The Potential Use of Metformin, Dipyridamole, N-Acetylcysteine and Statins as Adjunctive Therapy for Systemic Lupus Erythematosus



Abstract
:1. Introduction
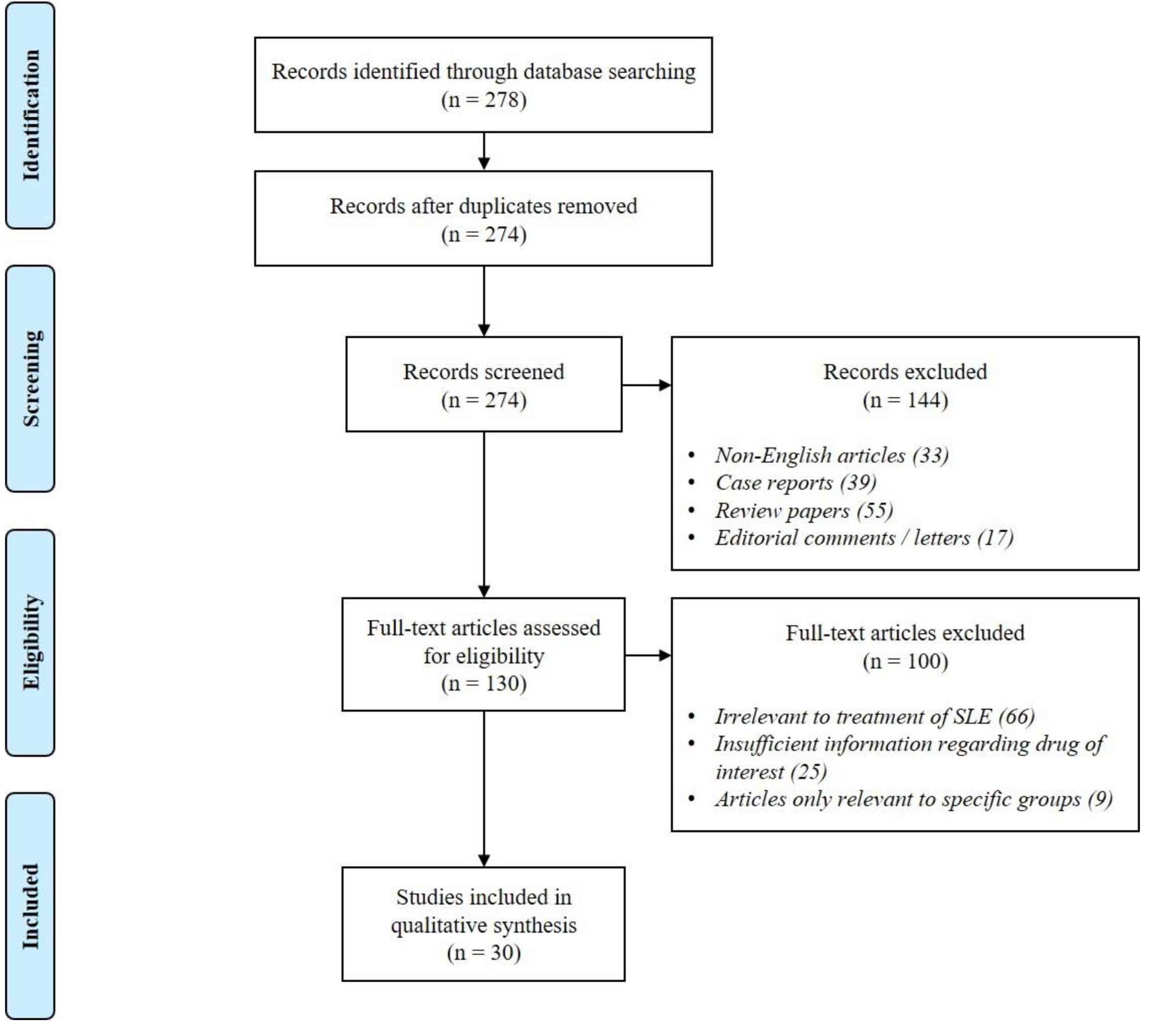
2. Methods
2.1. Literature Search
2.2. Study Eligibility Criteria
3. Results
4. Discussion
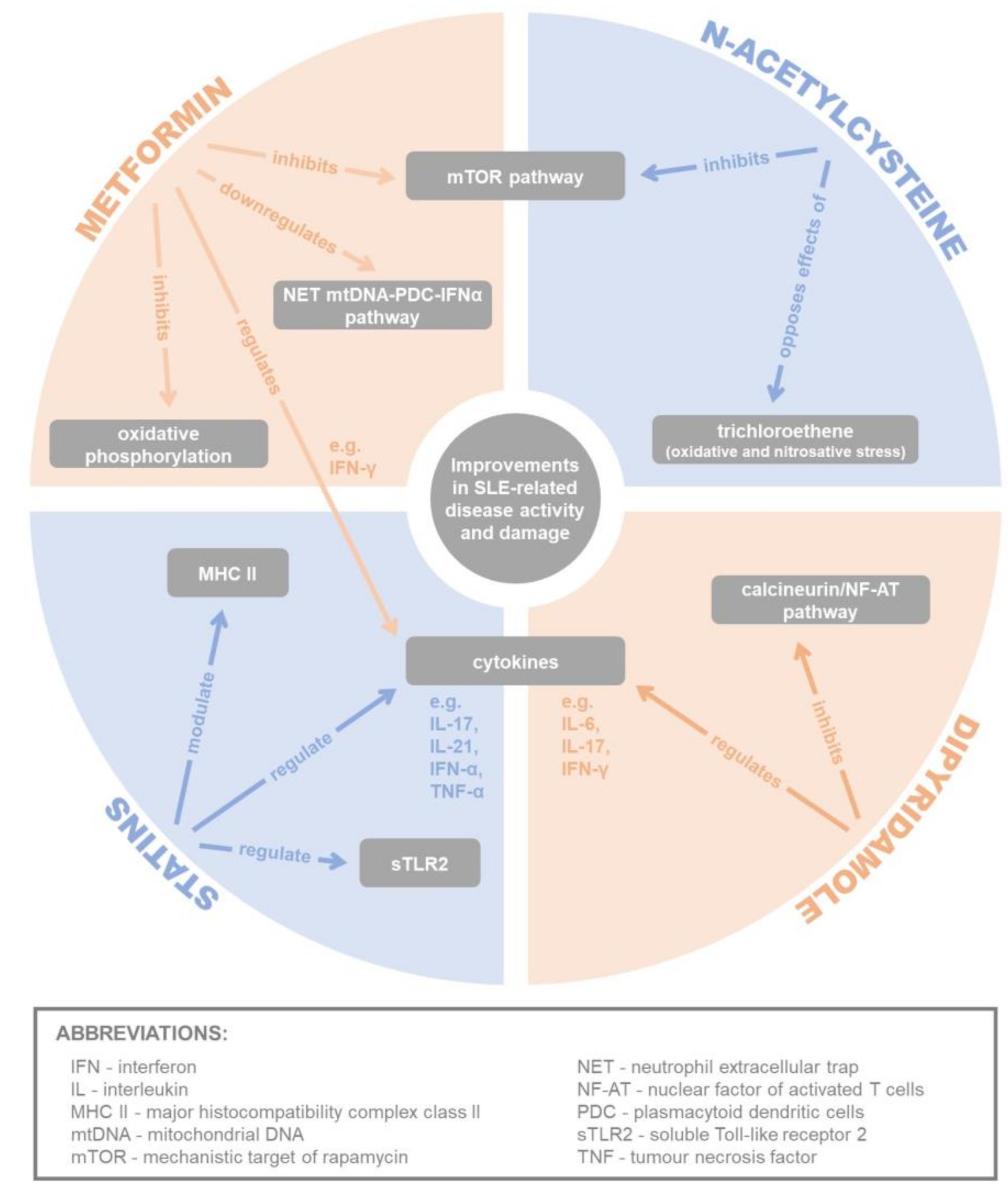
4.1. Metformin
4.1.1. Inhibition via AMPK/mTOR/STAT3 Pathway
4.1.2. Inhibition of Oxidative Phosphorylation
4.1.3. Downregulation of NET mtDNA-PDC-IFNα Pathway
4.2. Dipyridamole
Inhibition of Calcineurin/NF-AT Pathway
4.3. N-Acetylcysteine
4.3.1. Inhibition of mTOR Pathway
4.3.2. Reversal of Effects of ROS on F-Actin Polymerisation
4.3.3. Opposing the Effects of Trichloroethene
4.3.4. Improvement of Endothelial Dysfunction
4.4. Statins
4.4.1. Modulation of Cytokine Levels
4.4.2. Regulation of TLR Signalling
4.4.3. Modulation of MHC Class II Expression
4.4.4. Studies Portraying Contrasting Evidence
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fortuna, G.; Brennan, M.T. Systemic lupus erythematosus: Epidemiology, pathophysiology, manifestations, and management. Dent. Clin. N. Am. 2013, 57, 631–655. [Google Scholar] [CrossRef]
- Lau, C.S.; Mak, A. The socioeconomic burden of SLE. Nat. Rev. Rheumatol. 2009, 5, 400–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touma, Z.; Urowitz, M.B.; Gladman, D.D. Systemic lupus erythematosus: An update on current pharmacotherapy and future directions. Expert Opin. Biol. Ther. 2013, 13, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.; Martinez, L.; Isenberg, D.A.; Leandro, M.J.; Cambridge, G. Pragmatic treatment of patients with systemic lupus erythematosus with rituximab: Long-term effects on serum immunoglobulins. Arthritis Care Res. 2017, 69, 857–866. [Google Scholar]
- Srivastava, A. Belimumab in systemic lupus erythematosus. Indian J. Dermatol. 2016, 61, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Sciascia, S.; Radin, M.; Roccatello, D.; Sanna, G.; Bertolaccini, M.L. Recent advances in the management of systemic lupus erythematosus. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.A.; Hsieh, H.J.; et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase ii/iii systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010, 62, 222–233. [Google Scholar] [CrossRef]
- Rovin, B.H.; Furie, R.; Latinis, K.; Looney, R.J.; Fervenza, F.C.; Sanchez-Guerrero, J.; Maciuca, R.; Zhang, D.; Garg, J.P.; Brunetta, P.; et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: The lupus nephritis assessment with rituximab study. Arthritis Rheum. 2012, 64, 1215–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzova, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A phase iii, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits b lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef]
- Navarra, S.V.; Guzman, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.; Thomas, M.; Kim, H.Y.; Leon, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Shippey, E.A.; Wagler, V.D.; Collamer, A.N. Hydroxychloroquine: An old drug with new relevance. Clevel. Clin. J. Med. 2018, 85, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, T.; Chen, S.; Gu, Y.; Ye, S. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol. 2015, 67, 3190–3200. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type i ifn production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef] [PubMed]
- Ponticelli, C.; Moroni, G. Hydroxychloroquine in systemic lupus erythematosus (sle). Expert Opin. Drug Saf. 2017, 16, 411–419. [Google Scholar] [CrossRef]
- Wang, T.F.; Lim, W. What is the role of hydroxychloroquine in reducing thrombotic risk in patients with antiphospholipid antibodies? Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 714–716. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.; Tay, S.H. Outcome of lupus glomerulonephritis: The role of prospective observational cohort studies. Rheumatology 2016, 55, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Reprint—Preferred reporting items for systematic reviews and meta-analyses: The prisma statement. Phys. Ther. 2009, 89, 873–880. [Google Scholar]
- Schlender, L.; Martinez, Y.V.; Adeniji, C.; Reeves, D.; Faller, B.; Sommerauer, C.; Al Qur’an, T.; Woodham, A.; Kunnamo, I.; Sönnichsen, A.; et al. Efficacy and safety of metformin in the management of type 2 diabetes mellitus in older adults: A systematic review for the development of recommendations to reduce potentially inappropriate prescribing. BMC Geriatr. 2017, 17, 227. [Google Scholar] [CrossRef]
- Rojas, L.B.A.; Gomes, M.B. Metformin: An old but still the best treatment for type 2 diabetes. Diabetol. Metab. Syndr. 2013, 5, 6. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Nasri, H.; Rafieian-Kopaei, M. Metformin: Current knowledge. J. Res. Med. Sci. 2014, 19, 658–664. [Google Scholar]
- Li, L.; Jick, S.; Gopalakrishnan, C.; Heide-Jorgensen, U.; Norrelund, H.; Sorensen, H.T.; Christiansen, C.F.; Ehrenstein, V. Metformin use and risk of lactic acidosis in people with diabetes with and without renal impairment: A cohort study in Denmark and the UK. Diabet. Med. 2017, 34, 485–489. [Google Scholar] [CrossRef]
- Tankova, T. Current indications for metformin therapy. Rom. J. Intern. Med. 2003, 41, 215–225. [Google Scholar]
- Lee, S.Y.; Moon, S.J.; Kim, E.K.; Seo, H.B.; Yang, E.J.; Son, H.J.; Kim, J.K.; Min, J.K.; Park, S.H.; Cho, M.L. Metformin suppresses systemic autoimmunity in roquin(san/san) mice through inhibiting b cell differentiation into plasma cells via regulation of AMPK/MTOR/STAT3. J. Immunol. 2017, 198, 2661–2670. [Google Scholar] [CrossRef]
- Yin, Y.; Choi, S.C.; Xu, Z.; Zeumer, L.; Kanda, N.; Croker, B.P.; Morel, L. Glucose oxidation is critical for cd4+ t cell activation in a mouse model of systemic lupus erythematosus. J. Immunol. 2016, 196, 80–90. [Google Scholar] [CrossRef]
- Yin, Y.; Choi, S.C.; Xu, Z.; Perry, D.J.; Seay, H.; Croker, B.P.; Sobel, E.S.; Brusko, T.M.; Morel, L. Normalization of cd4+ t cell metabolism reverses lupus. Sci. Transl. Med. 2015, 7, 274ra218. [Google Scholar] [CrossRef]
- Inoki, K.; Kim, J.; Guan, K.L. Ampk and mtor in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef]
- Oaks, Z.; Perl, A. Metabolic control of the epigenome in systemic lupus erythematosus. Autoimmunity 2014, 47, 256–264. [Google Scholar] [CrossRef]
- Quinn, B.J.; Kitagawa, H.; Memmott, R.M.; Gills, J.J.; Dennis, P.A. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol. Metab. 2013, 24, 469–480. [Google Scholar] [CrossRef]
- Zhang, S.; Pruitt, M.; Tran, D.; Du Bois, W.; Zhang, K.; Patel, R.; Hoover, S.; Simpson, R.M.; Simmons, J.; Gary, J.; et al. B cell-specific deficiencies in mtor limit humoral immune responses. J. Immunol. 2013, 191, 1692–1703. [Google Scholar] [CrossRef]
- Deng, X.S.; Wang, S.; Deng, A.; Liu, B.; Edgerton, S.M.; Lind, S.E.; Wahdan-Alaswad, R.; Thor, A.D. Metformin targets stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle 2012, 11, 367–376. [Google Scholar] [CrossRef]
- Hirahara, K.; Ghoreschi, K.; Laurence, A.; Yang, X.P.; Kanno, Y.; O’Shea, J.J. Signal transduction pathways and transcriptional regulation in th17 cell differentiation. Cytokine Growth Factor Rev. 2010, 21, 425–434. [Google Scholar] [CrossRef]
- Diehl, S.A.; Schmidlin, H.; Nagasawa, M.; van Haren, S.D.; Kwakkenbos, M.J.; Yasuda, E.; Beaumont, T.; Scheeren, F.A.; Spits, H. Stat3-mediated up-regulation of blimp1 is coordinated with bcl6 down-regulation to control human plasma cell differentiation. J. Immunol. 2008, 180, 4805–4815. [Google Scholar] [CrossRef]
- Park, J.S.; Lim, M.A.; Cho, M.L.; Ryu, J.G.; Moon, Y.M.; Jhun, J.Y.; Byun, J.K.; Kim, E.K.; Hwang, S.Y.; Ju, J.H.; et al. P53 controls autoimmune arthritis via stat-mediated regulation of the th17 cell/TREG cell balance in mice. Arthritis Rheum. 2013, 65, 949–959. [Google Scholar] [CrossRef]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic regulation of t lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef]
- Harker, L.A.; Kadatz, R.A. Mechanism of action of dipyridamole. Thromb. Res. 1983, 4, 39–46. [Google Scholar] [CrossRef]
- Knabb, R.M.; Gidday, J.M.; Ely, S.W.; Rubio, R.; Berne, R.M. Effects of dipyridamole on myocardial adenosine and active hyperemia. Am. J. Physiol. 1984, 247, H804–H810. [Google Scholar] [CrossRef]
- Balakumar, P.; Nyo, Y.H.; Renushia, R.; Raaginey, D.; Oh, A.N.; Varatharajan, R.; Dhanaraj, S.A. Classical and pleiotropic actions of dipyridamole: Not enough light to illuminate the dark tunnel? Pharmacol. Res. 2014, 87, 144–150. [Google Scholar] [CrossRef]
- Kyttaris, V.C.; Zhang, Z.; Kampagianni, O.; Tsokos, G.C. Calcium signaling in systemic lupus erythematosus t cells: A treatment target. Arthritis Rheum. 2011, 63, 2058–2066. [Google Scholar] [CrossRef]
- Mokhtari, V.; Afsharian, P.; Shahhoseini, M.; Kalantar, S.M.; Moini, A. A review on various uses of n-acetyl cysteine. Cell J. 2017, 19, 11–17. [Google Scholar]
- Shahin, A.Y.; Hassanin, I.M.; Ismail, A.M.; Kruessel, J.S.; Hirchenhain, J. Effect of oral n-acetyl cysteine on recurrent preterm labor following treatment for bacterial vaginosis. Int. J. Gynaecol. Obstet. 2009, 104, 44–48. [Google Scholar] [CrossRef]
- Harada, M.; Kishimoto, K.; Furuhashi, T.; Naito, K.; Nakashima, Y.; Kawaguchi, Y.; Hiraoka, I. Infertility observed in reproductive toxicity study of N-acetyl-l-cysteine in rats. Biol. Reprod. 2003, 69, 242–247. [Google Scholar] [CrossRef]
- Nasr, A. Effect of n-acetyl-cysteine after ovarian drilling in clomiphene citrate-resistant PCOS women: A pilot study. Reprod. Biomed. Online 2010, 20, 403–409. [Google Scholar] [CrossRef]
- Lai, Z.W.; Hanczko, R.; Bonilla, E.; Caza, T.N.; Clair, B.; Bartos, A.; Miklossy, G.; Jimah, J.; Doherty, E.; Tily, H.; et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in t cells from systemic lupus erythematosus patients: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012, 64, 2937–2946. [Google Scholar] [CrossRef]
- Perl, A.; Hanczko, R.; Lai, Z.W.; Oaks, Z.; Kelly, R.; Borsuk, R.; Asara, J.M.; Phillips, P.E. Comprehensive metabolome analyses reveal n-acetylcysteine-responsive accumulation of kynurenine in systemic lupus erythematosus: Implications for activation of the mechanistic target of rapamycin. Metabolomics 2015, 11, 1157–1174. [Google Scholar] [CrossRef]
- Doherty, E.; Oaks, Z.; Perl, A. Increased mitochondrial electron transport chain activity at complex i is regulated by n-acetylcysteine in lymphocytes of patients with systemic lupus erythematosus. Antioxid. Redox Signal. 2014, 21, 56–65. [Google Scholar] [CrossRef]
- Jiang, X.; Chen, F. The effect of lipid peroxides and superoxide dismutase on systemic lupus erythematosus: A preliminary study. Clin. Immunol. Immunopathol. 1992, 63, 39–44. [Google Scholar] [CrossRef]
- Suwannaroj, S.; Lagoo, A.; Keisler, D.; McMurray, R.W. Antioxidants suppress mortality in the female nzb x nzw f1 mouse model of systemic lupus erythematosus (sle). Lupus 2001, 10, 258–265. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.; Luo, X.; Ansari, G.A.; Khan, M.F. Nitrosative stress and nitrated proteins in trichloroethene-mediated autoimmunity. PLoS ONE 2014, 9, e98660. [Google Scholar] [CrossRef]
- Kudaravalli, J. Improvement in endothelial dysfunction in patients with systemic lupus erythematosus with n-acetylcysteine and atorvastatin. Indian J. Pharmacol. 2011, 43, 311–315. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, Y.J.; Kim, K.J.; Choi, J.J.; Kim, W.U.; Cho, C.S. Osteoprotegerin causes apoptosis of endothelial progenitor cells by induction of oxidative stress. Arthritis Rheum. 2013, 65, 2172–2182. [Google Scholar] [CrossRef]
- Fernandez, D.; Bonilla, E.; Mirza, N.; Niland, B.; Perl, A. Rapamycin reduces disease activity and normalizes t cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2983–2988. [Google Scholar] [CrossRef]
- Fernandez, D.; Perl, A. Metabolic control of t-cell activation and death in sle. Autoimmunity Rev. 2009, 8, 184–189. [Google Scholar] [CrossRef]
- Shivakumar, S.; Tsokos, G.C.; Datta, S.K. T cell receptor α/β expressing double-negative (cd4-/cd8-) and cd4+ t helper cells in humans augment the production of pathogenic anti-DNA autoantibodies associated with lupus nephritis. J. Immunol. 1989, 143, 103–112. [Google Scholar]
- Lai, Z.W.; Borsuk, R.; Shadakshari, A.; Yu, J.; Dawood, M.; Garcia, R.; Francis, L.; Tily, H.; Bartos, A.; Faraone, S.V.; et al. Mechanistic target of rapamycin activation triggers il-4 production and necrotic death of double-negative t cells in patients with systemic lupus erythematosus. J. Immunol. 2013, 191, 2236–2246. [Google Scholar] [CrossRef]
- Shi, D.; Li, X.; Chen, H.; Che, N.; Zhou, S.; Lu, Z.; Shi, S.; Sun, L. High level of reactive oxygen species impaired mesenchymal stem cell migration via overpolymerization of f-actin cytoskeleton in systemic lupus erythematosus. Pathologie-Biologie 2014, 62, 382–390. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.; Ma, H.; Firoze Khan, M. Increased nitration and carbonylation of proteins in mrl +/+ mice exposed to trichloroethene: Potential role of protein oxidation in autoimmunity. Toxicol. Appl. Pharmacol. 2009, 237, 188–195. [Google Scholar] [CrossRef]
- Liu, Y.; Kaplan, M.J. Cardiovascular disease in systemic lupus erythematosus: An update. Curr. Opin. Rheumatol. 2018, 30, 441–448. [Google Scholar] [CrossRef]
- Mak, A.; Kow, N.Y.; Schwarz, H.; Gong, L.; Tay, S.H.; Ling, L.H. Endothelial dysfunction in systemic lupus erythematosus—A case-control study and an updated meta-analysis and meta-regression. Sci. Rep. 2017, 7, 7320. [Google Scholar] [CrossRef]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Bairey Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. 2013 acc/aha guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American college of cardiology/american heart association task force on practice guidelines. Circulation 2014, 129, S1–S45. [Google Scholar] [CrossRef]
- Plazak, W.; Gryga, K.; Dziedzic, H.; Tomkiewicz-Pajak, L.; Konieczynska, M.; Podolec, P.; Musial, J. Influence of atorvastatin on coronary calcifications and myocardial perfusion defects in systemic lupus erythematosus patients: A prospective, randomized, double-masked, placebo-controlled study. Arthritis Res. Ther. 2011, 13, R117. [Google Scholar] [CrossRef]
- Mok, C.C.; Wong, C.K.; To, C.H.; Lai, J.P.; Lam, C.S. Effects of rosuvastatin on vascular biomarkers and carotid atherosclerosis in lupus: A randomized, double-blind, placebo-controlled trial. Arthritis Care Res. 2011, 63, 875–883. [Google Scholar] [CrossRef] [Green Version]
- Castejon, R.; Castaneda, A.; Sollet, A.; Mellor-Pita, S.; Tutor-Ureta, P.; Jimenez-Ortiz, C.; Yebra-Bango, M. Short-term atorvastatin therapy improves arterial stiffness of middle-aged systemic lupus erythematosus patients with pathological pulse wave velocity. Lupus 2017, 26, 355–364. [Google Scholar] [CrossRef]
- Ruiz-Limon, P.; Barbarroja, N.; Perez-Sanchez, C.; Aguirre, M.A.; Bertolaccini, M.L.; Khamashta, M.A.; Rodriguez-Ariza, A.; Almaden, Y.; Segui, P.; Khraiwesh, H.; et al. Atherosclerosis and cardiovascular disease in systemic lupus erythematosus: Effects of in vivo statin treatment. Ann. Rheum. Dis. 2015, 74, 1450–1458. [Google Scholar] [CrossRef]
- Watanabe, T.; Oku, K.; Amengual, O.; Hisada, R.; Ohmura, K.; Nakagawa, I.; Shida, H.; Bohgaki, T.; Horita, T.; Yasuda, S.; et al. Effects of statins on thrombosis development in patients with systemic lupus erythematosus and antiphospholipid antibodies. Lupus 2018, 27, 225–234. [Google Scholar] [CrossRef]
- Yu, H.H.; Chen, P.C.; Yang, Y.H.; Wang, L.C.; Lee, J.H.; Lin, Y.T.; Chiang, B.L. Statin reduces mortality and morbidity in systemic lupus erythematosus patients with hyperlipidemia: A nationwide population-based cohort study. Atherosclerosis 2015, 243, 11–18. [Google Scholar] [CrossRef]
- Rozo, C.; Chinenov, Y.; Maharaj, R.K.; Gupta, S.; Leuenberger, L.; Kirou, K.A.; Bykerk, V.P.; Goodman, S.M.; Salmon, J.E.; Pernis, A.B. Targeting the rhoa-rock pathway to reverse t-cell dysfunction in sle. Ann. Rheum. Dis. 2017, 76, 740–747. [Google Scholar] [CrossRef]
- Aprahamian, T.; Bonegio, R.; Rizzo, J.; Perlman, H.; Lefer, D.J.; Rifkin, I.R.; Walsh, K. Simvastatin treatment ameliorates autoimmune disease associated with accelerated atherosclerosis in a murine lupus model. J. Immunol. 2006, 177, 3028–3034. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Schindler, U.; Smiley, S.T.; Grusby, M.J. Stat6 is required for mediating responses to il-4 and for development of th2 cells. Immunity 1996, 4, 313–319. [Google Scholar] [CrossRef]
- Takeda, K.; Tanaka, T.; Shi, W.; Matsumoto, M.; Minami, M.; Kashiwamura, S.; Nakanishi, K.; Yoshida, N.; Kishimoto, T.; Akira, S. Essential role of stat6 in il-4 signalling. Nature 1996, 380, 627–630. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Sun, Y.L.; Hoey, T.; Grusby, M.J. Impaired il-12 responses and enhanced development of th2 cells in stat4-deficient mice. Nature 1996, 382, 174–177. [Google Scholar] [CrossRef]
- Jacobson, N.G.; Szabo, S.J.; Weber-Nordt, R.M.; Zhong, Z.; Schreiber, R.D.; Darnell, J.E., Jr.; Murphy, K.M. Interleukin 12 signaling in t helper type 1 (th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (stat)3 and stat4. J. Exp. Med. 1995, 181, 1755–1762. [Google Scholar] [CrossRef]
- Jury, E.C.; Isenberg, D.A.; Mauri, C.; Ehrenstein, M.R. Atorvastatin restores LCK expression and lipid raft-associated signaling in t cells from patients with systemic lupus erythematosus. J. Immunol. 2006, 177, 7416–7422. [Google Scholar] [CrossRef]
- Janes, P.W.; Ley, S.C.; Magee, A.I.; Kabouridis, P.S. The role of lipid rafts in t cell antigen receptor (tcr) signalling. Semin. Immunol. 2000, 12, 23–34. [Google Scholar] [CrossRef]
- Edmonds, S.D.; Ostergaard, H.L. Dynamic association of cd45 with detergent-insoluble microdomains in t lymphocytes. J. Immunol. 2002, 169, 5036–5042. [Google Scholar] [CrossRef]
- Ferreira, G.A.; Teixeira, A.L.; Sato, E.I. Atorvastatin therapy reduces interferon-regulated chemokine cxcl9 plasma levels in patients with systemic lupus erythematosus. Lupus 2010, 19, 927–934. [Google Scholar] [CrossRef]
- Mach, F.; Sauty, A.; Iarossi, A.S.; Sukhova, G.K.; Neote, K.; Libby, P.; Luster, A.D. Differential expression of three t lymphocyte-activating cxc chemokines by human atheroma-associated cells. J. Clin. Investig. 1999, 104, 1041–1050. [Google Scholar] [CrossRef]
- Sheikh, A.M.; Ochi, H.; Manabe, A.; Masuda, J. Lysophosphatidylcholine posttranscriptionally inhibits interferon-gamma-induced ip-10, mig and i-tac expression in endothelial cells. Cardiovasc. Res. 2005, 65, 263–271. [Google Scholar]
- Amuro, H.; Ito, T.; Miyamoto, R.; Sugimoto, H.; Torii, Y.; Son, Y.; Nakamichi, N.; Yamazaki, C.; Hoshino, K.; Kaisho, T.; et al. Statins, inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme a reductase, function as inhibitors of cellular and molecular components involved in type i interferon production. Arthritis Rheum. 2010, 62, 2073–2085. [Google Scholar]
- Ferreira, G.A.; Teixeira, A.L.; Calderaro, D.C.; Sato, E.I. Atorvastatin reduced soluble receptors of TNF-α in systemic lupus erythematosus. Clin. Exp. Rheumatol. 2016, 34, 42–48. [Google Scholar] [PubMed]
- Green, A.; Dobias, S.B.; Walters, D.J.; Brasier, A.R. Tumor necrosis factor increases the rate of lipolysis in primary cultures of adipocytes without altering levels of hormone-sensitive lipase. Endocrinology 1994, 134, 2581–2588. [Google Scholar] [CrossRef]
- Ormseth, M.J.; Swift, L.L.; Fazio, S.; Linton, M.F.; Raggi, P.; Solus, J.F.; Oeser, A.; Bian, A.; Gebretsadik, T.; Shintani, A.; et al. Free fatty acids are associated with metabolic syndrome and insulin resistance but not inflammation in systemic lupus erythematosus. Lupus 2013, 22, 26–33. [Google Scholar] [CrossRef]
- Parameswaran, N.; Patial, S. Tumor necrosis factor-α signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Houssen, M.E.; El-Mahdy, R.H.; Shahin, D.A. Serum soluble toll-like receptor 2: A novel biomarker for systemic lupus erythematosus disease activity and lupus-related cardiovascular dysfunction. Int. J. Rheum. Dis. 2016, 19, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Xiao, Y.; Chen, G.; Fu, D.; Ye, Y.; Liang, L.; Fan, J.; Yang, X.; Sun, L.; Xu, H. Hmg-coa reductase inhibitor simvastatin suppresses toll-like receptor 2 ligand-induced activation of nuclear factor kappa b by preventing rhoa activation in monocytes from rheumatoid arthritis patients. Rheumatol. Int. 2011, 31, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Pope, R.M. Toll-like receptor signaling: A potential link among rheumatoid arthritis, systemic lupus, and atherosclerosis. J. Leukoc. Biol. 2010, 88, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M. Toll-like receptor—A potent driving force behind rheumatoid arthritis. J. Clin. Exp. Hematopathol. 2011, 51, 77–92. [Google Scholar] [CrossRef]
- Raby, A.C.; Le Bouder, E.; Colmont, C.; Davies, J.; Richards, P.; Coles, B.; George, C.H.; Jones, S.A.; Brennan, P.; Topley, N.; et al. Soluble tlr2 reduces inflammation without compromising bacterial clearance by disrupting tlr2 triggering. J. Immunol. 2009, 183, 506–517. [Google Scholar] [CrossRef]
- Lawman, S.; Mauri, C.; Jury, E.C.; Cook, H.T.; Ehrenstein, M.R. Atorvastatin inhibits autoreactive b cell activation and delays lupus development in New Zealand black/white f1 mice. J. Immunol. 2004, 173, 7641–7646. [Google Scholar] [CrossRef] [PubMed]
- van Leuven, S.I.; Mendez-Fernandez, Y.V.; Wilhelm, A.J.; Wade, N.S.; Gabriel, C.L.; Kastelein, J.J.; Stroes, E.S.; Tak, P.P.; Major, A.S. Mycophenolate mofetil but not atorvastatin attenuates atherosclerosis in lupus-prone ldlr(−/−) mice. Ann. Rheum. Dis. 2012, 71, 408–414. [Google Scholar] [CrossRef]
- Graham, K.L.; Lee, L.Y.; Higgins, J.P.; Steinman, L.; Utz, P.J.; Ho, P.P. Failure of oral atorvastatin to modulate a murine model of systemic lupus erythematosus. Arthritis Rheum. 2008, 58, 2098–2104. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, A.; Moosavi, M.; Sayedbonakdar, Z.; Farajzadegan, Z.; Kazemi, M.; Smiley, A. Atorvastatin effect on systemic lupus erythematosus disease activity: A double-blind randomized clinical trial. Clin. Rheumatol. 2014, 33, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.A.; Kiani, A.N.; Post, W.; Christopher-Stine, L.; Magder, L.S. Lupus atherosclerosis prevention study (laps). Ann. Rheum. Dis. 2011, 70, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Willis, R.; Seif, A.M.; McGwin, G., Jr.; Martinez-Martinez, L.A.; Gonzalez, E.B.; Doan, E.; Dang, N.; Papalardo, E.; Liu, J.; Vila, L.M.; et al. Effects of statins on proinflammatory/prothrombotic biomarkers and on disease activity scores in sle patients: Data from lumina (lxxvi), a multi-ethnic us cohort. Clin. Exp. Rheumatol. 2014, 32, 162–167. [Google Scholar] [PubMed]
- Schanberg, L.E.; Sandborg, C.; Barnhart, H.X.; Ardoin, S.P.; Yow, E.; Evans, G.W.; Mieszkalski, K.L.; Ilowite, N.T.; Eberhard, A.; Imundo, L.F.; et al. Use of atorvastatin in systemic lupus erythematosus in children and adolescents. Arthritis Rheum. 2012, 64, 285–296. [Google Scholar] [CrossRef]
- El Messaoudi, S.; Russel, F.G.; Colbers, A.; Bandell, C.C.; van den Broek, P.H.; Burger, D.M.; Rongen, G.A.; Riksen, N.P. The effect of dipyridamole on the pharmacokinetics of metformin: A randomized crossover study in healthy volunteers. Eur. J. Clin. Pharmacol. 2016, 72, 725–730. [Google Scholar] [CrossRef]
- Manzi, S.; Meilahn, E.N.; Rairie, J.E.; Conte, C.G.; Medsger, T.A., Jr.; Jansen-McWilliams, L.; D’Agostino, R.B.; Kuller, L.H. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: Comparison with the Framingham study. Am. J. Epidemiol. 1997, 145, 408–415. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Author(s) [Ref] | Year | Type of Study | Summary of Mechanism/Results |
|---|---|---|---|
| Metformin | |||
| Yin, Y.; et al. [27] | 2015 | Animal study |
|
| Yin, Y.; et al. [26] | 2016 | Animal study |
|
| Lee, S.Y.; et al. [25] | 2017 | Animal study |
|
| Wang, H.; et al. [12] | 2015 | Human study (NCT02741960; active, not recruiting) |
|
| Dipyridamole | |||
| Kyttaris, V.C.; et al. [41] | 2011 | Human study (NCT01781611; recruiting) |
|
| N-Acetylcysteine | |||
| Suwannaroj, S.; et al. [50] | 2001 | Animal study |
|
| Kudaravalli, J.; et al. [52] | 2011 | Animal study |
|
| Kim, J.Y.; et al. [53] | 2013 | Animal study |
|
| Wang, G.; et al. [51] | 2014 | Animal study |
|
| Shi, D.; et al. [58] | 2014 | Animal and human study |
|
| Lai, Z.W.; et al. [46] | 2012 | Human study (NCT00775476; suspended—funds exhausted) |
|
| Doherty, E.; et al. [48] | 2014 | Human study |
|
| Perl, A.; et al. [47] | 2015 | Human study (NCT00775476; suspended—funds exhausted) |
|
| Statins | |||
| Lawman, S.; et al. [91] | 2004 | Animal study |
|
| Aprahamian, T.; et al. [70] | 2006 | Animal study |
|
| Jury, E.C.; et al. [75] | 2006 | Human study |
|
| Ferreira, G.A.; et al. [78] | 2010 | Human study |
|
| Amuro, H.; et al. [81] | 2010 | Human study |
|
| Ruiz-Limon, P.; et al. [66] | 2015 | Human study |
|
| Ferreira, G.A.; et al. [82] | 2016 | Human study |
|
| Houssen, M.E.; et al. [86] | 2016 | Human study |
|
| Rozo, C.; et al. [69] | 2017 | Human study |
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, M.K.X.; Heng, T.Y.J.; Mak, A. The Potential Use of Metformin, Dipyridamole, N-Acetylcysteine and Statins as Adjunctive Therapy for Systemic Lupus Erythematosus. Cells 2019, 8, 323. https://doi.org/10.3390/cells8040323
Tan MKX, Heng TYJ, Mak A. The Potential Use of Metformin, Dipyridamole, N-Acetylcysteine and Statins as Adjunctive Therapy for Systemic Lupus Erythematosus. Cells. 2019; 8(4):323. https://doi.org/10.3390/cells8040323
Chicago/Turabian StyleTan, Marcus Kai Xuan, Thurston Yan Jia Heng, and Anselm Mak. 2019. "The Potential Use of Metformin, Dipyridamole, N-Acetylcysteine and Statins as Adjunctive Therapy for Systemic Lupus Erythematosus" Cells 8, no. 4: 323. https://doi.org/10.3390/cells8040323
APA StyleTan, M. K. X., Heng, T. Y. J., & Mak, A. (2019). The Potential Use of Metformin, Dipyridamole, N-Acetylcysteine and Statins as Adjunctive Therapy for Systemic Lupus Erythematosus. Cells, 8(4), 323. https://doi.org/10.3390/cells8040323