Comprehensive Analysis of the Characteristics and Differences in Adult and Newborn Brown Adipose Tissue (BAT): Newborn BAT Is a More Active/Dynamic BAT

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Care and In Vivo Experiment Procedures
2.2. Hematoxylin and Eosin Staining
2.3. Transmission Electron Microscopy
2.4. Scanning Electron Microscopy
2.5. Quantitative Real-Time Reverse-Transcription PCR
2.6. Western Blot Analysis
2.7. Analysis of Proteomic
2.8. Analysis of the Whole Transcriptome
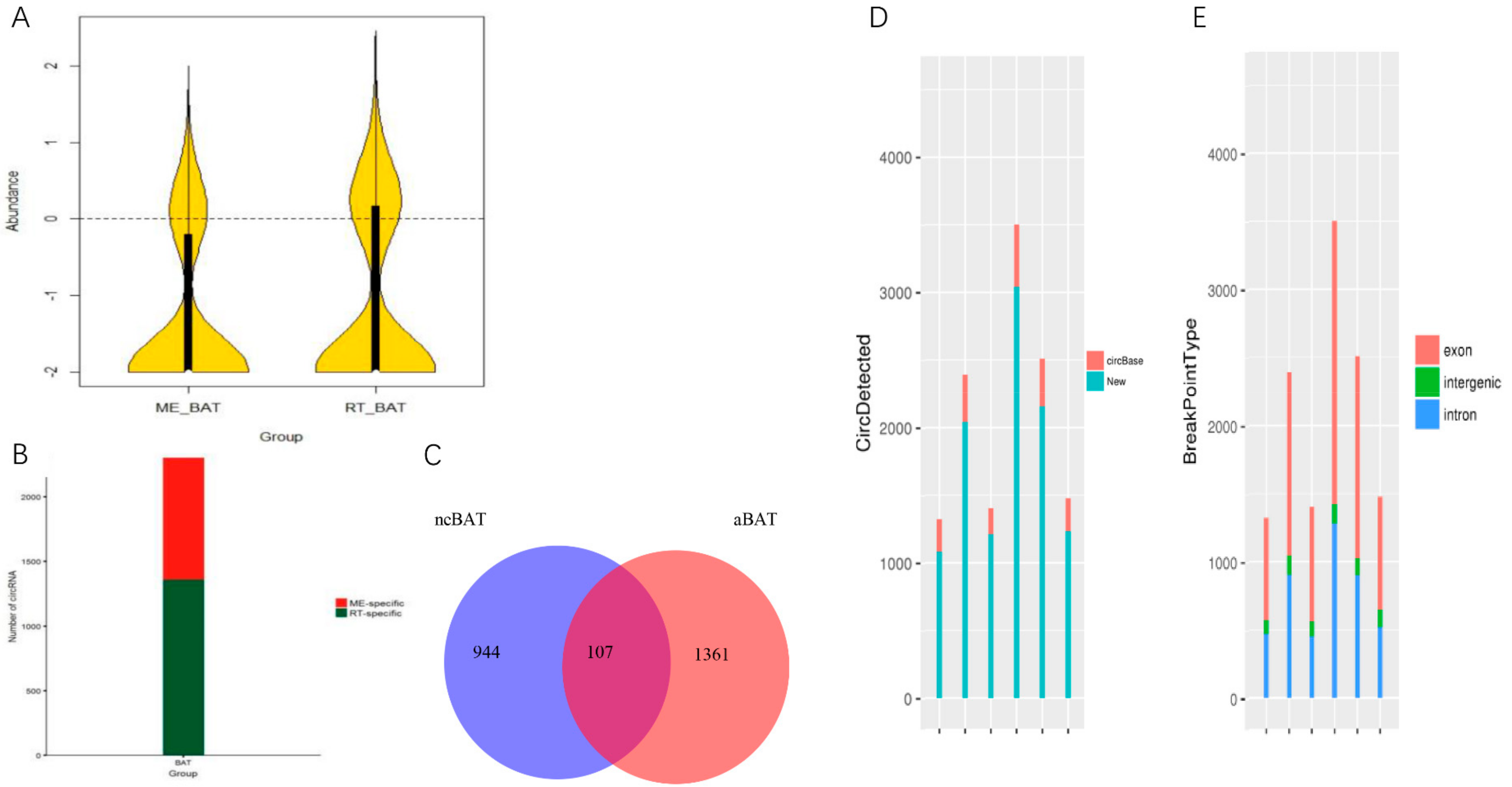
2.8.1. Transcriptions of lncRNA and circRNA
2.8.2. Transcriptions of microRNA
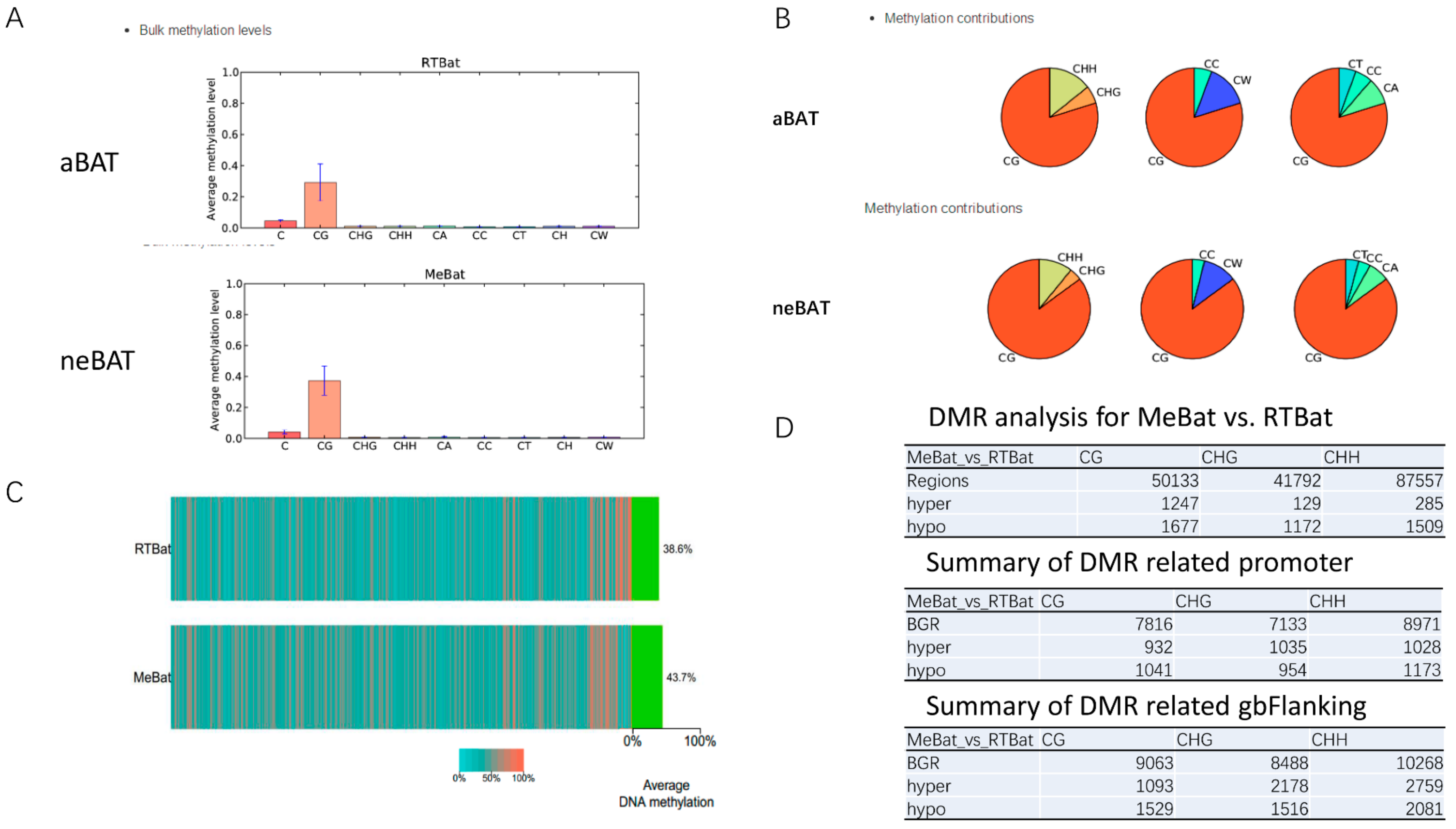
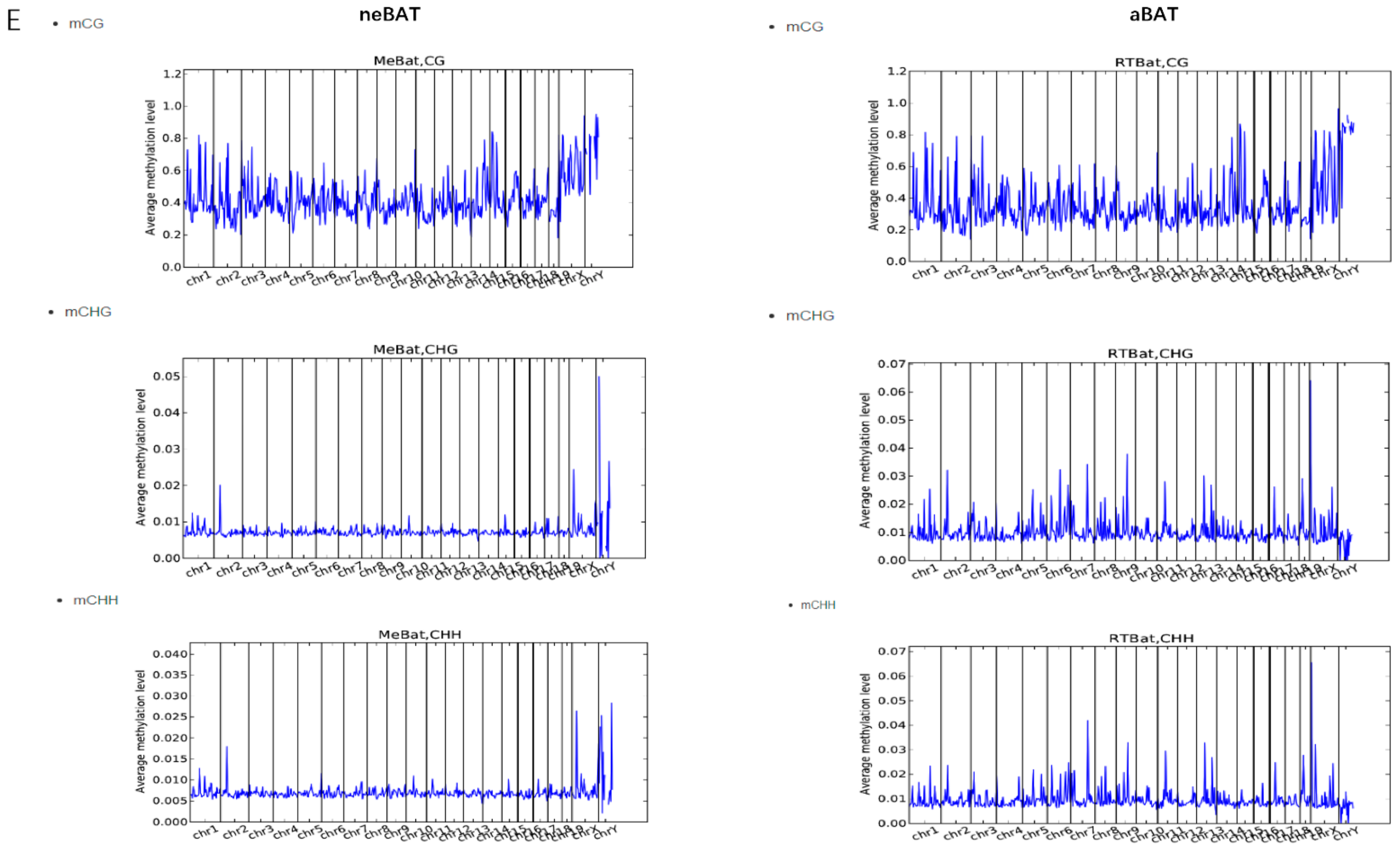
2.8.3. Transcriptions of DNA Methylation
2.9. Statistics
3. Results
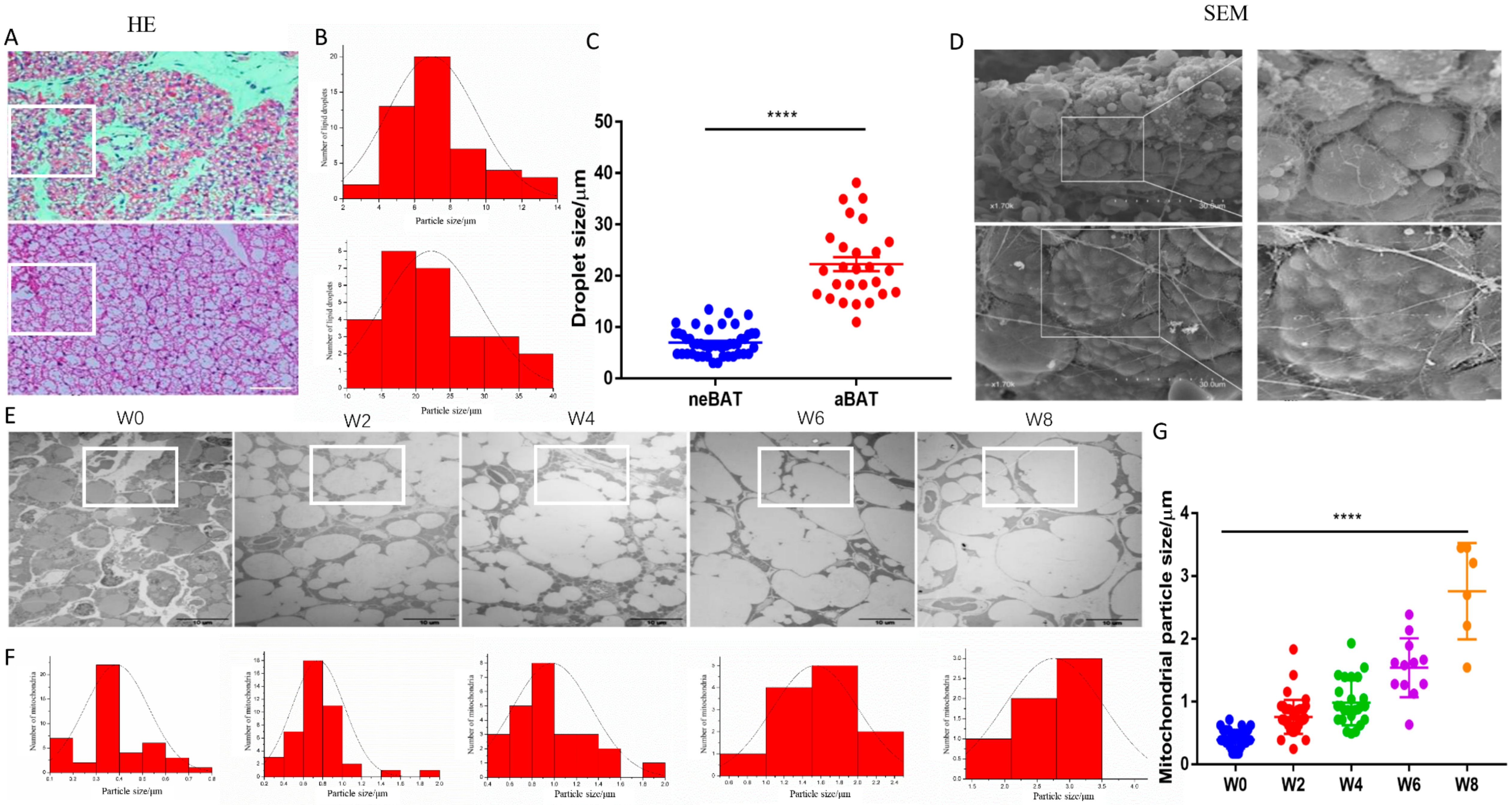
3.1. The Morphology of aBAT and neBAT
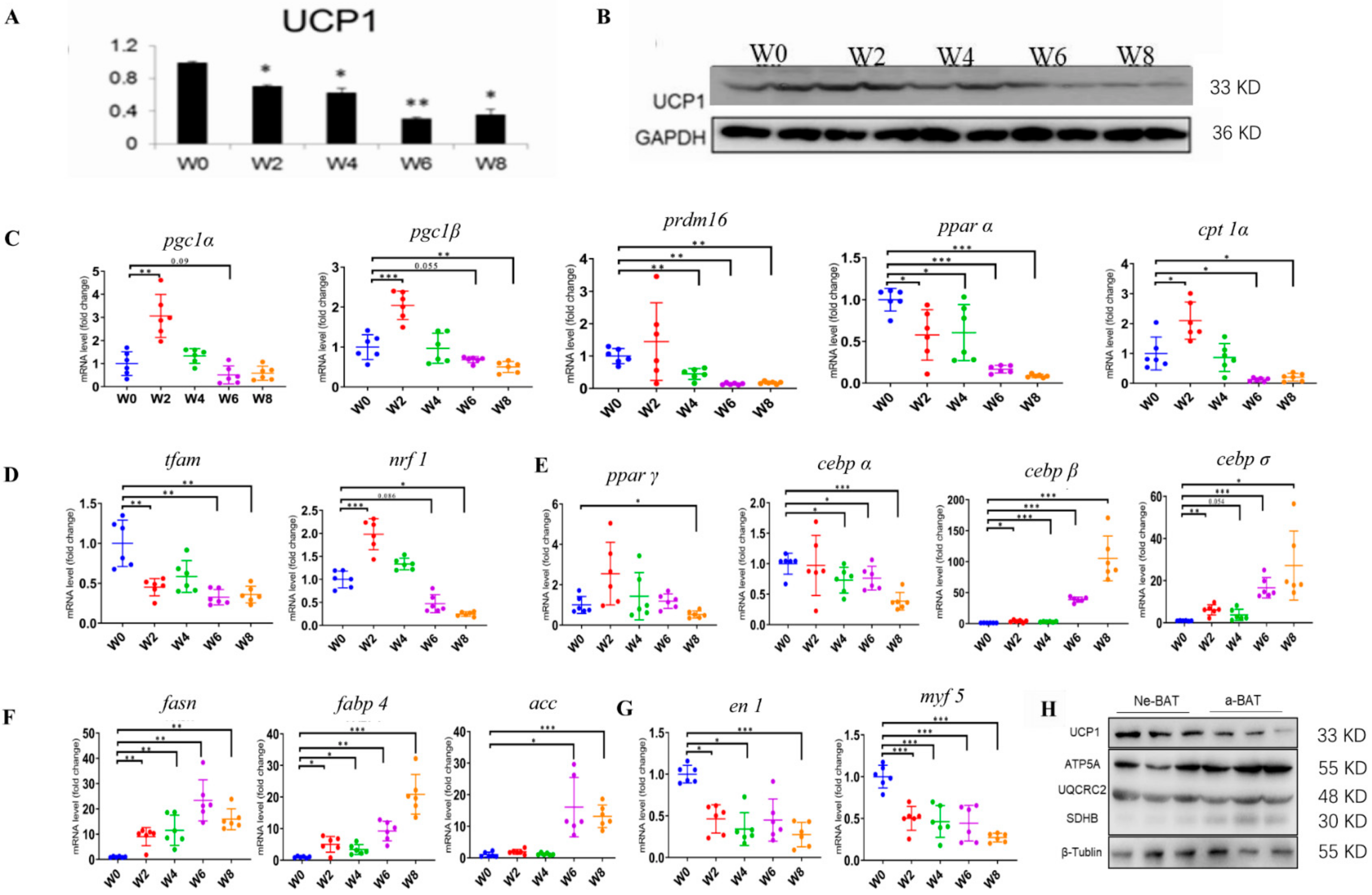
3.2. Brown Adipogenesis and Activation during Post-Delivery Development
3.3. NeBATs Are More Active Than Embryonic BAT in Mice
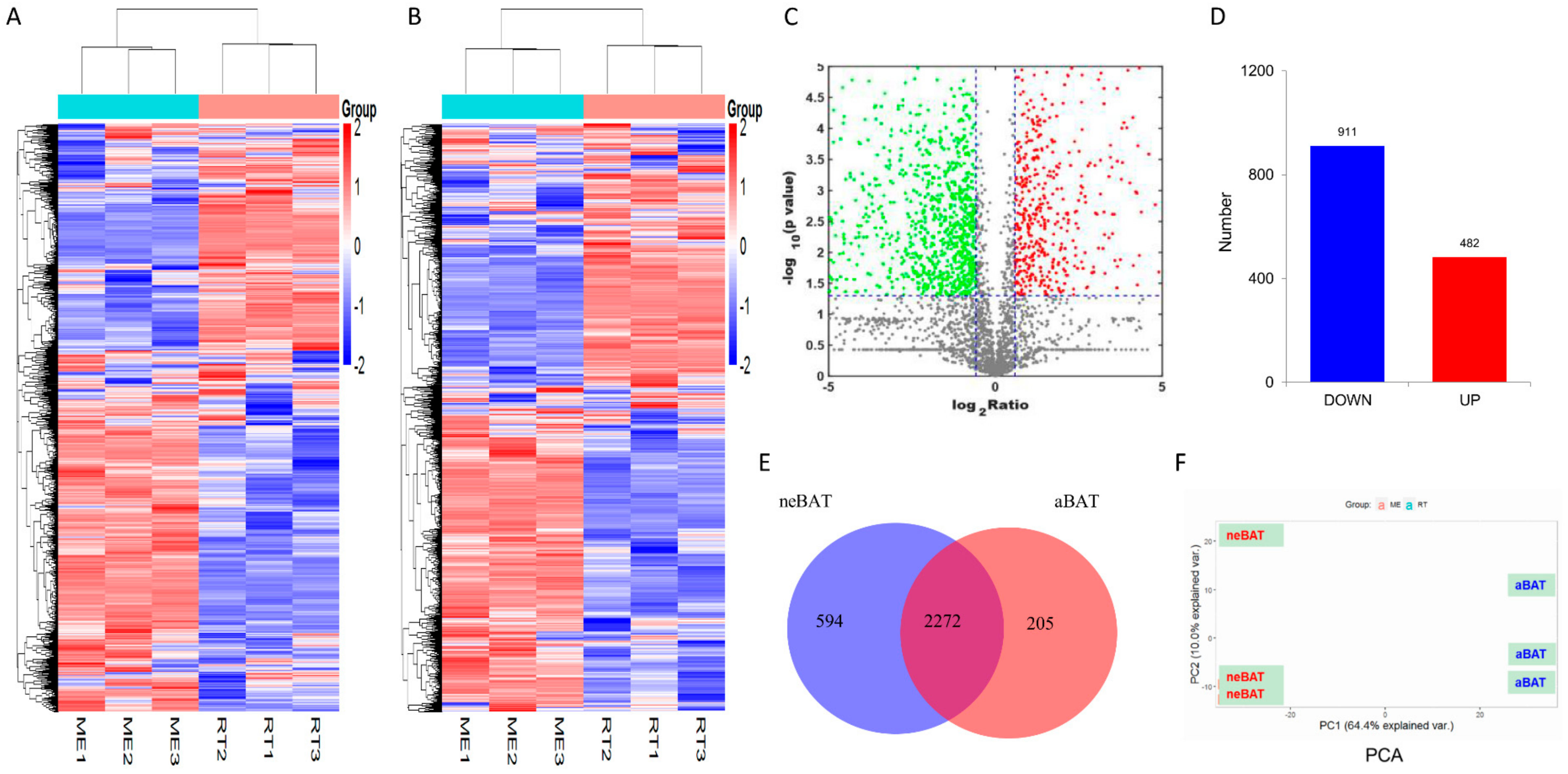
3.4. Analysis of Proteomic Data in neBAT and aBAT
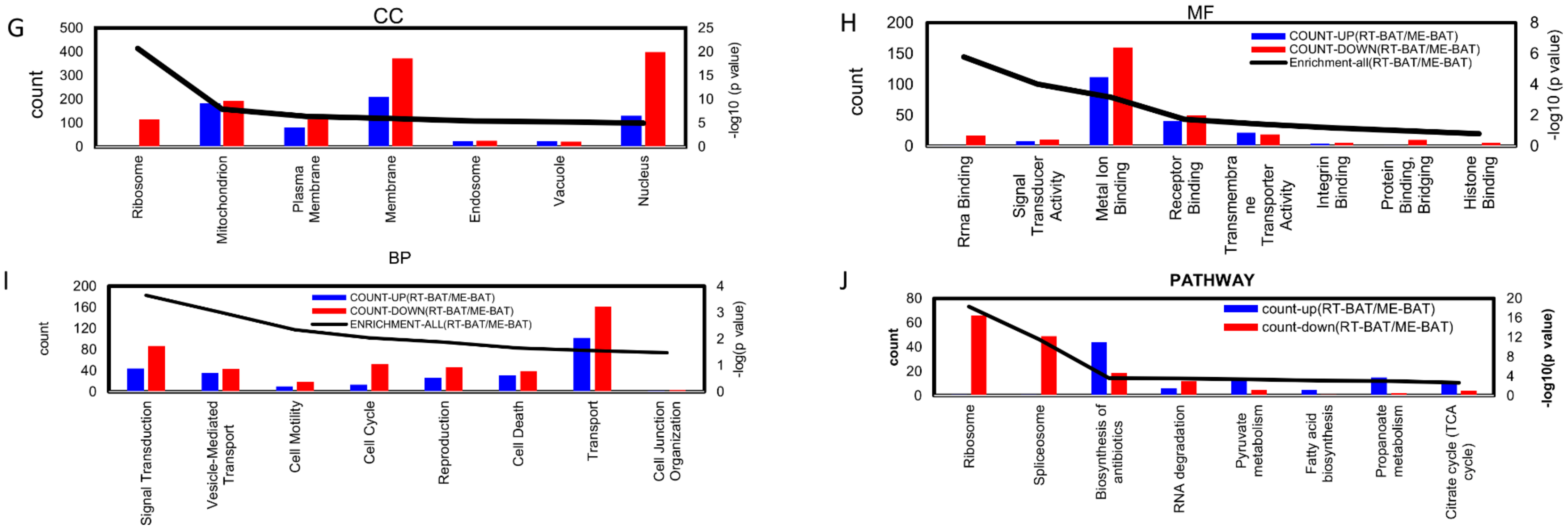
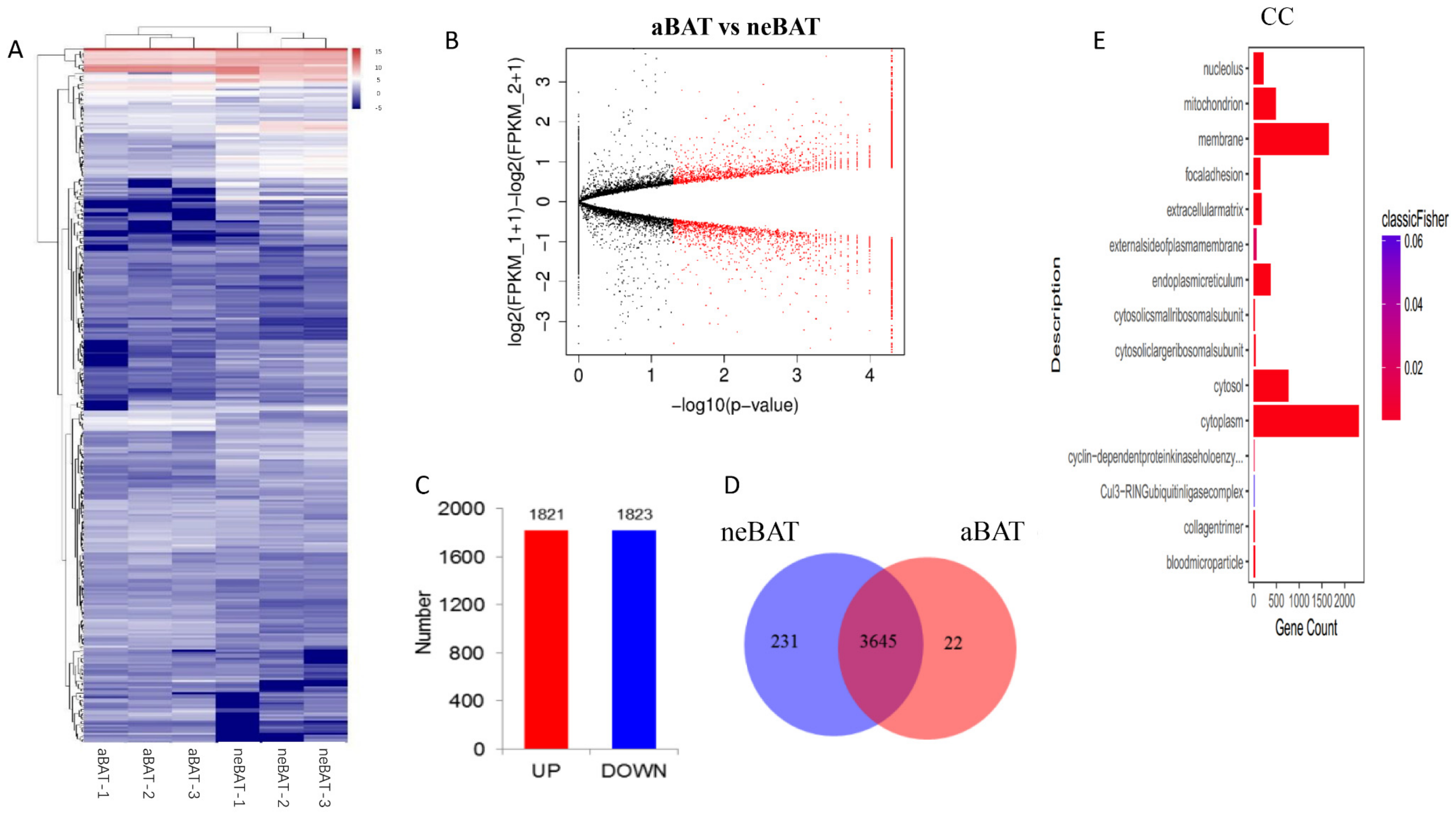
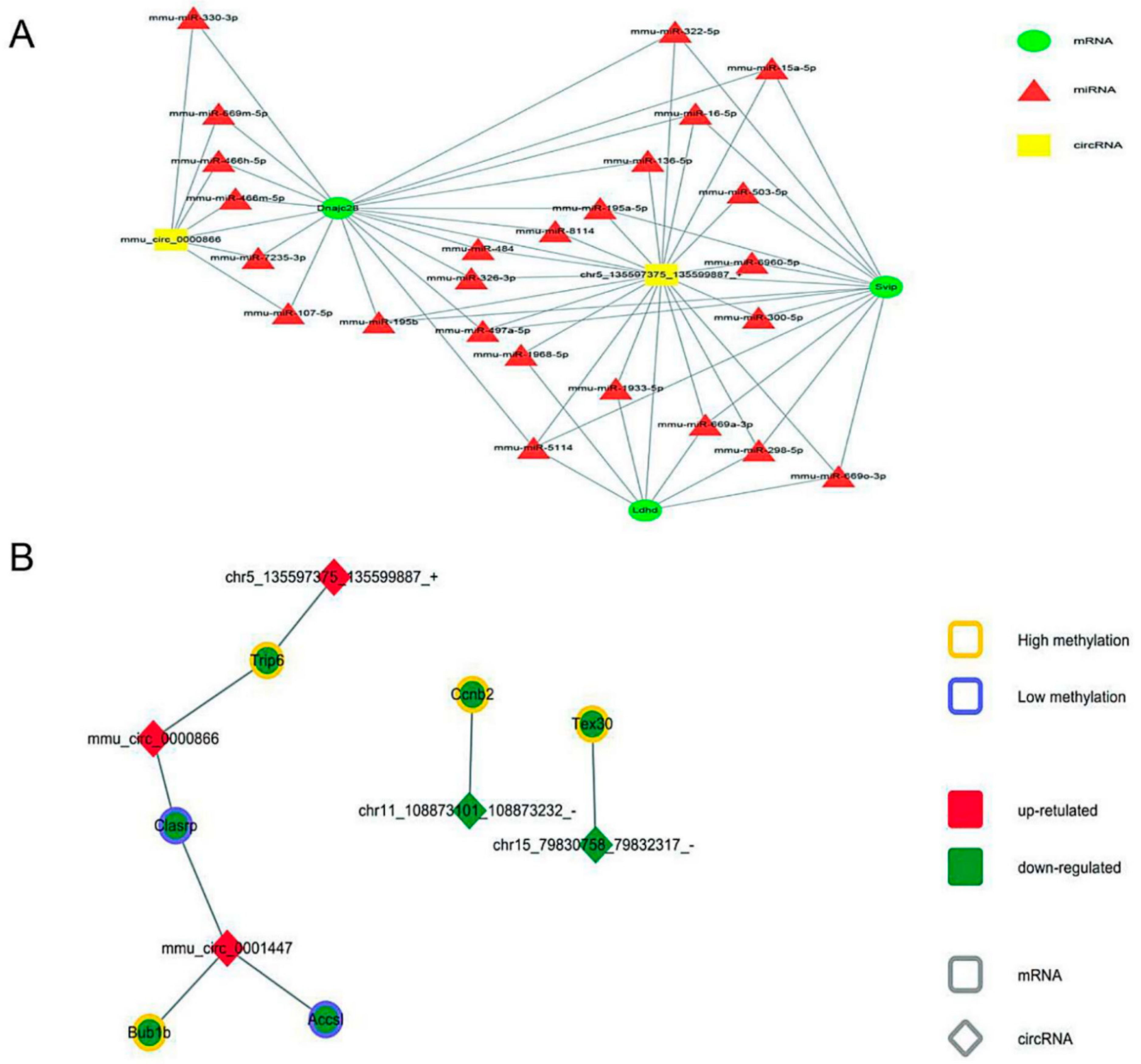
3.5. Comprehensive Analysis of Whole Transcriptome Data in neBAT and aBAT
3.6. Interaction among Transcriptome Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerback, S.; et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Symonds, M.E.; Aldiss, P.; Dellschaft, N.; Law, J.; Fainberg, H.P.; Pope, M.; Sacks, H.; Budge, H. Brown adipose tissue development and function and its impact on reproduction. J. Endocrinol. 2018, 238, R53–R62. [Google Scholar] [CrossRef] [Green Version]
- Farmer, S.R. Transcriptional control of adipocyte formation. Cell Metab. 2006, 4, 263–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, S.R. Molecular determinants of brown adipocyte formation and function. Genes Dev. 2008, 22, 1269–1275. [Google Scholar] [CrossRef] [Green Version]
- Kajimura, S.; Seale, P.; Spiegelman, B.M. Transcriptional control of brown fat development. Cell Metab. 2010, 11, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef]
- Seale, P.; Kajimura, S.; Spiegelman, B.M. Transcriptional control of brown adipocyte development and physiological function—Of mice and men. Genes Dev. 2009, 23, 788–797. [Google Scholar] [CrossRef] [Green Version]
- Vernochet, C.; McDonald, M.E.; Farmer, S.R. Brown adipose tissue: A promising target to combat obesity. Drug News Perspect. 2010, 23, 409–417. [Google Scholar] [CrossRef]
- Schulz, T.J.; Tseng, Y.H. Brown adipose tissue: Development, metabolism and beyond. Biochem. J. 2013, 453, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Spiegelman, B.M. Transcriptional activation of adipogenesis. Curr. Opin. Cell Biol. 1999, 11, 689–694. [Google Scholar] [CrossRef]
- Esterbauer, H.; Oberkofler, H.; Krempler, F.; Patsch, W. Human peroxisome proliferator activated receptor gamma coactivator 1 (PPARGC1) gene: cDNA sequence, genomic organization, chromosomal localization, and tissue expression. Genomics 1999, 62, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, S.; Seale, P.; Kubota, K.; Lunsford, E.; Frangioni, J.V.; Gygi, S.P.; Spiegelman, B.M. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature 2009, 460, 1154–1158. [Google Scholar] [CrossRef] [Green Version]
- Seale, P.; Kajimura, S.; Yang, W.; Chin, S.; Rohas, L.M.; Uldry, M.; Tavernier, G.; Langin, D.; Spiegelman, B.M. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007, 6, 38–54. [Google Scholar] [CrossRef] [Green Version]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scime, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar] [CrossRef] [Green Version]
- Cannon, B.; Nedergaard, J. Developmental biology: Neither fat nor flesh. Nature 2008, 454, 947–948. [Google Scholar] [CrossRef]
- Lepper, C.; Fan, C.M. Generating tamoxifen-inducible Cre alleles to investigate myogenesis in mice. In Myogenesis; Humana Press: Totowa, NJ, USA, 2012; Volume 798, pp. 297–308. [Google Scholar]
- Timmons, J.A.; Wennmalm, K.; Larsson, O.; Walden, T.B.; Lassmann, T.; Petrovic, N.; Hamilton, D.L.; Gimeno, R.E.; Wahlestedt, C.; Baar, K.; et al. Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc. Natl. Acad. Sci. USA 2007, 104, 4401–4406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joe, A.W.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Weng, Y.; Shi, F.; Jin, W. The Engrailed-1 Gene Stimulates Brown Adipogenesis. Stem Cells Int. 2016, 2016, 7369491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Li, B.; Li, M.; Niu, C.; Wang, G.; Li, T.; Krol, E.; Jin, W.; Speakman, J.R. Brown adipocytes can display a mammary basal myoepithelial cell phenotype in vivo. Mol. Metab. 2017, 6, 1198–1211. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Gurmaches, J.; Hung, C.M.; Sparks, C.A.; Tang, Y.; Li, H.; Guertin, D.A. PTEN loss in the Myf5 lineage redistributes body fat and reveals subsets of white adipocytes that arise from Myf5 precursors. Cell Metab. 2012, 16, 348–362. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Gurmaches, J.; Guertin, D.A. Adipocytes arise from multiple lineages that are heterogeneously and dynamically distributed. Nat. Commun. 2014, 5, 4099. [Google Scholar] [CrossRef] [Green Version]
- Atit, R.; Sgaier, S.K.; Mohamed, O.A.; Taketo, M.M.; Dufort, D.; Joyner, A.L.; Niswander, L.; Conlon, R.A. Beta-catenin activation is necessary and sufficient to specify the dorsal dermal fate in the mouse. Dev. Biol. 2006, 296, 164–176. [Google Scholar] [CrossRef] [Green Version]
- Konishi, M.; Mikami, T.; Yamasaki, M.; Miyake, A.; Itoh, N. Fibroblast growth factor-16 is a growth factor for embryonic brown adipocytes. J. Biol. Chem. 2000, 275, 12119–12122. [Google Scholar] [CrossRef] [Green Version]
- Longo, K.A.; Wright, W.S.; Kang, S.; Gerin, I.; Chiang, S.H.; Lucas, P.C.; Opp, M.R.; MacDougald, O.A. Wnt10b inhibits development of white and brown adipose tissues. J. Biol. Chem. 2004, 279, 35503–35509. [Google Scholar] [CrossRef] [Green Version]
- Pospisilik, J.A.; Schramek, D.; Schnidar, H.; Cronin, S.J.; Nehme, N.T.; Zhang, X.; Knauf, C.; Cani, P.D.; Aumayr, K.; Todoric, J.; et al. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell 2010, 140, 148–160. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 2002, 143, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.H.; Kokkotou, E.; Schulz, T.J.; Huang, T.L.; Winnay, J.N.; Taniguchi, C.M.; Tran, T.T.; Suzuki, R.; Espinoza, D.O.; Yamamoto, Y.; et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 2008, 454, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Yadav, H.; Quijano, C.; Kamaraju, A.K.; Gavrilova, O.; Malek, R.; Chen, W.; Zerfas, P.; Zhigang, D.; Wright, E.C.; Stuelten, C.; et al. Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell Metab. 2011, 14, 67–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadian, M.; Abbott, M.J.; Tang, T.; Hudak, C.S.; Kim, Y.; Bruss, M.; Hellerstein, M.K.; Lee, H.Y.; Samuel, V.T.; Shulman, G.I.; et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011, 13, 739–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Pan, R.; Pfeifer, A. Regulation of brown and beige fat by microRNAs. Pharmacol. Ther. 2017, 170, 1–7. [Google Scholar] [CrossRef]
- Wei, S.; Du, M.; Jiang, Z.; Hausman, G.J.; Zhang, L.; Dodson, M.V. Long noncoding RNAs in regulating adipogenesis: New RNAs shed lights on obesity. Cell. Mol. Life Sci. 2016, 73, 2079–2087. [Google Scholar] [CrossRef] [Green Version]
- Lo, K.A.; Huang, S.; Walet, A.C.E.; Zhang, Z.C.; Leow, M.K.; Liu, M.; Sun, L. Adipocyte Long-Noncoding RNA Transcriptome Analysis of Obese Mice Identified Lnc-Leptin, Which Regulates Leptin. Diabetes 2018, 67, 1045–1056. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Goff, L.A.; Trapnell, C.; Alexander, R.; Lo, K.A.; Hacisuleyman, E.; Sauvageau, M.; Tazon-Vega, B.; Kelley, D.R.; Hendrickson, D.G.; et al. Long noncoding RNAs regulate adipogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 3387–3392. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.Y.; Li, S.; Wang, G.X.; Yu, Q.; Lin, J.D. A long noncoding RNA transcriptional regulatory circuit drives thermogenic adipocyte differentiation. Mol. Cell 2014, 55, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Weiner, J.; Rohde, K.; Krause, K.; Zieger, K.; Kloting, N.; Kralisch, S.; Kovacs, P.; Stumvoll, M.; Bluher, M.; Bottcher, Y.; et al. Brown adipose tissue (BAT) specific vaspin expression is increased after obesogenic diets and cold exposure and linked to acute changes in DNA-methylation. Mol. Metab. 2017, 6, 482–493. [Google Scholar] [CrossRef]
- Son, M.J.; Kim, W.K.; Oh, K.J.; Park, A.; Lee da, S.; Han, B.S.; Lee, S.C.; Bae, K.H. Methyltransferase and demethylase profiling studies during brown adipocyte differentiation. BMB Rep. 2016, 49, 388–393. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Ge, K. Transcriptional and epigenetic regulation of PPARgamma expression during adipogenesis. Cell Biosci. 2014, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Wei, G.; You, Y.; Huang, Y.; Lee, H.J.; Dong, M.; Lin, J.; Hu, T.; Zhang, H.; Zhang, C. Rutin ameliorates obesity through brown fat activation. FASEB J. 2016, 31, 333–345. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.I.; Newbury, D.E.; Michael, J.R.; Ritchie, N.W.; Scott, J.H.J.; Joy, D.C. Scanning Electron Microscopy and X-Ray Microanalysis; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ji, J.; Yang, L.; Fang, Z.; Zhuang, M.; Zhang, Y.; Lv, H.; Liu, Y.; Li, Z. Complementary transcriptome and proteome profiling in cabbage buds of a recessive male sterile mutant provides new insights into male reproductive development. J. Proteom. 2018, 179, 80–91. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301. [Google Scholar] [CrossRef]
- Luo, H.; Bu, D.; Sun, L.; Fang, S.; Liu, Z.; Zhao, Y. Identification and function annotation of long intervening noncoding RNAs. Brief. Bioinform. 2016, 18, 789–797. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Guo, H.; Zhu, P.; Guo, F.; Li, X.; Wu, X.; Fan, X.; Wen, L.; Tang, F. Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing. Nat. Protoc. 2015, 10, 645. [Google Scholar] [CrossRef]
- Meröndun, J.; Murray, D.L.; Shafer, A.B. Genome-scale sampling suggests cryptic epigenetic structuring and insular divergence in Canada lynx. Mol. Ecol. 2019, 28, 3186–3196. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Lidell, M.E.; Betz, M.J.; Enerback, S. Brown adipose tissue and its therapeutic potential. J. Intern. Med. 2014, 276, 364–377. [Google Scholar] [CrossRef]
- Tanaka, T.; Yoshida, N.; Kishimoto, T.; Akira, S. Defective adipocyte differentiation in mice lacking the C/EBPbeta and/or C/EBPdelta gene. EMBO J. 1997, 16, 7432–7443. [Google Scholar] [CrossRef] [Green Version]
- Gilsanz, V.; Smith, M.L.; Goodarzian, F.; Kim, M.; Wren, T.A.; Hu, H.H. Changes in brown adipose tissue in boys and girls during childhood and puberty. J. Pediatrics 2012, 160, 604–609.e1. [Google Scholar] [CrossRef] [Green Version]
- Symonds, M.E.; Dellschaft, N.; Pope, M.; Birtwistle, M.; Alagal, R.; Keisler, D.; Budge, H. Developmental programming, adiposity, and reproduction in ruminants. Theriogenology 2016, 86, 120–129. [Google Scholar] [CrossRef]
- Labbe, S.M.; Caron, A.; Bakan, I.; Laplante, M.; Carpentier, A.C.; Lecomte, R.; Richard, D. In vivo measurement of energy substrate contribution to cold-induced brown adipose tissue thermogenesis. FASEB J. 2015, 29, 2046–2058. [Google Scholar] [CrossRef] [Green Version]
- Yoneshiro, T.; Wang, Q.; Tajima, K.; Matsushita, M.; Maki, H.; Igarashi, K.; Dai, Z.; White, P.J.; McGarrah, R.W.; Ilkayeva, O.R.; et al. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nature 2019, 572, 614–619. [Google Scholar] [CrossRef]
- Matsuzawa, Y.; Funahashi, T.; Kihara, S.; Shimomura, I. Adiponectin and metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 29–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reantragoon, R.; Corbett, A.J.; Sakala, I.G.; Gherardin, N.A.; Furness, J.B.; Chen, Z.; Eckle, S.B.; Uldrich, A.P.; Birkinshaw, R.W.; Patel, O. Antigen-loaded MR1 tetramers define T cell receptor heterogeneity in mucosal-associated invariant T cells. J. Exp. Med. 2013, 210, 2305–2320. [Google Scholar] [CrossRef]
- Ueda, N.; Tsuboi, K.; Uyama, T. N-acylethanolamine metabolism with special reference to N-acylethanolamine-hydrolyzing acid amidase (NAAA). Prog. Lipid Res. 2010, 49, 299–315. [Google Scholar] [CrossRef]
- Xue, H.; Zhang, J.; Guo, X.; Wang, J.; Li, J.; Gao, X.; Guo, X.; Li, T.; Xu, S.; Zhang, P. CREBRF is a potent tumor suppressor of glioblastoma by blocking hypoxia-induced autophagy via the CREB3/ATG5 pathway. Int. J. Oncol. 2016, 49, 519–528. [Google Scholar] [CrossRef]
- Quayle, S.N.; Lee, J.Y.; Cheung, L.W.T.; Ding, L.; Wiedemeyer, R.; Dewan, R.W.; Huang-Hobbs, E.; Zhuang, L.; Wilson, R.K.; Ligon, K.L. Somatic mutations of PIK3R1 promote gliomagenesis. PLoS ONE 2012, 7, e49466. [Google Scholar] [CrossRef]
- Pei, Y.F.; Zhang, Y.J.; Lei, Y.; Wu, D.W.; Ma, T.H.; Liu, X.Q. Hypermethylation of the CHRDL1 promoter induces proliferation and metastasis by activating Akt and Erk in gastric cancer. Oncotarget 2017, 8, 23155. [Google Scholar]
- Maldonado, L.; Brait, M.; Begum, S.; Chatterjee, A.; Loyo, M.; Barbosa, A.; Poeta, M.L.; Fazio, V.M.; Roberto, A.; Tarquini, E. GULP1, a Potential Tumor Suppressor Gene in Ovarian Tumors and Its Utility as a Biomarker; AACR: Philadelphia, PA, USA, 2010. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| ACC | AGCTGATCCTGCGAACCT | GCCAAGCGGATGTAAACT |
| AP2 | GAAGACTGCGAGGACCTC | GAAGTGCCGTAATCCCCACAC |
| CEBP/α | GCGGGAACGCAACAACATC | GTCACTGGTCAACTCCAGCAC |
| CEBP/β | TGACGCAACACACGTGTAACTG | AACAACCCCGCAGGAACAT |
| CEBP/δ | CGACTTCAGCGCCTACATTGA | GAAGAGGTCGGCGAAGAGTT |
| CPT1α | GACTCCGCTCGCTCATTCC | GACTGTGAACTGGAAGGCCA |
| CyclophilinA | CAAATGCTGGACCAAACACA | GCCATCCAGCCATTCAGTCT |
| EN1 | CTCACAGCAACCCCTAGTGT | CCGCTGCTCCGTGATATAG |
| Fasn | TAGAGGGAGCCAGAGAGACG | CCGACATACCGGCTATCACC |
| Myf5 | GCCTTCGGAGCACACAAAG | TGACCTTCTTCAGGCGTCTAC |
| NRF1 | CAACAGGGAAGAAACGGAAA | GCACCACATTCTCCAAAGGT |
| NRF2 | TAGATGACCATGAGTCGCTTGC | GCCAAACTTGCTCCATGTCC |
| PGC1α | ACCGCTTTCTGGGTGGATT | TGAGGACCGCTAGCAAGTTT |
| PGC1β | CGTATTTGAGGACAGCAGCA | TACTGGGTGGGCTCTGGTAG |
| PPARα | AGCCTCAGCCAAGGTTGAACT | TGGGGAGAGAGGACAGATGG |
| PPARγ2 | TCGCTGATGCACTGCCTATG | GAGAGGTCCACAGAGCTGATT |
| PRDM16 | GAAGTCACAGGAGGACACGG | CTCGCTCCTCAACACACCTC |
| Tfam | GTCCATAGGCACCGTATTGC | CCCATGCTGGAAAAACACTT |
| UCP1 | GGCAAAAACAGAAGGATTGC | TAAGCCGGCTGAGATCTTGT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Zhang, C.; Zhang, B.; Sheng, Y.; Xu, W.; Luo, Y.; He, X.; Huang, K. Comprehensive Analysis of the Characteristics and Differences in Adult and Newborn Brown Adipose Tissue (BAT): Newborn BAT Is a More Active/Dynamic BAT. Cells 2020, 9, 201. https://doi.org/10.3390/cells9010201
Liu J, Zhang C, Zhang B, Sheng Y, Xu W, Luo Y, He X, Huang K. Comprehensive Analysis of the Characteristics and Differences in Adult and Newborn Brown Adipose Tissue (BAT): Newborn BAT Is a More Active/Dynamic BAT. Cells. 2020; 9(1):201. https://doi.org/10.3390/cells9010201
Chicago/Turabian StyleLiu, Junyu, Chuanhai Zhang, Boyang Zhang, Yao Sheng, Wentao Xu, Yunbo Luo, Xiaoyun He, and Kunlun Huang. 2020. "Comprehensive Analysis of the Characteristics and Differences in Adult and Newborn Brown Adipose Tissue (BAT): Newborn BAT Is a More Active/Dynamic BAT" Cells 9, no. 1: 201. https://doi.org/10.3390/cells9010201