Highlights in Resistance Mechanism Pathways for Combination Therapy


Institute of Biomedical Sciences, Federal University of Rio de Janeiro, Rio de Janeiro 21941-902, Brazil
*
Author to whom correspondence should be addressed.
Cells 2019, 8(9), 1013; https://doi.org/10.3390/cells8091013
Submission received: 15 July 2019
/
Revised: 15 August 2019
/
Accepted: 20 August 2019
/
Published: 30 August 2019
(This article belongs to the Section Cell Signaling)
Abstract
:Combination chemotherapy has been a mainstay in cancer treatment for the last 60 years. Although the mechanisms of action and signaling pathways affected by most treatments with single antineoplastic agents might be relatively well understood, most combinations remain poorly understood. This review presents the most common alterations of signaling pathways in response to cytotoxic and targeted anticancer drug treatments, with a discussion of how the knowledge of signaling pathways might support and orient the development of innovative strategies for anticancer combination therapy. The ultimate goal is to highlight possible strategies of chemotherapy combinations based on the signaling pathways associated with the resistance mechanisms against anticancer drugs to maximize the selective induction of cancer cell death. We consider this review an extensive compilation of updated known information on chemotherapy resistance mechanisms to promote new combination therapies to be to discussed and tested.
1. Introduction
Cancer chemotherapy has evolved greatly since the first clinical trial using nitrogen mustard in 1942 [1]. This single-agent treatment, or monotherapy, gave rise to the study, screening, and development of several other small molecules as anticancer candidates. In the 1960s, the use of the combination of Vincristine, Amethopterin, 6-Mercaptopurine, and Prednisone (VAMP) for pediatric leukemia markedly changed the stigmatic status of cancer chemotherapy to “curable”, showing drastic increments in remissions and survival. It also made evident the superiority of drug combination over monotherapy [2]. However, the underlying mechanisms for the efficacy achieved with this combination therapy were unclear at the time and, in part, remain so today. Combination therapy aims to hamper cancer cell homeostasis/metabolism at multiple simultaneous targets to improve its therapeutic efficacy, reduce dosage, reduce side effects, and prevent or delay the development of acquired resistance [3]. However, acquired resistance will still eventually develop along with the treatment in response to the exposure to antineoplastic drugs.
Tumor cells can develop drug resistance due to intrinsic factors, such as mutations, translocations, epigenetic elements, and extrinsic factors, such as hypoxia, pH, hormones, cytokines from its microenvironment surroundings, and antineoplastic agents [4]. The tumor heterogeneity and mutational load have been directly associated with the emergence of acquired resistance to antitumor drugs [5,6]. Chemotherapeutic agents from diverse drug classifications have been combined to bypass multiple factors of drug resistance.
There is a growing concern with the chemical structure-based classification generally used by pharmacology texts and the restricted mindset it provides in combinatorial preclinical studies. The classification of drugs as cell cycle-dependent or independent was the first phenotype-based classification of antitumor agents [7,8]. Biological targets [9], biological activities [10], and structural prediction of antiproliferative activity [11] have also been proposed to facilitate insights into new drug combinations. Here we harvested the target information and biological activity to propose a rationale for drug combination.
Recent work with an RNAi-based high-throughput screening of combinations of antitumor drug libraries in cultures of almost every cancer type cell known [12] has reached a surprising conclusion. Pritchard and coauthors have claimed that most of the combination protocols approved since are, in fact, almost as good as single-agent treatments, with some few or almost negligible mechanistic contributions from single-drug components. Very few combinations exhibited real synergism. The authors suggested that each drug might affect different heterogeneous subpopulations. When the drug cocktail affects the same cell, the convergence of downstream signaling pathways related to cell death might limit the efficiency of the combined treatment [12].
The use of medications with different targets but with some overlap in their resistance-related signaling pathways might improve efficiency. Here we propose a strategy for combination studies that one of the drugs used in the antitumor cocktail should be an inhibitor of a known resistance signaling pathway of one or more drugs used in combination. The purpose of the discussion is to improve the discovery of drug combination studies. To improve the understanding of specific resistance mechanisms of widely used anticancer drugs is necessary. The information on resistance mechanisms, signaling pathways, and mechanisms of action for cytotoxic and target therapy is summarized in tables and discussed below.
2. Search Strategy
We highlighted the most clinically relevant information from selected articles (Pubmed), clinical trials (ClinicalTrials.gov), and the curated databases Drugbank [13] and SuperCYP/Transformer [14] relating to drug resistance mechanisms.
Drug names were used as keywords, as well as, but not restricted to target, off-target, mechanism of action, resistance, mechanism of resistance, signaling pathway, detoxification, elimination, metabolism, pharmacokinetics, pharmacodynamics, absorption, distribution, binding, clinical trial, combination, synthetic lethality, drug combination, Food and Drug Administration (FDA) approval, screening, combination screening, and drug screening.
Although extensive pharmacokinetic studies are required for the approval of a drug for human use, it is not mandatory to completely elucidate every single tissue or plasmatic detoxification enzyme, which can lead to some unclear or incomplete information [15]. In the absence of off-target ligands recognized, we have listed some off-target effects that might contribute to the development of innovative therapeutic strategies.
3. Resistance Mechanisms and Signaling Pathways
Cytotoxic Chemotherapy, at its most simplistic, means the use of chemical compounds to kill cancer cells more effectively than non-tumoral cells [2]. As the knowledge of cancer biology expands, drug development in cancer research could migrate from the discovery of natural compounds to the design of synthetic drug candidates that aim at exclusive or specific cancer targets in what is known as Targeted Therapy [2]. Targeted drug development has become an exciting field for academics and the pharmaceutical industry since imatinib approval in 1995. At the present time, there are twice as many targeted drugs (~120) approved by the FDA than cytotoxic small molecules (~60) (Supplementary Table S1; Table 2).
All FDA approved small molecule cytotoxic antineoplastic drugs available as at May 2019 are listed in Supplementary Table S1, and all available targeted antineoplastic agents approved by the FDA up to May 2019 are presented in Supplementary Table S2.
One of the most appealing differences between Cytotoxic and Targeted Therapy is in the development of specific acquired resistance mechanisms. If ATP-binding cassette (ABC) transporters, enzymatic detoxification, and DNA homeostasis proteins mostly affect cytotoxic drugs, the targeted antineoplastic agents do not. These conclusions can be reached by a close examination of Supplementary Tables S1 and S2. The summary of the most common specific resistance mechanisms for both cytotoxic and targeted therapies is presented in Table 1. The signaling pathways associated with resistance to antitumor small molecules and targeted therapies are briefly discussed below.
4. ABC Transporters
As seen in Table 1, the main cause of resistance to cytotoxic drugs is the overexpression of ABC transporters in the plasma membrane of cancer cells. Both intrinsic and acquired resistance can be specific to a certain drug, or nonspecific, covering a broad spectrum of drugs. When the spectrum of the resistance is so broad that it covers drugs with unrelated structures and targets in such a way that it is not possible to make a clear association between them, it is called multidrug resistance (MDR). ABC transporters were the first and most studied mechanism of resistance associated with clinical MDR [16,17]. MDR can be understood from either a cellular or a clinical point of view.
From a cellular perspective, resistance might be a consequence of the low intracellular concentration of the cytotoxic drug. ABC transporters can bind anticancer drugs either from the surroundings of the plasma membrane or intracellular vesicles, and transport them out of the cell directly to the external milieu or through exocytosis [18].
Clinically, MDR is the main cause of failure in the treatment with cytotoxic drugs [19], which can be explained in part, at least, by the extensive overexpression of several ABC transporters and its long list of substrates among antineoplastic agents (mitomycin, chlorambucil, methotrexate, pemetrexed, 5-fluorouracil, mercaptopurine, estramustine, docetaxel, paclitaxel, vinblastine, vincristine, vinorelbine, daunorubicin, doxorubicin, epirubicin, idarubicin, mitoxantrone, irinotecan, topotecan, dactinomycin, and etoposide—as seen in Supplementary Table S1).
For several years, detection and inhibition of a minimum panel of clinically relevant ABC transporters have been attempted [17,19]. However, it is clear that intra-tumor heterogeneity also affects the number of possible combinations of transporters regulated in different subpopulations. Some leukemia cohorts, for example, have shown overexpression of multiple ABC transporters among their patients [20,21,22]. The Cancer Genome Atlas (TCGA) data have also been used to show these expression level discrepancies among patients and cancer types [17].
The clinical use of ABC transporter inhibitors is long hampered by the severe toxicities presented. Numerous ABC transporter inhibitors have been clinically tested in the last forty years [17,19,23]. Recently, fourth-generation ABC transporter inhibitors have been developed, mostly based on natural and semi-synthetic compounds [24].
The development of reliable and validated methods for the adequate detection and quantification of ABC transporters is an ongoing challenge [17,19]. Nonetheless, the importance of these transporters in limiting drug delivery, affecting clinical outcomes, and the increased expression of ABC transporters in several cancers, justifies the considerable effort [19].
5. Enzymatic Detoxification
The second cause of resistance to cytotoxic drugs is the increased activity or expression of specific detoxification enzymes of each drug (Table 1). For example, Bleomycin, which can be used for cervical, head and neck, lymphomas, penile, testicular, and vulvar cancer [25], showed bleomycin hydrolase [26,27,28] as a mechanism of detoxification, and the increased activity of bleomycin hydrolase, N-acetylating enzymes, and bleomycin-binding proteins [27,28,29], as mechanisms of resistance (Supplementary Table S1).
In addition to specific detoxification enzymes of each drug, glutathione S-transferase (GST) shows a broad detoxification effect. GST participates in the detoxification of several antineoplastic drugs by binding a glutathione molecule to it, therefore promoting some inactivation and increasing the affinity to some ABC transporters, especially from ABCC and ABCG families [30,31,32].
6. DNA Homeostasis-Related Signaling Pathways and Cytoskeletal Disruptors
In addition to increased activity of detoxification, mutations, overexpression, and downregulation of key targets, namely topoisomerase I and tubulin isoforms, are the next most common alterations. These are alterations that affect two major families, the topoisomerase inhibitors and cytoskeletal disruptors, which are among the most used cytotoxic drugs. Tubulin directly participates in the formation of the metaphase spindle and separation of sister chromatids, and when this is not adequately completed, leads to cell death by mitotic catastrophe [33,34]. Changes in isotype expression [35], mutations in [35,36,37] and/or overexpression of tubulins [35], and post-translational modifications of tubulins have been observed in many cancers [38].
Since most cytotoxic drugs are genotoxic, it is not surprising to have proteins associated with DNA homeostasis among the most common resistance mechanisms (Table 1). Therefore, several common resistance mechanisms in response to cytotoxic drugs are associated with DNA homeostasis (topoisomerases, tubulin, dCK, NF-κB, MGMT, ALDH1, and TP53). These molecular pathways are crucial key points to cytotoxic treatment resistance and will be discussed briefly.
Topoisomerases are vital regulators of DNA topology during DNA replication, transcription, repair, and recombination. Type I or II topoisomerases produce reversible single- or double-strand breaks, respectively. Their current pharmacological inactivation is related to stalled replication fork and cytotoxic DNA fragmentation by the formation of irreparable covalently bound DNA–protein–drug ternary complexes. Several other classes of topoisomerase inhibitors are under development, but none have presented tolerable toxicity, acceptable specificity, and potency with proper pharmacokinetics [39,40].
dCK is a key enzyme, usually the rate-limiting one in the synthesis of deoxynucleosides in the salvage pathway, a crucial alternative compensatory pathway for deoxynucleotide synthesis when the de novo pathway is inhibited or downregulated [41,42]. dCK overexpression is a key component to resistance to nucleoside analogs (cladribine, clofarabine, cytarabine, decitabine, gemcitabine, nelarabine) (Supplementary Table S1).
Increased DNA methyltransferase (MGMT) protein levels are one of the most important mechanisms of resistance to alkylating agents because it can repair the cytotoxic O6-methylguanine DNA adduct and prevent its harmful effects, which affect therapy efficiency [43]. For example, when tumors showed methylation on the MGMT promoter, glioblastoma patients treated with the alkylating agent temozolomide and radiotherapy showed 21.7 months of median survival, compared to only 15.3 months among those who were assigned to radiotherapy only. In the absence of methylation of the MGMT promoter, there is no statistical difference in survival between the treatment groups, regardless of temozolomide treatment [44]. This suggested that patients with wild type MGMT have nearly no response to the alkylating agent. One may speculate that increasing the methylation status might have some benefit to enhance the cell death response induced by alkylating agent treatment in glioblastomas. After DNA damage, tumor suppressor TP53 is induced to regulate cell cycle arrest, DNA repair, and apoptotic cell death. Deletions, nonsense, and frameshift mutations that lead to its loss-of-function are common across a vast range of cancer cell type tumorigenesis. Gain-of-function oncogenic mutations are also common and represent a different mechanism for which TP53 can participate in tumorigenesis. When these oncogenic mutations are present in cancer, TP53 expression is usually associated with a more aggressive form of the disease, with increased tumor genome instability and metastatic potential (recently reviewed in [45]). Altogether, TP53 mutations are found in nearly 50% of human cancers, and they are one of the classically associated resistance mechanisms against cytotoxic therapy.
7. Activation of NF-κB
NF-κB is a small family of five proteins with two effector transcription factor complexes, p65–p50, and p52-RelB [46]. These complexes are respectively associated with the canonical and non-canonical signaling pathways. The canonical pathway is activated under the control of cell receptors to several pro-inflammatory cytokines, tumor necrosis factor (TNF), lipid polysaccharides, growth factors, and antigens, whereas, the non-canonical pathway is triggered by Lymphotoxin beta receptor (LTBR), cluster of differentiation 40 (CD40), B-cell activating factor receptor 3 (BR3), and RANK. Both pathways are limited by the initial participation of cytosolic inactivating complexes. The canonical pathway involves the IKB and IKK inhibitory proteins, and the non-canonical recruit only IKK proteins. The activation of the receptors for these pathways triggers a sequence of specific phosphorylations and ubiquitinations that rapidly and transiently release the active form of their respective transcription factor complexes, which translocate to the nucleus where they effectively activate hundreds of validated transcriptional targets [46]. Because of the huge plethora of target genes and our poor understanding of this complex orchestra, most clinically approved inhibitors have an incomplete description of their mechanisms of action. The NF-κB pathway participates in cellular immunity, inflammation, apoptosis, cell differentiation, proliferation, and response to stress. In cancer, activation of NF-κB rescues the cell from the apoptotic pathway, promoting its survival, preventing cell death, and promoting proliferation [46].
8. Increased Levels of ALDH1
Several types of cancer stem cell (CSC) populations have elevated aldehyde dehydrogenase activity (ALDH), which converts toxic aldehydes into carboxylic acids [47]. ALDH enzymatic activity supports cancer stem cell self-renewal, protection against oxidative stress, and participation in energetic metabolism as a reliable alternative source of nicotinamide adenine dinucleotide (NADH), a convenient substrate for ATP synthesis. They also confer resistance to selected anticancer agents by metabolic inactivation and have been implicated in every tumorigenic process, from initiation to metastasis [48,49]. Therefore, it is not surprising that an increased level of ALDH is one of the specific resistance mechanisms associated with cytotoxic therapy.
9. Signaling Pathways Associated with Targeted Therapies
Differently from cytotoxic drugs, the resistance mechanisms of target therapy are mostly specific key alterations in their signaling pathway targets, such as mitogen-activated protein kinase (MAPK) pathway (also known as RAS-RAF-MEK-ERK pathway), phosphoinositide 3-kinase pathway (PI3K-AKT-mTOR), epidermal growth factor (EGF), EGF receptor (EGFR), phosphatase and tensin homolog deleted on chromosome 10 (PTEN), insulin-like growth factors (IGFs), key regulators of apoptosis B-cell lymphoma 2 (BCL2) family, fibroblast growth factors (FGFs), and signal transducers and activators of transcription (STATs). Except for the overexpression of ABC transporters, which occurs but with less frequency than for cytotoxic drugs, the major mechanism of action of targeted therapy is key alterations in their specific signaling pathway targets (Table 1). Briefly, these signaling pathways can be associated with Survival (PI3K, AKT, mTOR, STAT), Proliferation (MAPK, STAT, growth factors—FGFs, EGFR, IGFs), and Cell Death (BCL2 family and PTEN).
Therefore, the initial excitement surrounding targeted therapy might be diminished by the nature of the therapy itself. From one perspective, single-agent targeted therapy inhibits specific dysregulated pathways. Such treatment would represent a strong but punctual selective pressure over cancer cell populations [50]. The strong specificity allows reduced toxicity and promotes efficient clinical response for those patients who might benefit from the treatment. On the other hand, targeted therapy is also largely limited by this strong specificity in the face of the multiple alterations cancer cells present [6], and which are necessary to circumvent the redundant signaling pathways that prevent tumorigenesis [51]. The efficacy of the therapy is also hampered by the large intra- and inter-tumor heterogeneity [52]. These limitations might be seen by the relatively small benefit some patients present from the intended targeted therapy and by the relative transient remission, accompanied by relapse, which is often and usually together with acquired resistance [53]. All these points have been the theme of some nice discussions in recent works [50,54,55,56].
One key aspect to circumvent the emergence of resistance might be to “spread” the selective pressure made by antineoplastic treatment attacking multiple pathways simultaneously, possibly by the combination of multiple drugs [19,56]. This must be performed according to already established guidelines such as (i) each single agent should be effective in monotherapy; (ii) use agents with different mechanisms of action, preferably on different subcellular structures and/or different phases of the cell cycle; (iii) avoid overlapping toxicities, particularly over vital organs or with life-threatening side effects; (iv) optimize dose and schedule of treatment and intervals to improve both efficacy and minimize toxicities; and, finally, (v) have a clear understanding of the mechanism of interaction among drugs [57,58,59,60]. The last of these is particularly poorly understood. This statement is supported by a relatively recent study [12].
10. Resistance Mechanisms can be Associated with the Hallmarks of Cancer Cells
When resistance mechanisms and signaling pathways are associated with specific hallmarks of cancer, it became evident that some hallmarks are mostly associated with oncogenes (genome instability, survival, proliferation, angiogenesis); meanwhile, others are almost tumor suppressor exclusives (evasion of inhibitory factors, cell death). Classic oncogene alterations are associated with gain-of-function in which one single genetic alteration is enough to over-activate the oncogenic signaling pathway; meanwhile, two-hit tumor suppressors are often associated with inactivating mutations and downregulation of key signaling elements of the pathway. It seems that the two-hit requirements for an effective intervention on a tumor suppressor pathway have limited the development of tumor suppressor-based therapies since most targeted therapies are aimed at oncogenes (Supplementary Table S2).
Cytotoxic and targeted therapies increased detoxification and specific signaling pathway mechanisms, respectively. Interestingly, both mechanisms can be associated with the hallmarks of cancer cells. Whereas increased detoxification mechanisms can be associated with altered metabolism of cancer cells, the signaling pathways induced by target therapy as resistance mechanisms can be associated with all the hallmarks. In Table 2, there is an association between the specific resistance mechanisms listed in Supplementary Tables S1 and S2 and the most affected signaling pathways in cancer cells, as well as their corresponding hallmarks of cancer.
11. Signaling Pathways Related to the Hallmark Evasion of Growth Suppression
PTEN is one of the most commonly affected tumor suppressors proteins during tumorigenesis [64]. Its gene encodes a tumor suppressor phosphatase protein that physiologically prevents the entry to the cell cycle, as well as G2/M transition and mitosis. Regulation of the cell cycle by PTEN has been recently reviewed elsewhere [65]. PTEN arrests cells at G1 by inactivating the PI3K/AKT pathway, thus inhibiting cell cycle progression through the regulation of several signaling molecules, including decreased levels of cyclin D and inactivation by phosphorylation of the retinoblastoma protein (RB). RB regulates the restriction point, the G1-late stage when cells become committed to proliferate [66]. Cell cycle progression relies on the phosphorylated state of RB, which is tightly controlled by cyclin-dependent kinases (CDKs).
Upon mitogenic stimulation, cyclin D-CDK4/CDK6 phosphorylates RB, thereby inactivating it and promoting the release of transcription factors of the E2F family, which activate the transcription of target genes required for cell cycle progression [67]. Dysregulation of this canonical RB function is central in cancer, with components of the CDK4/6-RB pathway often displaying mutations that will result in sustained cell proliferation; for instance, in tumors that retain RB expression, uncontrolled cell cycle activity may be due to the amplification of the cyclin D1 gene (CCND1) and CDK4, activating CDK4/6 mutations or silencing of CDK inhibitors (CKI) [67,68].
There are 13 members of the CDK family, from which only four—CDK1, CDK2, CDK4, and CDK6—are direct regulators of the cell cycle [69]. CDK4 and CDK6 have been implicated as drivers of oncogenesis in several cancers [70] for more than 20 years.
There are two major families of physiological CKI that regulate the cell cycle through cyclin-CDK activity in animals: INK4 and CIP/KIP families. They are both allosteric inhibitors of CDKs [71].
RB is also regulated by phosphorylation. It has been recently proposed that in a non-cycling cell, when RB is in its “active” form sequestering E2F family members, there are multiple mono-phosphorylated isoforms of RB (mP-RB) simultaneously present [72]. The authors showed functional differences between mP-RBs beyond the regulation of the cell cycle machinery. It is yet unknown which proteins participate in the complexes to each type of mP-RBs, which proteins are regulated by each isoform, in which situations these isoforms are selectively regulated, which isoforms are functionally distinct or redundant, and so on. There is also evidence for hypo- and hyper-phosphorylated isoforms, which have been largely studied by other groups [73,74].
12. CDK Inhibitors
Pharmacological specific inhibition of CDKs has been long pursued. The most promising early CDK inhibitor was the pan-inhibitor CDK flavopiridol (or alvocidib) [72,75,76,77]. Flavopiridol was tested in more than 60 clinical trials for 15 years, mostly with disappointing degrees of antitumor activity [76,78]. Flavopiridol did not obtain approval, which was attributed to limited clinical efficacy [79,80,81,82] and severe toxicities [80,82].
Selective CDK4/6 inhibitor compounds were developed, clinically tested, and recently approved for human use, namely, palbociclib (PD0332991, Pfizer, NY, USA), ribociblib (LEE011, Novartis, Basel, Switzerland) and abemaciclib (LY2835219, Eli Lilly, Indianapolis, USA). These specific CDK have raised much attention and excitement since their first approvals. Palbociclib was the first to receive its approval. It was indicated for first-line use among post-menopausal women with advanced hormone positive HER2 negative breast cancer who have not been previously treated with systemic chemotherapy [83]. In April 2019, its approval was also extended for the treatment of men with hormone positive HER2 negative metastatic breast cancer [84].
Recent reviews of the clinical trials with these selective CDK4/6 inhibitors and their impact on clinical outcomes have discussed great advances in median progression-free survival, and objective response ratio among hormone receptor positive breast cancer patients treated with all three approved CDK [85,86].
There are some similarities between the approvals of palbociclib [83], ribociclib [87], and abemaciclib [88] for breast cancer treatment. All three drugs were first approved for advanced hormone positive HER2 negative breast cancer patients in combination with hormone therapy (letrozole with palbociclib or ribociclib, and fulvestrant/abemaciclib), for which they presented synergistic growth inhibitory activity. The approvals were granted based mostly on progression-free survival and objective response ratio, without data on overall survival. The larger cohort among the three trials enrolled 165 women, a small cohort for survival purposes. Moreover, the three approvals are upfront treatments aiming mostly at disease control before systemic chemotherapy is given, which, although more aggressive, is also a more established curative approach.
The full potential of selective CDK is under investigation in many preclinical and clinical studies [85]. As target therapies, CDK treatment produces some specific resistances. Overcoming these predictive resistances might greatly improve clinical outcomes as recently discussed elsewhere [89].
Preclinical and clinical data support the notion that the rational treatment with cell cycle phase-specific antitumor agents might produce synergic cytotoxic effects depending on the choice and order of drugs to be administered. For example, sequential administration of CDK followed by DNA damaging cell cycle specific agents have been tested in a panel of TP53 mutant and wild-type breast and human colorectal cancer cell lines. The authors used roscovitine, a purine-based nonselective CDK, followed by doxorubicin [90]. The CDK inhibition increased apoptosis induced by doxorubicin only on TP53 null or mutant cell lines, highlighting the ability of wild type TP53 to prevent cell cycle synthetic lethality due to DNA damaging agents. The authors also reported superior benefits in terms of overall survival and decreased proliferation in human breast cancer xenografts treated with the sequential regime of roscovitine followed by doxorubicin over the concomitant combination (p < 0.0001) or either drug alone (p < 0.01) [90]. It is worth testing similar types of experiments with CDK4/6 selective inhibitors.
13. Signaling Pathways Related to Cell Death
In addition to sustained proliferation and evasion of growth suppression, resistance to cell death is one of the three pivotal drivers of tumorigenesis [91]. From analyzing Table 2, one can promptly realize that many of the same signaling disruptions are exploited by cancer cells and allow resistance to anticancer therapy. Importantly, programmed cell death (PCD), triggered by several external and internal stress signals, must be overcome if both tumorigenesis and drug resistance are to be successful. The best-characterized form of PCD is apoptosis, which is composed by two pathways—one activated by death receptor signaling and the other by intracellular, mitochondrial signaling—that converge at the level of effector proteases called caspases. These will execute an extensive intracellular proteolytic program, leading to apoptotic cell death. Disruptions in the mitochondrial (or “intrinsic”) apoptotic program are widely involved in cancer progression and therapy resistance [91]. The ultimate trigger to caspase activation in the intrinsic program is the release of mitochondrial factors, such as cytochrome c, through mitochondrial outer membrane permeabilization (MOMP). This process is tightly regulated by a dynamic balance among the members of the BCL2 protein family at the outer mitochondrial membrane (OMM): the pro-apoptotic BCL2 proteins, Bax and Bak, directly promote MOMP by forming pores at the OMM, which are usually inhibited by binding of the anti-apoptotic (e.g., BCL2, BCL2-like protein 1, and MCL1/BCL2L3) BCL2 proteins [92]. Additionally, BH3-only proteins (e.g., Bid, Bad, Bim, Noxa, and Puma) directly inhibit anti-apoptotic BCL2 proteins and/or directly activate Bax/Bak, thus inducing MOMP and triggering caspase activation. When activated, the tumor suppressor TP53, best known for its surveillance activity regarding DNA damage, promotes the expression of several genes regulating the intrinsic pathway, mainly Bax and PUMA, ultimately also leading to caspase activation [93]. Perhaps unsurprisingly, mutations leading to enhanced anti-apoptotic BCL2 signaling, downregulation of Bax and Bak, and p53 inactivation, promote drug resistance across many cancer types (Supplementary Tables S1 and S2).
14. Induction of Autophagy is a Common Cellular Phenomenon associated with Cell Death Resistance
Autophagy is an evolutionarily conserved process of elimination and recycling “damaged” or “old” cytoplasmic cell components, structures, and organelles in lysosomes [94]. Autophagy has been recently associated with several hallmarks of cancer [63]: sustained proliferation (energetic source); promotion of epithelial–mesenchymal transition; sustained survival during cell migration (evasion of anoikic cell death) and metastasis (facilitates tumor cell dormancy and quiescence, survival and proliferation during new tumor formation); pro-survival alternative energetic source of dysregulated aerobic metabolism; evasion of cell death (evasion of apoptosis, survival of residual cancer stem cells after chemotherapy); and, genome instability (sustains DNA damage repair system) [95,96].
Physiologically, basal autophagy is a recycling energetic system for the turnover of proteins and lipids, which might be increasingly activated in response to a stressor, like starvation. Therefore, it might be seen as a pro-survival process for any normal or cancerous cells. Under prolonged periods of starvation, a regulated programmed cell death (PCD) process known as autophagic cell death is observed [97]. As briefly discussed above, there are many possible opportunities for cancer therapy based on pharmacological modulation of autophagy and to promote increased cancer cell death. We can and should take advantage of these phenomena [98,99].
Autophagy plays its role in cancer by regulating several pathways which rule over cell life and death, such as BCL2, Class III and I PI3K (PI3K-I and PI3K-III), AKT, mTORC ½, and TP53 [100]. Despite the complexity of the mTOR signaling network, mTORC1 is a well-defined central autophagy inhibitor, acting as a point of convergence for many pathways: the pro-survival PI3K-I-AKT and MAPK pathways induce mTORC1, whereas AMP-activated protein kinase (AMPK) signaling inhibits it [101]. mTORC1 signaling is also induced by growth factors via the PI3K-I-AKT and MAPK pathways.
Furthermore, TP53 also presents a “Janus role” of its own in autophagy, both inducing (cytoplasmic p53) and inhibiting (nuclear p53, through AMPK) mTORC1 activity. Autophagy induction and enhanced PI3K-AKT-mTOR and MAPK signaling pathways are often related to resistance against a wide range of drugs in several cancer types, which is further evidence of its importance during tumorigenesis (Supplementary Table S2 and Table 2). Furthermore, it is important to have in mind the significant cross-talk between autophagy and apoptosis signaling [102]: As well as inhibiting autophagy, growth factor signaling, for example, it will also induce the JAK/STAT1/3 pathway, which will, in turn, induce anti-apoptotic BCL2 signaling.
The very nature of the network connection of the major signaling pathways, both affected during the initial tumorigenic process and therapy-induced, is very complex. The complexity is not only due to many connection points, but also because they affect various cellular processes related to all hallmarks of cancers. This sophistication makes the topic interesting and intriguing, as can be seen in Figure 1, where cell signaling is summarized according to cancer markers.
Another level of the additional complexity of the signaling network is evidenced by the fact that proteins can show multiple roles in the cell. For example, RB and β-catenin, both known to be involved in proliferation, have been described by regulating cell death according to the context of tumor cells [103,104].
15. Clinical Trials with Combination Regimens Containing Inhibitors of Signaling Pathways Related to Drug Resistance
The concept of increased cell death by blocking the resistance mechanisms that protect cancer cells against systemic chemotherapy is relatively simple and intuitive. Similarly, increased cell death is to be expected when combining anticancer drugs in a cocktail in which one of its drugs might act as an inhibitor of the signaling pathways related to the known resistance mechanisms against one or some other anticancer drugs in combination. The general simplest idea of combining cytotoxic and targeted chemotherapy is usually attributed for target therapy somehow sensitizing cells to increased cytotoxicity (Table 3). Meanwhile, the rationale for combining two targeted drugs might be much more variable and require a case-by-case evaluation (Table 4). Most combination studies between targeted therapies do not aim at the resistance mechanisms affected by the drugs in the combination. We have only found five examples that fit into this strategy. We searched among already registered clinical trials for examples of drug combinations in which a specific targeted drug might inhibit the signaling pathway-related to the resistance to the second targeted combined drug (Table 4). With this rationale, we found five ongoing phase I, six ongoing phase II, three phase I completed, and two phase II completed (Table 4).
Several clinical trials that investigate mechanism-based targeted drug combinations are ongoing. They present a rationale for dual inhibition of one or more pathways that could increase cell death (Table 4). Some combinations represent a dual blockade in pathways related to different hallmarks of cancer (Figure 1), such as the combination of bevacizumab and erlotinib for advanced liver cancer, which targets both tumor neovascularization and proliferative signaling (EGFR and vascular endothelial growth factor dual blockade, Table 4).
Another example of mechanism-based targeted drug combinations is ongoing for pancreatic tumors. Since the enhanced PI3K/mTOR activity confers CDK4/6 inhibitor resistance therapy, the inhibition of those pathways should be tested (Table 4). This is an interesting ongoing clinical trial that is currently evaluating the combination of ribociclib, a CDK4/6 inhibitor, and everolimus, an mTOR inhibitor in metastatic pancreatic adenocarcinoma patients refractory to 5-fluorouracil (5-FU) and gemcitabine-based chemotherapy [105]. Their cancers have either intrinsic or acquired resistances to first and second lines of treatment. The antiproliferative combination might synergize to increase cell death, which would be unlikely to happen with either targeted therapies alone. mTOR is one of the potent resistance mechanisms against CDK inhibitors (Supplementary Table S2). Its inhibition with everolimus is a strategy for inhibition of the cross-talk between the pro-survival PI3K-AKT-mTOR pathway and the proliferative RAS–RAF–MEK–ERK signaling pathway. This rationale has already presented some nice results in breast [106] and prostate cancers [107].
The dual-hit inhibition strategy has some proven benefits over single pathway inhibitors. Some dual hit combinations have been approved for different pathways and cancer types, such as dual inhibition of EGFR, HER2, and MAPK. These combinations trigger specific but distinct mechanisms of resistance. In EGFR and HER-2, for example, improved efficacy is seen without added or more severe toxicities [108].
16. Intrinsic Toxicity and Compensatory Mechanism of Inhibitors of Key Signaling Pathways of the Resistance Mechanisms
Many inhibitors of key signaling pathways of the resistance mechanisms can show high toxicity and compensatory mechanisms, as well as several potential targets of the same pathways. For example, somatic alterations in the MAPK pathway that are highly prevalent in human cancer and are also related to therapy resistance, show a wide range of targeted inhibitions (see Supplementary Table S2 and Table 1). Currently, there are no rat sarcoma protein (RAS) inhibitors available, but monotherapy of downstream selective MAPK inhibitors shows great promise, with some examples of already approved regimens.
Selective monotherapy with rapidly accelerated fibrosarcoma (RAF) inhibitors has greatly improved clinical progress in melanoma patients [168]. However, intra-pathway resistance mechanisms still arise, often related to RAF-independent extracellular signal–regulated kinases (ERK) activation. In this context, v-Raf murine sarcoma viral oncogene homolog B (BRAF) inhibitor, dabrafenib therapy has triggered rapidly-growing skin tumors, which can be partially mitigated by combination with trametinib, a mitogen-activated protein kinase kinase (MEK) inhibitor [169]. This downstream inhibition of the pathway reduces possibilities for ERK reactivation. As of May 2019, the FDA has approved a total of three BRAF/MEK combination regimens, with the dabrafenib and trametinib combination being indicated for the treatment of BRAF-V600-positive melanoma, non-small cell lung cancer and anaplastic thyroid cancer (Table 4).
There are other inhibition strategies targeting MAPK that aim to offer improved therapy. Early clinical studies of RAF/MEK inhibition combined with immunotherapy, for example, have faced interruption or patient discontinuation due to severe life-threatening toxicities [170], with evidence suggesting that the combination potentiates adverse effects of both strategies when used alone. Larger studies are expected to better define this strategy. This highlights the importance of avoiding accumulated overlapping toxicity in the rationale of the combination to be tested in trials, as seen with other targeted pathways. Importantly, while there are no FDA-approved selective ERK inhibitors, preclinical studies have already shown that ERK mutations and expression imbalances might arise as resistance mechanisms [171]. Recent clinical data suggest that ERK inhibitors therapy-related toxicities are not severe, which opens the possibility for combinations with upstream RAF/MEK inhibition and greater clinical success for the future of MAPK inhibition therapy [172].
The PI3K-AKT-mTOR pathway is one of the most frequently dysregulated pathways, not only in tumor development but as the mechanism of resistance after treatment (Table 1). Approximately 50 compounds targeting some of its key proteins have been in clinical development over the years [173]. Although these compounds have reached different stages of clinical trials, a promising response has not been observed as with other approved targeted therapies [174]. For a number of reasons, including high toxicity, only a few PI3K-AKT-mTOR pathway inhibitors have been approved by the FDA and indicated for cancer treatment: four PI3K inhibitors—idelalisib, copanlisib and, more recently, duvelisib and alpelisib (September 2018 and May 2019, respectively); one AKT inhibitor—miltefosine; and, two mTOR inhibitors—temsirolimus and everolimus.
Inhibition of this signaling pathway has proven to be a double-edged sword between potency and toxicity. Anti-PI3K and anti-mTOR monotherapies, although well-tolerated, have shown only modest efficacy in several clinical trials [173]. On the other hand, combination therapies targeting multiple PI3K-AKT-mTOR components are usually more effective but lead to a build-up of dose-limiting toxicities [173]. Most severe toxicities related to PI3K inhibitors might be explained by severe immune modulations in several organs. Many reviews have recently addressed this complex problem [175].
Generally, preclinical studies have shown that isoform-specific targeting of PI3K has better therapeutic efficacy and toxicity profiles than pan-inhibitors [174]. The same is true for direct AKT inhibition. Most AKT inhibitors in clinical development are pan-inhibitors, with many clinical trials suspended due to severe hyperglycemia [176]. The efficacy of the AKT inhibitors TCN, TCN-P, and edelfosine is limited due to their toxicity [177]. Miltefosine was the only AKT inhibitor approved by the FDA in 2014, indicated for visceral, cutaneous, and mucosal leishmaniasis. In cancer, miltefosine has limited use mainly due to its gastrointestinal and hemolytic toxicities. Therefore, it has been used as a topical formulation to treat cutaneous lesions caused by lymphoma and cutaneous breast cancer metastasis [178]. Given the central role of AKT signaling dysregulation in cancer, the development of safer AKT inhibitors is urgent.
Regarding mTOR inhibition, hematologic toxicities of various types were observed in the clinical trials that resulted in FDA approval of temsirolimus: 94% had hemoglobinemia, 53% had lymphocytopenia, 19% had neutropenia, and 40% had thrombocytopenia [179]. Both temsirolimus and everolimus display immunosuppressive activity, derived from B and T cell proliferation inhibition from rapamycin, the molecule of which they are analogs [180]. As such, their toxicity profiles also include infections, hypersensitivity reactions, angioedema, nephrotoxicity with proteinuria, kidney arterial and venous thrombosis, delays in wound healing, and increased risk of second tumors and increased risk of opportunistic infections [180]. These rare to severe side effects are dose-limiting, but rarely lead to discontinuation of mTOR inhibitor therapies because either adjusting the dose or support medications can overcome them.
17. Synthetic Lethality
The recognition of an effective chemotherapy regimen, either monotherapy or in combination, has been achieved in clinical trials, which are largely based on clinical and empirical experience, with a focus on therapeutic efficacy and tolerable toxicity. This is an expensive and time-consuming pipeline, for which most drugs fail [181]. Mechanism-based drug combinations have been mostly investigated at the preclinical stages. The search for better combinations has developed many screenings, such as those that aim at synthetic lethality and drug repurposing.
Synthetic lethality screenings have been thoroughly performed for the last 10 years in the search for gene and drug synergism. These screens have identified some combinations that might result in true drug interactions, from which one single combination has reached a place in the clinic, namely, the use of poly(ADP-ribose) polymerase (PARP) inhibitors in BRCA1 DNA repair associated (BRCA) gene mutated patients [182]. Inhibition of PARP activity induces synthetic lethality in mutated BRCA1/2 cancers by selectively targeting tumor cells that fail to repair DNA double-strand breaks [183].
Another promising example of the capabilities of synthetic lethality screening has been published for late ovarian cancer; the authors proposed a list of 84 new drug combinations to be tested in preclinical and clinical trials [184].
We strongly believe that the expansion in knowledge of signaling pathways and networks will present innovative opportunities for improved cancer therapies.
18. Drug Repurposing
Only 5% of anticancer drugs entering phase I clinical trials are ultimately granted FDA approval [185]. Drug repurposing aims to identify new applications for approved or investigational drugs that differ from their original clinical indication. This strategy allows for faster development at reduced costs since preclinical and clinical data might already be available for the repurposed drug [186]. Several analgesics and anesthetics, and antipsychotic, antibiotic and antiprotozoal drugs, among many other classes, have already been repurposed or are being tested on an oncology setting, based on modulation of numerous cell signaling pathways commonly disrupted in cancer [185]. Nitazoxanide (NTZ) is an antiprotozoal drug, which has been recently shown to inhibit autophagy in glioblastoma cells [187]. The combination with chloroquine (CQ), a well-known antimalarial autophagy inhibitor, had a synergistic effect, with CQ sensitizing the glioma cells to NTZ. Since NTZ has been shown to cross the blood–brain barrier (BBB) in mice, this highly lipophilic compound is a potential drug for reuse to treat gliomas [187].
Lipophilic antipsychotic drugs, known to cross the BBB effectively to bind to central dopamine D2 receptors and promote their therapeutic action, are able to modulate many cancer-associated intracellular signaling pathways, such as PI3K-AKT-mTOR, STAT3, and WNT [188], and may act as anti-cancer drugs. Chlorpromazine (CPZ), a dopamine D2 receptor antagonist, is able to cross the cell membrane and bind to FKBP-12, inhibiting the mTOR pathway, one of the most frequent points of dysregulation in cancer (Supplementary Table S3). This inhibition led to increased autophagic cell death in U-87 MG (glioblastoma) cells. Furthermore, CPZ inhibited tumor growth in human xenograft colon cancer and induced apoptosis in CRC cells in a p53-dependent manner mediated by c-Jun N-terminal Kinase (JNK) activation [189].
Other dopamine receptor-independent pharmacological activities of antipsychotics may be used as a basis for drug repurposing. Serotonin 5-hydroxytryptamine 7 (5-HT7) receptors are expressed by astrocytes and are commonly found overexpressed in glioblastoma, and are associated with apoptosis resistance, pro-survival signaling, and malignant transformation [188]. Thus, the use of antipsychotic drugs that also bind and inhibit 5-HT7, the antipsychotic risperidone (RIS), could be an attractive strategy for glioblastoma treatment. Other potential anticancer properties of RIS have already been demonstrated in both in vitro and in murine models. In resistant breast and colorectal cancer models, RIS is able to inhibit ABCG2 overexpression in a dose-dependent fashion, thus being able to reduce the impact of resistance mechanisms dependent on drug efflux that may arise from long-term chemotherapy [188].
Given the poor availability of efficient treatments in many cancer types (Supplementary Table S3), repurposing of such drugs given in combination with traditional chemotherapy and/or radiotherapy could improve cancer treatment and lead to lower doses and side effects, ultimately improving prognosis and quality of patients’ lives.
19. Conclusion and Final Considerations
Cytotoxic drugs are genotoxic, so the most common resistance mechanisms are associated with DNA homeostasis and increase detoxification as ABC transport. The resistance mechanisms of the targeted therapy are specific key changes in their targets of the signaling pathways, such as MAPK, PI3K, AKT, mTOR, and others. It is expected that the combination of chemotherapy and drugs intended to inhibit the mechanism of resistance induced by chemotherapy would increase cell death. This approach has the limitation that most inhibitors of ABC transport and MAPK receptors, PI3K, AKT, and mTOR, showed high toxicity and were not recommended for phase III clinical trial. The development of more selective inhibitors and repurposing of existing drugs are two different strategies that could be used to address this toxicity.
For the first strategy, a few examples have already been FDA approved. Initial clinical trials with pan-CDK inhibitors were very toxic and, as a consequence, did not reach phase III. Recently, three selective CDK4/6 inhibitors have been approved for metastatic breast cancer and are now being tested in various combinations for different tumors (Table 3 and Table 4). Many pre-clinical and clinical studies are necessary to understand when combinations of CDK inhibitors and other therapies may be promising since both antagonist and synergic effects can be achieved [190]. Even for the new generation of specific CDK4/6 inhibitors, the combination with first- and second-line cytotoxic therapy in glioblastomas showed antagonist and synergic effects depending on the combination. Abemaciclib increased cell death in temozolomide-treated groups, whereas it decreased carboplatin-induced cell death in glioblastoma cell lines (Hadju et al. unpublished data). In addition to new combinations of drugs given simultaneously, alternative regimes of sequential therapies should be explored depending on the possible biological effects that can be induced. Since CDK4/6 inhibitors induce transient cell cycle arrest, they might be suitable to be used in sequential cycles. Once the cell cycle blockage is released, cytotoxic drugs might maximize DNA damage-induced cell death of synchronized S-phase cells. Therefore, profound understandings of cell biology may help in designing new strategies.
Another possibility for dealing with the high toxicity of inhibiting key components of signaling pathways of resistance mechanisms is to explore less potent inhibitors in the form of safer drugs already in use for other purposes. For this, the careful off-target studies of drugs already in use for another proposal can be tested. For example, imatinib was originally designed to inhibit BCR-ABL tyrosine kinase, and it was identified that it also inhibits platelet-derived growth factor receptor (PDGFR) and KIT proto-oncogene tyrosine kinases. Since KIT and PDGFRA mutations are observed in 85% of gastrointestinal stromal tumors (GISTs), it was also tested and approved for GIST treatment [191]. The list of repurposed drugs can be found on the reproDB website [192]. It is urgent, however, that drug combinations are tested based on the mechanism of action, off-target, and resistance mechanisms, including drugs already designed for another purpose. This review collected the known off-target and resistance mechanism data of cytotoxic drugs (Supplementary Table S1) and anticancer-targeted therapies (Supplementary Table S2) that were FDA-approved up to May 2019, organized by drug classification. This effort hopefully will help build future experiments exploring new combinations of drugs, including known off-targets that interfere directly with resistance mechanisms of other drugs.
The effort of looking for new combinations that target the resistance mechanisms should not be limited to anticancer drugs but should also include repurposing drugs for other diseases. For example, lipophilic antipsychotic drugs and others that can inhibit the major mechanism of resistance of the target therapy, such as PI3K-AKT-mTOR and STAT3, [188] can be tested with a wide range of target therapies and cancers (Supplementary Table S2). Studies are particularly needed for tumors whose patients present a 5-year mean survival that is below 20% and have limited or even no FDA-approved combinations (Supplementary Table S3).
It is worth noting that the literature search for the resistance mechanisms discussed in this review was limited to the direct effectors of signaling pathways. However, let us not forget that resistance mechanisms might also be triggered by indirect modulation of signaling pathways, such as transcription factors and regulatory elements (enhancers and silencers, regulatory RNAs—lncRNA, miRNA). Moreover, most indirect pathways represent network-signaling interactions in which they might be tumor- or cell-type specific, with putative different triggers and modulations by specific co-repressors and/or co-activators. These also represent relevant targets for drug development, but more research will be needed, including high-throughput screening.
Bioinformatics and many preclinical studies are needed to understand the mechanisms of drug interactions for combination therapy. Since most signaling pathways are overlapping, with several points of interaction with other pathways, and show compensatory mechanisms, it is certainly a difficult task to predict this complex network, especially when particular mutations of tumor cells are also taken into account. One may wonder whether it is not more efficient to screen patients to the best protocols available instead of using a "fit the patient to the drug” approach, an unintended design of clinical trials for drug development. Would not it be beneficial, ethical, and possibly more economical for patients and drug development to avoid unnecessary exposure to inefficient protocols? We understand that both traditional trials and personalized therapy might (or should) be supported by individual, fast, and efficient targeting and/or drug screening, before any first-line treatments to identify intrinsic resistances and sensitivities. This might be achieved by designing faster high-throughput screenings with patient-derived tissue samples, such as first-generation organoid cultures [193]. Affordable protocols for organoid cultures of the major tissues affected by human cancers are urgently needed.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4409/8/9/1013/s1, Supplementary Table S1. Summary of FDA-approved anticancer cytotoxic drugs at May 2019. Supplementary Table S2. Summary of FDA-approved anticancer targeted therapies at May 2019. Supplementary Table S3. Approved drug combination options are limited, even when recommended in first-line therapy for the most prevalent and fatal cancer types.
Author Contributions
Conceptualization, J.M.A.D and H.L.B.; Validation, J.M.A.D., A.S.O.S. and L.C.M.S.; Writing—Original Draft Preparation, J.M.A.D., A.S.O.S., L.C.M.S. and H.L.B.; Writing—Review & Editing, J.M.A.D., L.C.M.S. and H.L.B.; Funding Acquisition, H.L.B.
Funding
This research was funded by the Brazilian funding agencies: National Council for Scientific and Technological Development (CNPq), Coordination for the Improvement of Higher Education Personnel (CAPES), and Foundation Carlos Chagas Filho Research Support of the State of Rio de Janeiro (FAPERJ).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gilman, A. The initial clinical trial of nitrogen mustard. Am. J. Surg. 1963, 105, 574–578. [Google Scholar] [CrossRef]
- DeVita, V.T.; Chu, E. A History of Cancer Chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2015, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Wu, W.; Wang, Y.; Alexander, P.B.; Sun, C.; Gong, Z.; Cheng, J.-N.; Sun, H.; Guan, Y.; Xia, X.; et al. Local mutational diversity drives intratumoral immune heterogeneity in non-small cell lung cancer. Nat. Commun. 2018, 9, 5361. [Google Scholar] [CrossRef]
- Bruce, W.R.; Meeker, B.E. Comparison of the sensitivity of hematopoietic colony-forming cells in different proliferative states to 5-fluorouracil. J. Natl. Cancer Inst. 1967, 38, 401–405. [Google Scholar]
- Hill, B.T.; Baserga, R. The cell cycle and its significance for cancer treatment. Cancer Treat. Rev. 1975, 2, 159–175. [Google Scholar] [CrossRef]
- Espinosa, E.; Zamora, P.; Feliu, J.; González Barón, M. Classification of anticancer drugs—A new system based on therapeutic targets. Cancer Treat. Rev. 2003, 29, 515–523. [Google Scholar] [CrossRef]
- Wu, X.-Z. A new classification system of anticancer drugs—Based on cell biological mechanisms. Med. Hypotheses 2006, 66, 883–887. [Google Scholar] [CrossRef]
- Dutt, R.; Madan, A.K. Classification Models for Anticancer Activity. Curr. Top. Med. Chem. 2013, 12, 2705–2726. [Google Scholar] [CrossRef]
- Pritchard, J.R.; Bruno, P.M.; Gilbert, L.A.; Capron, K.L.; Lauffenburger, D.A.; Hemann, M.T. Defining principles of combination drug mechanisms of action. Proc. Natl. Acad. Sci. 2013, 110, E170–E179. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Preissner, S.; Kroll, K.; Dunkel, M.; Senger, C.; Goldsobel, G.; Kuzman, D.; Guenther, S.; Winnenburg, R.; Schroeder, M.; Preissner, R. SuperCYP: a comprehensive database on Cytochrome P450 enzymes including a tool for analysis of CYP-drug interactions. Nucleic Acids Res. 2010, 38, D237–D243. [Google Scholar] [CrossRef] [PubMed]
- Mechanism matters. Nat. Med. 2010, 16, 347. [CrossRef] [PubMed]
- Juliano, R.L.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta - Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Stenke, L.; Varma, S.; Lindberg, M.L.; Björkholm, M.; Sjöberg, J.; Viktorsson, K.; Lewensohn, R.; Landgren, O.; Gottesman, M.M.; et al. Multidrug resistance in relapsed acute myeloid leukemia: Evidence of biological heterogeneity. Cancer 2013, 119, 3076–3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartholomae, S.; Gruhn, B.; Debatin, K.-M.; Zimmermann, M.; Creutzig, U.; Reinhardt, D.; Steinbach, D. Coexpression of Multiple ABC-Transporters is Strongly Associated with Treatment Response in Childhood Acute Myeloid Leukemia. Pediatr. Blood Cancer 2016, 63, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Marzac, C.; Garrido, E.; Tang, R.; Fava, F.; Hirsch, P.; De Benedictis, C.; Corre, E.; Lapusan, S.; Lallemand, J.-Y.; Marie, J.-P.; et al. ATP Binding Cassette transporters associated with chemoresistance: transcriptional profiling in extreme cohorts and their prognostic impact in a cohort of 281 acute myeloid leukemia patients. Haematologica 2011, 96, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Zhang, A.; Chen, M.; Liu, J. ABC Transporter Inhibitors in Reversing Multidrug Resistance to Chemotherapy. Curr. Drug Targets 2015, 16, 1356–1371. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Hoti, S.L. Development of Fourth Generation ABC Inhibitors from Natural Products: A Novel Approach to Overcome Cancer Multidrug Resistance. Anticancer. Agents Med. Chem. 2015, 15, 605–615. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute Bleomycin Sulfate. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/bleomycin (accessed on 15 June 2019).
- Zheng, W.; Johnston, S.A. The Nucleic Acid Binding Activity of Bleomycin Hydrolase Is Involved in Bleomycin Detoxification. Mol. Cell. Biol. 1998, 18, 3580–3585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazo, J.S.; Boland, C.J.; Schwartz, P.E. Bleomycin hydrolase activity and cytotoxicity in human tumors. Cancer Res. 1982, 42, 4026–4031. [Google Scholar] [PubMed]
- Lefterov, I.M.; Koldamova, R.P.; King, J.; Lazo, J.S. The C-terminus of human bleomycin hydrolase is required for protection against bleomycin-induced chromosomal damage. Mutat. Res. Mol. Mech. Mutagen. 1998, 421, 1–7. [Google Scholar] [CrossRef]
- Dortet, L.; Girlich, D.; Virlouvet, A.-L.; Poirel, L.; Nordmann, P.; Iorga, B.I.; Naas, T. Characterization of BRP MBL, the Bleomycin Resistance Protein Associated with the Carbapenemase NDM. Antimicrob. Agents Chemother. 2017, 61, e02413–e02416. [Google Scholar] [CrossRef]
- Ishikawa, T. ATP/Mg2+-dependent cardiac transport system for glutathione S-conjugates. A study using rat heart sarcolemma vesicles. J. Biol. Chem. 1989, 264, 17343–17348. [Google Scholar]
- Muller, M.; Meijer, C.; Zaman, G.J.; Borst, P.; Scheper, R.J.; Mulder, N.H.; de Vries, E.G.; Jansen, P.L. Overexpression of the gene encoding the multidrug resistance-associated protein results in increased ATP-dependent glutathione S-conjugate transport. Proc. Natl. Acad. Sci. 1994, 91, 13033–13037. [Google Scholar] [CrossRef]
- Brechbuhl, H.M.; Gould, N.; Kachadourian, R.; Riekhof, W.R.; Voelker, D.R.; Day, B.J. Glutathione Transport Is a Unique Function of the ATP-binding Cassette Protein ABCG2. J. Biol. Chem. 2010, 285, 16582–16587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-L.; Li, B.-Y.; Yang, R.; Xia, L.-Y.; Fan, A.-L.; Chu, Y.-C.; Wang, L.-J.; Wang, Z.-C.; Jiang, A.-Q.; Zhu, H.-L. A class of novel tubulin polymerization inhibitors exert effective anti-tumor activity via mitotic catastrophe. Eur. J. Med. Chem. 2019, 163, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, C.H.A.; Wu, S.-Y.; Lee, T.-R.; Chang, C.-Y.; Wu, J.-S.; Hsieh, H.-P.; Chang, J.-Y. Cancer Cells Acquire Mitotic Drug Resistance Properties Through Beta I-Tubulin Mutations and Alterations in the Expression of Beta-Tubulin Isotypes. PLoS ONE 2010, 5, e12564. [Google Scholar] [CrossRef] [PubMed]
- Hari, M.; Wang, Y.; Veeraraghavan, S.; Cabral, F. Mutations in alpha- and beta-tubulin that stabilize microtubules and confer resistance to colcemid and vinblastine. Mol. Cancer Ther. 2003, 2, 597–605. [Google Scholar] [PubMed]
- Yin, S.; Bhattacharya, R.; Cabral, F. Human Mutations That Confer Paclitaxel Resistance. Mol. Cancer Ther. 2010, 9, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binaschi, M.; Zunino, F.; Capranico, G. Mechanism of action of DNA topoisomerase inhibitors. Stem Cells 1995, 13, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Hevener, K.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar] [CrossRef] [PubMed]
- Sabini, E.; Hazra, S.; Konrad, M.; Lavie, A. Elucidation of Different Binding Modes of Purine Nucleosides to Human Deoxycytidine Kinase. J. Med. Chem. 2008, 51, 4219–4225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathanson, D.A.; Armijo, A.L.; Tom, M.; Li, Z.; Dimitrova, E.; Austin, W.R.; Nomme, J.; Campbell, D.O.; Ta, L.; Le, T.M.; et al. Co-targeting of convergent nucleotide biosynthetic pathways for leukemia eradication. J. Exp. Med. 2014, 211, 473–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkaria, J.N.; Kitange, G.J.; James, C.D.; Plummer, R.; Calvert, H.; Weller, M.; Wick, W. Mechanisms of Chemoresistance to Alkylating Agents in Malignant Glioma. Clin. Cancer Res. 2008, 14, 2900–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NF-kappa;B-signaling pathway in cancer. Onco. Targets. Ther. 2018, 11, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of Breast Cancer Stem Cells Identified by Aldehyde Dehydrogenase 1 Expression with Resistance to Sequential Paclitaxel and Epirubicin-Based Chemotherapy for Breast Cancers. Clin. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Lee, S.-H.; Hong, D.; Lee, J.-S.; Ahn, H.-S.; Ahn, J.-H.; Seong, T.W.; Lee, C.-H.; Jang, H.; Hong, K.M.; et al. Aldehyde dehydrogenase is used by cancer cells for energy metabolism. Exp. Mol. Med. 2016, 48, e272. [Google Scholar] [CrossRef]
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.D.; Vasiliou, V. Aldehyde Dehydrogenase Inhibitors: a Comprehensive Review of the Pharmacology, Mechanism of Action, Substrate Specificity, and Clinical Application. Pharmacol. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, S.; Swanton, C.; Taylor, B.S.; Costello, J.F. Treatment-Induced Mutagenesis and Selective Pressures Sculpt Cancer Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a026617. [Google Scholar] [CrossRef]
- Sever, R.; Brugge, J.S. Signal Transduction in Cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef]
- Katoh, M. Genomic testing, tumor microenvironment and targeted therapy of Hedgehog-related human cancers. Clin. Sci. 2019, 133, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Rothenstein, J.M.; Chooback, N. ALK inhibitors, resistance development, clinical trials. Curr. Oncol. 2018, 25, 59. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Dalin, S.; Hemann, M.T.; Lauffenburger, D.A.; Zhao, B. Differential selective pressure alters rate of drug resistance acquisition in heterogeneous tumor populations. Sci. Rep. 2016, 6, 36198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amirouchene-Angelozzi, N.; Swanton, C.; Bardelli, A. Tumor Evolution as a Therapeutic Target. Cancer Discov. 2017, 7, 805–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riviere, M.-K.; Le Tourneau, C.; Paoletti, X.; Dubois, F.; Zohar, S. Designs of drug-combination phase I trials in oncology: a systematic review of the literature. Ann. Oncol. 2015, 26, 669–674. [Google Scholar] [CrossRef]
- Day, D.; Siu, L.L. Approaches to modernize the combination drug development paradigm. Genome Med. 2016, 8, 115. [Google Scholar] [CrossRef]
- Mozgunov, P.; Jaki, T.; Paoletti, X. Randomized dose-escalation designs for drug combination cancer trials with immunotherapy. J. Biopharm. Stat. 2019, 29, 359–377. [Google Scholar] [CrossRef]
- Paller, C.J.; Bradbury, P.A.; Ivy, S.P.; Seymour, L.; LoRusso, P.M.; Baker, L.; Rubinstein, L.; Huang, E.; Collyar, D.; Groshen, S.; et al. Design of Phase I Combination Trials: Recommendations of the Clinical Trial Design Task Force of the NCI Investigational Drug Steering Committee. Clin. Cancer Res. 2014, 20, 4210–4217. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.A.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef]
- Singh, S.S.; Vats, S.; Chia, A.Y.-Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Huang, T.; Song, X.; Yang, Y.; Wan, X.; Alvarez, A.A.; Sastry, N.; Feng, H.; Hu, B.; Cheng, S.-Y. Autophagy and Hallmarks of Cancer. Crit. Rev. Oncog. 2018, 23, 247–267. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Brandmaier, A.; Hou, S.-Q.; Shen, W.H. Cell Cycle Control by PTEN. J. Mol. Biol. 2017, 429, 2265–2277. [Google Scholar] [CrossRef]
- Paramio, J.M.; Navarro, M.; Segrelles, C.; Gómez-Casero, E.; Jorcano, J.L. PTEN tumour suppressor is linked to the cell cycle control through the retinoblastoma protein. Oncogene 1999, 18, 7462–7468. [Google Scholar] [CrossRef] [Green Version]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017. [Google Scholar] [CrossRef]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef]
- Quereda, V.; Porlan, E.; Cañamero, M.; Dubus, P.; Malumbres, M. An essential role for Ink4 and Cip/Kip cell-cycle inhibitors in preventing replicative stress. Cell Death Differ. 2016, 23, 430–441. [Google Scholar] [CrossRef]
- Sanidas, I.; Morris, R.; Fella, K.A.; Rumde, P.H.; Boukhali, M.; Tai, E.C.; Ting, D.T.; Lawrence, M.S.; Haas, W.; Dyson, N.J. A Code of Mono-phosphorylation Modulates the Function of RB. Mol. Cell 2019, 73, 985–1000.e6. [Google Scholar] [CrossRef] [Green Version]
- Rigberg, D.A.; Kim, F.S.; Sebastian, J.L.; Kazanjian, K.K.; McFadden, D.W. Hypophosphorylated Retinoblastoma Protein Is Associated with G2Arrest in Esophageal Squamous Cell Carcinoma. J. Surg. Res. 1999, 84, 101–105. [Google Scholar] [CrossRef]
- Roesch, A.; Becker, B.; Meyer, S.; Hafner, C.; Wild, P.J.; Landthaler, M.; Vogt, T. Overexpression and hyperphosphorylation of retinoblastoma protein in the progression of malignant melanoma. Mod. Pathol. 2005, 18, 565–572. [Google Scholar] [CrossRef]
- Senderowicz, A.M. Flavopiridol: the first cyclin-dependent kinase inhibitor in human clinical trials. Invest. New Drugs 1999, 17, 313–320. [Google Scholar] [CrossRef]
- Deep, A.; Marwaha, R.K.; Marwaha, M.G.; Jyoti, J.; Nandal, R.; Sharma, A.K. Flavopiridol as cyclin dependent kinase (CDK) inhibitor: a review. New J. Chem. 2018, 42, 18500–18507. [Google Scholar] [CrossRef]
- Byrd, J.C.; Shinn, C.; Waselenko, J.K.; Fuchs, E.J.; Lehman, T.A.; Nguyen, P.L.; Flinn, I.W.; Diehl, L.F.; Sausville, E.; Grever, M.R. Flavopiridol induces apoptosis in chronic lymphocytic leukemia cells via activation of caspase-3 without evidence of bcl-2 modulation or dependence on functional p53. Blood 1998, 92, 3804–3816. [Google Scholar]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [Green Version]
- Aklilu, M. Phase II study of flavopiridol in patients with advanced colorectal cancer. Ann. Oncol. 2003, 14, 1270–1273. [Google Scholar] [CrossRef]
- Burdette-Radoux, S.; Tozer, R.G.; Lohmann, R.C.; Quirt, I.; Ernst, D.S.; Walsh, W.; Wainman, N.; Colevas, A.D.; Eisenhauer, E.A. Phase II trial of flavopiridol, a cyclin dependent kinase inhibitor, in untreated metastatic malignant melanoma. Invest. New Drugs 2004, 22, 315–322. [Google Scholar] [CrossRef]
- Lin, T.S.; Howard, O.M.; Neuberg, D.S.; Kim, H.H.; Shipp, M.A. Seventy-Two Hour Continuous Infusion Flavopiridol in Relapsed and Refractory Mantle Cell Lymphoma. Leuk. Lymphoma 2002, 43, 793–797. [Google Scholar] [CrossRef]
- Kouroukis, C.T.; Belch, A.; Crump, M.; Eisenhauer, E.; Gascoyne, R.D.; Meyer, R.; Lohmann, R.; Lopez, P.; Powers, J.; Turner, R.; et al. Flavopiridol in Untreated or Relapsed Mantle-Cell Lymphoma: Results of a Phase II Study of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2003, 21, 1740–1745. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Ibrance (palbociclib) FDA Approval History - Drugs.com. Available online: https://www.drugs.com/history/ibrance.html (accessed on 7 August 2019).
- Sobhani , N.; D’Angelo, A.; Pittacolo , M.; Roviello , G.; Miccoli , A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 Inhibitory Strategy and Combinations in Breast Cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef]
- Shah, M.; Nunes, M.R.; Stearns, V. CDK4/6 Inhibitors: Game Changers in the Management of Hormone Receptor–Positive Advanced Breast Cancer? Oncology (Williston Park). 2018, 32, 216–222. [Google Scholar] [PubMed]
- Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 2582. [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.-C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Portman, N.; Alexandrou, S.; Carson, E.; Wang, S.; Lim, E.; Caldon, C.E. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr. Relat. Cancer 2019, R15–R30. [Google Scholar] [CrossRef]
- Jabbour-Leung, N.A.; Chen, X.; Bui, T.; Jiang, Y.; Yang, D.; Vijayaraghavan, S.; McArthur, M.J.; Hunt, K.K.; Keyomarsi, K. Sequential Combination Therapy of CDK Inhibition and Doxorubicin Is Synthetically Lethal in p53-Mutant Triple-Negative Breast Cancer. Mol. Cancer Ther. 2016, 15, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef]
- Stegh, A.H. Targeting the p53 signaling pathway in cancer therapy—The promises, challenges and perils. Expert Opin. Ther. Targets 2012, 16, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Vessoni, A.T.; Filippi-Chiela, E.C.; Menck, C.F.; Lenz, G. Autophagy and genomic integrity. Cell Death Differ. 2013, 20, 1444–1454. [Google Scholar] [CrossRef]
- Hewitt, G.; Korolchuk, V.I. Repair, Reuse, Recycle: The Expanding Role of Autophagy in Genome Maintenance. Trends Cell Biol. 2017, 27, 340–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Levine, B. Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Kondapuram, S.K.; Sarvagalla, S.; Coumar, M.S. Targeting autophagy with small molecules for cancer therapy. J. Cancer Metastasis Treat. 2019, 2019. [Google Scholar] [CrossRef]
- Delou, J.M.A.; Biasoli, D.; Borges, H.L. The Complex Link between Apoptosis and Autophagy: a Promising New Role for RB. An. Acad. Bras. Cienc. 2016, 88, 2257–2275. [Google Scholar] [CrossRef] [Green Version]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.-M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- and pro-tumor functions of autophagy. Biochim. Biophys. Acta - Mol. Cell Res. 2009, 1793, 1524–1532. [Google Scholar] [CrossRef] [Green Version]
- Shang, L.; Wang, X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy 2011, 7, 924–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.-T.; Zhou, T.-T.; Liu, B.; Bao, J.-K. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Biasoli, D.; Kahn, S.A.; Cornélio, T.A.; Furtado, M.; Campanati, L.; Chneiweiss, H.; Moura-Neto, V.; Borges, H.L. Retinoblastoma protein regulates the crosstalk between autophagy and apoptosis, and favors glioblastoma resistance to etoposide. Cell Death Dis. 2013, 4, e767. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Soletti, R.C.; Sadarangani, A.; Sridevi, P.; Ramirez, M.E.; Eckmann, L.; Borges, H.L.; Wang, J.Y.J. Nuclear Expression of β-Catenin Promotes RB Stability and Resistance to TNF-Induced Apoptosis in Colon Cancer Cells. Mol. Cancer Res. 2013, 11, 207–218. [Google Scholar] [CrossRef] [PubMed]
- LEE011 Plus Everolimus in Patients With Metastatic Pancreatic Adenocarcinoma Refractory to Chemotherapy - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02985125?term=NCT02985125&rank=1 (accessed on 14 July 2019).
- Michaloglou, C.; Crafter, C.; Siersbaek, R.; Delpuech, O.; Curwen, J.O.; Carnevalli, L.S.; Staniszewska, A.D.; Polanska, U.M.; Cheraghchi-Bashi, A.; Lawson, M.; et al. Combined Inhibition of mTOR and CDK4/6 Is Required for Optimal Blockade of E2F Function and Long-term Growth Inhibition in Estrogen Receptor–positive Breast Cancer. Mol. Cancer Ther. 2018, 17, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Berrak, O.; Arisan, E.D.; Obakan-Yerlikaya, P.; Coker-Gürkan, A.; Palavan-Unsal, N. mTOR is a fine tuning molecule in CDK inhibitors-induced distinct cell death mechanisms via PI3K/AKT/mTOR signaling axis in prostate cancer cells. Apoptosis 2016, 21, 1158–1178. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Nowsheen, S.; Aziz, K.; Georgakilas, A.G. Toxicity and adverse effects of Tamoxifen and other anti-estrogen drugs. Pharmacol. Ther. 2013, 139, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Bosutinib in Combination With Pemetrexed in Patients With Selected Metastatic Solid Tumors - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03023319?term=bosutinib&draw=2&rank=2 (accessed on 15 July 2019).
- Secord, A.A.; Teoh, D.K.; Barry, W.T.; Yu, M.; Broadwater, G.; Havrilesky, L.J.; Lee, P.S.; Berchuck, A.; Lancaster, J.; Wenham, R.M. A Phase I Trial of Dasatinib, an Src-Family Kinase Inhibitor, in Combination with Paclitaxel and Carboplatin in Patients with Advanced or Recurrent Ovarian Cancer. Clin. Cancer Res. 2012, 18, 5489–5498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azacitidine and Venetoclax as Induction Therapy With Venetoclax Maintenance in the Elderly With AML. Available online: https://clinicaltrials.gov/show/NCT03466294 (accessed on 15 June 2019).
- Venetoclax and Chemotherapy as Frontline Therapy in Older Patients and Patients With Relapsed/Refractory ALL. Available online: https://clinicaltrials.gov/show/NCT03319901 (accessed on 15 June 2019).
- A Study of Abemaciclib (LY2835219) in Combination With Another Anti-cancer Drug in Participants With Lung Cancer (NSCLC). Available online: https://clinicaltrials.gov/show/NCT02079636 (accessed on 15 June 2019).
- INdividualized Screening Trial of Innovative Glioblastoma Therapy (INSIGhT). Available online: https://clinicaltrials.gov/show/NCT02977780 (accessed on 15 June 2019).
- Phase II Trial Evaluating the Efficacy of Palbociclib in Combination With Carboplatin for the Treatment of Unresectable Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. Available online: https://clinicaltrials.gov/show/NCT03194373 (accessed on 15 June 2019).
- Dose-Escalation Study Of Palbociclib + Nab-Paclitaxel In mPDAC. Available online: https://clinicaltrials.gov/show/NCT02501902 (accessed on 15 June 2019).
- Study Of Palbociclib Combined With Chemotherapy In Pediatric Patients With Recurrent/Refractory Solid Tumors. Available online: https://clinicaltrials.gov/show/NCT03709680 (accessed on 15 June 2019).
- LEE011 (Ribociclib) in Combination With Docetaxel Plus Prednisone in mCRPC. Available online: https://clinicaltrials.gov/show/NCT02494921 (accessed on 15 June 2019).
- SJDAWN: St. Jude Children’s Research Hospital Phase 1 Study Evaluating Molecularly-Driven Doublet Therapies for Children and Young Adults With Recurrent Brain Tumors. Available online: https://clinicaltrials.gov/show/NCT03434262 (accessed on 15 June 2019).
- Ribociclib (Ribociclib (LEE-011)) With Platinum-based Chemotherapy in Recurrent Platinum Sensitive Ovarian Cancer. Available online: https://clinicaltrials.gov/show/NCT03056833 (accessed on 15 June 2019).
- Niraparib and Temozolomide in Treating Patients With Extensive-Stage Small Cell Lung Cancer With a Complete or Partial Response to Platinum-Based First-Line Chemotherapy. Available online: https://clinicaltrials.gov/show/NCT03830918 (accessed on 15 June 2019).
- Olaparib Dose Escalating Trial + Concurrent RT With or Without Cisplatin in Locally Advanced NSCLC. Available online: https://clinicaltrials.gov/show/NCT01562210 (accessed on 15 June 2019).
- Rucaparib and Irinotecan in Cancers With Mutations in DNA Repair. Available online: https://clinicaltrials.gov/show/NCT03318445 (accessed on 15 June 2019).
- Berlin, J.; Ramanathan, R.K.; Strickler, J.H.; Subramaniam, D.S.; Marshall, J.; Kang, Y.-K.; Hetman, R.; Dudley, M.W.; Zeng, J.; Nickner, C.; et al. A phase 1 dose-escalation study of veliparib with bimonthly FOLFIRI in patients with advanced solid tumours. Br. J. Cancer 2018, 118, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Pommier, Y. Classification of PARP Inhibitors Based on PARP Trapping and Catalytic Inhibition, and Rationale for Combinations with Topoisomerase I Inhibitors and Alkylating Agents. In PARP Inhibitors for Cancer Therapy; Springer: Berlin, Germany, 2015; pp. 261–274. [Google Scholar]
- Veliparib and Temozolomide in Treating Patients With Acute Leukemia - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01139970 (accessed on 1 August 2019).
- Pishvaian, M.J.; Slack, R.S.; Jiang, W.; He, A.R.; Hwang, J.J.; Hankin, A.; Dorsch-Vogel, K.; Kukadiya, D.; Weiner, L.M.; Marshall, J.L.; et al. A phase 2 study of the PARP inhibitor veliparib plus temozolomide in patients with heavily pretreated metastatic colorectal cancer. Cancer 2018, 124, 2337–2346. [Google Scholar] [CrossRef] [PubMed]
- Veliparib and Topotecan With or Without Carboplatin in Treating Patients With Relapsed or Refractory Acute Leukemia, High-Risk Myelodysplasia, or Aggressive Myeloproliferative Disorders - Full Text View - ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00588991?term=veliparib%2C+topotecan&rank=2 (accessed on 14 July 2019).
- Study of Folfiri/Cetuximab in FcGammaRIIIa V/V Stage IV Colorectal Cancer Patients - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03874026?term=cetuximab&draw=2&rank=15 (accessed on 14 July 2019).
- Neoadjuvant Chemotherapy Combined With Cetuximab for EGFR Wild Type Locally Advanced Rectal Cancer - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03391843?term=cetuximab&draw=2&rank=4 (accessed on 14 July 2019).
- Necitumumab in the Neoadjuvant Setting With Gemcitabine in Surgically Resectable - 14X-US-I001 - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03574818?term=necitumumab&draw=2&rank=1 (accessed on 14 July 2019).
- A Study of Perioperative Chemotherapy Plus Panitumumab in Patients With Colorectal Cancer Liver Metastases - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01260415?term=panitumumab&draw=3 (accessed on 14 July 2019).
- Yoshino, T.; Uetake, H.; Tsuchihara, K.; Shitara, K.; Yamazaki, K.; Oki, E.; Sato, T.; Naitoh, T.; Komatsu, Y.; Kato, T.; et al. Rationale for and Design of the PARADIGM Study: Randomized Phase III Study of mFOLFOX6 Plus Bevacizumab or Panitumumab in Chemotherapy-naïve Patients With RAS ( KRAS/NRAS ) Wild-type, Metastatic Colorectal Cancer. Clin. Colorectal Cancer 2017, 16, 158–163. [Google Scholar] [CrossRef] [PubMed]
- DAPHNe: Paclitaxel/Trastuzumab/Pertuzumab in HER2-Positive BC - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03716180?term=pertuzumab&rank=7 (accessed on 14 July 2019).
- Copanlisib (BAY 80-6946) in Combination With Gemcitabine and Cisplatin in Advanced Cholangiocarcinoma - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02631590?term=copanlisib&draw=5&rank=32 (accessed on 14 July 2019).
- A Phase 1b/2 Study of IPI-145 Plus FCR in Previously Untreated, Younger Patients With CLL - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02158091?term=duvelisib&draw=4&rank=28 (accessed on 14 July 2019).
- Hauke, R.J.; Infante, J.R.; Rubin, M.S.; Shih, K.C.; Arrowsmith, E.R.; Hainsworth, J.D. Everolimus in combination with paclitaxel and carboplatin in patients with metastatic melanoma: a phase II trial of the Sarah Cannon Research Institute Oncology Research Consortium. Melanoma Res. 2013, 23, 468–473. [Google Scholar] [CrossRef]
- Everolimus With and Without Temozolomide in Adult Low Grade Glioma - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02023905?term=temozolomide%2C+pi3k&rank=1 (accessed on 14 July 2019).
- Zelenetz, A.D.; Barrientos, J.C.; Brown, J.R.; Coiffier, B.; Delgado, J.; Egyed, M.; Ghia, P.; Illés, Á.; Jurczak, W.; Marlton, P.; et al. Idelalisib or placebo in combination with bendamustine and rituximab in patients with relapsed or refractory chronic lymphocytic leukaemia: interim results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2017, 18, 297–311. [Google Scholar] [CrossRef] [Green Version]
- Dunn, L.A.; Fury, M.G.; Xiao, H.; Baxi, S.S.; Sherman, E.J.; Korte, S.; Pfister, C.; Haque, S.; Katabi, N.; Ho, A.L.; et al. A phase II study of temsirolimus added to low-dose weekly carboplatin and paclitaxel for patients with recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC). Ann. Oncol. 2017, 28, 2533–2538. [Google Scholar] [CrossRef] [PubMed]
- A Study of Avastin (Bevacizumab) and Xeloda (Capecitabine) in Patients With Advanced or Metastatic Liver Cancer - Full Text View - ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02013830?term=02013830&rank=1 (accessed on 14 July 2019).
- McWilliams, R.R.; Allred, J.B.; Slostad, J.A.; Katipamula, R.; Dronca, R.S.; Rumilla, K.M.; Erickson, L.A.; Bryce, A.H.; Joseph, R.W.; Kottschade, L.A.; et al. NCCTG N0879 (Alliance): A randomized phase 2 cooperative group trial of carboplatin, paclitaxel, and bevacizumab ± everolimus for metastatic melanoma. Cancer 2018, 124, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Onvansertib in Combination With FOLFIRI and Bevacizumab for Second Line Treatment of Metastatic Colorectal Cancer Patients With a Kras Mutation - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03829410?term=bevacizumab%2C+FOLFIRI&rank=3 (accessed on 14 July 2019).
- Van den Bent, M.J.; Klein, M.; Smits, M.; Reijneveld, J.C.; French, P.J.; Clement, P.; de Vos, F.Y.F.; Wick, A.; Mulholland, P.J.; Taphoorn, M.J.B.; et al. Bevacizumab and temozolomide in patients with first recurrence of WHO grade II and III glioma, without 1p/19q co-deletion (TAVAREC): A randomised controlled phase 2 EORTC trial. Lancet Oncol. 2018, 19, 1170–1179. [Google Scholar] [CrossRef]
- Lenvatinib and Weekly Paclitaxel for Patients With Recurrent Endometrial or Ovarian Cancer - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02788708?term=lenvatinib&rank=10 (accessed on 14 July 2019).
- Ramucirumab Plus FOLFIRI Versus Ramucirumab Plus Paclitaxel in Patients With Advanced or Metastatic Gastric Cancer, Who Failed One Prior Line of Palliative Chemotherapy - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03081143?term=ramucirumab%2C+folfiri&rank=2 (accessed on 14 July 2019).
- Regorafenib Combined With Irinotecan as Second-line in Patients With Metastatic Gastro-oesophageal Adenocarcinomas - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03722108?term=regorafenib&draw=2&rank=6 (accessed on 14 July 2019).
- Treating Relapsed/Recurrent/Refractory Pediatric Solid Tumors With Sorafenib in Combination With Irinotecan - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02747537?term=irinotecan%2C+vegfr&rank=3 (accessed on 14 July 2019).
- Santoro, A.; Su, W.-C.; Navarro, A.; Simonelli, M.; Yang, J.C.-H.; Ardizzoni, A.; Barlesi, F.; Kang, J.H.; Didominick, S.; Abdelhady, A.; et al. 1392PDose-determination results from a phase Ib/II study of ceritinib (CER) + ribociclib (RIB) in ALK-positive (ALK+) non-small cell lung cancer (NSCLC). Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Wood, A.C.; Krytska, K.; Ryles, H.T.; Infarinato, N.R.; Sano, R.; Hansel, T.D.; Hart, L.S.; King, F.J.; Smith, T.R.; Ainscow, E.; et al. Dual ALK and CDK4/6 Inhibition Demonstrates Synergy against Neuroblastoma. Clin. Cancer Res. 2017, 23, 2856–2868. [Google Scholar] [CrossRef] [PubMed]
- Next Generation Personalized Neuroblastoma Therapy (NEPENTHE). Available online: https://clinicaltrials.gov/ct2/show/NCT02780128?term=ceritinib%2C+ribociclib&rank=2 (accessed on 14 July 2019).
- Crizotinib in Combination With Enzalutamide in Metastatic Castration-resistant Prostate Cancer - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02207504?term=crizotinib&recrs=de&rank=4 (accessed on 14 July 2019).
- Tripathi, A.; Supko, J.G.; Gray, K.P.; Melnick, Z.; Taplin, M.-E.; Choudhury, A.D.; Pomerantz, M.; Bellmunt, J.; Yu, C.; Sun, Z.; et al. 852PPharmacokinetic (PK) analysis of concurrent administration of enzalutamide (enza) and crizotinib (crizo) in patients with metastatic castration resistant prostate cancer (CRPC). Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Kato, S.; Jardim, D.L.; Johnson, F.M.; Subbiah, V.; Piha-Paul, S.; Tsimberidou, A.M.; Falchook, G.S.; Karp, D.; Zinner, R.; Wheler, J.; et al. Phase I study of the combination of crizotinib (as a MET inhibitor) and dasatinib (as a c-SRC inhibitor) in patients with advanced cancer. Invest. New Drugs 2018, 36, 416–423. [Google Scholar] [CrossRef]
- Dasatinib and Crizotinib in Advanced Cancer - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01744652?term=crizotinib&recrs=de&rank=6 (accessed on 14 July 2019).
- Study of the Combination of Crizotinib and Dasatinib in Pediatric Research Participants With Diffuse Pontine Glioma (DIPG) and High-Grade Glioma (HGG) - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01644773?term=crizotinib&recrs=de&rank=22 (accessed on 14 July 2019).
- Michmerhuizen, N.L.; Leonard, E.; Matovina, C.; Harris, M.; Herbst, G.; Kulkarni, A.; Zhai, J.; Jiang, H.; Carey, T.E.; Brenner, J.C. Rationale for Using Irreversible Epidermal Growth Factor Receptor Inhibitors in Combination with Phosphatidylinositol 3-Kinase Inhibitors for Advanced Head and Neck Squamous Cell Carcinoma. Mol. Pharmacol. 2019, 95, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Copanlisib in Association With Cetuximab in Patients With Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinomas Harboring a PI3KCA Mutation/Amplification and/or a PTEN Loss - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02822482?term=copanlisib&rank=2 (accessed on 14 July 2019).
- Pazopanib and Everolimus in PI3KCA Mutation Positive/PTEN Loss Patients - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01430572 (accessed on 14 July 2019).
- Barr, P.M.; Saylors, G.B.; Spurgeon, S.E.; Cheson, B.D.; Greenwald, D.R.; OBrien, S.M.; Liem, A.K.D.; Mclntyre, R.E.; Joshi, A.; Abella-Dominicis, E.; et al. Phase 2 study of idelalisib and entospletinib: pneumonitis limits combination therapy in relapsed refractory CLL and NHL. Blood 2016, 127, 2411–2415. [Google Scholar] [CrossRef] [Green Version]
- A Study of Abemaciclib (LY2835219) Alone or in Combination With Other Agents in Participants With Previously Treated Pancreatic Ductal Adenocarcinoma - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02981342?term=NCT02981342&rank=1 (accessed on 14 July 2019).
- Keam, B.; Tahara, M.; Lin, J.-C.; Sacco, A.G.; Melichar, B.; Baney, T.; Hoffman, J.; Wang, D.D.; Wang, S.L.; Martini, J.-F.; et al. An international, multicenter, randomized, double-blind, placebo-controlled, parallel-group phase 2 study of palbociclib (an oral CDK4/6 inhibitor) plus cetuximab in patients with recurrent/metastatic (R/M) squamous cell carcinoma of the head and neck (SCCHN). J. Clin. Oncol. 2016, 34, TPS6102. [Google Scholar]
- Sullivan, R.J.; Amaria, R.N.; Lawrence, D.P.; Brennan, J.; Leister, C.; Singh, R.; Legos, J.; Thurm, H.; Yan, L.; Flaherty, K.T.; et al. Abstract PR06: Phase 1b dose-escalation study of trametinib (MEKi) plus palbociclib (CDK4/6i) in patients with advanced solid tumors. In Proceedings of the MAPK Pathways; American Association for Cancer Research: Philadelphia, PA, USA, 2015; p. PR06. [Google Scholar]
- A Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics, and Anti-Cancer Activity of Trametinib in Combination With Palbociclib in Subjects With Solid Tumors - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02065063 (accessed on 15 July 2019).
- Berghoff, A.S.; Preusser, M. Targeted Therapies for Melanoma Brain Metastases. Curr. Treat. Options Neurol. 2017, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Vemurafenib and Cobimetinib Combination in BRAF Mutated Melanoma With Brain Metastasis - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02537600?term=vemurafenib&rank=7 (accessed on 14 July 2019).
- A Study of Tarceva (Erlotinib) and Avastin (Bevacizumab) in Patients With Advanced or Metastatic Liver Cancer. - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00605722?term=00605722&rank=1 (accessed on 14 July 2019).
- Nissan, M.H.; Rosen, N.; Solit, D.B. ERK Pathway Inhibitors: How Low Should We Go? Cancer Discov. 2013, 3, 719–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK Inhibition in Melanoma with BRAF V600 Mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Pelster, M.S.; Amaria, R.N. Combined targeted therapy and immunotherapy in melanoma: a review of the impact on the tumor microenvironment and outcomes of early clinical trials. Ther. Adv. Med. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, B.S.; Durinck, S.; Stawiski, E.W.; Yin, J.; Wang, W.; Lin, E.; Moffat, J.; Martin, S.E.; Modrusan, Z.; Seshagiri, S. ERK Mutations and amplification confer resistance to ERK-inhibitor therapy. Clin. Cancer Res. 2018, 24, 4044–4055. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Lee Wong, D.J.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: Results of a phase I dose-escalation and expansion study. Cancer Discov. 2018, 8, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the clinical development of PI3K inhibitors: Strategies to improve their impact in solid tumors. Cancer Discov. 2019, 9. [Google Scholar] [CrossRef]
- Curigliano, G.; Shah, R.R. Safety and Tolerability of Phosphatidylinositol-3-Kinase (PI3K) Inhibitors in Oncology. Drug Saf. 2019, 42, 247–262. [Google Scholar] [CrossRef]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. AKt as a therapeutic target for cancer. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sampath, D.; Malik, A.; Plunkett, W.; Nowak, B.; Williams, B.; Burton, M.; Verstovsek, S.; Faderl, S.; Garcia-Manero, G.; List, A.F.; et al. Phase I clinical, pharmacokinetic, and pharmacodynamic study of the Akt-inhibitor triciribine phosphate monohydrate in patients with advanced hematologic malignancies. Leuk. Res. 2013, 37, 1461–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunyoto, T.; Potet, J.; Boelaert, M. Why miltefosine - A life-saving drug for leishmaniasis-is unavailable to people who need it the most. BMJ Glob. Heal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ghidini, M.; Petrelli, F.; Ghidini, A.; Tomasello, G.; Hahne, J.C.; Passalacqua, R.; Barni, S. Clinical development of mTor inhibitors for renal cancer. Expert Opin. Investig. Drugs 2017, 26, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Teng, Q.-X.; Ashar, Y.V.; Gupta, P.; Gadee, E.; Fan, Y.-F.; Reznik, S.E.; Wurpel, J.N.D.; Chen, Z.-S. Revisiting mTOR inhibitors as anticancer agents. Drug Discov. Today 2019. [Google Scholar] [CrossRef]
- Prasad, V.; De Jesús, K.; Mailankody, S. The high price of anticancer drugs: origins, implications, barriers, solutions. Nat. Rev. Clin. Oncol. 2017, 14, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Turk, A.A.; Wisinski, K.B. PARP inhibitors in breast cancer: Bringing synthetic lethality to the bedside. Cancer 2018, 124, 2498–2506. [Google Scholar] [CrossRef]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Heinzel, A.; Marhold, M.; Mayer, P.; Schwarz, M.; Tomasich, E.; Lukas, A.; Krainer, M.; Perco, P. Synthetic lethality guiding selection of drug combinations in ovarian cancer. PLoS ONE 2019, 14, e0210859. [Google Scholar] [CrossRef]
- Gupta, S.C.; Sung, B.; Prasad, S.; Webb, L.J.; Aggarwal, B.B. Cancer drug discovery by repurposing: Teaching new tricks to old dogs. Trends Pharmacol. Sci. 2013. [Google Scholar] [CrossRef]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology-patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, C.; Liu, Z.; Peng, F.; Chen, X.; Yang, G.; Zhang, D.; Yin, Z.; Ma, J.; Zheng, Z.; et al. Nitazoxanide, an antiprotozoal drug, inhibits late-stage autophagy and promotes ING1-induced cell cycle arrest in glioblastoma. Cell Death Dis. 2018, 9, 1032. [Google Scholar] [CrossRef] [PubMed]
- Hendouei, N.; Saghafi, F.; Shadfar, F.; Hosseinimehr, S.J. Molecular mechanisms of anti-psychotic drugs for improvement of cancer treatment. Eur. J. Pharmacol. 2019, 856, 172402. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-Y.; Lee, W.-T.; Cheng, C.-H.; Chen, K.-C.; Chou, C.-M.; Chung, C.-H.; Sun, M.-S.; Cheng, H.-W.; Ho, M.-N.; Lin, C.-W. Repositioning antipsychotic chlorpromazine for treating colorectal cancer by inhibiting sirtuin 1. Oncotarget 2015, 6, 27580–27595. [Google Scholar] [CrossRef] [PubMed]
- Soletti, R.C.; Biasoli, D.; Rodrigues, N.A.L.V.; Delou, J.M.A.; Maciel, R.; Chagas, V.L.A.; Martins, R.A.P.; Rehen, S.K.; Borges, H.L. Inhibition of pRB Pathway Differentially Modulates Apoptosis in Esophageal Cancer Cells. Transl. Oncol. 2017, 10, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Laurent, M.; Brahmi, M.; Dufresne, A.; Meeus, P.; Karanian, M.; Ray-Coquard, I.; Blay, J.-Y. Adjuvant therapy with imatinib in gastrointestinal stromal tumors (GISTs)—review and perspectives. Transl. Gastroenterol. Hepatol. 2019, 4, 24. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Patel, C.J. A standard database for drug repositioning. Sci. Data 2017, 4, 170029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aberle, M.R.; Burkhart, R.A.; Tiriac, H.; Olde Damink, S.W.M.; Dejong, C.H.C.; Tuveson, D.A.; van Dam, R.M. Patient-derived organoid models help define personalized management of gastrointestinal cancer. Br. J. Surg. 2018, 105, e48–e60. [Google Scholar] [CrossRef]
Figure 1.
Targets of approved or investigational pharmacological intervention on signaling pathways associated with resistance mechanisms against antineoplastic agents and their associations with the hallmarks of cancer. The hallmarks of cancer [91] are sustaining proliferative signaling, evasion of growth suppression, avoiding immune destruction, enabling replicative immortality, tumor-promoting inflammation, activation invasion and metastasis, inducing angiogenesis, genome instability and mutation, resisting cell death, and deregulating cellular energetics. The most commonly affected signaling pathways in response to cytotoxic or targeted chemotherapeutic agents related to specific resistance mechanisms are depicted. For more details, please consult the supplementary tables. Adapted from Hanahan and Weinberg (2011) [91].
Figure 1.
Targets of approved or investigational pharmacological intervention on signaling pathways associated with resistance mechanisms against antineoplastic agents and their associations with the hallmarks of cancer. The hallmarks of cancer [91] are sustaining proliferative signaling, evasion of growth suppression, avoiding immune destruction, enabling replicative immortality, tumor-promoting inflammation, activation invasion and metastasis, inducing angiogenesis, genome instability and mutation, resisting cell death, and deregulating cellular energetics. The most commonly affected signaling pathways in response to cytotoxic or targeted chemotherapeutic agents related to specific resistance mechanisms are depicted. For more details, please consult the supplementary tables. Adapted from Hanahan and Weinberg (2011) [91].


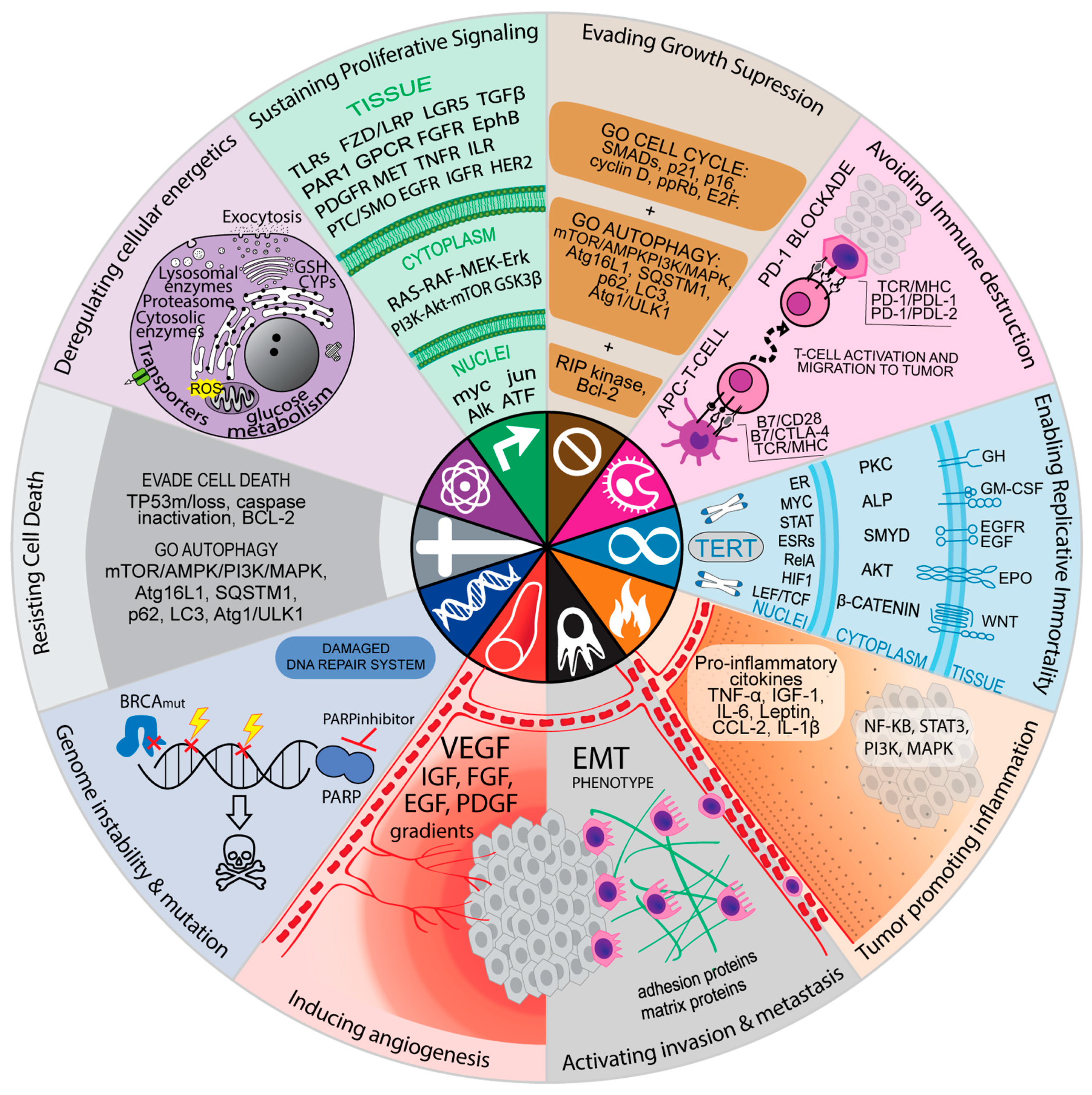{kind=link}
Table 1.
Most common specific resistance mechanisms and signaling pathways associated with cytotoxic and targeted therapies.
Table 1.
Most common specific resistance mechanisms and signaling pathways associated with cytotoxic and targeted therapies.
| Top | Cytotoxic Drugs (N = 59) | Drugs Affected | % | Targeted Drugs (N = 117) | Drugs Affected | % |
|---|---|---|---|---|---|---|
| 1 | ABC transporters | 21 | 36 | MAPK family | 34 | 29 |
| 2 | Enzymatic detoxification | 9 | 17 | PI3K-AKT-mTOR | 33 | 28 |
| 3 | Mutation in and/or downregulation of topoisomerases I/II | 7 | 12 | EGF and EGFR | 21 | 18 |
| 4 | Mutation in and/or overexpression of tubulins | 6 | 10 | PTEN | 14 | 12 |
| 5 | Decreased dCK | 6 | 8 | ABC transporters | 14 | 12 |
| 6 | Increased activity of GST | 5 | 8 | IGFs | 14 | 12 |
| 7 | Activation of NF-κB | 4 | 7 | JAK/STAT | 14 | 12 |
| 8 | Increased MGMT | 4 | 7 | BCL-2 family | 13 | 12 |
| 9 | Increased levels of ALDH1 | 3 | 5 | FGFs | 12 | 11 |
| 10 | Silencing or mutations in TP53 | 3 | 5 | ERBB2 (HER2) | 12 | 11 |
N = FDA approved antineoplastic drugs available as at May 2019. Abbreviations: ABC—ATP-binding cassette, dCK—deoxycytidine kinase, GST—glutathione s-transferase, NF-κB—factor nuclear kappa B, MGMT—O-6-methylguanine-DNA methyltransferase, ALDH1—aldehyde dehydrogenase 1 family, TP53—tumor protein p53, MAPK—mitogen activated protein kinases, PI3K—phosphoinositide 3-kinase, AKT—protein kinase B, mTOR—mammalian target of rapamycin, EGF—epithelial growth factor, EGFR—epithelial growth factor receptor, PTEN—phosphatase and tensin homolog, IGF—insulin-like growth factor 1, JAK—Janus kinase, STAT—signal transducer and activator of transcription, BCL2—B-cell lymphoma 2, FGF—fibroblast growth factor, ERBB2—erb-b2 receptor tyrosine kinase 2, HER2—human epidermal growth factor receptor 2.
Table 2.
Hallmarks of cancer and signaling pathways associated with specific resistance mechanisms.
| Top | Cytotoxic (N = 59) | Drugs Affected | % | Targeted therapies (N = 117) | Drugs Affected | % |
|---|---|---|---|---|---|---|
| 1 | Metabolism (Detoxification transporters and enzymes, protection from ROS, increased glucose metabolism) | 21 | 36 | Sustaining Proliferative Signaling (GF, Hedgehog, MAPK, PI3K, WNT, autophagy induction) | 65 | 56 |
| 2 | Sustaining Proliferative Signaling (GF, Hedgehog, MAPK, PI3K, WNT, autophagy induction) | 19 | 32 | Cell Death Evasion (BCL2 family, TP53, MDM2, PTEN, NF-KB, autophagy induction) | 41 | 35 |
| 3 | Cell Death Evasion (BCL2 family, TP53, MDM2, PTEN, NF-KB, autophagy induction) | 10 | 17 | Angiogenesis (EGF, IGF, FGF, VEGF, PDGF) | 31 | 26 |
| 4 | Genome instability (TP53, MDM2, NHEJ, RAD51, CHEK1/2, BRCA1/2, HDAC) | 5 | 8 | Metabolism (Detoxification transporters and enzymes, protection from ROS, increased glucose metabolism) | 21 | 18 |
| 5 | Evading growth suppressors (TP53, RB, cyclins, CDKs, p16, p18, p21) | 4 | 7 | Genome instability (TP53, MDM2, NHEJ, RAD51, CHEK1/2, BRCA1/2, HDAC, p21) | 18 | 15 |
| 6 | Inflammation (NF-kB) | 4 | 7 | Immune checkpoint (CD19, CD20, CTLA-4, NT5E, PCLP, PD-1, PD-L1) | 16 | 14 |
| 7 | Angiogenesis (EGF, IGF, FGF, VEGF, PDGF) | 2 | 3 | Evading growth suppressors (TP53, RB, cyclins, CDKs, p16, p18, p21) | 13 | 11 |
| 8 | Immune checkpoint (CD19, CD20, CTLA-4, NT5E, PCLP, PD-1, PD-L1) | 0 | 0 | EMT, invasion, and metastasis (EMT phenotype, integrin) | 13 | 11 |
| 9 | EMT, invasion, and metastasis (EMT phenotype, integrin) | 0 | 0 | Inflammation (NF-kB) | 12 | 10 |
| 10 | Replicative immortality (WNT, Hedgehog, TERT) | 0 | 0 | Replicative immortality (WNT, Hedgehog, TERT) | 8 | 7 |
N = FDA approved antineoplastic drugs available as at May 2019. Abbreviations: NF-κB—factor nuclear kappa B, TP53—tumor protein p53, MAPK—mitogen activated protein kinases, PI3K—phosphoinositide 3-kinase, EGF—epithelial growth factor, PTEN—phosphatase and tensin homolog, IGF—insulin-like growth factor 1, JAK—Janus kinase, STAT—signal transducer and activator of transcription, BCL2—B-cell lymphoma 2, FGF—fibroblast growth factor, ERBB2—erb-b2 receptor tyrosine kinase 2, HER2—human epidermal growth factor receptor 2, ROS—reactive oxygen species, GF—growth factor, NHEJ—non-homologous end joining, RAD51—RAD51 recombinase, CHEK1/2—checkpoint kinases 1 and 2, BRCA1/2—breast cancer type 1 susceptibility protein, and 2, HDAC—histone deacetylase, RB—retinoblastoma protein, CDKs—cyclin-dependent kinase, PDGF—platelet-derived growth factor, CTLA-4—cytotoxic T lymphocyte antigen 4, NT5E—ecto-5′-nucleotidase, PCLP—podocalyxin-like protein 1, PD-1—programmed cell death protein 1, PD-L1—programmed death-ligand 1, EMT—epithelial–mesenchymal transition, TERT—telomerase reverse transcriptase.
Table 3.
Clinical trials with combinations in which targeted therapies inhibit resistance mechanisms of cytotoxic drugs.
Table 3.
Clinical trials with combinations in which targeted therapies inhibit resistance mechanisms of cytotoxic drugs.
| Signalling Pathway Affected | Targeted Drug/Inhibitor Drug | Cytotoxic Drugs | Clinical Trial Phase | Indications | Obs | References |
|---|---|---|---|---|---|---|
| BCR-ABL | Bosutinib | Pemetrexed | Ongoing phase I | Bladder, cervical, NSCLC, ovarian | Recruiting | [109] |
| Dasatinib | Carboplatin + Paclitaxel | Phase I completed | Ovarian | Recommended for phase II | [110] | |
| BCL-2 | Venetoclax | Azacitidine | Ongoing phase II | AML | Elderly patients | [111] |
| Cyclophosphamide, etoposide, doxorubicin, methotrexate, 6-mercaptopurine, cytarabine | ALL | Ongoing phase II | Older patients with relapsed or refractory ALL | [112] | ||
| CDK4/6 | Abemaciclib | Pemetrexed, or gemcitabine | Ongoing phase I | NSCLC | For stage IV patients | [113] |
| Temozolomide | Ongoing phase II | Glioblastoma | [114] | |||
| Palbociclib | Carboplatin | Ongoing phase II | Metastatic head and neck squamous cell carcinoma | [115] | ||
| Nab-paclitaxel | Phase I completed | mPDAC | No results posted; last update in May 30, 2019. | [116] | ||
| Temozolomide + irinotecan | Ongoing phase I | Solid tumors, neuroblastoma, medulloblastoma | For children, adolescents and young adults | [117] | ||
| Ribociclib | Docetaxel + Prednisone | Ongoing phase Ib/II | mCRPC | [118] | ||
| Gemcitabine | Ongoing phase I | Malignant brain tumors | [119] | |||
| Paclitaxel + Carboplatin | Ongoing phase I | Ovarian cancer, fallopian tube cancer, peritoneal carcinoma | [120] | |||
| DNA DAMAGE REPAIR | Niraparib | Temozolomide | Ongoing phase Ib/II | SCLC | [121] | |
| Olaparib | Cisplatin | Ongoing phase I | Advanced NSCLC | [122] | ||
| Rucaparib | Irinotecan | Ongoing phase Ib | Solid tumors | For patients with DNA repair defects in solid tumors | [123] | |
| Veliparib | FOLFIRI | Ongoing phase II | Pancreatic (metastatic) | Second line therapy | [124] | |
| Phase I completed | Gastric | Recommended for further investigation | [124] | |||
| Irinotecan | Ongoing phase I | Breast, lung, ovarian, pancreatic, Hodgkin’s lymphoma | For cancer that is metastatic or cannot be removed | [125] | ||
| Temozolomide | Ongoing phase I | ALL | [126] | |||
| Phase II completed | CRC | Recommended for further investigation | [127] | |||
| Topotecan | Ongoing phase I | Acute leukemias | [128] | |||
| EGFR | Cetuximab | FOLFIRI | Ongoing phase II | CRC | For patients with FcγRIIIa polymorphism and wild-type KRAS, NRAS and BRAF | [129] |
| FOLFOXIRI | Ongoing phase II | Locally advanced rectal carcinoma | For EGFR wild type patients | [130] | ||
| Necitumumab | Gemcitabine + Cisplatin | Ongoing phase II | Stage IB, II or IIIA squamous NSCLC | Neoadjuvant therapy | [131] | |
| Panitumumab | FOLFOX/FOLFIRI | Phase II completed | Liver (metastatic) | For patients with wild-type KRAS; no results posted; last update May 14, 2019. | [132] | |
| mFOLFOX6 + bevacizumab/panitumumab | Ongoing phase III | Advanced/recurrent CRC | First-line therapy for patients with KRAS/NRAS wild-type tumors | [133] | ||
| Pertuzumab | Paclitaxel + Trastuzumab | Ongoing phase I | HER2-positive breast cancer | [134] | ||
| PI3K-AKT-MTOR | Copanlisib | Gemcitabine | Phase I completed | Cholangiocarcinoma | No results yet. | [135] |
| Duvelisib | Fludarabine + cyclophosphamide + rituximab (FCR) | Ongoing phase Ib/II | CLL | [136] | ||
| Everolimus | Carboplatin + Paclitaxel | Phase II completed | Melanoma | Everolimus failed to improve efficacy | [137] | |
| Temozolomide | Ongoing phase II | Low-grade glioma | [138] | |||
| Idelalisib | Bendamustine + Rituximab | Phase III completed | Relapsed or refractory CLL | Improved PFS but with serious adverse events and infections | [139] | |
| Temsirolimus | Carboplatin + Paclitaxel | Phase II completed | Recurrent or metastatic head and neck | Recommended further investigation for PI3K/mTOR mutations | [140] | |
| VEGF | Bevacizumab | Capecitabine | Phase II completed | Advanced or Metastatic Liver Cancer | "All patients presented serious adverse events; only 9.1% presented objective response" | [141] |
| Carboplatin + Paclitaxel (CPB) | Phase II completed | Melanoma | Recommended for phase III | [142] | ||
| Carboplatin + Paclitaxel + Everolimus (CPBE) | Phase II completed | Melanoma | Failed to improve PFS compared to CPB | |||
| FOLFIRI (+ Onvansertib) | Ongoing phase Ib/II | mCRC | Second line therapy for patients with KRAS mutation | [143] | ||
| Temozolomide | Phase II completed | Glioma (grade II/III) | Failed to improve 1-year OS | [144] | ||
| Lenvatinib | Paclitaxel | Ongoing phase I | Endometrial, ovarian, fallopian tube, or primary peritoneal cancer | [145] | ||
| Ramucirumab | FOLFIRI | Ongoing phase II | Gastric | For previous failed therapy | [146] | |
| Regorafenib | Irinotecan | Ongoing phase II | Metastatic gastro-esophageal adenocarcinomas | Second line therapy | [147] | |
| Sorafenib | Irinotecan | Ongoing phase II | Pediatric solid tumors | Patients with mutations in Raf, PDGFR, VEGFR, Flt-3, KIT, JAK, STAT, RAS, MEK, or ERK | [148] |
Abbreviations: OS—overall survival, FOLFIRI—folinic acid, 5-fluorouracil and irinotecan, FOLFIRINOX—folinic acid, 5-fluorouracil, irinotecan and oxaliplatin, CRC—colorectal cancer, mCRC—metastatic colorectal cancer, NSCLC—non-small cell lung cancer, SCLC—small cell lung cancer, ALL—acute lymphoblastic leukemia.
Table 4.
Clinical trials with combinations between targeted therapies in which the first inhibit resistance mechanisms of the second.
Table 4.
Clinical trials with combinations between targeted therapies in which the first inhibit resistance mechanisms of the second.
| Signalling Pathways | Targeted Therapy | Clinical Trial Phase | Type of Tumor | Rationale | References |
|---|---|---|---|---|---|
| ALK AND CDK4/6 | Ceritinib + Ribociclib | Phase I completed | NSCLC | ALK+ NSCLC tumors | [149] |
| Ongoing phase I | Neuroblastoma | In vitro synergy (lower phospho-RB1 levels only in ALK+ NB cells) | [150,151] | ||
| MET AND AR | Crizotinib + Enzalutamide | Ongoing phase I | mCRPC | AR inhibition upregulates MET (off-target effect for crizotinib) | [152,153] |
| PDGFR AND C-SRC | Crizotinib + Dasatinib | Ongoing phase I | Solid malignancies | Downstream effects of MET require c-Src, whose inhibition upregulates MET | [154,155] |
| Phase I completed | High-grade glioma | PDGFR upregulated in gliomas (off-target for dasatinib) | [156] | ||
| PI3K AND EGFR | Copanlisib + Cetuximab | Ongoing phase Ib/II | HNSCC | Aberrant PI3K signaling confers resistance to cetuximab and both pathways are upregulated in HNSCC | [157,158] |
| VEGFR AND MTOR | Pazopanib + Everolimus | Ongoing phase I | Solid tumors | mTOR pathway activation confers resistance to anti-VEGF therapy | [159] |
| BCR | Idelalisib + Entospletinib | Phase II completed | Lymphoid malignancies | Synergistic effect with simultaneous inhibition of multiple kinases in the BCR pathway (PI3K and Syk), but limited by severe life-threatening adverse effects. | [160] |
| CDK4/6 AND PI3K/MTOR | Abemaciclib + LY3023414 | Ongoing phase II | PDAC | Enhanced PI3K/mTOR activity confers resistance to CDKi therapy | [161] |
| Ribociclib + Everolimus | Ongoing phase II | mPDAC | [105] | ||
| CDK4/6 AND EGFR | Palbociclib + Cetuximab | Ongoing phase II | HNSCC | EGFR overexpression is an oncogenic driver and there is either a frequent loss of CDKN2A or amplification of CCND1 | [162] |
| CDK4/6 AND MEK | Palbociclib + Trametinib | Phase Ib completed | Advanced solid malignancies | CDK activity confers resistance to MEKi | [163,164] |
| BRAF AND MEK | Dabrafenib + Trametinib | Ongoing phase II | Melanoma and brain metastases | MAPK pathway reactivation confers resistance to BRAFi. Melanoma brain metastasis seem to lack ABCB1 expression. Combination can cross blood-brain barrier. | [165] |
| Vemurafenib + Cobimetinib | Ongoing phase II | [166] | |||
| VEGFR AND EGFR | Bevacizumab + Erlotinib | Phase II completed | Advanced or Metastatic Liver Cancer | Dual blockade of tumor neovascularization and proliferative signaling. | [167] |
Abbreviations: HNSCC - Head and neck squamous cell carcinoma, mCRPC - metastatic castration-resistant prostate cancer, mPDAC - metastatic pancreatic duct adenocarcinoma, NSCLC - non-small cell lung cancer, PDAC - Pancreatic duct adenocarcinoma.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Delou, J.M.A.; Souza, A.S.O.; Souza, L.C.M.; Borges, H.L. Highlights in Resistance Mechanism Pathways for Combination Therapy. Cells 2019, 8, 1013. https://doi.org/10.3390/cells8091013
AMA Style
Delou JMA, Souza ASO, Souza LCM, Borges HL. Highlights in Resistance Mechanism Pathways for Combination Therapy. Cells. 2019; 8(9):1013. https://doi.org/10.3390/cells8091013
Chicago/Turabian StyleDelou, João M. A., Alana S. O. Souza, Leonel C. M. Souza, and Helena L. Borges. 2019. "Highlights in Resistance Mechanism Pathways for Combination Therapy" Cells 8, no. 9: 1013. https://doi.org/10.3390/cells8091013
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.