Patient-Derived Glioma Models: From Patients to Dish to Animals

 ,
,
Abstract
1. Introduction
2. In Vitro GBM Model
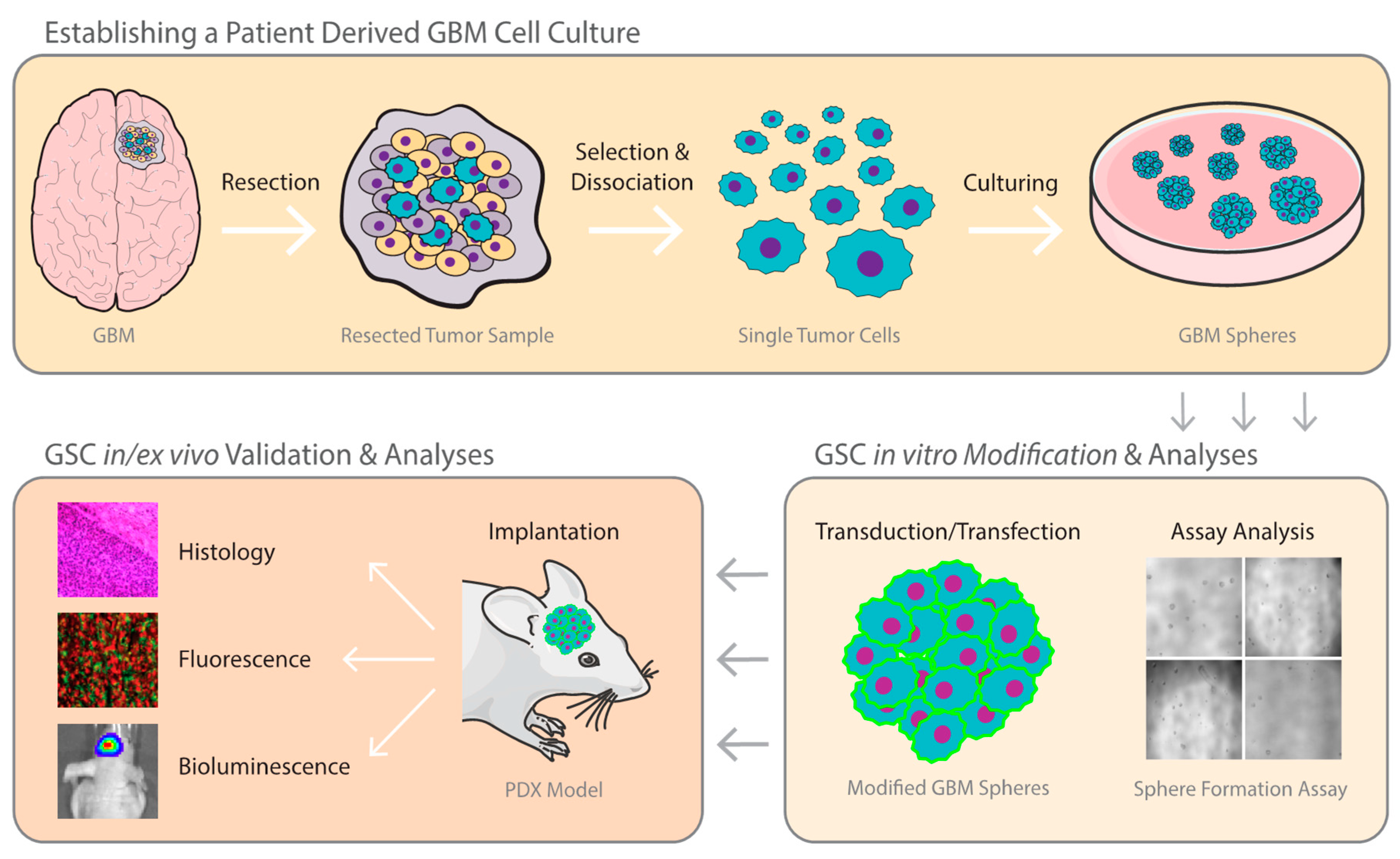
2.1. Establishing A GBM Stem Cell Line from Patient Tumor Tissue
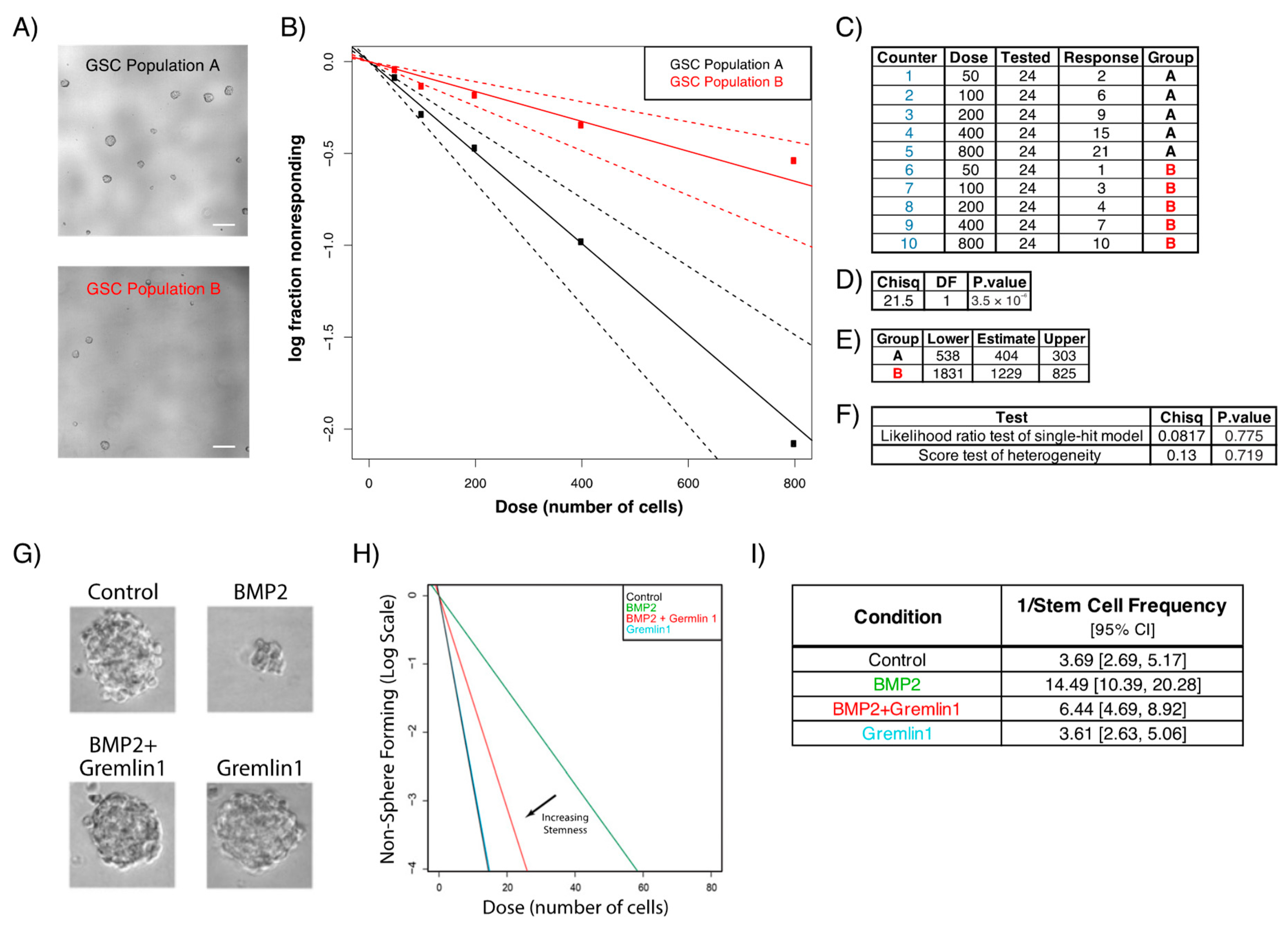
2.2. Sphere Formation Assays
3. In Vivo GBM Model
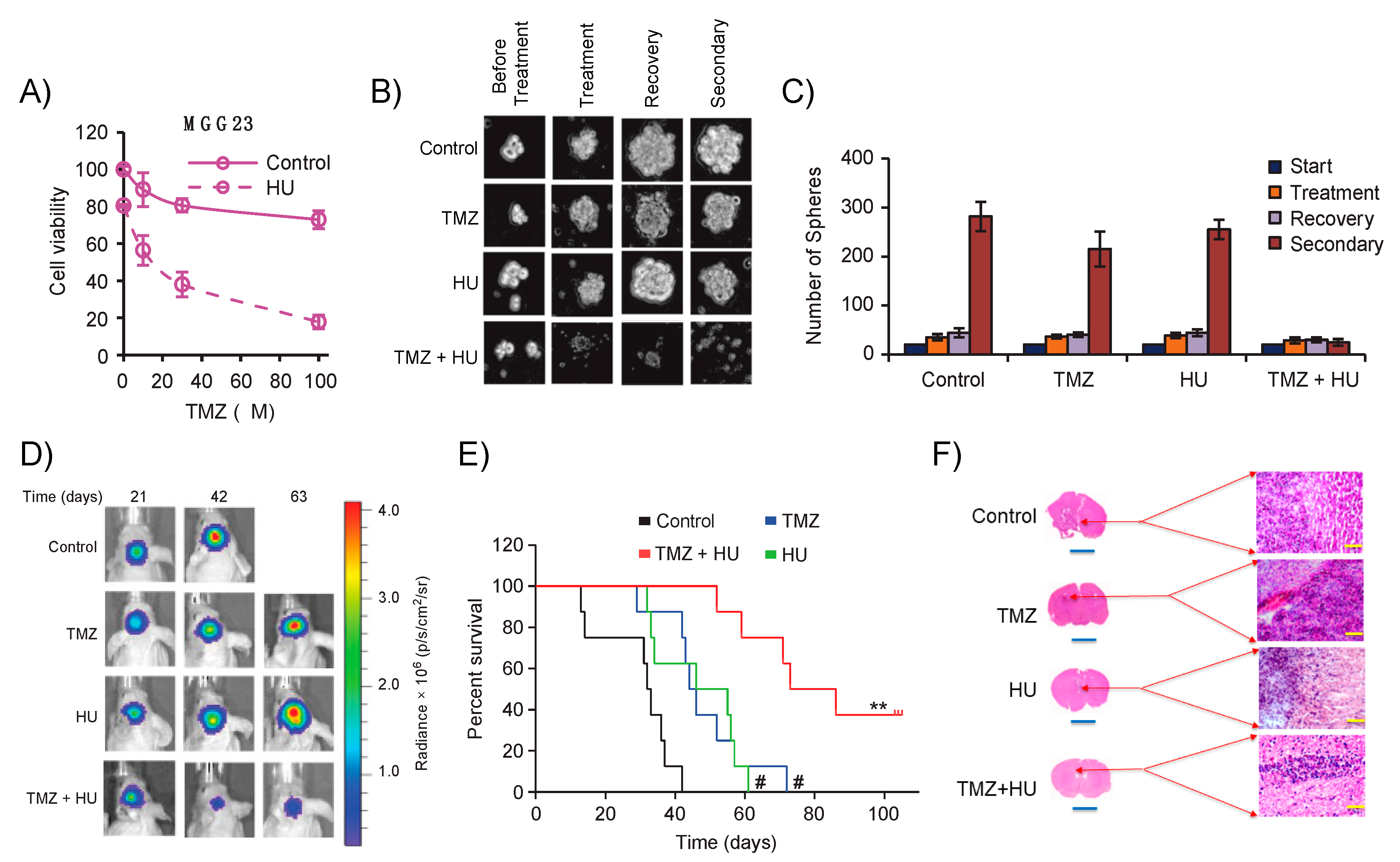
3.1. Establishing Patient-Derived Xenograft GBM Model
3.2. PDX Mirrors Hallmarks of Parental Tumor
3.3. Bioluminescence Imaging from In Vitro to In Vivo
Evaluating Novel Therapeutics against GSCs from Dish to Animals
3.4. Murine Cancer Models as Pre-Clinical Platforms for Cancer Studies and Drug Therapy
3.5. Phenocopying GBM Intratumoral Heterogeneity from Patients to Xenografts
3.6. A New Era for GBM Models?
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2015, 17, iv1–iv62. [Google Scholar] [PubMed]
- Ramirez, Y.P.; Weatherbee, J.L.; Wheelhouse, R.T.; Ross, A.H. Glioblastoma multiforme therapy and mechanisms of resistance. Pharmaceuticals 2013, 6, 1475–1506. [Google Scholar] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [PubMed]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Paulo, L.; Bruno, C.; Cláudia, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; Codon Publications: Brisbane, Australia, 2017; pp. 197–241. ISBN 9780323479745. [Google Scholar]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [PubMed]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Patrizii, M.; Bartucci, M.; Pine, S.R.; Sabaawy, H.E. Utility of Glioblastoma Patient-Derived Orthotopic Xenografts in Drug Discovery and Personalized Therapy. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Chalmers, A.J. Radioresistant glioma stem cells--therapeutic obstacle or promising target? DNA Repair (Amst) 2007, 6, 1391–1394. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.K.; Brothers, S.P.; Wahlestedt, C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol. Med. 2014, 6, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, R.; Inda, M.D.M.; Cavenee, W.K.; Furnari, F.B. Heterogeneity maintenance in glioblastoma: A social network. Cancer Res. 2011, 71, 4055–4060. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, U.; Geber, A.; Fligelman, B.; Leversha, M.; Wang, R.; Tabar, V.; Wilshire, J.; Hovinga, K.E.; Brennan, C.; Chadalavada, K. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; DeCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef]
- Bhat, K.P.L.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal Differentiation Mediated by NF-κB Promotes Radiation Resistance in Glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef]
- Wang, M.; Li, P.; Benos, P.V.; Santana-Santos, L.; Luthra, S.; Li, J.; Hu, B.B.; Cheng, S.-Y.; Smith, L.; Nakano, I.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Mahabir, R.; Tanino, M.; Elmansuri, A.; Wang, L.; Kimura, T.; Itoh, T.; Ohba, Y.; Nishihara, H.; Shirato, H.; Tsuda, M.; et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol. 2014, 16, 671–685. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef]
- Wislet-Gendebien, S. Regulation of neural markers nestin and GFAP expression by cultivated bone marrow stromal cells. J. Cell Sci. 2003, 116, 3295–3302. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.V.; Van Roosmalen, I.A.M.; Busschers, E.; Tomar, T.; Conroy, S.; Eggens-Meijer, E.; Fajardo, N.P.; Pore, M.M.; Balasubramanyian, V.; Wagemakers, M.; et al. Serum-induced differentiation of glioblastoma neurospheres leads to enhanced migration/invasion capacity that is associated with increased MMP9. PLoS ONE 2015, 10, e0145393. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniyan, V.; Vaillant, B.; Wang, S.; Gumin, J.; Elena Butalid, M.; Sai, K.; Mukheef, F.; Kim, S.H.; Boddeke, H.W.G.M.; Lang, F.; et al. Aberrant mesenchymal differentiation of glioma stem-like cells: Implications for therapeutic targeting. Oncotarget 2015, 6, 31007–31017. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Teng, J.; Da Hora, C.C.; Kantar, R.S.; Nakano, I.; Wakimoto, H.; Batchelor, T.T.; Antonio Chiocca, E.; Badr, C.E.; Tannous, B.A. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro Oncol. 2017, 19, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jeon, H.; Othmer, H. The Role of the Tumor Microenvironment in Glioblastoma: A Mathematical Model. IEEE Trans. Biomed. Eng. 2017, 64, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Manini, I.; Caponnetto, F.; Bartolini, A.; Ius, T.; Mariuzzi, L.; Di Loreto, C.; Beltrami, A.P.; Cesselli, D. Role of Microenvironment in Glioma Invasion: What We Learned from In Vitro Models. Int. J. Mol. Sci. 2018, 19, 147. [Google Scholar] [CrossRef] [PubMed]
- Colwell, N.; Larion, M.; Giles, A.J.; Seldomridge, A.N.; Sizdahkhani, S.; Gilbert, M.R.; Park, D.M. Hypoxia in the glioblastoma microenvironment: shaping the phenotype of cancer stem-like cells. Neuro Oncol. 2017, 19, 887–896. [Google Scholar] [CrossRef]
- Lv, D.; Hu, Z.; Lu, L.; Lu, H.; Xu, X. Three-dimensional cell culture: A powerful tool in tumor research and drug discovery. Oncol. Lett. 2017, 14, 6999–7010. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Azari, H.; Millette, S.; Ansari, S.; Rahman, M.; Deleyrolle, L.P.; Reynolds, B.A. Isolation and Expansion of Human Glioblastoma Multiforme Tumor Cells Using the Neurosphere Assay. JoVE 2011, 56, e3633. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Kesari, S.; Farrell, C.J.; Curry, W.T.; Zaupa, C.; Aghi, M.; Kuroda, T.; Stemmer-Rachamimov, A.; Shah, K.; Liu, T.C.; et al. Human glioblastoma-derived cancer stem cells: Establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009, 69, 3472–3481. [Google Scholar] [CrossRef] [PubMed]
- Uchida, N.; Buck, D.W.; He, D.; Reitsma, M.J.; Masek, M.; Phan, T.V.; Tsukamoto, A.S.; Gage, F.H.; Weissman, I.L. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14720–14725. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Kessler, J.D.; Read, T.A.; Kaiser, C.; Corbeil, D.; Huttner, W.B.; Johnson, J.E.; Wechsler-Reya, R.J. Isolation of neural stem cells from the postnatal cerebellum. Nat. Neurosci. 2005, 8, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sakariassen, P.; Tsinkalovsky, O.; Immervoll, H.; Bøe, S.O.; Svendsen, A.; Prestegarden, L.; Røsland, G.; Thorsen, F.; Stuhr, L.; et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int. J. Cancer 2008, 122, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Shmelkov, S.V.; Butler, J.M.; Hooper, A.T.; Hormigo, A.; Kushner, J.; Milde, T.; Clair, R.S.; Baljevic, M.; White, I.; Jin, D.K.; et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133– metastatic colon cancer cells initiate tumors. J. Clin. Investig. 2008. [Google Scholar] [CrossRef]
- Joo, K.M.; Kim, S.Y.; Jin, X.; Song, S.Y.; Kong, D.S.; Lee, J.I.; Jeon, J.W.; Kim, M.H.; Kang, B.G.; Jung, Y.; et al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Lab. Investig. 2008, 88, 808–815. [Google Scholar] [CrossRef]
- Lottaz, C.; Beier, D.; Meyer, K.; Kumar, P.; Hermann, A.; Schwarz, J.; Junker, M.; Oefner, P.J.; Bogdahn, U.; Wischhusen, J.; et al. Transcriptional profiles of CD133+ and CD133- glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Res. 2010, 70, 2030–2040. [Google Scholar] [CrossRef]
- Son, M.J.; Woolard, K.; Nam, D.H.; Lee, J.; Fine, H.A. SSEA-1 Is an Enrichment Marker for Tumor-Initiating Cells in Human Glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef]
- Senner, V.; Sturm, A.; Baur, I.; Schrell, U.H.M.; Distel, L.; Paulus, W. CD24 promotes invasion of glioma cells in vivo. J. Neuropathol. Exp. Neurol. 1999, 58, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Nishimura, M.C.; Bumbaca, S.M.; Kharbanda, S.; Forrest, W.F.; Kasman, I.M.; Greve, J.M.; Soriano, R.H.; Gilmour, L.L.; Rivers, C.S.; et al. A Hierarchy of Self-Renewing Tumor-Initiating Cell Types in Glioblastoma. Cancer Cell 2010, 17, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Cheray, M.; Bégaud, G.; Deluche, E.; Nivet, A.; Battu, S.; Lalloue, F.; Verdier, M.; Bessette, B. Cancer Stem-Like Cells in Glioblastoma. In Glioblastoma; Codon Publications: Brisbane, Australia, 2017; pp. 59–71. [Google Scholar]
- Dick, J.; Bonnet, D. Human Acute Myeloid Leukaemia is organised as a heirarchy that originates from the primitive haematopoetic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar]
- Clarke, M.F.; Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Insight Rev. Artic. 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Palm, T.; Schwamborn, J.C. Brain Tumor Stem Cells; Singh, S.K., Venugopal, C., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2010; Volume 391, ISBN 978-1-4939-8804-4. [Google Scholar]
- Taddei, M.; Giannoni, E.; Fiaschi, T.; Chiarugi, P. Anoikis: An emerging hallmark in health and diseases. J. Pathol. 2012, 226, 380–393. [Google Scholar] [CrossRef]
- Yan, K.; Wu, Q.; Yan, D.H.; Lee, C.H.; Rahim, N.; Tritschler, I.; DeVecchio, J.; Kalady, M.F.; Hjelmeland, A.B.; Rich, J.N. Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes Dev. 2014, 28, 1085–1100. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Teng, J.; Hejazi, S.; Hiddingh, L.; Carvalho, L.; De Gooijer, M.C.; Wakimoto, H.; Barazas, M.; Tannous, M.; Chi, A.S.; Noske, D.P.; et al. Recycling drug screen repurposes hydroxyurea as a sensitizer of glioblastomas to temozolomide targeting de novo DNA synthesis, irrespective of molecular subtype. Neuro Oncol. 2018, 20, 642–654. [Google Scholar] [CrossRef]
- Xu, Z.; Kader, M.; Sen, R.; Placantonakis, D.G. Orthotopic patient-derived glioblastoma xenografts in mice. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2018; Volume 1741, pp. 183–190. ISBN 978-1-4939-7659-1. [Google Scholar]
- Conroy, S.; Kruyt, F.A.E.; Wagemakers, M.; Bhat, K.P.L.; den Dunnen, W.F.A. IL-8 associates with a pro-angiogenic and mesenchymal subtype in glioblastoma. Oncotarget 2018, 9, 15721–15731. [Google Scholar] [CrossRef] [PubMed]
- Maguire, C.A.; Deliolanis, N.C.; Pike, L.; Niers, J.M.; Tjon-Kon-Fat, L.A.; Sena-Esteves, M.; Tannous, B.A. Gaussia luciferase variant for high-throughput functional screening applications. Anal. Chem. 2009, 81, 7102–7106. [Google Scholar] [CrossRef] [PubMed]
- Maguire, C.A.; Bovenberg, M.S.; Crommentuijn, M.H.H.; Niers, J.M.; Kerami, M.; Teng, J.; Sena-Esteves, M.; Badr, C.E.; Tannous, B.A. Triple bioluminescence imaging for in vivo monitoring of cellular processes. Mol. Ther. Nucleic Acids 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Bovenberg, M.S.S.; Degeling, M.H.; Hejazi, S.; Amante, R.J.; Van Keulen, M.; Jeuken, J.W.M.; Akbaripanahi, S.; Vleggeert-Lankamp, C.L.A.; Tannous, M.; Wesseling, P.; et al. Multiplex blood reporters for simultaneous monitoring of cellular processes. Anal. Chem. 2013, 85, 10205–10210. [Google Scholar] [CrossRef] [PubMed]
- Amante, R.J.; Badr, C.E. Cell-based bioluminescence screening assays. Methods Mol. Biol. 2014, 1098, 185–195. [Google Scholar]
- Tannous, B.A. Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat. Protoc. 2009, 4, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Kauer, T.M.; Figueiredo, J.-L.; Hingtgen, S.; Shah, K. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat. Neurosci. 2011, 15, 197–204. [Google Scholar] [CrossRef]
- Starossom, S.C.; Veremeyko, T.; Yung, A.W.Y.; Dukhinova, M.; Au, C.; Lau, A.Y.; Weiner, H.L.; Ponomarev, E.D. Platelets play differential role during the initiation and progression of autoimmune neuroinflammation. Circ. Res. 2015, 117, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Munthe, S.; Sørensen, M.D.; Thomassen, M.; Burton, M.; Kruse, T.A.; Lathia, J.D.; Poulsen, F.R.; Kristensen, B.W. Migrating glioma cells express stem cell markers and give rise to new tumors upon xenografting. J. Neurooncol. 2016, 130, 53–62. [Google Scholar] [CrossRef]
- Wurdinger, T.; Badr, C.; Pike, L.; de Kleine, R.; Weissleder, R.; Breakefield, X.O.; Tannous, B.A. A secreted luciferase for ex vivo monitoring of in vivo processes. Nat. Methods 2008, 5, 171–173. [Google Scholar] [CrossRef]
- Ding, H.; Roncari, L.; Shannon, P.; Wu, X.; Lau, N.; Karaskova, J.; Gutmann, D.H.; Squire, J.A.; Nagy, A.; Guha, A. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. 2001, 61, 3826–3836. [Google Scholar] [PubMed]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma Stem Cell Lines Expanded in Adherent Culture Have Tumor-Specific Phenotypes and Are Suitable for Chemical and Genetic Screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Von Baumgarten, L.; Brucker, D.; Tirniceru, A.; Kienast, Y.; Grau, S.; Burgold, S.; Herms, J.; Winkler, F. Bevacizumab has differential and dose-dependent effects on glioma blood vessels and tumor cells. Clin. Cancer Res. 2011, 17, 6192–6205. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Tran, A.; Nghiemphu, P.L.; Pope, W.B.; Solis, O.E.; Selch, M.; Filka, E.; Yong, W.H.; Mischel, P.S.; Liau, L.M.; et al. Phase II study of bevacizumab plus temozolomide during and after radiation therapy for patients with newly diagnosed glioblastoma multiforme. J. Clin. Oncol. 2011, 29, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Joo, K.M.; Kim, J.; Jin, J.; Kim, M.; Seol, H.J.; Muradov, J.; Yang, H.; Choi, Y.-L.; Park, W.Y.; Kong, D.S.; et al. Patient-Specific Orthotopic Glioblastoma Xenograft Models Recapitulate the Histopathology and Biology of Human Glioblastomas In Situ. Cell Rep. 2013, 3, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Green, A.L.; Ramkissoon, S.H.; McCauley, D.; Jones, K.; Perry, J.A.; Hsu, J.H.R.; Ramkissoon, L.A.; Maire, C.L.; Hubbell-Engler, B.; Knoff, D.S.; et al. Preclinical antitumor efficacy of selective exportin 1 inhibitors in glioblastoma. Neuro Oncol. 2015, 17, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Norton, L.; Albanell, J.; Kim, Y.M.; Mendelsohn, J. Recombinant humanized anti-HER2 antibody (herceptin(TM)) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts. Cancer Res. 1998, 58, 2825–2831. [Google Scholar]
- Sporn, J.R.; Bilgrami, S.A. Weekly paclitaxel plus Herceptin in metastatic breast cancer patients who relapse after stem-cell transplant. Ann. Oncol. 1999, 10, 1259–1260. [Google Scholar] [CrossRef]
- LeBlanc, R.; Catley, L.P.; Hideshima, T.; Lentzsch, S.; Mitsiades, C.S.; Mitsiades, N.; Neuberg, D.; Goloubeva, O.; Pien, C.S.; Adams, J.; et al. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 2002, 62, 4996–5000. [Google Scholar] [PubMed]
- Moreau, P.; Coiteux, V.; Hulin, C.; Leleu, X.; Van De Velde, H.; Acharya, M.; Harousseau, J.L. Prospective comparison of subcutaneous versus intravenous administration of bortezomib in patients with multiple myeloma. Haematologica 2008, 93, 1908–1911. [Google Scholar] [CrossRef]
- Mateos, M.V.; Hernández, J.M.; Hernández, M.T.; Gutiérrez, N.C.; Palomera, L.; Fuertes, M.; Díaz-Mediavilla, J.; Lahuerta, J.J.; De La Rubia, J.; Terol, M.J.; et al. Bortezomib plus melphalan and prednisone in elderly untreated patients with multiple myeloma: Results of a multicenter phase 1/2 study. Blood 2006, 108, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Mohapatra, G.; Kanai, R.; Curry, W.T.; Yip, S.; Nitta, M.; Patel, A.P.; Barnard, Z.R.; Stemmer-Rachamimov, A.O.; Louis, D.N.; et al. Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro Oncol. 2012, 14, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Merz, F.; Gaunitz, F.; Dehghani, F.; Renner, C.; Meixensberger, J.; Gutenberg, A.; Giese, A.; Schopow, K.; Hellwig, C.; Schäfer, M.; et al. Organotypic slice cultures of human glioblastoma reveal different susceptibilities to treatments. Neuro Oncol. 2013, 15, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Paw, I.; Carpenter, R.C.; Watabe, K.; Debinski, W.; Lo, H.W. Mechanisms regulating glioma invasion. Cancer Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef]
- Xie, Q.; Mittal, S.; Berens, M.E. Targeting adaptive glioblastoma: An overview of proliferation and invasion. Neuro Oncol. 2014, 16, 1575–1584. [Google Scholar] [CrossRef]
- Madsen, C.D.; Hooper, S.; Tozluoglu, M.; Bruckbauer, A.; Fletcher, G.; Erler, J.T.; Bates, P.A.; Thompson, B.; Sahai, E. STRIPAK components determine mode of cancer cell migration and metastasis. Nat. Cell Biol. 2015, 17, 68–80. [Google Scholar] [CrossRef]
- Khatau, S.B.; Bloom, R.J.; Bajpai, S.; Razafsky, D.; Zang, S.; Giri, A.; Wu, P.H.; Marchand, J.; Celedon, A.; Hale, C.M.; et al. The distinct roles of the nucleus and nucleus-cytoskeleton connections in three-dimensional cell migration. Sci. Rep. 2012, 2, 488. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Jung, S.; Kim, H.W.; Lee, J.H.; Kang, S.S.; Rhu, H.H.; Jeong, Y.I.; Yang, S.Y.; Chung, H.Y.; Bae, C.S.; Choi, C.; et al. Brain tumor invasion model system using organotypic brain-slice culture as an alternative to in vivo model. J. Cancer Res. Clin. Oncol. 2002, 128, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.L.; Wang, Y.; Wu, J.; Li, G.Y. Quantitative analysis of U251MG human glioma cells invasion in organotypic brain slice co-cultures. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2221–2229. [Google Scholar] [PubMed]
- Eisemann, T.; Costa, B.; Strelau, J.; Mittelbronn, M.; Angel, P.; Peterziel, H. An advanced glioma cell invasion assay based on organotypic brain slice cultures. BMC Cancer 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Knoblich, J.A.; Penninger, J.M.; Jackson, A.P.; Hurles, M.E.; Homfray, T.; Wenzel, D.; Bicknell, L.S.; Lancaster, M.A.; Renner, M.; Martin, C.-A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 2018, 15, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.G.; Jeong, Y.H.; Kim, Y.; Choi, Y.J.; Moon, H.E.; Park, S.H.; Kang, K.S.; Bae, M.; Jang, J.; Youn, H.; et al. A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat. Biomed. Eng. 2019, 3, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.M.J.; Seftor, E.A.; Bonde, G.; Cornell, R.A.; Hendrix, M.J.C. The fate of human malignant melanoma cells transplanted into zebrafish embryos: Assessment of migration and cell division in the absence of tumor formation. Dev. Dyn. 2005, 233, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
- Gansner, J.M.; Dang, M.; Ammerman, M.; Zon, L.I. Transplantation in zebrafish. Methods Cell Biol. 2017, 138, 629–647. [Google Scholar] [PubMed]
- Idilli, A.I.; Precazzini, F.; Mione, M.C.; Anelli, V. Zebrafish in translational cancer research: Insight into leukemia, melanoma, Glioma and endocrine tumor biology. Genes (Basel) 2017, 8, 236. [Google Scholar]
- Geiger, G.A.; Fu, W.; Kao, G.D. Temozolomide-mediated radiosensitization of human glioma cells in a zebrafish embryonic system. Cancer Res. 2008, 68, 3396–3404. [Google Scholar] [CrossRef]
- Lal, S.; La Du, J.; Tanguay, R.L.; Greenwood, J.A. Calpain 2 is required for the invasion of glioblastoma cells in the zebrafish brain microenvironment. J. Neurosci. Res. 2012, 90, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Wehmas, L.C.; Tanguay, R.L.; Punnoose, A.; Greenwood, J.A. Developing a Novel Embryo-Larval Zebrafish Xenograft Assay to Prioritize Human Glioblastoma Therapeutics. Zebrafish 2016, 13, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Konantz, M.; Balci, T.B.; Hartwig, U.F.; Dellaire, G.; André, M.C.; Berman, J.N.; Lengerke, C. Zebrafish xenografts as a tool for in vivo studies on human cancer. Ann. N. Y. Acad. Sci. 2012, 1266, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Vittori, M.; Motaln, H.; Turnšek, T.L. The Study of Glioma by Xenotransplantation in Zebrafish Early Life Stages. J. Histochem. Cytochem. 2015, 63, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Zon, L.I. Zebrafish tumor assays: The state of transplantation. Zebrafish 2009, 6, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Haldi, M.; Ton, C.; Seng, W.L.; McGrath, P. Human melanoma cells transplanted into zebrafish proliferate, migrate, produce melanin, form masses and stimulate angiogenesis in zebrafish. Angiogenesis 2006, 9, 139–151. [Google Scholar] [CrossRef]
- Corkery, D.P.; Dellaire, G.; Berman, J.N. Leukaemia xenotransplantation in zebrafish - chemotherapy response assay in vivo. Br. J. Haematol. 2011, 153, 786–789. [Google Scholar] [CrossRef]
- Pudelko, L.; Edwards, S.; Balan, M.; Nyqvist, D.; Al-Saadi, J.; Dittmer, J.; Almlöf, I.; Helleday, T.; Bräutigam, L. An orthotopic glioblastoma animal model suitable for high-throughput screenings. Neuro Oncol. 2018, 20, 1475–1484. [Google Scholar] [CrossRef]
- Filby, A.; Begum, J.; Jalal, M.; Day, W. Appraising the suitability of succinimidyl and lipophilic fluorescent dyes to track proliferation in non-quiescent cells by dye dilution. Methods 2015, 82, 29–37. [Google Scholar] [CrossRef]
- Vargas-Patron, L.A.; Agudelo-Dueñãs, N.; Madrid-Wolff, J.; Venegas, J.A.; González, J.M.; Forero-Shelton, M.; Akle, V. Xenotransplantation of human glioblastoma in Zebrafish larvae: In vivo imaging and proliferation assessment. Biol. Open 2019, 8. [Google Scholar] [CrossRef]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef] [PubMed]
- Zeng, A.; Ye, T.; Cao, D.; Huang, X.; Yang, Y.; Chen, X.; Xie, Y.; Yao, S.; Zhao, C. Identify a Blood-Brain Barrier Penetrating Drug-TNB using Zebrafish Orthotopic Glioblastoma Xenograft Model. Sci. Rep. 2017, 7, 14372. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.P.; Varma, S.; Gottesman, M.M. The clinical relevance of cancer cell lines. J. Natl. Cancer Inst. 2013, 105, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Allen, M.; Bjerke, M.; Edlund, H.; Nelander, S.; Westermark, B. Origin of the U87MG glioma cell line: Good news and bad news. Sci. Transl. Med. 2016, 8, 354re3. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Wang, J.; Sa, J.K.; Ladewig, E.; Lee, H.O.; Lee, I.H.; Kang, H.J.; Rosenbloom, D.S.; Camara, P.G.; Liu, Z.; et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat. Genet. 2017, 49, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 835–849. [Google Scholar] [CrossRef]
- Karpel-Massler, G.; Banu, M.A.; Shu, C.; Halatsch, M.-E.; Westhoff, M.-A.; Bruce, J.N.; Canoll, P.; Siegelin, M.D. Inhibition of deubiquitinases primes glioblastoma cells to apoptosis in vitro and in vivo. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- Nathanson, D.A.; Gini, B.; Mottahedeh, J.; Visnyei, K.; Koga, T.; Gomez, G.; Eskin, A.; Hwang, K.; Wang, J.; Masui, K.; et al. Targeted Therapy Resistance Mediated by Dynamic Regulation of Extrachromosomal Mutant EGFR DNA. Science (80-. ) 2014, 343, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Rath, P.; Liu, J.; Ryu, D.; Pei, L.; Noonepalle, S.K.; Shull, A.Y.; Feng, Q.; Litofsky, N.S.; Miller, D.C.; et al. Identification of Global DNA Methylation Signatures in Glioblastoma-Derived Cancer Stem Cells. J. Genet. Genomics 2015, 42, 355–371. [Google Scholar] [CrossRef]
- Tabatabai, G.; Weller, M. Glioblastoma stem cells. Cell Tissue Res. 2011, 343, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Gagne, L.M.; Boulay, K.; Topisirovic, I.; Huot, M.É.; Mallette, F.A. Oncogenic Activities of IDH1/2 Mutations: From Epigenetics to Cellular Signaling. Trends Cell Biol. 2017, 27, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; He, L.; Lugano, R.; Roodakker, K.; Bergqvist, M.; Smits, A.; Dimberg, A. IDH mutation status is associated with distinct vascular gene expression signatures in lower-grade gliomas. Neuro Oncol. 2018, 20, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Verbaan, D.; Lamba, S.; Zanon, C.; Jeuken, J.W.M.; Boots-Sprenger, S.H.E.; Wesseling, P.; Hulsebos, T.J.M.; Troost, D.; Van Tilborg, A.A.; et al. The combination of IDH1 mutations and MGMT methylation status predicts survival in glioblastoma better than either IDH1 or MGMT alone. Neuro Oncol. 2014, 16, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Zhang, W.; Wang, Y.; Peng, X.; Chen, B.; Qiu, X.; Li, G.; Li, S.; Wu, C.; Yao, K.; et al. IDH mutation and MGMT promoter methylation in glioblastoma: Results of a prospective registry. Oncotarget 2015, 6, 40896–40906. [Google Scholar] [CrossRef] [PubMed]
- Buqué, A.; Galluzzi, L. Modeling Tumor Immunology and Immunotherapy in Mice. Trends Cancer 2018, 4, 599–601. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cancer Stem Cell | Established Cell Line | |
|---|---|---|
| Advantages | Retains tumor heterogeneity | Highly time and cost effective |
| Mimics phenotype of original tumor | Easy to obtain and culture | |
| Recapitulates genetic, epigenetic, proteasomal, and transcriptomal make-up of original tumor | ||
| More likely to form adequate tumors in vivo | ||
| Disadvantages | Cost- and labor-intensive | Artificial selection in vitro |
| Only low number of passages recommended | Lack of tumor architecture and heterogeneity | |
| Potential low take and growth rate of patient-derived xenografts (PDX) | Does not mirror clinical response | |
| Does not show single-cell invasion, tumor necrosis, or microvascular proliferation | ||
| Does not mirror genotype of original tumor |
| GEM Models | Orthotopic PDX Models | |
|---|---|---|
| Advantages | Mice are immunocompetent | Better predictors of therapy efficacy in patients |
| Customized genetic modifications | Biomarkers for targeted therapy can be identified | |
| Tumor development can be followed over time | Heterogeneity is preserved | |
| Disadvantages | Mutations are limited, cannot reproduce heterogeneity | Cannot customize mutations |
| Mouse tumors are not a good predictable model for human therapy response | Mice are immunocompromised. Not suited to study immunotherapy |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Hora, C.C.; Schweiger, M.W.; Wurdinger, T.; Tannous, B.A. Patient-Derived Glioma Models: From Patients to Dish to Animals. Cells 2019, 8, 1177. https://doi.org/10.3390/cells8101177
da Hora CC, Schweiger MW, Wurdinger T, Tannous BA. Patient-Derived Glioma Models: From Patients to Dish to Animals. Cells. 2019; 8(10):1177. https://doi.org/10.3390/cells8101177
Chicago/Turabian Styleda Hora, Cintia Carla, Markus W. Schweiger, Thomas Wurdinger, and Bakhos A. Tannous. 2019. "Patient-Derived Glioma Models: From Patients to Dish to Animals" Cells 8, no. 10: 1177. https://doi.org/10.3390/cells8101177
APA Styleda Hora, C. C., Schweiger, M. W., Wurdinger, T., & Tannous, B. A. (2019). Patient-Derived Glioma Models: From Patients to Dish to Animals. Cells, 8(10), 1177. https://doi.org/10.3390/cells8101177