Conformational States of Exchange Protein Directly Activated by cAMP (EPAC1) Revealed by Ensemble Modeling and Integrative Structural Biology



Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Small-Angle X-ray Scattering (SAXS)
2.3. Ab Initio Molecular Shape Analysis
2.4. EPAC1 Homology Models for Rigid-Body Analysis
2.5. Rigid-Body Analysis
2.6. Polydispersity and Conformational Ensemble Analyses of EPAC1
3. Results
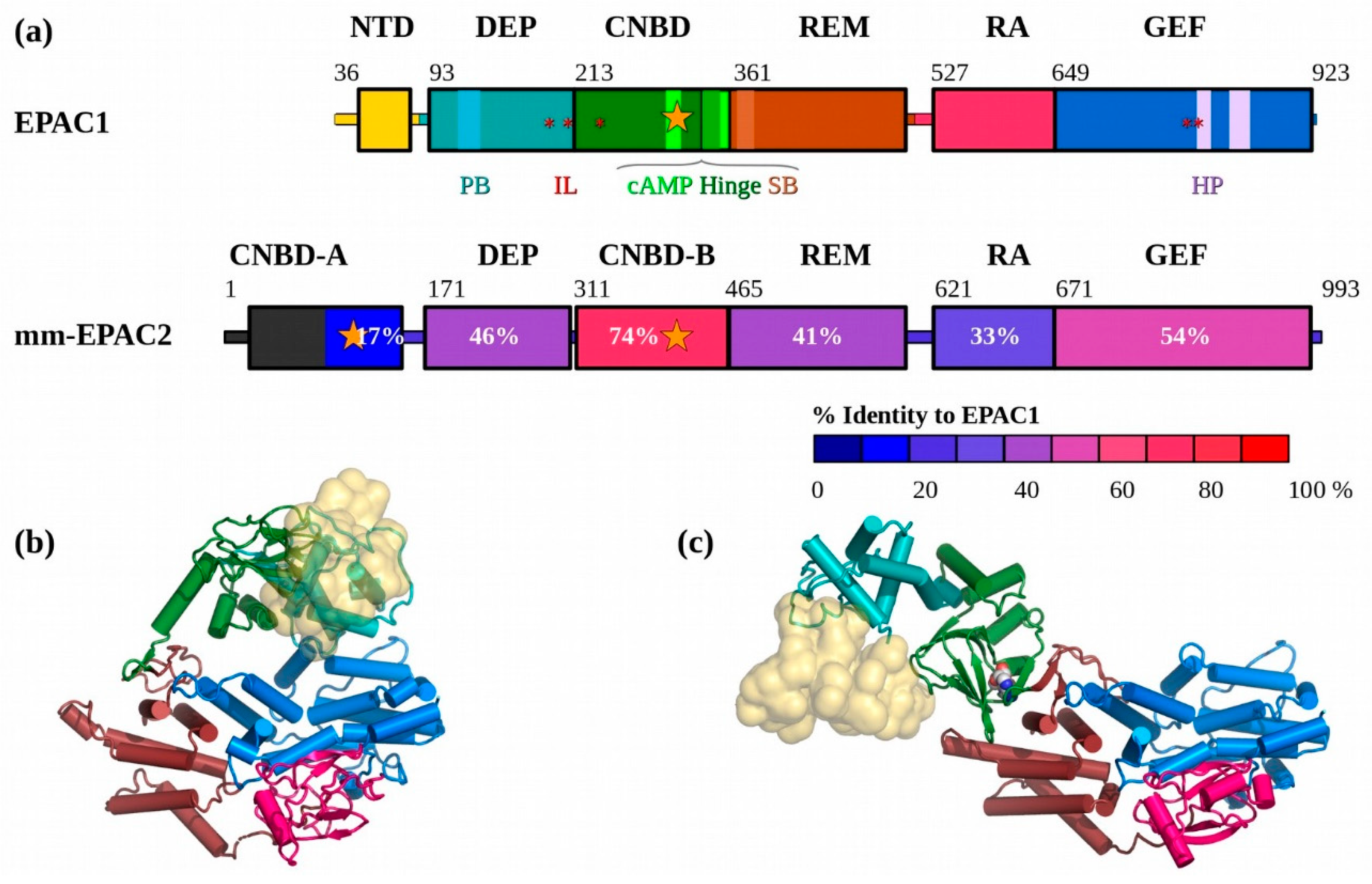
3.1. In Solution the apo-EPAC1 NTD Blocks the Effector Binding Site
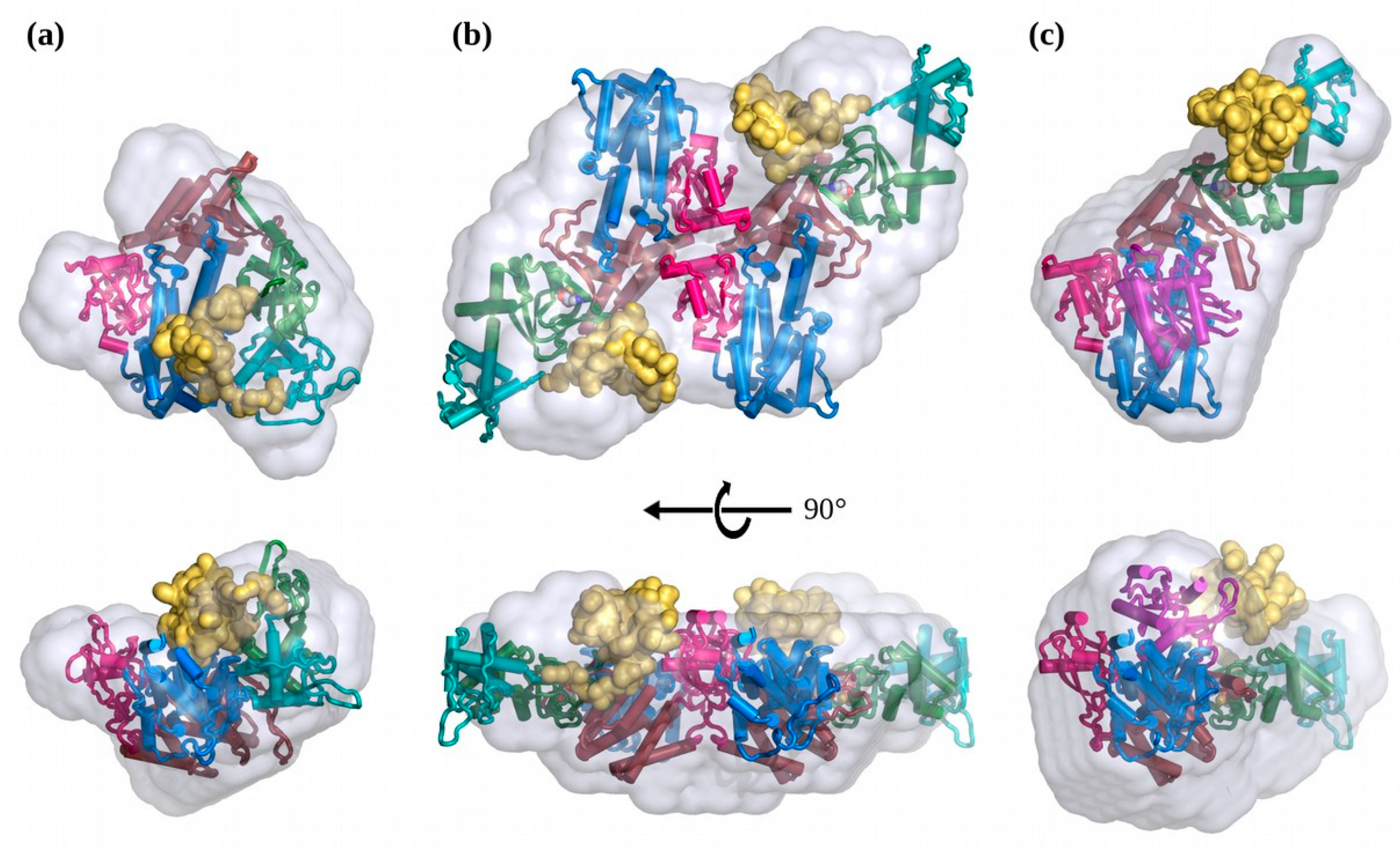
3.2. The Solution Structure of apo-EPAC1 is a Dynamic Mixture of Closed and Extended States
3.3. In Solution, cAMP-Bound EPAC1 Forms Dimers
3.4. The Solution Structure of the EPAC1:cAMP:Rap1 Ternary Complex
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A
SAXNS_ES Buffer Subtraction, MW Calibration, and MW Calculation
References
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. 2008, 40, 651–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robichaux, W.G.; Cheng, X. Intracellular cAMP Sensor EPAC: Physiology, Pathophysiology, and Therapeutics Development. Physiol. Rev. 2018, 98, 919–1053. [Google Scholar] [CrossRef] [PubMed]
- Alenkvist, I.; Gandasi, N.R.; Barg, S.; Tengholm, A. Recruitment of Epac2A to Insulin Granule Docking Sites Regulates Priming for Exocytosis. Diabetes 2017, 66, 2610–2622. [Google Scholar] [CrossRef] [Green Version]
- Niimura, M.; Miki, T.; Shibasaki, T.; Fujimoto, W.; Iwanaga, T.; Seino, S. Critical role of the N-terminal cyclic AMP-binding domain of Epac2 in its subcellular localization and function. J. Cell. Physiol. 2009, 219, 652–658. [Google Scholar] [CrossRef]
- Qiao, J.; Mei, F.C.; Popov, V.L.; Vergara, L.A.; Cheng, X. Cell cycle-dependent subcellular localization of exchange factor directly activated by cAMP. J. Biol. Chem. 2002, 277, 26581–26586. [Google Scholar] [CrossRef] [Green Version]
- Hochbaum, D.; Barila, G.; Ribeiro-Neto, F.; Altschuler, D.L. Radixin assembles cAMP effectors Epac and PKA into a functional cAMP compartment: Role in cAMP-dependent cell proliferation. J. Biol. Chem. 2011, 286, 859–866. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Dekker, F.J.; Maarsingh, H. Exchange protein directly activated by cAMP (epac): A multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol. Rev. 2013, 65, 670–709. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, U.; Cheng, X. Exchange protein directly activated by cAMP encoded by the mammalian rapgef3 gene: Structure, function and therapeutics. Gene 2015, 570, 157–167. [Google Scholar] [CrossRef]
- Kim, M.; Platt, M.J.; Shibasaki, T.; Quaggin, S.E.; Backx, P.H.; Seino, S.; Simpson, J.A.; Drucker, D.J. GLP-1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat. Med. 2013, 19, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Cheng, H.; Lao, D.H.; Na, L.; van Oort, R.J.; Brown, J.H.; Wehrens, X.H.T.; Chen, J.; Bers, D.M. Epac2 mediates cardiac β1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation 2013, 127, 913–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, S.; Fujita, T.; Cai, W.; Jin, M.; Namekata, I.; Mototani, Y.; Jin, H.; Ohnuki, Y.; Tsuneoka, Y.; Kurotani, R.; et al. Epac1-dependent phospholamban phosphorylation mediates the cardiac response to stresses. J. Clin. Investig. 2014, 124, 2785–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Robichaux, W.G.; Wang, Z.; Mei, F.C.; Cai, M.; Du, G.; Chen, J.; Cheng, X. Inhibition of Epac1 suppresses mitochondrial fission and reduces neointima formation induced by vascular injury. Sci Rep 2016, 6, 36552. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Heijnen, C.J.; van Velthoven, C.T.J.; Willemen, H.L.D.M.; Ishikawa, Y.; Zhang, X.; Sood, A.K.; Vroon, A.; Eijkelkamp, N.; Kavelaars, A. Balancing GRK2 and EPAC1 levels prevents and relieves chronic pain. J. Clin. Investig. 2013, 123, 5023–5034. [Google Scholar] [CrossRef] [Green Version]
- Singhmar, P.; Huo, X.; Eijkelkamp, N.; Berciano, S.R.; Baameur, F.; Mei, F.C.; Zhu, Y.; Cheng, X.; Hawke, D.; Mayor, F.; et al. Critical role for Epac1 in inflammatory pain controlled by GRK2-mediated phosphorylation of Epac1. Proc. Natl. Acad. Sci. USA 2016, 113, 3036–3041. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Mei, F.C.; Cheng, H.; Lao, D.H.; Hu, Y.; Wei, J.; Patrikeev, I.; Hao, D.; Stutz, S.J.; Dineley, K.T.; et al. Enhanced leptin sensitivity, reduced adiposity, and improved glucose homeostasis in mice lacking exchange protein directly activated by cyclic AMP isoform 1. Mol. Cell. Biol. 2013, 33, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Song, W.-J.; Mondal, P.; Li, Y.; Lee, S.E.; Hussain, M.A. Pancreatic β-cell response to increased metabolic demand and to pharmacologic secretagogues requires EPAC2A. Diabetes 2013, 62, 2796–2807. [Google Scholar] [CrossRef] [Green Version]
- Kai, A.K.L.; Lam, A.K.M.; Chen, Y.; Tai, A.C.P.; Zhang, X.; Lai, A.K.W.; Yeung, P.K.K.; Tam, S.; Wang, J.; Lam, K.S.; et al. Exchange protein activated by cAMP 1 (Epac1)-deficient mice develop β-cell dysfunction and metabolic syndrome. FASEB J. 2013, 27, 4122–4135. [Google Scholar] [CrossRef]
- Almahariq, M.; Mei, F.C.; Cheng, X. Cyclic AMP sensor EPAC proteins and energy homeostasis. Trends Endocrinol. Metab. 2014, 25, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Onodera, Y.; Nam, J.-M.; Bissell, M.J. Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J. Clin. Investig. 2014, 124, 367–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Robichaux, W.G.; Mei, F.C.; Kim, E.R.; Wang, H.; Tong, Q.; Jin, J.; Xu, M.; Chen, J.; Cheng, X. Role of Exchange Protein Directly Activated by Cyclic AMP Isoform 1 in Energy Homeostasis: Regulation of Leptin Expression and Secretion in White Adipose Tissue. Mol. Cell. Biol. 2016, 36, 2440–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehmann, H.; Das, J.; Knipscheer, P.; Wittinghofer, A.; Bos, J.L. Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature 2006, 439, 625–628. [Google Scholar] [CrossRef] [PubMed]
- Rehmann, H.; Arias-Palomo, E.; Hadders, M.A.; Schwede, F.; Llorca, O.; Bos, J.L. Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature 2008, 455, 124–127. [Google Scholar] [CrossRef]
- White, M.A.; Li, S.; Tsalkova, T.; Mei, F.C.; Liu, T.; Woods, V.L., Jr.; Cheng, X. Structural analyses of a constitutively active mutant of exchange protein directly activated by cAMP. PLoS ONE 2012, 7, e49932. [Google Scholar] [CrossRef] [Green Version]
- Tsalkova, T.; Blumenthal, D.K.; Mei, F.C.; White, M.A.; Cheng, X. Mechanism of Epac activation: Structural and functional analyses of Epac2 hinge mutants with constitutive and reduced activities. J. Biol. Chem. 2009, 284, 23644–23651. [Google Scholar] [CrossRef] [Green Version]
- Das, R.; Mazhab-Jafari, M.T.; Chowdhury, S.; SilDas, S.; Selvaratnam, R.; Melacini, G. Entropy-driven cAMP-dependent allosteric control of inhibitory interactions in exchange proteins directly activated by cAMP. J. Biol. Chem. 2008, 283, 19691–19703. [Google Scholar] [CrossRef] [Green Version]
- Selvaratnam, R.; Chowdhury, S.; VanSchouwen, B.; Melacini, G. Mapping allostery through the covariance analysis of NMR chemical shifts. Proc. Natl. Acad. Sci. USA 2011, 108, 6133–6138. [Google Scholar] [CrossRef] [Green Version]
- Mazhab-Jafari, M.T.; Das, R.; Fotheringham, S.A.; SilDas, S.; Chowdhury, S.; Melacini, G. Understanding cAMP-dependent allostery by NMR spectroscopy: Comparative analysis of the EPAC1 cAMP-binding domain in its apo and cAMP-bound states. J. Am. Chem. Soc. 2007, 129, 14482–14492. [Google Scholar] [CrossRef]
- VanSchouwen, B.; Selvaratnam, R.; Fogolari, F.; Melacini, G. Role of dynamics in the autoinhibition and activation of the exchange protein directly activated by cyclic AMP (EPAC). J. Biol. Chem. 2011, 286, 42655–42669. [Google Scholar] [CrossRef] [Green Version]
- Boulton, S.; Selvaratnam, R.; Blondeau, J.-P.; Lezoualc’h, F.; Melacini, G. Mechanism of Selective Enzyme Inhibition through Uncompetitive Regulation of an Allosteric Agonist. J. Am. Chem. Soc. 2018, 140, 9624–9637. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.M.; Wienk, H.; Wechselberger, R.W.; Bos, J.L.; Boelens, R.; Rehmann, H. Structural dynamics in the activation of Epac. J. Biol. Chem. 2008, 283, 6501–6508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walenta, E. Small Angle X-ray Scattering; Glatter, O., Kratky, O., Eds.; Academic Press: London, UK, 1982; ISBN 0-12-286280-5. [Google Scholar]
- Putnam, C.D.; Hammel, M.; Hura, G.L.; Tainer, J.A. X-ray solution scattering (SAXS) combined with crystallography and computation: Defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007, 40, 191–285. [Google Scholar] [CrossRef] [PubMed]
- Petoukhov, M.V.; Svergun, D.I. Applications of small-angle X-ray scattering to biomacromolecular solutions. Int. J. Biochem. Cell Biol. 2013, 45, 429–437. [Google Scholar] [CrossRef]
- Jacques, D.A.; Trewhella, J. Small-angle scattering for structural biology–expanding the frontier while avoiding the pitfalls. Protein Sci. 2010, 19, 642–657. [Google Scholar] [CrossRef] [Green Version]
- Brosey, C.A.; Tainer, J.A. Evolving SAXS versatility: Solution X-ray scattering for macromolecular architecture, functional landscapes, and integrative structural biology. Curr. Opin. Struct. Biol. 2019, 58, 197–213. [Google Scholar] [CrossRef]
- Hura, G.L.; Hodge, C.D.; Rosenberg, D.; Guzenko, D.; Duarte, J.M.; Monastyrskyy, B.; Grudinin, S.; Kryshtafovych, A.; Tainer, J.A.; Fidelis, K.; et al. Small angle X-ray scattering-assisted protein structure prediction in CASP13 and emergence of solution structure differences. Proteins 2019, 87, 1298–1314. [Google Scholar] [CrossRef] [Green Version]
- Almahariq, M.; Tsalkova, T.; Mei, F.C.; Chen, H.; Zhou, J.; Sastry, S.K.; Schwede, F.; Cheng, X. A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol. Pharmacol. 2013, 83, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Tsalkova, T.; Mei, F.C.; Cheng, X. A fluorescence-based high-throughput assay for the discovery of exchange protein directly activated by cyclic AMP (EPAC) antagonists. PLoS ONE 2012, 7, e30441. [Google Scholar] [CrossRef]
- Tsalkova, T.; Mei, F.C.; Li, S.; Chepurny, O.G.; Leech, C.A.; Liu, T.; Holz, G.G.; Woods, V.L., Jr.; Cheng, X. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc. Natl. Acad. Sci. USA 2012, 109, 18613–18618. [Google Scholar] [CrossRef] [Green Version]
- Mei, F.C.; Cheng, X. Interplay between exchange protein directly activated by cAMP (Epac) and microtubule cytoskeleton. Mol. Biosyst. 2005, 1, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Rehmann, H.; Schwede, F.; Døskeland, S.O.; Wittinghofer, A.; Bos, J.L. Ligand-mediated activation of the cAMP-responsive guanine nucleotide exchange factor Epac. J. Biol. Chem. 2003, 278, 38548–38556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dao, K.K.; Teigen, K.; Kopperud, R.; Hodneland, E.; Schwede, F.; Christensen, A.E.; Martinez, A.; Døskeland, S.O. Epac1 and cAMP-dependent protein kinase holoenzyme have similar cAMP affinity, but their cAMP domains have distinct structural features and cyclic nucleotide recognition. J. Biol. Chem. 2006, 281, 21500–21511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.A. Small Angle Scattering Group (SAXNS). Available online: https://xray.utmb.edu/SAXNS (accessed on 14 September 2012).
- Mylonas, E.; Svergun, D.I. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. Appl. Crystallogr. 2007, 40, s245–s249. [Google Scholar] [CrossRef] [Green Version]
- Rambo, R.P.; Tainer, J.A. Accurate assessment of mass, models and resolution by small-angle scattering. Nature 2013, 496, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Petoukhov, M.V.; Franke, D.; Shkumatov, A.V.; Tria, G.; Kikhney, A.G.; Gajda, M.; Gorba, C.; Mertens, H.D.T.; Konarev, P.V.; Svergun, D.I. New developments in the ATSAS program package for small-angle scattering data analysis. Appl. Crystallogr. 2012, 45, 342–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, S. BayesApp: A web site for indirect transformation of small-angle scattering data. Appl. Crystallogr. 2012, 45, 566–567. [Google Scholar] [CrossRef]
- Franke, D.; Svergun, D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. Appl. Crystallogr. 2009, 42, 342–346. [Google Scholar] [CrossRef] [Green Version]
- Fischer, H.; de Oliveira Neto, M.; Napolitano, H.B.; Polikarpov, I.; Craievich, A.F. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. Appl. Crystallogr. 2009, 43, 101–109. [Google Scholar] [CrossRef]
- Petoukhov, M.V.; Svergun, D.I. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 2005, 89, 1237–1250. [Google Scholar] [CrossRef] [Green Version]
- Tria, G.; Mertens, H.D.T.; Kachala, M.; Svergun, D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2015, 2, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernadó, P.; Mylonas, E.; Petoukhov, M.V.; Blackledge, M.; Svergun, D.I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007, 129, 5656–5664. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.; Barberato, C.; Koch, M.H.J. CRYSOL–A Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. Appl. Crystallogr. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Jacques, D.A.; Guss, J.M.; Svergun, D.I.; Trewhella, J. Publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 620–626. [Google Scholar] [CrossRef] [Green Version]
- Trewhella, J.; Duff, A.P.; Durand, D.; Gabel, F.; Guss, J.M.; Hendrickson, W.A.; Hura, G.L.; Jacques, D.A.; Kirby, N.M.; Kwan, A.H.; et al. 2017 publication guidelines for structural modelling of small-angle scattering data from biomolecules in solution: An update. Acta Crystallogr. D Struct. Biol. 2017, 73, 710–728. [Google Scholar] [CrossRef] [Green Version]
- Volkov, V.V.; Svergun, D.I. Uniqueness of ab initio shape determination in small-angle scattering. Appl. Crystallogr. 2003, 36, 860–864. [Google Scholar] [CrossRef] [Green Version]
- Kozin, M.B.; Svergun, D.I. Automated matching of high- and low-resolution structural models. Appl. Crystallogr. 2001, 34, 33–41. [Google Scholar] [CrossRef]
- Tuukkanen, A.T.; Kleywegt, G.J.; Svergun, D.I. Resolution of ab initio shapes determined from small-angle scattering. IUCrJ 2016, 3, 440–447. [Google Scholar] [CrossRef] [Green Version]
- Bordoli, L.; Schwede, T. Automated Protein Structure Modeling with SWISS-MODEL Workspace and the Protein Model Portal. Methods Mol. Biol. 2012, 857, 107–136. [Google Scholar]
- Kiefer, F.; Arnold, K.; Künzli, M.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009, 37, D387–D392. [Google Scholar] [CrossRef] [Green Version]
- Scott, E.E.; White, M.A.; He, Y.A.; Johnson, E.F.; Stout, C.D.; Halpert, J.R. Structure of mammalian cytochrome P450 2B4 complexed with 4-(4-chlorophenyl)imidazole at 1.9-A resolution: Insight into the range of P450 conformations and the coordination of redox partner binding. J. Biol. Chem. 2004, 279, 27294–27301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; White, M.A.; Ramana, K.V.; Petrash, J.M.; Watowich, S.J.; Bhatnagar, A.; Srivastava, S.K. Structure of a glutathione conjugate bound to the active site of aldose reductase. Proteins 2006, 64, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, A.; Erdin, S.; Lua, R.; Lichtarge, O. Evolutionary trace for prediction and redesign of protein functional sites. Methods Mol. Biol. 2012, 819, 29–42. [Google Scholar]
- Bressler, I.; Pauw, B.R.; Thünemann, A.F. McSAS: Software for the retrieval of model parameter distributions from scattering patterns. Appl. Crystallogr. 2015, 48, 962–969. [Google Scholar] [CrossRef] [Green Version]
- Bernadó, P.; Svergun, D.I. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol. Biosyst. 2012, 8, 151–167. [Google Scholar] [CrossRef]
- Kikhney, A.G.; Borges, C.R.; Molodenskiy, D.S.; Jeffries, C.M.; Svergun, D.I. SASBDB: Towards automatically curated and validated repository for biological scattering data. Protein Sci. 2019. [Google Scholar] [CrossRef] [Green Version]
- Valentini, E.; Kikhney, A.G.; Previtali, G.; Jeffries, C.M.; Svergun, D.I. SASBDB, a repository for biological small-angle scattering data. Nucleic Acids Res. 2015, 43, D357–D363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Tsalkova, T.; White, M.A.; Mei, F.C.; Liu, T.; Wang, D.; Woods, V.L., Jr.; Cheng, X. Mechanism of intracellular cAMP sensor Epac2 activation: cAMP-induced conformational changes identified by amide hydrogen/deuterium exchange mass spectrometry (DXMS). J. Biol. Chem. 2011, 286, 17889–17897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gloerich, M.; Ponsioen, B.; Vliem, M.J.; Zhang, Z.; Zhao, J.; Kooistra, M.R.; Price, L.S.; Ritsma, L.; Zwartkruis, F.J.; Rehmann, H.; et al. Spatial regulation of cyclic AMP-Epac1 signaling in cell adhesion by ERM proteins. Mol. Cell. Biol. 2010, 30, 5421–5431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consonni, S.V.; Gloerich, M.; Spanjaard, E.; Bos, J.L. cAMP regulates DEP domain-mediated binding of the guanine nucleotide exchange factor Epac1 to phosphatidic acid at the plasma membrane. Proc. Natl. Acad. Sci. USA 2012, 109, 3814–3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, T.; Umemura, M.; Yokoyama, U.; Okumura, S.; Ishikawa, Y. The role of Epac in the heart. Cell. Mol. Life Sci. 2017, 74, 591–606. [Google Scholar] [CrossRef]
- Lezoualc’h, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP Sensor EPAC Proteins and Their Role in Cardiovascular Function and Disease. Circ. Res. 2016, 118, 881–897. [Google Scholar] [CrossRef]
- Gong, B.; Shelite, T.; Mei, F.C.; Ha, T.; Hu, Y.; Xu, G.; Chang, Q.; Wakamiya, M.; Ksiazek, T.G.; Boor, P.J.; et al. Exchange protein directly activated by cAMP plays a critical role in bacterial invasion during fatal rickettsioses. Proc. Natl. Acad. Sci. USA 2013, 110, 19615–19620. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| apo-EPAC1 | cAMP: EPAC1 | cAMP: EPAC1: Rap1b | |
|---|---|---|---|
| q-range (Å−1) | 0.010–0.30 | 0.012–0.30 | 0.012–0.30 |
| Concentration (mg/mL) | 0.5 | 2.7 | 1.0 |
| Time (h) | |||
| Sample | 6 | 2 | 2.5 |
| Buffer | 6 | 3 | 11.5 |
| Rg (Å) | 33.7 ± 0.5 | 53.2 ± 0.7 | 40.5 ± 0.6 |
| Dmax (Å) | 110 | 185 | 142 |
| MW (kDa) | 100 | 100 | 119 |
| MW(Io) (kDa) {N} | 104 {1.0} | 178 {1.8} | 98 {0.8} |
| MW(Porod) (kDa) {N} | 113 {1.1} | 240 {2.4} | 124 {1.0} |
| MW(Rambo) (kDa) {N} | 97.8 {1.0} | 218 {2.2} | 126 {1.1} |
| DAMMIF NSD (10) | 0.8 ± 3 | 1.0 ± 8 | 0.9 ± 1 |
| SASRES(Å) | 40 ± 3 | 14 ± 2 | 42 ± 3 |
| Rigid Body Fitting | |||
| HM Crysol fit (X2) | 1.3 | NA | NA |
| CORAL fit (X2) | 1.0 | 1.0 | 1.0 |
| CORAL Model Rg (Å) | 32.7 | 41.5 1 | 40.5 |
| EOM:(X2) | 1.0 | 1.1 | 0.7 |
| Rg [FWHM] 2 {Area} | |||
| Peak (closed) | 32.9 [0.8] 2 {86%} | 0 | 0 |
| Peak (extended) | 38.5 [0.9] 2 {13%} | 0 | 0 |
| Peak (ternary) | 0 | 0 | 40.7 [0.7] 2 {100%} |
| Peak (dimer) | 0 | 51 [3] 2 {100%} | 0 |
| SASDB-id | SASDCQ6 | SASDCR6 | SASDCS6 |
| Analysis | Software: | Analysis | Software: |
| Image Data-processing: | SAXSLAB (Rigaku) | Buffer Subtraction: | SAXNS-ES 3 [45] |
| Data-analysis: | PRIMUS [48] | Real Space P(r): | GNOM [48], BayesApp [49] |
| Ab initio Models: | DAMMIF [50] | Molecular Weight: | SAXSMOW [51] |
| Rigid Body Refinement: | CORAL [52], BUNCH | Ensemble Analysis: | EOM [53], MES [54] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
White, M.A.; Tsalkova, T.; Mei, F.C.; Cheng, X. Conformational States of Exchange Protein Directly Activated by cAMP (EPAC1) Revealed by Ensemble Modeling and Integrative Structural Biology. Cells 2020, 9, 35. https://doi.org/10.3390/cells9010035
White MA, Tsalkova T, Mei FC, Cheng X. Conformational States of Exchange Protein Directly Activated by cAMP (EPAC1) Revealed by Ensemble Modeling and Integrative Structural Biology. Cells. 2020; 9(1):35. https://doi.org/10.3390/cells9010035
Chicago/Turabian StyleWhite, Mark Andrew, Tamara Tsalkova, Fang C. Mei, and Xiaodong Cheng. 2020. "Conformational States of Exchange Protein Directly Activated by cAMP (EPAC1) Revealed by Ensemble Modeling and Integrative Structural Biology" Cells 9, no. 1: 35. https://doi.org/10.3390/cells9010035