Mitogen-Activated Protein Kinases (MAPKs) and Cholangiocarcinoma: The Missing Link




Abstract
:1. Introduction
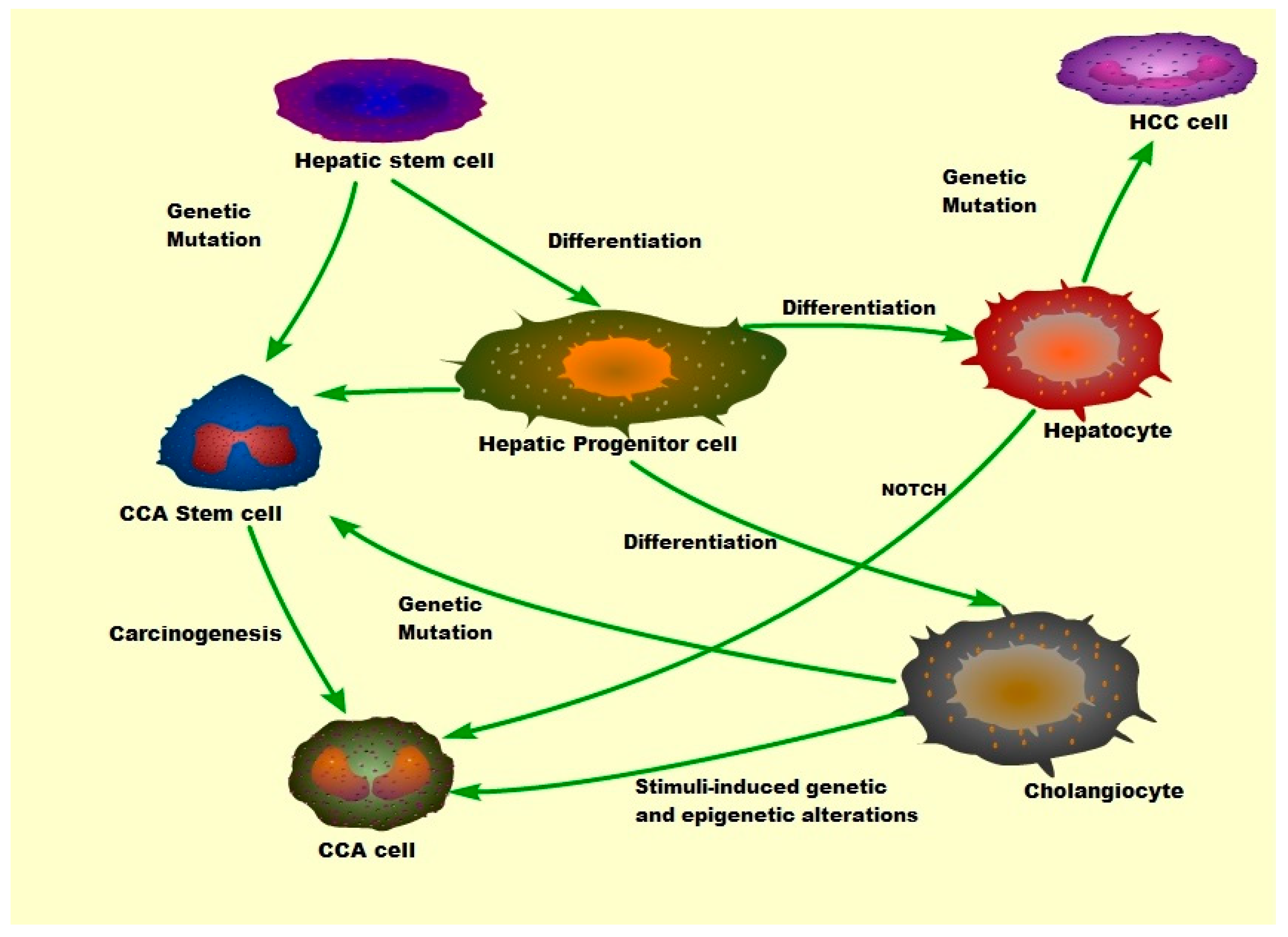
Cholangiocarcinoma (CCA)
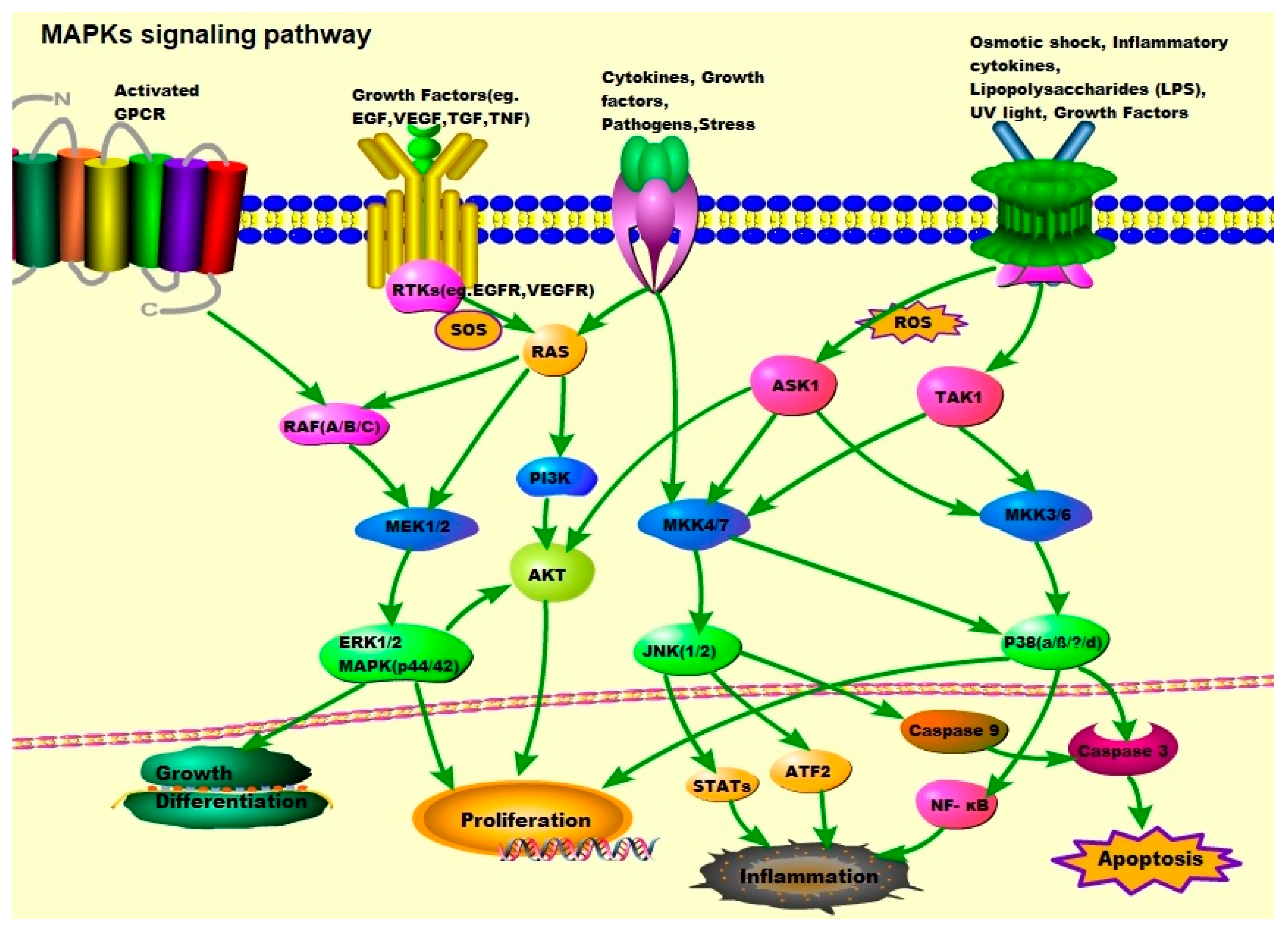
2. The MAPK Signaling Pathways: ERK1/2, JNK-1/2/3, and p38
2.1. JNK/MAPKs
2.2. p38/MAPKs
2.3. ERK/MAPKs
2.4. ‘Other’ Kinases
3. EMT Transition
4. Tumor-Associated Macrophages (TAMs) and Cancer Associated Fibroblasts (CAFs)
5. Biomarkers and Diagnosis
6. Therapy
6.1. Surgical Treatment
6.2. Non-Surgical Treatment: Targeted Therapies
Molecularly Targeted Therapy
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations (in Alphabetical Order)
| ALK | anaplastic lymphoma kinase |
| ARID1A | AT rich interactive domain 1A (SWI-like) |
| ASK1 | apoptosis signal-regulating kinase 1 |
| ATF4 | activated transcription factor 4 |
| ATO | arsenic trioxide |
| BAP1 | BRCA-associated protein 1 |
| BECs | biliary epithelial cells |
| BMDFs | bone marrow derived fibrocytes |
| BRAF | v-raf murine sarcoma viral oncogene homolog B1; |
| CA199 | carbohydrate antigen 19-9 |
| CAFs | cancer-associated fibroblasts |
| CCA | cholangiocarcinoma |
| CDKN2A | cyclin dependent kinase 2a/p16 |
| CDX2 | caudal type homeobox 2 |
| CEA | carcinoembryonic antigen |
| c-Met | tyrosine-protein kinase Met |
| CK-7 | cytokeratin 7 |
| CK-19 | cytokeratin 19 |
| CK-20 | cytokeratin 20 |
| CT | computed tomography |
| dCCA | distal cholangiocarcinoma |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| EGFR | epidermal growth factor receptor |
| eIF2a | eukaryotic initiation factor-α |
| EML4 | echinoderm microtubule-associated protein-like 4 |
| EMT | epithelial to mesenchymal transition |
| EMT-ATFs | EMT-activated transcription factors |
| ERK | extracellular signal-regulated kinase |
| ERS | endoplasmic reticulum stress |
| FAP | Fibroblast activation protein |
| 18FDG-PET | 18F-fluorodeoxyglucose positron emission tomography |
| FHC | ferritin heavy chain |
| FSP1 | fibroblast-specific protein 1 |
| GEF | guanine nucleotide exchange factor |
| GNAS | guanine nucleotide binding protein (G protein) alpha stimulating activity polypeptide 1 |
| GPCRs | G protein-coupled receptors |
| GRP78 | glucose-regulated protein 78 |
| HB-EGF | heparin binding EGF like growth factor |
| HCC | hepatocellular carcinoma |
| HNF | hepatocyte nuclear factor |
| hOCT1 | human organic cation transporter type 1 |
| HPCs | hepatic progenitor cells |
| HSCs | hepatic stellate cells |
| iCCA | intrahepatic cholangiocarcinoma |
| IDH1 | isocitrate dehydrogenase 1 |
| IDH2 | isocitrate dehydrogenase 2 |
| IKK | inhibitor of NF-κB kinase |
| IL6 | interleukin 6 |
| ILR | interleukin receptor |
| JNK | c-Jun NH2-terminal kinase |
| KC | Kupffer cell |
| KMT2C | lysine methyltransferase 2C |
| KRAS | V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog |
| LPC | liver parenchymal cells |
| LPS | lipopolysaccharide |
| MAPKs | mitogen-activated protein kinases |
| MAP2Ks | mitogen-activated protein kinase kinases |
| MAP3Ks | mitogen-activated protein kinase kinase kinases |
| MAP3K5 | mitogen-activated protein kinase kinase kinase 5 |
| MEK | mitogen-activated protein kinase |
| MEKK1 | mitogen-activated protein kinase kinase kinase 1 |
| MET | mesenchymal to epithelial transition |
| MKK3 | MAP kinase 3 |
| MKK4 | MAP kinase 4 |
| MKK6 | MAP kinase 6 |
| MKK7 | MAP kinase 7 |
| MLK3 | mitogen-activated protein kinase kinase kinase 11 |
| MMPs | Matrix metalloproteinases |
| MnSOD | manganese-superoxide dismutase |
| MRI | magnetic resonance imaging |
| mTOR | mammalian target of rapamycin |
| NF-κB | nuclear factor (NF)-κB |
| NLK | Nemo-like kinase |
| NRAS | neuroblastoma RAS viral (v-ras) oncogene homolog |
| OLT | orthotopic liver transplantation |
| PBC | primary biliary cholangitis |
| PBRM1 | polybromo 1 |
| pCCA | perihilar cholangiocarcinoma |
| PDGF | platelet-derived growth factor |
| PDGF-D | platelet derived growth factor D |
| PDGFR | platelet-derived growth factor receptor |
| PDGFR-β | platelet-derived growth factor receptor-β |
| PEG3 | paternally expressed 3 |
| PTEN | phosphatase and tensin homolog deleted on chromosome ten |
| PFS | progression-free survival |
| PFs | portal fibroblasts |
| PI3K | phosphatidylinositol 3-kinase |
| PIK3CA | phosphoinositide-3-kinase, catalytic, alpha polypeptide |
| PL | piperlongumine |
| PSC | primary sclerosing cholangitis |
| RNF43 | ring finger protein 43 |
| ROBO2 | roundabout guidance receptor 2 |
| ROS | reactive oxygen species |
| RTKs | receptor tyrosine kinases |
| SG | sulfated galactans |
| SMAD4 | sulfated galactans; mothers against decapentaplegic homolog 4 (Drosophila) |
| SOS | son of sevenless |
| α-SMA | α-smooth muscle actin |
| STAT3 | signal transducer and activator of transcription 3 |
| TACE | transcatheter arterial chemoembolization |
| TAK1 | transforming growth factor-β-activated kinase 1 |
| TAMs | tumor-associated macrophages |
| TNF | tumor necrosis factor |
| TNF-α | tumor necrosis factor alpha |
| TNFR | tumor necrosis factor receptor |
| TGF-β | transforming growth factor-β |
| TP53 | Tumor protein p53 |
| US | ultrasound |
| VEGF | vascular endothelial growth factor |
| VEGFR | vascular endothelial growth factor receptor |
References
- Shin, H.R.; Oh, J.K.; Lim, M.K.; Shin, A.; Kong, H.J.; Jung, K.W.; Won, Y.J.; Park, S.; Park, S.J.; Hong, S.T. Descriptive epidemiology of cholangiocarcinoma and clonorchiasis in Korea. J. Korean Med. Sci. 2010, 25, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- McLean, L.; Patel, T. Racial and ethnic variations in the epidemiology of intrahepatic cholangiocarcinoma in the United States. Liver Int. 2006, 26, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Tyson, G.L.; El-Serag, H.B. Risk factors for cholangiocarcinoma. Hepatology 2011, 54, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Davidson, B.R.; Goldin, R.D.; Heaton, N.; Karani, J.; Pereira, S.P.; Rosenberg, W.M.; Tait, P.; Taylor-Robinson, S.D.; Thillainayagam, A.V.; et al. Guidelines for the diagnosis and treatment of cholangiocarcinoma: An update. Gut 2012, 61, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaib, Y.H.; Davila, J.A.; McGlynn, K.; El-Serag, H.B. Rising incidence of intrahepatic cholangiocarcinoma in the United States: A true increase? J. Hepatol. 2004, 40, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Dodson, R.M.; Weiss, M.J.; Cosgrove, D.; Herman, J.M.; Kamel, I.; Anders, R.; Geschwind, J.F.; Pawlik, T.M. Intrahepatic cholangiocarcinoma: Management options and emerging therapies. J. Am. Coll. Surg. 2013, 217, 736–750 e734. [Google Scholar] [CrossRef]
- Labib, P.L.; Goodchild, G.; Pereira, S.P. Molecular pathogenesis of cholangiocarcinoma. BMC Cancer 2019, 19, 185. [Google Scholar] [CrossRef] [PubMed]
- Welzel, T.M.; Mellemkjaer, L.; Gloria, G.; Sakoda, L.C.; Hsing, A.W.; El Ghormli, L.; Olsen, J.H.; McGlynn, K.A. Risk factors for intrahepatic cholangiocarcinoma in a low-risk population: A nationwide case-control study. Int. J. Cancer 2007, 120, 638–641. [Google Scholar] [CrossRef]
- Welzel, T.M.; Graubard, B.I.; El-Serag, H.B.; Shaib, Y.H.; Hsing, A.W.; Davila, J.A.; McGlynn, K.A. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma in the United States: A population-based case-control study. Clin. Gastroenterol. Hepatol. 2007, 5, 1221–1228. [Google Scholar] [CrossRef]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European network for the study of cholangiocarcinoma (ens-cca). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Komuta, M.; Govaere, O.; Vandecaveye, V.; Akiba, J.; Van Steenbergen, W.; Verslype, C.; Laleman, W.; Pirenne, J.; Aerts, R.; Yano, H.; et al. Histological diversity in cholangiocellular carcinoma reflects the different cholangiocyte phenotypes. Hepatology 2012, 55, 1876–1888. [Google Scholar] [CrossRef] [PubMed]
- Aishima, S.; Oda, Y. Pathogenesis and classification of intrahepatic cholangiocarcinoma: Different characters of perihilar large duct type versus peripheral small duct type. J. Hepatobil. Pancreat. Sci. 2015, 22, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Gores, G.J. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 2013, 145, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Al-Bahrani, R.; Abuetabh, Y.; Zeitouni, N.; Sergi, C. Cholangiocarcinoma: Risk factors, environmental influences and oncogenesis. Ann. Clin. Lab. Sci. 2013, 43, 195–210. [Google Scholar]
- Isomoto, H.; Kobayashi, S.; Werneburg, N.W.; Bronk, S.F.; Guicciardi, M.E.; Frank, D.A.; Gores, G.J. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a stat3 pathway in cholangiocarcinoma cells. Hepatology 2005, 42, 1329–1338. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the mapks and their substrates, the mapk-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by erk, jnk, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed]
- Brancho, D.; Tanaka, N.; Jaeschke, A.; Ventura, J.J.; Kelkar, N.; Tanaka, Y.; Kyuuma, M.; Takeshita, T.; Flavell, R.A.; Davis, R.J. Mechanism of p38 map kinase activation in vivo. Genes Dev. 2003, 17, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Remy, G.; Risco, A.M.; Inesta-Vaquera, F.A.; Gonzalez-Teran, B.; Sabio, G.; Davis, R.J.; Cuenda, A. Differential activation of p38mapk isoforms by mkk6 and mkk3. Cell. Signal. 2010, 22, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Bubici, C.; Papa, S. Jnk signalling in cancer: In need of new, smarter therapeutic targets. Br. J. Pharmacol. 2014, 171, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Bravo, I.G.; Cheng, H.; Alonso, A. Multiple independent kinase cascades are targeted by hyperosmotic stress but only one activates stress kinase p38. Exp. Cell. Res. 2004, 292, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the raf-mek-erk mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Zhang, J.; Oo, C.; Min, R.W.M.; Kaplowitz, N. New insights into the role and mechanism of c-jun-n-terminal kinase signaling in the pathobiology of liver diseases. Hepatology 2018, 67, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Min, R.W.; Aghajan, M.; Kaplowitz, N. C-jun n-terminal kinase mediates mouse liver injury through a novel sab (sh3bp5)-dependent pathway leading to inactivation of intramitochondrial src. Hepatology 2016, 63, 1987–2003. [Google Scholar] [CrossRef] [PubMed]
- Mingo-Sion, A.M.; Marietta, P.M.; Koller, E.; Wolf, D.M.; Van Den Berg, C.L. Inhibition of jnk reduces g2/m transit independent of p53, leading to endoreduplication, decreased proliferation, and apoptosis in breast cancer cells. Oncogene 2004, 23, 596–604. [Google Scholar] [CrossRef]
- Vivanco, I.; Palaskas, N.; Tran, C.; Finn, S.P.; Getz, G.; Kennedy, N.J.; Jiao, J.; Rose, J.; Xie, W.; Loda, M.; et al. Identification of the jnk signaling pathway as a functional target of the tumor suppressor pten. Cancer Cell 2007, 11, 555–569. [Google Scholar] [CrossRef]
- Wang, J.; Tai, G. Role of c-jun n-terminal kinase in hepatocellular carcinoma development. Target. Oncol. 2016, 11, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Cubero, F.J.; Zhao, G.; Nevzorova, Y.A.; Hatting, M.; Al Masaoudi, M.; Verdier, J.; Peng, J.; Schaefer, F.M.; Hermanns, N.; Boekschoten, M.V.; et al. Haematopoietic cell-derived jnk1 is crucial for chronic inflammation and carcinogenesis in an experimental model of liver injury. J. Hepatol. 2015, 62, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Chen, F. Jnk-induced apoptosis, compensatory growth, and cancer stem cells. Cancer Res. 2012, 72, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Brenner, D.A.; Karin, M. A liver full of jnk: Signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 2012, 143, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Cazanave, S.C.; Mott, J.L.; Elmi, N.A.; Bronk, S.F.; Werneburg, N.W.; Akazawa, Y.; Kahraman, A.; Garrison, S.P.; Zambetti, G.P.; Charlton, M.R.; et al. Jnk1-dependent puma expression contributes to hepatocyte lipoapoptosis. J. Biol. Chem. 2009, 284, 26591–26602. [Google Scholar] [CrossRef]
- Qiu, W.; Wang, X.; Leibowitz, B.; Yang, W.; Zhang, L.; Yu, J. Puma-mediated apoptosis drives chemical hepatocarcinogenesis in mice. Hepatology 2011, 54, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Zhang, Y.; Beezhold, K.J.; Bhatia, D.; Zhao, H.; Chen, J.; Castranova, V.; Shi, X.; Chen, F. Sustained jnk1 activation is associated with altered histone h3 methylations in human liver cancer. J. Hepatol. 2009, 50, 323–333. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. Jnk signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef]
- Yuan, D.; Huang, S.; Berger, E.; Liu, L.; Gross, N.; Heinzmann, F.; Ringelhan, M.; Connor, T.O.; Stadler, M.; Meister, M.; et al. Kupffer cell-derived tnf triggers cholangiocellular tumorigenesis through jnk due to chronic mitochondrial dysfunction and ros. Cancer Cell 2017, 31, 771–789 e776. [Google Scholar] [CrossRef]
- Zhong, F.; Tong, Z.T.; Fan, L.L.; Zha, L.X.; Wang, F.; Yao, M.Q.; Gu, K.S.; Cao, Y.X. Guggulsterone-induced apoptosis in cholangiocarcinoma cells through ros/jnk signaling pathway. Am. J. Cancer Res. 2016, 6, 226–237. [Google Scholar]
- Feng, C.; He, K.; Zhang, C.; Su, S.; Li, B.; Li, Y.; Duan, C.Y.; Chen, S.; Chen, R.; Liu, Y.; et al. Jnk contributes to the tumorigenic potential of human cholangiocarcinoma cells through the mtor pathway regulated grp78 induction. PLoS ONE 2014, 9, e90388. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Xie, H.; Yang, F.; Shan, Q.; Dai, H.; Zhuo, J.; Wei, X.; Song, P.; Zhou, L.; Xu, X.; et al. Metformin potentiates the effect of arsenic trioxide suppressing intrahepatic cholangiocarcinoma: Roles of p38 mapk, erk3, and mtorc1. J. Hematol. Oncol. 2017, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Del Barco Barrantes, I.; Nebreda, A.R. Roles of p38 mapks in invasion and metastasis. Biochem. Soc. Trans. 2012, 40, 79–84. [Google Scholar] [CrossRef]
- Rousseau, S.; Dolado, I.; Beardmore, V.; Shpiro, N.; Marquez, R.; Nebreda, A.R.; Arthur, J.S.; Case, L.M.; Tessier-Lavigne, M.; Gaestel, M.; et al. Cxcl12 and c5a trigger cell migration via a pak1/2-p38alpha mapk-mapkap-k2-hsp27 pathway. Cell. Signal. 2006, 18, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Map kinase signaling pathways and hematologic malignancies. Blood 2003, 101, 4667–4679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagiwa, Y.; Marienfeld, C.; Tadlock, L.; Patel, T. Translational regulation by p38 mitogen-activated protein kinase signaling during human cholangiocarcinoma growth. Hepatology 2003, 38, 158–166. [Google Scholar] [CrossRef]
- Tan, F.L.; Ooi, A.; Huang, D.; Wong, J.C.; Qian, C.N.; Chao, C.; Ooi, L.; Tan, Y.M.; Chung, A.; Cheow, P.C.; et al. P38delta/mapk13 as a diagnostic marker for cholangiocarcinoma and its involvement in cell motility and invasion. Int. J. Cancer 2010, 126, 2353–2361. [Google Scholar] [CrossRef]
- Yoshikawa, D.; Ojima, H.; Iwasaki, M.; Hiraoka, N.; Kosuge, T.; Kasai, S.; Hirohashi, S.; Shibata, T. Clinicopathological and prognostic significance of egfr, vegf, and her2 expression in cholangiocarcinoma. Br. J. Cancer 2008, 98, 418–425. [Google Scholar] [CrossRef]
- Nakazawa, K.; Dobashi, Y.; Suzuki, S.; Fujii, H.; Takeda, Y.; Ooi, A. Amplification and overexpression of c-erbb-2, epidermal growth factor receptor, and c-met in biliary tract cancers. J. Pathol. 2005, 206, 356–365. [Google Scholar] [CrossRef]
- Dai, R.; Li, J.; Fu, J.; Chen, Y.; Wang, R.; Zhao, X.; Luo, T.; Zhu, J.; Ren, Y.; Cao, J.; et al. The tyrosine kinase c-met contributes to the pro-tumorigenic function of the p38 kinase in human bile duct cholangiocarcinoma cells. J. Biol. Chem. 2012, 287, 39812–39823. [Google Scholar] [CrossRef]
- Sia, D.; Tovar, V.; Moeini, A.; Llovet, J.M. Intrahepatic cholangiocarcinoma: Pathogenesis and rationale for molecular therapies. Oncogene 2013, 32, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, E.; Vasilaki, E.; Vorvis, C.; Iliopoulos, D.; Moustakas, A.; Kardassis, D.; Stournaras, C. Differential regulation of the two rhoa-specific gef isoforms net1/net1a by tgf-beta and mir-24: Role in epithelial-to-mesenchymal transition. Oncogene 2012, 31, 2862–2875. [Google Scholar] [CrossRef] [PubMed]
- Olieslagers, S.; Pardali, E.; Tchaikovski, V.; ten Dijke, P.; Waltenberger, J. Tgf-beta1/alk5-induced monocyte migration involves pi3k and p38 pathways and is not negatively affected by diabetes mellitus. Cardiovasc. Res. 2011, 91, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Chapnick, D.A.; Warner, L.; Bernet, J.; Rao, T.; Liu, X. Partners in crime: The tgfbeta and mapk pathways in cancer progression. Cell Biosci. 2011, 1, 42. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.; Yang, F.; Wise, C.E.; Meng, F.; Priester, S.; Munshi, M.K.; Guerrier, M.; Dostal, D.E.; Glaser, S.S. Simvastatin stimulates apoptosis in cholangiocarcinoma by inhibition of rac1 activity. Dig. Liver Dis. 2011, 43, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Cadamuro, M.; Nardo, G.; Indraccolo, S.; Dall’olmo, L.; Sambado, L.; Moserle, L.; Franceschet, I.; Colledan, M.; Massani, M.; Stecca, T.; et al. Platelet-derived growth factor-d and rho gtpases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 2013, 58, 1042–1053. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Biswas, S.; Liang, B.; Zou, X.; Shan, L.; Li, Y.; Fang, R.; Niu, J. Integrin beta6 serves as an immunohistochemical marker for lymph node metastasis and promotes cell invasiveness in cholangiocarcinoma. Sci. Rep. 2016, 6, 30081. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Harada, K.; Itatsu, K.; Ikeda, H.; Kakuda, Y.; Shimomura, S.; Shan Ren, X.; Yoneda, N.; Sasaki, M.; Nakanuma, Y. Epithelial-mesenchymal transition induced by transforming growth factor-{beta}1/snail activation aggravates invasive growth of cholangiocarcinoma. Am. J. Pathol. 2010, 177, 141–152. [Google Scholar] [CrossRef]
- Zen, Y.; Harada, K.; Sasaki, M.; Chen, T.C.; Chen, M.F.; Yeh, T.S.; Jan, Y.Y.; Huang, S.F.; Nimura, Y.; Nakanuma, Y. Intrahepatic cholangiocarcinoma escapes from growth inhibitory effect of transforming growth factor-beta1 by overexpression of cyclin d1. Lab. Investig. 2005, 85, 572–581. [Google Scholar] [CrossRef]
- Sritananuwat, P.; Sueangoen, N.; Thummarati, P.; Islam, K.; Suthiphongchai, T. Blocking erk1/2 signaling impairs tgf-beta1 tumor promoting function but enhances its tumor suppressing role in intrahepatic cholangiocarcinoma cells. Cancer Cell Int. 2017, 17, 85. [Google Scholar] [CrossRef]
- Yothaisong, S.; Dokduang, H.; Techasen, A.; Namwat, N.; Yongvanit, P.; Bhudhisawasdi, V.; Puapairoj, A.; Riggins, G.J.; Loilome, W. Increased activation of pi3k/akt signaling pathway is associated with cholangiocarcinoma metastasis and pi3k/mtor inhibition presents a possible therapeutic strategy. Tumour Biol. 2013, 34, 3637–3648. [Google Scholar] [CrossRef] [PubMed]
- Kokuryo, T.; Yokoyama, Y.; Nagino, M. Recent advances in cancer stem cell research for cholangiocarcinoma. J. Hepatobil. Pancreat. Sci. 2012, 19, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Utispan, K.; Sonongbua, J.; Thuwajit, P.; Chau-In, S.; Pairojkul, C.; Wongkham, S.; Thuwajit, C. Periostin activates integrin alpha5beta1 through a pi3k/aktdependent pathway in invasion of cholangiocarcinoma. Int. J. Oncol. 2012, 41, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Min, J.K.; Lee, J.W.; Kim, D.G.; Hong, H.J. Acquisition of chemoresistance in intrahepatic cholangiocarcinoma cells by activation of akt and extracellular signal-regulated kinase (erk)1/2. Biochem. Biophys. Res. Commun. 2011, 405, 333–337. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Su, Y.; Volpert, O.V.; Vande Woude, G.F. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive vegf and negative thrombospondin 1 regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 12718–12723. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, M.; Ojima, H.; Iwasaki, M.; Shimizu, H.; Kokubu, A.; Hiraoka, N.; Kosuge, T.; Yoshikawa, D.; Kono, T.; Furukawa, H.; et al. Prognostic significance of overexpression of c-met oncoprotein in cholangiocarcinoma. Br. J. Cancer 2011, 105, 131–138. [Google Scholar] [CrossRef]
- Terada, T.; Nakanuma, Y.; Sirica, A.E. Immunohistochemical demonstration of met overexpression in human intrahepatic cholangiocarcinoma and in hepatolithiasis. Hum. Pathol. 1998, 29, 175–180. [Google Scholar]
- Menakongka, A.; Suthiphongchai, T. Involvement of pi3k and erk1/2 pathways in hepatocyte growth factor-induced cholangiocarcinoma cell invasion. World J. Gastroenterol. 2010, 16, 713–722. [Google Scholar] [CrossRef]
- Rincon, M.; Davis, R.J. Regulation of the immune response by stress-activated protein kinases. Immunol. Rev. 2009, 228, 212–224. [Google Scholar] [CrossRef]
- Bettermann, K.; Vucur, M.; Haybaeck, J.; Koppe, C.; Janssen, J.; Heymann, F.; Weber, A.; Weiskirchen, R.; Liedtke, C.; Gassler, N.; et al. Tak1 suppresses a nemo-dependent but nf-kappab-independent pathway to liver cancer. Cancer Cell 2010, 17, 481–496. [Google Scholar] [CrossRef]
- Malato, Y.; Willenbring, H. The map3k tak1: A chock block to liver cancer formation. Hepatology 2010, 52, 1506–1509. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, S.; Aoyama, T.; Miura, K.; Osterreicher, C.H.; Kodama, Y.; Miyai, K.; Akira, S.; Brenner, D.A.; Seki, E. Disruption of tak1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, G.; Di Paolo, J.A.; Hammaker, D.; Boyle, D.L.; Budas, G.; Notte, G.T.; Mikaelian, I.; Barry, V.; Firestein, G.S. Regulation and function of apoptosis signal-regulating kinase 1 in rheumatoid arthritis. Biochem. Pharmacol. 2018, 151, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Fujino, G.; Noguchi, T.; Matsuzawa, A.; Yamauchi, S.; Saitoh, M.; Takeda, K.; Ichijo, H. Thioredoxin and traf family proteins regulate reactive oxygen species-dependent activation of ask1 through reciprocal modulation of the n-terminal homophilic interaction of ask1. Mol. Cell. Biol. 2007, 27, 8152–8163. [Google Scholar] [CrossRef] [PubMed]
- Hattori, K.; Naguro, I.; Runchel, C.; Ichijo, H. The roles of ask family proteins in stress responses and diseases. Cell Commun. Signal. 2009, 7, 9. [Google Scholar] [CrossRef]
- Glaser, S.S.; Gaudio, E.; Miller, T.; Alvaro, D.; Alpini, G. Cholangiocyte proliferation and liver fibrosis. Expert Rev. Mol. Med. 2009, 11, e7. [Google Scholar] [CrossRef] [PubMed]
- Roskams, T.; Desmet, V. Ductular reaction and its diagnostic significance. Semin. Diagn. Pathol. 1998, 15, 259–269. [Google Scholar] [PubMed]
- Diaz, R.; Kim, J.W.; Hui, J.J.; Li, Z.; Swain, G.P.; Fong, K.S.; Csiszar, K.; Russo, P.A.; Rand, E.B.; Furth, E.E.; et al. Evidence for the epithelial to mesenchymal transition in biliary atresia fibrosis. Hum. Pathol. 2008, 39, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Burns, W.C.; Kantharidis, P.; Thomas, M.C. The role of tubular epithelial-mesenchymal transition in progressive kidney disease. Cells Tissues Organs 2007, 185, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.; Ali, S.; McDonnell, B.J.; Burt, A.D.; Kirby, J.A. Chronic renal allograft dysfunction: The role of t cell-mediated tubular epithelial to mesenchymal cell transition. J. Am. Soc. Nephrol. 2004, 15, 390–397. [Google Scholar] [CrossRef]
- Roskams, T.A.; Theise, N.D.; Balabaud, C.; Bhagat, G.; Bhathal, P.S.; Bioulac-Sage, P.; Brunt, E.M.; Crawford, J.M.; Crosby, H.A.; Desmet, V.; et al. Nomenclature of the finer branches of the biliary tree: Canals, ductules, and ductular reactions in human livers. Hepatology 2004, 39, 1739–1745. [Google Scholar] [CrossRef]
- Alvaro, D.; Mancino, M.G.; Glaser, S.; Gaudio, E.; Marzioni, M.; Francis, H.; Alpini, G. Proliferating cholangiocytes: A neuroendocrine compartment in the diseased liver. Gastroenterology 2007, 132, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Heo, J.; Libbrecht, L.; Chu, I.S.; Kaposi-Novak, P.; Calvisi, D.F.; Mikaelyan, A.; Roberts, L.R.; Demetris, A.J.; Sun, Z.; et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med. 2006, 12, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Chen, X.P.; Zhang, W.; Dong, H.H.; Xiang, S.; Zhang, W.G.; Zhang, B.X. Combined hepatocellular cholangiocarcinoma originating from hepatic progenitor cells: Immunohistochemical and double-fluorescence immunostaining evidence. Histopathology 2008, 52, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Zhao, X.; Wang, X.W. Molecular carcinogenesis of hepatocellular carcinoma and intrahepatic cholangiocarcinoma: One step closer to personalized medicine? Cell Biosci. 2011, 1, 5. [Google Scholar] [CrossRef]
- Planas-Paz, L.; Orsini, V.; Boulter, L.; Calabrese, D.; Pikiolek, M.; Nigsch, F.; Xie, Y.; Roma, G.; Donovan, A.; Marti, P.; et al. The rspo-lgr4/5-znrf3/rnf43 module controls liver zonation and size. Nat. Cell Biol. 2016, 18, 467–479. [Google Scholar] [CrossRef]
- Guest, R.V.; Boulter, L.; Dwyer, B.J.; Forbes, S.J. Understanding liver regeneration to bring new insights to the mechanisms driving cholangiocarcinoma. NPJ Regener. Med. 2017, 2, 13. [Google Scholar] [CrossRef] [Green Version]
- Zong, Y.; Panikkar, A.; Xu, J.; Antoniou, A.; Raynaud, P.; Lemaigre, F.; Stanger, B.Z. Notch signaling controls liver development by regulating biliary differentiation. Development 2009, 136, 1727–1739. [Google Scholar] [CrossRef] [Green Version]
- Theise, N.D.; Yao, J.L.; Harada, K.; Hytiroglou, P.; Portmann, B.; Thung, S.N.; Tsui, W.; Ohta, H.; Nakanuma, Y. Hepatic ‘stem cell’ malignancies in adults: Four cases. Histopathology 2003, 43, 263–271. [Google Scholar] [CrossRef]
- Chiba, T.; Kita, K.; Zheng, Y.W.; Yokosuka, O.; Saisho, H.; Iwama, A.; Nakauchi, H.; Taniguchi, H. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology 2006, 44, 240–251. [Google Scholar] [CrossRef]
- Hay, E.D. Organization and fine structure of epithelium and mesenchyme in the developing chick embryo. Epithel. Mesenchymal Interact. 1968, 2, 31–35. [Google Scholar]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. Emt in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Agoston, A.T.; Pham, T.H.; Zhang, W.; Zhang, X.; Huo, X.; Peng, S.; Bajpai, M.; Das, K.; Odze, R.D.; et al. Acidic bile salts induce epithelial to mesenchymal transition via vegf signaling in non-neoplastic barrett’s cells. Gastroenterology 2019, 156, 130–144.e110. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. Emt, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, X.J.; Xing, J. Signal transduction pathways of emt induced by tgf-beta, shh, and wnt and their crosstalks. J. Clin. Med. 2016, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, J.; Guedj, N.; Claperon, A.; Nguyen Ho-Bouldoires, T.H.; Paradis, V.; Fouassier, L. Epithelial-mesenchymal transition in cholangiocarcinoma: From clinical evidence to regulatory networks. J. Hepatol. 2017, 66, 424–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakowlew, S.B. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 2006, 25, 435–457. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Landstrom, M.; Moustakas, A. Mechanism of tgf-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 2009, 21, 166–176. [Google Scholar] [CrossRef]
- Seoane, J. Escaping from the tgfbeta anti-proliferative control. Carcinogenesis 2006, 27, 2148–2156. [Google Scholar] [CrossRef]
- Sheahan, S.; Bellamy, C.O.; Dunbar, D.R.; Harrison, D.J.; Prost, S. Deficiency of g1 regulators p53, p21cip1 and/or prb decreases hepatocyte sensitivity to tgfbeta cell cycle arrest. BMC Cancer 2007, 7, 215. [Google Scholar] [CrossRef] [PubMed]
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of tgf-beta targeted cancer therapy. Int. J. Biol. Sci. 2012, 8, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; ten Dijke, P. Tgf-beta signalling from cell membrane to nucleus through smad proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Fairman, R.; Penry, J.; Shi, Y. Formation of a stable heterodimer between smad2 and smad4. J. Biol. Chem. 2001, 276, 20688–20694. [Google Scholar] [CrossRef] [PubMed]
- Thuault, S.; Tan, E.J.; Peinado, H.; Cano, A.; Heldin, C.H.; Moustakas, A. Hmga2 and smads co-regulate snail1 expression during induction of epithelial-to-mesenchymal transition. J. Biol. Chem. 2008, 283, 33437–33446. [Google Scholar] [CrossRef] [PubMed]
- Brandl, M.; Seidler, B.; Haller, F.; Adamski, J.; Schmid, R.M.; Saur, D.; Schneider, G. Ikk(alpha) controls canonical tgf(ss)-smad signaling to regulate genes expressing snail and slug during emt in panc1 cells. J. Cell Sci. 2010, 123, 4231–4239. [Google Scholar] [CrossRef] [PubMed]
- Chacko, B.M.; Qin, B.Y.; Tiwari, A.; Shi, G.; Lam, S.; Hayward, L.J.; De Caestecker, M.; Lin, K. Structural basis of heteromeric smad protein assembly in tgf-beta signaling. Mol. Cell 2004, 15, 813–823. [Google Scholar] [CrossRef]
- Lagna, G.; Hata, A.; Hemmati-Brivanlou, A.; Massague, J. Partnership between dpc4 and smad proteins in tgf-beta signalling pathways. Nature 1996, 383, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-smad pathways in tgf-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Yamamura, Y.; Hua, X.; Bergelson, S.; Lodish, H.F. Critical role of smads and ap-1 complex in transforming growth factor-beta-dependent apoptosis. J. Biol. Chem. 2000, 275, 36295–36302. [Google Scholar] [CrossRef]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. Traf6 mediates smad-independent activation of jnk and p38 by tgf-beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Koudelkova, P.; Dituri, F.; Mikulits, W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J. Hepatol. 2016, 65, 798–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, G. Stromal aspects of breast carcinoma. Exp. Mol. Pathol. 1979, 31, 248–260. [Google Scholar] [CrossRef]
- Kwa, M.Q.; Herum, K.M.; Brakebusch, C. Cancer-associated fibroblasts: How do they contribute to metastasis? Clin. Exp. Metastasis 2019, 36, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Zeltz, C.; Primac, I.; Erusappan, P.; Alam, J.; Noel, A.; Gullberg, D. Cancer-associated fibroblasts in desmoplastic tumors: Emerging role of integrins. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Santi, A.; Kugeratski, F.G.; Zanivan, S. Cancer associated fibroblasts: The architects of stroma remodeling. Proteomics 2018, 18, e1700167. [Google Scholar] [CrossRef]
- Okabe, H.; Beppu, T.; Hayashi, H.; Ishiko, T.; Masuda, T.; Otao, R.; Horlad, H.; Jono, H.; Ueda, M.; Shinriki, S.; et al. Hepatic stellate cells accelerate the malignant behavior of cholangiocarcinoma cells. Ann. Surg. Oncol. 2011, 18, 1175–1184. [Google Scholar] [CrossRef]
- Itou, R.A.; Uyama, N.; Hirota, S.; Kawada, N.; Wu, S.; Miyashita, S.; Nakamura, I.; Suzumura, K.; Sueoka, H.; Okada, T.; et al. Immunohistochemical characterization of cancer-associated fibroblasts at the primary sites and in the metastatic lymph nodes of human intrahepatic cholangiocarcinoma. Hum. Pathol. 2019, 83, 77–89. [Google Scholar] [CrossRef]
- Sirica, A.E.; Campbell, D.J.; Dumur, C.I. Cancer-associated fibroblasts in intrahepatic cholangiocarcinoma. Curr. Opin. Gastroenterol. 2011, 27, 276–284. [Google Scholar] [CrossRef]
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.J.; Alpaugh, R.K.; Palazzo, I.; Meropol, N.J.; Rogatko, A.; Xu, Z.; Hoffman, J.P.; Weiner, L.M.; Cheng, J.D. Fibroblast activation protein and its relationship to clinical outcome in pancreatic adenocarcinoma. Pancreas 2008, 37, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Takeya, M. Cafs and tams: Maestros of the tumour microenvironment. J. Pathol. 2017, 241, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, O.; Yoshida, M.; Koma, Y.; Yanai, T.; Hasegawa, D.; Kosaka, Y.; Nishimura, N.; Yokozaki, H. Collaboration of cancer-associated fibroblasts and tumour-associated macrophages for neuroblastoma development. J. Pathol. 2016, 240, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Techasen, A.; Loilome, W.; Namwat, N.; Dokduang, H.; Jongthawin, J.; Yongvanit, P. Cytokines released from activated human macrophages induce epithelial mesenchymal transition markers of cholangiocarcinoma cells. Asian Pac. J. Cancer Prev. 2012, 13, 115–118. [Google Scholar] [PubMed]
- Berasain, C.; Avila, M.A. Platelet-derived growth factor d: A new player in the complex cross-talk between cholangiocarcinoma cells and cancer-associated fibroblasts. Hepatology 2013, 58, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Raggi, C.; Invernizzi, P.; Andersen, J.B. Impact of microenvironment and stem-like plasticity in cholangiocarcinoma: Molecular networks and biological concepts. J. Hepatol. 2015, 62, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Bansal, R.; van Baarlen, J.; Storm, G.; Prakash, J. The interplay of the notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis. Sci. Rep. 2015, 5, 18272. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Chaurasia, B.; Goldau, J.; Vogt, M.C.; Ruud, J.; Nguyen, K.D.; Theurich, S.; Hausen, A.C.; Schmitz, J.; Bronneke, H.S.; et al. Signaling by il-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat. Immunol. 2014, 15, 423–430. [Google Scholar] [CrossRef]
- Hogdall, D.; Lewinska, M.; Andersen, J.B. Desmoplastic tumor microenvironment and immunotherapy in cholangiocarcinoma. Trends Cancer 2018, 4, 239–255. [Google Scholar] [CrossRef]
- Zhou, S.L.; Dai, Z.; Zhou, Z.J.; Chen, Q.; Wang, Z.; Xiao, Y.S.; Hu, Z.Q.; Huang, X.Y.; Yang, G.H.; Shi, Y.H.; et al. Cxcl5 contributes to tumor metastasis and recurrence of intrahepatic cholangiocarcinoma by recruiting infiltrative intratumoral neutrophils. Carcinogenesis 2014, 35, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Cadamuro, M.; Brivio, S.; Spirli, C.; Joplin, R.E.; Strazzabosco, M.; Fabris, L. Autocrine and paracrine mechanisms promoting chemoresistance in cholangiocarcinoma. Int. J. Mol. Sci. 2017, 18, 149. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Malato, Y.; Calvisi, D.F.; Naqvi, S.; Razumilava, N.; Ribback, S.; Gores, G.J.; Dombrowski, F.; Evert, M.; Chen, X.; et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Investig. 2012, 122, 2911–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekiya, S.; Suzuki, A. Intrahepatic cholangiocarcinoma can arise from notch-mediated conversion of hepatocytes. J. Clin. Investig. 2012, 122, 3914–3918. [Google Scholar] [CrossRef] [PubMed]
- Thongsom, S.; Suginta, W.; Lee, K.J.; Choe, H.; Talabnin, C. Piperlongumine induces g2/m phase arrest and apoptosis in cholangiocarcinoma cells through the ros-jnk-erk signaling pathway. Apoptosis 2017, 22, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Huntzicker, E.G.; Hotzel, K.; Choy, L.; Che, L.; Ross, J.; Pau, G.; Sharma, N.; Siebel, C.W.; Chen, X.; French, D.M. Differential effects of targeting notch receptors in a mouse model of liver cancer. Hepatology 2015, 61, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Greenhill, C. Liver cancer: Different effects of the notch receptors in liver cancer revealed. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 703. [Google Scholar] [PubMed]
- Wu, W.R.; Zhang, R.; Shi, X.D.; Zhu, M.S.; Xu, L.B.; Zeng, H.; Liu, C. Notch1 is overexpressed in human intrahepatic cholangiocarcinoma and is associated with its proliferation, invasiveness and sensitivity to 5-fluorouracil in vitro. Oncol. Rep. 2014, 31, 2515–2524. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Zhou, J.; Fu, J.; He, T.; Qin, J.; Wang, L.; Liao, L.; Xu, J. Phosphorylation of serine 68 of twist1 by mapks stabilizes twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 2011, 71, 3980–3990. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate mmp-3-induced emt and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Dolado, I.; Swat, A.; Ajenjo, N.; De Vita, G.; Cuadrado, A.; Nebreda, A.R. P38alpha map kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell 2007, 11, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Cuenda, A.; Rousseau, S. P38 map-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef]
- Papa, S.; Bubici, C.; Zazzeroni, F.; Pham, C.G.; Kuntzen, C.; Knabb, J.R.; Dean, K.; Franzoso, G. The nf-kappab-mediated control of the jnk cascade in the antagonism of programmed cell death in health and disease. Cell Death Differ. 2006, 13, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote tnfalpha-induced death and sustained jnk activation by inhibiting map kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. Integrating cell-signalling pathways with nf-kappab and ikk function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef]
- Saccani, S.; Pantano, S.; Natoli, G. P38-dependent marking of inflammatory genes for increased nf-kappa b recruitment. Nat. Immunol. 2002, 3, 69–75. [Google Scholar] [CrossRef]
- Kojima, T.; Fuchimoto, J.; Yamaguchi, H.; Ito, T.; Takasawa, A.; Ninomiya, T.; Kikuchi, S.; Ogasawara, N.; Ohkuni, T.; Masaki, T.; et al. C-jun n-terminal kinase is largely involved in the regulation of tricellular tight junctions via tricellulin in human pancreatic duct epithelial cells. J. Cell Physiol. 2010, 225, 720–733. [Google Scholar] [CrossRef]
- Xia, Y.; Karin, M. The control of cell motility and epithelial morphogenesis by jun kinases. Trends Cell Biol. 2004, 14, 94–101. [Google Scholar] [CrossRef]
- Kamiya, A.; Gonzalez, F.J. Tnf-alpha regulates mouse fetal hepatic maturation induced by oncostatin m and extracellular matrices. Hepatology 2004, 40, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.E.; Ashkenazi, A. Tumor necrosis factor: An apoptosis junkie? Cell 2004, 116, 491–497. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Reactive oxygen species in tnfalpha-induced signaling and cell death. Mol. Cells 2010, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Seubwai, W.; Wongkham, C.; Puapairoj, A.; Khuntikeo, N.; Pugkhem, A.; Hahnvajanawong, C.; Chaiyagool, J.; Umezawa, K.; Okada, S.; Wongkham, S. Aberrant expression of nf-kappab in liver fluke associated cholangiocarcinoma: Implications for targeted therapy. PLoS ONE 2014, 9, e106056. [Google Scholar]
- Sirica, A.E. The role of cancer-associated myofibroblasts in intrahepatic cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 9, 44–54. [Google Scholar] [CrossRef]
- Claperon, A.; Mergey, M.; Aoudjehane, L.; Ho-Bouldoires, T.H.; Wendum, D.; Prignon, A.; Merabtene, F.; Firrincieli, D.; Desbois-Mouthon, C.; Scatton, O.; et al. Hepatic myofibroblasts promote the progression of human cholangiocarcinoma through activation of epidermal growth factor receptor. Hepatology 2013, 58, 2001–2011. [Google Scholar] [CrossRef]
- Claperon, A.; Guedj, N.; Mergey, M.; Vignjevic, D.; Desbois-Mouthon, C.; Boissan, M.; Saubamea, B.; Paradis, V.; Housset, C.; Fouassier, L. Loss of ebp50 stimulates egfr activity to induce emt phenotypic features in biliary cancer cells. Oncogene 2012, 31, 1376–1388. [Google Scholar] [CrossRef]
- Endo, K.; Yoon, B.I.; Pairojkul, C.; Demetris, A.J.; Sirica, A.E. Erbb-2 overexpression and cyclooxygenase-2 up-regulation in human cholangiocarcinoma and risk conditions. Hepatology 2002, 36, 439–450. [Google Scholar] [CrossRef]
- Han, C.; Leng, J.; Demetris, A.J.; Wu, T. Cyclooxygenase-2 promotes human cholangiocarcinoma growth: Evidence for cyclooxygenase-2-independent mechanism in celecoxib-mediated induction of p21waf1/cip1 and p27kip1 and cell cycle arrest. Cancer Res. 2004, 64, 1369–1376. [Google Scholar] [CrossRef]
- Wehbe, H.; Henson, R.; Meng, F.; Mize-Berge, J.; Patel, T. Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res. 2006, 66, 10517–10524. [Google Scholar] [CrossRef]
- Sae-Lao, T.; Tohtong, R.; Bates, D.O.; Wongprasert, K. Sulfated galactans from red seaweed gracilaria fisheri target egfr and inhibit cholangiocarcinoma cell proliferation. Am. J. Chin. Med. 2017, 45, 615–633. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.K.; Subimerb, C.; Pairojkul, C.; Wongkham, S.; Cutcutache, I.; Yu, W.; McPherson, J.R.; Allen, G.E.; Ng, C.C.; Wong, B.H.; et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat. Genet. 2012, 44, 690–693. [Google Scholar] [CrossRef]
- Jiao, Y.; Pawlik, T.M.; Anders, R.A.; Selaru, F.M.; Streppel, M.M.; Lucas, D.J.; Niknafs, N.; Guthrie, V.B.; Maitra, A.; Argani, P.; et al. Exome sequencing identifies frequent inactivating mutations in bap1, arid1a and pbrm1 in intrahepatic cholangiocarcinomas. Nat. Genet. 2013, 45, 1470–1473. [Google Scholar] [CrossRef] [PubMed]
- Macias, R.I.R.; Banales, J.M.; Sangro, B.; Muntane, J.; Avila, M.A.; Lozano, E.; Perugorria, M.J.; Padillo, F.J.; Bujanda, L.; Marin, J.J.G. The search for novel diagnostic and prognostic biomarkers in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Borad, M.J.; Patel, T.; Gores, G.J. Cholangiocarcinoma: Molecular pathways and therapeutic opportunities. Semin. Liver Dis. 2014, 34, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Wang, K.; Gay, L.; Al-Rohil, R.; Rand, J.V.; Jones, D.M.; Lee, H.J.; Sheehan, C.E.; Otto, G.A.; Palmer, G.; et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 2014, 19, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct idh-mutant molecular profiles. Cell Rep. 2017, 18, 2780–2794. [Google Scholar] [CrossRef]
- Churi, C.R.; Shroff, R.; Wang, Y.; Rashid, A.; Kang, H.C.; Weatherly, J.; Zuo, M.; Zinner, R.; Hong, D.; Meric-Bernstam, F.; et al. Mutation profiling in cholangiocarcinoma: Prognostic and therapeutic implications. PLoS ONE 2014, 9, e115383. [Google Scholar] [CrossRef]
- Andersen, J.B.; Spee, B.; Blechacz, B.R.; Avital, I.; Komuta, M.; Barbour, A.; Conner, E.A.; Gillen, M.C.; Roskams, T.; Roberts, L.R.; et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012, 142, 1021–1031 e1015. [Google Scholar] [CrossRef]
- Chen, T.C.; Jan, Y.Y.; Yeh, T.S. K-ras mutation is strongly associated with perineural invasion and represents an independent prognostic factor of intrahepatic cholangiocarcinoma after hepatectomy. Ann. Surg. Oncol. 2012, 19 (Suppl. 3), S675–S681. [Google Scholar] [CrossRef]
- Isa, T.; Tomita, S.; Nakachi, A.; Miyazato, H.; Shimoji, H.; Kusano, T.; Muto, Y.; Furukawa, M. Analysis of microsatellite instability, k-ras gene mutation and p53 protein overexpression in intrahepatic cholangiocarcinoma. Hepatogastroenterology 2002, 49, 604–608. [Google Scholar] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Al-Diffhala, S.; Centeno, B.A. Primary liver cancers-part 1: Histopathology, differential diagnoses, and risk stratification. Cancer Control 2018, 25, 1073274817744625. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.S.; Yeh, M.M. The use of immunohistochemistry in liver tumors. Clin. Liver Dis. 2010, 14, 687–703. [Google Scholar] [CrossRef] [PubMed]
- Bioulac-Sage, P.; Cubel, G.; Taouji, S.; Scoazec, J.Y.; Leteurtre, E.; Paradis, V.; Sturm, N.; Nhieu, J.T.; Wendum, D.; Bancel, B.; et al. Immunohistochemical markers on needle biopsies are helpful for the diagnosis of focal nodular hyperplasia and hepatocellular adenoma subtypes. Am. J. Surg. Pathol. 2012, 36, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Han, C.P.; Hsu, J.D.; Koo, C.L.; Yang, S.F. Antibody to cytokeratin (ck8/ck18) is not derived from cam5.2 clone, and anticytokeratin cam5.2 (becton dickinson) is not synonymous with the antibody (ck8/ck18). Hum. Pathol. 2010, 41, 616–617, author reply 617. [Google Scholar] [CrossRef]
- Razumilava, N.; Gores, G.J. Notch-driven carcinogenesis: The merging of hepatocellular cancer and cholangiocarcinoma into a common molecular liver cancer subtype. J. Hepatol. 2013, 58, 1244–1245. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, H.; Kuroki, T.; Kitasato, A.; Adachi, T.; Tanaka, T.; Hirabaru, M.; Hirayama, T.; Kuroshima, N.; Hidaka, M.; Soyama, A.; et al. Sox9 expression in carcinogenesis and its clinical significance in intrahepatic cholangiocarcinoma. Dig. Liver Dis. 2015, 47, 1067–1075. [Google Scholar] [CrossRef]
- Ferrone, C.R.; Ting, D.T.; Shahid, M.; Konstantinidis, I.T.; Sabbatino, F.; Goyal, L.; Rice-Stitt, T.; Mubeen, A.; Arora, K.; Bardeesey, N.; et al. The ability to diagnose intrahepatic cholangiocarcinoma definitively using novel branched DNA-enhanced albumin rna in situ hybridization technology. Ann. Surg. Oncol. 2016, 23, 290–296. [Google Scholar] [CrossRef]
- Macias, R.I.R.; Kornek, M.; Rodrigues, P.M.; Paiva, N.A.; Castro, R.E.; Urban, S.; Pereira, S.P.; Cadamuro, M.; Rupp, C.; Loosen, S.H.; et al. Diagnostic and prognostic biomarkers in cholangiocarcinoma. Liver Int. 2019, 39, 108–122. [Google Scholar] [CrossRef] [Green Version]
- Forner, A.; Vidili, G.; Rengo, M.; Bujanda, L.; Ponz-Sarvise, M.; Lamarca, A. Clinical presentation, diagnosis and staging of cholangiocarcinoma. Liver Int. 2019, 39, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarnagin, W.R.; Fong, Y.; DeMatteo, R.P.; Gonen, M.; Burke, E.C.; Bodniewicz, B.J.; Youssef, B.M.; Klimstra, D.; Blumgart, L.H. Staging, resectability, and outcome in 225 patients with hilar cholangiocarcinoma. Ann. Surg. 2001, 234, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Nagino, M.; Ebata, T.; Yokoyama, Y.; Igami, T.; Sugawara, G.; Takahashi, Y.; Nimura, Y. Evolution of surgical treatment for perihilar cholangiocarcinoma: A single-center 34-year review of 574 consecutive resections. Ann. Surg. 2013, 258, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Endo, I.; Gonen, M.; Yopp, A.C.; Dalal, K.M.; Zhou, Q.; Klimstra, D.; D’Angelica, M.; DeMatteo, R.P.; Fong, Y.; Schwartz, L.; et al. Intrahepatic cholangiocarcinoma: Rising frequency, improved survival, and determinants of outcome after resection. Ann. Surg. 2008, 248, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.M.; Jarnagin, W.R.; Klimstra, D.; DeMatteo, R.P.; Fong, Y.; Blumgart, L.H. Intrahepatic cholangiocarcinoma: Resectability, recurrence pattern, and outcomes. J. Am. Coll. Surg. 2001, 193, 384–391. [Google Scholar] [CrossRef]
- Blechacz, B. Cholangiocarcinoma: Current knowledge and new developments. Gut Liver 2017, 11, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Darwish Murad, S.; Kim, W.R.; Therneau, T.; Gores, G.J.; Rosen, C.B.; Martenson, J.A.; Alberts, S.R.; Heimbach, J.K. Predictors of pretransplant dropout and posttransplant recurrence in patients with perihilar cholangiocarcinoma. Hepatology 2012, 56, 972–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, J.C.; Jones, C.M.; Duffy, J.P.; Petrowsky, H.; Farmer, D.G.; French, S.; Finn, R.; Durazo, F.A.; Saab, S.; Tong, M.J.; et al. Comparative analysis of resection and liver transplantation for intrahepatic and hilar cholangiocarcinoma: A 24-year experience in a single center. Arch. Surg. 2011, 146, 683–689. [Google Scholar] [CrossRef]
- Mansour, J.C.; Aloia, T.A.; Crane, C.H.; Heimbach, J.K.; Nagino, M.; Vauthey, J.N. Hilar cholangiocarcinoma: Expert consensus statement. HPB 2015, 17, 691–699. [Google Scholar]
- Song, S.C.; Choi, D.W.; Kow, A.W.; Choi, S.H.; Heo, J.S.; Kim, W.S.; Kim, M.J. Surgical outcomes of 230 resected hilar cholangiocarcinoma in a single centre. ANZ J. Surg. 2013, 83, 268–274. [Google Scholar] [CrossRef]
- Cardinale, V.; Carpino, G.; Reid, L.; Gaudio, E.; Alvaro, D. Multiple cells of origin in cholangiocarcinoma underlie biological, epidemiological and clinical heterogeneity. World J. Gastrointest. Oncol. 2012, 4, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Nakanuma, Y.; Sato, Y.; Harada, K.; Sasaki, M.; Xu, J.; Ikeda, H. Pathological classification of intrahepatic cholangiocarcinoma based on a new concept. World J. Hepatol. 2010, 2, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Kim, K.P.; Park, S.; Chang, H.M. Comparison of gemcitabine plus cisplatin versus capecitabine plus cisplatin as first-line chemotherapy for advanced biliary tract cancer. Asia Pac. J. Clin. Oncol. 2017, 13, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Lubner, S.J.; Mahoney, M.R.; Kolesar, J.L.; Loconte, N.K.; Kim, G.P.; Pitot, H.C.; Philip, P.A.; Picus, J.; Yong, W.P.; Horvath, L.; et al. Report of a multicenter phase ii trial testing a combination of biweekly bevacizumab and daily erlotinib in patients with unresectable biliary cancer: A phase ii consortium study. J. Clin. Oncol. 2010, 28, 3491–3497. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, S.H.; Chang, H.M.; Kim, J.S.; Choi, H.J.; Lee, M.A.; Jang, J.S.; Jeung, H.C.; Kang, J.H.; Lee, H.W.; et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2012, 13, 181–188. [Google Scholar] [CrossRef]
- Crane, C.H.; Koay, E.J. Solutions that enable ablative radiotherapy for large liver tumors: Fractionated dose painting, simultaneous integrated protection, motion management, and computed tomography image guidance. Cancer 2016, 122, 1974–1986. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.S.; Wo, J.Y.; Yeap, B.Y.; Ben-Josef, E.; McDonnell, E.I.; Blaszkowsky, L.S.; Kwak, E.L.; Allen, J.N.; Clark, J.W.; Goyal, L.; et al. Multi-institutional phase ii study of high-dose hypofractionated proton beam therapy in patients with localized, unresectable hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J. Clin. Oncol. 2016, 34, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Ragab, O.; Kamrava, M. Another solution that enables ablative radiotherapy for large liver tumors: Percutaneous interstitial high-dose rate brachytherapy. Cancer 2016, 122, 2766. [Google Scholar] [CrossRef]
- Mukewar, S.; Gupta, A.; Baron, T.H.; Gores, G.; Furutani, K.; Haddock, M.G.; Hallemeier, C.L. Endoscopically inserted nasobiliary catheters for high dose-rate brachytherapy as part of neoadjuvant therapy for perihilar cholangiocarcinoma. Endoscopy 2015, 47, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase ii study of bgj398 in patients with fgfr-altered advanced cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Guagnano, V.; Kauffmann, A.; Wohrle, S.; Stamm, C.; Ito, M.; Barys, L.; Pornon, A.; Yao, Y.; Li, F.; Zhang, Y.; et al. Fgfr genetic alterations predict for sensitivity to nvp-bgj398, a selective pan-fgfr inhibitor. Cancer Discov. 2012, 2, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Yamada, D.; Hirsova, P.; Bronk, S.F.; Werneburg, N.W.; Krishnan, A.; Salim, W.; Zhang, L.; Trushina, E.; Truty, M.J.; et al. A hippo and fibroblast growth factor receptor autocrine pathway in cholangiocarcinoma. J. Biol. Chem. 2016, 291, 8031–8047. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Bahleda, R.; Dienstmann, R.; Infante, J.R.; Mita, A.; Italiano, A.; Calvo, E.; Moreno, V.; Adamo, B.; Gazzah, A.; et al. Phase i dose-escalation study of jnj-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 3401–3408. [Google Scholar] [CrossRef]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Ewald, F.; Norz, D.; Grottke, A.; Hofmann, B.T.; Nashan, B.; Jucker, M. Dual inhibition of pi3k-akt-mtor- and raf-mek-erk-signaling is synergistic in cholangiocarcinoma and reverses acquired resistance to mek-inhibitors. Investig. New Drugs 2014, 32, 1144–1154. [Google Scholar] [CrossRef]
- Finn, R.S.; Ahn, D.H.; Javle, M.M.; Tan, B.R., Jr.; Weekes, C.D.; Bendell, J.C.; Patnaik, A.; Khan, G.N.; Laheru, D.; Chavira, R.; et al. Phase 1b investigation of the mek inhibitor binimetinib in patients with advanced or metastatic biliary tract cancer. Investig. New Drugs 2018, 36, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Song, X.; Utpatel, K.; Shang, R.; Yang, Y.M.; Xu, M.; Zhang, J.; Che, L.; Gordan, J.; Cigliano, A.; et al. Mek inhibition suppresses k-ras wild-type cholangiocarcinoma in vitro and in vivo via inhibiting cell proliferation and modulating tumor microenvironment. Cell Death Dis. 2019, 10, 120. [Google Scholar] [CrossRef]
- Goyal, L.; Zheng, H.; Yurgelun, M.B.; Abrams, T.A.; Allen, J.N.; Cleary, J.M.; Knowles, M.; Regan, E.; Reardon, A.; Khachatryan, A.; et al. A phase 2 and biomarker study of cabozantinib in patients with advanced cholangiocarcinoma. Cancer 2017, 123, 1979–1988. [Google Scholar] [CrossRef] [Green Version]
- Shroff, R.T.; Yarchoan, M.; O’Connor, A.; Gallagher, D.; Zahurak, M.L.; Rosner, G.; Ohaji, C.; Sartorius-Mergenthaler, S.; Parkinson, R.; Subbiah, V.; et al. The oral vegf receptor tyrosine kinase inhibitor pazopanib in combination with the mek inhibitor trametinib in advanced cholangiocarcinoma. Br. J. Cancer 2017, 116, 1402–1407. [Google Scholar] [CrossRef]
- Yokoi, K.; Kobayashi, A.; Motoyama, H.; Kitazawa, M.; Shimizu, A.; Notake, T.; Yokoyama, T.; Matsumura, T.; Takeoka, M.; Miyagawa, S.I. Survival pathway of cholangiocarcinoma via akt/mtor signaling to escape raf/mek/erk pathway inhibition by sorafenib. Oncol. Rep. 2018, 39, 843–850. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Rankin, C.; Siegel, A.B.; Iqbal, S.; Gong, I.Y.; Micetich, K.C.; Kayaleh, O.R.; Lenz, H.J.; Blanke, C.D. S0941: A phase 2 swog study of sorafenib and erlotinib in patients with advanced gallbladder carcinoma or cholangiocarcinoma. Br. J. Cancer 2014, 110, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Lozano, E.; Macias, R.I.R.; Monte, M.J.; Asensio, M.; Del Carmen, S.; Sanchez-Vicente, L.; Alonso-Pena, M.; Al-Abdulla, R.; Munoz-Garrido, P.; Satriano, L.; et al. Causes of hoct1-dependent cholangiocarcinoma resistance to sorafenib and sensitization by tumor-selective gene therapy. Hepatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.G.; Lozano, E.; Herraez, E.; Asensio, M.; Di Giacomo, S.; Romero, M.R.; Briz, O.; Serrano, M.A.; Efferth, T.; Macias, R.I.R. Chemoresistance and chemosensitization in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, C.J.; Lafuente-Barquero, J.; Andersen, J.B. Epigenome remodeling in cholangiocarcinoma. Trends Cancer 2019, 5, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (idh)1 and idh2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted inhibition of mutant idh2 in leukemia cells induces cellular differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Dong, Q.; Zhang, C.; Kuan, P.F.; Liu, Y.; Jeck, W.R.; Andersen, J.B.; Jiang, W.; Savich, G.L.; Tan, T.X.; et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013, 32, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant idh1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef]
- Burris, H.; Mellinghoff, I.; Maher, E.; Wen, P.; Beeram, M.; Touat, M.; Faris, J.; Azad, N.; Cloughesy, T.; Gore, L.; et al. The first reported results of ag.120, a first.In.Class, potent inhibitor of the idh1 mutant protein, in a phase 1 study of patients with advanced idh1.Mutant solid tumors, including gliomas. Mol. Cancer. Ther. 2015, 14, PL04–05. [Google Scholar]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of ag-120 (ivosidenib): A first-in-class mutant idh1 inhibitor for the treatment of idh1 mutant cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct idh-mutant molecular profiles. Cell Rep. 2017, 19, 2878–2880. [Google Scholar] [CrossRef] [PubMed]
- Gani, F.; Nagarajan, N.; Kim, Y.; Zhu, Q.; Luan, L.; Bhaijjee, F.; Anders, R.A.; Pawlik, T.M. Program death 1 immune checkpoint and tumor microenvironment: Implications for patients with intrahepatic cholangiocarcinoma. Ann. Surg. Oncol. 2016, 23, 2610–2617. [Google Scholar] [CrossRef] [PubMed]
- Fontugne, J.; Augustin, J.; Pujals, A.; Compagnon, P.; Rousseau, B.; Luciani, A.; Tournigand, C.; Cherqui, D.; Azoulay, D.; Pawlotsky, J.M.; et al. Pd-l1 expression in perihilar and intrahepatic cholangiocarcinoma. Oncotarget 2017, 8, 24644–24651. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Doi, T.; De Braud, F.; Piha-Paul, S.; Hollebecque, A.; Razak, A.A.; Lin, C.C.; Ott, P.A.; He, A.R.; Yuan, S.S.; et al. Safety and efficacy of pembrolizumab (mk.3475) in patients (pts) with advanced biliary tract cancer: Interim results of keynote.028 [abstract]. Eur. J. Cancer 2015, 51 (Suppl. 3), S122. [Google Scholar] [CrossRef]
- Fan, B.; Mellinghoff, I.K.; Wen, P.Y.; Lowery, M.A.; Goyal, L.; Tap, W.D.; Pandya, S.S.; Manyak, E.; Jiang, L.; Liu, G.; et al. Clinical pharmacokinetics and pharmacodynamics of ivosidenib, an oral, targeted inhibitor of mutant idh1, in patients with advanced solid tumors. Investig. New Drugs 2019, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Valade, E.; Dosne, A.G.; Xie, H.; Kleiman, R.; Li, L.Y.; Perez-Ruixo, J.J.; Ouellet, D. Assessment of the effect of erdafitinib on cardiac safety: Analysis of ecgs and exposure-qtc in patients with advanced or refractory solid tumors. Cancer Chemother. Pharmacol. 2019, 84, 621–633. [Google Scholar] [CrossRef]
- Borad, M.J.; Gores, G.J.; Roberts, L.R. Fibroblast growth factor receptor 2 fusions as a target for treating cholangiocarcinoma. Curr. Opin. Gastroenterol. 2015, 31, 264–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R., Jr. Ros1 protein-tyrosine kinase inhibitors in the treatment of ros1 fusion protein-driven non-small cell lung cancers. Pharmacol. Res. 2017, 121, 202–212. [Google Scholar] [CrossRef]
- Sigal, D.; Tartar, M.; Xavier, M.; Bao, F.; Foley, P.; Luo, D.; Christiansen, J.; Hornby, Z.; Maneval, E.C.; Multani, P. Activity of entrectinib in a patient with the first reported ntrk fusion in neuroendocrine cancer. J. Natl. Compr. Cancer Netw. 2017, 15, 1317–1322. [Google Scholar] [CrossRef]
- Huang, C.Y.; Hsieh, F.S.; Wang, C.Y.; Chen, L.J.; Chang, S.S.; Tsai, M.H.; Hung, M.H.; Kuo, C.W.; Shih, C.T.; Chao, T.I.; et al. Palbociclib enhances radiosensitivity of hepatocellular carcinoma and cholangiocarcinoma via inhibiting ataxia telangiectasia-mutated kinase-mediated DNA damage response. Eur. J. Cancer 2018, 102, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.C.; Ros, W.; Delord, J.P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and safety of pembrolizumab in previously treated advanced cervical cancer: Results from the phase ii keynote-158 study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhang, B.; Liang, H.; Lu, Y.; Ai, X.; Zhang, B.; Chen, X. Jnk inhibitor sp600125 enhances tgf-beta-induced apoptosis of rbe human cholangiocarcinoma cells in a smad-dependent manner. Mol. Med. Rep. 2013, 8, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Waetzig, V.; Herdegen, T. Context-specific inhibition of jnks: Overcoming the dilemma of protection and damage. Trends Pharmacol. Sci. 2005, 26, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Bost, F.; Charbono, W.; Dean, N.; McKay, R.; Rhim, J.S.; Depatie, C.; Mercola, D. C-jun nh(2)-terminal kinase mediates proliferation and tumor growth of human prostate carcinoma. Clin. Cancer Res. 2003, 9, 391–401. [Google Scholar]
- Chromik, A.M.; Muller, A.M.; Korner, J.; Belyaev, O.; Holland-Letz, T.; Schmitz, F.; Herdegen, T.; Uhl, W.; Mittelkotter, U. Genetic deletion of jnk1 and jnk2 aggravates the dss-induced colitis in mice. J. Investig. Surg. 2007, 20, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Harris, R.; Coloff, J.L.; Jin, J.Y.; Leshin, B.; Miliani de Marval, P.; Tao, S.; Rathmell, J.C.; Hall, R.P.; Zhang, J.Y. The c-jun nh2-terminal kinase 2 plays a dominant role in human epidermal neoplasia. Cancer Res. 2010, 70, 3080–3088. [Google Scholar] [CrossRef]
- Yoon, C.H.; Kim, M.J.; Kim, R.K.; Lim, E.J.; Choi, K.S.; An, S.; Hwang, S.G.; Kang, S.G.; Suh, Y.; Park, M.J.; et al. C-jun n-terminal kinase has a pivotal role in the maintenance of self-renewal and tumorigenicity in glioma stem-like cells. Oncogene 2012, 31, 4655–4666. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Oncogenes | Tumor Suppressor Genes | Chromatin-Remodeling Genes | Gain of Function of Oncogenes |
|---|---|---|---|
| MLL3 | TP53 | ARID1A | KRAS |
| ROBO2 | PTEN | ARID1B | BRAF |
| RNF43 | BAP1 | PIK3CA | |
| PEG3 | PBRM1 | ||
| GNAS | |||
| NRAS | |||
| PTPN3 | |||
| CDKN2A | |||
| SMAD4 | |||
| IDH1/2 |
| DRUG | Target | Phase | Identifier |
|---|---|---|---|
| AG-120 [226] | IDH1 | I | NCT02073994 |
| Enasidenib [12] | IDH2 | I/II | NCT02273739 |
| JNJ-42756493 [227] | FGFR | I | NCT01703481 |
| BGJ398+ PD173072 [165,202,205] | FGFR | I | NCT01004224 |
| NVP-BGJ398 [201,202] | FGFR | II | NCT02150967 |
| Ponatinib [228] | FGFR | II | NCT02265341 |
| Ceritinib [229] | ROS1-ALK | II | NCT02638909 |
| LDK378 (Ceritinib) [229] | ROS2-ALK | II | NCT02374489 |
| Entrectinib [230] | ROS3-ALK | II | NCT02568267 |
| Binimetinib (MEK162) [207] | MEK1/2 | I | NCT00959127 |
| Trametinib and Pazopanib [210] | MEK/VEGFR | II | NCT01438554 |
| Cabozantinib [209] | c-MET-VEGFR2 | II | NCT01954745 |
| Cisplatin + gemcitabine [193] | EGFR | III | NCT00262769 |
| Gemcitabine/Oxaliplatin + Erlotinib [196] | EGFR | III | NCT01149122 |
| Sorafenib [212] | VEGFR-PDGFR-BRAF | II | NCT00238212 |
| Anetumab ravtansine [12] | anti-mesothelin antibody-drug conjugate | I | NCT03102320 |
| Palbociclib [231] | CDK4/6 Inhibitor | I/II | NCT03065062 |
| Pembrolizumab + GM-CSF [224] | PD1 | II | NCT02703714 |
| Pembrolizumab [232] | PD1 | II | NCT02628067 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Nelson, L.J.; Ávila, M.A.; Cubero, F.J. Mitogen-Activated Protein Kinases (MAPKs) and Cholangiocarcinoma: The Missing Link. Cells 2019, 8, 1172. https://doi.org/10.3390/cells8101172
Chen C, Nelson LJ, Ávila MA, Cubero FJ. Mitogen-Activated Protein Kinases (MAPKs) and Cholangiocarcinoma: The Missing Link. Cells. 2019; 8(10):1172. https://doi.org/10.3390/cells8101172
Chicago/Turabian StyleChen, Chaobo, Leonard J. Nelson, Matías A. Ávila, and Francisco Javier Cubero. 2019. "Mitogen-Activated Protein Kinases (MAPKs) and Cholangiocarcinoma: The Missing Link" Cells 8, no. 10: 1172. https://doi.org/10.3390/cells8101172
APA StyleChen, C., Nelson, L. J., Ávila, M. A., & Cubero, F. J. (2019). Mitogen-Activated Protein Kinases (MAPKs) and Cholangiocarcinoma: The Missing Link. Cells, 8(10), 1172. https://doi.org/10.3390/cells8101172