Large-Scale Assessment of Bioinformatics Tools for Lysine Succinylation Sites


1
Department of Bioscience and Bioinformatics, Kyushu Institute of Technology, 680–4 Kawazu, Iizuka, Fukuoka 820-8502, Japan
2
Biomedical Informatics R&D Center, Kyushu Institute of Technology, 680-4 Kawazu, Iizuka, Fukuoka 820-8502, Japan
*
Author to whom correspondence should be addressed.
Cells 2019, 8(2), 95; https://doi.org/10.3390/cells8020095
Submission received: 26 December 2018
/
Revised: 24 January 2019
/
Accepted: 24 January 2019
/
Published: 28 January 2019
(This article belongs to the Special Issue Bioinformatics and Computational Biology 2019)
Abstract
:Lysine succinylation is a form of posttranslational modification of the proteins that play an essential functional role in every aspect of cell metabolism in both prokaryotes and eukaryotes. Aside from experimental identification of succinylation sites, there has been an intense effort geared towards the development of sequence-based prediction through machine learning, due to its promising and essential properties of being highly accurate, robust and cost-effective. In spite of these advantages, there are several problems that are in need of attention in the design and development of succinylation site predictors. Notwithstanding of many studies on the employment of machine learning approaches, few articles have examined this bioinformatics field in a systematic manner. Thus, we review the advancements regarding the current state-of-the-art prediction models, datasets, and online resources and illustrate the challenges and limitations to present a useful guideline for developing powerful succinylation site prediction tools.
1. Introduction
Lysine succinylation is an evolutionarily conserved posttranslational modification (PTM) known to be involved in the regulation of diverse cellular process [1,2,3,4,5,6,7]. The succinylation process modifies a target protein with a succinyl group (–CO–CH2–CH2–CO2H), which is transmitted from succinyl-CoA to the specific α-amino group of a lysine residue [8,9,10,11,12]. The succinylation firstly was discovered in histone protein [13], and its regulatory role has been examined through the gene expression regarding chromatin reorganization [14,15,16]. Nevertheless, the published studies have provided little information regarding the enzyme which catalyzes histone lysine succinylation [17,18,19]. In fact, it is unclear whether this reaction is enzymatic or not [8,9,20]. In addition to histones, the succinylated proteins were found in the cytoplasm, nucleus, and mitochondria [7,21,22,23,24], indicating that lysine succinylation controls a variety of biological functions [14,18,25,26]. Lysine succinylation in HeLa cells induced different diseases via histone proteins, including UV-induced stress and cancer [12,27,28,29,30,31,32,33,34]. Therefore, identification of succinylation sites is a key to understanding the functional proteins.
A few years ago lysine succinylation was identified as a protein modification [2,3,25]. This modification can make notable alterations in protein function and structure regulation [3,13,35,36,37]. It can also participate in regulating many biological processes such as calorie restriction and metabolisms [38,39,40,41,42,43,44]. The identification of protein succinylation sites is a crucial topic in cellular pathology and physiology, which may provide valuable information for biomedical research and drug development. In recent years, high-throughput methods with mass spectrometry and succinylation enrichment analysis have been extensively implemented to identify lysine succinylation in several organisms [1,2,7,22,25,37,45,46,47,48,49]. A large-scale protein lysine-succinylated sites have been verified by experimentally in both prokaryotes [7,24,50,51] and eukaryotes [2,24,25,47]. Despite great advances through experimental investigation, the conventional experimental approaches are still difficult and time-consuming tasks [5,7,44,52,53]. Computational methods for succinylation site prediction are highly needed before experimental validation.
Our objective is to provide the useful and practical guidelines for the prediction of protein succinylation and to illustrate which predictor performs the best, whether the existing prediction model can be improved, and which features significantly contribute to prediction accuracy. We have assessed the performance of two different statistical methods: support vector machine (SVM) and random forest (RF) with five major types of descriptors. We also assess the performances of the individual and combined features with statistical significance tests, illustrating their contribution to the prediction accuracy. A synopsis of the existing computational approaches for lysine succinylation prediction is presented in Figure 1.
2. Existing Prediction Models
Nowadays, several machine learning-based predictors have been employed to identify succinylation sites [54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70]. The SucPred [54] is the first succinylation site predictor, which was established by Zhao et al. in 2015 through different encoding descriptors, including position amino acids weight composition, van der Waals volume normalized, grouped weight-based encoding, and auto-correlation functions, via SVM. By using SVM, Xu et al. developed iSuc-PseAAC [55] that implemented a composition of pseudo-amino acids (PseAAC) scheme. The SuccFind [56] predictor was established by Xu et al. which considered several amino acid-based composition encodings, including amino acid composition (AAC), k-space amino acid pairs (CKSAAP), and amino acid index (AAindex) through a feature selection algorithm. Two prediction tools of iSuc-PseOpt [70] and pSuc-Lys [61] were constructed by Jea et al., based on the PseAAC descriptor via a RF classifier. The SucStruct [58] and Success [67] predictors were developed by Lopez et al. based on the secondary structure-based features (SF) with decision trees (DT) algorithm. Dehzang et al. constructed two prediction tools of PSSM-Suc [57] and SSEvol-Suc [66] with a DT classifier by using evolutionary- and sequence-based features [67,68]. Hasan et al. developed the SuccinSite [59], SuccinSite2.0 [62], and GPSuc [65] predictors with the RF classifiers by integrating multiple sequence features. The SuccinSite2.0 [62] and GPSuc [65] predictors implemented different species-specific classifiers and integrated them. Until now, the GPSuc is one of the most updated predictors. On the other hand, abovementioned existing methods differ in various aspects, such as training and test datasets used, sliding window sizes and algorithms preferred, a ratio of positive versus negative samples, categories of sequence features encoded, and generality of whether the predictive classifiers are universal or species-specific. In addition, there have been distinct differences in terms of practical aspects of the web server implementation, adjustability of prediction inflexibility thresholds, support of batch predictions and computational efficiency. With various succinylation site predictors becoming available, comprehensive comparison of the strengths and weaknesses of them are essential. This comparison may reveal difficulties and guide improvement toward efficient succinylation site predictors.
A lot of focus has been placed on research of protein succinylation with an increase in databases [59,71,72]. The SuccinSite database records 4411 experimentally identified succinylation proteins with 12,456 lysine succinylation sites for different species [59]. It should, however, be noted that the succinylation proteins overlap with other modifications due to some exhibiting dual properties. Recently many studies have suggested that lysine succinylation extensively overlaps with acetylation [25,27,42,63,68,73,74,75,76].
To date, 12 methods were analyzed, i.e., SucPred [54], iSuc-PseAAC [55], SuccFind [5,6], iSuc-PseOpt [70], pSuc-Lys [61], SucStruct [58], PSSM-Suc [57], SuccinSite [59], SSEvol-Suc [66], SuccinSite2.0 [62], Success [67], and GPSuc [65] (Table 1., The SucPred used highly unbalanced (i.e., 1436 positive and 18,958 negative samples) training datasets, derived from the CPLM (http://cplm.biocuckoo.org) database [71]. For testing models, they used 250 positive samples but did not consider any negative samples. The pSuc-Lys, iSuc-PseAAC, and iSuc-PseOpt used 1167 positive and 3553 negative samples as the training dataset from the CPLM database but did not consider any independent datasets. The SucFind used 2713 positive and 23,598 negative samples as the training dataset from the CPLM database but did not consider any independent sets. The PSSM-Suc used 1782 positive and 1872 negative samples as the training dataset but did not consider any independent samples. The Success [67], SucStruct [58] and SSEvol-Suc [66] used a balanced training dataset (1782 positive and 1872 negative samples) from the CPLM database but did not consider any independent samples. In addition, few existing predictors have updated the latest datasets [59,65].
3. Datasets Collection and Preparation
Positive and Negative Samples
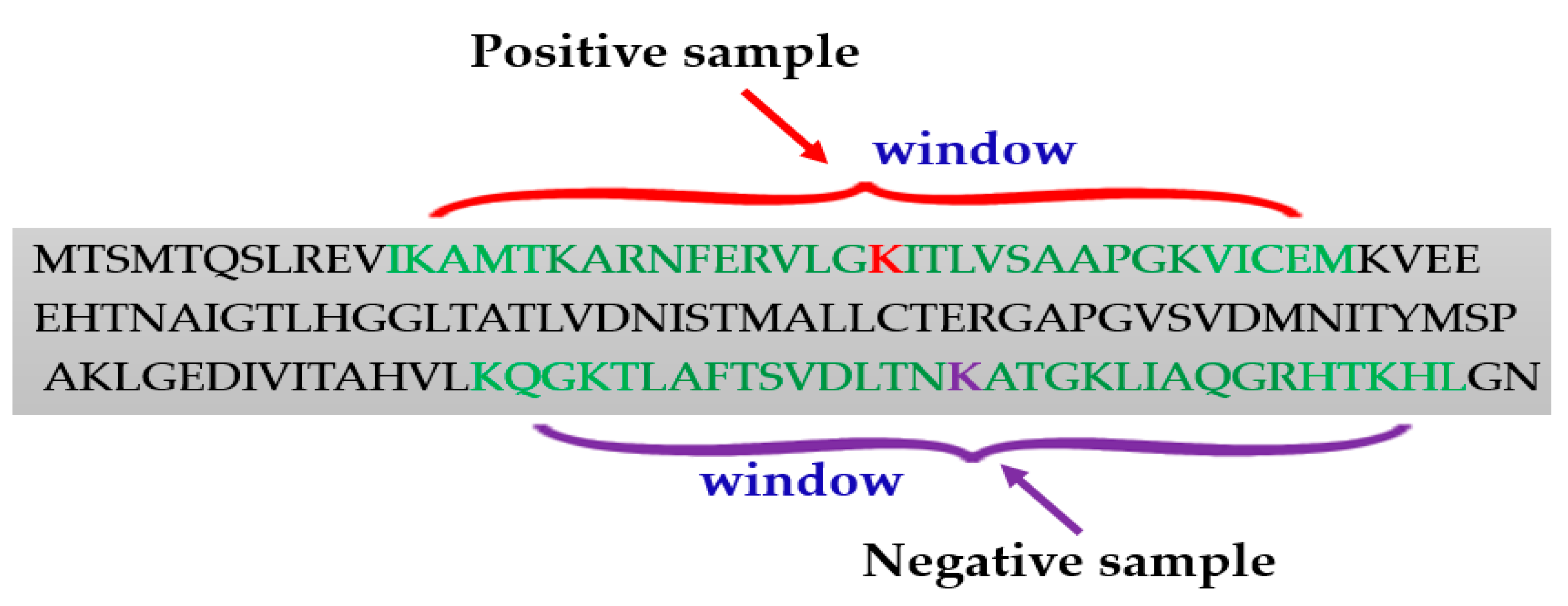
Generating the positive and negative samples from the protein sequences is an important step for lysine succinylation sites prediction. Usually, the positive samples were collected based on the experimentally verified lysine (K) residues. The sequence window strategy was applied to construct the positive samples. The fragment windows were the sequences of the peptide with a lysine residue to be succinylated in the center. To accurately predict succinylation sites, analysis of flanking residues in the window fragment is important, because a very small number of residues would miss valuable evidence and a large number of them may introduce unavoidable redundancy. For example, to select the window fragments of 31 (±15), the length of the full sequence of proteins was inputted; for the fragment window model, a window size of 31 was fixed so that the lysine residue is centered (Figure 2). Most of the researchers have tested different window fragments to enhance predictive performance in succinylation site prediction (Table 1).
To generate a set of fragment windows that are regarded as negative samples are very challenging. There is no standard method to generate the negative samples. Researchers typically considered the experimentally identified succinylated lysines as positive samples, while they regarded all the remaining lysine residues as negative instances. Nonetheless, some negative samples may be positive are generated by experimental errors, which decreases prediction accuracy.
Recently thousands of succinylated proteins and their sites have been identified experimentally from diverse species including Homo sapiens (H. sapiens), Saccharomyces cerevisiae (S. cerevisiae), Mus musculus (M. musculus), Toxoplasma gondii (T. gondii), Histoplasma capsulatum (H. capsulatum), Mycobacterium tuberculosis (M. tuberculosis), Escherichia coli (E. coli), Solanum lycopersicum (S. lycopersicum), and Triticum aestivum (T. aestivum) [7,22,37,47,59]. To examine the species-specific datasets, we collected the datasets of nine species and removed redundant sequences with a 30% similarity cutoff using CD-HIT [77] and recorded them at http://kurata14.bio.kyutech.ac.jp/GPSuc [65]. A statistic of the training and independent datasets is shown in Table 2.
4. Algorithms of Predicting Lysine Succinylation Site
Many machine learning algorithms such as RF, SVM, adaptive boosting (AdaBoost), and DT have been employed to predict succinylation sites, while the two machine learning algorithms of SVM and RF are intensively used (Table 1). Employed machine learning algorithms are briefly explained as follows.
4.1. Random Forest
In protein bioinformatics research, RF is a well-established and extensively used machine learning algorithm [62,65,78,79]. RF works as a collective and supervised decision classifier, which ‘votes’ for one of the two classes, either positive or negative samples. The RF algorithm is very straightforward and does not produce any bias results. However, it is necessary to select the optimum number of decision trees. In this review, to examine the selected, individual descriptors, we used 1000 decision trees via 5-fold cross-validation (CV) test to validate the method performances by using a package of R software (https://cran.r-project.org/web/packages/randomForest/).
4.2. Support Vector Machine
SVM is another machine learning algorithm and broadly used in protein bioinformatics research [54,55,56,57,80]. Various kernel function including the linear/polynomial/sigmoid and Gaussian radial basis function were used to develop SVM models. A critical point is the optimization of parameters. Prior to model construction, it is recommended to optimize SVM parameters, which affect the prediction performance dramatically. In this review, we used the SVMlight (http://svmlight.joachims.org) package to examine the individual features with default parameters.
4.3. Adaptive Boosting
AdaBoost works as a meta-classifier that is frequently used to classify binary samples [66]. This algorithm iteratively adjusts weight values to decrease the misclassified samples until the weight values do not change.
4.4. Decision Trees
5. Motif Conservation of Species-Specific and Generic Succinylation Sites
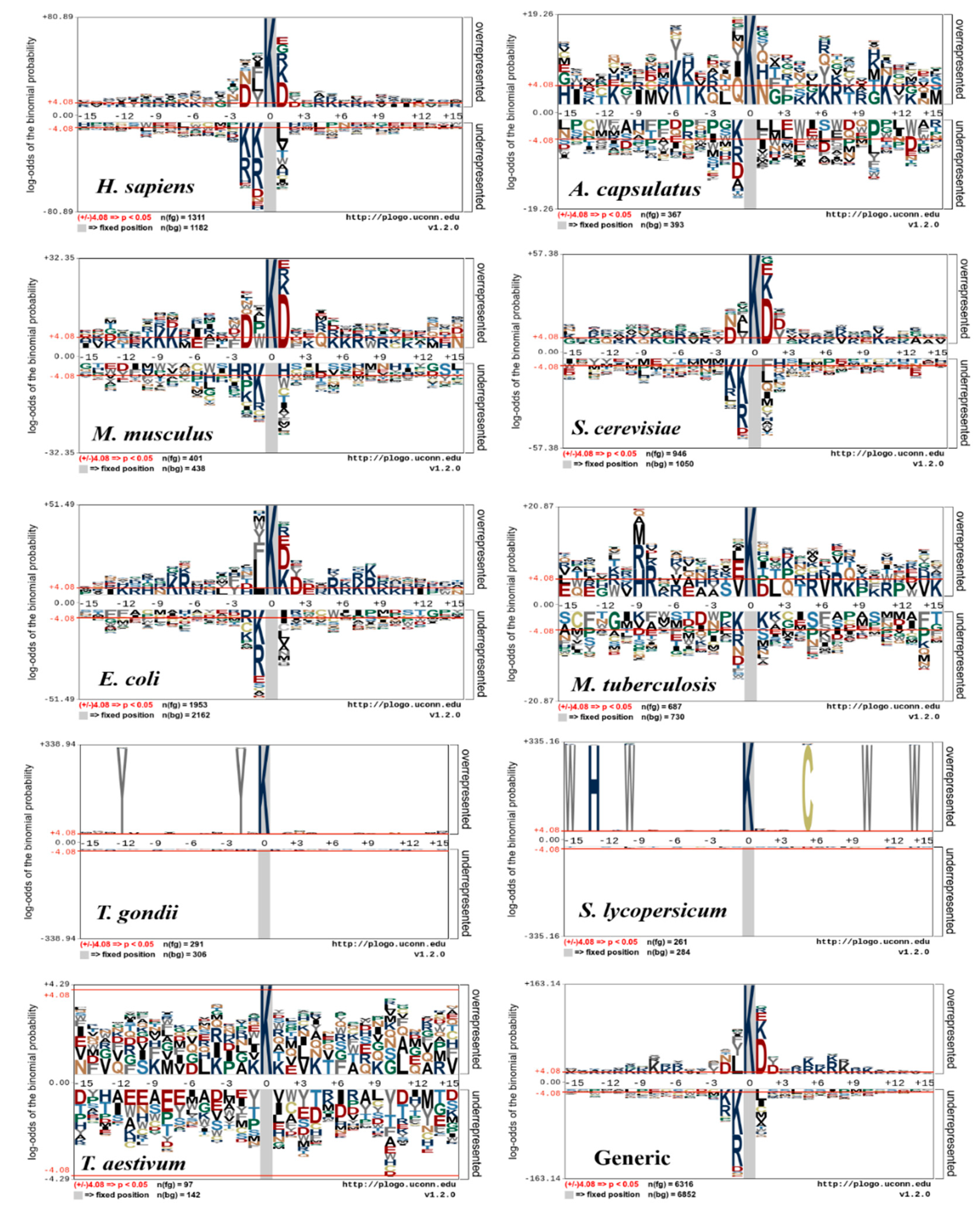
The sequence motif conservation surrounding the succinylation sites could partly be illustrated for the different species datasets. To reveal succinylation site sequences of 9 different species, a pLogo (https://plogo.uconn.edu/) software was used as shown in Figure 3 [81], which classifies and displays significant differences of succinylated vs non-succinylated sites by position-specific amino acid compositions on the sequence fragments (±15). At each position of pLogo graphs, over- or under- X-axis amino acids were plotted, where X denotes each amino acid residue [59,65,78]. The height of the corresponding residue letter of positive (if over-represented) or negative samples (if under-represented) were harbored. The cumulative percentages of these over-/under-represented residues were reported in the label of Y-axis. Consequently, the amino acids above the X-axis indicated frequently detected residues around succinylation sites. In Figure 3, the upper portion displays a set of positive samples and the middle portion displays consistent residues, while the lower portion shows depleted amino acids.
Since the sequence motifs for H. sapiens, S. cerevisiae, and M. musculus resembled each other (Figure 3), an H. sapiens succinylation site tool could identify succinylation sites for M. musculus, and S. cerevisiae and the reverse is also true. The sequence patterns of succinylated proteins around H. sapiens, M. musculus, H. capsulatum, S. cerevisiae, and E. coli are widely distributed than the other four species. It was observed that charged amino acids (K, R, and D) were significantly enriched at positions (−10, −9, −8, −7, −6, −5 −2; +2, +4, +5, +6, +7, and +10) for H. sapiens, M. musculus, H. capsulatum, S. cerevisiae, and E. coli models. In S. lycopersicum, M. tuberculosis, and T. aestivum species, the neutral amino acids (C, F, G, and S) were significantly depleted. In S. cerevisiae and T. gondi, some of the charged residues (D, K, and R) were over- and under-represented. In addition, neutral amino acids (S, Q, and C) were frequently distributed around the succinylation sites and most of the specific amino acid positions were not significantly enriched/depleted except for S. lycopersicum, T. gondii, and T. aestivum. While the generic model seems to have some sequence motifs, it is clearly shown that the sequence motifs are species-specific. Therefore, the generic model may result in incomplete or erroneous information to a query sequence. Hasan et al. suggested that the surrounding succinylation sites vary, depending on species [65] and the species-specific classifiers are necessary to identify the succinylation sites, as well as developers of other PTM site predictors for ubiquitination [82], acetylation [83,84], methylation [85], phosphorylation [86,87], and malonylation [88].
6. Important Descriptors for Predicting Succinylation Sites
Feature extraction is one of the most important and challenging steps, enabling the accurate prediction of lysine succinylation sites. Ideally, the features can clearly distinguish succinylated sites from random lysine sites. In previous studies, different types of features were adopted to distinguish the succinylated sites from non-succinylated sites. The frequently used features are AAindex, ACF, EBGW, VDWV, WAAC, AAC, CKSAAP, PseAAC, Binary, SF, PSSM, pCKSAAP and some structural features (SFs) (Table 3). These major feature types include (1) protein sequence features, (2) evolutionary features, (3) protein physicochemical properties, (4) structural features, and (5) binary profile annotations.
To develop a statistical predictor, an effective mathematical expression is needed to formulate the protein or peptide samples [89,90,91,92]. Composition analysis of proteome-wide amino acids can describe the particular information of a specified organism, since the organism manages to reduce the protein synthesis cost by adjusting their residue contents under specific growth conditions [19,93]. Therefore, sequence information was valuable to develop species-specific succinylation predictors. To transform protein or fragment sequences into numeric vectors, orthogonal binary coding [59,62], AAindex [65], PseAAC [55,61,70] were measured. To accesses the positional information of amino acids around the positive and negative samples, the WAAC [54], ACF [54], and VDW [54] were introduced. Moreover, to introduce the amino acids frequency information in fragment sequences, the pCKSAAP [62,65] and CKSAAP [56,59] schemes were used. To fix the length of the sequence, AAindex encoding is particularly suitable [59,62,65]. To identify the conserved residues at the specific sequence, evolutionary information is an important characteristic [57,65], because the conserved residues are always functionally relevant [62]. Since the SF is far more conserved than the sequence, SF encoding could be a valuable indicator to identify the function of succinylation proteins [58]. To make an effective prediction model, optimization of incorporative feature methods is typically crucial. The SuccinSite used a linear combination of different features with weight values [59]. Recently, the outputs of distinct features have been combined using a logistic regression (LR) algorithm [65,94]. These two models can be integrated for further enhancement of accuracy of succinylation site prediction.
7. Features Assessment of Species-specific Succinylation Sites
To classify the succinylation and non-succinylation samples, machine learning algorithms have been effectively employed (Table 1). A majority of succinylation site predictors used conditional RFs [57,58,59,61,62,70], while a few of them used SVM classifiers [54,55,56]. Therefore, we chose these two machine learning algorithms due to their successful implementation. We also measured the area under the ROC curve (AUC). Table 4 summarizes the optimal performances with respect to 31 window sequences by the RF and SVM classification algorithms.
Twelve types of feature descriptors were employed in the previous succinylation predictors (Table 3). We investigated whether they are effective in prediction of the nine species-specific models and selected five major descriptors of CKSAAP, AAindex, Binary, PseAAC, and pCKSAAP (the other seven descriptors were not effectively used). A five-fold CV test on the training dataset and a test on the independent dataset were performed to assess the prediction performance by the five selected feature descriptors (Table 4), where the employed datasets are shown in Table 2. The top two features for H. sapiens, M. musculus, H. capsulatum, and E. coli were pCKSAAP and CKSAAP for training dataset. On the other hand, in the independent dataset, the AAindex and binary performed better. For the M. tuberculosis dataset, the top two features were pCKSAAP and CKSAAP in both of training and independent datasets. In the S. cerevisiae dataset, the top descriptor was pCKSAAP. In the T. gondii and T. aestivum datasets, CKSAAP, pKSAAP, and PseAAC encoding schemes were important. It is intriguing that, in the S. lycopersicum dataset, positional encodings of Binary, AAindex, and PseAAC were essential for the independent test. The pCKSAAP was an effective encoding feature that describes long- and short-range interfaces of amino acids within a protein or a sequence window [95,96,97,98], achieving best prediction results on M. tuberculosis, H. sapiens, M. musculus, H. capsulatum, S. cerevisiae, E. coli, and T. aestivum species for training datasets. The performance comparison indicated that the RF algorithm was the best for almost all the species datasets, followed by the SVM.
8. Comparative Analysis of Different Predictors
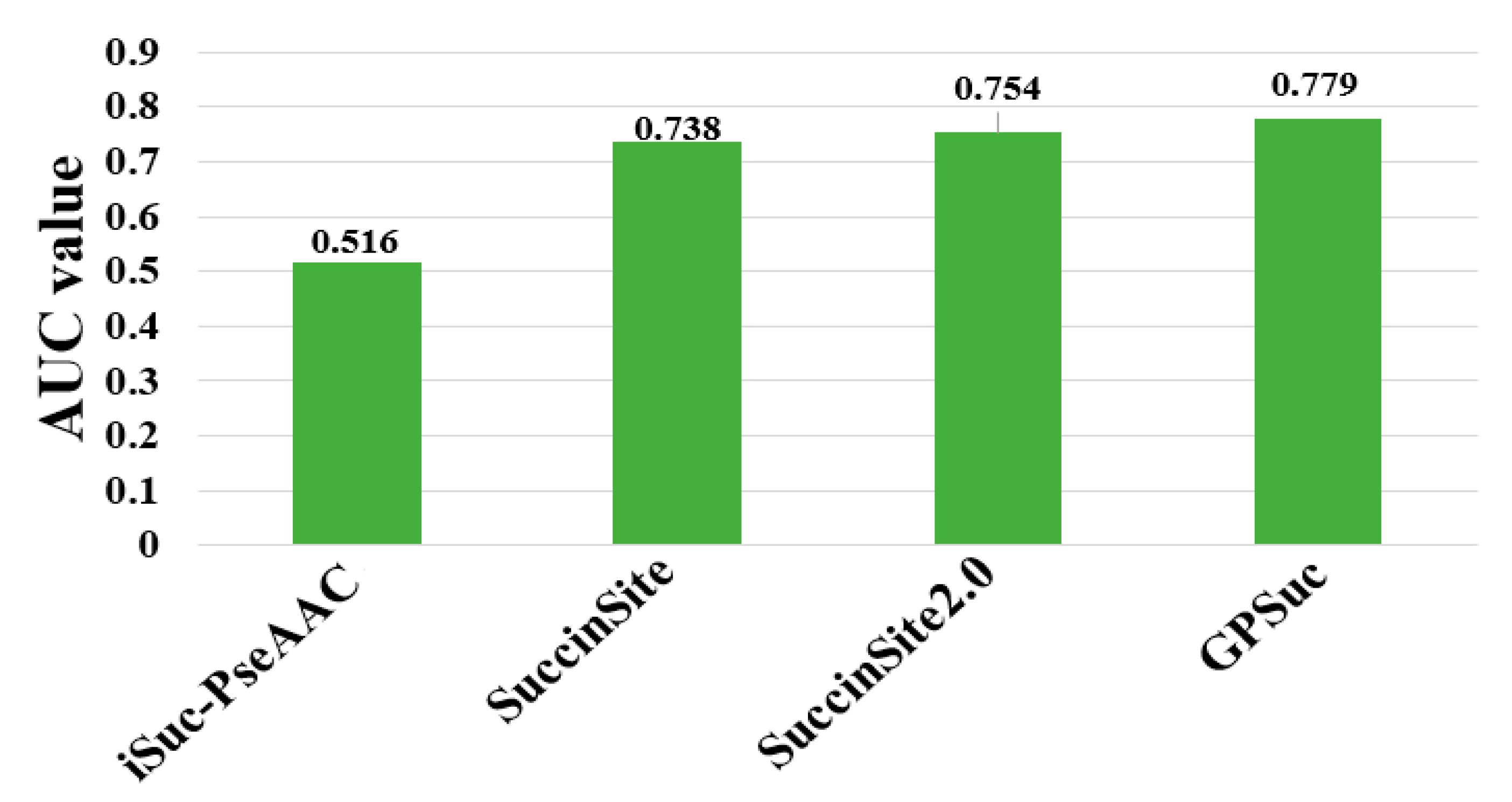
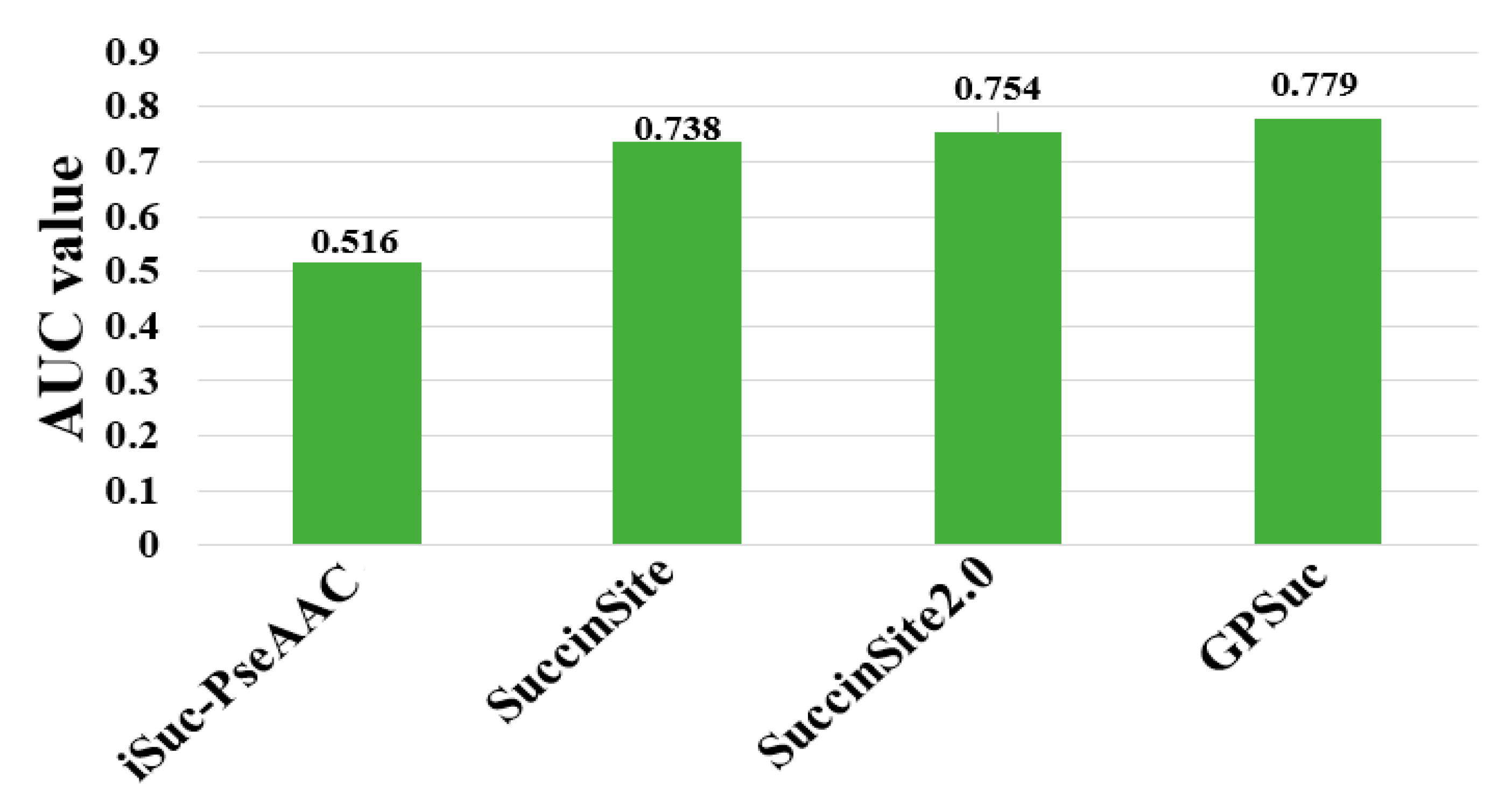
The performances of existing tools were compared by using different criteria as shown in Table 1. Note that it is difficult to exhaustively compare the analytical results obtained from different algorithms, because they use diverse assessment procedures for training and independent datasets and ratios of positive and negative samples. Although many predictors are not publicly accessible, including Success, SSEvol-Suc, SucPred, SucPred, pSuc-Lys, iSuc-PseOpt, SuccFind, SucStruct [58], and PSSM-Suc [57], only four of succinylation predictors of iSuc-PseAAC, SuccinSite, SuccinSite2.0, and GPSuc are publicly available and user-friendly. An independent dataset was constructed to make a fair comparison based on our previously published articles [65]. The dataset consisted of 254 positive and 2977 negative samples (http://kurata14.bio.kyutech.ac.jp/GPSuc) [65]. Figure 4 shows that the prediction performance of the four predictors with respect to 124 proteins. The top-performing SuccinSite2.0 and GPSuc with the AUC value of 0.754 and 0.779, respectively.
Recently the GPSuc and SuccinSite2.0 predictors have made an effort to establish the species-specific classifiers [62], while the others combined the data of each species into a generic model. Many predictors other than SuccinSite [59], SuccinSite2.0 [62], and GPSuc [65] were not validated by using independent data (Table 1).
9. The Online Employment Services
For biologists, web application or a standalone software package is required. There were 12 web services developed along with research publication; however, most of them are not available for public. The exiting tools were compared under the following conditions: (i) whether the existing web employment supports batch prediction; (ii) whether the scheme has the binary or probability scores; In Table 1, comprehensive information was summarized for all the existing tools. Among all the implementations, Success, PSSM-Suc and SucStruct did not provide web-services to implement their prediction models. The pSuc-Lys, SSEvol-Suc, and Suc-PseOpt predictors did not fulfill some criteria regarding sequence fragment position, prediction scores, and thresholds information. On the other hand, users cannot submit more than 100 sequences to the pSuc-Lys and Suc-PseOpt servers. The iSuc-PseAAC and Success servers did not attach the all prediction succinylation scores in the final output page. Users can get more satisfactory results from the SuccinSite, SuccinSite2.0, and GPSuc in a FASTA format. In the GPSuc user can select classifiers for nine species and their combined species. The GPSuc includes nine examined species classifiers and illustrated better performances than the SuccinStie2.0. The prediction output of the GPSuc, SuccinSite, and SuccinSite2.0 contains four items: protein name, predicted lysine position, expectation score, and explanation of succinylation sites. In the viewpoint of users, the prediction model should contain at least the position of the anticipated succinylation sites, sequence fragments, and probability scores, or assessment of the predicted result. In addition, it is obligatory that the predictor should provide flexibility modification to the output page of the provided stand-alone software or online servers. Particularly user control of the prediction stringency is essential for spreading predictors because users are interested in the prediction scores with an assured threshold.
10. Perceptions for Prediction Models
Sequence redundancy is an essential problem to consider prior to model assembly since the performance of the predictive models might be overestimated by overfitting of the training dataset and lead to poor scalability and performances on independent datasets. In succinylation prediction, most of the developers conducted the redundancy of sequence prior to model assembly. The CD-HIT (http://weizhongli-lab.org/cd-hit) [77,99] and BLAST algorithm (blastclust) (http://nebc.nox.ac.uk/bioinformatics/docs/blastclust) [100] are extensively used to eliminate data redundancy. The CD-HIT software is very popular for deleting the homolog sequences; however, this framework is a heuristic, i.e., it can have biases on the redundancy level model [101]. Recently, Martin and Johannes introduced the Linclust software (https://github.com/soedinglab/mmseqs2) [102] to reduce the compositional bias correction on the sequences, while advanced algorithms are still necessary. To reflect the ratio of succinylation and non-succinylation samples in the training data set is another problem. Usually, non- succinylation sites expressively outnumber the succinylation sites. Hence, a succinylation training dataset should be generated by using reliable and nonbiased methods. To choose the ratio of non-succinylation ratio samples to positive samples, a random selection procedure is often piloted.
Some prediction tools use small datasets to train their simulations, resulting in poor estimate performance when verified with the independent dataset [59,62]. For instance, an early study of the iSuc-PseAAC did not achieve good performance on the independent test dataset due to the limited training dataset (Figure 4). Through the developments in high-throughput sequencing with mass spectrometry analysis, a large number of succinylation sites are being identified and their associated databases are frequently updated. Many succinylation sites that were overlooked by previous studies are now experimentally verified as positive samples, i.e., the old versions of the database include a number of false negative samples. This indicates that the prediction models developed based on the old version database can be improved by using up-to-date succinylation samples. To extrapolate future unknown data, we should increase the number of non-redundant succinylation samples and use them as an independent dataset to validate the prediction models.
The motifs of succinylation proteins may significantly differ in diverse species, as shown in Figure 3. Nevertheless, all the existing predictors other than SuccinSite2.0 and GPSuc ignored the differences among species and combined all species models into a generic one. From now on, a computational method should consider species-specific classifiers. The current prediction tools are established individually based on sequence or secondary structural information. In future analysis, with an increase in tertiary structural information of succinylation samples, it is effective to employ such a structural descriptor [103]. Finally, it is required to present software applications or web servers so that users can easily access prediction models.
To reveal the significant information on the PTMs, graphical logos are widely used that give position-specific information (i.e., conserved patterns or motifs information) of amino acids. Several software packages are implemented to visualize the sequence motifs, such as pLogo [81], WebLogo [104], and iceLogo [105]. The existing algorithms highlighted the characters of amino acids that are enriched (i.e., occur more frequently than expected) and depleted (i.e., occur less than expected). However, the resulting plots sometimes suffered visual disorder, which makes principal sequence patterns ambiguous. Therefore, the next generation sequence logo needs to generate more suitable models for the efficient visualization of sequence motifs.
11. Conclusions
To assess the currently available succinylation site prediction tools, we comprehensively compared the predictor performances using an independent dataset. The predictive capabilities of combinations of different descriptors were evaluated to explore the optimal combination. In living cells, combining experimental and computational approaches will accelerate the buildup of our understanding on protein succinylation and hence support exploration of the consistent controlling networks. This review has designated that a large volume of lysine-succinylation site analyses is being carried out and explained the details in the employed datasets, motif conservation, encoding schemes, and machine learning algorithms. Moreover, we described limitations of current methodologies for prediction of lysine succinylation and provided perceptions into dataset assembly processes, model updates, and performance improvements.
Author Contributions
M.M.H., and H.K. designed and conceived the study. M.M.H. and M.S.K. performed the analyses. M.M.H., M.S.K., and H.K. wrote the manuscript. All authors discussed the prediction results and commented on the manuscript.
Funding
This work was supported by the Grant-in-Aid for Challenging Exploratory Research with JSPS KAKENHI Grant Number 17K20009 and partially supported by the developing key technologies for discovering and manufacturing pharmaceuticals used for next-generation treatments and diagnoses both from the Ministry of Economy, Trade and Industry, Japan (METI) and from Japan Agency for Medical Research and Development (AMED).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, X.; Hu, X.; Wan, Y.; Xie, G.; Li, X.; Chen, D.; Cheng, Z.; Yi, X.; Liang, S.; Tan, F. Systematic identification of the lysine succinylation in the protozoan parasite Toxoplasma gondii. J. Proteome Res. 2014, 13, 6087–6095. [Google Scholar] [CrossRef] [PubMed]
- Colak, G.; Xie, Z.; Zhu, A.Y.; Dai, L.; Lu, Z.; Zhang, Y.; Wan, X.; Chen, Y.; Cha, Y.H.; Lin, H.; et al. Identification of lysine succinylation substrates and the succinylation regulatory enzyme CobB in Escherichia coli. Mol. Cell Proteomics 2013, 12, 3509–3520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Ariyantoro, A.R.; Katsuno, N.; Nishizu, T. Effects of dual modification with succinylation and annealing on physicochemical, thermal and morphological properties of corn starch. Foods 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Alleyn, M.; Breitzig, M.; Lockey, R.; Kolliputi, N. The dawn of succinylation: a posttranslational modification. Am. J. Physiol., Cell Physiol. 2018, 314, C228–C232. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xie, L.; Yang, Z.; Zhou, J.; Xie, J. Lysine succinylation of Mycobacterium tuberculosis isocitrate lyase (ICL) fine-tunes the microbial resistance to antibiotics. J. Biomol. Struct. Dyn. 2017, 35, 1030–1041. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, G.; Song, L.; Mu, P.; Wang, S.; Liang, W.; Lin, Q. Global analysis of protein lysine succinylation profiles in common wheat. BMC Genomics 2017, 18, 309. [Google Scholar] [CrossRef]
- Yokoyama, A.; Katsura, S.; Sugawara, A. Biochemical analysis of histone succinylation. Biochem. Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Xu, X.; Liu, T.; Yang, J.; Chen, L.; Liu, B.; Wei, C.; Wang, L.; Jin, Q. The first succinylome profile of Trichophyton rubrum reveals lysine succinylation on proteins involved in various key cellular processes. BMC Genomics 2017, 18. [Google Scholar] [CrossRef]
- Sadhukhan, S.; Liu, X.; Ryu, D.; Nelson, O.D.; Stupinski, J.A.; Li, Z.; Chen, W.; Zhang, S.; Weiss, R.S.; Locasale, J.W.M.; et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 4320–4325. [Google Scholar] [CrossRef]
- Moin, A.; Ali, T.M.; Hasnain, A. Effect of succinylation on functional and morphological properties of starches from broken kernels of Pakistani Basmati and Irri rice cultivars. Food Chem. 2016, 191, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Nagano-Shoji, M.; Kubo, S.; Kawamura, Y.; Yoshida, A.; Kawasaki, H.; Nishiyama, M.; Yoshida, M.; Kosono, S. Altered acetylation and succinylation profiles in Corynebacterium glutamicum in response to conditions inducing glutamate overproduction. Microbiol. Open 2016, 5, 152–173. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Dai, J.; Dai, L.; Tan, M.; Cheng, Z.; Wu, Y.; Boeke, J.D.; Zhao, Y. Lysine succinylation and lysine malonylation in histones. Mol. Cell Proteomics 2012, 11, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R.; Payne, R.M. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 2013, 288, 29036–29045. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Lu, Z.; Xie, Z.; Cheng, Z.; Chen, Y.; Tan, M.; Luo, H.; Zhang, Y.; He, W.; Yang, K.; et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell Proteomics 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Lawal, O.S.; Adebowale, K.O. Effect of acetylation and succinylation on solubility profile, water absorption capacity, oil absorption capacity and emulsifying properties of mucuna bean (Mucuna pruriens) protein concentrate. Nahrung 2004, 48, 129–136. [Google Scholar] [CrossRef]
- Zaghloul, M.; Prakash, V. Effect of succinylation on the functional and physicochemical properties of alpha-globulin, the major protein fraction from Sesamum indicum L. Nahrung 2002, 46, 364–369. [Google Scholar] [CrossRef]
- Xu, C.; Ge, L.; Zhang, Y.; Dehmer, M.; Gutman, I. Prediction of therapeutic peptides by incorporating q-Wiener index into Chou’s general PseAAC. J. Biomed. Inform. 2017. [Google Scholar] [CrossRef]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Li, J.; Deng, W.; Yu, Z.; Fang, W.; Chen, M.; Liao, W.; Xie, J.; Pan, W. Proteomic analysis of lysine succinylation of the human pathogen Histoplasma capsulatum. J. Proteomics 2017, 154, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Sankari, E.S.; Manimegalai, D. Predicting membrane protein types using various decision tree classifiers based on various modes of general PseAAC for imbalanced datasets. J. Theor. Biol. 2017, 435, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Chen, R.; Li, C.; Li, W.; Ye, Z. Global analysis of protein lysine succinylation profiles and their overlap with lysine acetylation in the marine bacterium Vibrio parahemolyticus. J. Proteome Res. 2015, 14, 4309–4318. [Google Scholar] [CrossRef] [PubMed]
- Weinert, B.T.; Scholz, C.; Wagner, S.A.; Iesmantavicius, V.; Su, D.; Daniel, J.A.; Choudhary, C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013, 4, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Dennison, J.B.; Ayres, M.L.; Kaluarachchi, K.; Plunkett, W.; Gandhi, V. Intracellular succinylation of 8-chloroadenosine and its effect on fumarate levels. J. Biol. Chem. 2010, 285, 8022–8030. [Google Scholar] [CrossRef]
- Xu, H.; Chen, X.; Xu, X.; Shi, R.; Suo, S.; Cheng, K.; Zheng, Z.; Wang, M.; Wang, L.; Zhao, Y.; et al. Lysine acetylation and succinylation in hela cells and their essential roles in response to UV-induced stress. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- He, D.; Wang, Q.; Li, M.; Damaris, R.N.; Yi, X.; Cheng, Z.; Yang, P. Global proteome analyses of lysine acetylation and succinylation reveal the widespread involvement of both modification in metabolism in the embryo of germinating rice seed. J. Proteome Res. 2016, 15, 879–890. [Google Scholar] [CrossRef]
- Chen, Y. Quantitative analysis of the Sirt5-regulated lysine succinylation proteome in mammalian cells. Methods Mol. Biol. 2016, 1410, 23–37. [Google Scholar] [CrossRef]
- Bontemps-Gallo, S.; Madec, E.; Robbe-Masselot, C.; Souche, E.; Dondeyne, J.; Lacroix, J.M. The opgC gene is required for OPGs succinylation and is osmoregulated through RcsCDB and EnvZ/OmpR in the phytopathogen Dickeya dadantii. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Atila, M.; Katselis, G.; Chumala, P.; Luo, Y. Characterization of N-succinylation of l-lysylphosphatidylglycerol in Bacillus subtilis using tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Tayyab, S.; Qasim, M.A. A correlation between changes in conformation and molecular properties of bovine serum albumin upon succinylation. J. Biochem. 1986, 100, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Jou, Y.H.; Johnson, G.; Pressman, D. Succinylation of hapten-protein conjugates facilitates coupling to erythrocytes by water soluble carbodiimide: Preparation of stable and sensitive target cells for use in hemolytic assays. J. Immunol. Methods 1981, 42, 79–92. [Google Scholar] [CrossRef]
- Thuy, L.P.; Brown, J.E.; Baugh, R.F.; Hougie, C. Effects of succinylation and dodecyl, succinylation on bovine factor VIII/von Willebrand factor complex. Thromb. Res. 1980, 18, 305–313. [Google Scholar] [CrossRef]
- Rosen, R.; Becher, D.; Buttner, K.; Biran, D.; Hecker, M.; Ron, E.Z. Probing the active site of homoserine trans-succinylase. FEBS Lett. 2004, 577, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Bochmann, S.M.; Spiess, T.; Kotter, P.; Entian, K.D. Synthesis and succinylation of subtilin-like lantibiotics are strongly influenced by glucose and transition state regulator AbrB. Appl. Environ. Microbiol. 2015, 81, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, J.; Cheng, Z.; Gao, P.; Sun, J.; Chen, X.; Chen, C.; Wang, Y.; Wang, Z. Quantitative global proteome and lysine succinylome analyses provide insights into metabolic regulation and lymph node metastasis in gastric cancer. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.L.; Xing, J.; Chong, C.K.; Liu, H.; Corke, H. Determination of the degree of succinylation in diverse modified starches by raman spectroscopy. J. Agric. Food Chem. 2000, 48, 5105–5108. [Google Scholar] [CrossRef] [PubMed]
- Alcalde, M.; Plou, F.J.; Teresa Martin, M.; Valdes, I.; Mendez, E.; Ballesteros, A. Succinylation of cyclodextrin glycosyltransferase from Thermoanaerobacter sp. 501 enhances its transferase activity using starch as donor. J. Biotechnol. 2001, 86, 71–80. [Google Scholar] [CrossRef]
- Wan, Y.; Liu, J.; Guo, S. Effects of succinylation on the structure and thermal aggregation of soy protein isolate. Food Chem. 2018, 245, 542–550. [Google Scholar] [CrossRef]
- Smestad, J.; Erber, L.; Chen, Y.; Maher, L.J., III. Chromatin succinylation correlates with active gene expression and is perturbed by defective TCA cycle metabolism. iScience 2018, 2, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Yang, M.; Yue, Y.; Ge, F.; Li, Y.; Guo, X.; Zhang, J.; Zhang, F.; Nie, X.; Wang, S. Lysine succinylation contributes to aflatoxin production and pathogenicity in Aspergillus flavus. Mol. Cell. Proteomics 2018, 17, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Mujahid, H.; Meng, X.; Xing, S.; Peng, X.; Wang, C.; Peng, Z. Malonylome analysis in developing rice (Oryza sativa) seeds suggesting that protein lysine malonylation is well-conserved and overlaps with acetylation and succinylation substantially. J. Proteomics 2018, 170, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.Q.; Li, G.Y.; Xie, Q.T.; Zhang, B.; Li, X.M.; Pan, Y.; Chen, H.Q. Evaluation studies on the combined effect of hydrothermal treatment and octenyl succinylation on the physic-chemical, structural and digestibility characteristics of sweet potato starch. Food Chem. 2018, 256, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, Y.; Chen, Y.; Cheng, Z.; Gu, J.; Deng, J.; Bi, L.; Chen, C.; Mo, R.; Wang, X.; et al. Succinylome analysis reveals the involvement of lysine succinylation in metabolism in pathogenic Mycobacterium tuberculosis. Mol. Cell. Proteomics 2015, 14, 796–811. [Google Scholar] [CrossRef]
- Jin, W.; Wu, F. Proteome-wide identification of lysine succinylation in the proteins of tomato (Solanum lycopersicum). PLoS ONE 2016, 11, e0147586. [Google Scholar] [CrossRef]
- Komine-Abe, A.; Nagano-Shoji, M.; Kubo, S.; Kawasaki, H.; Yoshida, M.; Nishiyama, M.; Kosono, S. Effect of lysine succinylation on the regulation of 2-oxoglutarate dehydrogenase inhibitor, OdhI, involved in glutamate production in Corynebacterium glutamicum. Biosci. Biotechnol. Biochem. 2017, 81, 2130–2138. [Google Scholar] [CrossRef]
- Feng, S.; Jiao, K.; Guo, H.; Jiang, M.; Hao, J.; Wang, H.; Shen, C. Succinyl-proteome profiling of Dendrobium officinale, an important traditional Chinese orchid herb, revealed involvement of succinylation in the glycolysis pathway. BMC Genomics 2017, 18. [Google Scholar] [CrossRef]
- Okanishi, H.; Kim, K.; Fukui, K.; Yano, T.; Kuramitsu, S.; Masui, R. Proteome-wide identification of lysine succinylation in thermophilic and mesophilic bacteria. Biochim. Biophys. Acta 2017, 1865, 232–242. [Google Scholar] [CrossRef]
- Xie, L.; Liu, W.; Li, Q.; Chen, S.; Xu, M.; Huang, Q.; Zeng, J.; Zhou, M.; Xie, J. First succinyl-proteome profiling of extensively drug-resistant Mycobacterium tuberculosis revealed involvement of succinylation in cellular physiology. J. Proteome Res. 2015, 14, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Liu, Z.; Tian, G.; Bao, X.; Ishibashi, T.; Li, X.D. Site-specific installation of succinyl lysine analog into histones reveals the effect of h2bk34 succinylation on nucleosome dynamics. Cell Chem. Biol. 2018, 25, 166–174.e7. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, K.A.; Abraham, D.M.; Liu, J.; Locasale, J.W.; Grimsrud, P.A.; Hirschey, M.D. Ablation of Sirtuin5 in the postnatal mouse heart results in protein succinylation and normal survival in response to chronic pressure overload. J. Biol. Chem. 2018, 293, 10630–10645. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ning, Q.; Chai, H.; Ma, Z. Accurate in silico identification of protein succinylation sites using an iterative semi-supervised learning technique. J. Theor. Biol. 2015, 374, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ding, Y.X.; Ding, J.; Lei, Y.H.; Wu, L.Y.; Deng, N.Y. iSuc-PseAAC: predicting lysine succinylation in proteins by incorporating peptide position-specific propensity. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.D.; Shi, S.P.; Wen, P.P.; Qiu, J.D. SuccFind: A novel succinylation sites online prediction tool via enhanced characteristic strategy. Bioinformatics 2015. [Google Scholar] [CrossRef] [PubMed]
- Dehzangi, A.; Lopez, Y.; Lal, S.P.; Taherzadeh, G.; Michaelson, J.; Sattar, A.; Tsunoda, T.; Sharma, A. PSSM-Suc: Accurately predicting succinylation using position specific scoring matrix into bigram for feature extraction. J. Theor. Biol. 2017, 425, 97–102. [Google Scholar] [CrossRef]
- Lopez, Y.; Dehzangi, A.; Lal, S.P.; Taherzadeh, G.; Michaelson, J.; Sattar, A.; Tsunoda, T.; Sharma, A. SucStruct: Prediction of succinylated lysine residues by using structural properties of amino acids. Anal. Biochem. 2017, 527, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Yang, S.; Zhou, Y.; Mollah, M.N. SuccinSite: a computational tool for the prediction of protein succinylation sites by exploiting the amino acid patterns and properties. Mol. Biosyst. 2016, 12, 786–795. [Google Scholar] [CrossRef]
- Thanamani, B.; Thanamani Selvados, A. Feature Selection based on Information Gain. Int. J. Innov. Technol. Expl. Eng. 2013, 2, 2278–3075. [Google Scholar]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.C. pSuc-Lys: Predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. J. Theor. Biol. 2016, 394, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Khatun, M.S.; Mollah, M.N.; Cao, Y.; Guo, D. A systematic identification of species-specific protein succinylation sites using joint element features information. Int. J. Nanomed. 2017, 12, 6303–6315. [Google Scholar] [CrossRef] [PubMed]
- Ning, Q.; Zhao, X.; Bao, L.; Ma, Z.; Zhao, X. Detecting succinylation sites from protein sequences using ensemble support vector machine. BMC Bioinform. 2018, 19, 237. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, M.; Wang, Y.; Chen, Z.; Zhang, J.; Lin, X.; Ge, F.; Zhao, J. Effects of PSII manganese-stabilizing protein succinylation on photosynthesis in the model cyanobacterium Synechococcus sp. PCC 7002. Plant Cell Physiol. 2018, 59, 1466–1482. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Kurata, H. GPSuc: Global prediction of generic and species-specific succinylation sites by aggregating multiple sequence features. PloS One 2018, 13, e0200283. [Google Scholar] [CrossRef] [PubMed]
- Dehzangi, A.; Lopez, Y.; Lal, S.P.; Taherzadeh, G.; Sattar, A.; Tsunoda, T.; Sharma, A. Improving succinylation prediction accuracy by incorporating the secondary structure via helix, strand and coil, and evolutionary information from profile bigrams. PLoS ONE 2018, 13, e0191900. [Google Scholar] [CrossRef] [PubMed]
- Lopez, Y.; Sharma, A.; Dehzangi, A.; Lal, S.P.; Taherzadeh, G.; Sattar, A.; Tsunoda, T. Success: evolutionary and structural properties of amino acids prove effective for succinylation site prediction. BMC Genomics 2018, 19. [Google Scholar] [CrossRef]
- Gaviard, C.; Broutin, I.; Cosette, P.; De, E.; Jouenne, T.; Hardouin, J. Lysine succinylation and acetylation in Pseudomonas aeruginosa. J. Proteome Res. 2018, 17, 2449–2459. [Google Scholar] [CrossRef]
- Ai, H.; Wu, R.; Zhang, L.; Wu, X.; Ma, J.; Hu, H.; Huang, L.; Chen, W.; Zhao, J.; Liu, H. pSuc-PseRat: Predicting lysine succinylation in proteins by exploiting the ratios of sequence coupling and properties. J. Comput. Biol. 2017, 24, 1050–1059. [Google Scholar] [CrossRef]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.C. iSuc-PseOpt: Identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal. Biochem. 2016, 497, 48–56. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Gao, T.; Pan, Z.; Cheng, H.; Yang, Q.; Cheng, Z.; Guo, A.; Ren, J.; Xue, Y. CPLM: a database of protein lysine modifications. Nucleic Acids Res. 2014, 42, D531–D536. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhou, J.; Lin, S.; Deng, W.; Zhang, Y.; Xue, Y. PLMD: An updated data resource of protein lysine modifications. J. Genet. Genomics 2017, 44, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Chen, X.; Xu, X.; Shi, R.; Suo, S.; Cheng, K.; Zheng, Z.; Wang, M.; Wang, L.; Zhao, Y.; et al. Corrigendum: Lysine acetylation and succinylation in hela cells and their essential roles in response to UV-induced stress. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Yang, Q.; Li, P.; Wen, Y.; Li, S.; Chen, J.; Liu, X.; Wang, L.; Li, X. Cadmium inhibits lysine acetylation and succinylation inducing testicular injury of mouse during development. Toxicol. Lett. 2018, 291, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Meyer, J.G.; Schilling, B. quantification of site-specific protein lysine acetylation and succinylation stoichiometry using data-independent acquisition mass spectrometry. J. Vis. Exp. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Deng, X.; Wang, J.; Zhu, G.; Cao, H.; Yuan, L.; Yan, Y. First comprehensive proteome analyses of lysine acetylation and succinylation in seedling leaves of Brachypodium distachyon L. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Khatun, M.S.; Mollah, M.N.H.; Yong, C.; Guo, D. NTyroSite: Computational identification of protein nitrotyrosine sites using sequence evolutionary features. Molecules 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Khatun, M.S.; Kurata, H. Computational modeling of lysine post-translational modification: An overview. Curr. Synthetic Sys. Biol. 2018, 6. [Google Scholar] [CrossRef]
- Hasan, M.M; Khatun, M.S. Prediction of protein post-translational modification sites: An overview. Ann. Proteom. Bioinform. 2018, 2, 49–57. [Google Scholar] [CrossRef]
- O’Shea, J.P.; Chou, M.F.; Quader, S.A.; Ryan, J.K.; Church, G.M.; Schwartz, D. pLogo: A probabilistic approach to visualizing sequence motifs. Nat. Methods 2013, 10, 1211–1212. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qiu, J.D.; Shi, S.P.; Suo, S.B.; Huang, S.Y.; Liang, R.P. Incorporating key position and amino acid residue features to identify general and species-specific ubiquitin conjugation sites. Bioinformatics 2013, 29, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Wuyun, Q.; Zheng, W.; Zhang, Y.; Ruan, J.; Hu, G. Improved species-specific lysine acetylation site prediction based on a large variety of features set. PLoS ONE 2016, 11, e0155370. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, M.; Wang, H.; Tan, H.; Zhang, Z.; Webb, G.I.; Song, J. Accurate in silico identification of species-specific acetylation sites by integrating protein sequence-derived and functional features. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.P.; Shi, S.P.; Xu, H.D.; Wang, L.N.; Qiu, J.D. Accurate in silico prediction of species-specific methylation sites based on information gain feature optimization. Bioinformatics 2016, 32, 3107–3115. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.D.; Lee, T.Y.; Tzeng, S.W.; Horng, J.T. KinasePhos: A web tool for identifying protein kinase-specific phosphorylation sites. Nucleic Acids Res. 2005, 33, W226–W229. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wang, H.; Wang, J.; Leier, A.; Marquez-Lago, T.; Yang, B.; Zhang, Z.; Akutsu, T.; Webb, G.I.; Daly, R.J. PhosphoPredict: A bioinformatics tool for prediction of human kinase-specific phosphorylation substrates and sites by integrating heterogeneous feature selection. Sci. Rep. 2017, 7, 6862. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.N.; Shi, S.P.; Xu, H.D.; Wen, P.P.; Qiu, J.D. Computational prediction of species-specific malonylation sites via enhanced characteristic strategy. Bioinformatics 2017, 33, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.P.; Xu, H.D.; Wen, P.P.; Qiu, J.D. Progress and challenges in predicting protein methylation sites. Mol. Biosyst. 2015, 11, 2610–2619. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, X.; Li, F.; Li, C.; Marquez-Lago, T.; Leier, A.; Akutsu, T.; Webb, G.I.; Xu, D.; Smith, A.I.; et al. Large-scale comparative assessment of computational predictors for lysine post-translational modification sites. Brief. Bioinform. 2018. [Google Scholar] [CrossRef]
- Khatun, M.S.; Hasan, M.M.; Mollah, M.N.H.; Kurata, H. SIPMA: A systematic identification of protein-protein interactions in Zea mays using autocorrelation features in a machine-learning framework. In Proceedings of the IEEE 18th International Conference on Bioinformatics and Bioengineering, Taichung, Taiwan, 29–31 October 2018; IEEE: Piscataway, NJ, USA, 2018; pp. 122–125. [Google Scholar]
- Hasan, M.M.; Kurata, H. iLMS, Computational Identification of lysine-malonylation sites by combining multiple sequence features. In Proceedings of the IEEE 18th International Conference on Bioinformatics and Bioengineering, Taichung, Taiwan, 29–31 October 2018; IEEE: Piscataway, NJ, USA, 2018; pp. 356–359. [Google Scholar]
- Siu, M.; Thompson, L.U. Effect of succinylation on the protein quality and urinary excretion of bound and free amino acids. J. Agric. Food Chem. 1982, 30, 1179–1183. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhou, Y.; Zhang, Z.; Song, J. Towards more accurate prediction of ubiquitination sites: A comprehensive review of current methods, tools and features. Brief. Bioinform. 2015, 16, 640–657. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M; Khatun, M.S.; Kurata, H. A comprehensive review of in silico analysis for protein S-sulfenylation sites. Protein Pept. Lett. 2018, 25, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Guo, D.; Kurata, H. Computational identification of protein S-sulfenylation sites by incorporating the multiple sequence features information. Mol. Biosyst. 2017, 13, 2545–2550. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Zhou, Y.; Lu, X.; Li, J.; Song, J.; Zhang, Z. Computational Identification of protein pupylation sites by using profile-based composition of k-spaced amino acid pairs. PLoS ONE 2015, 10, e0129635. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Khatun, M.S.T. Recent progress and challenges for protein pupylation sites prediction. EC Proteomics Bioinform. 2017, 2, 36–45. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Li, W.; Fu, L.; Niu, B.; Wu, S.; Wooley, J. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief. Bioinform. 2012, 13, 656–668. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. Clustering huge protein sequence sets in linear time. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Radi, R. Protein tyrosine nitration: Biochemical mechanisms and structural basis of functional effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Colaert, N.; Helsens, K.; Martens, L.; Vandekerckhove, J.; Gevaert, K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 2009, 6, 786–787. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
An overview of current computational prediction algorithms of succinylation sites.

Figure 2.
Window selection procedure for generating positive and negative samples.
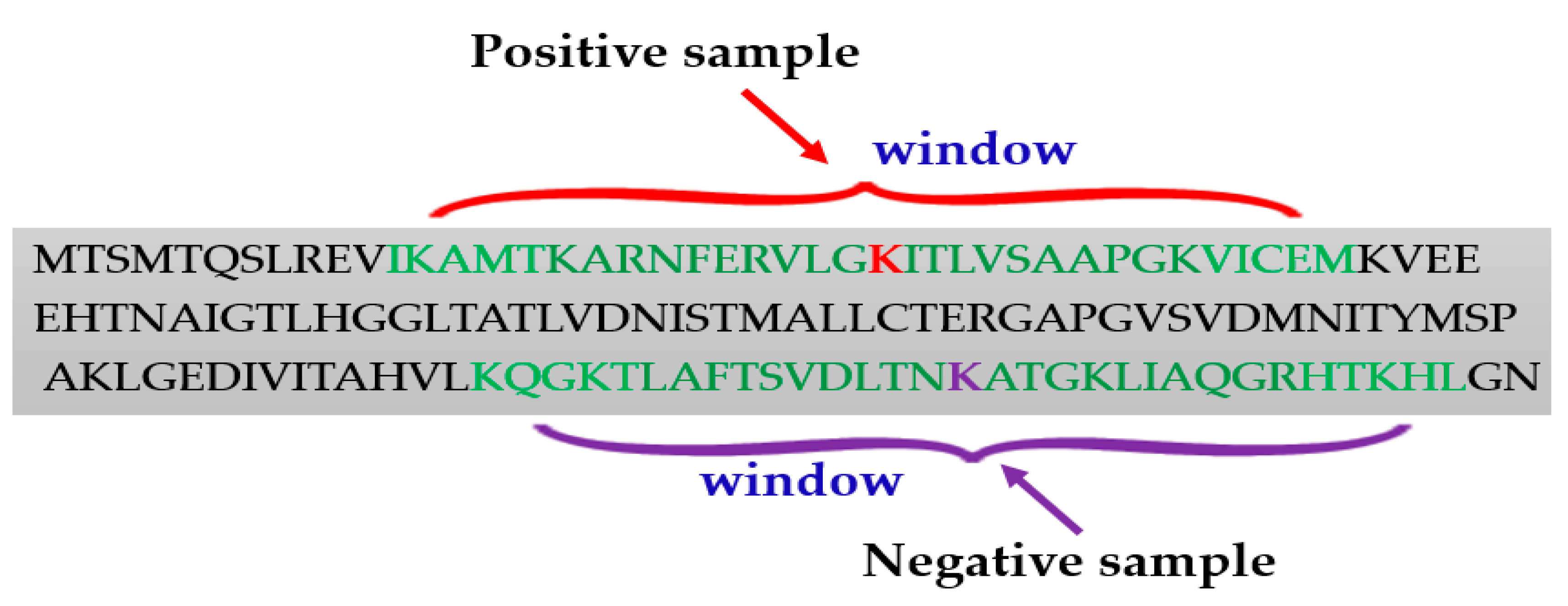
Figure 3.
pLogo graphs of the sequences with the centered succinylation sites. Nine species-specific datasets of H. sapiens, H. capsulatum, M. musculus, E. coli, M. tuberculosis, S. cerevisiae, T. gondii, S. lycopersicum and T. aestivum (https://plogo.uconn.edu/) and their combined (generic) datasets are used. The significantly enriched/depleted amino acid residues (student t-test, p < 0.05) are shown.
Figure 3.
pLogo graphs of the sequences with the centered succinylation sites. Nine species-specific datasets of H. sapiens, H. capsulatum, M. musculus, E. coli, M. tuberculosis, S. cerevisiae, T. gondii, S. lycopersicum and T. aestivum (https://plogo.uconn.edu/) and their combined (generic) datasets are used. The significantly enriched/depleted amino acid residues (student t-test, p < 0.05) are shown.

Figure 4.
Performance comparison of generic succinylation site prediction models on an independent dataset.
Figure 4.
Performance comparison of generic succinylation site prediction models on an independent dataset.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of the reviewed predictors for lysine succinylation sites.
| Tools | SucPred | iSuc-PseAAC | SuccFind | iSuc-PseOpt | pSuc-Lys | SucStruct | PSSM-Suc | SuccinSite | SuccinSite2.0 | SSEvol-Suc | Success | GPSuc |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Species | Generic | Generic | Generic | Generic | Generic | Generic | Generic | Generic | Generic and Species-specific | Generic | Generic | Generic and Species-specific |
| Web-server link | http://59.73.198.144:8088/SucPred/ | http://app.aporc.org/iSuc-PseAAC/ | http://bioinfo.ncu.edu.cn/SuccFind.aspx | http://www.jci-bioinfo.cn/iSuc-PseOpt | http://www.jci-bioinfo.cn/pSuc-Lys | https://github.com/YosvanyLopez/ | https://github.com/YosvanyLopez/PSSM-Suc | http://systbio.cau.edu.cn/SuccinSite/ | https://biocomputer.bio.cuhk.edu.hk/SuccinSite2.0/ | https://github.com/YosvanyLopez/SSEvol-Suc | https://github.com/YosvanyLopez/Success | http://kurata14.bio.kyutech.ac.jp/GPSuc/ |
| Working server | No | Yes | No | No | No | No | No | Yes | Yes | No | No | Yes |
| Machine learning | SVM | SVM | SVM | RF | RF | DT | DT | RF | RF | AdaBoost | SVM | RF and LR |
| Dataset size (Protein/succinylated) | 897/2511 | 896/2521 | 1044/2938 | 896/2521 | 896/2521 | 670/1782 | 670 / 1782 | 2322/5004 | 2322/5004 | 670/1782 | 670/1782 | 2322/5004 |
| Training (Pos/Neg) | 1436/18,958 | 1167/3553 | 2713/23598 | 1167/3553 | 1167/3553 | 1782/1872 | 1782/1643 | 4750/9500 | 4750/9500 | 1782/1872 | 1782/1872 | 4750/9500 |
| Independent (Pos/Neg) | 250/- | - | - | - | - | - | - | 254/2977 | 254/2977 | - | - | 254/2977 |
| Homolog redundancy | 35% | 40% | 30% | 40% | 40% | 40% | 40% | 30% | 30% | 40% | 40% | 30% |
| Window size | from −9 to +9 | from −7 to +7 | from −10 to +10 | from −15 to +15 | from −15 to +15 | from −15 to +15 | from −15 to +15 | from −13 to +13 | from −20 to +20 | from −15 to +15 | from −15 to +15 | from −20 to +20 |
| Adjusted batch prediction | NO | No | No | No | No | No | No | Yes | Yes | No | No | Yes |
| Processing time for a protein | - | Within 20 s | - | - | - | - | - | Within 20 s | Within 5 min | - | - | Within 5 min |
Table 2.
Statistics of the positive and negative samples of nine species-specific datasets used in this study.
Table 2.
Statistics of the positive and negative samples of nine species-specific datasets used in this study.
| Species | Datasets | Positive Samples | Negative Samples |
|---|---|---|---|
| H. sapiens | Training | 1351 | 2702 |
| Independent | 54 | 2004 | |
| M. musculus | Training | 414 | 828 |
| Independent | 24 | 679 | |
| E. coli | Training | 1942 | 3884 |
| Independent | 289 | 1381 | |
| M. tuberculosis | Training | 699 | 1398 |
| Independent | 61 | 242 | |
| S. cerevisiae | Training | 961 | 1922 |
| Independent | 90 | 1423 | |
| T. gondii | Training | 282 | 564 |
| Independent | 26 | 261 | |
| S. lycopersicum | Training | 242 | 484 |
| Independent | 33 | 274 | |
| A. capsulatus | Training | 332 | 664 |
| Independent | 50 | 591 | |
| T. aestivum | Training | 113 | 226 |
| Independent | 32 | 309 |
Table 3.
Statistics of feature encoding schemes used in the aforementioned succinylation site prediction tools.
Table 3.
Statistics of feature encoding schemes used in the aforementioned succinylation site prediction tools.
| Encoding Types | Genetic Explanation | References |
|---|---|---|
| AAindex | Based on the AAindex indices database, the encoding scheme of AAindex reveals the biochemical properties of the sequences. | [56,59,62] |
| ACF | The auto correlation function features for surrounding succinylation sequences. | [54] |
| EBGW | Coding based on grouped weight of physicochemical properties of sequences surrounding succinylation sites. | [54] |
| VDWV | Van der Waals volume properties of surrounding succinylation sequences. | [54] |
| WAAC | Position weight amino acid composition of surrounding succinylation sequences. | [54] |
| AAC | The amino acid composition characterizes the specific state of the surrounding succinylation sequences. | [65] |
| CKSAAP | The CKSAAP encoding represents the short sequence motif information in surrounding succinylation sites. | [56,59] |
| PseAAC | The pseudo amino acid composition reflects a vectorized sequence-coupling model of surrounding succinylation sites. | [56,61,70] |
| SF | The predicted structural feature reflects the structural properties of protein in surrounding succinylation sites. | [66] |
| Binary | The position-specific information measured by binary profile for the curated sequences. | [59,62,65] |
| PSSM | The PSSM exposes the evolutionary information from the sequences. | [57] |
| pCKSAAP | The pCKSAAP reflects the sequence patterns and evolutionary information from the query sequences. | [62,65] |
Data of Table 1 is used.
Table 4.
Performance of five major types of features for the training and independent datasets.
| Methods | Training | Independent | |||
|---|---|---|---|---|---|
| H. sapiens | RF | SVM | RF | SVM | |
| pCKSAAP | 0.856 | 0.838 | 0.695 | 0.691 | |
| CKSAAP | 0.816 | 0.831 | 0.677 | 0.663 | |
| AAindex | 0.739 | 0.728 | 0.759 | 0.755 | |
| Binary | 0.767 | 0.754 | 0.822 | 0.809 | |
| PseAAC | 0.819 | 0.822 | 0.658 | 0.649 | |
| H. capsulatum | pCKSAAP | 0.789 | 0.792 | 0.638 | 0.634 |
| CKSAAP | 0.788 | 0.783 | 0.619 | 0.607 | |
| AAindex | 0.712 | 0.722 | 0.658 | 0.666 | |
| Binary | 0.713 | 0.698 | 0.665 | 0.647 | |
| PseAAC | 0.759 | 0.743 | 0.612 | 0.614 | |
| M. musculus | pCKSAAP | 0.801 | 0.788 | 0.637 | 0.634 |
| CKSAAP | 0.777 | 0.767 | 0.646 | 0.651 | |
| AAindex | 0.648 | 0.655 | 0.679 | 0.672 | |
| Binary | 0.639 | 0.641 | 0.677 | 0.659 | |
| PseAAC | 0.711 | 0.722 | 0.609 | 0.611 | |
| E. coli | pCKSAAP | 0.769 | 0.761 | 0.679 | 0.684 |
| CKSAAP | 0.773 | 0.782 | 0.646 | 0.631 | |
| AAindex | 0.719 | 0.721 | 0.633 | 0.619 | |
| Binary | 0.689 | 0.674 | 0.619 | 0.607 | |
| PseAAC | 0.733 | 0.734 | 0.608 | 0.603 | |
| M. tuberculosis | pCKSAAP | 0.708 | 0.712 | 0.688 | 0.679 |
| CKSAAP | 0.689 | 0.675 | 0.664 | 0.671 | |
| AAindex | 0.667 | 0.658 | 0.656 | 0.655 | |
| Binary | 0.629 | 0.617 | 0.639 | 0.634 | |
| PseAAC | 0.643 | 0.634 | 0.629 | 0.617 | |
| S. cerevisiae | pCKSAAP | 0.882 | 0.869 | 0.776 | 0.772 |
| CKSAAP | 0.879 | 0.863 | 0.752 | 0.744 | |
| AAindex | 0.742 | 0.733 | 0.759 | 0.749 | |
| Binary | 0.741 | 0.745 | 0.798 | 0.787 | |
| PseAAC | 0.790 | 0.768 | 0.699 | 0.675 | |
| T. gondii | pCKSAAP | 0.834 | 0.836 | 0.657 | 0.666 |
| CKSAAP | 0.826 | 0.822 | 0.655 | 0.638 | |
| AAindex | 0.726 | 718 | 0.663 | 0.647 | |
| Binary | 0.744 | 0.745 | 0.679 | 0.671 | |
| PseAAC | 0.801 | 0.788 | 0.678 | 0.664 | |
| S. lycopersicum | pCKSAAP | 0.842 | 0.836 | 0.649 | 0.642 |
| CKSAAP | 0.833 | 0.824 | 0.648 | 0.637 | |
| AAindex | 0.753 | 0.765 | 0.644 | 0.629 | |
| Binary | 0.729 | 0.722 | 0.637 | 0.631 | |
| PseAAC | 0.801 | 0.783 | 0.678 | 0.658 | |
| T. aestivum | pCKSAAP | 0.822 | 0.826 | 0.649 | 0.654 |
| CKSAAP | 0.821 | 0.811 | 0.638 | 0.634 | |
| AAindex | 0.736 | 0.734 | 0.604 | 0.611 | |
| Binary | 0.726 | 0.719 | 0.612 | 0.596 | |
| PseAAC | 0.778 | 0.769 | 0.632 | 0.628 | |
AUC values are used to assess the prediction performance.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hasan, M.M.; Khatun, M.S.; Kurata, H. Large-Scale Assessment of Bioinformatics Tools for Lysine Succinylation Sites. Cells 2019, 8, 95. https://doi.org/10.3390/cells8020095
AMA Style
Hasan MM, Khatun MS, Kurata H. Large-Scale Assessment of Bioinformatics Tools for Lysine Succinylation Sites. Cells. 2019; 8(2):95. https://doi.org/10.3390/cells8020095
Chicago/Turabian StyleHasan, Md. Mehedi, Mst. Shamima Khatun, and Hiroyuki Kurata. 2019. "Large-Scale Assessment of Bioinformatics Tools for Lysine Succinylation Sites" Cells 8, no. 2: 95. https://doi.org/10.3390/cells8020095
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.