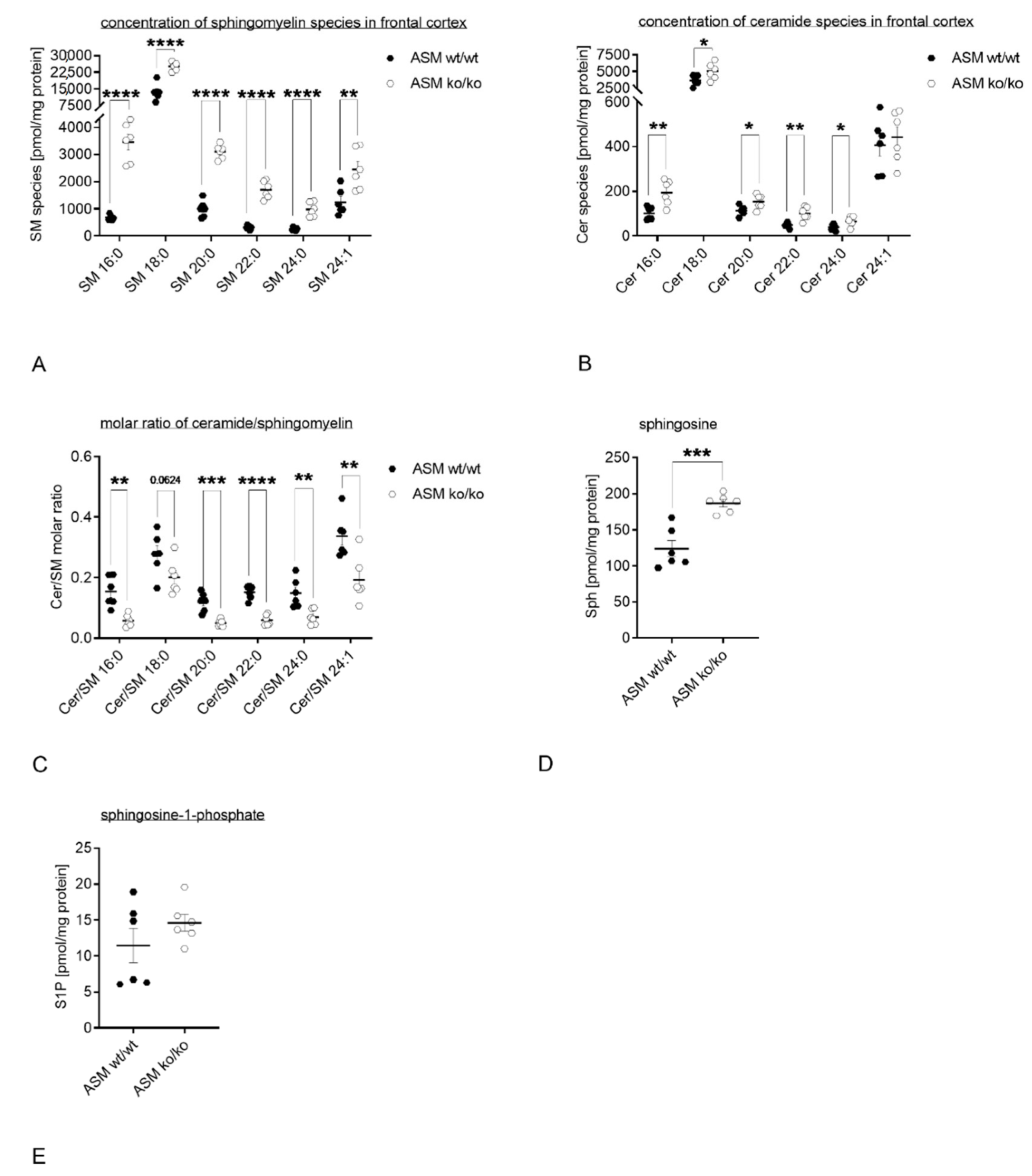
3.2. Genetic ASM Deficiency Impacts the Sphingolipidome in the Murine Frontal Cortex
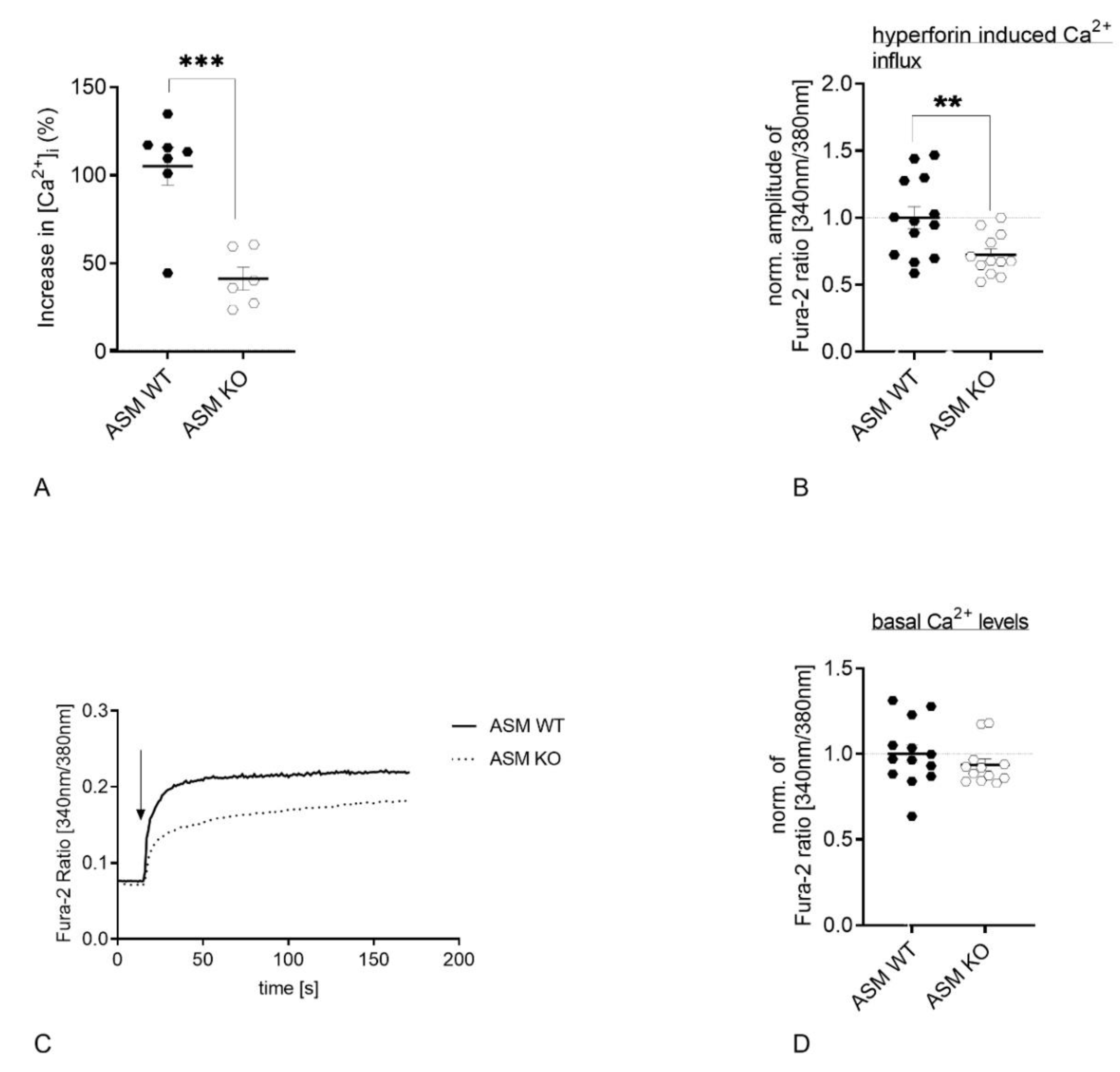
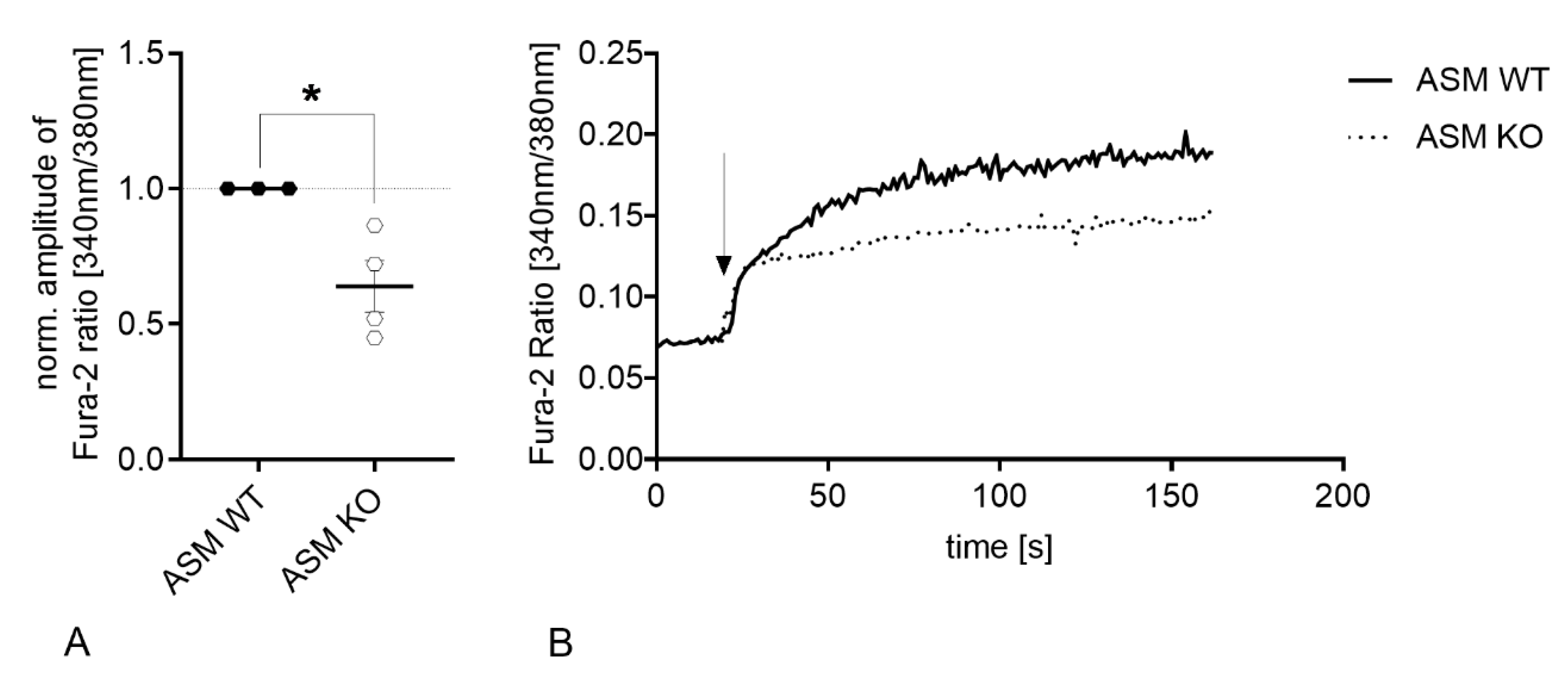
3.3. Genetic ASM Deficiency Decreases Hyperforin-Induced Ca2+ Influx in Primary Murine Neuronal Cells and Synaptosomes
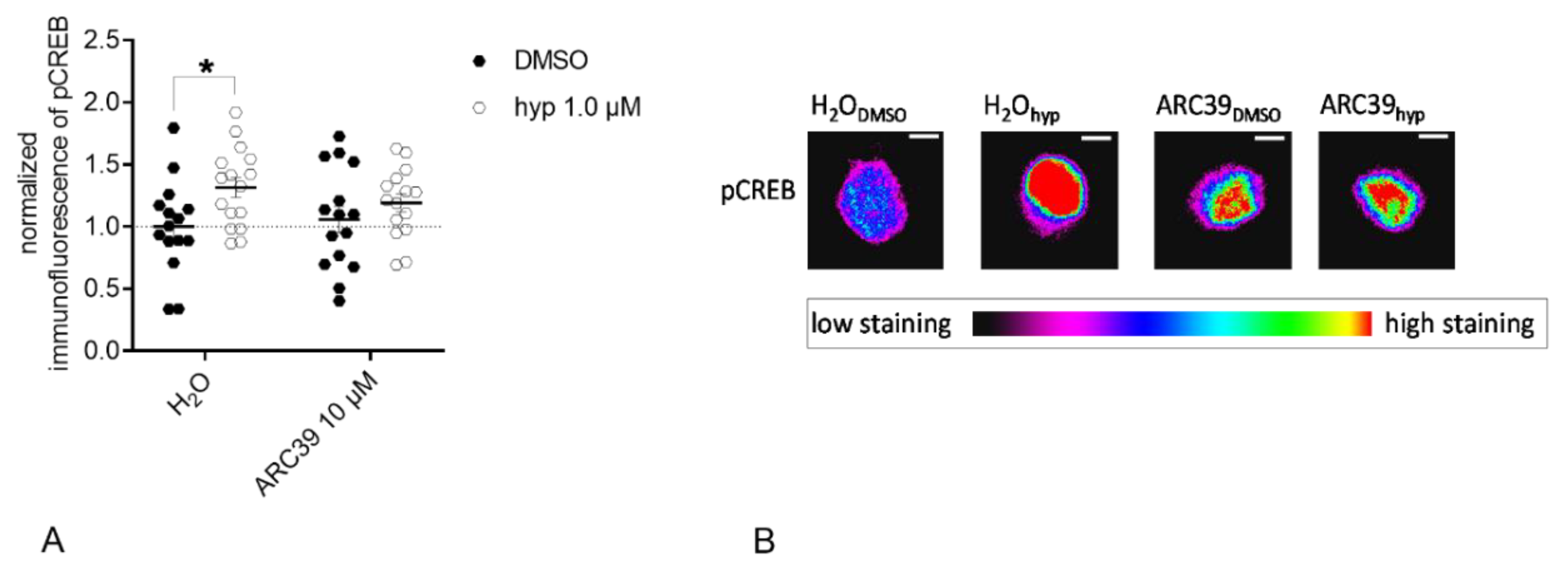
3.4. ASM Activity Inhibition Prevents Hyperforin-Induced CREB Phosphorylation in Primary Rat Neuronal Cells
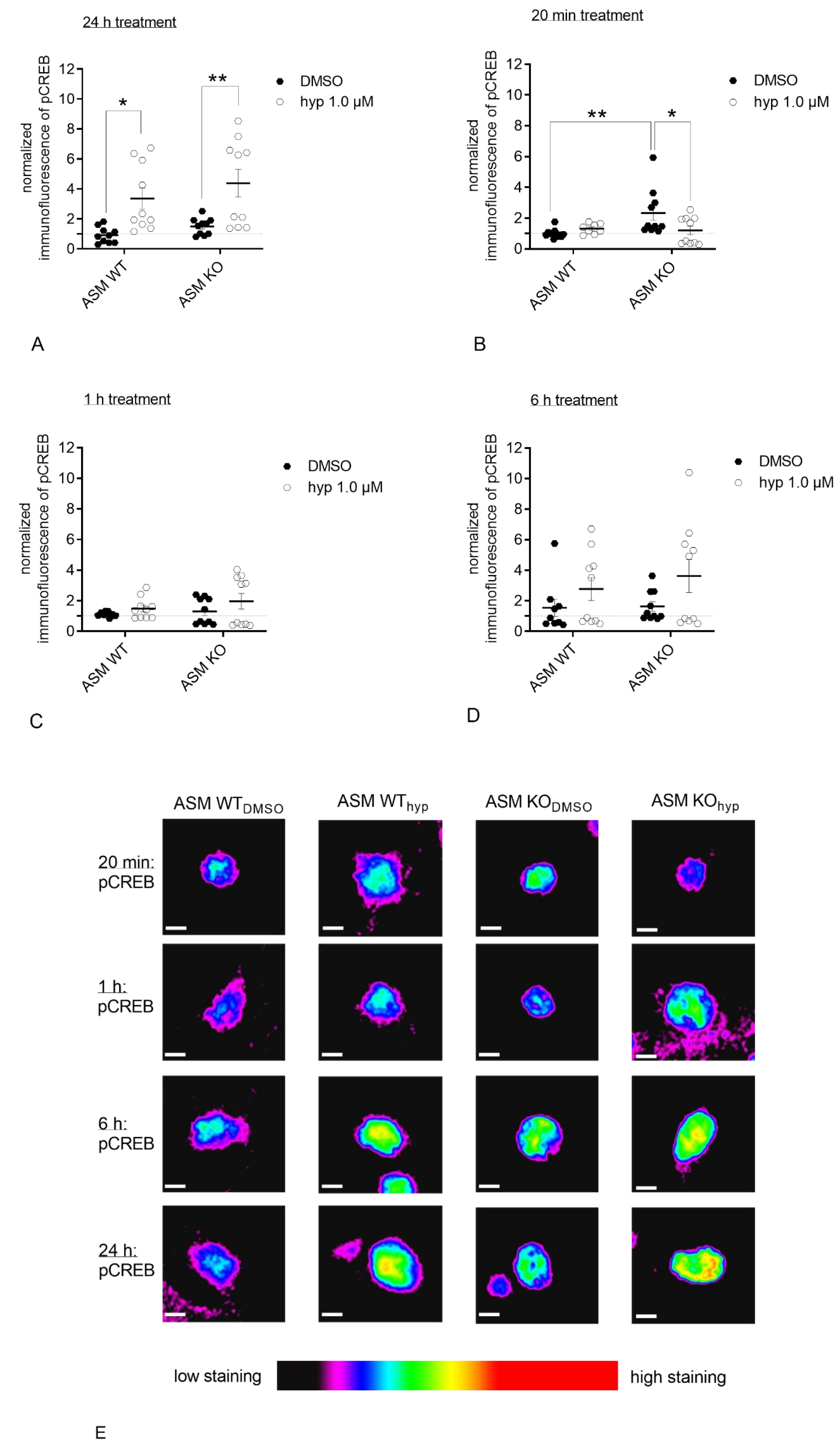
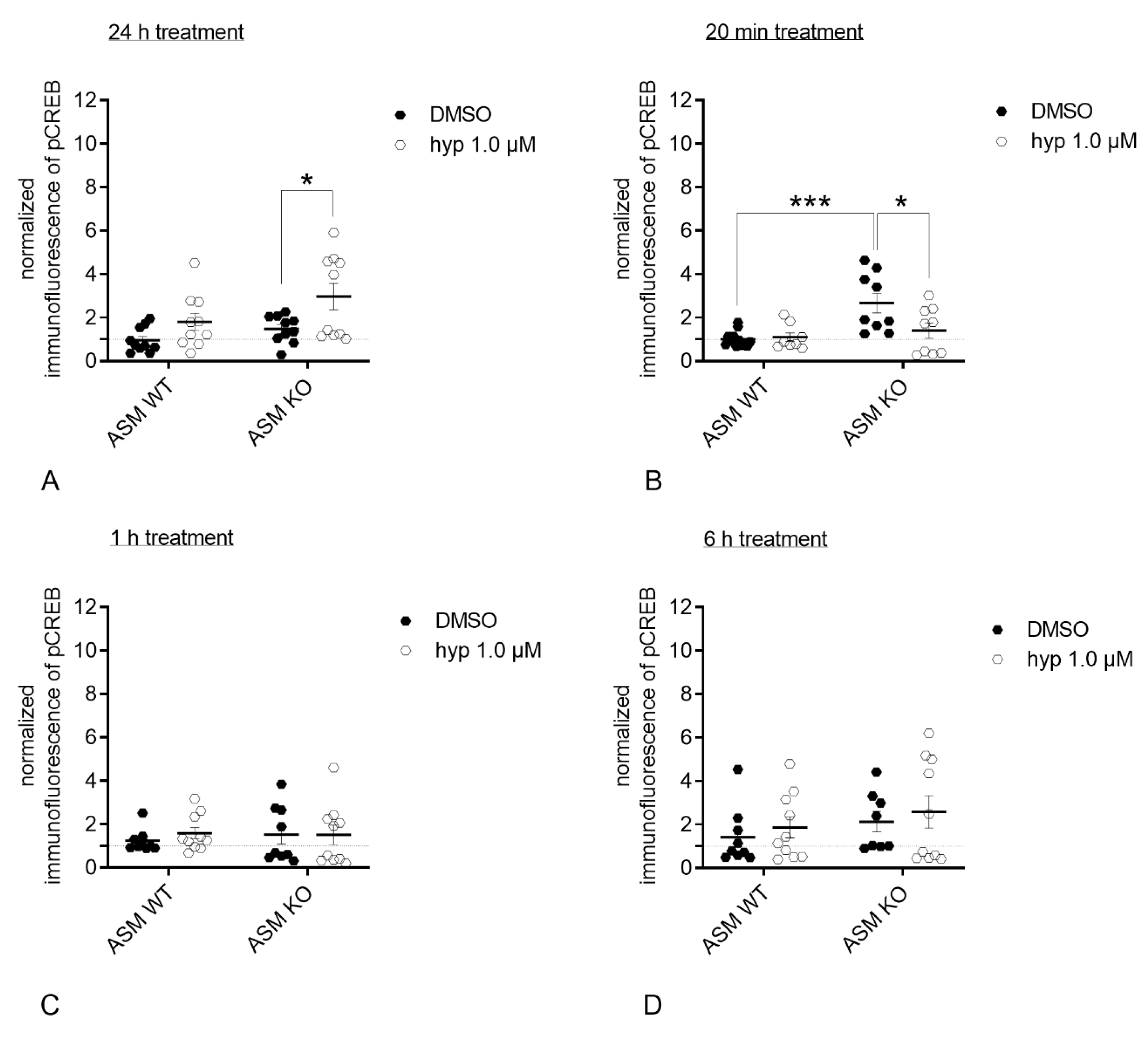
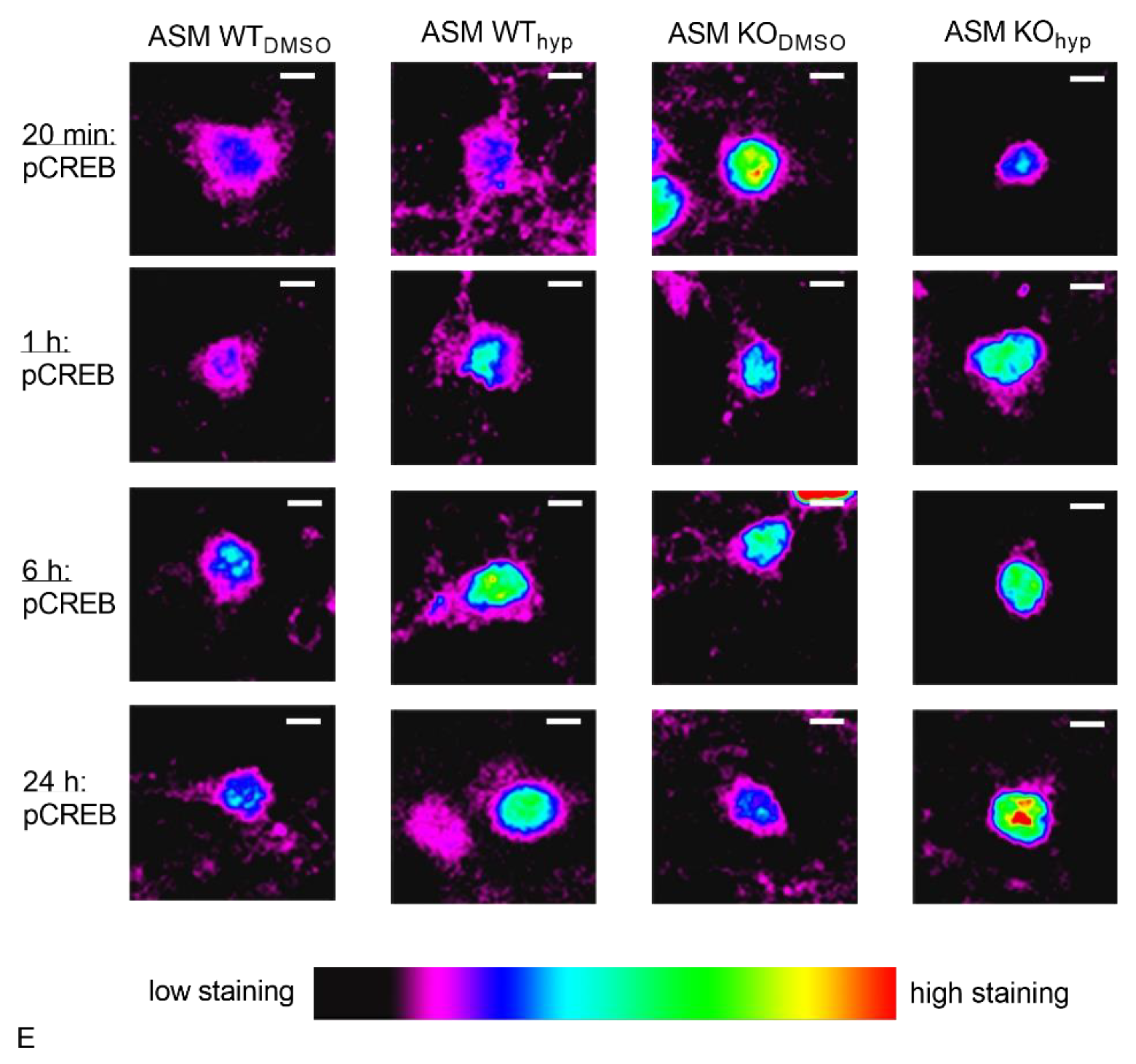
3.5. Genetic ASM Deficiency Impacts Hyperforin-Induced CREB-Phosphorylation in Primary Murine Neuronal Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Full Name |
| Anti/anti | antibiotic/antimycotic |
| ARC39 | C10 bisphosphonate 1-aminodecane-1,1-bisphosphonic acid |
| ASM | acid sphingomyelinase |
| ASM KO | ASM-knockout |
| ASM WT | ASM-wildtype |
| Cer | ceramide |
| CREB | cAMP response element-binding protein |
| DAG | diacylglycerol |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DIV | days in vitro |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| DMSO | dimethyl sulfoxide |
| E 18 | embryonic day 18 |
| EGTA | ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| FCS | fetal calf serum |
| Fura-2-AM | Fura-2-acetoxymethylester |
| GAD2/65 | glutamic acid decarboxylase 2/65 |
| gp | guinea pig |
| HBSS-/- | Hank’s balanced salt solution without calcium and magnesium |
| HBSS+/+ | Hank’s balanced salt solution with calcium and magnesium |
| HPLC | high-performance liquid chromatography |
| hyp | hyperforin |
| MAP 2 | microtubule associated protein 2 |
| MDD | major depressive disorder |
| MQ | Millipore water |
| ms | mouse |
| MS | mass spectrometry |
| NB | Neurobasal |
| NBA | Neurobasal A |
| P | postnatal age |
| PBS | phosphate buffered saline |
| PC12 | pheochromocytoma 12 cell line |
| PCR | polymerase chain reaction |
| pCREB | phospho-CREB (serine 133) |
| PPD-mix | papain-dispase-DNase mixture |
| rb | rabbit |
| RT | room temperature (20–22 °C) |
| S1P | sphingosine 1-phosphate |
| SRM | selected reaction monitoring |
| SDS | sodium dodecylsulfate |
| SM | sphingomyelin |
| Sph | sphingosine |
| TBST | TRIS buffered saline + 1% (v/v) Tween |
| TRIS | tris(hydroxymethyl)-aminomethan |
| TRPC6 | canonical transient receptor potential channel 6 |
| USA | United States of America |
| UV | ultraviolet light |
| vs. | versus |
References
- Gulbins, E.; Palmada, M.; Reichel, M.; Lüth, A.; Böhmer, C.; Amato, D.; Müller, C.P.; Tischbirek, C.H.; Groemer, T.W.; Tabatabai, G.; et al. Acid sphingomyelinase–ceramide system mediates effects of antidepressant drugs. Nat. Med. 2013, 19, 934–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornhuber, J.; Medlin, A.; Bleich, S.; Jendrossek, V.; Henkel, A.W.; Wiltfang, J.; Gulbins, E. High activity of acid sphingomyelinase in major depression. J. Neural Transm. 2005, 112, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Kornhuber, J.; Müller, C.P.; Becker, K.A.; Reichel, M.; Gulbins, E. The ceramide system as a novel antidepressant target. Trends Pharmacol. Sci. 2014, 35, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.B.; Kennedy, E.P. Sphingomyelinase in normal human spleens and in spleens from subjects with Niemann-Pick disease. J. Lipid Res. 1967, 8, 202–209. [Google Scholar] [PubMed]
- Grassmé, H.; Riethmüller, J.; Gulbins, E. Biological aspects of ceramide-enriched membrane domains. Prog. Lipid Res. 2007, 46, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Stancevic, B.; Kolesnick, R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 2010, 584, 1728–1740. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, X.; Becker, K.A.; Gulbins, E. Ceramide-enriched membrane domains—Structure and function. Biochim. Biophys. Acta Biomembr. 2009, 1788, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Lahiri, S.; Futerman, A.H. The metabolism and function of sphingolipids and glycosphingolipids. Cell. Mol. Life Sci. 2007, 64, 2270–2284. [Google Scholar] [CrossRef]
- Goñi, F.M.; Alonso, A. Sphingomyelinases: Enzymology and membrane activity. FEBS Lett. 2002, 531, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Stoffel, W.; Jenke, B.; Blöck, B.; Zumbansen, M.; Koebke, J. Neutral sphingomyelinase 2 (smpd3) in the control of postnatal growth and development. Proc. Natl. Acad. Sci. USA 2005, 102, 4554–4559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassmé, H.; Jekle, A.; Riehle, A.; Schwarz, H.; Berger, J.; Sandhoff, K.; Kolesnick, R.; Gulbins, E. CD95 Signaling via Ceramide-rich Membrane Rafts. J. Biol. Chem. 2001, 276, 20589–20596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Gulbins, E.; Zhang, Y. Oxidative stress triggers Ca-dependent lysosome trafficking and activation of acid sphingomyelinase. Cell. Physiol. Biochem. 2012, 30, 815–826. [Google Scholar] [CrossRef]
- Verderio, C.; Gabrielli, M.; Giussani, P. Role of sphingolipids in the biogenesis and biological activity of extracellular vesicles. J. Lipid Res. 2018, 59, 1325–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schissel, S.L.; Keesler, G.A.; Schuchman, E.H.; Williams, K.J.; Tabas, I. The Cellular Trafficking and Zinc Dependence of Secretory and Lysosomal Sphingomyelinase, Two Products of the Acid Sphingomyelinase Gene. J. Biol. Chem. 1998, 273, 18250–18259. [Google Scholar] [CrossRef] [Green Version]
- Schissel, S.L.; Schuchman, E.H.; Williams, K.J.; Tabas, I. Zn2+-stimulated Sphingomyelinase is Secreted by Many Cell Types and is a Product of the Acid Sphingomyelinase Gene. J. Biol. Chem. 1996, 271, 18431–18436. [Google Scholar] [CrossRef] [Green Version]
- Kornhuber, J.; Muehlbacher, M.; Trapp, S.; Pechmann, S.; Friedl, A.; Reichel, M.; Mühle, C.; Terfloth, L.; Groemer, T.W.; Spitzer, G.M.; et al. Identification of Novel Functional Inhibitors of Acid Sphingomyelinase. PLoS ONE 2011, 6, e23852. [Google Scholar] [CrossRef] [Green Version]
- Kornhuber, J.; Tripal, P.; Reichel, M.; Terfloth, L.; Bleich, S.; Wiltfang, J.; Gulbins, E. Identification of New Functional Inhibitors of Acid Sphingomyelinase Using a Structure—Property—Activity Relation Model. J. Med. Chem. 2008, 51, 219–237. [Google Scholar] [CrossRef]
- Kölzer, M.; Werth, N.; Sandhoff, K. Interactions of acid sphingomyelinase and lipid bilayers in the presence of the tricyclic antidepressant desipramine. FEBS Lett. 2004, 559, 96–98. [Google Scholar] [CrossRef] [Green Version]
- Olsen, A.S.B.; Færgeman, N.J. Sphingolipids: Membrane microdomains in brain development, function and neurological diseases. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Emir, T.L.R. TRP Channels in the Brain: What Are They There For? In The Neurobiological Basis of Suicide; Informa UK Limited: Colchester, UK, 2017; pp. 295–322. [Google Scholar]
- Just, S.; Chenard, B.L.; Ceci, A.; Strassmaier, T.; Chong, J.A.; Blair, N.T.; Gallaschun, R.J.; Del Camino, D.; Cantin, S.; D’Amours, M.; et al. Treatment with HC-070, a potent inhibitor of TRPC4 and TRPC5, leads to anxiolytic and antidepressant effects in mice. PLoS ONE 2018, 13, e0191225. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jia, Y.-C.; Cui, K.; Li, N.; Zheng, Z.-Y.; Wang, Y.-Z.; Yuan, X.-B. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nat. Cell Biol. 2005, 434, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Leuner, K.; Li, W.; Amaral, M.D.; Rudolph, S.; Calfa, G.; Schuwald, A.M.; Harteneck, C.; Inoue, T.; Pozzo-Miller, L. Hyperforin modulates dendritic spine morphology in hippocampal pyramidal neurons by activating Ca2+-permeable TRPC6 channels. Hippocampus 2013, 23, 40–52. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.; Feng, S.; Ge, R.; Du, W.; Zhang, X.; He, Z.; Wang, Y. TRPC6 channels promote dendritic growth via the CaMKIV-CREB pathway. J. Cell Sci. 2008, 121, 2301–2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Du, W.; Zhou, K.; Tai, Y.; Yao, H.; Jia, Y.; Ding, Y.; Wang, Y. Critical role of TRPC6 channels in the formation of excitatory synapses. Nat. Neurosci. 2008, 11, 741–743. [Google Scholar] [CrossRef]
- Du, W.; Huang, J.; Yao, H.; Zhou, K.; Duan, B.; Wang, Y. Inhibition of TRPC6 degradation suppresses ischemic brain damage in rats. J. Clin. Investig. 2010, 120, 3480–3492. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Zhang, J.-C.; Fu, J.; Chen, F.; Wang, J.; Wu, Z.-L.; Yuan, S.-Y. Hyperforin Attenuates Brain Damage Induced by Transient Middle Cerebral Artery Occlusion (MCAO) in Rats via Inhibition of TRPC6 Channels Degradation. Br. J. Pharmacol. 2013, 33, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Heiser, J.H.; Schuwald, A.M.; Sillani, G.; Ye, L.; Müller, W.E.; Leuner, K. TRPC6 channel-mediated neurite outgrowth in PC12 cells and hippocampal neurons involves activation of RAS/MEK/ERK, PI3K, and CAMKIV signaling. J. Neurochem. 2013, 127, 303–313. [Google Scholar] [CrossRef]
- Leuner, K.; Kazanski, V.; Muller, M.; Essin, K.; Henke, B.; Gollasch, M.; Harteneck, C.; Müller, W.E. Hyperforin—A key constituent of St. John’s wort specifically activates TRPC6 channels. FASEB J. 2007, 21, 4101–4111. [Google Scholar] [CrossRef] [Green Version]
- Samapati, R.; Yang, Y.; Yin, J.; Stoerger, C.; Arenz, C.; Dietrich, A.; Gudermann, T.; Adam, D.; Wu, S.; Freichel, M.; et al. Lung Endothelial Ca2+ and Permeability Response to Platelet-Activating Factor Is Mediated by Acid Sphingomyelinase and Transient Receptor Potential Classical. Am. J. Respir. Crit. Care Med. 2012, 185, 160–170. [Google Scholar] [CrossRef]
- Tabeling, C.; Yu, H.; Wang, L.; Ranke, H.; Goldenberg, N.M.; Zabini, D.; Noe, E.; Krauszman, A.; Gutbier, B.; Yin, J.; et al. CFTR and sphingolipids mediate hypoxic pulmonary vasoconstriction. Proc. Natl. Acad. Sci. USA 2015, 112, E1614–E1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitler, S.; Ye, L.; Andreyeva, A.; Schumacher, F.; Monti, J.; Nürnberg, B.; Nowak, G.; Kleuser, B.; Reichel, M.; Fejtová, A.; et al. Acid sphingomyelinase—A regulator of canonical transient receptor potential channel 6 (TRPC6) activity. J. Neurochem. 2019, 150, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Horinouchi, K.; Erlich, S.; Perl, D.P.; Ferlinz, K.; Bisgaier, C.L.; Sandhoff, K.; Desnick, R.J.; Stewart, C.L.; Schuchman, E.H. Acid sphingomyelinase deficient mice: A model of types A and B Niemann-Pick disease. Nat. Genet. 1995, 10, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Frischknecht, R.; Heine, M.; Perrais, D.; Seidenbecher, C.I.; Choquet, D.; Gundelfinger, E.D. Brain extracellular matrix affects AMPA receptor lateral mobility and short-term synaptic plasticity. Nat. Neurosci. 2009, 12, 897–904. [Google Scholar] [CrossRef]
- Facci, L.; Skaper, S.D. Culture of Rodent Cortical, Hippocampal, and Striatal Neurons. Recent Results Cancer Res. 2017, 1727, 39–47. [Google Scholar] [CrossRef]
- Deusser, J.; Schmidt, S.; Ettle, B.; Plötz, S.; Huber, S.; Müller, C.P.; Masliah, E.; Winkler, J.; Ekohl, Z. Serotonergic dysfunction in the A53T alpha-synuclein mouse model of Parkinson’s disease. J. Neurochem. 2015, 135, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Davydova, D.; Marini, C.; King, C.; Klueva, J.; Bischof, F.; Romorini, S.; Montenegro-Venegas, C.; Heine, M.; Schneider, R.; Schröder, M.S.; et al. Bassoon Specifically Controls Presynaptic P/Q-type Ca2+ Channels via RIM-Binding Protein. Neuron 2014, 82, 181–194. [Google Scholar] [CrossRef] [Green Version]
- Kaech, S.; Banker, G. Culturing hippocampal neurons. Nat. Protoc. 2006, 1, 2406–2415. [Google Scholar] [CrossRef]
- Mühle, C.; Kornhuber, J. Assay to measure sphingomyelinase and ceramidase activities efficiently and safely. J. Chromatogr. A 2017, 1481, 137–144. [Google Scholar] [CrossRef]
- Gulbins, A.; Schumacher, F.; Becker, K.A.; Wilker, B.; Soddemann, M.; Boldrin, F.; Müller, C.P.; Edwards, M.J.; Goodman, M.; Caldwell, C.C.; et al. Antidepressants act by inducing autophagy controlled by sphingomyelin-ceramide. Mol. Psychiatry 2018, 23, 2324–2346. [Google Scholar] [CrossRef] [Green Version]
- Kachler, K.; Bailer, M.; Heim, L.; Schumacher, F.; Reichel, M.; Holzinger, C.D.; Trump, S.; Mittler, S.; Monti, J.; Trufa, D.; et al. Enhanced Acid Sphingomyelinase Activity Drives Immune Evasion and Tumor Growth in Non–Small Cell Lung Carcinoma. Cancer Res. 2017, 77, 5963–5976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leuner, K.; Heiser, J.H.; Derksen, S.; Mladenov, M.I.; Fehske, C.J.; Schubert, R.; Gollasch, M.; Schneider, G.; Harteneck, C.; Chatterjee, S.S.; et al. Simple 2,4-Diacylphloroglucinols as Classic Transient Receptor Potential-6 Activators—Identification of a Novel Pharmacophore. Mol. Pharmacol. 2009, 77, 368–377. [Google Scholar] [CrossRef] [Green Version]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [PubMed]
- Roth, A.G.; Drescher, D.; Yang, Y.; Redmer, S.; Uhlig, S.; Arenz, C. Potent and Selective Inhibition of Acid Sphingomyelinase by Bisphosphonates. Angew. Chem. Int. Ed. 2009, 48, 7560–7563. [Google Scholar] [CrossRef] [PubMed]
- Naser, E.; Kadow, S.; Schumacher, F.; Mohamed, Z.H.; Kappe, C.; Hessler, G.; Pollmeier, B.; Kleuser, B.; Arenz, C.; Becker, K.A.; et al. Characterization of the small molecule ARC39, a direct and specific inhibitor of acid sphingomyelinase in vitro. J. Lipid Res. 2020, 61, 896–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ImageJ-Image Processing and Analysis in Java. Available online: https://imagej.nih.gov/ij/download.html (accessed on 17 November 2020).
- Villani, M.; Subathra, M.; Im, Y.-B.; Choi, Y.; Signorelli, P.; Del Poeta, M.; Luberto, C. Sphingomyelin synthases regulate production of diacylglycerol at the Golgi. Biochem. J. 2008, 414, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Galvan, C.; Camoletto, P.G.; Cristofani, F.; Van Veldhoven, P.P.; Ledesma, M.D. Anomalous Surface Distribution of Glycosyl Phosphatidyl Inositol–anchored Proteins in Neurons Lacking Acid Sphingomyelinase. Mol. Biol. Cell 2008, 19, 509–522. [Google Scholar] [CrossRef]
- Buccinnà, B.; Piccinini, M.; Prinetti, A.; Scandroglio, F.; Prioni, S.; Valsecchi, M.; Votta, B.; Grifoni, S.; Lupino, E.; Ramondetti, C.; et al. Alterations of myelin-specific proteins and sphingolipids characterize the brains of acid sphingomyelinase-deficient mice, an animal model of Niemann-Pick disease type A. J. Neurochem. 2009, 109, 105–115. [Google Scholar] [CrossRef]
- Contreras, F.-X.; Ernst, A.M.; Haberkant, P.; Björkholm, P.; Lindahl, E.; Gönen, B.; Tischer, C.; Elofsson, A.; Von Heijne, G.; Thiele, C.; et al. Molecular recognition of a single sphingolipid species by a protein’s transmembrane domain. Nat. Cell Biol. 2012, 481, 525–529. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, S.; Birod, K.; Männich, J.; Eberle, M.; Wegner, M.-S.; Wanger, R.; Hartmann, D.; Ferreirós, N.; Geisslinger, G.; Grösch, S. Ceramide metabolism in mouse tissue. Int. J. Biochem. Cell Biol. 2013, 45, 1886–1894. [Google Scholar] [CrossRef]
- Ledesma, M.D.; Prinetti, A.; Sonnino, S.; Schuchman, E.H. Brain pathology in Niemann Pick disease type A: Insights from the acid sphingomyelinase knockout mice. J. Neurochem. 2011, 116, 779–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scandroglio, F.; Venkata, J.K.; Loberto, N.; Prioni, S.; Schuchman, E.H.; Chigorno, V.; Prinetti, A.; Sonnino, S. Lipid content of brain, brain membrane lipid domains, and neurons from acid sphingomyelinase deficient mice. J. Neurochem. 2008, 107, 329–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonze, B.E.; Ginty, D.D. Function and Regulation of CREB Family Transcription Factors in the Nervous System. Neuron 2002, 35, 605–623. [Google Scholar] [CrossRef] [Green Version]
- Gibon, J.; Deloulme, J.-C.; Chevallier, T.; Ladevèze, E.; Abrous, D.N.; Bouron, A. The antidepressant hyperforin increases the phosphorylation of CREB and the expression of TrkB in a tissue-specific manner. Int. J. Neuropsychopharmacol. 2013, 16, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornhauser, J.M.; Cowan, C.W.; Shaywitz, A.J.; Dolmetsch, R.E.; Griffith, E.C.; Hu, L.S.; Haddad, C.; Xia, Z.; Greenberg, M.E. CREB Transcriptional Activity in Neurons Is Regulated by Multiple, Calcium-Specific Phosphorylation Events. Neuron 2002, 34, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.-Y.; Deisseroth, K.; Tsien, R.W. Activity-dependent CREB phosphorylation: Convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 2808–2813. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Bhattacharya, S.; Wonnemann, M.; Singer, A.; Müller, W. Hyperforin as a possible antidepressant component of hypericum extracts. Life Sci. 1998, 63, 499–510. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K. Synaptic Dysfunction in Depression: Potential Therapeutic Targets. Science 2012, 338, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, A.I.; Camoletto, P.G.; Morando, L.; Sassoe-Pognetto, M.; Giustetto, M.; Van Veldhoven, P.P.; Schuchman, E.H.; Ledesma, M.D. Pharmacological reversion of sphingomyelin-induced dendritic spine anomalies in a Niemann Pick disease type A mouse model. EMBO Mol. Med. 2014, 6, 398–413. [Google Scholar] [CrossRef] [Green Version]
- Camoletto, P.G.; Vara, H.; Morando, L.; Connell, E.; Marletto, F.P.; Giustetto, M.; Sassoé-Pognetto, M.; Van Veldhoven, P.P.; Ledesma, M.D. Synaptic Vesicle Docking: Sphingosine Regulates Syntaxin1 Interaction with Munc18. PLoS ONE 2009, 4, e5310. [Google Scholar] [CrossRef]
- Popova, D.; Castrén, E.; Taira, T. Chronic fluoxetine administration enhances synaptic plasticity and increases functional dynamics in hippocampal CA3-CA1 synapses. Neuropharmacology 2017, 126, 250–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesca, F.; Baldelli, P.; Valtorta, F.; Benfenati, F. The synapsins: Key actors of synapse function and plasticity. Prog. Neurobiol. 2010, 91, 313–348. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeitler, S.; Schumacher, F.; Monti, J.; Anni, D.; Guhathakurta, D.; Kleuser, B.; Friedland, K.; Fejtová, A.; Kornhuber, J.; Rhein, C. Acid Sphingomyelinase Impacts Canonical Transient Receptor Potential Channels 6 (TRPC6) Activity in Primary Neuronal Systems. Cells 2020, 9, 2502. https://doi.org/10.3390/cells9112502
Zeitler S, Schumacher F, Monti J, Anni D, Guhathakurta D, Kleuser B, Friedland K, Fejtová A, Kornhuber J, Rhein C. Acid Sphingomyelinase Impacts Canonical Transient Receptor Potential Channels 6 (TRPC6) Activity in Primary Neuronal Systems. Cells. 2020; 9(11):2502. https://doi.org/10.3390/cells9112502
Chicago/Turabian StyleZeitler, Stefanie, Fabian Schumacher, Juliana Monti, Daniela Anni, Debarpan Guhathakurta, Burkhard Kleuser, Kristina Friedland, Anna Fejtová, Johannes Kornhuber, and Cosima Rhein. 2020. "Acid Sphingomyelinase Impacts Canonical Transient Receptor Potential Channels 6 (TRPC6) Activity in Primary Neuronal Systems" Cells 9, no. 11: 2502. https://doi.org/10.3390/cells9112502