Mitochondrial Involvement in the Adaptive Response to Chronic Exposure to Environmental Pollutants and High-Fat Feeding in a Rat Liver and Testis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
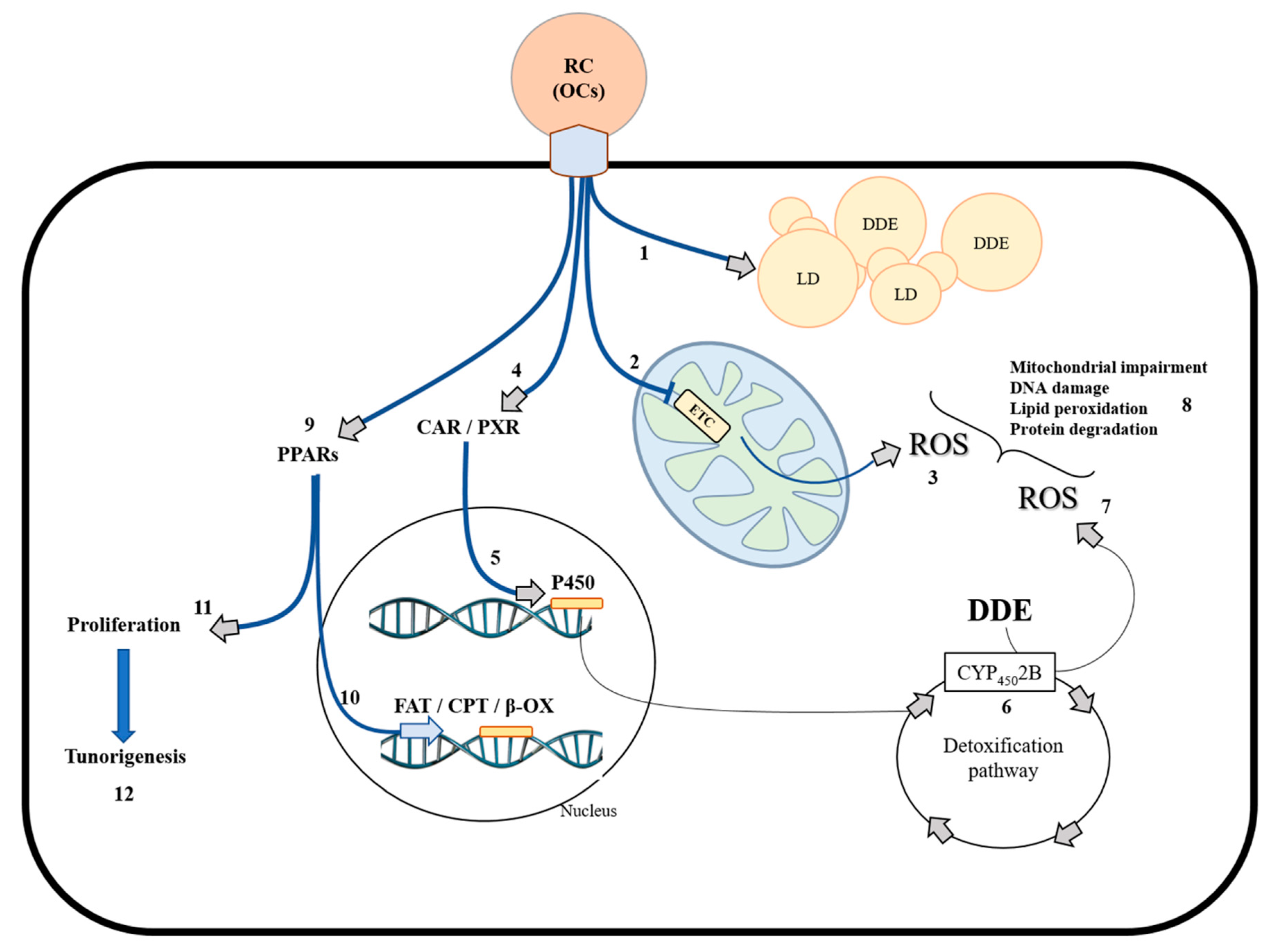
2. Mitochondria and Oxidative Stress Generation in Biological Systems
3. Physiological Adaption to HFD and DDE in the Liver
3.1. HFD Induces Hepatic Fat Accumulation and Cellular Stress Targeting Mitochondria
3.2. DDE Exposure: Hepatic Mechanisms Used to Counteract Liver Damage
3.3. Dietary Fats and DDE Showed No Synergic Effect in Hepatic Stress Generation and Damage
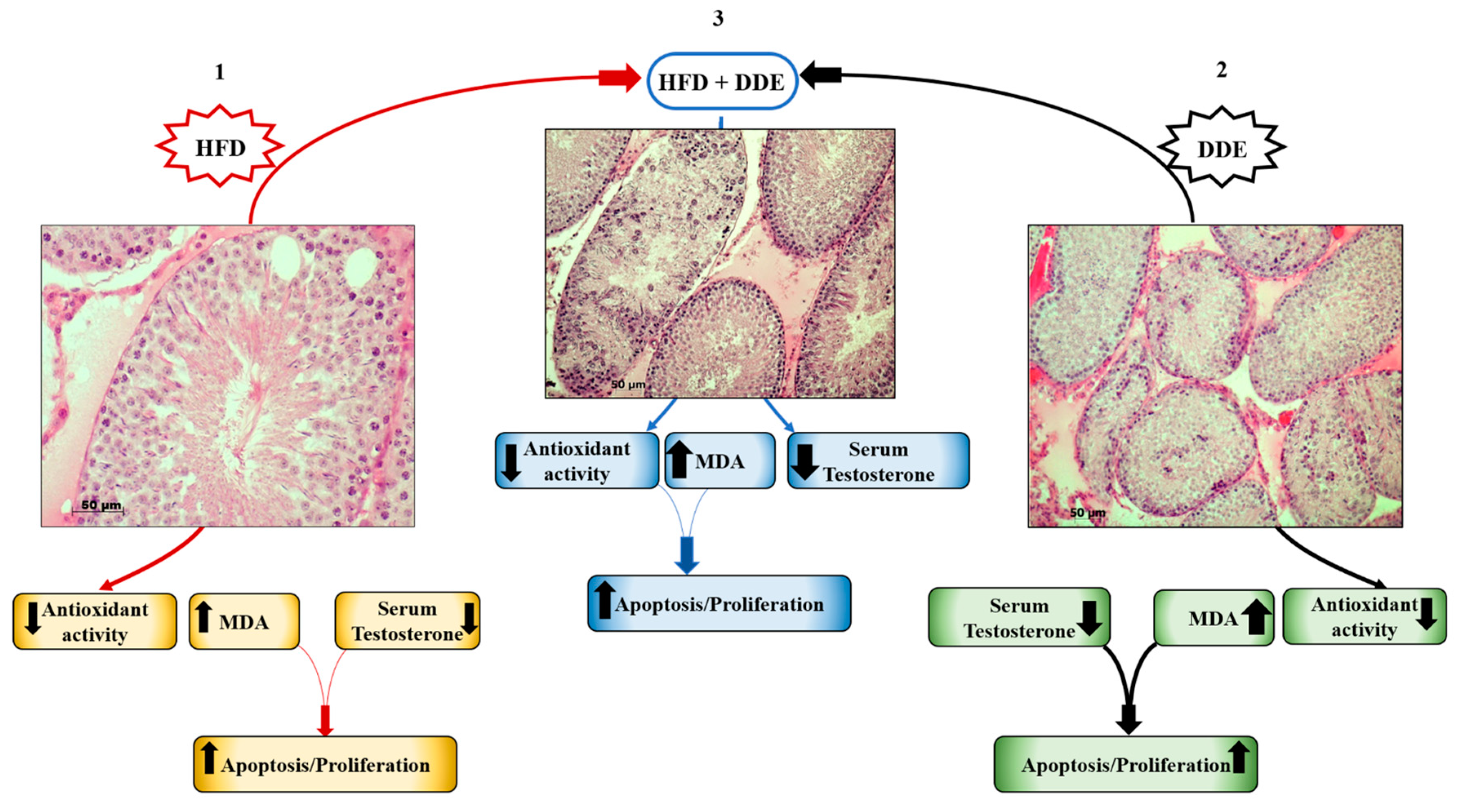
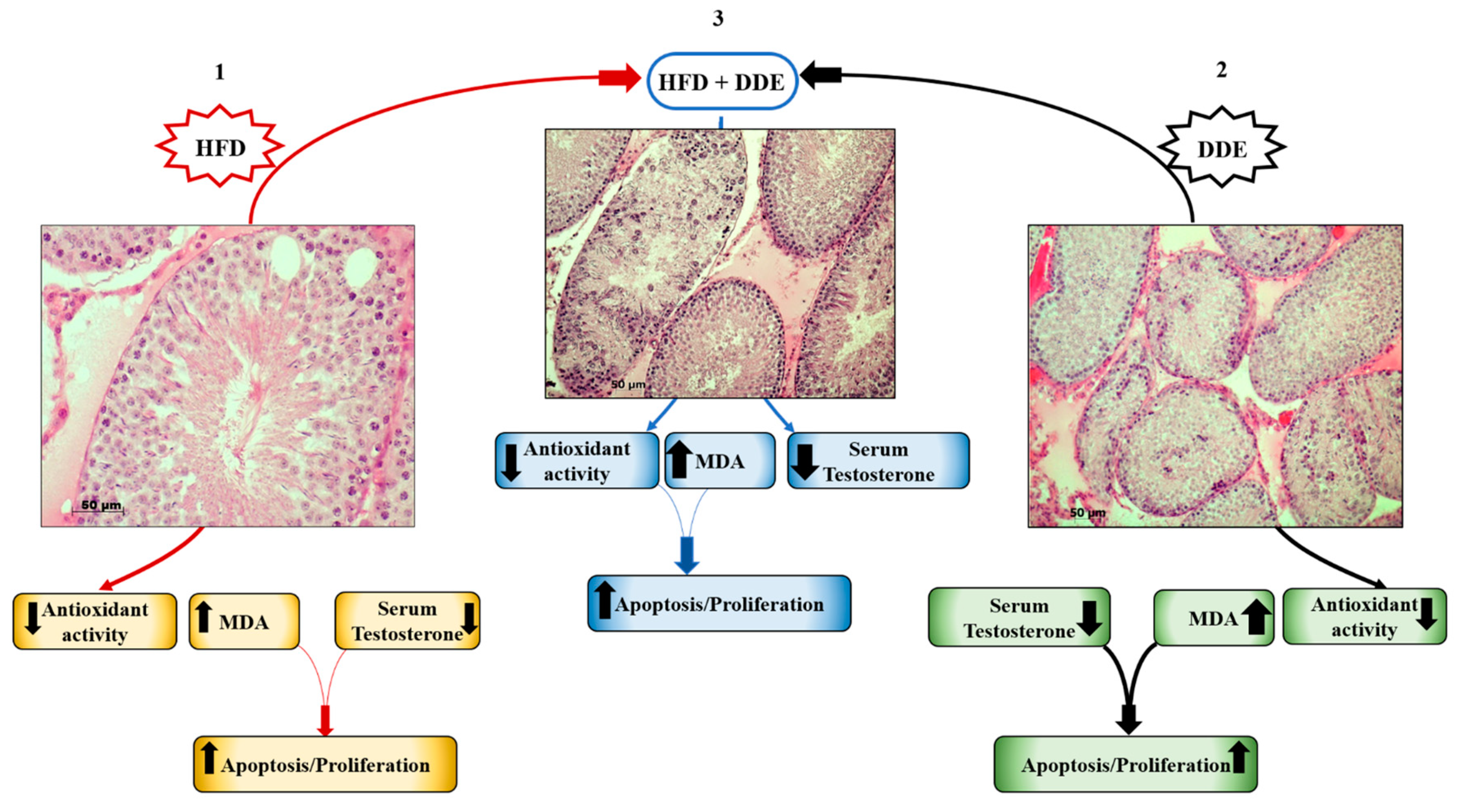
4. Physiological Adaption to HFD and DDE in the Testis
4.1. HFD Alters Testicular Function and Affects Hormonal Homeostasis
4.2. DDE Negatively Affects Spermatogenesis and Testicular Function
4.3. Simultaneous Exposure to HFD + DDE did not Show a Synergic Effect on Testicular Function
5. Conclusions
Funding
Conflicts of Interest
References
- Kannan, K.; Tanabe, S.; Williams, R.J.; Tatsukawa, R. Persistent organochlorine residues in foodstuffs from Australia, Papua New Guinea and the Solomon Islands: Contamination levels and human dietary exposure. Sci. Total Environ. 1994, 153, 29–49. [Google Scholar] [CrossRef]
- Grimalt, J.O.; van Drooge, B.L.; Ribes, A.; Vilanova, R.M.; Fernandez, P.; Appleby, P. Persistent organochlorine compounds in soils and sediments of European high altitude mountain lakes. Chemosphere 2004, 54, 1549–1561. [Google Scholar] [CrossRef] [PubMed]
- Carrera, G.; Fernández, P.; Vilanova, R.M.; Grimalt, J.O. Persistent organic pollutants in snow from European high mountain areas. Sci. Total Environ. 1994, 153, 29–49. [Google Scholar] [CrossRef]
- Bishopp, F.C. Insect Problems in World War II with Special References to the Insecticide DDT. Am. J. Public Health Nations Health 1945, 35, 373–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davison, K.L.; Sell, J.L. DDT thins shells of eggs from mallard ducks maintained on ad libitum or controlled-feeding regimens. Arch. Environ. Contam. Toxicol. 1974, 2, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Speich, S.M.; Calambokidas, J.; Shea, D.W.; Peard, J.; Witter, M.; Fry, D.M. Eggshell Thinning and Organochlorine Contaminants in Western Washington Waterbirds. Colon. Waterbirds 1992, 15, 103–112. [Google Scholar] [CrossRef]
- Fry, D.M. Reproductive effects in birds exposed to pesticides and industrial chemicals. Environ. Health Perspect. 1995, 103, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Ware, G.W. Effects of DDT on reproduction in higher animals. Residue Rev. 1975, 59, 119–140. [Google Scholar] [PubMed]
- Rehwagen, R. WHO recommends DDT to control malaria. BMJ 2006, 333, 622. [Google Scholar] [CrossRef] [PubMed]
- Wania, F.; Mackay, D. Tracking the Distribution of Persistent Organic Pollutants Control strategies for these contaminants will require a better understanding of how they move around the globe. Environ. Sci. Technol. 1996, 30, 390A–396A. [Google Scholar] [CrossRef] [PubMed]
- Streit, B. Bioaccumulation processes in ecosystems. Cell. Mol. Life Sci. 1992, 48, 955–970. [Google Scholar] [CrossRef] [PubMed]
- Kelce, W.R.; Stone, C.R.; Laws, S.C.; Gray, L.E.; Kemppainen, J.A.; Wilson, E.M. Persistent DDT metabolite p,p’–DDE is a potent androgen receptor antagonist. Nature 1995, 375, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Patisaul, H.B.; Adewale, H.B. Long-Term Effects of Environmental Endocrine Disruptors on Reproductive Physiology and Behavior. Front. Behav. Neurosci. 2009, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 2017, 68, 3–33. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liang, X.; Hu, Y.; Wang, Y.; Yu, H.; Yang, K. p,p′-DDE induces mitochondria-mediated apoptosis of cultured rat Sertoli cells. Toxicology 2008, 253, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Marouani, N.; Hallegue, D.; Sakly, M.; Benkhalifa, M.; Rhouma, K.B.; Tebourbi, O. p,p’-DDT induces testicular oxidative stress-induced apoptosis in adult rats. Reprod. Biol. Endocrinol. 2017, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Hariri, N.; Thibault, L. High-fat diet-induced obesity in animal models. Nutr. Res. Rev. 2010, 23, 270–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Q.; Yeung, S.C.; Ip, M.S.M.; Mak, J.C.W. Dysregulation of cardiac lipid parameters in high-fat high-cholesterol diet-induced rat model. Lipids Health Dis. 2018, 17, 255. [Google Scholar] [CrossRef] [PubMed]
- Mollica, M.P.; Lionetti, L.; Putti, R.; Cavaliere, G.; Gaita, M.; Barletta, A. From chronic overfeeding to hepatic injury: Role of endoplasmic reticulum stress and inflammation. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, L.; Mollica, M.P.; Lombardi, A.; Cavaliere, G.; Gifuni, G.; Barletta, A. From chronic overnutrition to insulin resistance: The role of fat-storing capacity and inflammation. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Pataky, M.W.; Wang, H.; Yu, C.S.; Arias, E.B.; Ploutz-Snyder, R.J.; Zheng, X.; Cartee, G.D. High-Fat Diet-Induced Insulin Resistance in Single Skeletal Muscle Fibers is Fiber Type Selective. Sci. Rep. 2017, 7, 13642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Patil, I.Y.; Jiang, T.; Sancheti, H.; Walsh, J.P.; Stiles, B.L.; Yin, F.; Cadenas, E. High-fat diet induces hepatic insulin resistance and impairment of synaptic plasticity. PLoS ONE 2015, 10, e0128274. [Google Scholar] [CrossRef] [PubMed]
- Soltis, A.R.; Kennedy, N.J.; Xin, X.; Zhou, F.; Ficarro, S.B.; Yap, Y.S.; Matthews, B.J.; Lauffenburger, D.A.; White, F.M.; Marto, J.A.; et al. Hepatic Dysfunction Caused by Consumption of a High-Fat Diet. Cell Rep. 2017, 21, 3317–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koek, G.H.; Liedorp, P.R.; Bast, A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin. Chim. Acta 2011, 412, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 8713–8742. [Google Scholar] [CrossRef] [PubMed]
- Iossa, S.; Lionetti, L.; Mollica, M.P.; Crescenzo, R.; Botta, M.; Barletta, A.; Liverini, G. Effect of high-fat feeding on metabolic efficiency and mitochondrial oxidative capacity in adult rats. Br. J. Nutr. 2003, 90, 953–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lionetti, L.; Mollica, M.P.; Crescenzo, R.; D’Andrea, E.; Ferraro, M.; Bianco, F.; Liverini, G.; Iossa, S. Skeletal muscle subsarcolemmal mitochondrial dysfunction in high-fat fed rats exhibiting impaired glucose homeostasis. Int. J. Obes. (Lond.) 2007, 31, 1596–1604. [Google Scholar] [CrossRef] [Green Version]
- Iossa, S.; Mollica, M.P.; Lionetti, L.; Crescenzo, R.; Botta, M.; Liverini, G. Skeletal muscle oxidative capacity in rats fed high-fat diet. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lionetti, L.; Mollica, M.P.; Donizzetti, I.; Gifuni, G.; Sica, R.; Pignalosa, A.; Cavaliere, G.; Gaita, M.; De Filippo, C.; Zorzano, A.; et al. High-lard and high-fish-oil diets differ in their effects on function and dynamic behaviour of rat hepatic mitochondria. PLoS ONE 2014, 9, e92753. [Google Scholar] [CrossRef] [PubMed]
- Putti, R.; Sica, R.; Migliaccio, V.; Lionetti, L. Diet impact on mitochondrial bioenergetics and dynamics. Front. Physiol. 2015, 6, 109. [Google Scholar] [CrossRef]
- Matsuzawa-Nagata, N.; Takamura, T.; Ando, H.; Nakamura, S.; Kurita, S.; Misu, H.; Ota, T.; Yokoyama, M.; Honda, M.; Miyamoto, K.; et al. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism 2008, 57, 1071–1077. [Google Scholar] [CrossRef]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Migliaccio, V.; Sica, R.; Scudiero, R.; Simoniello, P.; Putti, R.; Lionetti, L. Physiological Adaptation to Simultaneous Chronic Exposure to High-Fat Diet and Dichlorodipheniletylhene (DDE) in Wistar Rat Testis. Cells 2019, 8, 443. [Google Scholar] [CrossRef]
- Migliaccio, V.; Scudiero, R.; Sica, R.; Lionetti, L.; Putti, R. Oxidative stress and mitochondrial uncoupling protein 2 expression in hepatic steatosis induced by exposure to xenobiotic DDE and high fat diet in male Wistar rats. PLoS ONE 2019, 14, e0215955. [Google Scholar] [CrossRef]
- Yeung, B.H.; Wan, H.T.; Law, A.Y.; Wong, C.K. Endocrine disrupting chemicals: Multiple effects on testicular signaling and spermatogenesis. Spermatogenesis 2011, 1, 231–239. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Grivell, L.A. Nucleo-mitochondrial interactions in mitochondrial gene expression. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 121–164. [Google Scholar] [CrossRef]
- Poyton, R.O.; McEwen, J.E. Crosstalk between nuclear and mitochondrial genomes. Annu. Rev. Biochem. 1996, 65, 563–607. [Google Scholar] [CrossRef]
- Fakouri, N.B.; Hansen, T.L.; Desler, C.; Anugula, S.; Rasmussen, L.J. From Powerhouse to Perpetrator-Mitochondria in Health and Disease. Biology 2019, 8, 35. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Moghaddas, S.; Tandler, B.; Kerner, J.; Hoppel, C.L. Mitochondrial dysfunction in cardiac disease: Ischemia–reperfusion, aging, and heart failure. J. Mol. Cell. Cardiol. 2001, 33, 1065–1089. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial defects in cardiomyopathy and neuromuscular disease. Am. Heart J. (Suppl.) 2000, 139, 70–85. [Google Scholar] [CrossRef]
- Barja, G. Mitochondrial oxygen radical generation and leak: Sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J. Bioenerg. Biomembr. 1999, 31, 347–366. [Google Scholar] [CrossRef]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef]
- Bleier, L.; Dröse, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Yang, W.; Hekimi, S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010, 8, e1000556. [Google Scholar] [CrossRef]
- Lee, S.J.; Hwang, A.B.; Kenyon, C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 2010, 20, 2131–2136. [Google Scholar] [CrossRef]
- Schaar, C.E.; Dues, D.J.; Spielbauer, K.K.; Machiela, E.; Cooper, J.F.; Senchuk, M.; Hekimi, S.; Van Raamsdonk, J.M. Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 2015, 11, e1004972. [Google Scholar] [CrossRef]
- Hrycay, E.G.; Bandiera, S.M. Involvement of Cytochrome P450 in Reactive Oxygen Species Formation and Cancer. Adv. Pharmacol. 2015, 74, 35–84. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Day, R.M.; Suzuki, Y.J. Cell proliferation, reactive oxygen and cellular glutathione. Dose Response 2006, 3, 425–442. [Google Scholar] [CrossRef]
- Roberts, C.K.; Sindhu, K.K. Oxidative stress and metabolic syndrome. Life Sci. 2009, 84, 705–712. [Google Scholar] [CrossRef]
- Mahjoub, S.; Masrour-Roudsari, J. Role of oxidative stress in pathogenesis of metabolic syndrome. Casp. J. Intern. Med. 2012, 3, 386–396. [Google Scholar]
- Renaud, H.J.; Rutter, A.; Winn, L.M. Assessment of xenobiotic biotransformation including reactive oxygen species generation in the embryo using benzene as an example. Methods Mol. Biol. 2012, 889, 253–263. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Klotz, L.O.; Steinbrenner, H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biol. 2017, 13, 646–654. [Google Scholar] [CrossRef]
- Kesh, S.B.; Sarkar, D.; Manna, K. High-fat diet-induced oxidative stress and its impact on metabolic syndrome: A review. Asian J. Pharm. Clin. Res. 2016, 9, 47–52. [Google Scholar]
- Terra, L.F.; Lobba, A.R. High-fat diet and fish oil affect adipocyte metabolism in a depot-specific manner. J. Physiol. 2017, 595, 1859–1860. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Yang, Y.; Hu, Y.; Sun, Y.; Zhang, S.; Peng, W.; Zhong, Y.; Huang, X.; Kong, W. A long-term high-fat diet increases oxidative stress, mitochondrial damage and apoptosis in the inner ear of D-galactose-induced aging rats. Hear Res. 2012, 287, 15–24. [Google Scholar] [CrossRef]
- Migliaccio, V.; Sica, R.; Di Gregorio, I.; Putti, R.; Lionetti, L. High-Fish Oil and High-Lard Diets Differently Affect Testicular Antioxidant Defense and Mitochondrial Fusion/Fission Balance in Male Wistar Rats: Potential Protective Effect of ω3 Polyunsaturated Fatty Acids Targeting Mitochondria Dynamics. Int. J. Mol. Sci. 2019, 20, 3110. [Google Scholar] [CrossRef]
- Klop, B.; Elte, J.W.; Cabezas, M.C. Dyslipidemia in Obesity: Mechanisms and Potential Targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, L.; Mollica, M.P.; Sica, R.; Donizzetti, I.; Gifuni, G.; Pignalosa, A.; Cavaliere, G.; Putti, R. Differential effects of high-fish oil and high-lard diets on cells and cytokines involved in the inflammatory process in rat insulin-sensitive tissues. Int. J. Mol. Sci. 2014, 15, 3040–3063. [Google Scholar] [CrossRef]
- Shimobayashi, M.; Albert, V.; Woelnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C.; Clement, N.; Moes, S.; Colombi, M.; Meier, J.A.; et al. Insulin resistance causes inflammation in adipose tissue. J. Clin. Investig. 2018, 128, 1538–1550. [Google Scholar] [CrossRef]
- Jeukendrup, A.E. Modulation of carbohydrate and fat utilization by diet, exercise and environment. Biochem. Soc. Trans. 2003, 31, 1270–1273. [Google Scholar] [CrossRef] [Green Version]
- Fisher, E.C.; Evans, W.J.; Phinney, S.D.; Blackburn, G.L.; Bistrian, B.R.; Young, V.R. Changes in skeletal muscle metabolism induced by a eucaloric ketogenic diet. In Biochemistry of Exercise; Knuttgen, H.G., Vogel, J.A., Poortmans, J., Eds.; Human Kinetics Publishers Inc.: Champaign, IL, USA, 1983; pp. 497–507. [Google Scholar]
- Kiens, B.; Helge, J.W. Effect of high-fat diets on exercise performance. Proc. Nutr. Soc. 1998, 57, 73–75. [Google Scholar] [CrossRef] [Green Version]
- Samuel, V.T.; Liu, Z.X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef]
- Szalowska, E.; Tesfay, H.A.; van Hijum, S.A.; Kersten, S. Transcriptomic signatures of peroxisome proliferator-activated receptor alpha (PPARalpha) in different mouse liver models identify novel aspects of its biology. BMC Genom. 2014, 15, 1106. [Google Scholar] [CrossRef]
- Rakhshandehroo, M.; Knoch, B.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 2010, 612089. [Google Scholar] [CrossRef]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef]
- Cotter, D.G.; Ercal, B.; Huang, X.; Leid, J.M.; d’Avignon, D.A.; Graham, M.J.; Dietzen, D.J.; Brunt, E.M.; Patti, G.J.; Crawford, P.A. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Investig. 2014, 124, 5175–5190. [Google Scholar] [CrossRef] [Green Version]
- Liss, K.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef]
- Villarroya, F.; Iglesias, R.; Giralt, M. PPARs in the Control of Uncoupling Proteins Gene Expression. PPAR Res. 2007, 2007, 74364. [Google Scholar] [CrossRef]
- Nakatani, T.; Tsuboyama-Kasaoka, N.; Takahashi, M.; Miura, S.; Ezaki, O. Mechanism for Peroxisome Proliferator-activated Receptor-Activator-induced Up-regulation of UCP2 mRNA in Rodent Hepatocytes. J. Biol. Chem. 2002, 277, 9562–9569. [Google Scholar] [CrossRef]
- Auestad, N.; Korsak, R.A.; Morrow, J.W.; Edmond, J. Fatty acid oxidation and ketogenesis by astrocytes in primary culture. J. Neurochem. 1991, 56, 1376–1386. [Google Scholar] [CrossRef]
- Yamasaki, M.; Hasegawa, S.; Imai, M.; Takahashi, N.; Fukui, T. High-fat diet-induced obesity stimulates ketone body utilization in osteoclasts of the mouse bone. Biochem. Biophys. Res. Commun. 2016, 473, 654–661. [Google Scholar] [CrossRef] [Green Version]
- Sunny, N.E.; Satapati, S.; Fu, X.; He, T.; Mehdibeigi, R.; Spring-Robinson, C.; Duarte, J.; Potthoff, M.J.; Browning, J.D.; Burgess, S.C. Progressive adaptation of hepatic ketogenesis in mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1226–E1235. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial Oxidative Stress and Antioxidants Balance in Fatty Liver Disease. Hepatol. Commun. 2018, 2, 1425–1439. [Google Scholar] [CrossRef] [Green Version]
- Mollica, M.P.; Lionetti, L.; Moreno, M.; Lombardi, A.; De Lange, P.; Antonelli, A.; Lanni, A.; Cavaliere, G.; Barletta, A.; Goglia, F. 3,5-diiodo-l-thyronine, by modulating mitochondrial functions, reverses hepatic fat accumulation in rats fed a high-fat diet. J. Hepatol. 2009, 51, 363–370. [Google Scholar] [CrossRef]
- Iossa, S.; Lionetti, L.; Mollica, M.P.; Crescenzo, R.; Barletta, A.; Liverini, G. Effect of long-term high-fat feeding on energy balance and liver oxidative activity in rats. Br. J. Nutr. 2000, 84, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, L.; Iossa, S.; Brand, M.D.; Liverini, G. Relationship between membrane potential and respiration rate in isolated liver mitochondria from rats fed an energy dense diet. Mol. Cell. Biochem. 1996, 158, 133–138. [Google Scholar]
- Migliaccio, V.; Lionetti, L.; Putti, R.; Sica, R.; Scudiero, R. Combined effects of DDE and hyperlipidic diet on metallothionein expression and synthesis in rat tissues. Environ. Toxicol. 2019, 34, 283–293. [Google Scholar] [CrossRef]
- Viarengo, A.; Burlando, B.; Ceratto, N.; Panfoli, I. Antioxidant role of metallothioneins: A comparative overview. Cell. Mol. Biol. (Noisy-le-Grand) 2000, 46, 407–417. [Google Scholar]
- Takahashi, Y.; Ogra, Y.; Suzuki, K.T. Nuclear trafficking of metallothionein requires oxidation of a cytosolic partner. J. Cell. Physiol. 2005, 202, 563–569. [Google Scholar] [CrossRef]
- Chubatsu, L.S.; Meneghini, R. Metallothionein protects DNA from oxidative damage. Biochem. J. 1993, 291, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Vukovic, V.; Pheng, S.R.; Stewart, A.; Vik, C.H.; Hedley, D.W. Protection from radiation-induced DNA single-strand breaks by induction of nuclear Metallothionein. Int. J. Radiat. Biol. 2000, 76, 757–762. [Google Scholar] [CrossRef]
- Abel, J.; de Ruiter, N. Inhibition of hydroxyl-radical-generated DNA degradation by metallothionein. Toxicol. Lett. 1989, 47, 191–196. [Google Scholar] [CrossRef]
- Chiaverini, N.; De Ley, M. Protective effect of metallothionein on oxidative stress-induced DNA damage. Free Radic. Res. 2010, 44, 605–613. [Google Scholar] [CrossRef]
- Ledesma, A.; de Lacoba, M.G.; Rial, E. The mitochondrial uncoupling proteins. Genome Biol. Prot. Fam. Rev. 2002, 3, 1–9. [Google Scholar]
- Boss, O.; Samec, S.; Paoloni-Giacobino, A.; Rossier, C.; Dulloo, A.; Seydoux, J.; Muzzin, P.; Giacobino, J.P. Uncoupling protein-3: A new member of the mitochondrial carrier family with tissue specific expression. FEBS Lett. 1997, 408, 39–42. [Google Scholar] [CrossRef]
- Ramsden, D.B.; Ho, P.W.L.; Ho, J.W.M.; Liu, H.F.; So, D.H.F.; Tse, H.M.; Chan, K.H.; Ho, S.L. Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): Structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain Behav. 2012, 2, 468–478. [Google Scholar] [CrossRef]
- Sreedhar, A.; Zhao, Y. Uncoupling Protein 2 and Metabolic Diseases. Mitochondrion 2017, 34, 135–140. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Hirtz, C.; Carrera, G.; Cazenave, R.; Troly, M.; Salvayre, R.; Pénicaud, L.; Casteilla, L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J. 1999, 11, 809–815. [Google Scholar] [CrossRef]
- Horimoto, M.; Fulop, P.; Derdak, Z.; Resnick, M.; Wands, J.R.; Baffy, G. Uncoupling protein-2 deficiency promotes oxidant stress and delays liver regeneration in mice. Hepatology 2004, 39, 386–392. [Google Scholar] [CrossRef]
- Wahlang, B.; Beier, J.I.; Clair, H.B.; Bellis-Jones, H.J.; Falkner, K.C.; McClain, C.J.; Cave, M.C. Toxicant-associated steatohepatitis. Toxicol. Pathol. 2013, 41, 343–360. [Google Scholar] [CrossRef]
- Schwingel, P.A.; Cotrim, H.P.; Salles, B.R.; Almeida, C.E.; dos Santos, C.R., Jr.; Nachef, B.; Andrade, A.R.; Zoppi, C.C. Anabolic-androgenic steroids: A possible new risk factor of toxicant-associated fatty liver disease. Liver Int. 2011, 31, 348–353. [Google Scholar] [CrossRef]
- Le Magueresse-Battistoni, B.; Vidal, H.; Naville, D. Environmental Pollutants and Metabolic Disorders: The Multi-Exposure Scenario of Life. Front. Endocrinol. (Lausanne) 2018, 9, 582. [Google Scholar] [CrossRef] [Green Version]
- Kourouma, A.; Quan, C.; Duan, P.; Qi, S.; Yu, T.; Wang, Y.; Yang, K. Bisphenol A Induces Apoptosis in Liver Cells through Induction of ROS. Adv. Toxicol. 2015, 2015, 901983. [Google Scholar] [CrossRef]
- Yavaşoğlu, N.Ü.; Köksal, C.; Dağdeviren, M.; Aktuğ, H.; Yavaşoğlu, A. Induction of oxidative stress and histological changes in liver by subacute doses of butyl cyclohexyl phthalate. Environ. Toxicol. 2014, 29, 345–353. [Google Scholar] [CrossRef]
- Arciello, M.; Gori, M.; Maggio, R.; Barbaro, B.; Tarocchi, M.; Galli, A.; Balsano, C. Environmental pollution: A tangible risk for NAFLD pathogenesis. Int. J. Mol. Sci. 2013, 14, 22052–22066. [Google Scholar] [CrossRef]
- Laing, S.; Wang, G.; Briazova, T.; Zhang, C.; Wang, A.; Zheng, Z.; Gow, A.; Chen, A.F.; Rajagopalan, S.; Chen, L.C.; et al. Airborne particulate matter selectively activates endoplasmic reticulum stress response in the lung and liver tissues. Am. J. Physiol. Cell Physiol. 2010, 299, C736–C749. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.-H.; Fiel, M.I.; Sun, Q.; Guo, J.; Gordon, R.E.; Chen, L.C.; Friedman, S.L.; Odin, J.A.; Allina, J. Kupffer cell activation by ambient air particulate matter exposure may exacerbate non-alcoholic fatty liver disease. J. Immunotoxicol. 2009, 6, 266–275. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Chen, Q. Mediating Roles of PPARs in the Effects of Environmental Chemicals on Sex Steroids. PPAR Res. 2017, 2017, 3203161. [Google Scholar] [CrossRef]
- Yáñez, L.; Borja-Aburto, V.H.; Rojas, E.; de la Fuente, H.; González-Amaro, R.; Gómez, H.; Jongitud, A.A.; Díaz-Barriga, F. DDT induces DNA damage in blood cells. Studies in vitro and in women chronically exposed to this insecticide. Environ. Res. 2004, 94, 18–24. [Google Scholar] [CrossRef]
- Binelli, A.; Riva, C.; Cogni, D.; Provini, A. Genotoxic effects of p,p’-DDT (1,1,1-trichloro-2,2-bis-(chlorophenyl)ethane) and its metabolites in Zebra mussel (D. polymorpha) by SCGE assay and micronucleus test. Environ. Mol. Mutagen. 2008, 49, 406–415. [Google Scholar] [CrossRef]
- Song, Y.; Yang, K.D. Effect of p, p’-DDE on DNA damage and expression of FasL gene of rat Sertoli cell in vitro. Wei Sheng Yan Jiu 2006, 35, 261–263. [Google Scholar]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Fan, M.; Candas, D.; Jiang, G.; Papadopoulos, S.; Tian, L.; Woloschak, G.; Grdina, D.J.; Li, J.J. CDK1 Enhances Mitochondrial Bioenergetics for Radiation-Induced DNA Repair. Cell Rep. 2015, 13, 2056–2063. [Google Scholar] [CrossRef] [Green Version]
- Brace, L.E.; Vose, S.C.; Stanya, K.; Gathungu, R.M.; Marur, V.R.; Longchamp, A.; Treviño-Villarreal, H.; Mejia, P.; Vargas, D.; Inouye, K.; et al. Increased oxidative phosphorylation in response to acute and chronic DNA damage. NPJ Aging Mech. Dis. 2016, 2, 16022. [Google Scholar] [CrossRef]
- Nims, R.W.; Lubet, R.A. Induction of cytochrome P-450 in the Norway rat, Rattus norvegicus, following exposure to potential environmental contaminants. J. Toxicol. Environ. Health 1995, 46, 271–292. [Google Scholar] [CrossRef]
- Wyde, M.E.; Bartolucci, E.; Ueda, A.; Zhang, H.; Yan, B.; Negishi, M.; You, L. The environmental pollutant 1,1-dichloro-2,2-bis (p-chlorophenyl) ethylene induces rat hepatic cytochrome P450 2B and 3A expression through the constitutive androstane receptor and pregnane X receptor. Mol. Pharmacol. 2003, 64, 474–481. [Google Scholar] [CrossRef]
- Mota, P.C.; Cordeiro, M.; Pereira, S.P.; Oliveira, P.J.; Moreno, A.J.; Ramalho-Santos, J. Differential effects of p,p′-DDE on testis and liver mitochondria: Implications for reproductive toxicology. Reprod. Toxicol. 2011, 31, 80–85. [Google Scholar] [CrossRef]
- Ferreira, F.M.; Madeira, V.M.; Moreno, A.J. Interactions of 2,2-bis(p-chlorophenyl)-1,1-dichloroethylene with mitochondrial oxidative phosphorylation. Biochem. Pharmacol. 1997, 53, 299–308. [Google Scholar] [CrossRef]
- Vidal, J.D.; Whitney, K.M. Morphologic manifestations of testicular and epididymal toxicity. Spermatogenesis 2014, 4, 1–17. [Google Scholar] [CrossRef]
- Siddighi, S.; Patton, W.C.; Jacobson, J.D.; King, A.; Chan, P.J. Correlation of sperm parameters with apoptosis assessed by dual fluorescence dna integrity assay. Arch. Androl. 2009, 50, 311–314. [Google Scholar] [CrossRef]
- França, L.R.; Avelar, G.F.; Almeida, F.F. Spermatogenesis and sperm transit through the epididymis in mammals with emphasis on pigs. Theriogenology 2005, 63, 300–318. [Google Scholar] [CrossRef]
- Palmer, N.O.; Bakos, H.W.; Fullston, T.; Lane, M. Impact of obesity on male fertility, sperm function and molecular composition. Spermatogenesis 2012, 2, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Katib, A. Mechanisms linking obesity to male infertility. Cent. Eur. J. Urol. 2015, 68, 79–85. [Google Scholar] [CrossRef]
- Kort, H.I.; Massey, J.B.; Elsner, C.W.; Mitchell-Leef, D.; Shapiro, D.B.; Witt, M.A.; Roudebush, W.E. Impact of body mass index values on sperm quantity and quality. J. Androl. 2006, 27, 450–452. [Google Scholar] [CrossRef]
- MacDonald, A.A.; Herbison, G.P.; Showell, M.; Farquhar, C.M. The impact of body mass index on semen parameters and reproductive hormones in human males: A systematic review with meta-analysis. Hum. Reprod. Update 2010, 16, 293–311. [Google Scholar] [CrossRef]
- Vested, A.; Giwercman, A.; Bonde, J.P.; Toft, G. Persistent organic pollutants and male reproductive health. Asian J. Androl. 2014, 16, 71–80. [Google Scholar] [CrossRef]
- Bonefeld-Jørgensen, E.C.; Andersen, H.R.; Rasmussen, T.H.; Vinggaard, A.M. Effect of highly bioaccumulated polychlorinated biphenyl congeners on estrogen and androgen receptor activity. Toxicology 2001, 158, 141–153. [Google Scholar] [CrossRef]
- Penna-Videau, S.; Bustos-Obregon, E.; Cermeno-Vivas, J.R.; Chirino, D. Malathion affects spermatogenic proliferation in mouse. Int. J. Morphol. 2012, 30, 1399–1407. [Google Scholar] [CrossRef]
- Lebda, M.; Gad, S.; Gaafar, H. Effects of lipoic acid on acrylamide induced testicular damage. Mater. Sociomed. 2014, 26, 208–212. [Google Scholar] [CrossRef]
- Türedi, S.; Yuluğ, E.; Alver, A.; Kutlu, O.; Kahraman, C. Effects of resveratrol on doxorubicin induced testicular damage in rats. Exp. Toxicol. Pathol. 2015, 67, 229–235. [Google Scholar] [CrossRef]
- Song, L.; Liu, J.; Jin, X.; Li, Z.; Zhao, M.; Liu, W. p,p’-Dichlorodiphenyldichloroethylene induces colorectal adenocarcinoma cell proliferation through oxidative stress. PLoS ONE 2014, 9, e112700. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Migliaccio, V.; Di Gregorio, I.; Putti, R.; Lionetti, L. Mitochondrial Involvement in the Adaptive Response to Chronic Exposure to Environmental Pollutants and High-Fat Feeding in a Rat Liver and Testis. Cells 2019, 8, 834. https://doi.org/10.3390/cells8080834
Migliaccio V, Di Gregorio I, Putti R, Lionetti L. Mitochondrial Involvement in the Adaptive Response to Chronic Exposure to Environmental Pollutants and High-Fat Feeding in a Rat Liver and Testis. Cells. 2019; 8(8):834. https://doi.org/10.3390/cells8080834
Chicago/Turabian StyleMigliaccio, Vincenzo, Ilaria Di Gregorio, Rosalba Putti, and Lillà Lionetti. 2019. "Mitochondrial Involvement in the Adaptive Response to Chronic Exposure to Environmental Pollutants and High-Fat Feeding in a Rat Liver and Testis" Cells 8, no. 8: 834. https://doi.org/10.3390/cells8080834