Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism



1
Laboratory of Biochemistry, Faculty of Medicine, University of Thessaly, BIOPOLIS, 41500 Larissa, Greece
2
Gerald Bronfman Department of Oncology, Faculty of Medicine, McGill University, Montreal, QC H4A 3T2, Canada
3
Laboratory of Physiology, Faculty of Medicine, University of Thessaly, BIOPOLIS, 41500 Larissa, Greece
*
Authors to whom correspondence should be addressed.
Cells 2019, 8(3), 214; https://doi.org/10.3390/cells8030214
Submission received: 5 February 2019
/
Revised: 24 February 2019
/
Accepted: 26 February 2019
/
Published: 3 March 2019
Abstract
:Oxygen deprivation or hypoxia characterizes a number of serious pathological conditions and elicits a number of adaptive changes that are mainly mediated at the transcriptional level by the family of hypoxia-inducible factors (HIFs). The HIF target gene repertoire includes genes responsible for the regulation of metabolism, oxygen delivery and cell survival. Although the involvement of HIFs in the regulation of carbohydrate metabolism and the switch to anaerobic glycolysis under hypoxia is well established, their role in the control of lipid anabolism and catabolism remains still relatively obscure. Recent evidence indicates that many aspects of lipid metabolism are modified during hypoxia or in tumor cells in a HIF-dependent manner, contributing significantly to the pathogenesis and/or progression of cancer and metabolic disorders. However, direct transcriptional regulation by HIFs has been only demonstrated in relatively few cases, leaving open the exact and isoform-specific mechanisms that underlie HIF-dependency. This review summarizes the evidence for both direct and indirect roles of HIFs in the regulation of genes involved in lipid metabolism as well as the involvement of HIFs in various diseases as demonstrated by studies with transgenic animal models.
1. Oxygen Sensing and Hypoxia-Inducible Factor (HIF) Regulation
Insufficient oxygen availability in cells and tissues (hypoxia), a consequence of an imbalance between oxygen supply and metabolic demand, is encountered both physiologically (i.e., during intense exercise or embryogenesis) and in pathological conditions such as cancer, ischemia and metabolism related diseases. Response to hypoxia comprises reduction of oxygen consumption, via metabolic adjustments, and intensification of mechanisms responsible for oxygen transport to cells such as upregulation of erythropoiesis and angiogenesis. These adaptations require extensive reprogramming of gene expression, coordination of which is achieved by the hypoxia-inducible factors (HIFs) [1].
HIFs are heterodimeric transcription factors that consist of an oxygen regulated alpha subunit and a constitutively expressed beta subunit, also known as ARNT (aryl hydrocarbon receptor nuclear translocator), both members of the basic helix-loop-helix (bHLH) proteins of the PER-ARNT-SIM (PAS) DNA binding protein family. The active heterodimer binds to hypoxia-response elements (HREs) on the promoter or enhancer regions of target genes, causing their transcriptional activation [2]. Three HIF-α isoforms have been identified to date. HIF-1α is expressed ubiquitously in cells and tissues, while, HIF-2α (termed also EPAS1) is tissue specific [3,4]. The third and least studied HIF-α isoform, HIF-3α, exists in multiple splice variants most of which act as dominant-negative regulators of HIF activity [5,6].
Under physiological oxygen conditions, HIF-α isoforms are constantly produced and destroyed in a process that involves hydroxylation at two proline residues within a conserved HIF-α region termed oxygen dependent degradation domain (ODDD). This modification is catalyzed by three prolyl-hydroxylases (PHDs), enzymes that act as “oxygen sensors” in the cell, as their catalytic activity requires oxygen as a substrate. Following hydroxylation, HIF-α is recognized by the von Hippel–Lindau (VHL) tumor suppressor protein, an E3 ubiquitin ligase complex member, resulting to HIF-1α ubiquitination, targeting to the proteasome and degradation [7] (Figure 1). Another oxygen-sensitive enzyme, the asparaginyl hydroxylase FIH (factor-inhibiting HIF) modifies HIF-α subunits at the C-terminal transactivation domain and disrupts the interaction between HIF-α and the transcriptional co-activators p300/CBP thereby impairing residual HIF transcriptional activity [8].
In addition to oxygen tension, HIF-1 expression and activity are also controlled by oxygen-independent mechanisms regulating gene transcription, mRNA translation, protein–protein interactions and post-translational modification of the HIF-1α subunit. Transcriptional upregulation of the HIF-1α gene (HIF1A) in response to inflammation is achieved in a NF-κB-dependent manner [9,10,11]. Transcription of HIF1A also involves STAT3 (signal transducer and activator of transcription 3) [12] and Sp1 [13]. Moreover, activation of the PI-3K/AKT pathway by growth factors leads to increased HIF-1α mRNA and protein synthesis (reviewed in [2]). HIF-1α is also regulated through its association with other proteins. To mention only few examples, HIF-1α interaction with the molecular chaperone HSP90 results in its stabilization, whereas binding to RACK1, has the opposite effect [14,15,16]. Post-translationally, in addition to hydroxylation, HIF-1α is subject to SUMOylation [17,18,19,20], acetylation [21,22], deacetylation [23] and S-nitrosylation [24], although the impact of these modifications on HIF-1α stability and/or activity has not yet been adequately clarified. In contrast, direct phosphorylation by several kinases is important for HIF-1α regulation and is extensively studied (reviewed in [25]) (Figure 2).
Phosphorylation by GSK3 (glycogen synthase kinase 3) at three residues within the N-terminal transactivation domain causes degradation of HIF-1α in a VHL-independent manner [26]. A similar role has also been proposed for Plk3 (Polo-like kinase 3)-mediated phosphorylation of HIF-1α [27]. On the other hand, direct modifications of HIF-1α by ATM [28], CDK1 [29] or PKA [30] have been shown that stabilize HIF-1α by inhibiting its degradation. Downstream of its stabilization, transcriptional activity of HIF-1α also depends of its efficient accumulation inside the nucleus, a process regulated by ERK1/2-dependent phosphorylation. Translocation of HIF-1α inside the nucleus appears to be constitutive and is mediated by multiple import receptors; the importin α/β family, which recognize a nuclear localization signal (NLS) at the C-terminal domain of HIF-1α [31,32,33], as well as importins 4/7, which interact with the N-terminal part of HIF-1α [34]. However, CRM1-dependent nuclear export of HIF-1α depends in its modification by ERK1/2, which phosphorylates HIF-1α at sites adjacent to an atypical hydrophobic nuclear export signal (NES), thereby preventing CRM1 binding and increasing the nuclear concentration and activity of HIF-1α [35,36]. Inhibition of ERK-mediated phosphorylation of HIF-1α tips the balance in favor of nuclear export and cytoplasmic localization of a major pool of HIF-1α, which is bound by mortalin and targeted to the mitochondrial surface, where it participates to the formation of an anti-apoptotic complex [37].
Finally, phosphorylation by CK1δ (casein kinase 1δ) within the PAS domain impairs HIF-1α association with ARNT, hinders the formation of a functional heterodimer and thus, decreases HIF-1 transcriptional activity [38]. Interestingly, the association between HIF-1α and ARNT can also be inhibited by interaction of HIF-1α with MgcRacGAP (male germ cell RacGTPase Activating Protein) in cancer cells [39,40] or after treatment of human bronchial smooth muscle cells with the proinflammatory factor TNF-α [11]. Much less is known regarding the oxygen-independent mechanisms and post-translational modifications that regulate HIF-2α. The few examples include deacetylation by Sirt1, which enhances HIF-2α activity [41] and phosphorylation by CK1δ, which, in contrast to HIF-1α, promotes HIF-2α nuclear activity [42]. The crosstalk between the signaling pathways, that result to modification and regulation of the HIF-α isoforms, with those controlling metabolic homeostasis may ultimately define the exact role of HIFs in the metabolic adaptation of cells to hypoxia.
2. The Involvement of HIFs in the Regulation of Lipid Metabolism
When oxygen is sparse, cells adapt to hypoxia by reprogramming the expression of a number of genes involved in energy metabolism. The role of HIF-1 in the activation of genes encoding for proteins involved in carbohydrate metabolism has long been established (reviewed in [43,44]). HIF-1 not only promotes glucose uptake by activating the transcription of transporters GLUT1 and GLUT3, but also enhances anaerobic energy production, as it upregulates most of the glycolytic enzymes (including HK1/2, ENO1, PGK1 and PKM2) and proteins that facilitate the synthesis and excretion of lactate (LDH and MCT4). Moreover, in order to reduce mitochondrial function for decreasing consumption of oxygen and ROS production, HIF-1 stimulates the expression of pyruvate dehydrogenase kinase (PDK1) and BNIP3 [45,46,47]. PDK inhibits the pyruvate dehydrogenase complex and blocks the conversion of pyruvate, the glycolytic end product, to acetyl-CoA, which normally feeds into TCA cycle by producing citrate. Therefore, the flow of pyruvate into the mitochondria is decreased, fueling the production of lactate by LDH in the cytoplasm. On the other hand, BNIP3 triggers mitochondrial autophagy, further reducing mitochondrial metabolic processes.
Despite the extensive literature on HIF-dependent regulation of carbohydrate metabolism, the effects of hypoxia and HIFs on lipid metabolism have only recently become the focus of closer examination (Figure 3). Fatty acids (FAs), provided either by exogenous FA uptake or de novo synthesis, are used as substrates for oxidation and energy production, membrane synthesis, energy storage in form of triacylglycerols (TAGs) and production of signaling molecules and, therefore, are essential for cell survival and proliferation both under normoxia and hypoxia. However, as FA oxidation takes place inside mitochondria and requires oxygen, FA metabolism has to be modified under hypoxia in order to serve mainly processes other than energy production. Furthermore, as conversion of glucose into citrate—the major source of cytoplasmic acetyl-CoA and FA precursor—is prohibited under hypoxia due to the inhibition of the TCA cycle, alternative sources of FA precursors have to be exploited. In tumor cells, which usually have to grow in a hypoxic microenvironment, these hypoxia-mediated changes in lipid metabolism are especially important in order to maintain the high proliferation rate that characterizes cancer cells.
Uptake of extracellular FA and TAG synthesis are promoted under hypoxia by transcription factor PPARγ, the gene of which is a directly activated by HIF-1 [48]. Extracellular FA influx and lipogenesis under hypoxia are also enhanced via HIF-1-mediated induction of the expression of FABP (fatty acid binding protein) 3 and 7 in cancer cells [49] and FABP4 in primary mouse hepatocytes [50]. In addition, HIF-1 can promote the endocytosis of lipoproteins, by upregulating the expression of low-density lipoprotein receptor–related protein (LRP1), the receptor that internalizes LDL in vascular smooth muscle cells [51], as well as the expression of VLDL receptor (VLDLR) in cardiomyocytes [52].
To also maintain de novo FA synthesis under hypoxia, production of FA precursors is supported in human renal cell carcinoma (RCC) as well as other cancer cells through HIF-dependent stimulation of reductive glutamine metabolism [53,54]. This proceeds via conversion of glutamine to α-ketoglutarate and its subsequent reductive carboxylation that produces citrate, in a reversion of the TCA cycle reaction catalyzed by IDH (isocitrate dehydrogenase). This may be an indirect result of the HIF-mediated decrease of intracellular citrate levels (due to upregulation of PDK1) but IDH1 or 2 may also actively contribute to the preservation of citrate levels under hypoxia [55,56,57]. Moreover, HIF-1 increases the amount of α-ketoglutarate, which can be used as substrate for citrate synthesis and FA/lipid production, by inducing the expression of GLS1 (glutaminase 1) [58], as well as, by inducing the E3 ubiquitin ligase SIAH2, which in turn mediates the proteolysis of the E1 subunit (OGDH2) of the α-ketoglutarate dehydrogenase complex (αKGDH) [57]. Adequate FA supply is further supported by Akt- and HIF-1-dependent activation of SREBP-1, which in turn upregulates the expression of FASN (fatty acid synthase), an essential lipogenic enzyme, the activity of which is correlated with cancer progression and hypoxia induced chemoresistance [59].
As FA catabolism is impaired under hypoxia, an excess of intracellularly accumulated free FAs could cause lipotoxicity. To avoid this, cells can convert FAs to neutral TAGs, that are stored in lipid droplets (LDs) and can serve as the main form of energy depots [60,61]. Two enzymes of the TAG biosynthesis pathway, AGPAT2 (acylglycerol-3-phosphate acyltransferase 2) [62] and lipin-1 [63], have been shown to mediate hypoxia-induced LD accumulation. AGPAT2, or else LPAATβ (lysophosphatidic acid acyltransferase β), catalyzes the conversion of lysophosphatidic acid (LPA) to phosphatidic acid (PA). Interestingly AGPAT2, which is a direct target of HIF-1 [62], is one of the genes mutated in patients with congenital generalized lipodystrophy, and is upregulated in biopsies from cancer patients. Likewise, HIF-1 also directly upregulates the expression of lipin-1, a phosphatidic acid (PA) phosphatase that catalyzes the conversion of PA to diacylglycerol (DAG) in TAG synthesis [63]. AGPAT2 and lipin-1 upregulation is necessary for LD accumulation and increased viability and chemoresistance under hypoxia [62,63,64]. The importance of the hypoxic upregulation of AGPAT2 and lipin-1 may extend beyond the formation of lipid droplets. The products of their catalytic activity LPA and PA can either be used as precursors of TAGs or as precursors for the synthesis of phospholipids, which are important blocks for new membrane formation [61]. Formation of lipid droplets under hypoxia is further favored by the hypoxic induction of essential constituents of LD membranes. Stimulation of the LD coat protein adipophilin/perilipin 2 (PLIN2) expression by HIF-2 promotes RCC lipid storage, ER homeostasis and viability [65], and the induction of HIG2/HILPDA (Hypoxia-inducible protein 2/hypoxia-inducible lipid droplet associated) by HIF-1 increases lipid accumulation in both cancer and normal cells [66,67]. Furthermore, HIG2 upregulation under hypoxia inhibits the adipose triglyceride lipase (ATGL) and impairs intracellular lipolysis in various cancer cells [68].
Finally, lipid accumulation under hypoxia is additionally supported by the inhibition of enzymes involved in fatty acid degradation. Under low oxygen concentration, fatty acid β-oxidation is actively reduced by HIF-1- and HIF-2-dependent downregulation of the transcriptional coactivator of β-oxidation enzyme PGC-1α (proliferator-activated receptor-γ coactivator-1α) [69] and carnitine palmitoyltransferase 1A (CPT1A), the limiting component of mitochondrial fatty acid transport, in both hepatoma and RCC cells [69,70] as well as by the HIF-1-mediated decreased expression of MCAD and LCAD (medium- and long-chain acyl-CoA dehydrogenases) in hepatoma cells, which depends on the hypoxic inhibition of PGC-1β, a transcription factor involved in mitochondrial regulation [71]. As HIFs have not been shown to possess intrinsic transcription repressor activity, downregulation of these enzymes may be mediated by the action of HIF-1 target genes that remain, in most cases, to be identified. In summary, hypoxia overall causes enhanced lipogenesis by HIF-dependent induction of genes involved in FA uptake, synthesis and storage (Table 1). Importantly, as discussed below, induction of these genes and subsequent lipid accumulation are indispensable for cancer cell proliferation under hypoxia.
3. HIF-Dependent Regulation of Lipid Metabolism and Cancer Cell Proliferation
Hypoxia develops in tumors as a consequence of the high proliferation rate of cancer cells and aberrant angiogenesis. Activation of the hypoxia response pathway helps cancer cells to adapt and survive by affecting multiple metabolic pathways [72]. Enhanced esterification of free FAs to neutral TAGs and storage in expanded LDs, protects cancer cells from lipotoxicity [73]. In addition, segregation of free FAs in LDs protects solid tumor cancer cells that are exposed to intermittent hypoxia from the lethal formation of free radicals during cycles of hypoxia and reoxygenation [49,65,74,75].
Besides their role in sequestering potential harmful FAs, LDs serve as energy stores and reservoirs of building blocks for the production of the essential sterol esters and phospholipids required in proliferating cells for the biogenesis of new membranes (reviewed in [76]). A connection between HIF-induced TG synthesis and cell proliferation is supported by metabolic profiling analysis of cancer cells kept under hypoxia, which has shown that the concentration of TAGs and derivative phospholipids PC and PE is substantially increased in a HIF-1α-dependent manner [77]. It has to be pointed out that, in many cancer types, silencing of HIFs or interfering with the expression of its target genes required for lipid accumulation, results in reduction of proliferation potential and chemoresistance under hypoxia [49,55,57,59,62,63,65,68,70,78,79]. Moreover, the overexpression of HIF-regulated genes involved in lipid metabolism has been correlated with malignant subtypes of human cancers or poor patient prognosis [70,79]. Intervening with HIF-dependent reprogramming of lipid metabolism can indeed suppress effectively cancer cell proliferation. Systemic administration of glutaminase inhibitors suppressed the growth of RCC cells as xenografts in mice [54], while recent studies have shown that modulation of HIF-1α phosphorylation can regulate LD accumulation and cancer cell growth, specifically under hypoxia [64,80]. Modification of HIF-1α by CK1δ reduced induction of lipin-1 and restricted lipid droplet formation and cell proliferation under hypoxia in a HIF-1 and lipin-1-dependent manner [64]. In addition, inhibition of ERK-mediated phosphorylation of HIF-1α by transduced recombinant HIF-1α-derived peptides abolished induction of lipin-1 expression, reduced lipid droplet accumulation and triggered apoptosis in cancer cells grown under hypoxia [80].
4. HIF-Dependent Regulation of Lipid Metabolism in Obesity and Metabolic Syndrome
HIF-dependent regulation of lipid metabolism in response to hypoxia, or other stimuli including diet, has been implicated in disorders affecting organs involved in lipid processing and storage, such as the adipose tissue and the liver. In obesity, enlargement of adipocytes beyond the oxygen diffusion limit and their distancing from the vasculature, leads to the development of local hypoxia (reviewed in [81]). Accordingly, visceral adipose tissue from obese human subjects is characterized by increased expression of HIF-1α [82]. Hypoxia has been shown to increase liver lipid contents via induction of HIFs in mice [69] and hepatocellular carcinoma cells [63], while a theoretical model of hepatic lipid accumulation, suggests that hypoxia is contributing to lipid accumulation and steatosis [83]. In addition, several studies have shown that liver HIF stabilization after hepatocyte-specific VHL deletion increased liver lipid accumulation [45,84,85]. In one case, liver specific overexpression of constitutively active forms of both HIF-1α and HIF-2α was required to phenocopy the VHL deletion suggesting that both isoforms are involved in the accumulation of lipids in the liver [45]. On the other hand, experiments in which HIF-1α, HIF-2α or both isoforms were deleted concomitantly with VHL indicated that liver lipid accumulation was mediated predominantly via activation of HIF-2α [84,85]. A number of animal studies based on the deletion or overexpression of HIFs or other components of the hypoxia-response network, suggest that HIF activation can be either beneficial or detrimental in terms of metabolic disease. As this subject has recently been reviewed [86], only a brief overview will be presented.
4.1. HIFs as Suppressors of Obesity
A number of studies have shown that obesity is increased by inhibition of HIFs and decreased by HIF activation. In an in vitro adipocyte differentiation study, hypoxia inhibited adipogenesis via HIF-1-dependent upregulation of DEC1/Stra13 and subsequent repression of PPARγ2 expression [87]. Accordingly, transgenic mice overexpressing an adipose tissue-selective dominant negative HIF-1α mutant that decreased HIF-1 activity, developed increased obesity after high-fat diet treatment and accumulated enlarged adipocyte LDs [88]. Similar phenotypes were also observed after HIF activation in adipose tissue specific PHD2 knockout (KO) mice [89] or global FIH KO mice [90], which in both cases protected from high-fat diet-induced obesity. Interestingly, the effects of neuron-specific FIH knockouts resembled those of the global null mutants, suggesting that the nervous system is implicated in the FIH-driven regulation of metabolism [90].
4.2. HIFs as Promoters of Obesity
In contrast to the above, there have been studies showing that HIF activation induces obesity. Adipocyte specific KO of HIF1A [82,91,92,93], inhibition of HIF-1 by acriflavine [91] or PX-478 [94], or adipocyte specific ARNT KO [93,95] decreased obesity and insulin resistance in mice fed with high-fat diet. In agreement, adipose specific ablation of the PHD2 gene caused HIF-1-dependent reduction of lipolysis and enhanced adiposity in mice [96]. This effect can be correlated with the capacity of HIF-1 to downregulate FA oxidation in adipose tissue [82]. Resistance of mice with depletion of adipocyte HIF-1α to insulin has also been linked to the downregulation of adiponectin expression via HIF-1-mediated regulation of the SOCS3-STAT3 signal transduction pathway [91].
4.3. HIFs and Non-Alcoholic Fatty Liver Disease (NAFLD)
Hypoxia and obesity are also linked to liver diseases, such as non-alcoholic fatty liver disease (NAFLD), characterized by inflammation, fibrosis and steatosis. Diet, adipokines and stress are significant contributing factors in NALFD [97]. Although the causality and molecular mechanisms that underlie NAFLD are not completely understood, the development of hypoxia in the liver is implicated in the pathogenesis of the disease. Hepatocyte-specific HIF-1 activation has been shown to promote alcohol-induced hepatomegaly and hepatic lipid accumulation, while hepatocyte-specific deletion of HIF-1α protected mice from alcohol- and lipopolysaccharide (LPS)-induced liver damage, hepatomegaly and lipid accumulation [98]. In a contrasting study, hepatocyte-specific HIF-1α-null mice exposed to ethanol-containing liquid diet exhibited enhanced accumulation of lipids in the liver, via inactivation of the HIF-1-regulated transcriptional repressor DEC1 [99]. Similarly, HIF-1α liver KO enhanced lipid accumulation in choline deprivation-induced NAFLD [100]. Interestingly in this case, liver lipid accumulation was inhibited by overexpression of lipin-1, the direct target of HIF-1 mediating TAG biosynthesis. This was attributed to a non-catalytic nuclear function of lipin-1 and regulation of the PPARα target genes controlling peroxisomal fatty acid oxidation [100]. Finally, it appears that extrahepatic expression of HIFs may also affect liver lipid metabolism. A recent study has shown that mice with intestine-specific disruption of HIF-2α had substantially lower high-fat-diet-induced hepatic steatosis and obesity compared to control animals [101]. This effect was also reproduced when mice were treated with PT2385, a HIF-2α-specific inhibitor. Subsequent analysis suggested that hepatic steatosis developed as a result of decreased ceramide production in the intestine, a process involving a direct gene target of HIF-2, Neu3 (neuraminidase 3) [101].
In conclusion, the animal studies that have investigated the involvement of HIFs in obesity and other metabolic disorders are often conflicting and do not clarify their exact role in the dysregulation of metabolism that contributes to the onset of these disorders. These discrepancies may reflect differences in the genetic background, age and diet of the mice used in these studies or the level of inhibition of HIF activity achieved with the different genetic or pharmaceutical approaches. In addition, they may also result from the complexity of systemic metabolic regulation in combination with the multifaceted roles of HIFs in cellular functions that extend further than lipid metabolism.
5. HIF-Dependent Regulation of Lipid Metabolism in Cardiovascular Disease
Deregulation of the adipose tissue function and ectopic lipid accumulation is a primary factor for the development of cardiovascular disease. A number of studies indicate that many of the HIF-target genes involved in lipid metabolism can contribute to cardiovascular pathogenesis. Upregulation of LRP1 by HIF-1 contributes to the deposition of lipids in atherosclerotic plaques in human vascular smooth muscle cells, while vascular cell LRP1 and HIF-1α co-localize in immunohistochemical samples of human advanced atherosclerotic plaques [51]. Another HIF-1 target gene, HIG2/Hilpda, stimulates lesion formation and development of atherosclerosis, as the expression of various atherosclerotic pathogenic markers was decreased by conditional Hilpda KO in macrophages of ApoE-/- mice [67]. This is in line with older in vitro studies showing hypoxia-dependent formation of cytosolic lipid LDs in macrophages [102]. Concerning the direct effects of hypoxia on cardiac function, experiments with ventricular HIF-1α KO mice have shown that HIF-1-induced PPARγ activation contributes to metabolic reprogramming and development of contractile dysfunction under pathological stress [48]. Similarly, VHL-null hearts, in which HIFs were activated, developed a number of features associated with human heart failure, including lipid accumulation, myofibril rarefaction, altered nuclear morphology, myocyte loss, and fibrosis, resulting in premature death [103]. These pathogenic features were prevented by the simultaneous cardiac ablation of both VHL and HIF-1α, strongly suggesting the involvement of HIF-1. Interestingly, deletion of VHL specifically in mice adipocytes also caused the development of lethal cardiac hypertrophy, which was, however rescued by genetic deletion of HIF-2α but not HIF-1α [104]. In contrast to the harmful effects of VHL deletion, inhibition of PHDs that also leads to HIF activation has been suggested to play a protective role in cardiovascular disease. In atherosclerotic mice due to LDLR (low-density lipoprotein receptor) KO, deletion of PHD1 [105] or PHD inhibition [106] resulted to reduced atherosclerotic plaque development.
On the other hand, genetic deletion of PHD2 in endothelial and hematopoietic mouse cells induced severe pulmonary vascular remodeling and right ventricular hypertrophy, characteristic features of clinical pulmonary arterial hypertension [107]. Although the phenotypes caused by PHD KO cannot be necessarily attributed to HIF activity, since PHDs may also have additional substrates or partners [108], pulmonary hypertension has been long known to be linked to HIF activation, since exposure to chronic hypoxia can indeed cause pulmonary arterial smooth muscle cell proliferation, migration and hypertrophy leading to pulmonary vascular remodeling and eventually pulmonary hypertension [109]. Many studies with both human subjects and animal models have implicated HIFs in the response of the pulmonary vasculature to hypoxia and also revealed the involvement of HIFs in forms of pulmonary hypertension not directly caused by hypoxia (reviewed in [110]). Pulmonary vascular remodeling is supported by extensive metabolic reprogramming, affecting both glucose and lipid metabolism, many aspects of which may be mediated by HIFs [111,112]. The importance of this reprogramming is illustrated by the fact that deficiency of malonyl-CoA decarboxylase, a key regulatory enzyme for fatty acid oxidation, in mice can attenuate the vasoconstriction and vascular remodeling caused by hypoxia [113,114]. Recent metabolomics studies in a murine model of pulmonary arterial hypertension have indeed shown changes in lung tissue lipid composition compatible with HIF-dependent metabolic reprogramming [115]. However, whether any of the HIF targets listed Table 1 is directly involved in pulmonary vascular remodeling remains to be shown.
6. Conclusions
Recent information gathered from investigations in cell lines, animals and patient biopsy samples signify the importance of hypoxia and HIF activation in the regulation of lipid metabolism, and their contribution to the development and progression of cancer and other pathological conditions associated with the accumulation of lipids in various types of cells and organs. A number of HIF inhibitors are currently being tested, along with conventional therapies, for the treatment of different types of cancer [116], as cancer cells depend on HIF function, including HIF-mediated stimulation of lipid synthesis, for survival, proliferation and metastasis. As many studies have also shown that HIF inactivation by deletion, silencing or chemical inhibition can revert the effects of lipid accumulation in various mouse models, targeting of HIF function may also represent a valid therapeutic approach in metabolic diseases. However, as the repertoire of direct HIF targets involved in the complex regulation of lipid metabolism is far from exhausted, further detailed investigation is required to reveal the exact steps controlled by HIFs, especially in terms of HIF-α isoform and tissue specificity.
Funding
This work was partially supported by a grant provided by the Research Committee of the University of Thessaly (Contract Nr 5309.17.03) to E.P.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Bardos, J.I.; Ashcroft, M. Negative and positive regulation of HIF-1: A complex network. Biochim. Biophys. Acta 2005, 1755, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Poon, E.; Harris, A.L.; Ashcroft, M. Targeting the hypoxia-inducible factor (HIF) pathway in cancer. Expert Rev. Mol. Med. 2009, 11, e26. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Duan, C. Hypoxia-inducible factor 3 biology: Complexities and emerging themes. Am. J. Physiol. Cell Physiol. 2016, 310, C260–C269. [Google Scholar] [CrossRef] [PubMed]
- Ravenna, L.; Salvatori, L.; Russo, M.A. HIF3alpha: The little we know. FEBS J. 2016, 283, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Ratcliffe, P.J. Signalling hypoxia by HIF hydroxylases. Biochem. Biophys. Res. Commun. 2005, 338, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliffe, P.J. New horizons in hypoxia signaling pathways. Exp. Cell Res. 2017, 356, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Belaiba, R.S.; Bonello, S.; Zahringer, C.; Schmidt, S.; Hess, J.; Kietzmann, T.; Gorlach, A. Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol. Biol. Cell 2007, 18, 4691–4697. [Google Scholar] [CrossRef] [PubMed]
- Tsapournioti, S.; Mylonis, I.; Hatziefthimiou, A.; Ioannou, M.G.; Stamatiou, R.; Koukoulis, G.K.; Simos, G.; Molyvdas, P.A.; Paraskeva, E. TNFalpha induces expression of HIF-1alpha mRNA and protein but inhibits hypoxic stimulation of HIF-1 transcriptional activity in airway smooth muscle cells. J. Cell Physiol. 2013, 228, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, A.I.; Paraskeva, E.; Peidis, P.; Muaddi, H.; Li, S.; Raptis, L.; Pantopoulos, K.; Simos, G.; Koromilas, A.E. eIF2α Kinase PKR modulates the hypoxic response by Stat3-dependent transcriptional suppression of HIF-1α. Cancer Res. 2010, 70, 7820–7829. [Google Scholar] [CrossRef] [PubMed]
- Vlaminck, B.; Toffoli, S.; Ghislain, B.; Demazy, C.; Raes, M.; Michiels, C. Dual effect of echinomycin on hypoxia-inducible factor-1 activity under normoxic and hypoxic conditions. FEBS J. 2007, 274, 5533–5542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Liu, Y.V.; McDonald, K.R.; Wesley, J.B.; Zhang, H.; Semenza, G.L. Spermidine/spermine N(1)-acetyltransferase-1 binds to hypoxia-inducible factor-1alpha (HIF-1alpha) and RACK1 and promotes ubiquitination and degradation of HIF-1alpha. J. Biol. Chem. 2007, 282, 33358–33366. [Google Scholar] [CrossRef] [PubMed]
- Amir, S.; Wang, R.; Simons, J.W.; Mabjeesh, N.J. SEPT9_v1 up-regulates hypoxia-inducible factor 1 by preventing its RACK1-mediated degradation. J. Biol. Chem. 2009, 284, 11142–11151. [Google Scholar] [CrossRef] [PubMed]
- Berta, M.A.; Mazure, N.; Hattab, M.; Pouyssegur, J.; Brahimi-Horn, M.C. SUMOylation of hypoxia-inducible factor-1alpha reduces its transcriptional activity. Biochem. Biophys. Res. Commun. 2007, 360, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Carbia-Nagashima, A.; Gerez, J.; Perez-Castro, C.; Paez-Pereda, M.; Silberstein, S.; Stalla, G.K.; Holsboer, F.; Arzt, E. RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1alpha during hypoxia. Cell 2007, 131, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Jeong, J.W.; Park, J.A.; Kim, S.H.; Bae, M.K.; Choi, S.J.; Kim, K.W. Sumoylation increases HIF-1alpha stability and its transcriptional activity. Biochem. Biophys. Res. Commun. 2004, 324, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Kang, X.; Zhang, S.; Yeh, E.T. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell 2007, 131, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Wei, W.; Yu, X.D. Hypoxia-inducible factors: Crosstalk between their protein stability and protein degradation. Cancer Lett. 2007, 257, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sonveaux, P.; Rabbani, Z.N.; Liu, S.; Yan, B.; Huang, Q.; Vujaskovic, Z.; Dewhirst, M.W.; Li, C.Y. Regulation of HIF-1alpha stability through S-nitrosylation. Mol. Cell 2007, 26, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Mennerich, D.; Dimova, E.Y. Hypoxia-Inducible Factors (HIFs) and Phosphorylation: Impact on Stability, Localization, and Transactivity. Front. Cell Dev. Biol. 2016, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Flugel, D.; Gorlach, A.; Michiels, C.; Kietzmann, T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol. Cell Biol. 2007, 27, 3253–3265. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Yao, Y.; Lu, L.; Costa, M.; Dai, W. Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of HIF-1alpha. J. Biol. Chem. 2010, 285, 38944–38950. [Google Scholar] [CrossRef] [PubMed]
- Cam, H.; Easton, J.B.; High, A.; Houghton, P.J. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1alpha. Mol. Cell 2010, 40, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Warfel, N.A.; Dolloff, N.G.; Dicker, D.T.; Malysz, J.; El-Deiry, W.S. CDK1 stabilizes HIF-1alpha via direct phosphorylation of Ser668 to promote tumor growth. Cell Cycle 2013, 12, 3689–3701. [Google Scholar] [CrossRef] [PubMed]
- Bullen, J.W.; Tchernyshyov, I.; Holewinski, R.J.; DeVine, L.; Wu, F.; Venkatraman, V.; Kass, D.L.; Cole, R.N.; Van Eyk, J.; Semenza, G.L. Protein kinase A-dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci. Signal. 2016, 9, ra56. [Google Scholar] [CrossRef] [PubMed]
- Depping, R.; Steinhoff, A.; Schindler, S.G.; Friedrich, B.; Fagerlund, R.; Metzen, E.; Hartmann, E.; Kohler, M. Nuclear translocation of hypoxia-inducible factors (HIFs): Involvement of the classical importin alpha/beta pathway. Biochim. Biophys. Acta 2008, 1783, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Kallio, P.J.; Okamoto, K.; O’Brien, S.; Carrero, P.; Makino, Y.; Tanaka, H.; Poellinger, L. Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha. EMBO J. 1998, 17, 6573–6586. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.C.; Shibuya, M. A variant of nuclear localization signal of bipartite-type is required for the nuclear translocation of hypoxia inducible factors (1alpha, 2alpha and 3alpha). Oncogene 2001, 20, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Chachami, G.; Paraskeva, E.; Mingot, J.M.; Braliou, G.G.; Gorlich, D.; Simos, G. Transport of hypoxia-inducible factor HIF-1alpha into the nucleus involves importins 4 and 7. Biochem. Biophys. Res. Commun 2009, 390, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Chachami, G.; Samiotaki, M.; Panayotou, G.; Paraskeva, E.; Kalousi, A.; Georgatsou, E.; Bonanou, S.; Simos, G. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J. Biol. Chem. 2006, 281, 33095–33106. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Chachami, G.; Paraskeva, E.; Simos, G. Atypical CRM1-dependent nuclear export signal mediates regulation of hypoxia-inducible factor-1alpha by MAPK. J. Biol. Chem. 2008, 283, 27620–27627. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Kourti, M.; Samiotaki, M.; Panayotou, G.; Simos, G. Mortalin-mediated and ERK-controlled targeting of HIF-1alpha to mitochondria confers resistance to apoptosis under hypoxia. J. Cell Sci. 2017, 130, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Kalousi, A.; Mylonis, I.; Politou, A.S.; Chachami, G.; Paraskeva, E.; Simos, G. Casein kinase 1 regulates human hypoxia-inducible factor HIF-1. J. Cell Sci. 2010, 123, 2976–2986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyberopoulou, A.; Venieris, E.; Mylonis, I.; Chachami, G.; Pappas, I.; Simos, G.; Bonanou, S.; Georgatsou, E. MgcRacGAP interacts with HIF-1alpha and regulates its transcriptional activity. Cell Physiol. Biochem. 2007, 20, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- Lyberopoulou, A.; Mylonis, I.; Papachristos, G.; Sagris, D.; Kalousi, A.; Befani, C.; Liakos, P.; Simos, G.; Georgatsou, E. MgcRacGAP, a cytoskeleton regulator, inhibits HIF-1 transcriptional activity by blocking its dimerization. Biochim. Biophys. Acta 2013, 1833, 1378–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dioum, E.M.; Chen, R.; Alexander, M.S.; Zhang, Q.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science 2009, 324, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Pangou, E.; Befani, C.; Mylonis, I.; Samiotaki, M.; Panayotou, G.; Simos, G.; Liakos, P. HIF-2alpha phosphorylation by CK1delta promotes erythropoietin secretion in liver cancer cells under hypoxia. J. Cell Sci. 2016, 129, 4213–4226. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Semenza, G.L. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Simon, M.C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 2017, 292, 16825–16832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.Y.; Safran, M.; Buckley, M.R.; Ebert, B.L.; Glickman, J.; Bosenberg, M.; Regan, M.; Kaelin, W.G., Jr. Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation in vivo. EMBO J. 2006, 25, 4650–4662. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, L.; Liu, H.; Prabhakaran, K.; Zhang, X.; Borowitz, J.L.; Isom, G.E. HIF-1alpha activation by a redox-sensitive pathway mediates cyanide-induced BNIP3 upregulation and mitochondrial-dependent cell death. Free Radic. Biol. Med. 2007, 43, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, J.; Suter, M.; Windak, R.; Krebs, T.; Felley, A.; Montessuit, C.; Tokarska-Schlattner, M.; Aasum, E.; Bogdanova, A.; Perriard, E.; et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009, 9, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, Y.; Garbacz, W.G.; Jiang, M.; Xu, M.; Huang, H.; Tsung, A.; Billiar, T.R.; Ramakrishnan, S.K.; Shah, Y.M.; et al. Fatty acid binding protein-4 (FABP4) is a hypoxia inducible gene that sensitizes mice to liver ischemia/reperfusion injury. J. Hepatol. 2015, 63, 855–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, J.; Aledo, R.; Sendra, J.; Costales, P.; Juan-Babot, O.; Badimon, L.; Llorente-Cortes, V. Hypoxia stimulates low-density lipoprotein receptor-related protein-1 expression through hypoxia-inducible factor-1alpha in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Perman, J.C.; Bostrom, P.; Lindbom, M.; Lidberg, U.; StAhlman, M.; Hagg, D.; Lindskog, H.; Scharin Tang, M.; Omerovic, E.; Mattsson Hulten, L.; et al. The VLDL receptor promotes lipotoxicity and increases mortality in mice following an acute myocardial infarction. J. Clin. Investig. 2011, 121, 2625–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gameiro, P.A.; Yang, J.; Metelo, A.M.; Perez-Carro, R.; Baker, R.; Wang, Z.; Arreola, A.; Rathmell, W.K.; Olumi, A.; Lopez-Larrubia, P.; et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013, 17, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Mou, J.; Shao, B.; Wei, Y.; Liang, H.; Takano, N.; Semenza, G.L.; Xie, G. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 2019, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Furuta, E.; Pai, S.K.; Zhan, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Airola, M.V.; Reue, K. How lipid droplets “TAG” along: Glycerolipid synthetic enzymes and lipid storage. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Siniossoglou, S. Phospholipid metabolism and nuclear function: Roles of the lipin family of phosphatidic acid phosphatases. Biochim. Biophys. Acta 2013, 1831, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Triantafyllou, E.A.; Georgatsou, E.; Mylonis, I.; Simos, G.; Paraskeva, E. Expression of AGPAT2, an enzyme involved in the glycerophospholipid/triacylglycerol biosynthesis pathway, is directly regulated by HIF-1 and promotes survival and etoposide resistance of cancer cells under hypoxia. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Sembongi, H.; Befani, C.; Liakos, P.; Siniossoglou, S.; Simos, G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J. Cell Sci. 2012, 125, 3485–3493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourti, M.; Ikonomou, G.; Giakoumakis, N.N.; Rapsomaniki, M.A.; Landegren, U.; Siniossoglou, S.; Lygerou, Z.; Simos, G.; Mylonis, I. CK1delta restrains lipin-1 induction, lipid droplet formation and cell proliferation under hypoxia by reducing HIF-1alpha/ARNT complex formation. Cell. Signal. 2015. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Ackerman, D.; Sanchez, D.J.; Li, B.; Ochocki, J.D.; Grazioli, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; Simon, M.C. HIF2alpha-Dependent Lipid Storage Promotes Endoplasmic Reticulum Homeostasis in Clear-Cell Renal Cell Carcinoma. Cancer Discov. 2015, 5, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Gimm, T.; Wiese, M.; Teschemacher, B.; Deggerich, A.; Schodel, J.; Knaup, K.X.; Hackenbeck, T.; Hellerbrand, C.; Amann, K.; Wiesener, M.S.; et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 2010, 24, 4443–4458. [Google Scholar] [CrossRef] [PubMed]
- Maier, A.; Wu, H.; Cordasic, N.; Oefner, P.; Dietel, B.; Thiele, C.; Weidemann, A.; Eckardt, K.U.; Warnecke, C. Hypoxia-inducible protein 2 Hig2/Hilpda mediates neutral lipid accumulation in macrophages and contributes to atherosclerosis in apolipoprotein E-deficient mice. FASEB J. 2017, 31, 4971–4984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Saarinen, A.M.; Hitosugi, T.; Wang, Z.; Wang, L.; Ho, T.H.; Liu, J. Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ma, Z.; Zhao, C.; Wang, Y.; Wu, G.; Xiao, J.; McClain, C.J.; Li, X.; Feng, W. HIF-1α and HIF-2α are critically involved in hypoxia-induced lipid accumulation in hepatocytes through reducing PGC-1α-mediated fatty acid β-oxidation. Toxicol. Lett. 2014, 226, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Zhang, L.; Brett-Morris, A.; Aguila, B.; Kerner, J.; Hoppel, C.L.; Puchowicz, M.; Serra, D.; Herrero, L.; Rini, B.I.; et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 2017, 8, 1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Li, T.; Li, X.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Yoo, W.; Noh, K.H.; Ahn, J.H.; Yu, J.H.; Seo, J.A.; Kim, S.G.; Choi, K.M.; Baik, S.H.; Choi, D.S.; Kim, T.W.; et al. HIF-1alpha expression as a protective strategy of HepG2 cells against fatty acid-induced toxicity. J. Cell. Biochem. 2014, 115, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, D.; Tumanov, S.; Qiu, B.; Michalopoulou, E.; Spata, M.; Azzam, A.; Xie, H.; Simon, M.C.; Kamphorst, J.J. Triglycerides Promote Lipid Homeostasis during Hypoxic Stress by Balancing Fatty Acid Saturation. Cell Rep. 2018, 24, 2596–2605. [Google Scholar] [CrossRef] [PubMed]
- Young, R.M.; Ackerman, D.; Quinn, Z.L.; Mancuso, A.; Gruber, M.; Liu, L.; Giannoukos, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; et al. Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev. 2013, 27, 1115–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petan, T.; Jarc, E.; Jusovic, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Valli, A.; Rodriguez, M.; Moutsianas, L.; Fischer, R.; Fedele, V.; Huang, H.L.; Van Stiphout, R.; Jones, D.; McCarthy, M.; Vinaxia, M.; et al. Hypoxia induces a lipogenic cancer cell phenotype via HIF1alpha-dependent and -independent pathways. Oncotarget 2015, 6, 1920–1941. [Google Scholar] [CrossRef] [PubMed]
- Han, J.S.; Lee, J.H.; Kong, J.; Ji, Y.; Kim, J.; Choe, S.S.; Kim, J.B. Hypoxia Restrains Lipid Utilization via Protein Kinase A and ATGL Downregulation through Hypoxia Inducible Factor. Mol. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Kato, H.; Kitajima, S.; Lee, K.L.; Gradin, K.; Okamoto, T.; Poellinger, L. Interaction between von Hippel-Lindau Protein and Fatty Acid Synthase Modulates Hypoxia Target Gene Expression. Sci. Rep. 2017, 7, 7190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagiota, A.; Kourti, M.; Simos, G.; Mylonis, I. HIF-1alpha-derived cell-penetrating peptides inhibit ERK-dependent activation of HIF-1 and trigger apoptosis of cancer cells under hypoxia. Cell Mol. Life Sci. 2018. [Google Scholar] [CrossRef]
- Ichiki, T.; Sunagawa, K. Novel roles of hypoxia response system in glucose metabolism and obesity. Trends Cardiovasc. Med. 2014, 24, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, J.; Danzer, C.; Simka, T.; Ukropec, J.; Walter, K.M.; Kumpf, S.; Mirtschink, P.; Ukropcova, B.; Gasperikova, D.; Pedrazzini, T.; et al. Dietary obesity-associated Hif1alpha activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 2012, 26, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, J.; Guthke, R.; Dahmen, U.; Dirsch, O.; Holzhuetter, H.G.; Schuster, S. A theoretical study of lipid accumulation in the liver-implications for nonalcoholic fatty liver disease. Biochim. Biophys. Acta 2014, 1841, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Qu, A.; Taylor, M.; Xue, X.; Matsubara, T.; Metzger, D.; Chambon, P.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-inducible transcription factor 2alpha promotes steatohepatitis through augmenting lipid accumulation, inflammation, and fibrosis. Hepatology 2011, 54, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Rha, J.; Selak, M.A.; Unger, T.L.; Keith, B.; Liu, Q.; Haase, V.H. Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol. Cell Biol. 2009, 29, 4527–4538. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J.; Xie, C.; Jiang, C. The role of hypoxia-inducible factors in metabolic diseases. Nat. Rev. Endocrinol. 2018, 15, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Yun, Z.; Maecker, H.L.; Johnson, R.S.; Giaccia, A.J. Inhibition of PPAR gamma 2 gene expression by the HIF-1-regulated gene DEC1/Stra13: A mechanism for regulation of adipogenesis by hypoxia. Dev. Cell 2002, 2, 331–341. [Google Scholar] [CrossRef]
- Zhang, X.; Lam, K.S.; Ye, H.; Chung, S.K.; Zhou, M.; Wang, Y.; Xu, A. Adipose tissue-specific inhibition of hypoxia-inducible factor 1{alpha} induces obesity and glucose intolerance by impeding energy expenditure in mice. J. Biol. Chem. 2010, 285, 32869–32877. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, H.; Ichiki, T.; Inoue, E.; Nomura, M.; Miyazaki, R.; Hashimoto, T.; Ikeda, J.; Takayanagi, R.; Fong, G.H.; Sunagawa, K. Prolyl hydroxylase domain protein 2 plays a critical role in diet-induced obesity and glucose intolerance. Circulation 2013, 127, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Fu, Z.; Linke, S.; Chicher, J.; Gorman, J.J.; Visk, D.; Haddad, G.G.; Poellinger, L.; Peet, D.J.; Powell, F.; et al. The asparaginyl hydroxylase factor inhibiting HIF-1alpha is an essential regulator of metabolism. Cell Metab. 2010, 11, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Kim, J.H.; Li, F.; Qu, A.; Gavrilova, O.; Shah, Y.M.; Gonzalez, F.J. Hypoxia-inducible factor 1alpha regulates a SOCS3-STAT3-adiponectin signal transduction pathway in adipocytes. J. Biol. Chem. 2013, 288, 3844–3857. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, J.W.; Osborne, O.; Oh da, Y.; Sasik, R.; Schenk, S.; Chen, A.; Chung, H.; Murphy, A.; Watkins, S.M.; et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell 2014, 157, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Qu, A.; Matsubara, T.; Chanturiya, T.; Jou, W.; Gavrilova, O.; Shah, Y.M.; Gonzalez, F.J. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 2011, 60, 2484–2495. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Halberg, N.; Khan, M.; Magalang, U.J.; Scherer, P.E. Selective inhibition of hypoxia-inducible factor 1alpha ameliorates adipose tissue dysfunction. Mol. Cell Biol. 2013, 33, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Gesta, S.; Boucher, J.; Wang, X.L.; Kahn, C.R. The differential role of Hif1beta/Arnt and the hypoxic response in adipose function, fibrosis, and inflammation. Cell Metab. 2011, 14, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, Z.; Morton, N.M.; Moreno Navarrete, J.M.; West, C.C.; Stewart, K.J.; Fernandez-Real, J.M.; Schofield, C.J.; Seckl, J.R.; Ratcliffe, P.J. Adipocyte pseudohypoxia suppresses lipolysis and facilitates benign adipose tissue expansion. Diabetes 2015, 64, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, B.; Levin, I.; Csak, T.; Petrasek, J.; Mueller, C.; Kodys, K.; Catalano, D.; Mandrekar, P.; Szabo, G. Hepatocyte-specific hypoxia-inducible factor-1alpha is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology 2011, 53, 1526–1537. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Goda, N.; Kanai, M.; Niwa, D.; Osanai, K.; Yamamoto, Y.; Senoo-Matsuda, N.; Johnson, R.S.; Miura, S.; Kabe, Y.; et al. HIF-1alpha induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J. Hepatol. 2012, 56, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Tanaka, M.; Goda, N. HIF-1-dependent lipin1 induction prevents excessive lipid accumulation in choline-deficient diet-induced fatty liver. Sci. Rep. 2018, 8, 14230. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Yagai, T.; Luo, Y.; Liang, X.; Chen, T.; Wang, Q.; Sun, D.; Zhao, J.; Ramakrishnan, S.K.; Sun, L.; et al. Activation of intestinal hypoxia-inducible factor 2alpha during obesity contributes to hepatic steatosis. Nat. Med. 2017, 23, 1298–1308. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, P.; Magnusson, B.; Svensson, P.A.; Wiklund, O.; Boren, J.; Carlsson, L.M.; Stahlman, M.; Olofsson, S.O.; Hulten, L.M. Hypoxia converts human macrophages into triglyceride-loaded foam cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1871–1876. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Mason, S.; Liu, D.; Huang, Y.; Marks, C.; Hickey, R.; Jovin, I.S.; Pypaert, M.; Johnson, R.S.; Giordano, F.J. Hypoxia-inducible factor-dependent degeneration, failure, and malignant transformation of the heart in the absence of the von Hippel-Lindau protein. Mol. Cell Biol. 2008, 28, 3790–3803. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Huang, Y.; Booth, C.J.; Haase, V.H.; Johnson, R.S.; Celeste Simon, M.; Giordano, F.J.; Yun, Z. Activation of hypoxia-inducible factor-2 in adipocytes results in pathological cardiac hypertrophy. J. Am. Heart Assoc. 2013, 2, e000548. [Google Scholar] [CrossRef] [PubMed]
- Marsch, E.; Demandt, J.A.; Theelen, T.L.; Tullemans, B.M.; Wouters, K.; Boon, M.R.; van Dijk, T.H.; Gijbels, M.J.; Dubois, L.J.; Meex, S.J.; et al. Deficiency of the oxygen sensor prolyl hydroxylase 1 attenuates hypercholesterolaemia, atherosclerosis, and hyperglycaemia. Eur. Heart J. 2016, 37, 2993–2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahtu-Korpela, L.; Maatta, J.; Dimova, E.Y.; Horkko, S.; Gylling, H.; Walkinshaw, G.; Hakkola, J.; Kivirikko, K.I.; Myllyharju, J.; Serpi, R.; et al. Hypoxia-Inducible Factor Prolyl 4-Hydroxylase-2 Inhibition Protects Against Development of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 608–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Z.; Li, M.; Wharton, J.; Zhu, M.M.; Zhao, Y.Y. Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans Through Hypoxia-Inducible Factor-2alpha. Circulation 2016, 133, 2447–2458. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kaelin, W.G., Jr. The EGLN-HIF O2-Sensing System: Multiple Inputs and Feedbacks. Mol. Cell 2017, 66, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Semenza, G.L. HIF and the lung: Role of hypoxia-inducible factors in pulmonary development and disease. Am. J. Respir. Crit. Care Med. 2011, 183, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Laurie, S.S. HIF and pulmonary vascular responses to hypoxia. J. Appl. Physiol. 2014, 116, 867–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Alessandro, A.; El Kasmi, K.C.; Plecita-Hlavata, L.; Jezek, P.; Li, M.; Zhang, H.; Gupte, S.A.; Stenmark, K.R. Hallmarks of Pulmonary Hypertension: Mesenchymal and Inflammatory Cell Metabolic Reprogramming. Antioxid. Redox Signal. 2018, 28, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Michelakis, E.D. The metabolic basis of pulmonary arterial hypertension. Cell Metab. 2014, 19, 558–573. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Bonnet, S.; Rochefort, G.; Haromy, A.; Folmes, K.D.; Lopaschuk, G.D.; Dyck, J.R.; Michelakis, E.D. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci. Transl. Med. 2010, 2, 44ra58. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J. Metabolic dysfunction in the pathogenesis of pulmonary hypertension. Cell Metab. 2010, 12, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Garcia, J.L.; Arias, T.; Rojas, Y.; Garcia-Ruiz, V.; Santos, A.; Martin-Puig, S.; Ruiz-Cabello, J. Metabolic Reprogramming in the Heart and Lung in a Murine Model of Pulmonary Arterial Hypertension. Front. Cardiovasc. Med. 2018, 5, 110. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Pharmacologic Targeting of Hypoxia-Inducible Factors. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 379–403. [Google Scholar] [CrossRef] [PubMed]
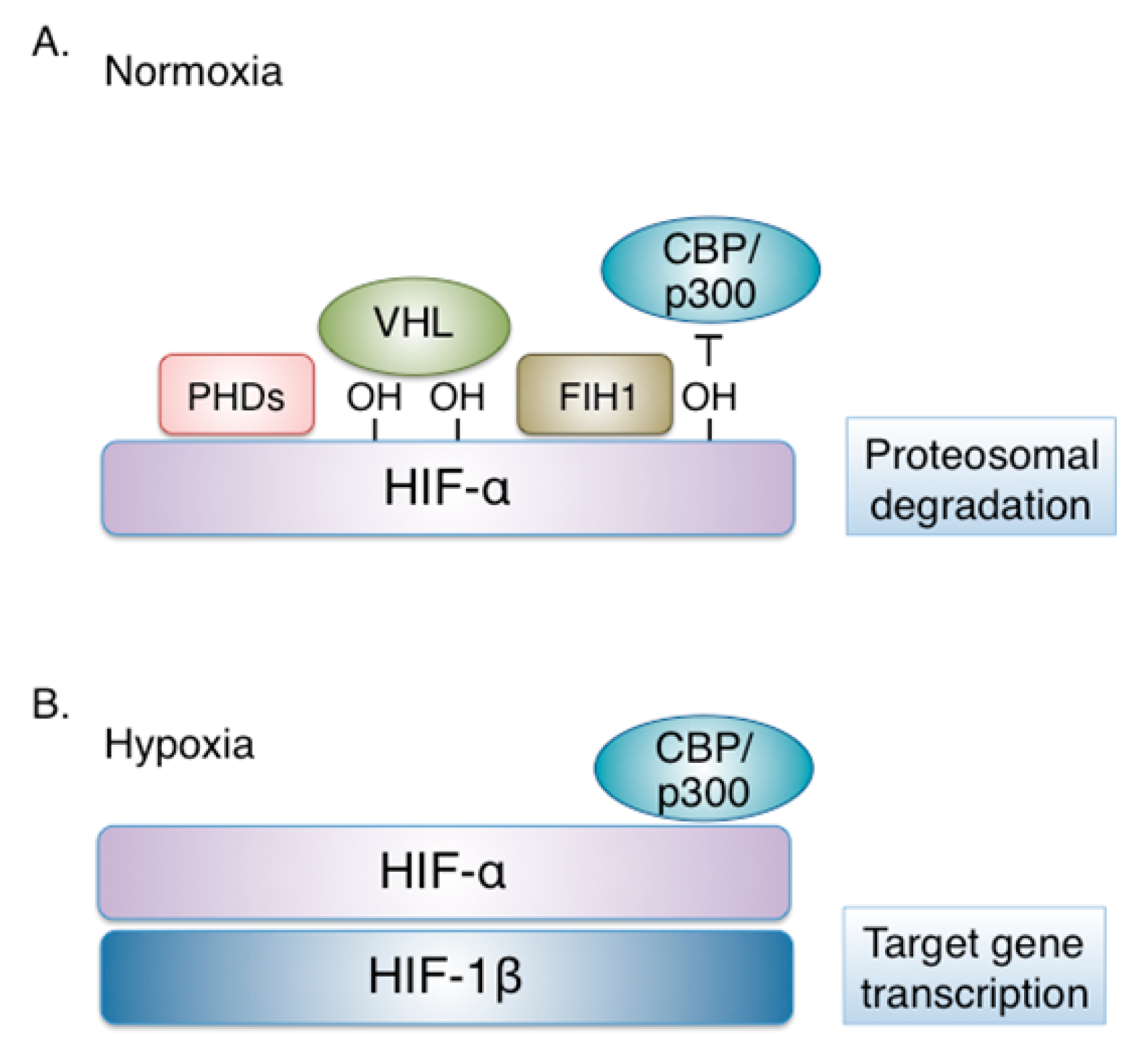
Figure 1.
Regulation of hypoxia-inducible factor (HIF) by oxygen. (A) Under physiological oxygen concentration (Normoxia), HIF-α isoforms are modified by oxygen-dependent prolyl-hydroxylases (PHDs), recognized by the von Hippel–Lindau (VHL) tumor suppressor protein, ubiquitinated and targeted to the proteasome for degradation. In addition, HIF-α modification by FIH (factor-inhibiting HIF), an oxygen-sensitive asparaginyl hydroxylase, disrupts interaction with the transcriptional co-activators p300/CBP and impairs residual HIF transcriptional activity. (B) When oxygen becomes limited (Hypoxia), PHDs and FIH are inactive. The non-hydroxylated HIF-α is stable and dimerizes with HIF-1β. The HIF heterodimer interacts with p300/CBP and activates the transcription of HIF target genes.
Figure 1.
Regulation of hypoxia-inducible factor (HIF) by oxygen. (A) Under physiological oxygen concentration (Normoxia), HIF-α isoforms are modified by oxygen-dependent prolyl-hydroxylases (PHDs), recognized by the von Hippel–Lindau (VHL) tumor suppressor protein, ubiquitinated and targeted to the proteasome for degradation. In addition, HIF-α modification by FIH (factor-inhibiting HIF), an oxygen-sensitive asparaginyl hydroxylase, disrupts interaction with the transcriptional co-activators p300/CBP and impairs residual HIF transcriptional activity. (B) When oxygen becomes limited (Hypoxia), PHDs and FIH are inactive. The non-hydroxylated HIF-α is stable and dimerizes with HIF-1β. The HIF heterodimer interacts with p300/CBP and activates the transcription of HIF target genes.

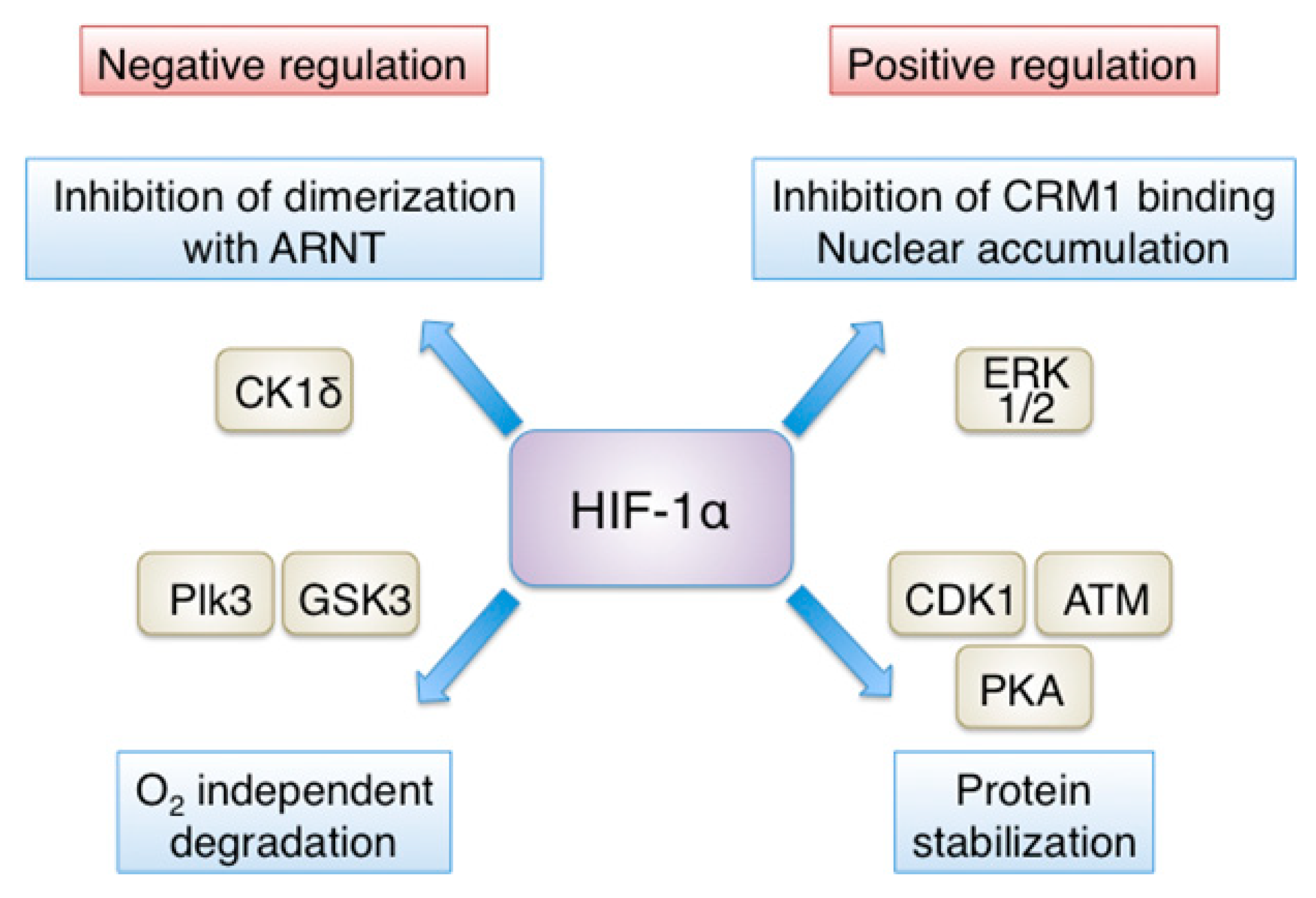
Figure 2.
Positive and negative regulation of HIF-1α by phosphorylation. Direct phosphorylation by several kinases is important for HIF-1α regulation. Positive regulation: ERK1/2-dependent phosphorylation inhibits binding of the exportin CRM1 and promotes nuclear accumulation of HIF-1α, while phosphorylation by ATM, CDK1 or PKA inhibits HIF-1α degradation. Negative regulation: phosphorylation by casein CK1δ impairs HIF-1α association with ARNT and thus, decreases HIF-1 transcriptional activity, while phosphorylation by GSK3 or Plk3 results in VHL-independent degradation of HIF-1α. See text for details and references.
Figure 2.
Positive and negative regulation of HIF-1α by phosphorylation. Direct phosphorylation by several kinases is important for HIF-1α regulation. Positive regulation: ERK1/2-dependent phosphorylation inhibits binding of the exportin CRM1 and promotes nuclear accumulation of HIF-1α, while phosphorylation by ATM, CDK1 or PKA inhibits HIF-1α degradation. Negative regulation: phosphorylation by casein CK1δ impairs HIF-1α association with ARNT and thus, decreases HIF-1 transcriptional activity, while phosphorylation by GSK3 or Plk3 results in VHL-independent degradation of HIF-1α. See text for details and references.

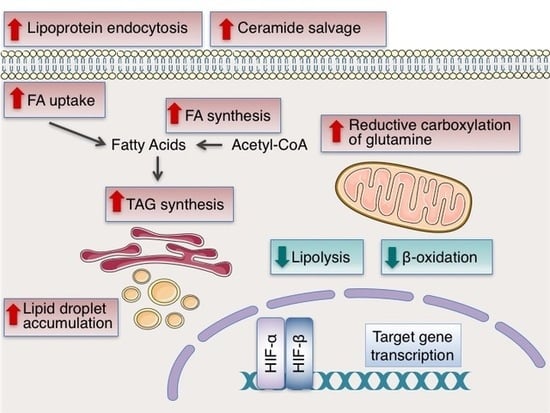
Figure 3.
Reprogramming of lipid metabolism under hypoxia. Hypoxia enhances lipogenesis by HIF-dependent modulation of proteins involved in fatty acid (FA) uptake, synthesis, storage and usage. Uptake of extracellular FA is promoted under hypoxia by activation of the transcription factor PPARγ and the increased expression of FABPs 3, 4 and 7. Endocytosis of lipoproteins is enhanced by the upregulation of LRP1 and VLDLR, while ceramide levels are increased by upregulation of NEU3. To maintain de novo FA synthesis under hypoxia, preservation of citrate levels and synthesis of acetyl-CoA is achieved by stimulation of reductive glutamine metabolism, mediated, at least in part, by induction of GLS1 and proteolysis of the OGDH2 subunit of the α-ketoglutarate dehydrogenase complex (αKGDH) by SIAH2. Adequate FA supply is further supported by activation of SREBP-1, which in turn upregulates the expression of FASN. To avoid lipotoxicity and/or replete lipid stores, FAs are converted to neutral triacylglycerols (TAGs), which are stored in lipid droplets (LDs). Formation of LDs under hypoxia is favored by the upregulation of the TAG biosynthesis pathway enzymes AGPAT2 and lipin-1, and the LD membrane proteins PLIN2 and HIG2. Finally, lipid accumulation under hypoxia is additionally supported by the inhibition of β-oxidation through downregulation of PGC-1α, CPT1A, PGC-1β, MCAD and LCAD. The proteins upregulated or activated under hypoxia are shown in red and the proteins downregulated or inhibited under hypoxia are shown in green. See text for details and references.
Figure 3.
Reprogramming of lipid metabolism under hypoxia. Hypoxia enhances lipogenesis by HIF-dependent modulation of proteins involved in fatty acid (FA) uptake, synthesis, storage and usage. Uptake of extracellular FA is promoted under hypoxia by activation of the transcription factor PPARγ and the increased expression of FABPs 3, 4 and 7. Endocytosis of lipoproteins is enhanced by the upregulation of LRP1 and VLDLR, while ceramide levels are increased by upregulation of NEU3. To maintain de novo FA synthesis under hypoxia, preservation of citrate levels and synthesis of acetyl-CoA is achieved by stimulation of reductive glutamine metabolism, mediated, at least in part, by induction of GLS1 and proteolysis of the OGDH2 subunit of the α-ketoglutarate dehydrogenase complex (αKGDH) by SIAH2. Adequate FA supply is further supported by activation of SREBP-1, which in turn upregulates the expression of FASN. To avoid lipotoxicity and/or replete lipid stores, FAs are converted to neutral triacylglycerols (TAGs), which are stored in lipid droplets (LDs). Formation of LDs under hypoxia is favored by the upregulation of the TAG biosynthesis pathway enzymes AGPAT2 and lipin-1, and the LD membrane proteins PLIN2 and HIG2. Finally, lipid accumulation under hypoxia is additionally supported by the inhibition of β-oxidation through downregulation of PGC-1α, CPT1A, PGC-1β, MCAD and LCAD. The proteins upregulated or activated under hypoxia are shown in red and the proteins downregulated or inhibited under hypoxia are shown in green. See text for details and references.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Representative HIF direct or indirect target genes that mediate reprogramming of lipid metabolism under hypoxia.
Table 1.
Representative HIF direct or indirect target genes that mediate reprogramming of lipid metabolism under hypoxia.
| Functional Category /Protein Name | HIF Isoform & Effect | Outcome & Experimental Evidence | Ref. |
|---|---|---|---|
| FA & Lipoprotein Uptake | |||
| PPARγ | HIF-1 Positive | Increased expression HIF-1 binds to the promoter of PPARγ and activates its transcription | [48] |
| FABP3 | HIF-1 Positive | Increased expression HIF-1α depletion inhibits the induction of FABP3 under hypoxia | [49] |
| FABP4 | HIF-1 Positive | Increased expression HIF-1 binds to the promoter of FABP4 and activates its transcription | [50] |
| FABP7 | HIF-1 Positive | Increased expression HIF-1α depletion inhibits the induction of FABP7 under hypoxia | [49] |
| LRP1 | HIF-1 Positive | Increased expression HIF-1α binds to the LRP1 promoter and activates its transcription | [51] |
| VDLR | HIF-1 Positive | Increased expression HIF-1α depletion inhibits activation of VDLR promoter under hypoxia | [52] |
| Reductive Carboxylation of Glutamine | |||
| GLS1 | HIF-1 Positive | Increased expression HIF-1α depletion inhibits the induction of GLS1 under hypoxia | [58] |
| OGDH2 | HIF-1 Negative | Increased proteolysis SIAH2 (a HIF-1 target) mediates proteolysis of OGDH2 | [57] |
| Ceramide Salvage | |||
| NEU3 | HIF-2 Positive | Increased expression HIF-2α binds to the NEU3 promoter and activates its transcription | [101] |
| FA Synthesis | |||
| SREBP-1 | HIF-1 Positive | Up-regulation Inhibition of HIF-1 impairs phospho-SREBP-1 increase under hypoxia | [59,69] |
| FASN | HIF-1 Positive | Increased expression Inhibition of HIF-1 impairs the induction of FASN under hypoxia Increased binding of SREBP-1 to the FASN promoter under hypoxia | [59] |
| TG Synthesis | |||
| AGPAT2 | HIF-1 Positive | Increased expression HIF-1 binds to the promoter of AGPAT2 and activates its transcription | [62] |
| Lipin-1 | HIF-1 Positive | Increased expression HIF-1 binds to the promoter of LPIN1 and activates its transcription | [63] |
| LD Accumulation | |||
| PLIN2 | HIF-2 Positive | Increased expression HIF-2α depletion inhibits the induction of PLIN2 under hypoxia | [65] |
| HIG2 | HIF-1 Positive | Increased expression HIF-1 binds to the promoter of HIG2 and activates its transcription | [66] |
| β-Oxidation | |||
| PGC-1α | HIF-1 & HIF-2 Negative | Reduced expression HIF-1α or HIF-2α depletion inhibits reduction of PGC-1α expression under hypoxia | [69] |
| CPT1A | HIF-1 & HIF-2 Negative | Reduced expression HIF-1α or HIF-2α depletion inhibit reduction of CPT1A expression under hypoxia | [69,70] |
| MCAD | HIF-1 Negative | Reduced expression HIF-1α depletion inhibits reduction of MCAD expression under hypoxia | [71] |
| LCAD | HIF-1 Negative | Reduced expression HIF-1α depletion inhibits reduction of LCAD expression under hypoxia | [71] |
| PGC-1β | HIF-1 Negative | Reduced expression HIF-1α depletion inhibits reduction of PGC-1β expression under hypoxia | [71] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. https://doi.org/10.3390/cells8030214
AMA Style
Mylonis I, Simos G, Paraskeva E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells. 2019; 8(3):214. https://doi.org/10.3390/cells8030214
Chicago/Turabian StyleMylonis, Ilias, George Simos, and Efrosyni Paraskeva. 2019. "Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism" Cells 8, no. 3: 214. https://doi.org/10.3390/cells8030214
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.