Adenosine Depletion as A New Strategy to Decrease Glioblastoma Stem-Like Cells Aggressiveness

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Adenosine Quantification
2.3. Western Blot
2.4. RT-qPCR
2.5. Protein Stability
2.6. Cell Adhesion
2.7. Cell Migration and Invasion
2.8. Zymography Assay
2.9. MRPs Activity
2.10. Cell Viability Assay
2.11. Soft Agar Colony Formation Assay
2.12. Statistical Analysis
3. Results
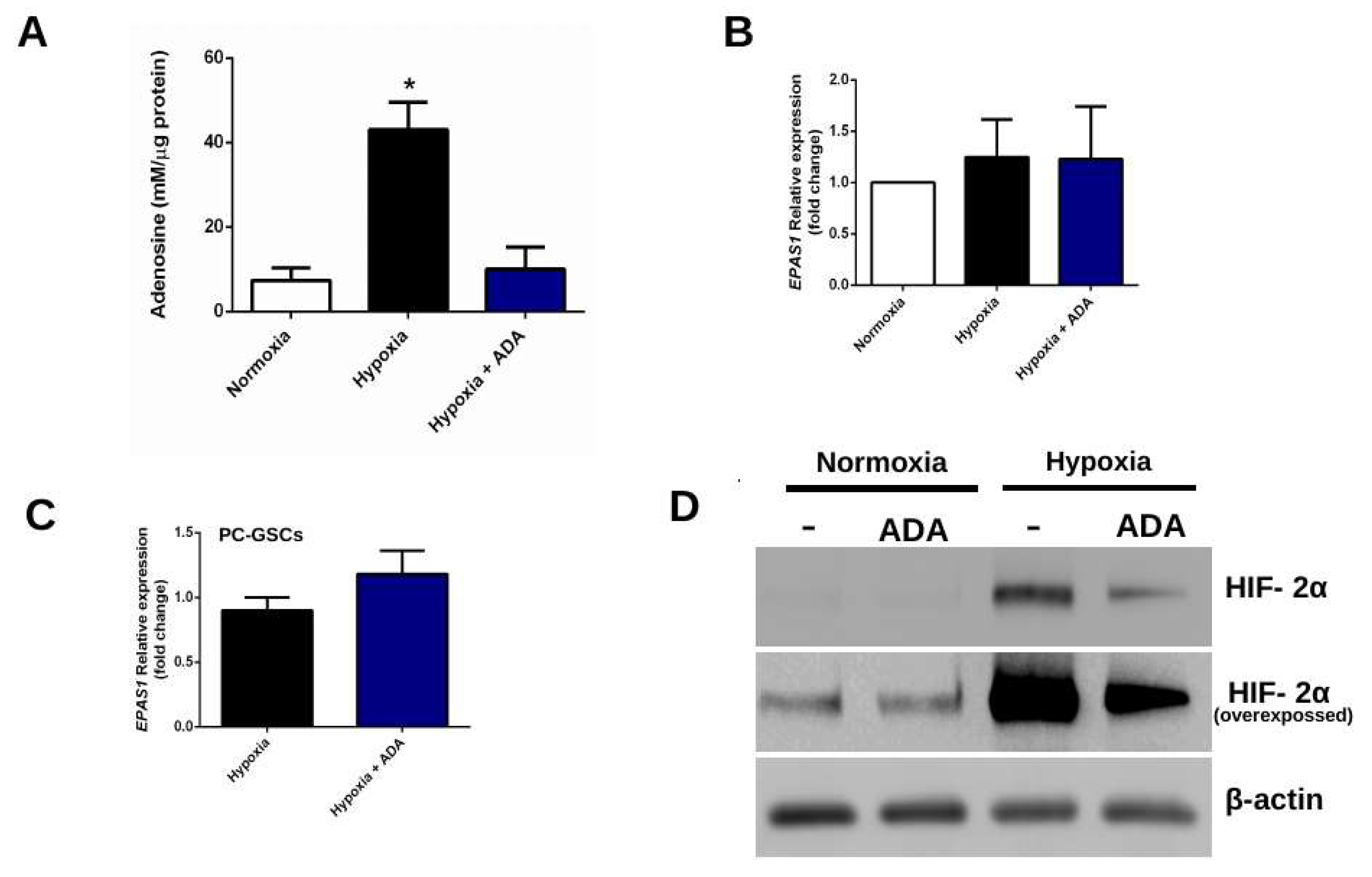
3.1. Adenosine Depletion Decreases HIF-2α Levels Under Hypoxia
3.1.1. Adenosine Deaminase Decrease Extracellular Adenosine Levels
3.1.2. Adenosine Deaminase Decreases HIF-2α Protein But Not mRNA Levels
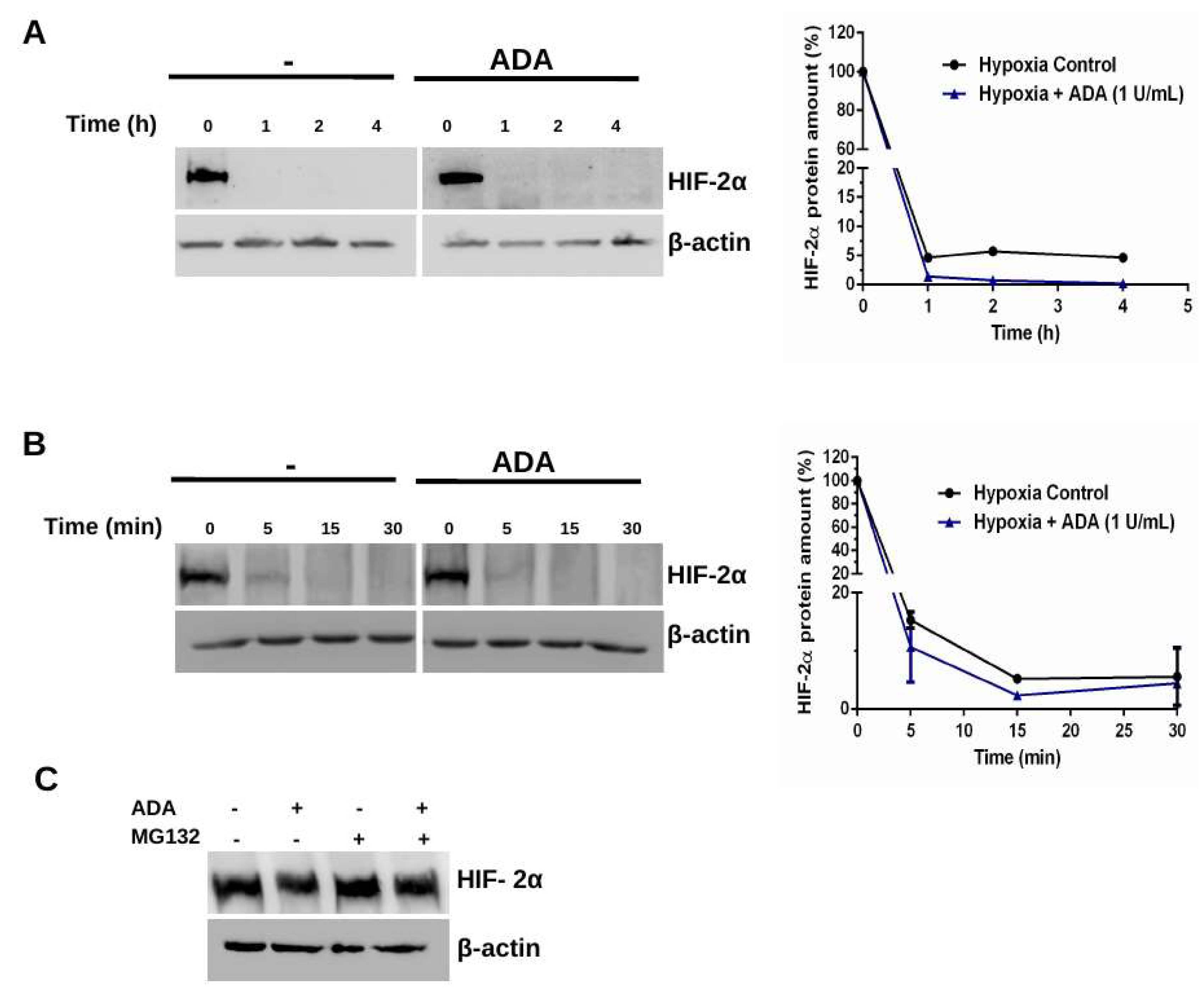
3.2. Adenosine Depletion Inhibits HIF-2α Stability Under Hypoxia
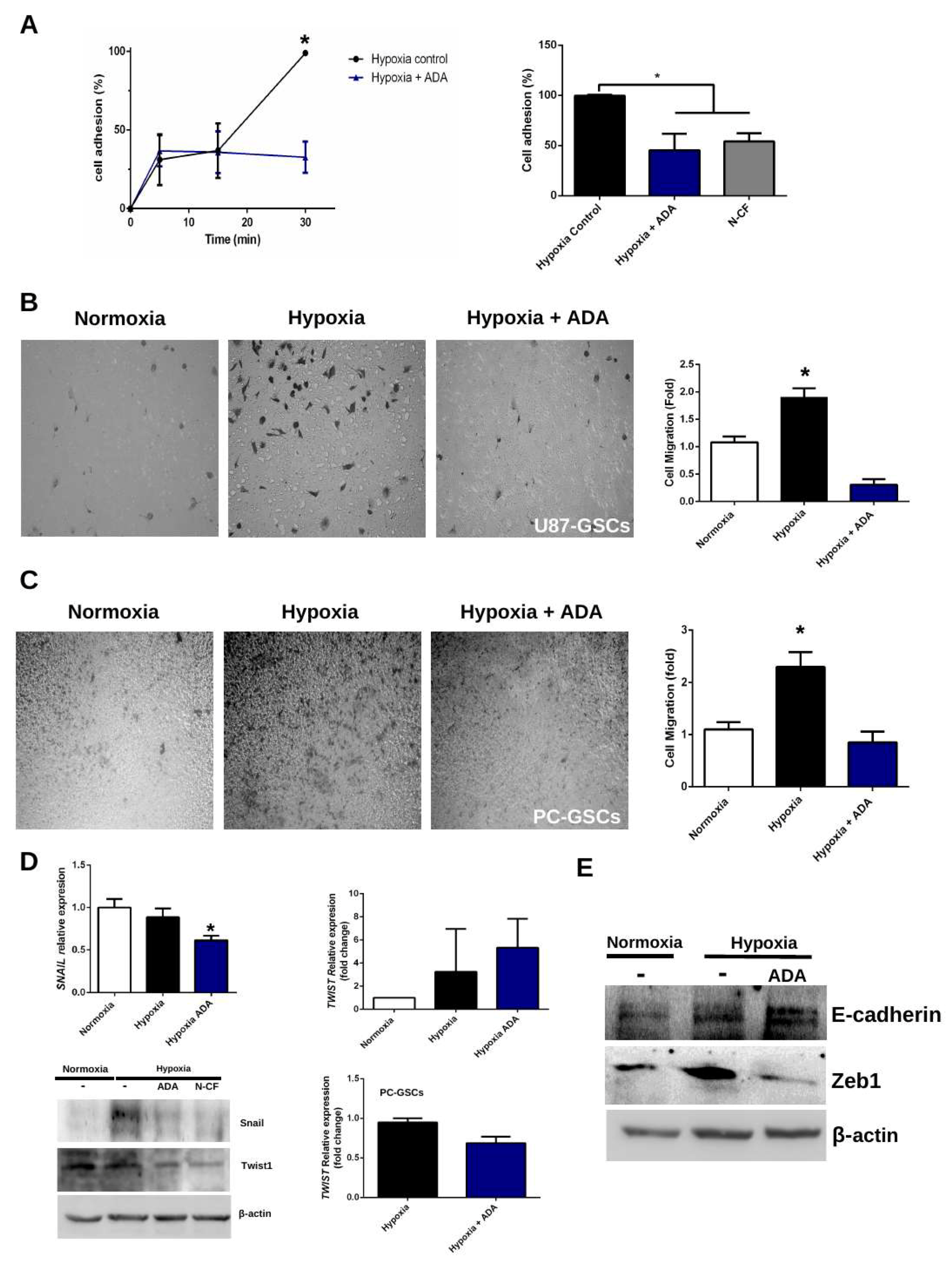
3.3. Adenosine Depletion Decreases GSCs Hypoxia-Dependent Cell Adhesion and Migration
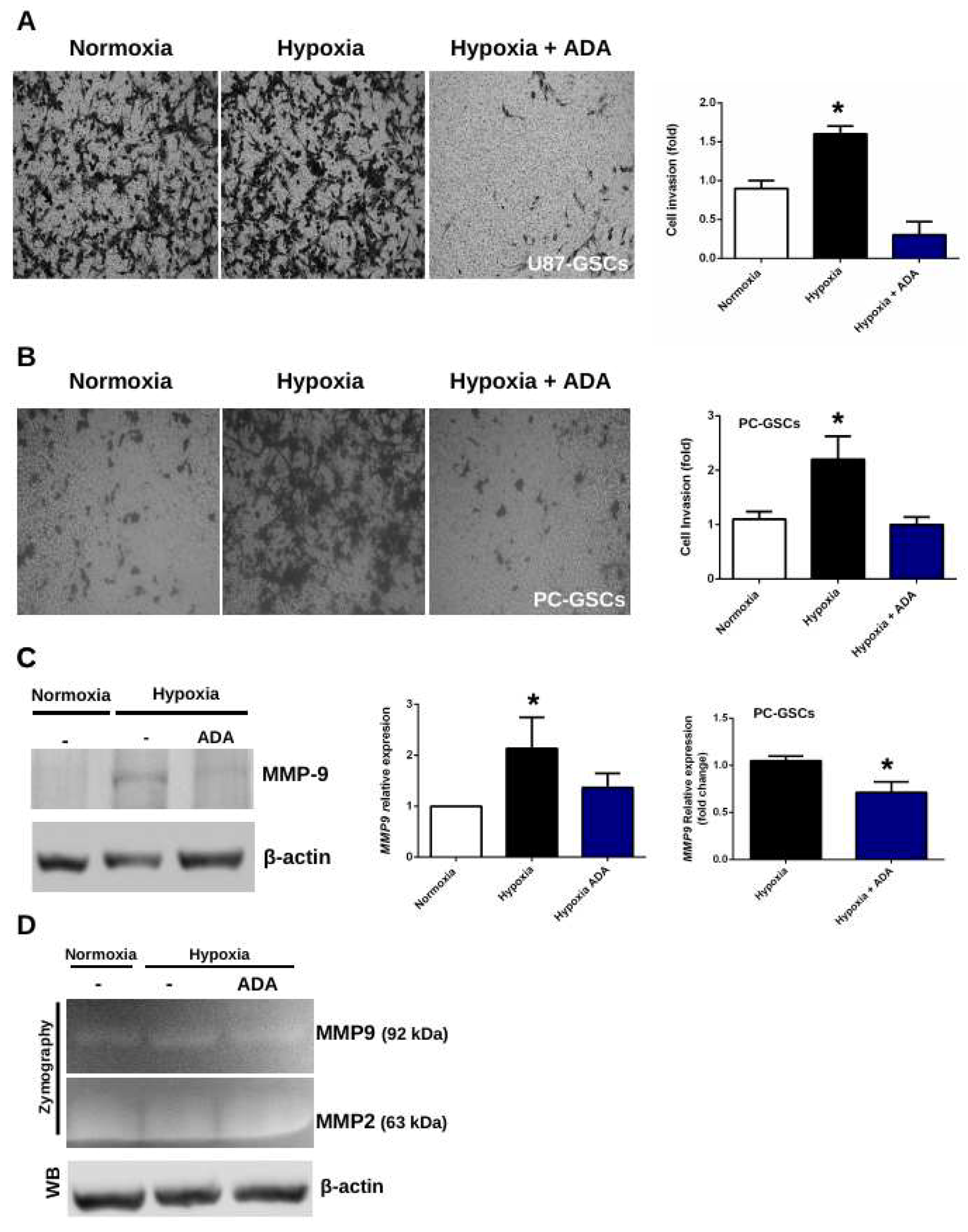
3.4. Adenosine Depletion Decreases GSCs Hypoxia-Dependent Cell Invasion
3.5. Adenosine Depletion Decreases GSCs In Vitro Tumorigenicity and Vincristine Chemoresistance
4. Discussion
4.1. Adenosine Deaminase and Hypoxia-Dependent HIF-2α Stability
4.2. Adenosine Deaminase in GSCs Invasive Phenotype Under Hypoxia
4.3. Adenosine Deaminase in GSCs Chemoresistance and Clonogenicity
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Jiang, T. Understanding high grade glioma: Molecular mechanism, therapy and comprehensive management. Cancer Lett. 2013, 331, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A. Glioblastoma and Other Malignant Gliomas. JAMA 2013, 310, 1842. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.; Kesari, S. Malignant Gliomas in Adults—NEJM. Malig. Gliomas Adults 2008, 492–507. [Google Scholar]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Vtorushin, S.V.; Khristenko, K.Y.; Zavyalova, M.V.; Perelmuter, V.M.; Litviakov, N.V.; Denisov, E.V.; Dulesova, A.Y.; Cherdyntseva, N.V. The Phenomenon of Multi-Drug Resistance in the Treatment of Malignant Tumors. Exp. Oncol. 2014, 36, 144–156. [Google Scholar]
- Bambury, R.M.; Morris, P.G. The search for novel therapeutic strategies in the treatment of recurrent glioblastoma multiforme. Expert Rev. Anticancer Ther. 2014, 14, 955–964. [Google Scholar] [CrossRef]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar] [CrossRef]
- Campos, B.; Olsen, L.R.; Urup, T.; Poulsen, H.S. A comprehensive profile of recurrent glioblastoma. Oncogene 2016, 35, 5819–5825. [Google Scholar] [CrossRef]
- Torres, A.; Vargas, Y.; Uribe, D.; Jaramillo, C.; Gleisner, A.; Salazar-Onfray, F.; López, M.N.; Melo, R.; Oyarzún, C.; San Martín, R.; et al. Adenosine A3 receptor elicits chemoresistance mediated by multiple resistance-associated protein-1 in human glioblastoma stem-like cells. Oncotarget 2016, 7, 67373–67386. [Google Scholar] [CrossRef]
- Uribe, D.; Torres, Á.; Rocha, J.D.; Niechi, I.; Oyarzún, C.; Sobrevia, L.; San Martín, R.; Quezada, C. Multidrug resistance in glioblastoma stem-like cells: Role of the hypoxic microenvironment and adenosine signaling. Mol. Aspects Med. 2017, 55, 140–151. [Google Scholar] [CrossRef]
- Torres, Á.; Erices, J.I.; Sanchez, F.; Ehrenfeld, P.; Turchi, L.; Virolle, T.; Uribe, D.; Niechi, I.; Spichiger, C.; Rocha, J.D.; et al. Extracellular adenosine promotes cell migration/invasion of Glioblastoma Stem-like Cells through A3 Adenosine Receptor activation under hypoxia. Cancer Lett. 2019, 446, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Maugeri-Saccà, M.; Di Martino, S.; De Maria, R. Biological and clinical implications of cancer stem cells in primary brain tumors. Front. Oncol. 2013, 3, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappellari, A.R.; Rockenbach, L.; Dietrich, F.; Clarimundo, V.; Glaser, T.; Braganhol, E.; Abujamra, A.L.; Roesler, R.; Ulrich, H.; Liveira Battastini, A.M. Characterization of Ectonucleotidases in Human Medulloblastoma Cell Lines: Ecto-5′NT/CD73 in Metastasis as Potential Prognostic Factor. PLoS ONE 2012, 7, 1–11. [Google Scholar] [CrossRef]
- Stagg, J.; Divisekera, U.; McLaughlin, N.; Sharkey, J.; Pommey, S.; Denoyer, D.; Dwyer, K.M.; Smyth, M.J. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1547–1552. [Google Scholar] [CrossRef] [Green Version]
- Booth, C.; Gaspar, H.B. Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID). Biologics 2009, 3, 349–358. [Google Scholar]
- Flinn, A.M.; Gennery, A.R. Adenosine deaminase deficiency: A review. Orphanet J. Rare Dis. 2018, 13, 65. [Google Scholar] [CrossRef]
- Pérez-Aguilar, M.C.; Goncalves, L.; Bonfante-Cabarcas, R. Adenosine deaminase in severe combined immunodeficiency syndrome. Investig. Clínica 2012, 53, 315–324. [Google Scholar]
- Gessi, S.; Merighi, S.; Varani, K.; Leung, E.; Mac Lennan, S.; Borea, P.A. The A3 adenosine receptor: An enigmatic player in cell biology. Pharmacol. Ther. 2008, 117, 123–140. [Google Scholar] [CrossRef]
- Quezada, C.; Garrido, W.; Oyarzún, C.; Fernández, K.; Segura, R.; Melo, R.; Casanello, P.; Sobrevia, L.; San Martín, R. 5′-Ectonucleotidase Mediates Multiple-Drug Resistance in Glioblastoma Multiforme Cells. J. Cell. Physiol. 2013, 228, 602–608. [Google Scholar] [CrossRef]
- Stagg, J.; Divisekera, U.; Duret, H.; Sparwasser, T.; Teng, M.W.L.; Darcy, P.K.; Smyth, M.J. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. 2011, 71, 2892–2900. [Google Scholar] [CrossRef] [PubMed]
- Zhi, X.; Chen, S.; Zhou, P.; Shao, Z.; Wang, L.; Ou, Z.; Yin, L. RNA interference of ecto-5′-nucleotidase (CD73) inhibits human breast cancer cell growth and invasion. Clin. Exp. Metastasis 2007, 24, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Huang, P. Adenosine A2B Receptor: From Cell Biology to Human Diseases. Front. Chem. 2016, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B. Adenosine - A physiological or pathophysiological agent? J. Mol. Med. 2014, 92, 201–206. [Google Scholar] [CrossRef]
- Stagg, J.; Smyth, M.J. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 2010, 29, 5346–5358. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Wang, X.; Bai, Y.; Liao, H.; Qiu, S.; Yang, Y.; Yan, X.-H.; Chen, J.; Guo, H.; Zhang, S. The HIF-2alpha dependent induction of PAP and adenosine synthesis regulates glioblastoma stem cell function through the A2B adenosine receptor. Int. J. Biochem. Cell Biol. 2014, 49, 8–16. [Google Scholar] [CrossRef]
- Gessi, S.; Merighi, S.; Sacchetto, V.; Simioni, C.; Borea, P.A. Adenosine receptors and cancer. Biochim. Biophys. Acta 2011, 1808, 1400–1412. [Google Scholar] [CrossRef] [Green Version]
- Bar, E.E.; Lin, A.; Mahairaki, V.; Matsui, W.; Eberhart, C.G. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am. J. Pathol. 2010, 177, 1491–1502. [Google Scholar] [CrossRef]
- Chouaib, S.; Messai, Y.; Couve, S.; Escudier, B.; Hasmim, M.; Noman, M.Z. Hypoxia promotes tumor growth in linking angiogenesis to immune escape. Front. Immunol. 2012, 3, 1–10. [Google Scholar] [CrossRef]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef]
- McCord, A.M.; Jamal, M.; Shankavarum, U.T.; Lang, F.F.; Camphausen, K.; Tofilon, P.J. Physiologic Oxygen Concentration Enhances the Stem-Like Properties of CD133+ Human Glioblastoma Cells In vitro. Mol. Cancer Res. 2009, 7, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2010, 29, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; von Stechow, L.; Schänzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.-T.; Nistér, M.; et al. A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 2010, 133, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Lemos, R.; Liu, X.; Powis, G. The hypoxia-associated factor switches cells from HIF-1α- to HIF-2α-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef] [PubMed]
- Marín-Hernández, A.; Gallardo-Pérez, J.C.; Ralph, S.J.; Rodríguez-Enríquez, S.; Moreno-Sánchez, R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev. Med. Chem. 2009, 9, 1084–1101. [Google Scholar] [CrossRef] [PubMed]
- Petrella, B.L.; Lohi, J.; Brinckerhoff, C.E. Identification of membrane type-1 matrix metalloproteinase as a target of hypoxia-inducible factor-2 alpha in von Hippel-Lindau renal cell carcinoma. Oncogene 2005, 24, 1043–1052. [Google Scholar] [CrossRef]
- Shin, B.; Burkhardt, J.-K.; Boockvar, J. Glioblastoma multiforme stem-like cells and hypoxia: The novel role of HAF. Neurosurgery 2011, 69, N21. [Google Scholar] [CrossRef]
- Kolenda, J.; Jensen, S.S.; Aaberg-Jessen, C.; Christensen, K.; Andersen, C.; Brünner, N.; Kristensen, B.W. Effects of hypoxia on expression of a panel of stem cell and chemoresistance markers in glioblastoma-derived spheroids. J. Neurooncol. 2011, 103, 43–58. [Google Scholar] [CrossRef]
- Jin, F.; Zhao, L.; Guo, Y.-J.; Zhao, W.-J.; Zhang, H.; Wang, H.-T.; Shao, T.; Zhang, S.-L.; Wei, Y.-J.; Feng, J.; et al. Influence of Etoposide on anti-apoptotic and multidrug resistance-associated protein genes in CD133 positive U251 glioblastoma stem-like cells. Brain Res. 2010, 1336, 103–111. [Google Scholar] [CrossRef]
- Peigñan, L.; Garrido, W.; Segura, R.; Melo, R.; Rojas, D.; Cárcamo, J.G.; San Martín, R.; Quezada, C. Combined use of anticancer drugs and an inhibitor of multiple drug resistance-associated protein-1 increases sensitivity and decreases survival of glioblastoma multiforme cells in vitro. Neurochem. Res. 2011, 36, 1397–1406. [Google Scholar] [CrossRef]
- Rocha, R.; Torres, Á.; Ojeda, K.; Uribe, D.; Rocha, D.; Erices, J.; Niechi, I.; Ehrenfeld, P.; San Martín, R.; Quezada, C. The Adenosine A3 Receptor Regulates Differentiation of Glioblastoma Stem-Like Cells to Endothelial Cells under Hypoxia. Int. J. Mol. Sci. 2018, 19, 1228. [Google Scholar] [CrossRef] [PubMed]
- Vieira, C.; Magalhães-Cardoso, M.T.; Ferreirinha, F.; Silva, I.; Dias, A.S.; Pelletier, J.; Sévigny, J.; Correia-De-Sá, P. Feed-forward inhibition of cd73 and upregulation of adenosine deaminase contribute to the loss of adenosine neuromodulation in postinflammatory ileitis. Mediat. Inflamm. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Chamboredon, S.; Ciais, D.; Desroches-Castan, A.; Savi, P.; Bono, F.; Feige, J.-J.; Cherradi, N. Hypoxia-inducible factor-1α mRNA: A new target for destabilization by tristetraprolin in endothelial cells. Mol. Biol. Cell 2011, 22, 3366–3378. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Bar-Yehuda, S.; Synowitz, M.; Powell, J.D.; Klotz, K.N.; Gessi, S.; Borea, P.A. Adenosine receptors and cancer. Handb. Exp. Pharmacol. 2009, 399–441. [Google Scholar] [CrossRef]
- Koeppen, M.; Eckle, T.; Eltzschig, H.K. Interplay of hypoxia and A2B adenosine receptors in tissue protection. Adv. Pharmacol. 2011, 61, 145–186. [Google Scholar]
- Yan, A.; Joachims, M.L.; Thompson, L.F.; Miller, A.D.; Canoll, P.D.; Bynoe, M.S. CD73 Promotes Glioblastoma Pathogenesis and Enhances Its Chemoresistance via A2B Adenosine Receptor Signaling. J. Neurosci. 2019, 39, 4387–4402. [Google Scholar] [CrossRef]
- Torres, Á.; Arriagada, V.; Erices, J.I.; Toro, M.d.L.Á.; Rocha, J.D.; Niechi, I.; Carrasco, C.; Oyarzún, C.; Quezada, C. FK506 Attenuates the MRP1-Mediated Chemoresistant Phenotype in Glioblastoma Stem-Like Cells. Int. J. Mol. Sci. 2018, 19, 2697. [Google Scholar] [CrossRef]
- Benyahia, B.; Huguet, S.; Declèves, X.; Mokhtari, K.; Crinière, E.; Bernaudin, J.F.; Scherrmann, J.M.; Delattre, J.Y. Multidrug resistance-associated protein MRP1 expression in human gliomas: Chemosensitization to vincristine and etoposide by indomethacin in human glioma cell lines overexpressing MRP1. J. Neurooncol. 2004, 66, 65–70. [Google Scholar] [CrossRef]
- Tivnan, A.; Zakaria, Z.; O’Leary, C.; Kögel, D.; Pokorny, J.L.; Sarkaria, J.N.; Prehn, J.H.M. Inhibition of multidrug resistance protein 1 (MRP1) improves chemotherapy drug response in primary and recurrent glioblastoma multiforme. Front. Neurosci. 2015, 9, 1–10. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niechi, I.; Uribe-Ojeda, A.; Erices, J.I.; Torres, Á.; Uribe, D.; Rocha, J.D.; Silva, P.; Richter, H.G.; San Martín, R.; Quezada, C. Adenosine Depletion as A New Strategy to Decrease Glioblastoma Stem-Like Cells Aggressiveness. Cells 2019, 8, 1353. https://doi.org/10.3390/cells8111353
Niechi I, Uribe-Ojeda A, Erices JI, Torres Á, Uribe D, Rocha JD, Silva P, Richter HG, San Martín R, Quezada C. Adenosine Depletion as A New Strategy to Decrease Glioblastoma Stem-Like Cells Aggressiveness. Cells. 2019; 8(11):1353. https://doi.org/10.3390/cells8111353
Chicago/Turabian StyleNiechi, Ignacio, Atenea Uribe-Ojeda, José Ignacio Erices, Ángelo Torres, Daniel Uribe, José Dellis Rocha, Pamela Silva, Hans G. Richter, Rody San Martín, and Claudia Quezada. 2019. "Adenosine Depletion as A New Strategy to Decrease Glioblastoma Stem-Like Cells Aggressiveness" Cells 8, no. 11: 1353. https://doi.org/10.3390/cells8111353
APA StyleNiechi, I., Uribe-Ojeda, A., Erices, J. I., Torres, Á., Uribe, D., Rocha, J. D., Silva, P., Richter, H. G., San Martín, R., & Quezada, C. (2019). Adenosine Depletion as A New Strategy to Decrease Glioblastoma Stem-Like Cells Aggressiveness. Cells, 8(11), 1353. https://doi.org/10.3390/cells8111353