H3K18Ac as a Marker of Cancer Progression and Potential Target of Anti-Cancer Therapy

 ,
,
Abstract
:1. Introduction
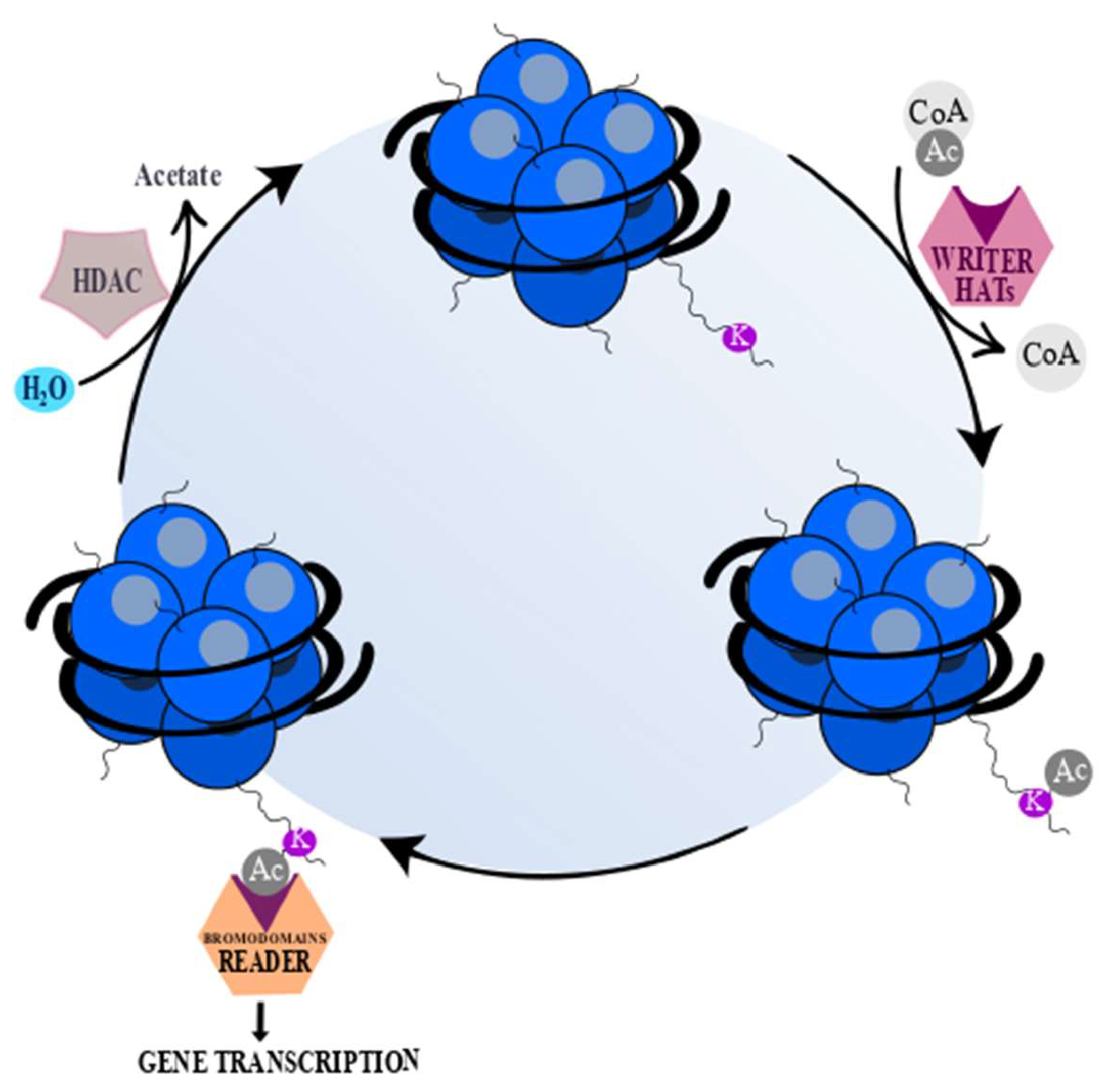
2. Histone Acetyltransferases and Histone Deacetylases
2.1. HAT Classification
2.2. HDAC Classification and Their Inhibitors
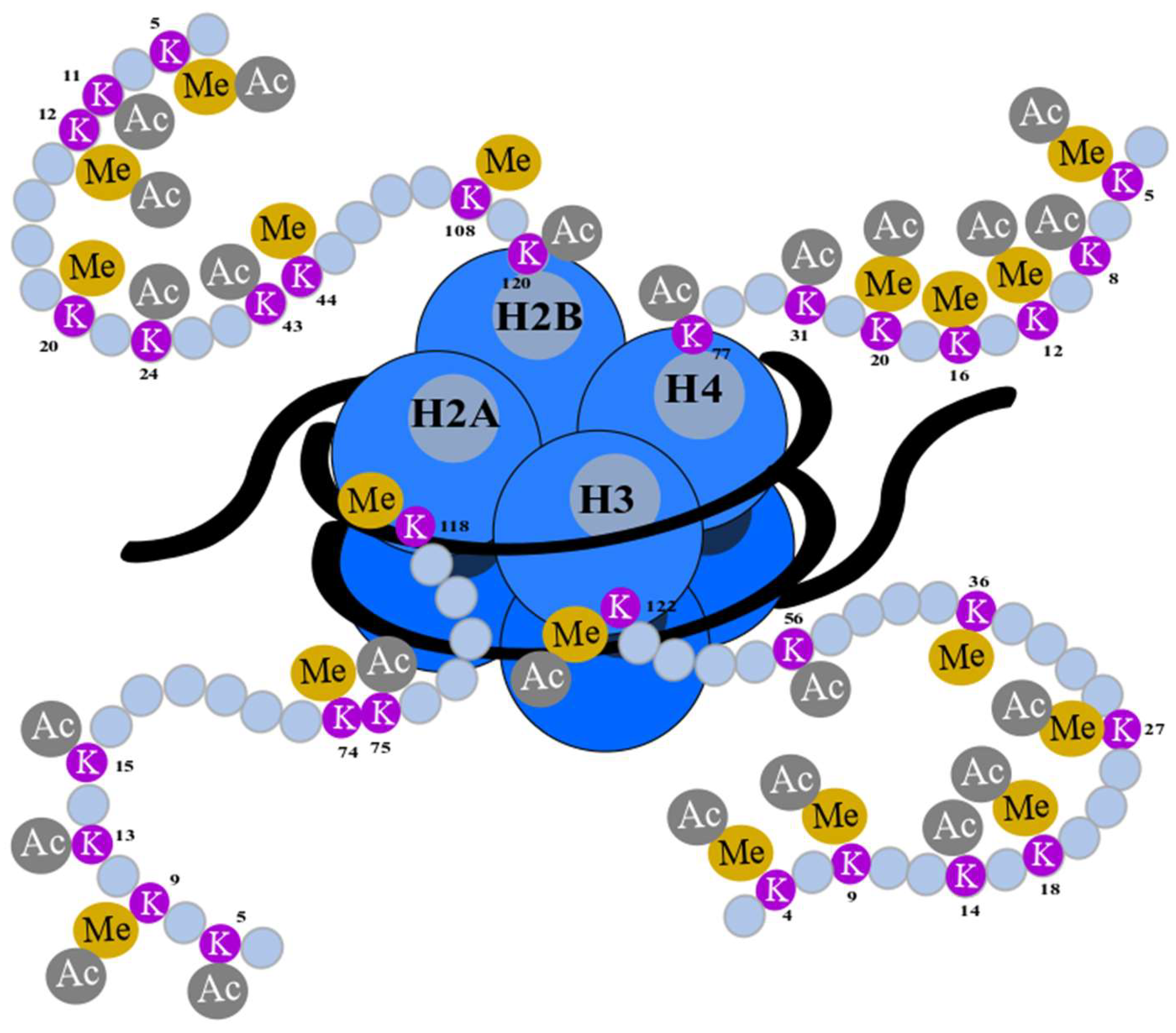
3. Histone 3 Modifications as Biomarkers of Cancer Progression
4. H3K18Ac as a Biomarker in Cancer Progression
4.1. Prostate Cancer
4.2. Pancreatic Cancer
4.3. Colon Cancer
4.4. Breast Cancer
4.5. Hepatocellular Carcinoma
4.6. Lung Cancer
4.7. Thyroid Cancer
5. Alteration of H3K18 Acetylation via Viral Oncoprotein: Adenovirus E1A
6. The Role of H3K18Ac in Regulation of Cancer Hallmarks
6.1. H3K18 Deacetylation by SIRT6 and SIRT7
6.2. H3K18Ac Deacetylation by HDAC1
7. Clinical Aspects of Global Histone Modification, including Acetylation and Methylation
8. Potential Strategies for Pharmacological Targeting H3K18
9. Future Perspectives on Epigenetic Anti-Cancer Therapy
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
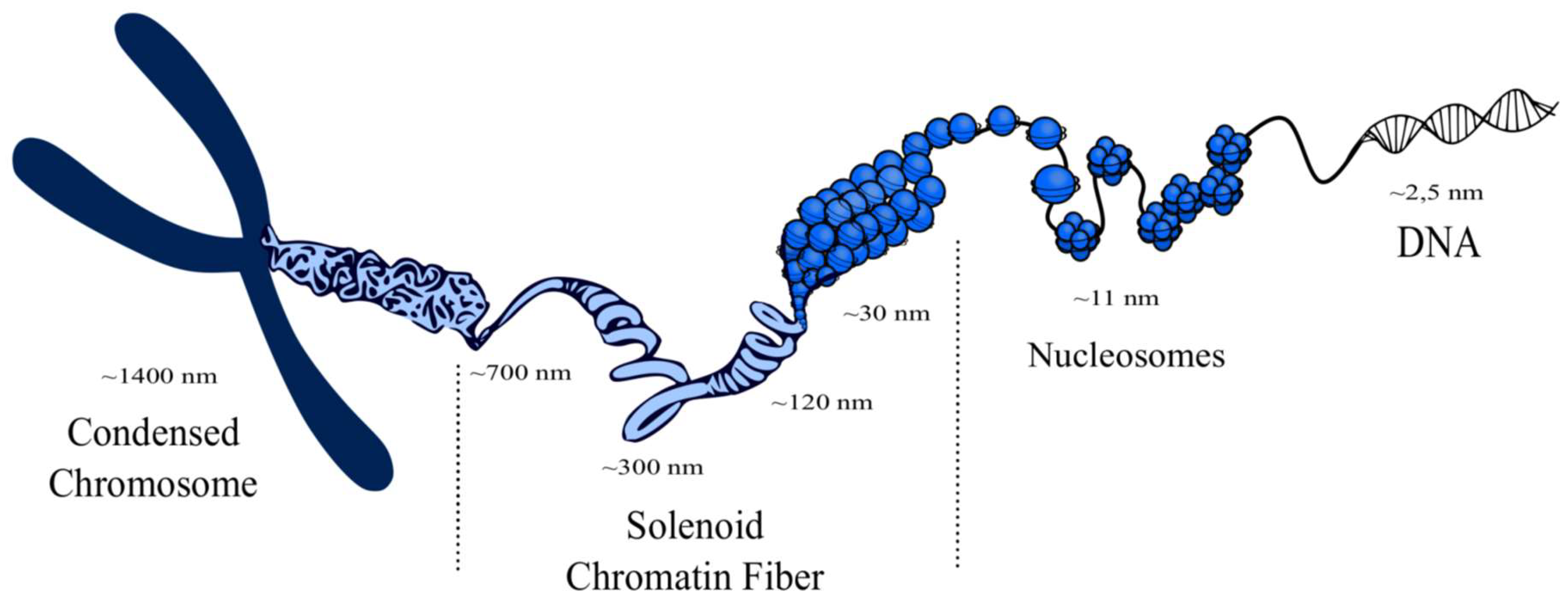
- Perišić, O.; Schlick, T. Computational strategies to address chromatin structure problems. Phys. Biol. 2016, 13, 35006. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.L.; Watson, M.; Wilkins, O.G.; Cato, L.; Travers, A.; Thomas, J.O.; Stott, K. Highly disordered histone H1−DNA model complexes and their condensates. Proc. Natl. Acad. Sci. USA 2018, 115, 11964–11969. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.J.; Rhodes, D. Structure of the “30nm” chromatin fibre: A key role for the linker histone. Curr. Opin. Struct. Biol. 2006, 16, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-M.; Kim, B.J.; Norwood Toro, L.; Skoultchi, A.I. H1 linker histone promotes epigenetic silencing by regulating both DNA methylation and histone H3 methylation. Proc. Natl. Acad. Sci. USA 2013, 110, 1708–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, G.; Shanmuganandam, K.; Bendre, A.; Mujumdar, D.; Goel, A.; Shiras, A.; Shiras, A. Epigenetic regulation of DNA methyltransferases: DNMT1 and DNMT3B in gliomas. J. Neurooncol. 2011, 104, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Shahid, Z.; Simpson, B.; Singh, G. Genetics, Histone Code; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Schwartz, U.; Németh, A.; Diermeier, S.; Exler, J.H.; Hansch, S.; Maldonado, R.; Heizinger, L.; Merkl, R.; Längst, G. Characterizing the nuclease accessibility of DNA in human cells to map higher order structures of chromatin. Nucleic Acids Res. 2018, 47, 1239. [Google Scholar] [CrossRef] [PubMed]
- Judes, G.; Dubois, L.; Rifaï, K.; Idrissou, M.; Mishellany, F.; Pajon, A.; Besse, S.; Daures, M.; Degoul, F.; Bignon, Y.-J.; et al. TIP60: An actor in acetylation of H3K4 and tumor development in breast cancer. Epigenomics 2018, 10, 1415–1430. [Google Scholar] [CrossRef]
- Hansen, J.C.; Connolly, M.; McDonald, C.J.; Pan, A.; Pryamkova, A.; Ray, K.; Seidel, E.; Tamura, S.; Rogge, R.; Maeshima, K. The 10-nm chromatin fiber and its relationship to interphase chromosome organization. Biochem. Soc. Trans. 2018, 46, 67–76. [Google Scholar] [CrossRef]
- DNA Packaging: Nucleosomes and Chromatin | Learn Science at Scitable. Available online: https://www.nature.com/scitable/topicpage/dna-packaging-nucleosomes-and-chromatin-310 (accessed on 22 April 2019).
- Eukaryotic Epigenetic Gene Regulation | Biology for Majors I. Available online: https://courses.lumenlearning.com/wm-biology1/chapter/reading-eukaryotic-epigenetic-gene-regulation/ (accessed on 22 April 2019).
- How Do Histone Proteins Help in the Coiling of DNA - Pediaa.Com. Available online: https://pediaa.com/how-do-histone-proteins-help-in-the-coiling-of-dna/ (accessed on 22 April 2019).
- Ou, H.D.; Phan, S.; Deerinck, T.J.; Thor, A.; Ellisman, M.H.; O’Shea, C.C. ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 2017, 357, eaag0025. [Google Scholar] [CrossRef]
- Di Martile, M.; Del Bufalo, D.; Trisciuoglio, D. The multifaceted role of lysine acetylation in cancer: Prognostic biomarker and therapeutic target. Oncotarget 2016, 7, 55789–55810. [Google Scholar] [CrossRef]
- Kaypee, S.; Sudarshan, D.; Shanmugam, M.K.; Mukherjee, D.; Sethi, G.; Kundu, T.K. Aberrant lysine acetylation in tumorigenesis: Implications in the development of therapeutics. Pharmacol. Ther. 2016, 162, 98–119. [Google Scholar] [CrossRef]
- Hammond, C.M.; Strømme, C.B.; Huang, H.; Patel, D.J.; Groth, A. Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell Biol. 2017, 18, 141–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, J.; Daoud, A.; Eblen, S.T. Targeting Chromatin Remodeling for Cancer Therapy. Curr. Mol. Pharmacol. 2019, 12. [Google Scholar] [CrossRef]
- Tang, L.; Nogales, E.; Ciferri, C. Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Prog. Biophys. Mol. Biol. 2010, 102, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Schvartzman, J.M.; Thompson, C.B.; Finley, L.W.S. Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 2018, 217, 2247–2259. [Google Scholar] [CrossRef]
- Izzo, A.; Schneider, R. Chatting histone modifications in mammals. Brief. Funct. Genomics 2010, 9, 429. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Braun, S.; Madhani, H.D. Shaping the landscape: Mechanistic consequences of ubiquitin modification of chromatin. EMBO Rep. 2012, 13, 619–630. [Google Scholar] [CrossRef]
- Lopez, R.; Sarg, B.; Lindner, H.; Bartolomé, S.; Ponte, I.; Suau, P.; Roque, A. Linker histone partial phosphorylation: Effects on secondary structure and chromatin condensation. Nucleic Acids Res. 2015, 43, 4463–4476. [Google Scholar] [CrossRef]
- Tran, T.Q.; Lowman, X.H.; Kong, M. Molecular Pathways: Metabolic Control of Histone Methylation and Gene Expression in Cancer. Clin. Cancer Res. 2017, 23, 4004–4009. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.E.; Gozani, O. An unexpected journey: Lysine methylation across the proteome. Biochim. Biophys. Acta 2014, 1839, 1395–1403. [Google Scholar] [CrossRef] [Green Version]
- McGrath, J.; Trojer, P. Targeting histone lysine methylation in cancer. Pharmacol. Ther. 2015, 150, 1–22. [Google Scholar] [CrossRef]
- Marmorstein, R.; Zhou, M.-M. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef]
- Simó-Riudalbas, L.; Esteller, M. Targeting the histone orthography of cancer: Drugs for writers, erasers and readers. Br. J. Pharmacol. 2015, 172, 2716–2732. [Google Scholar] [CrossRef]
- Ntranos, A.; Casaccia, P. Bromodomains: Translating the words of lysine acetylation into myelin injury and repair. Neurosci. Lett. 2016, 625, 4–10. [Google Scholar] [CrossRef]
- Protamine Kinase—An Overview | ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/medicine-and-dentistry/protamine-kinase (accessed on 22 April 2019).
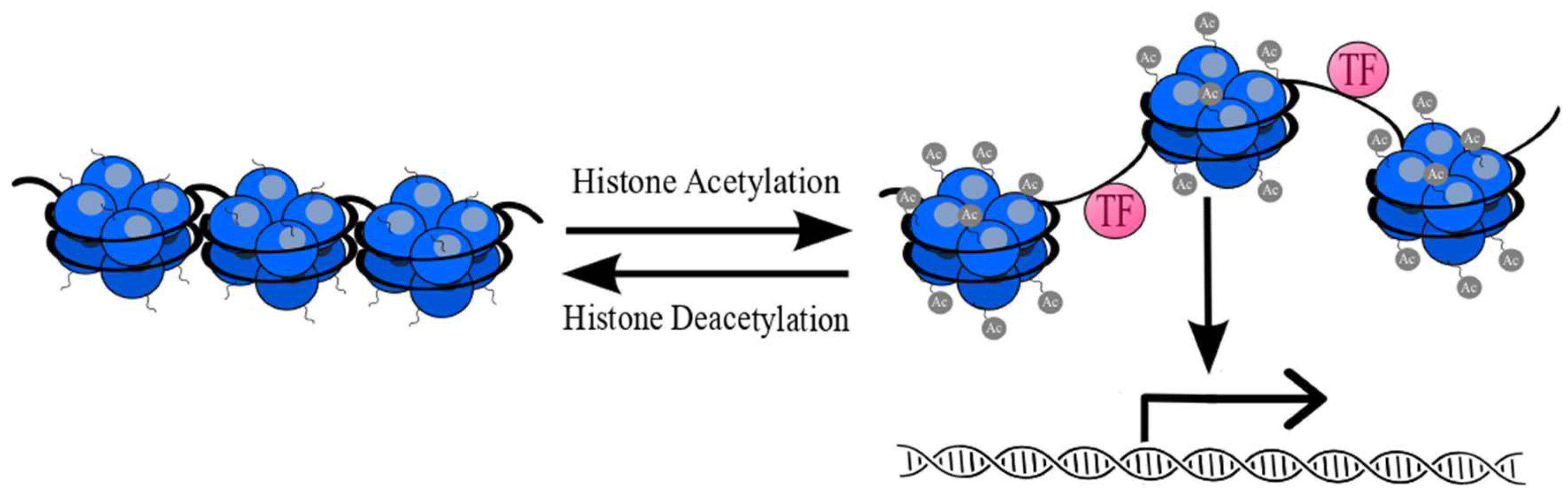
- Steunou, A.-L.; Rossetto, D.; Côté, J. Regulating Chromatin by Histone Acetylation. In Fundamentals of Chromatin; Springer: New York, NY, USA, 2014; pp. 147–212. [Google Scholar]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Yang, X.-J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef]
- Trisciuoglio, D.; Di Martile, M.; Del Bufalo, D. Emerging Role of Histone Acetyltransferase in Stem Cells and Cancer. Stem. Cells Int. 2018, 2018, 8908751. [Google Scholar] [CrossRef]
- Barnes, C.E.; English, D.M.; Cowley, S.M. Acetylation & Co: An expanding repertoire of histone acylations regulates chromatin and transcription. Essays Biochem. 2019, EBC20180061. [Google Scholar]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef]
- Koprinarova, M.; Schnekenburger, M.; Diederich, M. Role of Histone Acetylation in Cell Cycle Regulation. Curr. Top. Med. Chem. 2015, 16, 732–744. [Google Scholar] [CrossRef]
- Jian, W.; Yan, B.; Huang, S.; Qiu, Y. Histone deacetylase 1 activates PU.1 gene transcription through regulating TAF9 deacetylation and transcription factor IID assembly. FASEB J. 2017, 31, 4104–4116. [Google Scholar] [CrossRef] [Green Version]
- Greer, C.B.; Tanaka, Y.; Kim, Y.J.; Xie, P.; Zhang, M.Q.; Park, I.-H.; Kim, T.H. Histone Deacetylases Positively Regulate Transcription through the Elongation Machinery. Cell Rep. 2015, 13, 1444–1455. [Google Scholar] [CrossRef] [Green Version]
- Angiolilli, C.; Kabala, P.A.; Grabiec, A.M.; Van Baarsen, I.M.; Ferguson, B.S.; García, S.; Malvar Fernandez, B.; McKinsey, T.A.; Tak, P.P.; Fossati, G.; et al. Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann. Rheum. Dis. 2017, 76, 277–285. [Google Scholar] [CrossRef]
- Li, W.; Sun, Z. Mechanism of Action for HDAC Inhibitors-Insights from Omics Approaches. Int. J. Mol. Sci. 2019, 20, 1616. [Google Scholar] [CrossRef]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef]
- Brochier, C.; Dennis, G.; Rivieccio, M.A.; McLaughlin, K.; Coppola, G.; Ratan, R.R.; Langley, B. Specific Acetylation of p53 by HDAC Inhibition Prevents DNA Damage-Induced Apoptosis in Neurons. J. Neurosci. 2013, 33, 8621–8632. [Google Scholar] [CrossRef]
- Lanzillotta, A.; Pignataro, G.; Branca, C.; Cuomo, O.; Sarnico, I.; Benarese, M.; Annunziato, L.; Spano, P.; Pizzi, M. Targeted acetylation of NF-kappaB/RelA and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol. Dis. 2013, 49, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.J.; Lee, J.E.; Lee, A.S.; Kang, K.P.; Lee, S.; Park, S.K.; Lee, S.Y.; Han, M.K.; Kim, D.H.; Kim, W. SIRT1 overexpression decreases cisplatin-induced acetylation of NF-κB p65 subunit and cytotoxicity in renal proximal tubule cells. Biochem. Biophys. Res. Commun. 2012, 419, 206–210. [Google Scholar] [CrossRef]
- Cheng, M.-H.; Wong, Y.-H.; Chang, C.-M.; Yang, C.-C.; Chen, S.-H.; Yuan, C.-L.; Kuo, H.-M.; Yang, C.-Y.; Chiu, H.-F. B1, a novel HDAC inhibitor, induces apoptosis through the regulation of STAT3 and NF-κB. Int. J. Mol. Med. 2017, 39, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Emori, T.; Kitamura, K.; Okazaki, K. Nuclear Smad7 Overexpressed in Mesenchymal Cells Acts as a Transcriptional Corepressor by Interacting with HDAC-1 and E2F to Regulate Cell Cycle. Biol. Open 2012, 1, 247–260. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. Histone Deacetylase Inhibitor-Induced Autophagy in Tumor Cells: Implications for p53. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef]
- Zhao, Y.; Lu, S.; Wu, L.; Chai, G.; Wang, H.; Chen, Y.; Sun, J.; Yu, Y.; Zhou, W.; Zheng, Q.; et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol. Cell. Biol. 2006, 26, 2782–2790. [Google Scholar] [CrossRef]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.B.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Bisson, W.H.; Löhr, C.V.; Williams, D.E.; Ho, E.; Dashwood, R.H.; Rajendran, P. Histone and Non-Histone Targets of Dietary Deacetylase Inhibitors. Curr. Top. Med. Chem. 2016, 16, 714–731. [Google Scholar] [CrossRef]
- Clayton, A.L.; Hazzalin, C.A.; Mahadevan, L.C. Enhanced Histone Acetylation and Transcription: A Dynamic Perspective. Mol. Cell 2006, 23, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.J.; Kim, S.H.; Yang, W.I.; Ko, Y.H.; Yoon, S.O. Long Non-coding RNA HOTAIR Expression in Diffuse Large B-Cell Lymphoma: In Relation to Polycomb Repressive Complex Pathway Proteins and H3K27 Trimethylation. J. Pathol. Transl. Med. 2016, 50, 369–376. [Google Scholar] [CrossRef]
- Ellinger, J.; Schneider, A.-C.; Bachmann, A.; Kristiansen, G.; Müller, S.C.; Rogenhofer, S. Evaluation of Global Histone Acetylation Levels in Bladder Cancer Patients. Anticancer Res. 2016, 36, 3961–3964. [Google Scholar] [PubMed]
- Liu, B.-L.; Cheng, J.-X.; Zhang, X.; Wang, R.; Zhang, W.; Lin, H.; Xiao, X.; Cai, S.; Chen, X.-Y.; Cheng, H. Global Histone Modification Patterns as Prognostic Markers to Classify Glioma Patients. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 2888–2896. [Google Scholar] [CrossRef]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global Histone Modifications in Breast Cancer Correlate with Tumor Phenotypes, Prognostic Factors, and Patient Outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [Green Version]
- Lutz, L.; Fitzner, I.C.; Ahrens, T.; Geißler, A.-L.; Makowiec, F.; Hopt, U.T.; Bogatyreva, L.; Hauschke, D.; Werner, M.; Lassmann, S. Histone modifiers and marks define heterogeneous groups of colorectal carcinomas and affect responses to HDAC inhibitors in vitro. Am. J. Cancer Res. 2016, 6, 664–676. [Google Scholar]
- Nakazawa, T.; Kondo, T.; Ma, D.; Niu, D.; Mochizuki, K.; Kawasaki, T.; Yamane, T.; Iino, H.; Fujii, H.; Katoh, R. Global histone modification of histone H3 in colorectal cancer and its precursor lesions. Hum. Pathol. 2012, 43, 834–842. [Google Scholar] [CrossRef]
- Ye, C.; Han, K.; Lei, J.; Zeng, K.; Zeng, S.; Ju, H.; Yu, L. Inhibition of histone deacetylase 7 reverses concentrative nucleoside transporter 2 repression in colorectal cancer by up-regulating histone acetylation state. Br. J. Pharmacol. 2018, 175, 4209–4217. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhao, M.; Yin, N.; He, B.; Wang, B.; Yuan, Y.; Yu, F.; Hu, J.; Yin, B.; Lu, Q. Abnormal Histone Acetylation and Methylation Levels in Esophageal Squamous Cell Carcinomas. Cancer Investig. 2011, 29, 548–556. [Google Scholar] [CrossRef]
- Calcagno, D.Q.; Wisnieski, F.; da Silva Mota, E.R.; Maia de Sousa, S.B.; Costa da Silva, J.M.; Leal, M.F.; Gigek, C.O.; Santos, L.C.; Rasmussen, L.T.; Assumpção, P.P.; et al. Role of histone acetylation in gastric cancer: Implications of dietetic compounds and clinical perspectives. Epigenomics 2019, 11, 349–362. [Google Scholar] [CrossRef]
- Park, Y.S.; Jin, M.Y.; Kim, Y.J.; Yook, J.H.; Kim, B.S.; Jang, S.J. The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann. Surg. Oncol. 2008, 15, 1968–1976. [Google Scholar] [CrossRef]
- Feng, G.-W.; Dong, L.-D.; Shang, W.-J.; Pang, X.-L.; Li, J.-F.; Liu, L.; Wang, Y. HDAC5 promotes cell proliferation in human hepatocellular carcinoma by up-regulating Six1 expression. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 811–816. [Google Scholar]
- Mi, W.; Guan, H.; Lyu, J.; Zhao, D.; Xi, Y.; Jiang, S.; Andrews, F.H.; Wang, X.; Gagea, M.; Wen, H.; et al. YEATS2 links histone acetylation to tumorigenesis of non-small cell lung cancer. Nat. Commun. 2017, 8, 1088. [Google Scholar] [CrossRef]
- Manuyakorn, A.; Paulus, R.; Farrell, J.; Dawson, N.A.; Tze, S.; Cheung-Lau, G.; Hines, O.J.; Reber, H.; Seligson, D.B.; Horvath, S.; et al. Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: Results from RTOG 9704. J. Clin. Oncol. 2010, 28, 1358–1365. [Google Scholar] [CrossRef]
- Valdés-Mora, F.; Gould, C.M.; Colino-Sanguino, Y.; Qu, W.; Song, J.Z.; Taylor, K.M.; Buske, F.A.; Statham, A.L.; Nair, S.S.; Armstrong, N.J.; et al. Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer. Nat. Commun. 2017, 8, 1346. [Google Scholar] [CrossRef] [PubMed]
- Webber, L.P.; Wagner, V.P.; Curra, M.; Vargas, P.A.; Meurer, L.; Carrard, V.C.; Squarize, C.H.; Castilho, R.M.; Martins, M.D. Hypoacetylation of acetyl-histone H3 (H3K9ac) as marker of poor prognosis in oral cancer. Histopathology 2017, 71, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Ganai, S.A.; Banday, S.; Farooq, Z.; Altaf, M. Modulating epigenetic HAT activity for reinstating acetylation homeostasis: A promising therapeutic strategy for neurological disorders. Pharmacol. Ther. 2016, 166, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Friedmann, D.R.; Marmorstein, R. Structure and mechanism of non-histone protein acetyltransferase enzymes. FEBS J. 2013, 280, 5570–5581. [Google Scholar] [CrossRef] [Green Version]
- Salah Ud-Din, A.I.M.; Tikhomirova, A.; Roujeinikova, A. Structure and Functional Diversity of GCN5-Related N-Acetyltransferases (GNAT). Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef]
- Avvakumov, N.; Côté, J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 2007, 26, 5395–5407. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; Wang, F.; Cai, Y.; Jin, J. The Functional Analysis of Histone Acetyltransferase MOF in Tumorigenesis. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef]
- Ngo, L.; Brown, T.; Zheng, Y.G. Bisubstrate inhibitors to target histone acetyltransferase 1. Chem. Biol. Drug Des. 2019. [Google Scholar] [CrossRef]
- Dyson, H.J.; Wright, P.E. Role of Intrinsic Protein Disorder in the Function and Interactions of the Transcriptional Coactivators CREB-binding Protein (CBP) and p300. J. Biol. Chem. 2016, 291, 6714–6722. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Bishayee, A.; Pandey, A. Targeting Histone Deacetylases with Natural and Synthetic Agents: An Emerging Anticancer Strategy. Nutrients 2018, 10, 731. [Google Scholar] [CrossRef]
- De Ruijter, A.J.M.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B.P. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Khan, D.H.; He, S.; Yu, J.; Winter, S.; Cao, W.; Seiser, C.; Davie, J.R. Protein Kinase CK2 Regulates the Dimerization of Histone Deacetylase 1 (HDAC1) and HDAC2 during Mitosis. J. Biol. Chem. 2013, 288, 16518–16528. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.D.W.; Cowley, S.M. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem. Soc. Trans. 2013, 41, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, T.; Nakayama, J.-I. Physiological roles of class I HDAC complex and histone demethylase. J. Biomed. Biotechnol. 2011, 2011, 129383. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef] [Green Version]
- Lane, A.A.; Chabner, B.A. Histone Deacetylase Inhibitors in Cancer Therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Shang, Y.-P.; Chen, H.; Li, J. Histone deacetylases function as novel potential therapeutic targets for cancer. Hepatol. Res. 2017, 47, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Hess-Stumpp, H. Histone deacetylase inhibitors and cancer: From cell biology to the clinic. Eur. J. Cell Biol. 2005, 84, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Wawruszak, A.; Kalafut, J.; Okon, E.; Czapinski, J.; Halasa, M.; Przybyszewska, A.; Miziak, P.; Okla, K.; Rivero-Muller, A.; Stepulak, A. Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells. Cancers 2019, 11, 148. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Poklepovic, A.; Kirkwood, J.; Dent, P. HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget 2017, 8, 83155–83170. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Liu, J.; Chen, D.; Yan, L.; Zheng, W. Sirtuin Inhibition: Strategies, Inhibitors, and Therapeutic Potential. Trends Pharmacol. Sci. 2017, 38, 459–472. [Google Scholar] [CrossRef]
- Villalba, J.M.; Alcaín, F.J. Sirtuin activators and inhibitors. BioFactors 2012, 38, 349–359. [Google Scholar] [CrossRef]
- Spiegelman, N.A.; Price, I.R.; Jing, H.; Wang, M.; Yang, M.; Cao, J.; Hong, J.Y.; Zhang, X.; Aramsangtienchai, P.; Sadhukhan, S.; et al. Direct Comparison of SIRT2 Inhibitors: Potency, Specificity, Activity-Dependent Inhibition, and On-Target Anticancer Activities. ChemMedChem 2018, 13, 1890–1894. [Google Scholar] [CrossRef]
- Liu, N.; Li, S.; Wu, N.; Cho, K.-S. Acetylation and deacetylation in cancer stem-like cells. Oncotarget 2017, 8, 89315–89325. [Google Scholar] [CrossRef] [Green Version]
- Kamińska, K.; Nalejska, E.; Kubiak, M.; Wojtysiak, J.; Żołna, Ł.; Kowalewski, J.; Lewandowska, M.A. Prognostic and Predictive Epigenetic Biomarkers in Oncology. Mol. Diagn. Ther. 2019, 23, 83–95. [Google Scholar] [CrossRef]
- Messier, T.L.; Gordon, J.A.R.; Boyd, J.R.; Tye, C.E.; Browne, G.; Stein, J.L.; Lian, J.B.; Stein, G.S. Histone H3 lysine 4 acetylation and methylation dynamics define breast cancer subtypes. Oncotarget 2016, 7, 5094. [Google Scholar] [CrossRef]
- Yi, S.-J.; Hwang, S.Y.; Oh, M.-J.; Kim, Y.-H.; Ryu, H.; Rhee, S.-K.; Jhun, B.H.; Kim, K. Oncogenic N-Ras Stimulates SRF-Mediated Transactivation via H3 Acetylation at Lysine 9. BioMed Res. Int. 2018, 2018, 5473725. [Google Scholar] [CrossRef]
- Charlop-Powers, Z.; Zeng, L.; Zhang, Q.; Zhou, M.-M. Structural insights into selective histone H3 recognition by the human Polybromo bromodomain 2. Cell Res. 2010, 20, 529–538. [Google Scholar] [CrossRef]
- Liao, L.; Alicea-Velázquez, N.L.; Langbein, L.; Niu, X.; Cai, W.; Cho, E.-A.; Zhang, M.; Greer, C.B.; Yan, Q.; Cosgrove, M.S.; et al. High affinity binding of H3K14ac through collaboration of bromodomains 2, 4 and 5 is critical for the molecular and tumor suppressor functions of PBRM1. Mol. Oncol. 2019, 13, 811–828. [Google Scholar] [CrossRef]
- Zhang, E.; Han, L.; Yin, D.; He, X.; Hong, L.; Si, X.; Qiu, M.; Xu, T.; De, W.; Xu, L.; et al. H3K27 acetylation activated-long non-coding RNA CCAT1 affects cell proliferation and migration by regulating SPRY4 and HOXB13 expression in esophageal squamous cell carcinoma. Nucleic Acids Res. 2017, 45, 3086–3101. [Google Scholar] [CrossRef]
- Chen, F.; Qi, S.; Zhang, X.; Wu, J.; Yang, X.; Wang, R. lncRNA PLAC2 activated by H3K27 acetylation promotes cell proliferation and invasion via the activation of Wnt/β-catenin pathway in oral squamous cell carcinoma. Int. J. Oncol. 2019, 54, 1183–1194. [Google Scholar] [CrossRef]
- Dong, H.; Hu, J.; Zou, K.; Ye, M.; Chen, Y.; Wu, C.; Chen, X.; Han, M. Activation of LncRNA TINCR by H3K27 acetylation promotes Trastuzumab resistance and epithelial-mesenchymal transition by targeting MicroRNA-125b in breast Cancer. Mol. Cancer 2019, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, R.; Li, L.-W.; Liu, X.; Wang, Y.-F.; Wang, Q.-X.; Zhang, Q. Long non-coding RNA HOTAIR mediates the switching of histone H3 lysine 27 acetylation to methylation to promote epithelial-to-mesenchymal transition in gastric cancer. Int. J. Oncol. 2018, 54, 77–86. [Google Scholar] [CrossRef]
- Vadla, R.; Haldar, D. Mammalian target of rapamycin complex 2 (mTORC2) controls glycolytic gene expression by regulating Histone H3 Lysine 56 acetylation. Cell Cycle 2018, 17, 110–123. [Google Scholar] [CrossRef]
- Saidi, D.; Cheray, M.; Osman, A.M.; Stratoulias, V.; Lindberg, O.R.; Shen, X.; Blomgren, K.; Joseph, B. Glioma-induced SIRT1-dependent activation of hMOF histone H4 lysine 16 acetyltransferase in microglia promotes a tumor supporting phenotype. Oncoimmunology 2018, 7, e1382790. [Google Scholar] [CrossRef]
- Xu, Q.; Liu, X.; Zhu, S.; Hu, X.; Niu, H.; Zhang, X.; Zhu, D.; Nesa, E.U.; Tian, K.; Yuan, H. Hyper-acetylation contributes to the sensitivity of chemo-resistant prostate cancer cells to histone deacetylase inhibitor Trichostatin A. J. Cell. Mol. Med. 2018, 22, 1909–1922. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ao, X.; Ding, W.; Ponnusamy, M.; Wu, W.; Hao, X.; Yu, W.; Wang, Y.; Li, P.; Wang, J. Critical role of FOXO3a in carcinogenesis. Mol. Cancer 2018, 17, 104. [Google Scholar] [CrossRef]
- Nucleosomes and Histone Proteins | amsbio. Available online: http://www.amsbio.com/nucleosomes-and-histone-proteins.aspx (accessed on 22 April 2019).
- Lee, J.-H.; Yang, B.; Lindahl, A.J.; Damaschke, N.; Boersma, M.D.; Huang, W.; Corey, E.; Jarrard, D.F.; Denu, J.M. Identifying Dysregulated Epigenetic Enzyme Activity in Castrate-Resistant Prostate Cancer Development. ACS Chem. Biol. 2017, 12, 2804–2814. [Google Scholar] [CrossRef]
- Damodaran, S.; Damaschke, N.; Gawdzik, J.; Yang, B.; Shi, C.; Allen, G.O.; Huang, W.; Denu, J.; Jarrard, D. Dysregulation of Sirtuin 2 (SIRT2) and histone H3K18 acetylation pathways associates with adverse prostate cancer outcomes. BMC Cancer 2017, 17, 874. [Google Scholar] [CrossRef]
- Haider, R.; Massa, F.; Kaminski, L.; Clavel, S.; Djabari, Z.; Robert, G.; Laurent, K.; Michiels, J.-F.; Durand, M.; Ricci, J.-E.; et al. Sirtuin 7: A new marker of aggressiveness in prostate cancer. Oncotarget 2017, 8, 77309–77316. [Google Scholar] [CrossRef]
- Juliano, C.N.; Izetti, P.; Pereira, M.P.; dos Santos, A.P.; Bravosi, C.P.; Abujamra, A.L.; Prolla, P.A.; Osvaldt, A.B.; Edelweiss, M.I.A. H4K12 and H3K18 Acetylation Associates With Poor Prognosis in Pancreatic Cancer. Appl. Immunohistochem. Mol. Morphol. 2016, 24, 337–344. [Google Scholar] [CrossRef]
- Ashktorab, H.; Belgrave, K.; Hosseinkhah, F.; Brim, H.; Nouraie, M.; Takkikto, M.; Hewitt, S.; Lee, E.L.; Dashwood, R.H.; Smoot, D. Global Histone H4 Acetylation and HDAC2 Expression in Colon Adenoma and Carcinoma. Dig. Dis. Sci. 2009, 54, 2109–2117. [Google Scholar] [CrossRef]
- Bardhan, K.; Paschall, A.V.; Yang, D.; Chen, M.R.; Simon, P.S.; Bhutia, Y.D.; Martin, P.M.; Thangaraju, M.; Browning, D.D.; Ganapathy, V.; et al. IFN Induces DNA Methylation-Silenced GPR109A Expression via pSTAT1/p300 and H3K18 Acetylation in Colon Cancer. Cancer Immunol. Res. 2015, 3, 795–805. [Google Scholar] [CrossRef]
- Pandey, V.; Kumar, V. Stabilization of SIRT7 deacetylase by viral oncoprotein HBx leads to inhibition of growth restrictive RPS7 gene and facilitates cellular transformation. Sci. Rep. 2015, 5, 14806. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.S.; Jung, W.; Lee, E.; Chang, H.; Choi, J.H.; Kim, H.G.; Kim, A.; Kim, B. SIRT7, H3K18ac, and ELK4 Immunohistochemical Expression in Hepatocellular Carcinoma. J. Pathol. Transl. Med. 2016, 50, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Chen, H.; Yin, M.; Ye, X.; Chen, G.; Zhou, X.; Yin, L.; Zhang, C.; Ding, B. MiR-376a and Histone Deacetylation 9 Form A Regulatory Circuitry in Hepatocellular Carcinoma. Cell. Physiol. Biochem. 2015, 35, 729–739. [Google Scholar] [CrossRef]
- Zhang, T.; Meng, J.; Liu, X.; Zhang, X.; Peng, X.; Cheng, Z.; Zhang, F. ING5 differentially regulates protein lysine acetylation and promotes p300 autoacetylation. Oncotarget 2018, 9, 1617–1629. [Google Scholar] [CrossRef]
- Puppin, C.; Passon, N.; Lavarone, E.; Di Loreto, C.; Frasca, F.; Vella, V.; Vigneri, R.; Damante, G. Levels of histone acetylation in thyroid tumors. Biochem. Biophys. Res. Commun. 2011, 411, 679–683. [Google Scholar] [CrossRef]
- Ferrari, R.; Berk, A.J.; Kurdistani, S.K. Viral manipulation of the host epigenome for oncogenic transformation. Nat. Rev. Genet. 2009, 10, 290–294. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Chen, M.; Pettersson, U. A new look at adenovirus splicing. Virology 2014, 456–457, 329–341. [Google Scholar] [CrossRef]
- Hsu, E.; Pennella, M.A.; Zemke, N.R.; Eng, C.; Berk, A.J. Adenovirus E1A Activation Domain Regulates H3 Acetylation Affecting Varied Steps in Transcription at Different Viral Promoters. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Radko, S.; Jung, R.; Olanubi, O.; Pelka, P. Effects of Adenovirus Type 5 E1A Isoforms on Viral Replication in Arrested Human Cells. PLoS ONE 2015, 10, e0140124. [Google Scholar] [CrossRef]
- Ferrari, R.; Gou, D.; Jawdekar, G.; Johnson, S.A.; Nava, M.; Su, T.; Yousef, A.F.; Zemke, N.R.; Pellegrini, M.; Kurdistani, S.K.; et al. Adenovirus Small E1A Employs the Lysine Acetylases p300/CBP and Tumor Suppressor Rb to Repress Select Host Genes and Promote Productive Virus Infection. Cell Host Microbe 2014, 16, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Haberz, P.; Arai, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Mapping the interactions of adenoviral E1A proteins with the p160 nuclear receptor coactivator binding domain of CBP. Protein Sci. 2016, 25, 2256–2267. [Google Scholar] [CrossRef]
- King, C.R.; Zhang, A.; Tessier, T.M.; Gameiro, S.F.; Mymryk, J.S. Hacking the Cell: Network Intrusion and Exploitation by Adenovirus E1A. MBio 2018, 9, e00390-18. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, D.; Ogryzko, V.; Kao, H.Y.; Nash, A.; Chen, H.; Nakatani, Y.; Evans, R.M. A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell 1999, 96, 393–403. [Google Scholar] [CrossRef]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577. [Google Scholar]
- Giotopoulos, G.; Chan, W.-I.; Horton, S.J.; Ruau, D.; Gallipoli, P.; Fowler, A.; Crawley, C.; Papaemmanuil, E.; Campbell, P.J.; Göttgens, B.; et al. The epigenetic regulators CBP and p300 facilitate leukemogenesis and represent therapeutic targets in acute myeloid leukemia. Oncogene 2016, 35, 279–289. [Google Scholar] [CrossRef]
- Gao, Y.; Geng, J.; Hong, X.; Qi, J.; Teng, Y.; Yang, Y.; Qu, D.; Chen, G. Expression of p300 and CBP is associated with poor prognosis in small cell lung cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 760–767. [Google Scholar]
- Ferrari, R.; Su, T.; Li, B.; Bonora, G.; Oberai, A.; Chan, Y.; Sasidharan, R.; Berk, A.J.; Pellegrini, M.; Kurdistani, S.K. Reorganization of the host epigenome by a viral oncogene. Genome Res. 2012, 22, 1212–1221. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.-J.; Loewenstein, P.M.; Green, M. The adenovirus E1A oncoprotein N-terminal transcriptional repression domain enhances p300 autoacetylation and inhibits histone H3 Lys18 acetylation. Genes Cancer 2015, 6, 30. [Google Scholar] [PubMed]
- Horwitz, G.A.; Zhang, K.; McBrian, M.A.; Grunstein, M.; Kurdistani, S.K.; Berk, A.J. Adenovirus Small e1a Alters Global Patterns of Histone Modification. Science 2008, 321, 1084–1085. [Google Scholar] [CrossRef] [Green Version]
- Barber, M.F.; Michishita-Kioi, E.; Xi, Y.; Tasselli, L.; Kioi, M.; Moqtaderi, Z.; Tennen, R.I.; Paredes, S.; Young, N.L.; Chen, K.; et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 2012, 487, 114–118. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.W.; Zeng, Y.; Wu, B.; Deiters, A.; Liu, W.R. A Chemical Biology Approach to Reveal Sirt6-targeted Histone H3 Sites in Nucleosomes. ACS Chem. Biol. 2016, 11, 1973–1981. [Google Scholar] [CrossRef] [Green Version]
- Karim, M.F.; Yoshizawa, T.; Sato, Y.; Sawa, T.; Tomizawa, K.; Akaike, T.; Yamagata, K. Inhibition of H3K18 deacetylation of Sirt7 by Myb-binding protein 1a (Mybbp1a). Biochem. Biophys. Res. Commun. 2013, 441, 157–163. [Google Scholar] [CrossRef]
- Paredes, S.; Villanova, L.; Chua, K.F. Molecular Pathways: Emerging Roles of Mammalian Sirtuin SIRT7 in Cancer. Clin. Cancer Res. 2014, 20, 1741–1746. [Google Scholar] [CrossRef] [Green Version]
- Mahalingaiah, P.K.S.; Ponnusamy, L.; Singh, K.P. Oxidative stress-induced epigenetic changes associated with malignant transformation of human kidney epithelial cells. Oncotarget 2017, 8, 11127–11143. [Google Scholar] [CrossRef]
- Senese, S.; Zaragoza, K.; Minardi, S.; Muradore, I.; Ronzoni, S.; Passafaro, A.; Bernard, L.; Draetta, G.F.; Alcalay, M.; Seiser, C.; et al. Role for Histone Deacetylase 1 in Human Tumor Cell Proliferation. Mol. Cell. Biol. 2007, 27, 4784–4795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, M.; Park, P.-H.; Jackson, D.; Shukla, S.D. Evidence for the role of oxidative stress in the acetylation of histone H3 by ethanol in rat hepatocytes. Alcohol 2010, 44, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Jana, S.; Patra, K.; Jana, J.; Mandal, D.P.; Bhattacharjee, S. Nrf-2 transcriptionally activates P21 Cip/WAF1 and promotes A549 cell survival against oxidative stress induced by H2O2. Chem. Biol. Interact. 2018, 285, 59–68. [Google Scholar] [CrossRef]
- Cichon, M.A.; Radisky, D.C. ROS-induced epithelial-mesenchymal transition in mammary epithelial cells is mediated by NF-kB-dependent activation of Snail. Oncotarget 2014, 5, 2827–2838. [Google Scholar] [CrossRef]
- Wisnieski, F.; Leal, M.F.; Calcagno, D.Q.; Santos, L.C.; Gigek, C.O.; Chen, E.S.; Artigiani, R.; Demachki, S.; Assumpção, P.P.; Lourenço, L.G.; et al. BMP8B Is a Tumor Suppressor Gene Regulated by Histone Acetylation in Gastric Cancer. J. Cell. Biochem. 2017, 118, 869–877. [Google Scholar] [CrossRef]
- Watanabe, T.; Morinaga, S.; Akaike, M.; Numata, M.; Tamagawa, H.; Yamamoto, N.; Shiozawa, M.; Ohkawa, S.; Kameda, Y.; Nakamura, Y.; et al. The cellular level of histone H3 lysine 4 dimethylation correlates with response to adjuvant gemcitabine in Japanese pancreatic cancer patients treated with surgery. Eur. J. Surg. Oncol. 2012, 38, 1051–1057. [Google Scholar] [CrossRef]
- Barlési, F.; Giaccone, G.; Gallegos-Ruiz, M.I.; Loundou, A.; Span, S.W.; Lefesvre, P.; Kruyt, F.A.E.; Rodriguez, J.A. Global Histone Modifications Predict Prognosis of Resected Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2007, 25, 4358–4364. [Google Scholar] [CrossRef]
- Van Den Broeck, A.; Brambilla, E.; Moro-Sibilot, D.; Lantuejoul, S.; Brambilla, C.; Eymin, B.; Khochbin, S.; Gazzeri, S. Loss of Histone H4K20 Trimethylation Occurs in Preneoplasia and Influences Prognosis of Non-Small Cell Lung Cancer. Clin. Cancer Res. 2008, 14, 7237–7245. [Google Scholar] [CrossRef] [PubMed]
- Viotti, M.; Wilson, C.; McCleland, M.; Koeppen, H.; Haley, B.; Jhunjhunwala, S.; Klijn, C.; Modrusan, Z.; Arnott, D.; Classon, M.; et al. SUV420H2 is an epigenetic regulator of epithelial/mesenchymal states in pancreatic cancer. J. Cell Biol. 2018, 217, 763–777. [Google Scholar] [CrossRef]
- Yokoyama, M.; Chiba, T.; Zen, Y.; Oshima, M.; Kusakabe, Y.; Noguchi, Y.; Yuki, K.; Koide, S.; Tara, S.; Saraya, A.; et al. Histone lysine methyltransferase G9a is a novel epigenetic target for the treatment of hepatocellular carcinoma. Oncotarget 2017, 8, 21315–21326. [Google Scholar] [CrossRef]
- Mito, J.K.; Qian, X.; Doyle, L.A.; Hornick, J.L.; Jo, V.Y. Role of Histone H3K27 Trimethylation Loss as a Marker for Malignant Peripheral Nerve Sheath Tumor in Fine-Needle Aspiration and Small Biopsy Specimens. Am. J. Clin. Pathol. 2017, 148, 179–189. [Google Scholar] [CrossRef]
- Pekmezci, M.; Cuevas-Ocampo, A.K.; Perry, A.; Horvai, A.E. Significance of H3K27me3 loss in the diagnosis of malignant peripheral nerve sheath tumors. Mod. Pathol. 2017, 30, 1710. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Xiong, H.; Chen, Y.; Wang, C.; Zhang, H.; Xu, P.; Han, J.; Xiao, S.; Ding, H.; Chen, Z.; et al. Discovery and biological evaluation of thiobarbituric derivatives as potent p300/CBP inhibitors. Bioorg. Med. Chem. 2018, 26, 5397–5407. [Google Scholar] [CrossRef]
- Zucconi, B.E.; Luef, B.; Xu, W.; Henry, R.A.; Nodelman, I.M.; Bowman, G.D.; Andrews, A.J.; Cole, P.A. Modulation of p300/CBP Acetylation of Nucleosomes by Bromodomain Ligand I-CBP112. Biochemistry 2016, 55, 3727–3734. [Google Scholar] [CrossRef] [Green Version]
- Hiraoka, N.; Kikuchi, J.; Koyama, D.; Wada, T.; Mori, S.; Nakamura, Y.; Furukawa, Y. Alkylating agents induce histone H3K18 hyperacetylation and potentiate HDAC inhibitor-mediated global histone acetylation and cytotoxicity in mantle cell lymphoma. Blood Cancer J. 2013, 3, e169. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.-Y.; Li, G.; Deng, Z.-J.; Liu, L.-Y.; Chen, L.; Tang, J.-Z.; Wang, Y.-Q.; Cao, S.-T.; Fang, Y.-X.; Wen, F.; et al. Dicer interacts with SIRT7 and regulates H3K18 deacetylation in response to DNA damaging agents. Nucleic Acids Res. 2016, 44, 3629–3642. [Google Scholar] [CrossRef]
- Li, S.; Wu, B.; Zheng, W. Cyclic tripeptide-based potent human SIRT7 inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 461–465. [Google Scholar] [CrossRef]
- Kim, J.-H.; Kim, D.; Cho, S.J.; Jung, K.-Y.; Kim, J.-H.; Lee, J.M.; Jung, H.J.; Kim, K.R. Identification of a novel SIRT7 inhibitor as anticancer drug candidate. Biochem. Biophys. Res. Commun. 2019, 508, 451–457. [Google Scholar] [CrossRef]
- Baxter, E.; Windloch, K.; Gannon, F.; Lee, J.S. Epigenetic regulation in cancer progression. Cell Biosci. 2014, 4, 45. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Origin | Experimental Model | Marker Expression | References |
|---|---|---|---|
| Brain | BV2 microglia cells C6 glioma cells GL261 glioma cells in vitro | H4K16Ac ↑ in BV2 microglia upon coculture with C6 glioma cells, GL261 glioma cells, or murine primary glioma tumorspheres | [102] |
| U87glioblastoma cells in vitro | H3K56Ac ↓ on the promoters of glycolytic genes after knockdown of mTORC2 | [101] | |
| Breast | MCF10A (non-cancerous cell line with characteristics of epithelial hyperplasia) MCF7 (mature luminal subtype of breast cancer cell line expressing estrogen and progesterone receptors) MDA-MB-231 (triple negative (ESR1-, PGR-, HER2-) invasive metastatic adenocarcinoma cell line) in vitro | Global H3K4Ac ↑ in cancer cell lines (MCF7, MDA-MB-231) and H3K4Me3 in MDA-MB-231 metastatic cell line compared to a normal breast epithelial cell line (MCF10A) | [93] |
| MDA-MB-231 (triple negative) MCF-7 (ER+) with transfected TIP60 grafted to athymic Balb-c mice in vivo | H3K4Ac ↑ (in cells with TIP60) H3K4Ac ↓ (in cells without TIP60) | [8] | |
| Cervix | HeLa cervical carcinoma cells | H3K56Ac ↑ after EGF treatment | [101] |
| Esophagus | ESCC cell lines Eca-109 and TE-1 in vitro | H3K27Ac ↑ at the promoter of CCAT1 in ESCC cells (Eca-109) compared with normal human esophageal epithelial cells (HET-1A) | [97] |
| Oral cavity | OSCC cell lines SCC-9 and CAL-27 in vitro Male nude mice in vivo | H3K27Ac ↑ at the promoter of PLAC2 in OSCC cells and normal epithelial HOK cells | [98] |
| Prostate | PC3 prostate cancer cells PC3/Doc docetaxel-resistant PC3 cells in vitro | ↑ acetylated H3 and H4 in resistant cells compared to PC3 cells. TSA, an HDAC inhibitor, ↑ acetylation of histones H3K9, H4K8, and H4K16 | [103] |
| Tumor Localization | Type of Cancer | Assay | Marker | Expression in Tumor Cells | Patients Survival | Reference |
|---|---|---|---|---|---|---|
| Bladder | 271 urothelial bladder cancers | TMA | H3Ac H4Ac H3K18Ac | Lower H3Ac levels in cancer than in normal urothelial tissue but similar levels of H4Ac and H3K18Ac | No correlation of histone acetylation and PFS or CSS | [55] |
| Blood | 231 diffuse large B-cell lymphomas (DLBCLs) | TMA | H3K27Me3 | High expression in 35.7% of cases | Lower survival for patients with high expression of H3K27Me3 (independent predictor) | [54] |
| Brain | 230 gliomas (WHO grades 1–4) | IHC | H3K9Ac | Broad distribution of staining (mean 70% of tumor cells) | Lower survival for patients with grade 1–2 astrocytomas with H3K9Ac in less than 88% of tumor cells | [56] |
| Breast | 880 breast cancers of different histology | TMA | H3K9Ac H3K18Ac H4K12Ac H4K16Ac H3K4Me2 H4K20Me3 | Low score for H4K16Ac in 78.9% of, and high scores for H3K18Ac and H4K20Me3 in 81.4% and 69.8% of breast tumors, respectively. About 50%/50% of low/high scores for other markers (H3K9Ac, H4K12Ac, H4R3Me2, and H3K4Me2) | Lower BCSS and metastatic-specific survival for patients with low (<100) marker scores (except H4K20Me3). Lower DFS for patients with low scores for H3K18Ac, H4R3Me2, and H3K9Ac. | [57] |
| Deep soft tissues | 131 sarcomas including 82 SS, 39 MPNST, and 10 FDP | IHC and TMA | H3K27Me3 | Broad distribution of staining | No association between H3K27Me3 expression in MPNST and PFS or OS | [148] |
| Lung | 285 lung cancers (stages 1–4) | TMA | H3K18Ac H3K4Me2 | Broad distribution of staining for both H3K18Ac and H3K4Me2 | Lower 15-year survival for patients with stage 1 lung adenocarcinomas with lower expression of both modifications (independent predictor) | [57] |
| 138 early-stage NSCLCs (stage I to IIIA) | IHC | H2AK5Ac | Broad distribution of staining (mean 68% of tumor cells) | Lower survival for patients with stage II NSCLCs with H2AK5Ac in less than 5% of tumor cells | [143] | |
| IHC | H3K9Ac | Broad distribution of staining (mean 42% of tumor cells) | Lower survival for patients with stage I adenocarcinomas with H3K9Ac in more than 68% of tumor cells | |||
| IHC | H3K4Me2 | Broad distribution of staining (mean 57% of tumor cells) | Lower survival for patients with stage I large-cell or squamous cell carcinomas with H3K4Me2 in less than 85% of tumor cells | |||
| 157 stage I–IV lung cancers (50 squamous cell carcinomas and 107 adenocarcinomas) | IHC | H4K20Me3 | H4K20Me3 score between 0 and 100 in 70% of the tumor samples | Lower survival for patients with stage I adenocarcinomas with a low (≤100) H4K20Me3 score | [144] | |
| Pancreas | 229 pancreatic adenocarcinomas | TMA | H3K18Ac H3K4Me2 H3K9Me2 | Broad distribution of staining for each modification | Low levels of H3K4Me2, H3K9Me2, and H3K18Ac (<60%, <25%, and <35% of tumor cells, respectively) were each independent predictors of lower OS for patients with stages I and II of the disease. Lower OS for patients treated with postoperative chemoradiotherapy with low H3K4Me2 or H3K9Me2 staining (each was an independent predictor) | [66] |
| 119 pancreatic cancers (stage I–IV) | H3K9Ac H3K18Ac H4K12Ac | Median percentage of stained cells for H3K9Ac (80%), H3K18Ac (65%), and H4K12Ac (60%) | High expression (H score ≥100) of H3K18Ac and H4K12Ac were both independently associated with shorter OS | [109] | ||
| 61 pancreatic cancers (stage IB–III) | TMA | H3K9Ac H3K18Ac H3K4Me2 H3K4Me3 H3K9Me2 | The median H-scores were: H3K9me2, 158; H3K9ac, 140; H3K4me2, 142; H3K4me3, 160; H3K18ac, 162. | None of histone modifications influenced OS or DFS. | [109] | |
| Stomach | 261 gastric carcinomas (stage I–IV) | TMA | H3K9Ac H4K16Ac H4K20Me3 | Strong staining (score = 4) for H3K6Ac, H4K16Ac, and H4K20Me3 in most cases (49%, 60.9%, and 54.4%, respectively). Strong staining for H3K9Me3 in only 33% of the cases. | No influence of H3K9Ac, H4K16Ac, or H4K20Me3 expression on survival. Lower survival for patients with strong H3K9Me3 staining (independent predictor). | [63] |
| Oral | 33 OL and 86 OSCC | IHC | H3K9Ac | Staining in 81% NOM cells, 94.4% OL cells, and 78.2% OSCC cells | Lower survival for patients with low expression | [68] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hałasa, M.; Wawruszak, A.; Przybyszewska, A.; Jaruga, A.; Guz, M.; Kałafut, J.; Stepulak, A.; Cybulski, M. H3K18Ac as a Marker of Cancer Progression and Potential Target of Anti-Cancer Therapy. Cells 2019, 8, 485. https://doi.org/10.3390/cells8050485
Hałasa M, Wawruszak A, Przybyszewska A, Jaruga A, Guz M, Kałafut J, Stepulak A, Cybulski M. H3K18Ac as a Marker of Cancer Progression and Potential Target of Anti-Cancer Therapy. Cells. 2019; 8(5):485. https://doi.org/10.3390/cells8050485
Chicago/Turabian StyleHałasa, Marta, Anna Wawruszak, Alicja Przybyszewska, Anna Jaruga, Małgorzata Guz, Joanna Kałafut, Andrzej Stepulak, and Marek Cybulski. 2019. "H3K18Ac as a Marker of Cancer Progression and Potential Target of Anti-Cancer Therapy" Cells 8, no. 5: 485. https://doi.org/10.3390/cells8050485