Neuroinflammatory Reactions in the Brain of 1,2-DCE-Intoxicated Mice during Brain Edema

Abstract
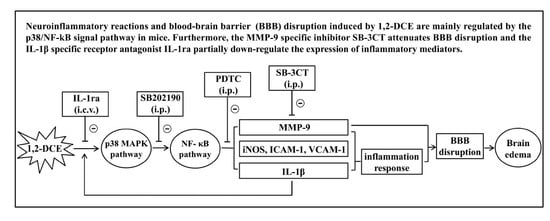
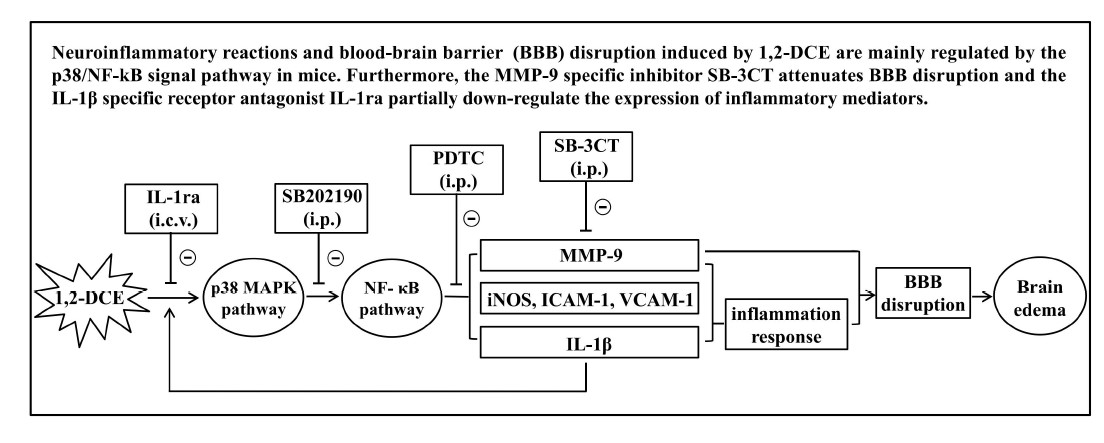
1. Introduction
2. Materials and Methods
2.1. Animal Care and Use Statement
2.2. Reagents
2.3. Experimental Procedures and Treatment
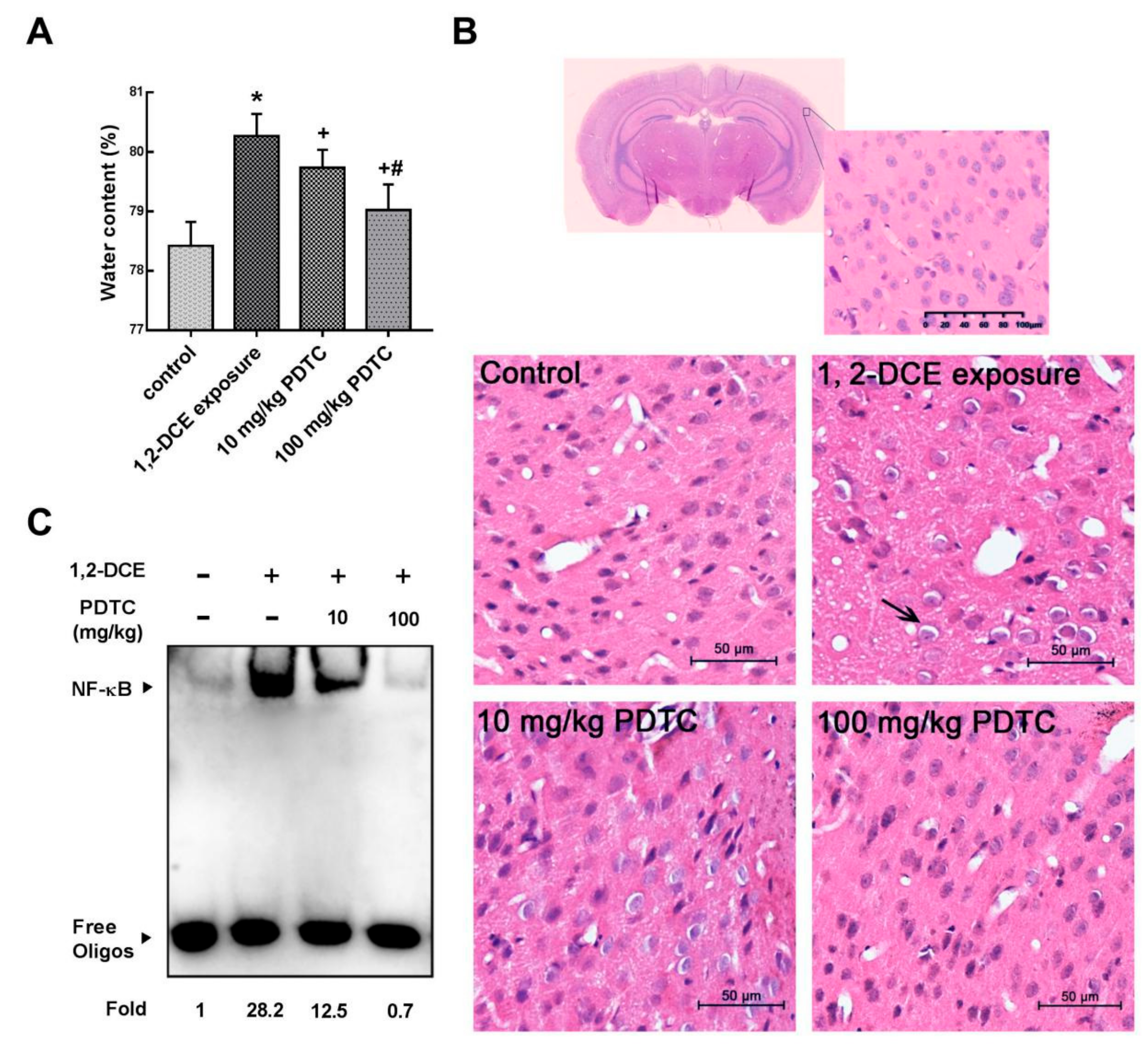
2.4. Brain Water Content
2.5. Histological Observation
2.6. Immunostaining
2.7. Western Blotting
2.8. Quantitative Real-Time (RT)-PCR
2.9. Electrophoretic Mobility Shift Assay (EMSA)
2.10. ELISA
2.11. Statistical Analysis
3. Results
3.1. Involvement of NF-κB in 1,2-DCE-Induced Brain Edema in Mice
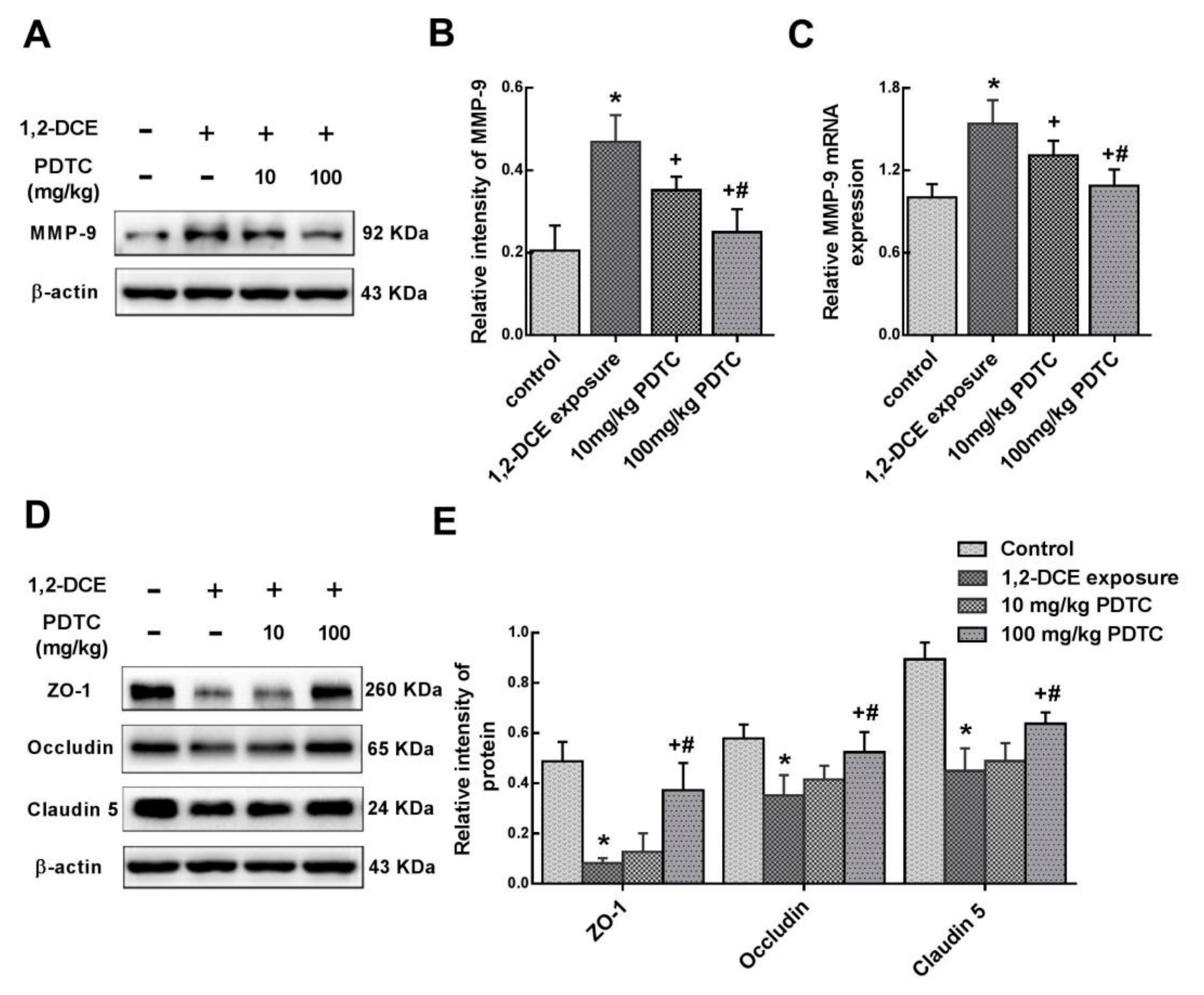
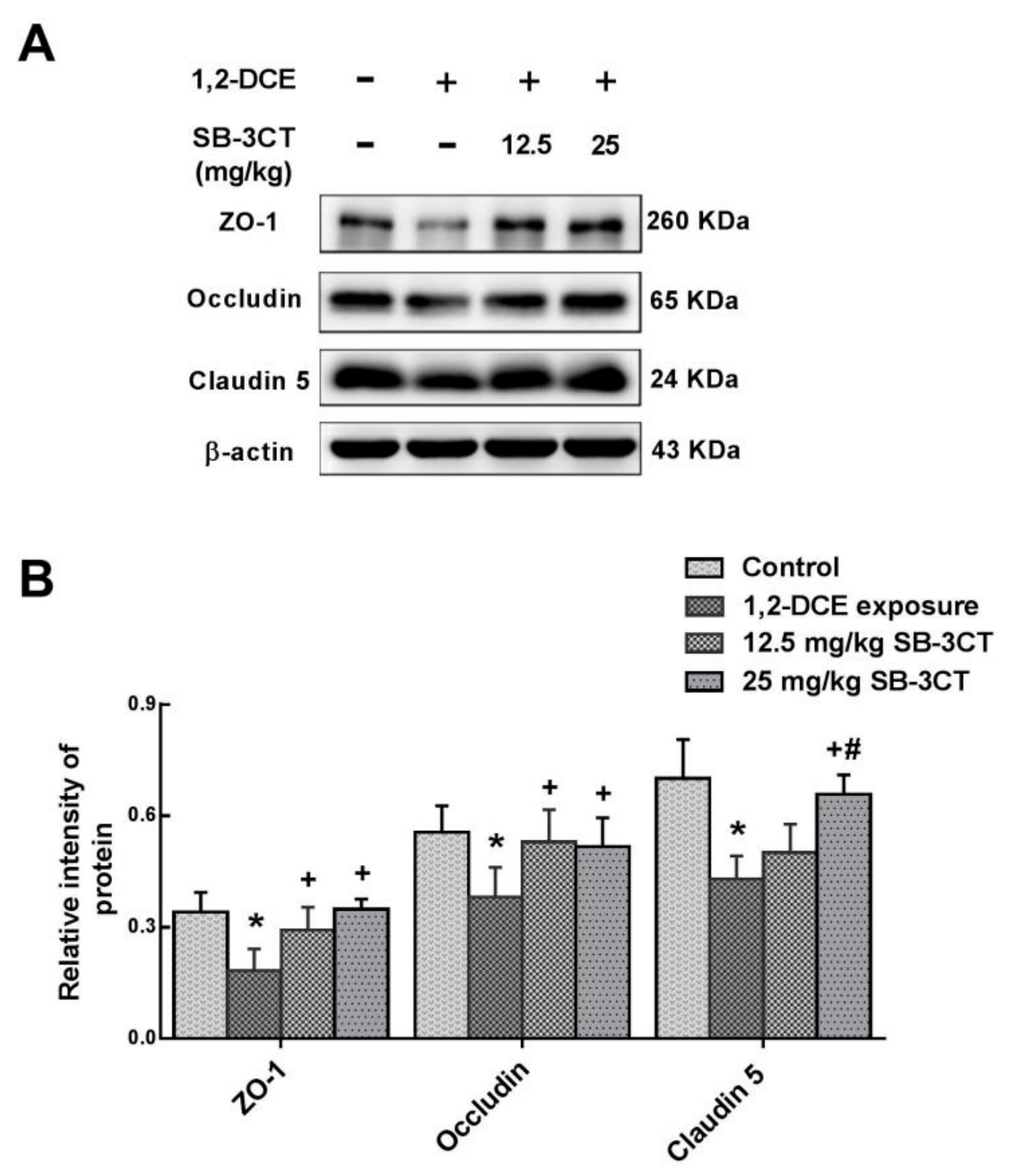
3.2. MMP-9 Involvement in 1,2-DCE-Induced BBB Disruption in Mice
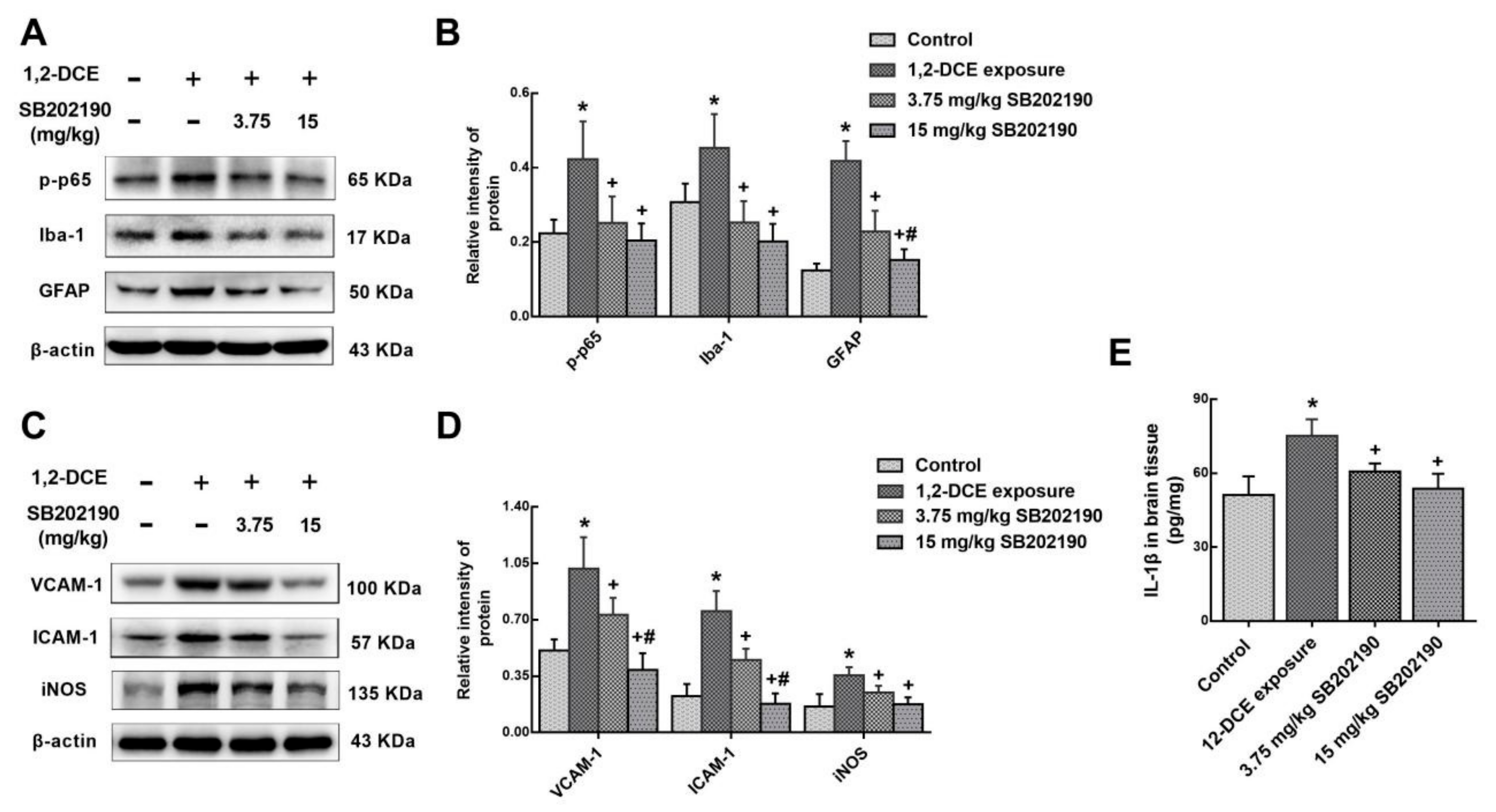
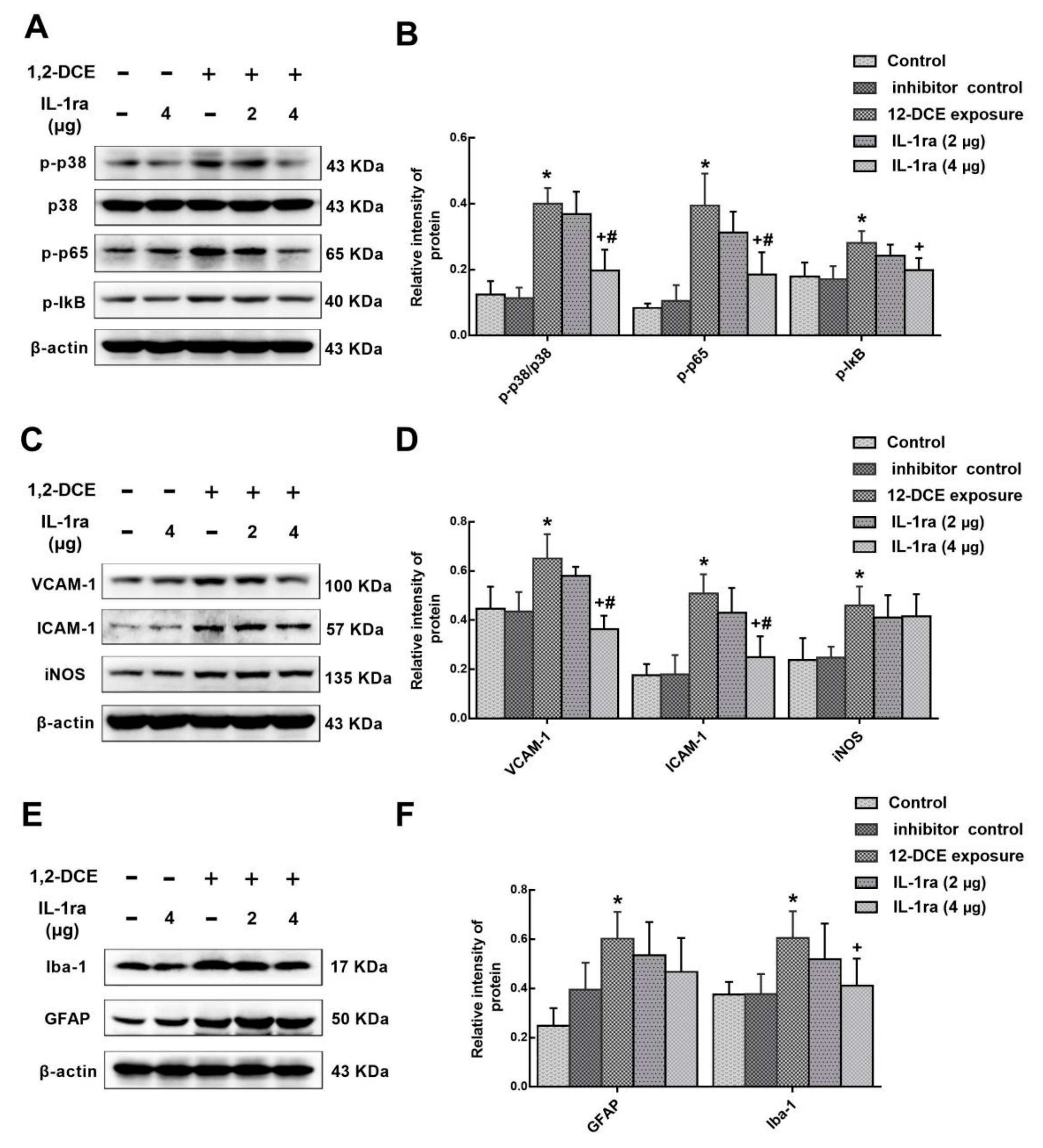
3.3. p38 MAPK Signaling Pathway Involvement in the Inflammatory Reaction During the Course of 1,2-DCE-Induced Brain Edema in Mice
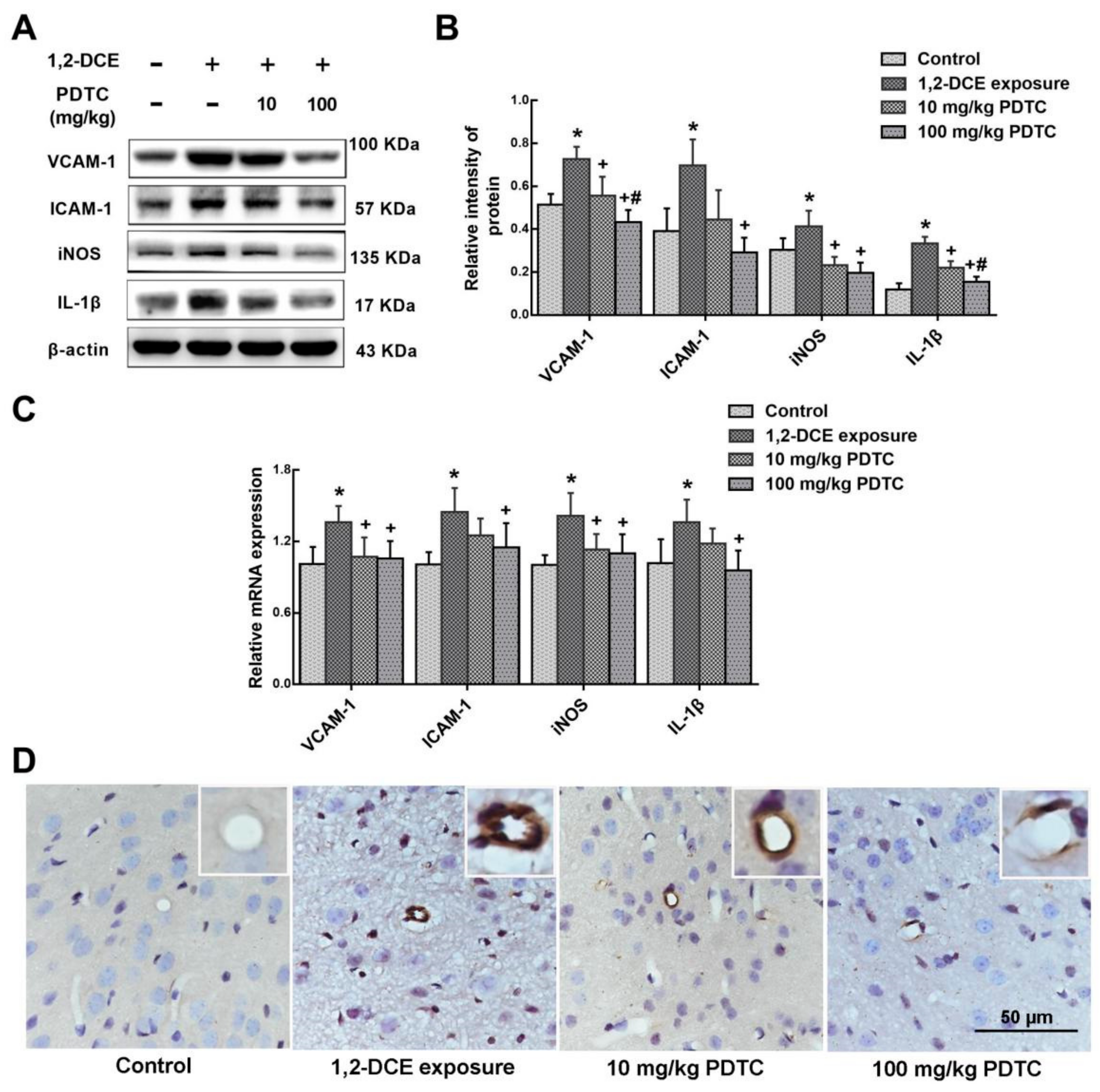
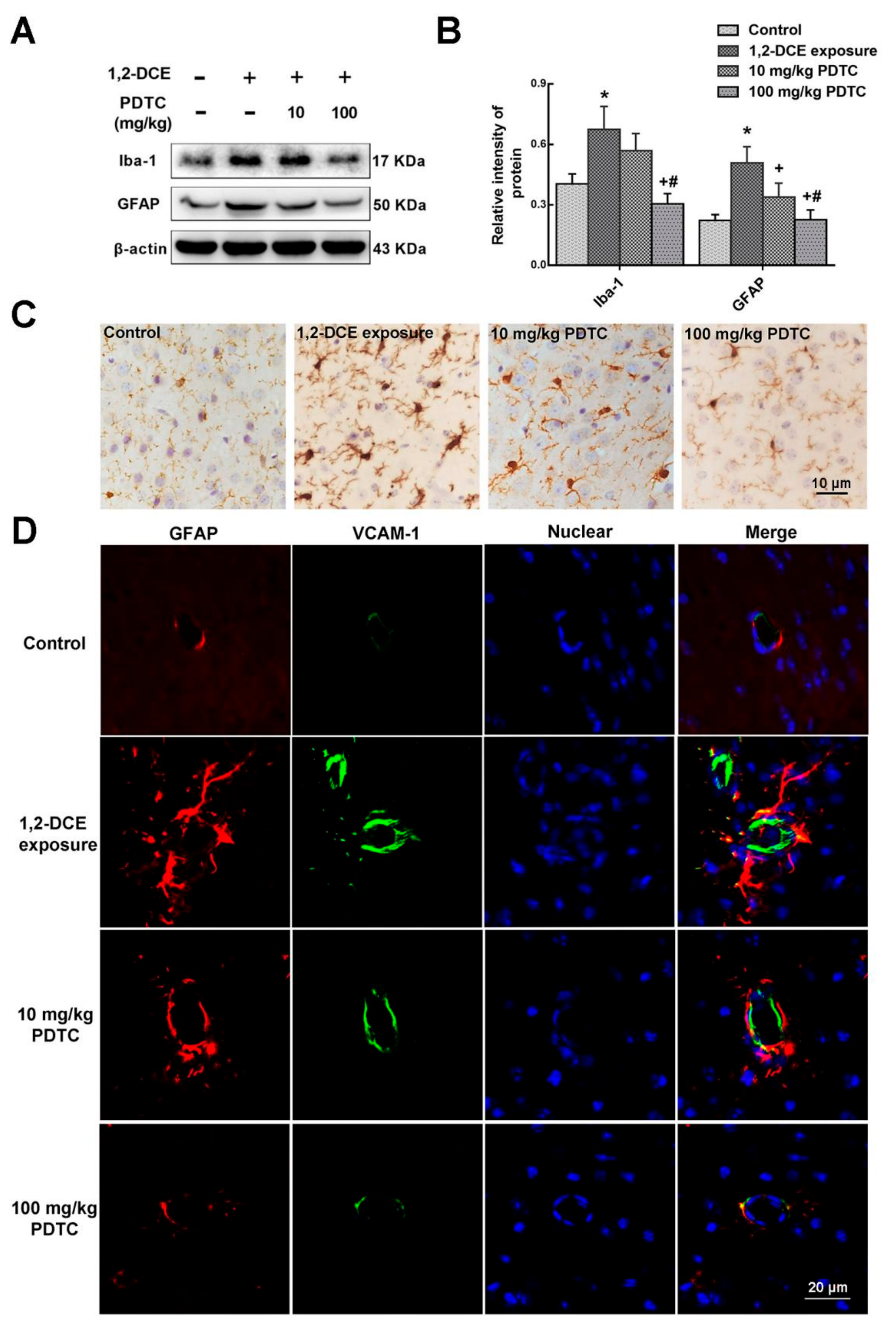
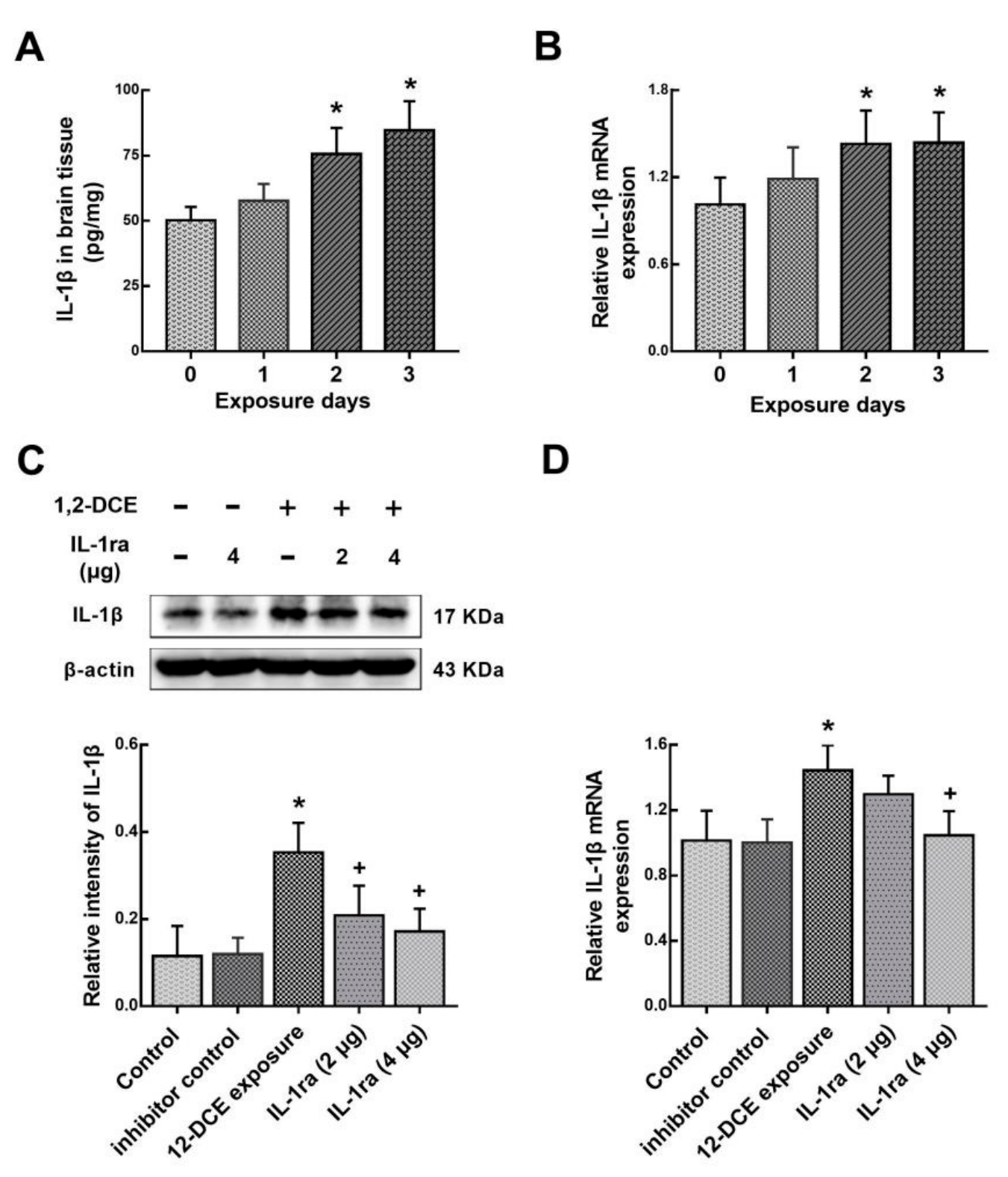
3.4. IL-1β Involvement in the Neuroinflammatory Reaction During the Course of 1,2-DCE-Induced Brain Edema in Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Take, M.; Matsumoto, M.; Takeuchi, T.; Haresaku, M.; Kondo, H.; Senoh, H.; Umeda, Y.; Takamura-Enya, T.; Fukushima, S. Inhalation exposure to 1,2-dichloropropane: Distribution of blood and tissue concentrations of 1,2-dichloropropane in rats during and after exposure. J. Environ. Sci. Health A Tox. Hazard Subst. Environ. Eng. 2014, 49, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhan, F.; Zheng, W.; Liu, L.; Kong, L. Diagnosis and prognosis evaluation of 1,2-dichloroethane encephalopathy--magnetic resonance imaging combined with diffusion tensor imaging and magnetic resonance spectroscopy study. Neurol. India 2011, 59, 108–110. [Google Scholar] [PubMed]
- Liu, J.R.; Fang, S.; Ding, M.P.; Chen, Z.C.; Zhou, J.J.; Sun, F.; Jiang, B.; Huang, J. Toxic encephalopathy caused by occupational exposure to 1, 2-Dichloroethane. J. Neurol. Sci. 2010, 292, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.B.; Hu, H.T.; Zhang, Y.; Song, S.J. Clinical and cranial MRI analysis on five cases of toxic encephalopathy induced by dichloroethane. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi 2009, 27, 744–746. [Google Scholar] [PubMed]
- Zhou, X.; Zhou, W.; Zhou, J.; Long, L.; Xiao, B. 1,2-Dichloroethane-induced toxic leukoencephalopathy with a brain biopsy. Neurol. Sci. 2015, 36, 817–819. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Liao, Y.; Tan, X.; Guo, J.; Wang, G.; Zhao, F.; Jin, Y. Involvement of the p38 MAPK signaling pathway in overexpression of matrix metalloproteinase-9 during the course of brain edema in 1,2-dichloroethane-intoxicated mice. Neurotoxicology 2018, 69, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yuan, Y.; Zhang, J.; Gao, L.; Tan, X.; Yang, G.; Lv, X.; Jin, Y. Roles of aquaporins and matrix metalloproteinases in mouse brain edema formation induced by subacute exposure to 1,2-dichloroethane. Neurotoxicol. Teratol. 2014, 44, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Chen, S.; Zhang, C.; Meng, F.; Wu, W.; Hu, R.; Hadass, O.; Lehmidi, T.; Blair, G.J.; Lee, M.; et al. Inhibition of MMP-9 by a selective gelatinase inhibitor protects neurovasculature from embolic focal cerebral ischemia. Mol. Neurodegener. 2012, 7. [Google Scholar] [CrossRef]
- Pan, C.; Wang, C.; Zhang, L.; Song, L.; Chen, Y.; Liu, B.; Liu, W.T.; Hu, L.; Pan, Y. Procyanidins attenuate neuropathic pain by suppressing matrix metalloproteinase-9/2. J. Neuroinflammation 2018, 15, 187. [Google Scholar] [CrossRef]
- Rana, A.; Musto, A.E. The role of inflammation in the development of epilepsy. J. Neuroinflammation 2018, 15. [Google Scholar] [CrossRef]
- Sun, J.; Nan, G. The Mitogen-Activated Protein Kinase (MAPK) Signaling Pathway as a Discovery Target in Stroke. J. Mol. Neurosci. 2016, 59, 90–98. [Google Scholar] [CrossRef]
- Zhao, Y.W.; Pan, Y.Q.; Tang, M.M.; Lin, W.J. Blocking p38 Signaling Reduces the Activation of Pro-inflammatory Cytokines and the Phosphorylation of p38 in the Habenula and Reverses Depressive-Like Behaviors Induced by Neuroinflammation. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Tao, X.G.; Shi, J.H.; Hao, S.Y.; Chen, X.T.; Liu, B.Y. Protective Effects of Calpain Inhibition on Neurovascular Unit Injury through Downregulating Nuclear Factor-kappaB-related Inflammation during Traumatic Brain Injury in Mice. Chin. Med. J. 2017, 130, 187–198. [Google Scholar]
- Zhang, H.; Liang, B.; Li, T.; Zhou, Y.; Shang, D.; Du, Z. Orexin A Suppresses Oxidized LDL Induced Endothelial Cell Inflammation via MAPK p38 and NF-kappaB Signaling Pathway. IUBMB Life 2018, 70, 961–968. [Google Scholar] [CrossRef]
- Kim, S.K.; Choe, J.Y.; Park, K.Y. Rebamipide Suppresses Monosodium Urate Crystal-Induced Interleukin-1beta Production Through Regulation of Oxidative Stress and Caspase-1 in THP-1 Cells. Inflammation 2016, 39, 473–482. [Google Scholar] [CrossRef]
- Tseng, H.C.; Lee, I.T.; Lin, C.C.; Chi, P.L.; Cheng, S.E.; Shih, R.H.; Hsiao, L.D.; Yang, C.M. IL-1beta promotes corneal epithelial cell migration by increasing MMP-9 expression through NF-kappaB- and AP-1-dependent pathways. PLoS ONE 2013, 8, e57955. [Google Scholar] [CrossRef]
- Yang, C.M.; Hsieh, H.L.; Yu, P.H.; Lin, C.C.; Liu, S.W. IL-1beta Induces MMP-9-Dependent Brain Astrocytic Migration via Transactivation of PDGF Receptor/NADPH Oxidase 2-Derived Reactive Oxygen Species Signals. Mol. Neurobiol. 2015, 52, 303–317. [Google Scholar] [CrossRef]
- Barnes, P.J.; Adcock, I.M. NF-kB:a pivotal role in asthma and a new target for therapy. Trends Pharmacol. Sci. 1997, 18, 46–50. [Google Scholar] [CrossRef]
- Thawkar, B.S.; Kaur, G. Inhibitors of NF-kappaB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol. 2019, 326, 62–74. [Google Scholar] [CrossRef]
- Fang, W.; Sha, L.; Kodithuwakku, N.D.; Wei, J.; Zhang, R.; Han, D.; Mao, L.; Li, Y. Attenuated Blood-Brain Barrier Dysfunction by XQ-1H Following Ischemic Stroke in Hyperlipidemic Rats. Mol. Neurobiol. 2015, 52, 162–175. [Google Scholar] [CrossRef]
- Yang, J.T.; Lee, T.H.; Lee, I.N.; Chung, C.Y.; Kuo, C.H.; Weng, H.H. Dexamethasone inhibits ICAM-1 and MMP-9 expression and reduces brain edema in intracerebral hemorrhagic rats. Acta Neurochir. 2011, 153, 2197–2203. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Sharma, N.; Jamwal, S.; Kumar, P. Beneficial effect of antidepressants against rotenone induced Parkinsonism like symptoms in rats. Pathophysiology 2016, 23, 123–134. [Google Scholar] [CrossRef]
- Yan, J.Q.; Sun, J.C.; Zhai, M.M.; Cheng, L.N.; Bai, X.L.; Feng, C.L. Lovastatin induces neuroprotection by inhibiting inflammatory cytokines in 6-hydroxydopamine treated microglia cells. Int. J. Clin. Exp. Med. 2015, 8, 9030–9037. [Google Scholar]
- Jin, X.; Liao, Y.; Tan, X.; Wang, G.; Zhao, F.; Jin, Y. Involvement of CYP2E1 in the Course of Brain Edema Induced by Subacute Poisoning With 1,2-Dichloroethane in Mice. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Swaroop, S.; Sengupta, N.; Suryawanshi, A.R.; Adlakha, Y.K.; Basu, A. HSP60 plays a regulatory role in IL-1beta-induced microglial inflammation via TLR4-p38 MAPK axis. J. Neuroinflammation 2016, 13. [Google Scholar] [CrossRef]
- Allan, S.M.; Rothwell, N.J. Inflammation in central nervous system injury. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 1669–1677. [Google Scholar] [CrossRef]
- Luo, X.G.; Chen, S.D. The changing phenotype of microglia from homeostasis to disease. Transl. Neurodegener. 2012, 1. [Google Scholar] [CrossRef]
- Correale, J. The role of microglial activation in disease progression. Mult. Scler. 2014, 20, 1288–1295. [Google Scholar] [CrossRef]
- Hernandez-Ontiveros, D.G.; Tajiri, N.; Acosta, S.; Giunta, B.; Tan, J.; Borlongan, C.V. Microglia activation as a biomarker for traumatic brain injury. Front. Neurol. 2013, 4. [Google Scholar] [CrossRef]
- Klein, B.; Mrowetz, H.; Barker, C.M.; Lange, S.; Rivera, F.J.; Aigner, L. Age Influences Microglial Activation After Cuprizone-Induced Demyelination. Front. Aging Neurosci. 2018, 10. [Google Scholar] [CrossRef]
- Varnum, M.M.; Ikezu, T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch. Immunol. Ther. Exp. 2012, 60, 251–266. [Google Scholar] [CrossRef]
- Von Bernhardi, R.; Eugenin-von Bernhardi, L.; Eugenin, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7. [Google Scholar] [CrossRef]
- Waisman, A.; Liblau, R.S.; Becher, B. Innate and adaptive immune responses in the CNS. Lancet Neurol. 2015, 14, 945–955. [Google Scholar] [CrossRef]
- Brambilla, R. The contribution of astrocytes to the neuroinflammatory response in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. 2019, 137, 757–783. [Google Scholar] [CrossRef]
- Sajja, V.S.S.S.; Hlavac, N.; VandeVord, P.J. Role of Glia in Memory Deficits Following Traumatic Brain Injury: Biomarkers of Glia Dysfunction. Front. Integr. Neurosci. 2016, 10. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Park, H.Y.; Han, M.H.; Park, C.; Jin, C.Y.; Kim, G.Y.; Choi, I.W.; Kim, N.D.; Nam, T.J.; Kwon, T.K.; Choi, Y.H. Anti-inflammatory effects of fucoidan through inhibition of NF-kappaB, MAPK and Akt activation in lipopolysaccharide-induced BV2 microglia cells. Food Chem. Toxicol. 2011, 49, 1745–1752. [Google Scholar] [CrossRef]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef]
- Li, Y.F.; Xu, X.B.; Chen, X.H.; Wei, G.; He, B.; Wang, J.D. The nuclear factor-kappaB pathway is involved in matrix metalloproteinase-9 expression in RU486-induced endometrium breakdown in mice. Hum. Reprod. 2012, 27, 2096–2106. [Google Scholar] [CrossRef]
- Vafadari, B.; Salamian, A.; Kaczmarek, L. MMP-9 in translation: From molecule to brain physiology, pathology, and therapy. J. Neurochem. 2016, 139, 91–114. [Google Scholar] [CrossRef]
- Perez-Hernandez, M.; Fernandez-Valle, M.E.; Rubio-Araiz, A.; Vidal, R.; Gutierrez-Lopez, M.D.; O’Shea, E.; Colado, M.I. 3,4-Methylenedioxymethamphetamine (MDMA, ecstasy) produces edema due to BBB disruption induced by MMP-9 activation in rat hippocampus. Neuropharmacology 2017, 118, 157–166. [Google Scholar] [CrossRef]
- Chen, F.; Ohashi, N.; Li, W.; Eckman, C.; Nguyen, J.H. Disruptions of occludin and claudin-5 in brain endothelial cells in vitro and in brains of mice with acute liver failure. Hepatology 2009, 50, 1914–1923. [Google Scholar] [CrossRef]
- Ma, C.; Zou, L.; Xia, Y.; Tu, Y.; Xue, D.; Yang, Y.; Liu, D.; Liu, Y.; Wu, H.; Dan, H.; et al. Extracts of Coleus forskohlii relieves cough and asthma symptoms via modulating inflammation and the extracellular matrix. J. Cell Biochem. 2018, 120, 9648–9655. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A review: Inflammatory process in Alzheimer’s disease, role of cytokines. Sci. World J. 2012, 2012, 756357. [Google Scholar] [CrossRef]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef]
- Kitic, M.; Hochmeister, S.; Wimmer, I.; Bauer, J.; Misu, T.; Mader, S.; Reindl, M.; Fujihara, K.; Lassmann, H.; Bradl, M. Intrastriatal injection of interleukin-1 beta triggers the formation of neuromyelitis optica-like lesions in NMO-IgG seropositive rats. Acta Neuropathol. Commun. 2013, 1. [Google Scholar] [CrossRef]
- Thornton, P.; McColl, B.W.; Cooper, L.; Rothwell, N.J.; Allan, S.M. Interleukin-1 drives cerebrovascular inflammation via MAP kinase-independent pathways. Curr. Neurovasc. Res. 2010, 7, 330–340. [Google Scholar] [CrossRef]
- Wu, C.Y.; Hsieh, H.L.; Jou, M.J.; Yang, C.M. Involvement of p42/p44 MAPK, p38 MAPK, JNK and nuclear factor-kappa B in interleukin-1beta-induced matrix metalloproteinase-9 expression in rat brain astrocytes. J. Neurochem. 2004, 90, 1477–1488. [Google Scholar] [CrossRef]
- Edlinger, C.; Lichtenauer, M.; Wernly, B.; Pistulli, R.; Paar, V.; Prodinger, C.; Krizanic, F.; Thieme, M.; Kammler, J.; Jung, C.; et al. Disease-specific characteristics of vascular cell adhesion molecule-1 levels in patients with peripheral artery disease. Heart Vessels 2018, 34, 976–983. [Google Scholar] [CrossRef]
- De Alba, J.; Clayton, N.M.; Collins, S.D.; Colthup, P.; Chessell, I.; Knowles, R.G. GW274150, a novel and highly selective inhibitor of the inducible isoform of nitric oxide synthase (iNOS), shows analgesic effects in rat models of inflammatory and neuropathic pain. Pain 2006, 120, 170–181. [Google Scholar] [CrossRef]
- Kim, D.W.; Im, S.H.; Kim, J.Y.; Kim, D.E.; Oh, G.T.; Jeong, S.W. Decreased brain edema after collagenase-induced intracerebral hemorrhage in mice lacking the inducible nitric oxide synthase gene. Laboratory investigation. J. Neurosurg. 2009, 111, 995–1000. [Google Scholar] [CrossRef]
- Thiel, V.E.; Audus, K.L. Nitric oxide and blood-brain barrier integrity. Antioxid. Redox Signal. 2001, 3, 273–278. [Google Scholar] [CrossRef]
- Hsieh, H.L.; Yang, C.M. Role of redox signaling in neuroinflammation and neurodegenerative diseases. Biomed Res. Int. 2013, 2013, 484613. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer (5′→3′) | Primer Sequences | Length (bp) |
|---|---|---|---|
| Mmp9 (MMP-9) | Sense | GAAGGCTCTGCTGTTCAG | 129 |
| Antisense | AAGATGTCGTGTGAGTTCC | ||
| Vcam1 (VCAM-1) | Sense | CTGTTCCAGCGAGGGTCTAC | 287 |
| Antisense | CACAGCCAATAGCAGCACAC | ||
| Icam1 (ICAM-1) | Sense | GTGGGTCGAAGGTGGTTCTT | 168 |
| Antisense | GCAGTTCCAGGGTCTGGTTT | ||
| Nos2 (iNOS) | Sense | GGGTCACAACTTTACAGGGAGT | 149 |
| Antisense | GAGTGAACAAGACCCAAGCG | ||
| Il1β (IL-1β) | Sense | GAAATGCCACCTTTTGACAGTG | 116 |
| Antisense | TGGATGCTCTCATCAGGACAG | ||
| GAPDH | Sense | CAATGTGTCCGTCGTGGATCT | 124 |
| Antisense | GTCCTCAGTGTAGCCCAAGATG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, X.; Wang, T.; Liao, Y.; Guo, J.; Wang, G.; Zhao, F.; Jin, Y. Neuroinflammatory Reactions in the Brain of 1,2-DCE-Intoxicated Mice during Brain Edema. Cells 2019, 8, 987. https://doi.org/10.3390/cells8090987
Jin X, Wang T, Liao Y, Guo J, Wang G, Zhao F, Jin Y. Neuroinflammatory Reactions in the Brain of 1,2-DCE-Intoxicated Mice during Brain Edema. Cells. 2019; 8(9):987. https://doi.org/10.3390/cells8090987
Chicago/Turabian StyleJin, Xiaoxia, Tong Wang, Yingjun Liao, Jingjing Guo, Gaoyang Wang, Fenghong Zhao, and Yaping Jin. 2019. "Neuroinflammatory Reactions in the Brain of 1,2-DCE-Intoxicated Mice during Brain Edema" Cells 8, no. 9: 987. https://doi.org/10.3390/cells8090987
APA StyleJin, X., Wang, T., Liao, Y., Guo, J., Wang, G., Zhao, F., & Jin, Y. (2019). Neuroinflammatory Reactions in the Brain of 1,2-DCE-Intoxicated Mice during Brain Edema. Cells, 8(9), 987. https://doi.org/10.3390/cells8090987