yRACK1/Asc1 proxiOMICs—Towards Illuminating Ships Passing in the Night

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid Construction
2.2. Yeast Strains and Growth Conditions
2.3. Western Blot Analysis
2.4. BioID Experiments
2.5. Liquid Chromatography-Mass Spectrometry (LC-MS) Analysis
2.6. Phenotypic Growth Tests
2.7. Accession Number
3. Results
3.1. Expression of Rps2-BirA* in S. cerevisiae Strains of the Σ1278b Genetic Background
3.2. Rps2-BirA* Specifically Biotinylates Def1 and Stm1
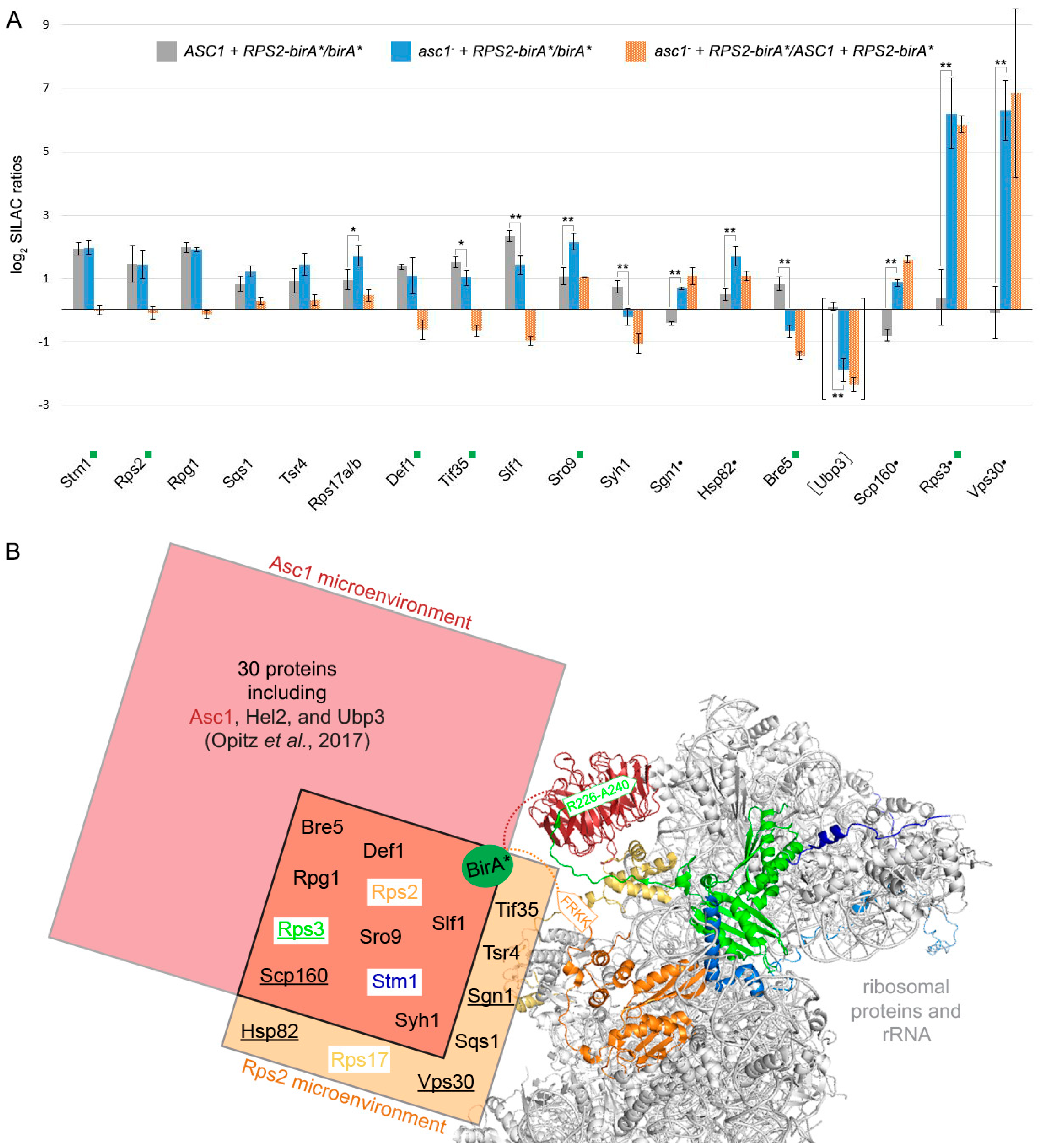
3.3. Asc1 Inversely Affects Rps2-BirA*-Mediated Biotinylation of the mRNA-Binding Paralogs Sro9 and Slf1
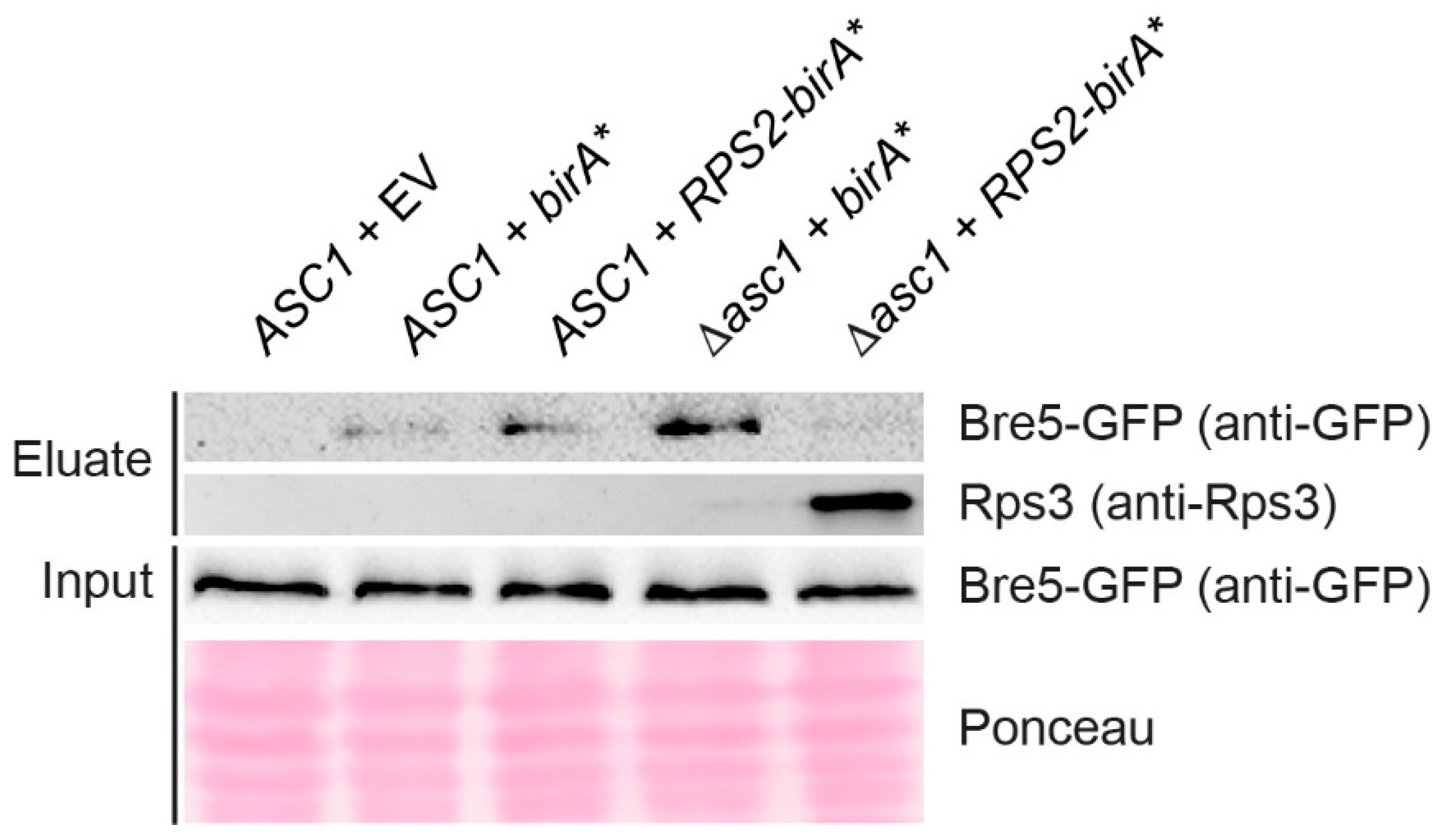
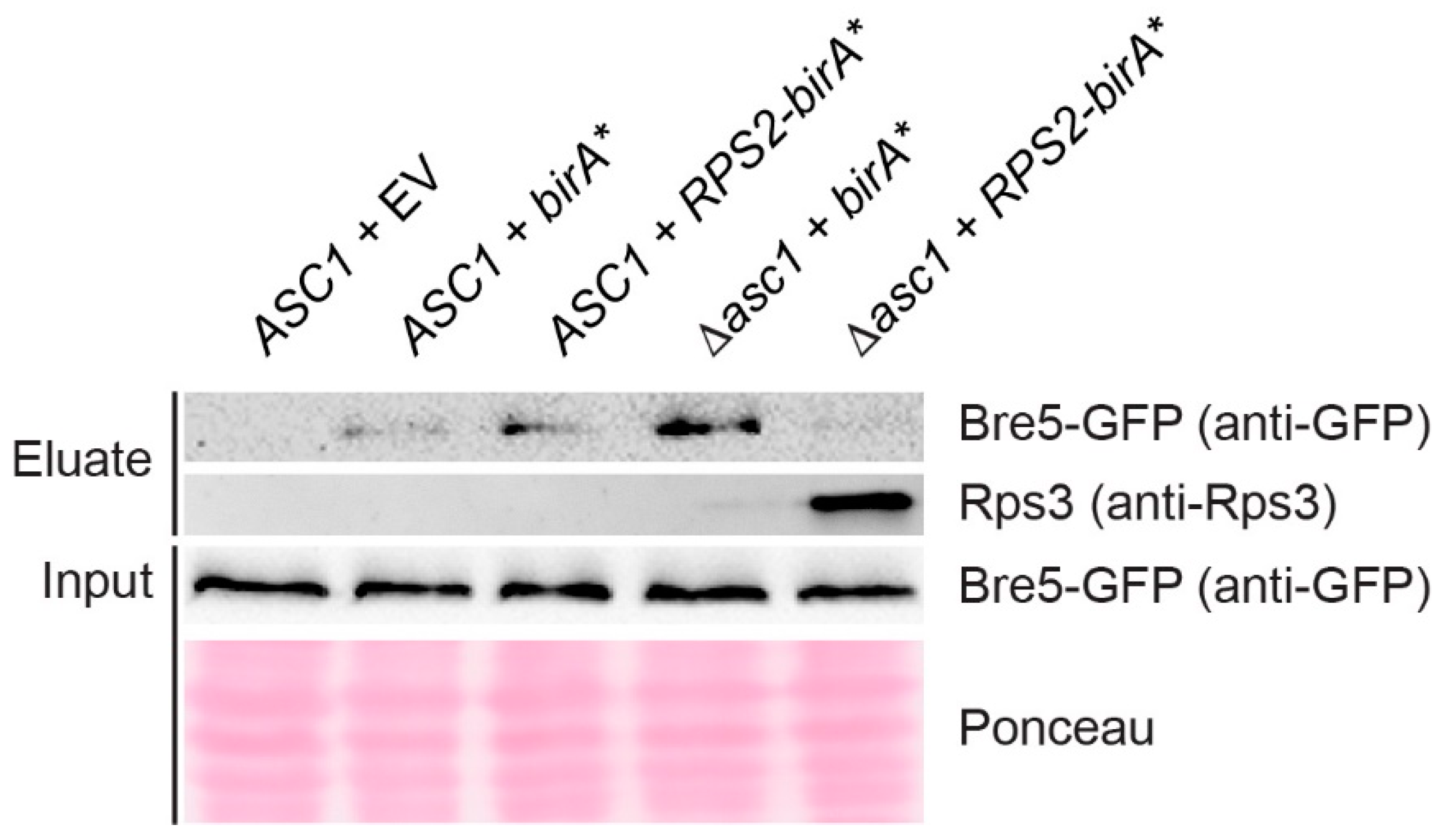
3.4. Asc1 Inversely Affects Rps2-BirA*-Mediated Biotin-Enrichment of Bre5 on the One Hand and Rps3 and Vps30 on the Other Hand
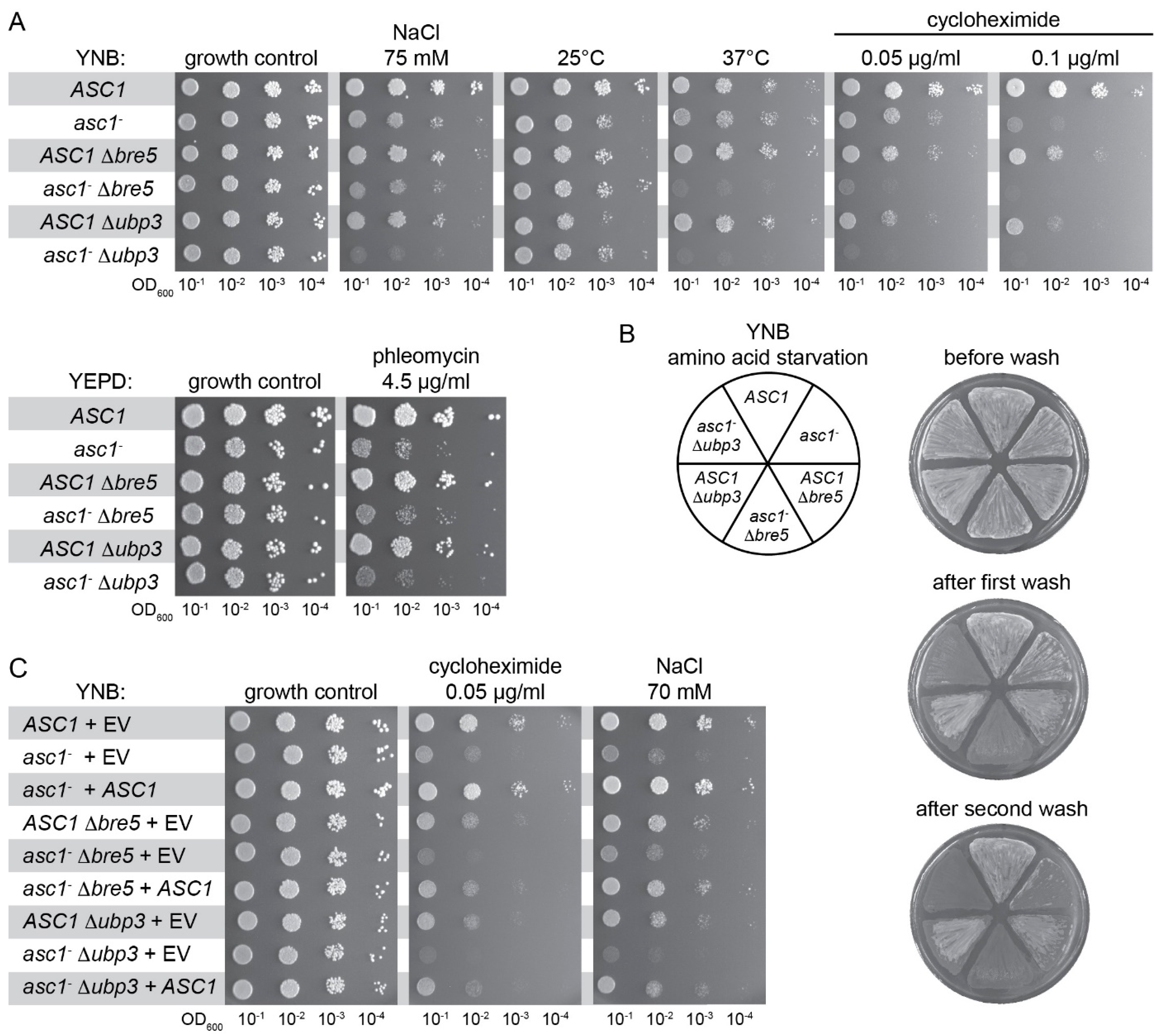
3.5. ASC1 Genetically Interacts with UBP3/BRE5 to Protect Cells Against Heat, Starvation, and Translational Stress
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferreira-Cerca, S.; Pöll, G.; Kühn, H.; Neueder, A.; Jakob, S.; Tschochner, H.; Milkereit, P. Analysis of the in vivo assembly pathway of eukaryotic 40S ribosomal proteins. Mol. Cell 2007, 28, 446–457. [Google Scholar] [CrossRef]
- Heuer, A.; Thomson, E.; Schmidt, C.; Berninghausen, O.; Becker, T.; Hurt, E.; Beckmann, R. Cryo-EM structure of a late pre-40S ribosomal subunit from Saccharomyces cerevisiae. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Rachfall, N.; Schmitt, K.; Bandau, S.; Smolinski, N.; Ehrenreich, A.; Valerius, O.; Braus, G.H. RACK1/Asc1p, a ribosomal node in cellular signaling. Mol. Cell. Proteom. 2013, 12, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Valerius, O.; Kleinschmidt, M.; Rachfall, N.; Schulze, F.; López Marín, S.; Hoppert, M.; Streckfuss-Bömeke, K.; Fischer, C.; Braus, G.H. The Saccharomyces homolog of mammalian RACK1, Cpc2/Asc1p, is required for FLO11-dependent adhesive growth and dimorphism. Mol. Cell. Proteom. 2007, 6, 1968–1979. [Google Scholar] [CrossRef] [PubMed]
- Jannot, G.; Bajan, S.; Giguère, N.J.; Bouasker, S.; Banville, I.H.; Piquet, S.; Hutvagner, G.; Simard, M.J. The ribosomal protein RACK1 is required for microRNA function in both C. elegans and humans. EMBO Rep. 2011, 12, 581–586. [Google Scholar] [CrossRef]
- Li, S.; Esterberg, R.; Lachance, V.; Ren, D.; Radde-Gallwitz, K.; Chi, F.; Parent, J.L.; Fritz, A.; Chen, P. Rack1 is required for Vangl2 membrane localization and planar cell polarity signaling while attenuating canonical Wnt activity. Proc. Natl. Acad. Sci. USA 2011, 108, 2264–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volta, V.; Beugnet, A.; Gallo, S.; Magri, L.; Brina, D.; Pesce, E.; Calamita, P.; Sanvito, F.; Biffo, S. RACK1 depletion in a mouse model causes lethality, pigmentation deficits and reduction in protein synthesis efficiency. Cell. Mol. Life Sci. 2012, 70, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Wehner, P.; Shnitsar, I.; Urlaub, H.; Borchers, A. RACK1 is a novel interaction partner of PTK7 that is required for neural tube closure. Development 2011, 138, 1321–1327. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J.; Xie, D. RACK1, a versatile hub in cancer. Oncogene 2015, 34, 1890–1898. [Google Scholar] [CrossRef]
- Schmitt, K.; Smolinski, N.; Neumann, P.; Schmaul, S.; Hofer-Pretz, V.; Braus, G.H.; Valerius, O. Asc1p/RACK1 Connects Ribosomes to Eukaryotic Phosphosignaling. Mol. Cell. Biol. 2017, 37, e00279-16. [Google Scholar] [CrossRef]
- Dudley, A.M.; Janse, D.M.; Tanay, A.; Shamir, R.; Church, G.M. A global view of pleiotropy and phenotypically derived gene function in yeast. Mol. Syst. Biol. 2005, 1, 2005.0001. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lee, I.; Moradi, E.; Hung, N.J.; Johnson, A.W.; Marcotte, E.M. Rational extension of the ribosome biogenesis pathway using network-guided genetics. PLoS Biol. 2009, 7, e1000213. [Google Scholar] [CrossRef] [PubMed]
- Tigano, M.; Ruotolo, R.; Dallabona, C.; Fontanesi, F.; Barrientos, A.; Donnini, C.; Ottonello, S. Elongator-dependent modification of cytoplasmic tRNALysUUU is required for mitochondrial function under stress conditions. Nucleic Acids Res. 2015, 43, 8368–8380. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Jaafar, L.; Cashikar, A.; Flores-Rozas, H. Identification of genes required for protection from doxorubicin by a genome-wide screen in Saccharomyces cerevisiae. Cancer Res. 2007, 67, 11411–11418. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shem, A.; Garreau de Loubresse, N.; Melnikov, S.; Jenner, L.; Yusupova, G.; Yusupov, M. The structure of the eukaryotic ribosome at 3.0 A resolution. Science 2011, 334, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Lomakin, I.B.; Steitz, T.A. The initiation of mammalian protein synthesis and mRNA scanning mechanism. Nature 2013, 500, 307–311. [Google Scholar] [CrossRef] [Green Version]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Opitz, N.; Schmitt, K.; Hofer-Pretz, V.; Neumann, B.; Krebber, H.; Braus, G.H.; Valerius, O. Capturing the Asc1p/Receptor for Activated C Kinase 1 (RACK1) Microenvironment at the Head Region of the 40S Ribosome with Quantitative BioID in Yeast. Mol. Cell. Proteom. 2017, 16, 2199–2218. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.D.; Harreman, M.; Taschner, M.; Reid, J.; Walker, J.; Erdjument-Bromage, H.; Tempst, P.; Svejstrup, J.Q. Proteasome-mediated processing of Def1, a critical step in the cellular response to transcription stress. Cell 2013, 154, 983–995. [Google Scholar] [CrossRef]
- Ikeuchi, K.; Izawa, T.; Inada, T. Recent Progress on the Molecular Mechanism of Quality Controls Induced by Ribosome Stalling. Front. Genet. 2019, 9, 743. [Google Scholar] [CrossRef] [Green Version]
- Sitron, C.S.; Park, J.H.; Brandman, O. Asc1, Hel2, and Slh1 couple translation arrest to nascent chain degradation. RNA 2017, 23, 798–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeuchi, K.; Inada, T. Ribosome-associated Asc1/RACK1 is required for endonucleolytic cleavage induced by stalled ribosome at the 3′ end of nonstop mRNA. Sci. Rep. 2016, 6, 28234. [Google Scholar] [CrossRef] [PubMed]
- Kuroha, K.; Akamatsu, M.; Dimitrova, L.; Ito, T.; Kato, Y.; Shirahige, K.; Inada, T. Receptor for activated C kinase 1 stimulates nascent polypeptide-dependent translation arrest. EMBO Rep. 2010, 11, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Sitron, C.S.; Brandman, O. CAT tails drive degradation of stalled polypeptides on and off the ribosome. Nat. Struct. Mol. Biol. 2019, 26, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Simms, C.L.; Kim, K.Q.; Yan, L.L.; Qiu, J.; Zaher, H.S. Interactions between the mRNA and Rps3/uS3 at the entry tunnel of the ribosomal small subunit are important for no-go decay. PLoS Genet. 2018, 14, e1007818. [Google Scholar] [CrossRef]
- Simms, C.L.; Yan, L.L.; Zaher, H.S. Ribosome Collision Is Critical for Quality Control during No-Go Decay. Mol. Cell 2017, 68, 361–373. [Google Scholar] [CrossRef]
- Jung, Y.; Kim, H.D.; Yang, H.W.; Kim, H.J.; Jang, C.Y.; Kim, J. Modulating cellular balance of Rps3 mono-ubiquitination by both Hel2 E3 ligase and Ubp3 deubiquitinase regulates protein quality control. Exp. Mol. Med. 2017, 49, e390. [Google Scholar] [CrossRef]
- Sundaramoorthy, E.; Leonard, M.; Mak, R.; Liao, J.; Fulzele, A.; Bennett, E.J. ZNF598 and RACK1 Regulate Mammalian Ribosome-Associated Quality Control Function by Mediating Regulatory 40S Ribosomal Ubiquitylation. Mol. Cell 2017, 65, 751–760. [Google Scholar] [CrossRef]
- Kraft, C.; Deplazes, A.; Sohrmann, M.; Peter, M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol. 2008, 10, 602–610. [Google Scholar] [CrossRef]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef]
- Schopp, I.M.; Amaya Ramirez, C.C.; Debeljak, J.; Kreibich, E.; Skribbe, M.; Wild, K.; Béthune, J. Split-BioID a conditional proteomics approach to monitor the composition of spatiotemporally defined protein complexes. Nat. Commun. 2017, 8, 15690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, T.J. RACK1 research—Ships passing in the night? FEBS Lett. 2012, 586, 2787–2789. [Google Scholar] [CrossRef] [PubMed]
- Mumberg, D.; Müller, R.; Funk, M. Regulatable promoters of Saccharomyces cerevisiae: Comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 1994, 22, 5767–5768. [Google Scholar] [CrossRef] [PubMed]
- Gueldener, U.; Heinisch, J.; Koehler, G.J.; Voss, D.; Hegemann, J.H. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002, 30, e23. [Google Scholar] [CrossRef]
- Huh, W.K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O′Shea, E.K. Global analysis of protein localization in budding yeast. Nature 2003, 425, 686–691. [Google Scholar] [CrossRef]
- Kushnirov, V.V. Rapid and reliable protein extraction from yeast. Yeast 2000, 16, 857–860. [Google Scholar] [CrossRef]
- Wessel, D.; Flügge, U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Csordas, A.; Sun, Z.; Jarnuczak, A.; Perez-Riverol, Y.; Ternent, T.; Campbell, D.S.; Bernal-Llinares, M.; Okuda, S.; Kawano, S.; et al. The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45, D1100–D1106. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Wilson, K.P.; Shewchuk, L.M.; Brennan, R.G.; Otsuka, A.J.; Matthews, B.W. Escherichia coli biotin holoenzyme synthetase/bio repressor crystal structure delineates the biotin- and DNA-binding domains. Proc. Natl. Acad. Sci. USA 1992, 89, 9257–9261. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.L.; Wagner, S.; Herrmannová, A.; Burela, L.; Zhang, F.; Saini, A.K.; Valásek, L.; Hinnebusch, A.G. The C-terminal region of eukaryotic translation initiation factor 3a (eIF3a) promotes mRNA recruitment, scanning, and, together with eIF3j and the eIF3b RNA recognition motif, selection of AUG start codons. Mol. Cell. Biol. 2010, 30, 4415–4434. [Google Scholar] [CrossRef]
- Cuchalová, L.; Kouba, T.; Herrmannová, A.; Dányi, I.; Chiu, W.L.; Valásek, L. The RNA recognition motif of eukaryotic translation initiation factor 3g (eIF3g) is required for resumption of scanning of posttermination ribosomes for reinitiation on GCN4 and together with eIF3i stimulates linear scanning. Mol. Cell. Biol. 2010, 30, 4671–4686. [Google Scholar] [CrossRef]
- Black, J.J.; Musalgaonkar, S.; Johnson, A.W. Tsr4 Is a Cytoplasmic Chaperone for the Ribosomal Protein Rps2 in Saccharomyces cerevisiae. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef]
- Rössler, I.; Embacher, J.; Pillet, B.; Murat, G.; Liesinger, L.; Hafner, J.; Unterluggauer, J.J.; Birner-Gruenberger, R.; Kressler, D.; Pertschy, B. Tsr4 and Nap1, two novel members of the ribosomal protein chaperOME. Nucleic Acids Res. 2019, 47, 6984–7002. [Google Scholar] [CrossRef]
- Van Dyke, N.; Pickering, B.F.; Van Dyke, M.W. Stm1p alters the ribosome association of eukaryotic elongation factor 3 and affects translation elongation. Nucleic Acids Res. 2009, 37, 6116–6125. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, H.; Nagai, R.; Abe, T.; Wada, M.; Ito, K.; Takeuchi-Tomita, N. Tight interaction of eEF2 in the presence of Stm1 on ribosome. J. Biochem. 2018, 163, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Bolger, G.B. The RNA-binding protein SERBP1 interacts selectively with the signaling protein RACK1. Cell. Signal. 2017, 35, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Röther, S.; Burkert, C.; Brünger, K.M.; Mayer, A.; Kieser, A.; Strässer, K. Nucleocytoplasmic shuttling of the La motif-containing protein Sro9 might link its nuclear and cytoplasmic functions. RNA 2010, 16, 1393–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobel, S.G.; Wolin, S.L. Two yeast La motif-containing proteins are RNA-binding proteins that associate with polyribosomes. Mol. Biol. Cell 1999, 10, 3849–3862. [Google Scholar] [CrossRef]
- Kershaw, C.J.; Costello, J.L.; Castelli, L.M.; Talavera, D.; Rowe, W.; Sims, P.F.; Ashe, M.P.; Hubbard, S.J.; Pavitt, G.D.; Grant, C.M. The yeast La related protein Slf1p is a key activator of translation during the oxidative stress response. PLoS Genet. 2015, 11, e1004903. [Google Scholar] [CrossRef]
- Higashio, H.; Sato, K.; Nakano, A. Smy2p participates in COPII vesicle formation through the interaction with Sec23p/Sec24p subcomplex. Traffic 2008, 9, 79–93. [Google Scholar] [CrossRef]
- Bilsland, E.; Hult, M.; Bell, S.D.; Sunnerhagen, P.; Downs, J.A. The Bre5/Ubp3 ubiquitin protease complex from budding yeast contributes to the cellular response to DNA damage. DNA Repair 2007, 6, 1471–1484. [Google Scholar] [CrossRef]
- Baum, S.; Bittins, M.; Frey, S.; Seedorf, M. Asc1p, a WD40-domain containing adaptor protein, is required for the interaction of the RNA-binding protein Scp160p with polysomes. Biochem. J. 2004, 380, 823–830. [Google Scholar] [CrossRef] [Green Version]
- Coyle, S.M.; Gilbert, W.V.; Doudna, J.A. Direct link between RACK1 function and localization at the ribosome in vivo. Mol. Cell. Biol. 2009, 29, 1626–1634. [Google Scholar] [CrossRef]
- Lan, C.; Lee, H.C.; Tang, S.; Zhang, L. A novel mode of chaperone action: Heme activation of Hap1 by enhanced association of Hsp90 with the repressed Hsp70-Hap1 complex. J. Biol. Chem. 2004, 279, 27607–27612. [Google Scholar] [CrossRef]
- Chantrel, Y.; Gaisne, M.; Lions, C.; Verdière, J. The transcriptional regulator Hap1p (Cyp1p) is essential for anaerobic or heme-deficient growth of Saccharomyces cerevisiae: Genetic and molecular characterization of an extragenic suppressor that encodes a WD repeat protein. Genetics 1998, 148, 559–569. [Google Scholar] [PubMed]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O2-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef]
- Müller, M.; Kötter, P.; Behrendt, C.; Walter, E.; Scheckhuber, C.Q.; Entian, K.D.; Reichert, A.S. Synthetic quantitative array technology identifies the Ubp3-Bre5 deubiquitinase complex as a negative regulator of mitophagy. Cell Rep. 2015, 10, 1215–1225. [Google Scholar] [CrossRef]
- Ossareh-Nazari, B.; Bonizec, M.; Cohen, M.; Dokudovskaya, S.; Delalande, F.; Schaeffer, C.; Van Dorsselaer, A.; Dargemont, C. Cdc48 and Ufd3, new partners of the ubiquitin protease Ubp3, are required for ribophagy. EMBO Rep. 2010, 11, 548–554. [Google Scholar] [CrossRef] [Green Version]
- Nostramo, R.; Varia, S.N.; Zhang, B.; Emerson, M.M.; Herman, P.K. The Catalytic Activity of the Ubp3 Deubiquitinating Protease Is Required for Efficient Stress Granule Assembly in Saccharomyces cerevisiae. Mol. Cell. Biol. 2015, 36, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, P.; Wagner, S.A.; Weinert, B.T.; Sharma, S.; Bacinskaja, G.; Rehman, M.; Juffer, A.H.; Walther, T.C.; Lisby, M.; Choudhary, C. Proteome-wide analysis of lysine acetylation suggests its broad regulatory scope in Saccharomyces cerevisiae. Mol. Cell. Proteom. 2012, 11, 1510–1522. [Google Scholar] [CrossRef]
- Swaney, D.L.; Beltrao, P.; Starita, L.; Guo, A.; Rush, J.; Fields, S.; Krogan, N.J.; Villén, J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 2013, 10, 676–682. [Google Scholar] [CrossRef]
- Weinert, B.T.; Scholz, C.; Wagner, S.A.; Iesmantavicius, V.; Su, D.; Daniel, J.A.; Choudhary, C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep. 2013, 4, 842–851. [Google Scholar] [CrossRef]
- Dong, J.; Aitken, C.E.; Thakur, A.; Shin, B.S.; Lorsch, J.R.; Hinnebusch, A.G. Rps3/uS3 promotes mRNA binding at the 40S ribosome entry channel and stabilizes preinitiation complexes at start codons. Proc. Natl. Acad. Sci. USA 2017, 114, E2126–E2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juszkiewicz, S.; Chandrasekaran, V.; Lin, Z.; Kraatz, S.; Ramakrishnan, V.; Hegde, R.S. ZNF598 Is a Quality Control Sensor of Collided Ribosomes. Mol. Cell 2018, 72, 469–481.e467. [Google Scholar] [CrossRef] [PubMed]
- Winz, M.L.; Peil, L.; Turowski, T.W.; Rappsilber, J.; Tollervey, D. Molecular interactions between Hel2 and RNA supporting ribosome-associated quality control. Nat. Commun. 2019, 10, 563. [Google Scholar] [CrossRef] [PubMed]
- Limoncelli, K.A.; Merrikh, C.N.; Moore, M.J. ASC1 and RPS3: New actors in 18S nonfunctional rRNA decay. RNA 2017, 23, 1946–1960. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, K.; Tesina, P.; Matsuo, Y.; Sugiyama, T.; Cheng, J.; Saeki, Y.; Tanaka, K.; Becker, T.; Beckmann, R.; Inada, T. Collided ribosomes form a unique structural interface to induce Hel2-driven quality control pathways. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Yatime, L.; Hein, K.L.; Nilsson, J.; Nissen, P. Structure of the RACK1 dimer from Saccharomyces cerevisiae. J. Mol. Biol. 2011, 411, 486–498. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Q.; Qiu, G.; Zhou, S.; Jing, Z.; Wang, J.; Wang, W.; Cao, J.; Han, K.; Cheng, Q.; et al. RACK1 Promotes Autophagy by Enhancing the Atg14L-Beclin 1-Vps34-Vps15 Complex Formation upon Phosphorylation by AMPK. Cell Rep. 2015, 13, 1407–1417. [Google Scholar] [CrossRef] [Green Version]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid Name | Description | Reference |
|---|---|---|
| pME2783 | pRS416MET25 with MET25Prom, CYC1Term, URA3, CEN/ARS | [33] |
| pME2787 | pRS426MET25 with MET25Prom, CYC1Term, URA3, 2 μm | [33] |
| pME4364 | CYC1Term, URA3, CEN/ARS, ASC1 with its native promoter (+500 bp) | [10] |
| pME4480 | MET25Prom, CYC1Term, URA3, 2 μm, birA* | [18] |
| pME4478 | MET25Prom, CYC1Term, URA3, 2 μm, ASC1-birA* | [18] |
| pME4799 | MET25Prom, CYC1Term, URA3, 2 μm, RPS2-birA* | This study |
| pUG73 | LEU2 deletion cassette | [34] |
| Strain Name | Description | Background | Reference |
|---|---|---|---|
| RH2817 | MATα, ura3-52, trp1::hisG | Σ1278b | [4] |
| RH3510 | MATα, ura3-52, trp1::hisG, asc1-loxP SNR24 | Σ1278b | [3] |
| RH3493 | MATα, ura3-52, trp1::hisG, Δarg4::loxP, Δlys1::loxP | Σ1278b | [10] |
| RH3520 | MATα, ura3-52, trp1::hisG, asc1-loxP SNR24, ∆arg4::loxP, ∆lys1::loxP | Σ1278b | [10] |
| Y06078 | MATa; ura3Δ0; leu2Δ0; his3Δ1; met15Δ0; BRE5::kanMX4 | S288c | Euroscarf |
| Y06148 | MATa; ura3Δ0; leu2Δ0; his3Δ1; met15Δ0; UBP3::kanMX4 | S288c | Euroscarf |
| RH3789 | MATα, ura3-52, trp1::hisG, ∆bre5::kanMX4 | Σ1278b | This study |
| RH3790 | MATα, ura3-52, trp1::hisG, asc1-loxP SNR24, ∆bre5::kanMX4 | Σ1278b | This study |
| RH3791 | MATα, ura3-52, trp1::hisG, ∆ubp3::kanMX4 | Σ1278b | This study |
| RH3792 | MATα, ura3-52, trp1::hisG, asc1-loxP SNR24, ∆ubp3::kanMX4 | Σ1278b | This study |
| BRE5-GFP | MATa, ura3Δ0, leu2Δ0, his3Δ1, met15Δ0, BRE5-GFP_HIS3MX6 | S288c | Invitrogen, [35] |
| RH3793 | MATa, ura3Δ0, leu2Δ0, his3Δ1, MET15, BRE5-GFP_HIS3MX6 | S288c | This study |
| RH3794 | MATa, ura3Δ0, leu2Δ0, his3Δ1, MET15, BRE5-GFP_HIS3MX6, ∆asc1::LEU2 | S288c | This study |
| No. | Command | Description |
|---|---|---|
| 1 | Generic matrix upload | proteinGroups.txt normalized ratios, etc. |
| 2.1 | Filter rows based on categorical columns | Remove rows with “+” in reverse column |
| 2.2 | Remove rows with “+” in potential contaminant column | |
| 2.3 | Remove rows with “+” in only identified by site column | |
| 3 | Transform | Inverse ratios (1/x), ratios are reported as follows: ASC1 RPS2-birA*/birA* asc1− RPS2-birA*/birA* asc1− RPS2-birA*/ASC1 RPS2-birA* |
| 4 | Transform | log2(x) |
| 5 | Filter rows based on valid values | 9 valid values in total, Reduce matrix (431 proteins remained) |
| 6.1 | Categorical annotation rows | Group biological replicates 1: ASC1 RPS2-birA*/birA* |
| 6.2 | 2: asc1− RPS2-birA*/birA* | |
| 6.3 | 3: asc1− RPS2-birA*/ASC1 RPS2-birA* | |
| 6.4 | 4: ASC1 RPS2-birA*/birA* and asc1− RPS2-birA*/birA* (for two sample t-test) | |
| 7.1 7.2 | Two-sample tests | Select the two groups in 4 (see 6.4), p-value threshold: 0.05 p-value threshold: 0.01 |
| Filter for proteins enriched from ASC1 RPS2-birA* | ||
| 8 | Filter rows based on valid values | 3 valid values in group 1 greater than or equal to 0.485 (approximately 40% enrichment), reduce matrix |
| 9 | Average groups | Calculate mean and standard deviation |
| Filter for proteins enriched from asc1− RPS2-birA* but not from ASC1 RPS2-birA* | ||
| 10 | Filter rows based on valid values | 3 valid values in group 3 outside -0.485 and 0.485 Reduce matrix |
| 11 | Filter rows based on valid values | 3 valid values in group 2 greater than or equal to 0.485, Reduce matrix |
| 12 | Average groups | Calculate mean and standard deviation |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmitt, K.; Valerius, O. yRACK1/Asc1 proxiOMICs—Towards Illuminating Ships Passing in the Night. Cells 2019, 8, 1384. https://doi.org/10.3390/cells8111384
Schmitt K, Valerius O. yRACK1/Asc1 proxiOMICs—Towards Illuminating Ships Passing in the Night. Cells. 2019; 8(11):1384. https://doi.org/10.3390/cells8111384
Chicago/Turabian StyleSchmitt, Kerstin, and Oliver Valerius. 2019. "yRACK1/Asc1 proxiOMICs—Towards Illuminating Ships Passing in the Night" Cells 8, no. 11: 1384. https://doi.org/10.3390/cells8111384