The Effects of Divalent Cation-Chelated Prion Fibrils on the Immune Response of EOC 13.31 Microglia Cells


1
Department of Biomedical Sciences, National Chung Cheng University, 168 University Road, Min-Hsiung Chia-Yi 62102, Taiwan
2
Center for Nano Bio-Detections, National Chung Cheng University, Min-Hsiung Chia-Yi 62102, Taiwan
3
Center for Innovative Research on Aging Society (CIRAS), National Chung Cheng University, Min-Hsiung Chia-Yi 62102, Taiwan
*
Author to whom correspondence should be addressed.
Cells 2020, 9(10), 2285; https://doi.org/10.3390/cells9102285
Submission received: 31 August 2020
/
Revised: 29 September 2020
/
Accepted: 12 October 2020
/
Published: 13 October 2020
(This article belongs to the Special Issue Regulation of Cytokine Signaling in Immunity)
Abstract
:Transmissible spongiform encephalopathies (TSEs) are epidemic neurodegenerative diseases caused by prion proteins; in particular, they are induced by misfolded prion proteins (PrPSc). PrPSc tend to aggregate into insoluble amyloid prion fibrils (fPrPWT), resulting in apoptosis of neuron cells and sequential neurodegeneration. Previous studies indicate that microglia cells play an important role in the innate immune system, and that these cells have good neuroprotection and delay the onset of TSEs. However, microglia can be a double-sided blade. For example, both Cu2+ and Mn2+ can induce microglia activation and secrete many inflammatory cytokines that are fatal to neuron cells. Unfortunately, PrP have cation binding sites at the N-terminus. When PrPSc accumulate during microglial phagocytosis, microglia may change the phenotype to secrete pro-inflammation cytokines, which increases the severity of the disease. Some studies have revealed an increase in the concentration of Mn2+ in the brains of patients. In this study, we treated microglia with fPrPWT and cations and determined IκBα and IL-1β expression by Western blotting and quantitative polymerase chain reaction. The results showed that Mn–fPrPWT decreased IκBα levels and dramatically increased IL-1β mRNA expression. In addition, competing binding between Cu2+ and Mn2+ can decrease the effect of Mn–fPrPWT on IκBα and IL-1β. The effects of divalent cations and fPrPWT in microglia inflammation are also discussed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Transmissible spongiform encephalopathies (TSEs), also called prion diseases, are neurodegenerative diseases. Mammals can become infected with TSEs through inherent, transmitted, and sporadic conditions [1]. In humans, the population that contracts TSEs through sporadic prion conditions is higher (more than 85% of patients) than the transmitted and inherent populations [2,3,4]. Transmitted prion diseases, such as the variant Creutzfeldt–Jakob disease (vCJD), have drawn more attention and are mainly ascribed to infection through blood or viscera [5,6]. Scrapie prion proteins (PrPSc), abnormal isoforms of cellular prion proteins (PrPC), are pathogens of prion diseases. Structurally, PrPC are rich in α-helices, whereas PrPSc are rich in β-sheets. Once PrPSc present in the central nervous system (CNS), PrPSc can serve as a template for PrPC to process a structural conversion from an α-helix-rich structure to a β-sheet-rich structure [7,8]. The accumulation of misfolded PrPSc can induce brain inflammation, neuron degeneration, and plaque aggregation [9,10]. Patients carrying prion diseases rapidly develop progressive dementia with behavioral abnormalities, ataxia, and myoclonus [9].
Prion proteins contain a flexible N-terminal domain and a structured C-terminal domain. In the N-terminal domain, there are five contiguous copies of octapeptides (P(H/Q)GG(G/S/T)WGQ) called octapeptide repeats (ORs) [11]. Among these octapeptides, four of them contain histidine residues to bind divalent cations such as copper and manganese ions. The repetition of ORs affects the age of onset in patients [12]. High concentrations of divalent cations in the CNS can accelerate fibril conversion [13] and can, therefore, enhance the toxicity in neuron cells [14]. According to a blood investigation, patients with neurodegenerative diseases have a high concentration of heavy metal ions [13]. Sporadic Creutzfeldt–Jakob disease (sCJD) patients have higher concentrations of copper and manganese ions, while patients with Alzheimer’s disease have higher concentrations of copper ions, and patients with Parkinson’s disease have higher concentrations of iron and zinc ions [15]. However, an analysis of TSE-infected mouse/human brain homogenates using X-ray photoelectron emission microscopy indicated reduced levels of copper and increased levels of manganese [16,17]. Furthermore, Cu2+-binding in the ORs of PrPC can protect cells against oxidative stress. This protection is lost when PrPC are converted into PrPSc [18]. Therefore, the divalent cations in the CNS are important and complicated in neurodegenerative diseases.
Microglia are the primary immune cells in the CNS. Microglia are responsible for maintaining the brain’s homeostasis and play an important role in prion diseases. Microglia occupy approximately 10% of healthy brain and spinal cord cells [19], and constantly patrol brain parenchyma and sensitively respond to environmental changes in the brain [20]. Upon abnormal changes in the environment, microglia in the homeostasis state (M0) are activated into a pro-inflammatory state (M1) or an anti-inflammatory state (M2). The physiological response in this microglia activation includes killing abnormal cells, pathogen clearance, and cytokine secretion. In neurodegenerative diseases, microglia cells are turned into activated states (i.e., M1 and M2) [19]. The M1 state secretes inflammatory cytokines, such as IL-1β and tumor necrosis factor-α (TNF-α), to cause apoptosis of neuron cells [21]. The M2 state secretes anti-inflammatory cytokines, such as TGF-β and IL-10, to protect cells and to inhibit the production of pro-inflammation cytokines. The switch between the M0 and M1/M2 states depends on the factors in milieu and stimulated microglia possess states of classical activation, alternative activation, and acquired deactivation [22,23]. Classical activation can induce pro-inflammatory cytokines, such as TNF-α, IL-1β, and NO [24,25]. Alternative activation is associated with the M2 state and is only activated by IL-4 and IL-13 [23]. Acquired deactivation can alleviate acute inflammation and can be induced by uptake of apoptosis cells, i.e., IL-10 and transforming growth factor-β (TGF-β) [22,26].
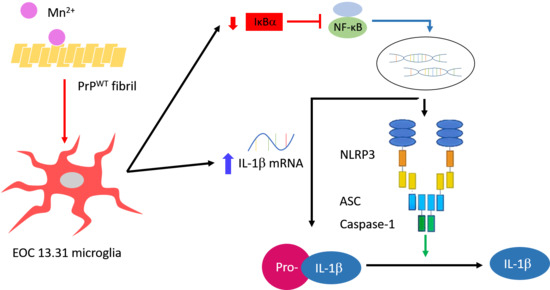
Activation of the nucleotide-binding and oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome indicates chronic inflammation in a variety of inflammatory cells [27]. As shown in Figure 1, the main components of the NLRP3 inflammasome are NLRP3 protein, apoptosis-associated speck-like protein containing a CARD (ASC), and pro-caspase-1 [28]. The activity of the NLRP3 inflammasome requires upstream signals of priming and activation in sequence [29]. Priming, the first signal, can be triggered by microbial molecules (e.g., lipopolysaccharide (LPS)) or by endogenous cytokines (e.g., TNF-α). This priming signal can increase the activity of NF-κB, resulting in the high expression of NLRP3 and pro-IL-1β. Activation, the second signal, can be triggered by multiple damage-associated molecular patterns (DAMPs) and pathogen-associated molecular pattern molecules (PAMPs). Moreover, K+-efflux, Ca2+, reactive oxygen species (ROS), and lysosomal ruptures participate in the mechanism of the NLRP3 inflammasome activation [29]. Pro-caspase-1 is recruited to the NLRP3 inflammasome and processed into caspase-1 via autocatalysis. Caspase-1 can cleave pro-inflammatory cytokine such as pro-IL-1β and pro-IL18 [28]. In addition, Mn2+ induces activation of the NLRP3 inflammasome, resulting in pro-caspase-1 cleavage [30]. These sequential signals can promote pro-IL-1β cleavage, can secrete more IL-1β, and can induce inflammation in neurons.
As aforementioned, recent studies have revealed the correlation between neurodegenerative diseases and microglia. However, the effect of metal-chelated prion fibrils in microglia remains ambiguous. In this research, we prepared prion proteins with or without ORs and their fibrils. We treated microglia cells with fibrils chelated with copper and/or manganese ions to study inflammation-associated mRNA. The effect of Cu2+- and Mn2+-chelated prion fibrils were investigated.
2. Materials and Methods
2.1. Prion Purification
The wild-type mouse prion plasmid pET101 with insertion of PrP23–231 (PrPWT) and PrPΔOct genes [31] were transformed to BL21 Escherichia coli (E. coli.) individually. After protein overexpression induced by isopropyl β-D-1-thiogalactopyranoside, E. coli was disrupted by French press or lysozyme treatment. Inclusion bodies were collected by centrifugation and then dissolved in urea- and glutathione-containing buffer at pH 8.5 according to a previously described procedure [32]. For PrP23–231, the soluble inclusion bodies were mixed with nickel-chelating Sepharose for further experimentation with immobilized metal affinity chromatography. Prion proteins were eluted with urea-containing buffer at pH 4. For PrPΔOct, the protein was purified by sulphopropyl (SP) Sepharose cation exchange chromatography. Fractions containing prion proteins were refolded in glutathion disulfide overnight. The refolded prion proteins were further purified with a C4 reversed-phase high-performance liquid chromatography (HPLC) column under an elution of acetonitrile/water gradient. After lyophilization, the purified protein powders were stored at −80 °C. The purity of the prion was confirmed by SDS-PAGE.
2.2. Fibril Conversion
Fibril conversion starting from 0.25 μg/μL PrPWT or PrPΔOct was performed in buffer solution containing 50 mM of 2-(N-morpholino) ethanesulfonic acid (MES, pH 6.5), 2 M of GdnHCl, and 2.5 ng/μL of PrPWT or PrPΔOct fibril seeds at 37 °C under shaking as described previously [33]. The progress of fibril formation was monitored by fluorescence intensity of thioflavin T (ThT) for its binding to the cross-β-sheets in the fibrils. The fibrils converted from PrPWT and PrPΔOct are denoted as fPrPWT or fPrPΔOct, respectively. The morphology of fPrPWT was examined by transmission electron microscopy (TEM) according to a previously described procedure [33]. The fibril samples were dialyzed against water and then mixed with CuCl2 or MnCl2 for further experiments. The binding of copper ions to fPrPWT was examined using a Copper Assay Kit (Sigma-Aldrich, St. Louis, MO, USA).
2.3. Cell Culture
The immortalized mouse brain EOC 13.31 microglia cells (ATCC CRL-2468) were purchased from the Bioresources Collection and Research Center (BCRC, Hsinchu, Taiwan). The condition medium containing CSF-1 for EOC 13.31 culture was collected from mouse bone marrow cell LADMAC (ATCC CRL-2420) after five to seven days of culture. The culture medium of EOC 13.31 cells contained Dulbecco’s modified Eagle medium (DMEM) supplemented with 4 mM of L-glutamine, 1.5 g/L of sodium bicarbonate, and 4.5 g/L of glucose at pH 7.4. After filtering the DMEM with a 0.22 μM filter cup, the condition medium and fetal bovine serum (FBS) were added to the DMEM to reach final concentrations at 20% and 10%, respectively. The LADMAC was cultured in Eagle’s Minimum Essential Medium (EMEM) supplemented with 1.5 g/L of sodium bicarbonate, 0.1 mM of non-essential amino acids, 1.0 mM of sodium pyruvate, and 10% FBS.
2.4. Detection of Cell Viability and Cellular Reactive Oxygen Species
EOC 13.31 cells were cultured in 96-well plates and incubated overnight. In the cell viability assays, EOC 13.31 cells were treated with fPrP and cations for 24 h, and then 10 μL Cell Counting Kit 8 (CCK8) reagent was added for 40 min. Cell viability was determined based on the decrease of OD450 recorded by a plate reader (Thermo Fisher Scientific, Waltham, MA, USA). To detect the cellular ROS in the EOC 13.31 cells, the microglia cells were cultured in 96-well black plates and incubated overnight. After treatment of fPrP and cations for 4 h, 50 μM of 2′,7′-dichlorofluorescin diacetate (DCFDA) was added and incubated for 40 min. Fluorescence data were recorded via a fluorescence plate reader (BMG LABTECH, Offenburger, Germany).
2.5. Western Blot
A radioimmunoprecipitation assay (RIPA) lysis buffer with protease and a phosphatase inhibitor was used to collect cell lysate after washing with PBS. The cells were lysed with RIPA lysis buffer for 15 min on ice and then collected by cell scrapers. The collected supernatant was centrifuged at 12,000× g for 10 min and stored at −20 °C. The cell lysate was diluted by 2× sample buffer and then heated at 95 °C for 5 min. Gel electrophoresis was performed with 12.5% SDS-PAGE. Afterward, wet polyvinylidene fluoride (PVDF) membranes were soaked in methanol for at least 15 min. Transfer buffer containing 0.37 M of glycine, 46.2 mM of Tris-base, and 20% methanol (v/v) was used to transfer proteins onto the PVDF membrane under 400 mA for 2 h at 4 °C. The membranes were blocked with 5% skimmed milk in phosphate buffered saline with Tween® 20 (PBST) for 1 h and then washed with PBST. Afterward, primary antibody diluted 1000 fold with 5% skimmed milk in PBST was used to interact with the membrane under shaking at room temperature for 1 h. Four primary antibodies were used: ab9722, ab263899, and ab32518 from Abcam for IL-1β, NLRP3, and IκBα, respectively, and A5441 from Sigma for β-actin. After washing with PBST, secondary antibodies (A9044 and A0545 from Sigma for anti-mouse IgG and anti-rabbit IgG, respectively) diluted 100,000 fold with 5% skimmed milk in PBST were used for further incubation for 1 h at room temperature. The Western blot membranes were imaged by a luminescence imaging system (TopBio, Czechia) and analyzed using Image J software (National Institutes of Health, MD, USA).
2.6. Extraction of mRNA
Cells were dissociated from culture dishes by 0.05% trypsin. Subsequently, the medium was removed by centrifugation. The cell pellets were washed twice by PBS. Cells were lysed by incubation with 1 mL of TRIzol® reagent for 5–10 min. Afterward, 200 μL chloroform was added for mRNA extraction. The extracted mRNA was precipitated by 50% isopropanol. The mRNA precipitate was then washed with 75% ethanol (diluted with diethyl pyrocarbonate-treated water) and then spun down. Finally, the mRNA was air-dried and stored at −20 °C until further measurements.
2.7. Quantitative PCR
Total mRNA was treated with DNase (Invitrogen, Carlsbad, CA, USA) to remove residual DNA, then converted to cDNA with Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT, Invitrogen, Carlsbad, CA, USA) for 50 min at 37 °C. The reaction was terminated by heating polymerase at 70 °C for 10 min. The Power SYBR Green PCR Master Mix Kit (Applied Biosystems, Foster City, CA, USA) was used to run quantitative PCR (qPCR). Each well contained 20 ng of cDNA, 10 μL of SYBR green mix, 10 μM of forward primer, and 10 μM of reverse primer. In addition, diethyl pyrocarbonate (DEPC)-treated water was added to the total sample volume of 20 μL. The primers used for the qPCR were IL-1β forward, 5′-TCC TCT CCA GCC AAG CTT CC-3′, reverse, 5′-GGT TTG GAA GCA GCC CTT CAT-3′; GAPDH forward, 5′-AGG TCG GTG TGA ACG GAT TTG-3′, reverse, 5′-TGT AGA CCA TGT AGT TGA GGT CA-3′. In the qPCR, the temperatures were set at 60 °C and 72 °C for annealing and elongation, respectively. The relative amount of IL-1β mRNA was determined using the comparative Ct (ΔΔCt) method by normalizing the target mRNA Ct values to GAPDH. All reported data were obtained from triplicate measurements.
2.8. Statistical Analysis
All reported data were collected from three independent experiments, and values were analyzed by the Student’s t-test using GraphPad 5.0 software. The images of Western blotting were analyzed by Image J software. Statistical significance is shown using asterisks (ns: p > 0.05; *: p < 0.05; **: p < 0.005; ***: p < 0.0005).
3. Results
3.1. The Effect of Divalent Cations and Fprpwt on the Production of Cellular Ros in Eoc 13.31 Microglia Cells
The kinetics of fPrPWT formation is shown in the time course of ThT fluorescence, and the morphology of fPrPWT is shown in a TEM image (Figure S1). To examine the effect of divalent cations and fPrPWT on the cellular ROS production in microglia, we determined the production of cellular ROS based on the fluorescence of DCFDA in EOC 13.31 cells. As shown in Figure 2a, cellular ROS increased when treated with copper and manganese ions in a dose-dependent manner. In contrast, cellular ROS production was not related to the dose of fPrPWT (Figure 2b). These results indicate that copper or manganese ions, rather than fPrPWT, can induce ROS in microglia cells. In Figure 2c, one can see that fPrPWT did not increase ROS levels with the addition of copper, but it slightly increased the levels of ROS with the addition of manganese. It is known that PrPC can decrease manganese uptake and ROS level in cells [34]. Therefore, we propose that the PrPC to PrPSc conversion caused the loss of PrPC function. As a result, the cellular manganese and ROS levels were enhanced. In addition to the ROS assay, the viability of the cells after treatment with fPrPWT and cations for 24 h was tested. As shown in Figure S2, cation-bound fPrPWT caused weak cell death. The cells were in good condition for ROS determination.
3.2. The Effect of Divalent Cations on the Expression of nlrp3 and IκBα in Fprpwt-Treated Microglia Cells
Cellular ROS cause mitochondrial damage and stimulate NLRP3 inflammasome activation [30,35]. Although fPrPWT cannot induce cellular ROS in EOC 13.31 cells, it can slightly increase the level of NLRP3 (Figure 3b). According to Figure 1, fPrPWT might activate NF-κB in microglia. IκBα is an inhibitor of NF-κB. When IκBα is phosphorylated and degraded, the active NF-κB is released and enters the nucleus to promote target gene expression such as NLRP3, IL-1β, or IL-18. Treatment with Mn–fPrPWT can severely decrease IκBα protein levels (Figure 3a,b). This indicates that NF-κB activity is increased in Mn–fPrPWT treatment. In contrast, Cu2+-chelated fPrPWT (Cu–fPrPWT) does not activate NF-κB in microglia, and treatment with Cu2+ or Mn2+ does not decrease IκBα protein levels (Figure 3c,d). Therefore, the increase of NF-κB activity is ascribed to Mn–fPrPWT rather than free Mn2+. There is no difference between the control and 10 μM of Cu2+ or Mn2+ treatment in NLRP3 levels. Notably, Mn–fPrPWT increases NF-κB activity, but it does not affect NLRP3 expression.
3.3. The Effect of Divalent Cations on the Expression of IL-1β Mrna in Fprpwt-Treated Microglia Cells
Previous results showed that Mn–fPrPWT increases NF-κB activity. Therefore, we tested the mRNA level of IL-1β, another NF-κB downstream protein, by qPCR. Mn–fPrPWT was able to upregulate the mRNA levels of IL-1β, but copper chelation had no such effect on the microglia cells (Figure 4a). Notably, IL-1β mRNA levels were not affected by treatment with fPrPWT or Cu2+ (Figure 4a,b). This result shows that only Mn–fPrPWT can promote the priming signal in the NLRP3 inflammasome by inducing NF-κB activity and the sequential upregulation of IL-1β mRNA in microglia.
3.4. The Effect of Octapeptide Repeat of Prions on the Expression of Inflammatory Cytokines in Microglia Cells
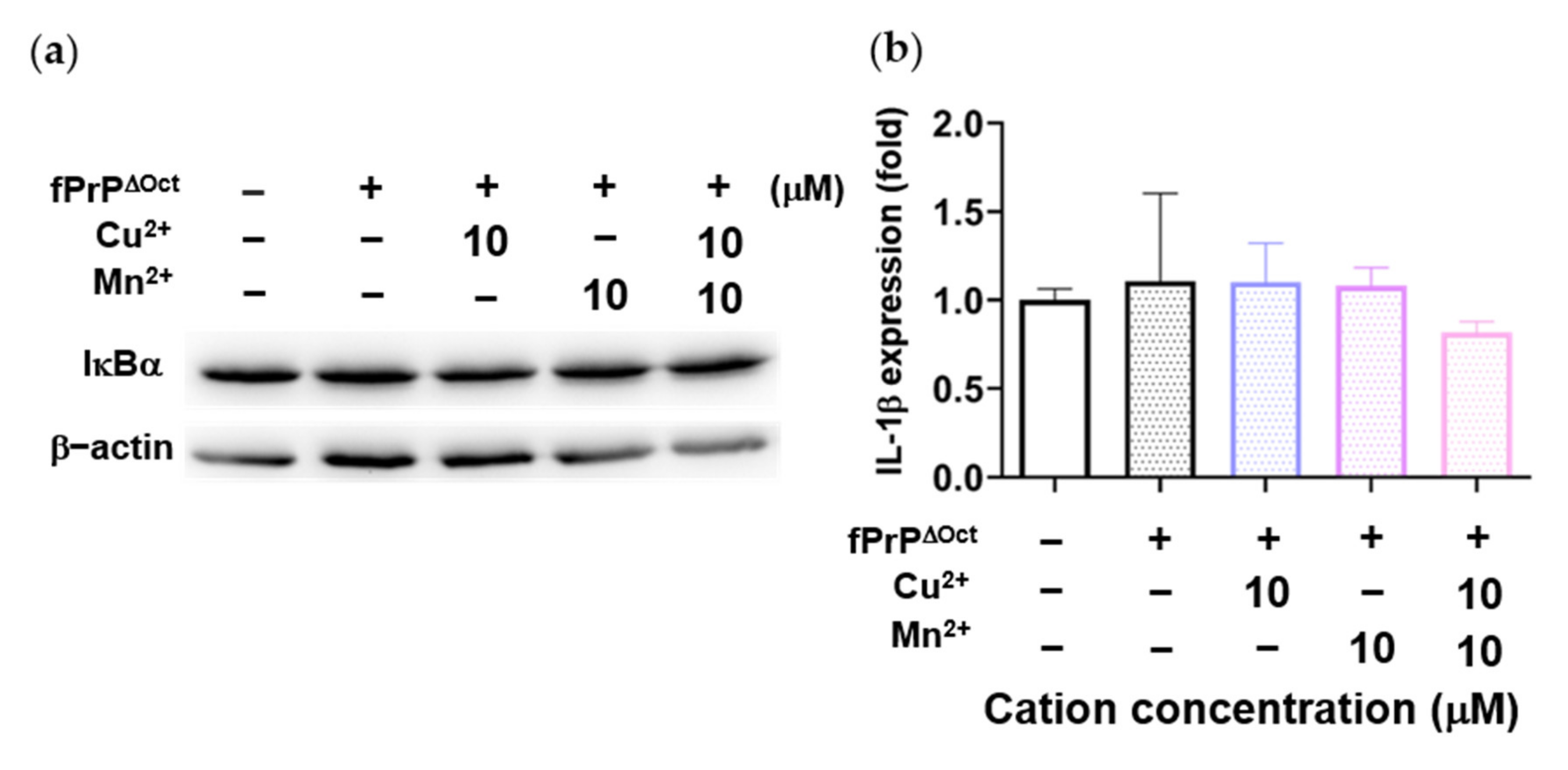
As Figure 3 and Figure 4 indicate, Mn–fPrPWT rather than fPrPWT was able to activate NF-κB, the metal binding sites of prions critical in microglia inflammation. The binding of copper ions in fPrPWT is shown in Figure S3. As divalent cations bound to octapeptide repeat regions of prion proteins, we used fibrils converted from octapeptide-deleted prions (fPrPΔOct) to perform the above experiments. Mn–fPrPΔOct treatment did not change the expression of IκBα (Figure 5a) and IL-1β (Figure 5b). This result indicates that Mn–fPrPΔOct does not activate NF-κB as Mn–fPrPWT does. Conclusively, the binding of manganese ions on prion fibrils is critical for inducing the priming signal in the NLRP3 inflammasome.
4. Discussion
Prion proteins are a well-known Cu2+-binding proteins with four strong binding sites [36]. Prion proteins can bind Mn2+ with moderate affinity and are considered a manganese-binding protein [37]. Manganese ions have been found to be neurotoxic to the human brain and cause cognitive dysfunction and motor impairment resembling Parkinson’s disease [38]. The concentration of manganese ions in the brain of prion disease patients is higher than that in healthy brains [16], especially in accumulated plaques [15]. Other than copper and manganese ions, zinc and iron ions are also essential metal ions in humans. In terms of iron, PrPC are considered to modulate iron homeostasis with the Zrt, Irt-like protein (ZIP) family [39], but the role of iron in the inflammation associated with prion disease is ambiguous. In regards to zinc, there are many studies focusing on zinc as an antagonist with copper [40], and PrPC can induce the uptake of zinc in neurons [41]. In addition, an increase in zinc levels decreases the activity of the NLRP3 inflammasome [42,43]. Therefore, a zinc replacement in PrPSc can potentially inhibit inflammation in microglia.
In previous research, fPrPWT was shown to activate the NLRP3 inflammasome and IL-1β cleavage, but was not able to induce activation of NF-κB [44]. Therein, to activate the priming signal, LPS was added prior to the treatment of fPrPWT. In our results, a low concentration of Mn–fPrPWT severely increased IL-1β mRNA levels by activating NF-κB in microglia cells. Conclusively, Mn–fPrPWT can initiate the priming signal but fPrPWT can initiate the activation signal in the NLRP3 inflammasome. Taking Mn–fPrPWT and fPrPWT in sequence can produce IL-1β in microglia.
Our results reveal that manganese ions enable fPrPWT to activate NF-κB through their binding to ORs. This effect may come from microglia phagocyte PrPSc that uptake manganese and increase the concentration in cells. In addition, octapeptide repeat numbers can affect the age of onset of patients [12]. Enlargement of the ORs copy number may increase the amount of manganese on ORs and then increase the sensitivity of manganese in microglia.
We also observed that NF-κB activity and IL-1β mRNA levels were slightly lower when both Mn2+ and Cu2+ were added into the fPrPWT. As reported previously, the binding affinity of Cu2+ is higher than that of Mn2+ [37]. Cu2+ may compete with Mn2+ in ORs; therefore, the effect of Mn–fPrPWT on NF-κB activation was weakened by the Cu2+ addition. Overall, the binding of manganese ions on prion fibrils is critical for inducing the priming signal in the NLRP3 inflammasome.
PrPSc are notorious for increasing IL-1β and for inducing neurotoxicity [45]. Herein, we propose that PrPSc have a positive effect as a double-edged sword in microglia activation. This activation can decrease prion infectivity and progression by phagocytosis [46]. Therefore, it is not always an advantage to indiscriminately inhibit the activation of microglia.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4409/9/10/2285/s1, Figure S1: The kinetics and TEM image of fPrP. Figure S2: The cell viability of EOC 13.31 treated with fPrPWT, Cu2+, Mn2+ and cation-bound fPrPWT. Figure S3: The determination of copper level of Cu-fPrP.
Author Contributions
Conceptualization, H.-I.J. and C.-I.L.; Methodology, H.-I.J.; Investigation, H.-I.J., Z.-Y.L., J.-X.G. and C.-I.L.; Analysis, H.-I.J. and C.-I.L.; Writing, H.-I.J. and C.-I.L.; Editing, C.-I.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Ministry of Science and Technology (MOST-108-2113-M-194-004 and MOST 109-2113-M-194-002) and the Center for Nano Bio-Detection from The Featured Research Areas College Development Plan of National Chung Cheng University. The APC was funded by Center for Innovative Research on Aging Society (CIRAS).
Acknowledgments
We thank Cheng-Liang Huang in National Chia-Yi University for technical support in TEM imaging.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Soto, C.; Satani, N. The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol. Med. 2011, 17, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Gambetti, P.; Parchi, P.; Chen, S.G. Hereditary Creutzfeldt-Jakob disease and fatal familial insomnia. Clin. Lab. Med. 2003, 23, 43–64. [Google Scholar] [CrossRef]
- Will, R.G. Acquired prion disease: Iatrogenic CJD, variant CJD, kuru. Br. Med. Bull. 2003, 66, 255–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Cell biology. A unifying role for prions in neurodegenerative diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wroe, S.J.; Pal, S.; Siddique, D.; Hyare, H.; Macfarlane, R.; Joiner, S.; Linehan, J.M.; Brandner, S.; Wadsworth, J.D.F.; Hewitt, P.; et al. Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: A case report. Lancet 2006, 368, 2061–2067. [Google Scholar] [CrossRef]
- Llewelyn, C.A.; Hewitt, P.E.; Knight, R.S.G.; Amar, K.; Cousens, S.; Mackenzie, J.; Will, R.G. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 2004, 363, 417–421. [Google Scholar] [CrossRef]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363. [Google Scholar] [CrossRef] [Green Version]
- Bradford, B.M.; Mabbott, N.A. Prion disease and the innate immune system. Viruses 2012, 4, 3389–3419. [Google Scholar] [CrossRef] [Green Version]
- Geschwind, M.D. Prion Diseases. Continuum 2015, 21, 1612–1638. [Google Scholar] [CrossRef]
- Aguzzi, A.; Nuvolone, M.; Zhu, C. The immunobiology of prion diseases. Nat. Rev. Immunol. 2013, 13, 888–902. [Google Scholar] [CrossRef]
- Moore, R.A.; Herzog, C.; Errett, J.; Kocisko, D.A.; Arnold, K.M.; Hayes, S.F.; Priola, S.A. Octapeptide repeat insertions increase the rate of protease-resistant prion protein formation. Protein Sci. 2006, 15, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Croes, E.A.; Theuns, J.; Houwing-Duistermaat, J.J.; Dermaut, B.; Sleegers, K.; Roks, G.; Van den Broeck, M.; van Harten, B.; van Swieten, J.C.; Cruts, M.; et al. Octapeptide repeat insertions in the prion protein gene and early onset dementia. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Deloncle, R.; Fauconneau, B.; Guillard, O.; Delaval, J.; Lesage, G.; Pineau, A. Copper brain protein protection against free radical-induced neuronal death: Survival ratio in SH-SY5Y neuroblastoma cell cultures. J. Trace Elem. Med. Biol. 2017, 39, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.F.; Harischandra, D.S.; Kanthasamy, A.; Sivasankar, S. Copper-induced structural conversion templates prion protein oligomerization and neurotoxicity. Sci. Adv. 2016, 2, e1600014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesketh, S.; Sassoon, J.; Knight, R.; Brown, D.R. Elevated manganese levels in blood and CNS in human prion disease. Mol. Cell Neurosci. 2008, 37, 590–598. [Google Scholar] [CrossRef]
- Johnson, C.J.; Gilbert, P.U.; Abrecht, M.; Baldwin, K.L.; Russell, R.E.; Pedersen, J.A.; Aiken, J.M.; McKenzie, D. Low copper and high manganese levels in prion protein plaques. Viruses 2013, 5, 654–662. [Google Scholar] [CrossRef] [Green Version]
- Scheiber, I.F.; Mercer, J.F.B.; Dringen, R. Metabolism and functions of copper in brain. Prog. Neurobiol. 2014, 116, 33–57. [Google Scholar] [CrossRef]
- Singh, N.; Singh, A.; Das, D.; Mohan, M.L. Redox control of prion and disease pathogenesis. Antioxid. Redox Signal. 2010, 12, 1271–1294. [Google Scholar] [CrossRef] [Green Version]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflammation 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colton, C.; Wilcock, D.M. Assessing activation states in microglia. Cns Neurol. Disord. Drug Targets 2010, 9, 174–191. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A. Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharm. 2009, 4, 399–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, W.; Rowe, D.; Xie, W.; Ortiz, I.; He, Y.; Appel, S.H. Microglial activation and dopaminergic cell injury: An in vitro model relevant to Parkinson’s disease. J. Neurosci. 2001, 21, 8447–8455. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Huang, Y.G.; Fang, D.; Le, W.D. (-)-Epigallocatechin gallate inhibits lipopolysaccharide-induced microglial activation and protects against inflammation-mediated dopaminergic neuronal injury. J. Neurosci. Res. 2004, 78, 723–731. [Google Scholar] [CrossRef]
- Sawada, M.; Suzumura, A.; Hosoya, H.; Marunouchi, T.; Nagatsu, T. Interleukin-10 inhibits both production of cytokines and expression of cytokine receptors in microglia. J. Neurochem. 1999, 72, 1466–1471. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Duncan, B.; Li, X.; Shi, J. The role of NLRP3 inflammasome in infection-related, immune-mediated and autoimmune skin diseases. J. Derm. Sci. 2020, 98, 146–151. [Google Scholar] [CrossRef]
- Sarkar, S.; Rokad, D.; Malovic, E.; Luo, J.; Harischandra, D.S.; Jin, H.; Anantharam, V.; Huang, X.; Lewis, M.; Kanthasamy, A.; et al. Manganese activates NLRP3 inflammasome signaling and propagates exosomal release of ASC in microglial cells. Sci. Signal. 2019, 12, 563. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.H.; Huang, M.Y.; Lee, Y.R.; Lin, Y.K.; Chen, H.R.; Lee, C.I. The effect of octapeptid repeats on prion folding and misfolding. 2020. in preparation. [Google Scholar]
- Bocharova, O.V.; Breydo, L.; Parfenov, A.S.; Salnikov, V.V.; Baskakov, I.V. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc). J. Mol. Biol. 2005, 346, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J.; Yu, K.H.; Wu, J.R.; Lee, C.F.; Jheng, C.P.; Chen, H.R.; Lee, C.I. Liberation of GPI-anchored prion from phospholipids accelerates amyloidogenic conversion. Int. J. Mol. Sci. 2013, 14, 17943–17957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.J.; Anantharam, V.; Saetveit, N.J.; Houk, R.S.; Kanthasamy, A.; Kanthasamy, A.G. Normal cellular prion protein protects against manganese-induced oxidative stress and apoptotic cell death. Toxicol. Sci. 2007, 98, 495–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minutoli, L.; Puzzolo, D.; Rinaldi, M.; Irrera, N.; Marini, H.; Arcoraci, V.; Bitto, A.; Crea, G.; Pisani, A.; Squadrito, F.; et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxid. Med. Cell Longev. 2016, 2016, 2183026. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R. Metallic prions. Biochem. Soc. Symp. 2004, 193–202. [Google Scholar]
- Brazier, M.W.; Davies, P.; Player, E.; Marken, F.; Viles, J.H.; Brown, D.R. Manganese binding to the prion protein. J. Biol. Chem. 2008, 283, 12831–12839. [Google Scholar] [CrossRef] [Green Version]
- Peres, T.V.; Schettinger, M.R.C.; Chen, P.; Carvalho, F.; Avila, D.S.; Bowman, A.B.; Aschner, M. Manganese-induced neurotoxicity: A review of its behavioral consequences and neuroprotective strategies. BMC Pharm. Toxicol. 2016, 17, 57. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Asthana, A.; Baksi, S.; Desai, V.; Haldar, S.; Hari, S.; Tripathi, A.K. The prion-ZIP connection: From cousins to partners in iron uptake. Prion 2015, 9, 420–428. [Google Scholar] [CrossRef] [Green Version]
- Stahl, J.L.; Greger, J.L.; Cook, M.E. Zinc, copper and iron utilisation by chicks fed various concentrations of zinc. Br. Poult. Sci. 1989, 30, 123–134. [Google Scholar] [CrossRef]
- Watt, N.T.; Taylor, D.R.; Kerrigan, T.L.; Griffiths, H.H.; Rushworth, J.V.; Whitehouse, I.J.; Hooper, N.M. Prion protein facilitates uptake of zinc into neuronal cells. Nat. Commun. 2012, 3, 1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summersgill, H.; England, H.; Lopez-Castejon, G.; Lawrence, C.B.; Luheshi, N.M.; Pahle, J.; Mendes, P.; Brough, D. Zinc depletion regulates the processing and secretion of IL-1β. Cell Death Dis. 2014, 5, e1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muroi, M.; Tanamoto, K. Zinc- and oxidative property-dependent degradation of pro-caspase-1 and NLRP3 by ziram in mouse macrophages. Toxicol. Lett. 2015, 235, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Hafner-Bratkovič, I.; Benčina, M.; Fitzgerald, K.A.; Golenbock, D.; Jerala, R. NLRP3 inflammasome activation in macrophage cell lines by prion protein fibrils as the source of IL-1β and neuronal toxicity. Cell Mol. Life Sci. 2012, 69, 4215–4228. [Google Scholar]
- Stoll, G.; Jander, S.; Schroeter, M. Cytokines in CNS disorders: Neurotoxicity versus neuroprotection. J. Neural. Transm. Suppl. 2000, 59, 81–89. [Google Scholar]
- Beringue, V.; Demoy, M.; Lasmézas, C.I.; Gouritin, B.; Weingarten, C.; Deslys, J.P.; Andreux, J.P.; Couvreur, P.; Dormont, D. Role of spleen macrophages in the clearance of scrapie agent early in pathogenesis. J. Pathol. 2000, 190, 495–502. [Google Scholar] [CrossRef]
Figure 1.
Activity of the NLRP3 inflammasome is controlled by two signal pathways: Priming and activation in sequence [29]. PAMPs, pathogen-associated molecular pattern molecules; DAMPs, damage-associated molecular patterns; ROS, reactive oxygen species; NLRP3, nucleotide-binding and oligomerization domain-like receptor family pyrin domain-containing 3.
Figure 1.
Activity of the NLRP3 inflammasome is controlled by two signal pathways: Priming and activation in sequence [29]. PAMPs, pathogen-associated molecular pattern molecules; DAMPs, damage-associated molecular patterns; ROS, reactive oxygen species; NLRP3, nucleotide-binding and oligomerization domain-like receptor family pyrin domain-containing 3.
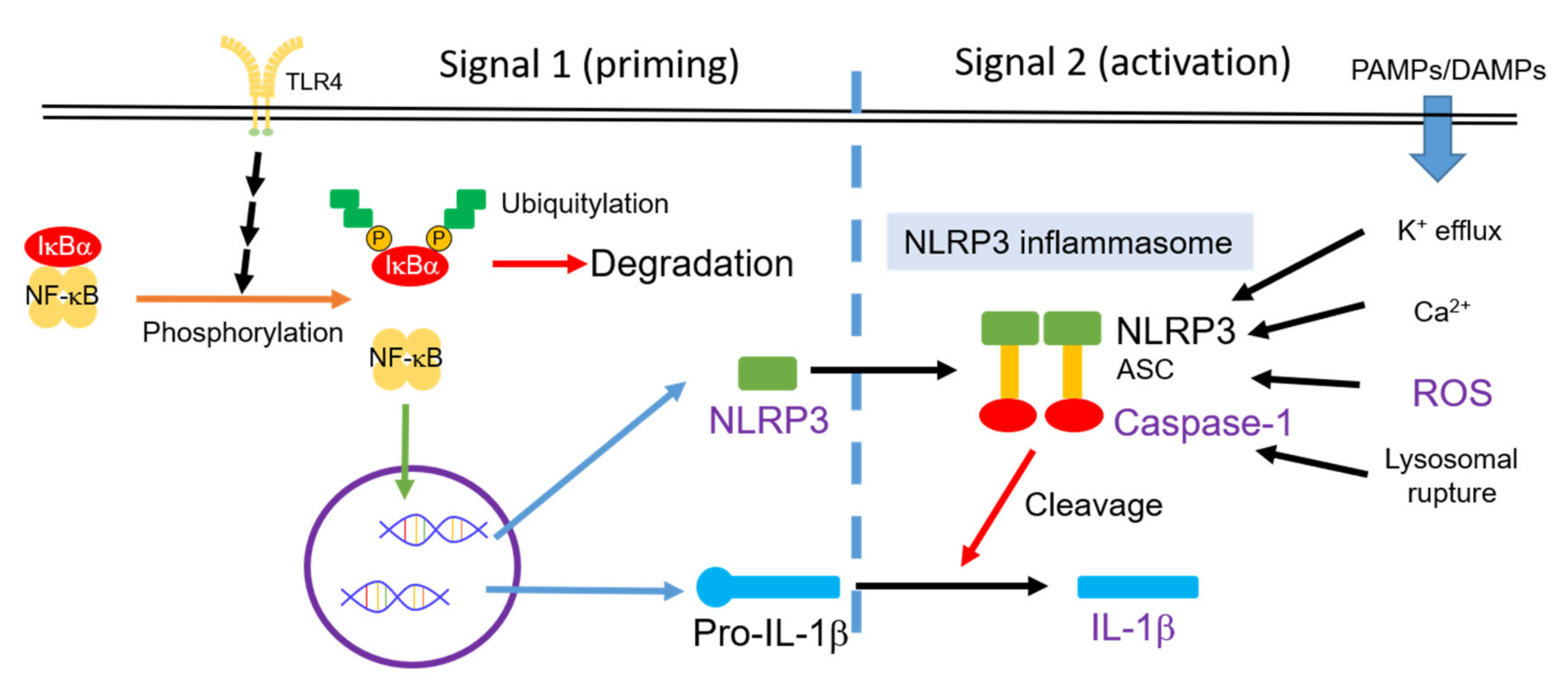
Figure 2.
Determination of cellular ROS induced by fPrPWT or by cations based on 2′,7′-dichlorofluorescin diacetate (DCFDA) fluorescence. (a) EOC 13.31 microglia treated with copper and manganese ions. (b) EOC 13.31 microglia treated with fPrPWT and 100 μM of H2O2. (c) EOC 13.31 microglia treated with pre-mixed 2 μM of fPrPWT and 10 μM of copper or manganese ions. Statistical significance is shown using asterisks (ns: p > 0.05; **: p < 0.005; ***: p < 0.0005).
Figure 2.
Determination of cellular ROS induced by fPrPWT or by cations based on 2′,7′-dichlorofluorescin diacetate (DCFDA) fluorescence. (a) EOC 13.31 microglia treated with copper and manganese ions. (b) EOC 13.31 microglia treated with fPrPWT and 100 μM of H2O2. (c) EOC 13.31 microglia treated with pre-mixed 2 μM of fPrPWT and 10 μM of copper or manganese ions. Statistical significance is shown using asterisks (ns: p > 0.05; **: p < 0.005; ***: p < 0.0005).

Figure 3.
Determination of NLRP3 and IκBα protein levels in EOC 13.31 microglia cell lysate by Western blotting. (a) A Western blot image of NLRP3 and IκBα proteins collected after treating EOC 13.31 cells with 2 μM of fPrPWT and/or 10 μM of Cu2+ and/or Mn2+ for 24 h. (b) Quantification of NLRP3 and IκBα protein levels shown in (a). (c) A Western blot image of NLRP3 and IκBα proteins collected after treating EOC 13.31 cells with copper and manganese ions for 24 h. (d) Quantification of NLRP3 and IκBα protein levels shown in (c). Statistical significance is shown using asterisks (ns: p > 0.05; *: p < 0.05; **: p < 0.005; ***: p < 0.0005).
Figure 3.
Determination of NLRP3 and IκBα protein levels in EOC 13.31 microglia cell lysate by Western blotting. (a) A Western blot image of NLRP3 and IκBα proteins collected after treating EOC 13.31 cells with 2 μM of fPrPWT and/or 10 μM of Cu2+ and/or Mn2+ for 24 h. (b) Quantification of NLRP3 and IκBα protein levels shown in (a). (c) A Western blot image of NLRP3 and IκBα proteins collected after treating EOC 13.31 cells with copper and manganese ions for 24 h. (d) Quantification of NLRP3 and IκBα protein levels shown in (c). Statistical significance is shown using asterisks (ns: p > 0.05; *: p < 0.05; **: p < 0.005; ***: p < 0.0005).
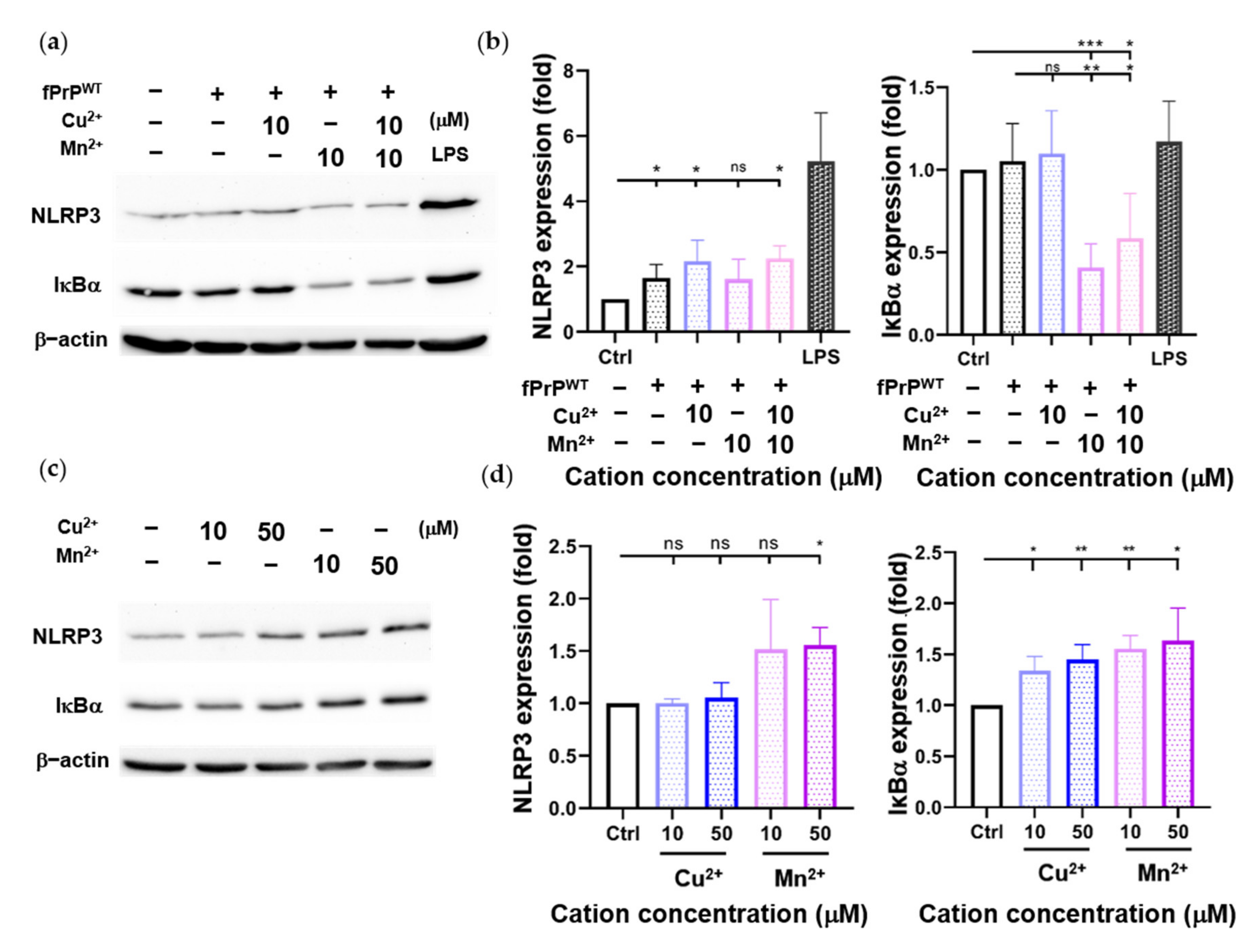
Figure 4.
Expression of IL-1β mRNA in EOC 13.31 microglia cells determined by qPCR. The cells were treated with (a) fPrPWT chelated with 10 μM of copper and/or manganese ions and (b) 10 μM and 50 μM of copper or manganese ions without fPrPWT for 24 h. Statistical significance is shown using asterisks (***: p < 0.0005).
Figure 4.
Expression of IL-1β mRNA in EOC 13.31 microglia cells determined by qPCR. The cells were treated with (a) fPrPWT chelated with 10 μM of copper and/or manganese ions and (b) 10 μM and 50 μM of copper or manganese ions without fPrPWT for 24 h. Statistical significance is shown using asterisks (***: p < 0.0005).
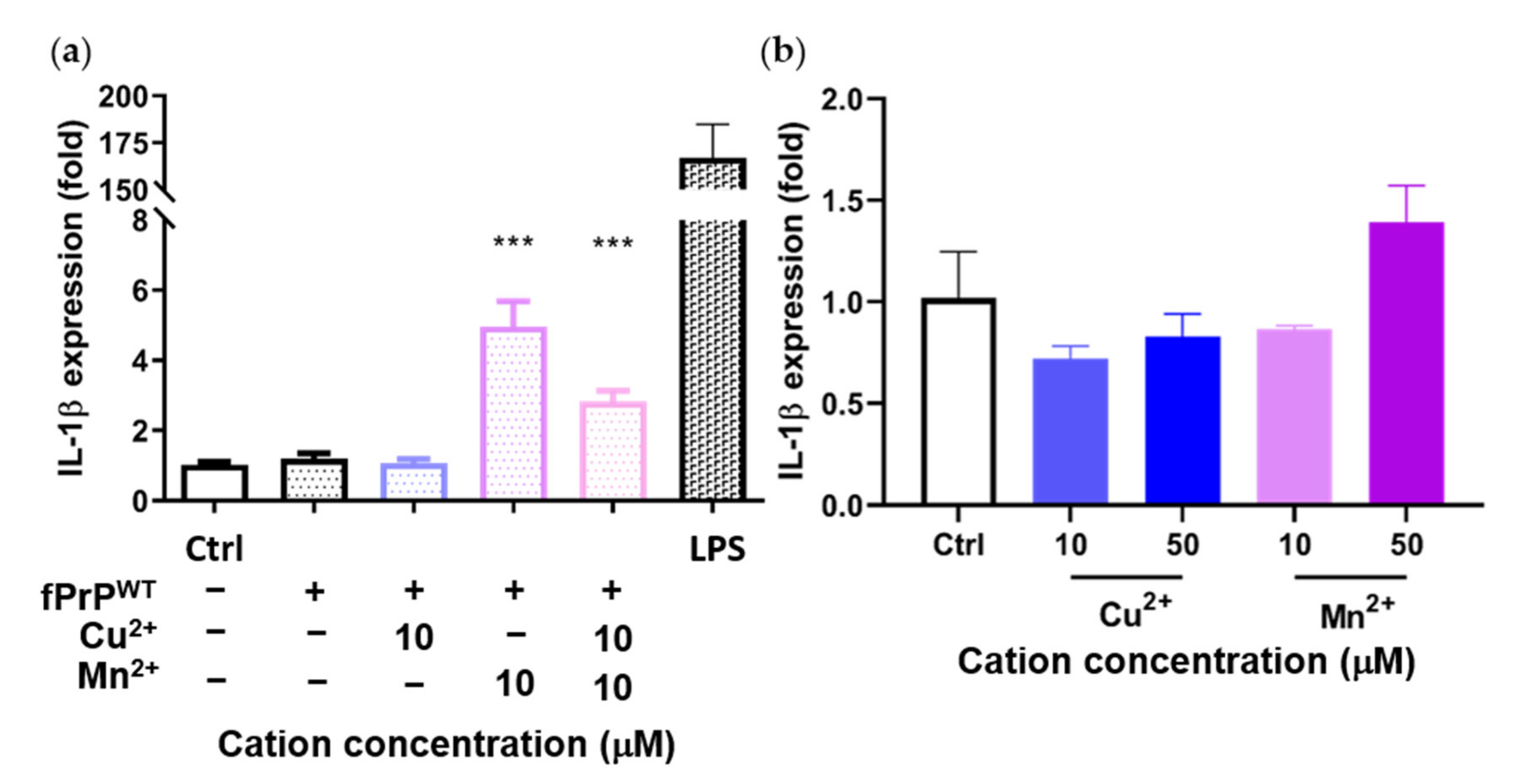
Figure 5.
IκBα levels and IL-1β mRNA expression in EOC 13.31 cells treated with fibrils converted from octapeptide-deleted prions (fPrPΔOct) with copper and manganese ions. (a) IκBα protein levels in 2 μM of fPrPΔOct and 10 μM of cation-treated microglia. (b) IL-1β mRNA expression in 2 μM of fPrPΔOct and 10 μM of cation-treated microglia.
Figure 5.
IκBα levels and IL-1β mRNA expression in EOC 13.31 cells treated with fibrils converted from octapeptide-deleted prions (fPrPΔOct) with copper and manganese ions. (a) IκBα protein levels in 2 μM of fPrPΔOct and 10 μM of cation-treated microglia. (b) IL-1β mRNA expression in 2 μM of fPrPΔOct and 10 μM of cation-treated microglia.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jen, H.-I.; Lin, Z.-Y.; Guo, J.-X.; Lee, C.-I. The Effects of Divalent Cation-Chelated Prion Fibrils on the Immune Response of EOC 13.31 Microglia Cells. Cells 2020, 9, 2285. https://doi.org/10.3390/cells9102285
AMA Style
Jen H-I, Lin Z-Y, Guo J-X, Lee C-I. The Effects of Divalent Cation-Chelated Prion Fibrils on the Immune Response of EOC 13.31 Microglia Cells. Cells. 2020; 9(10):2285. https://doi.org/10.3390/cells9102285
Chicago/Turabian StyleJen, Huan-I, Zih-You Lin, Jin-Xun Guo, and Cheng-I Lee. 2020. "The Effects of Divalent Cation-Chelated Prion Fibrils on the Immune Response of EOC 13.31 Microglia Cells" Cells 9, no. 10: 2285. https://doi.org/10.3390/cells9102285
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.