New Frontiers: ARID3a in SLE


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
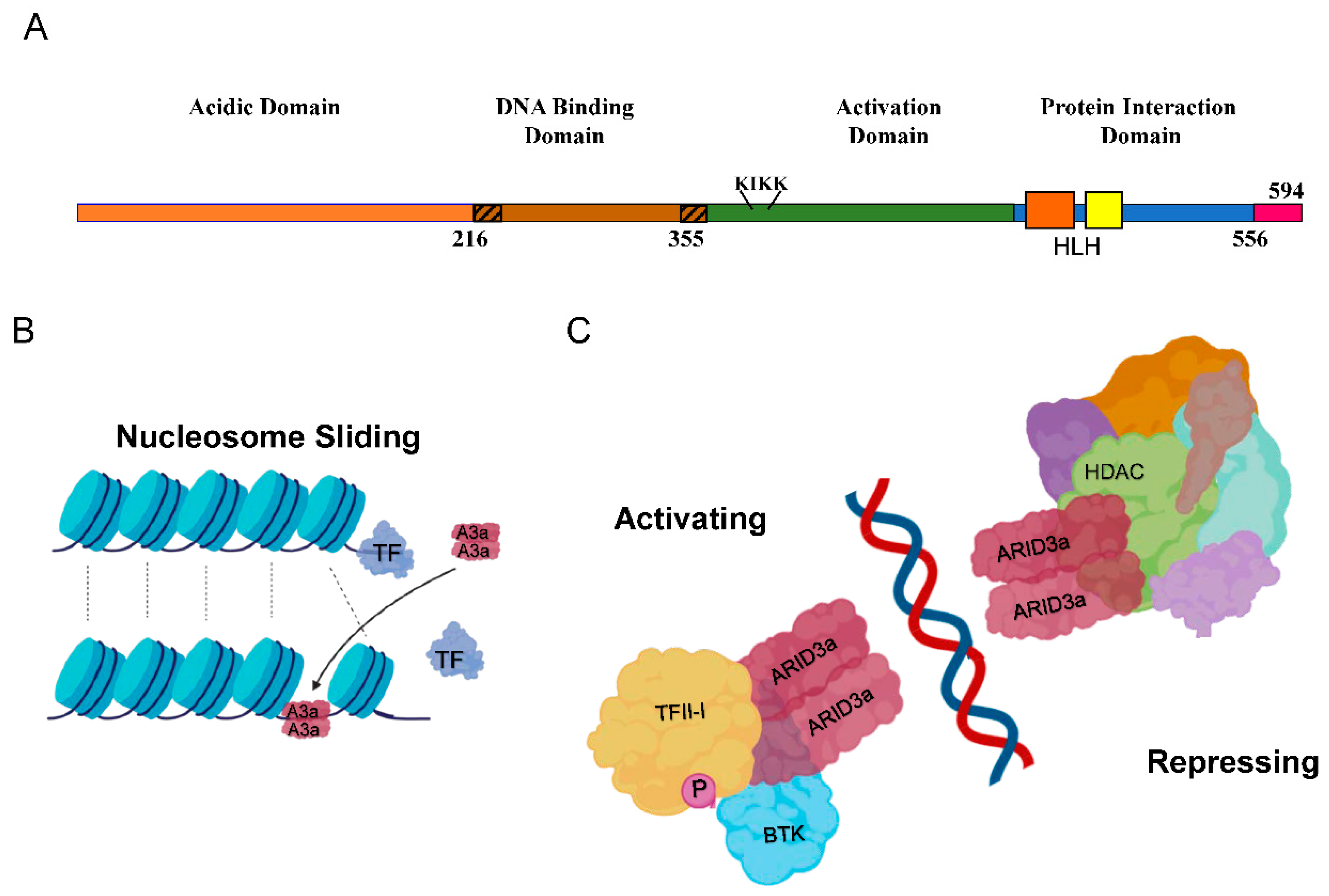
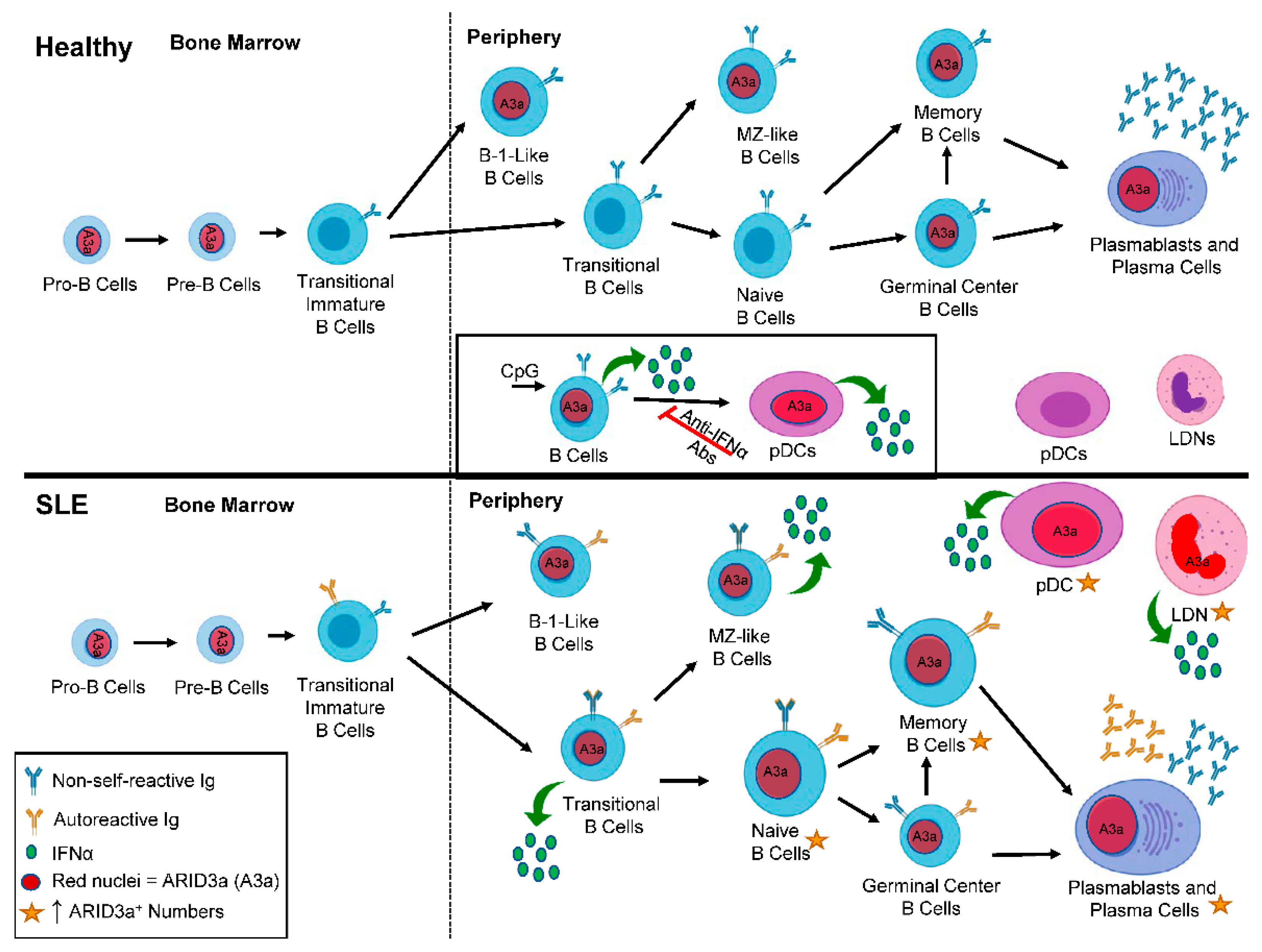
2. ARID3a and Autoantibodies in SLE
3. ARID3a and IFNα in SLE
4. ARID3a and Nephritis
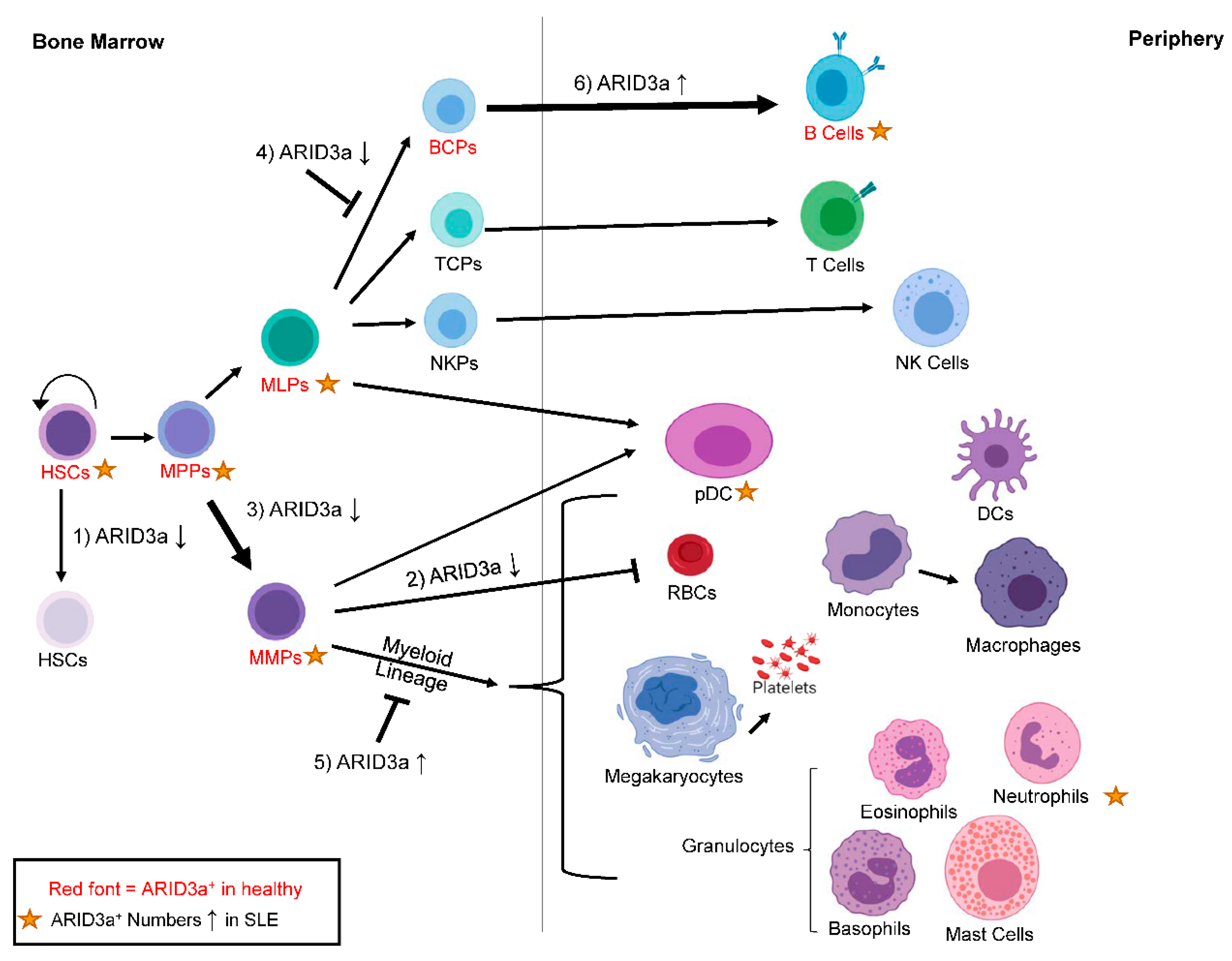
5. ARID3a and Hematopoiesis
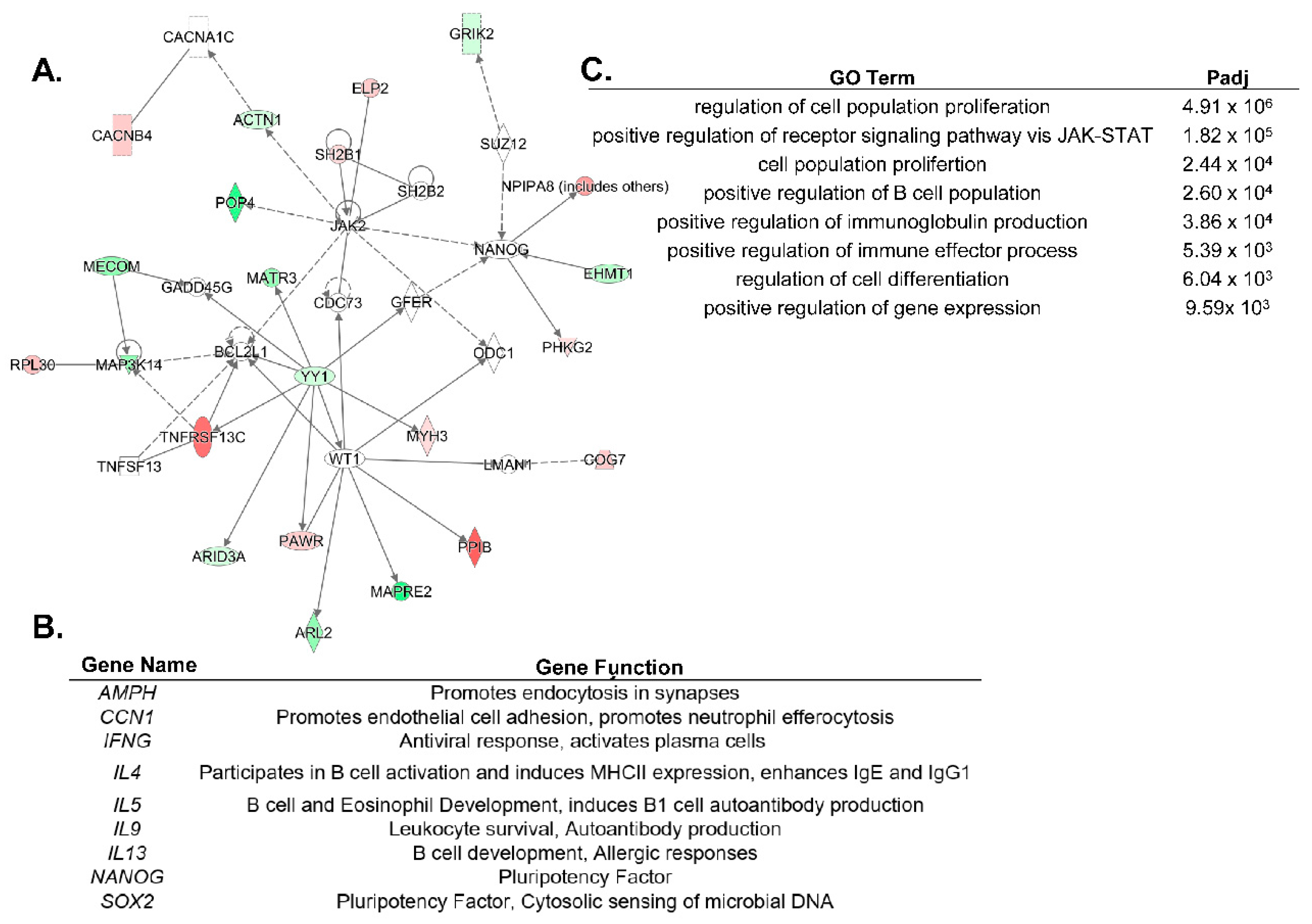
6. Regulation of ARID3a Expression
7. Clinical Implications
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carter, E.E.; Barr, S.G.; Clarke, A.E. The global burden of SLE: Prevalence, health disparities and socioeconomic impact. Nat. Rev. Rheumatol. 2016, 12, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Bombardier, C.; Gladman, D.D.; Urowitz, M.B.; Caron, D.; Chang, C.H. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum. 1992, 35, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.D.; Ibanez, D.; Urowitz, M.B. Systemic lupus erythematosus disease activity index 2000. J. Rheumatol. 2002, 29, 288–291. [Google Scholar] [PubMed]
- Golbus, J.; McCune, W.J. Lupus nephritis. Classification, prognosis, immunopathogenesis, and treatment. Rheum. Dis. Clin. N. Am. 1994, 20, 213–242. [Google Scholar]
- Contreras, G.; Lenz, O.; Pardo, V.; Borja, E.; Cely, C.; Iqbal, K.; Nahar, N.; de La Cuesta, C.; Hurtado, A.; Fornoni, A.; et al. Outcomes in African Americans and Hispanics with lupus nephritis. Kidney Int. 2006, 69, 1846–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lech, M.; Anders, H.J. The pathogenesis of lupus nephritis. J. Am. Soc. Nephrol. 2013, 24, 1357–1366. [Google Scholar] [CrossRef]
- Webb, C.F.; Das, C.; Eneff, K.L.; Tucker, P.W. Identification of a matrix-associated region 5′ of an immunoglobulin heavy chain variable region gene. Mol. Cell. Biol. 1991, 11, 5206–5211. [Google Scholar] [CrossRef]
- Webb, C.F.; Das, C.; Eaton, S.; Calame, K.; Tucker, P.W. Novel protein-DNA interactions associated with increased immunoglobulin transcription in response to antigen plus interleukin-5. Mol. Cell. Biol. 1991, 11, 5197–5205. [Google Scholar] [CrossRef]
- Webb, C.; Zong, R.T.; Lin, D.; Wang, Z.; Kaplan, M.; Paulin, Y.; Smith, E.; Probst, L.; Bryant, J.; Goldstein, A.; et al. Differential regulation of immunoglobulin gene transcription via nuclear matrix-associated regions. Cold Spring Harb. Symp. Quant. Biol. 1999, 64, 109–118. [Google Scholar] [CrossRef]
- Lin, D.; Ippolito, G.C.; Zong, R.T.; Bryant, J.; Koslovsky, J.; Tucker, P. Bright/ARID3A contributes to chromatin accessibility of the immunoglobulin heavy chain enhancer. Mol. Cancer 2007, 6, 23. [Google Scholar] [CrossRef]
- Herrscher, R.F.; Kaplan, M.H.; Lelsz, D.L.; Das, C.; Scheuermann, R.; Tucker, P.W. The immunoglobulin heavy-chain matrix-associating regions are bound by Bright: A B cell-specific trans-activator that describes a new DNA-binding protein family. Genes Dev. 1995, 9, 3067–3082. [Google Scholar] [CrossRef] [PubMed]
- Rajaiya, J.; Nixon, J.C.; Ayers, N.; Desgranges, Z.P.; Roy, A.L.; Webb, C.F. Induction of immunoglobulin heavy chain transcription through the transcription factor Bright requires TFII-I. Mol. Cell. Biol. 2006, 26, 4758–4768. [Google Scholar] [CrossRef] [PubMed]
- Rajaiya, J.; Hatfield, M.; Nixon, J.C.; Rawlings, D.J.; Webb, C.F. Bruton’s tyrosine kinase regulates immunoglobulin promoter activation in association with the transcription factor Bright. Mol. Cell. Biol. 2006, 25, 2073–2084. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.F.; Das, C.; Coffman, R.L.; Tucker, P.W. Induction of immunoglobulin mu mRNA in a B cell transfectant stimulated with interleukin-5 and a T-dependent antigen. J. Immunol. 1989, 143, 3934–3939. [Google Scholar]
- Kortschak, R.D.; Tucker, P.W.; Saint, R. ARID proteins come in from the desert. Trends Biochem. Sci. 2000, 25, 294–299. [Google Scholar] [CrossRef]
- Wilsker, D.; Patsialou, A.; Dallas, P.B.; Moran, E. ARID proteins: A diverse family of DNA binding proteins implicated in the control of cell growth, differentiation, and development. Cell Growth Differ. 2002, 13, 95–106. [Google Scholar] [PubMed]
- Wilsker, D.; Probst, L.; Wain, H.M.; Maltais, L.; Tucker, P.W.; Moran, E. Nomenclature of the ARID family of DNA-binding proteins. Genomics 2005, 86, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Popowski, M.; Templeton, T.D.; Lee, B.K.; Rhee, C.; Li, H.; Miner, C.; Dekker, J.D.; Orlanski, S.; Bergman, Y.; Iyer, V.R.; et al. Bright/Arid3A acts as a barrier to somatic cell reprogramming through direct regulation of Oct4, Sox2, and Nanog. Stem Cell Rep. 2014, 2, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Ratliff, M.L.; Templeton, T.D.; Ward, J.M.; Webb, C.F. The Bright Side of Hematopoiesis: Regulatory Roles of ARID3a/Bright in Human and Mouse Hematopoiesis. Front. Immunol. 2014, 5, 113. [Google Scholar] [CrossRef]
- Nixon, J.C.; Rajaiya, J.B.; Ayers, N.; Evetts, S.; Webb, C.F. The transcription factor, Bright, is not expressed in all human B lymphocyte subpopulations. Cell. Immunol. 2004, 228, 42–53. [Google Scholar] [CrossRef]
- Hayakawa, K.; Li, Y.S.; Shinton, S.A.; Bandi, S.R.; Formica, A.M.; Brill-Dashoff, J.; Hardy, R.R. Crucial Role of Increased Arid3a at the Pre-B and Immature B Cell Stages for B1a Cell Generation. Front. Immunol. 2019, 10, 457. [Google Scholar] [CrossRef] [PubMed]
- Meffre, E. The establishment of early B cell tolerance in humans: Lessons from primary immunodeficiency diseases. Ann. N. Y. Acad. Sci. 2011, 1246, 1–10. [Google Scholar] [CrossRef] [PubMed]
- De Groof, A.; Hemon, P.; Mignen, O.; Pers, J.O.; Wakeland, E.K.; Renaudineau, Y.; Lauwerys, B.R. Dysregulated Lymphoid Cell Populations in Mouse Models of Systemic Lupus Erythematosus. Clin. Rev. Allergy Immunol. 2017, 53, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Nixon, J.C.; Ferrell, S.; Miner, C.; Oldham, A.L.; Hochgeschwender, U.; Webb, C.F. Transgenic mice expressing dominant-negative bright exhibit defects in B1 B cells. J. Immunol. 2008, 181, 6913–6922. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.F.; Smith, E.A.; Medina, K.L.; Buchanan, K.L.; Smithson, G.; Dou, S. Expression of Bright at two distinct stages of B lymphocyte development. J. Immunol. 1998, 160, 4747–4754. [Google Scholar] [PubMed]
- Zhou, Y.; Li, Y.S.; Bandi, S.R.; Tang, L.; Shinton, S.A.; Hayakawa, K.; Hardy, R.R. Lin28b promotes fetal B lymphopoiesis through the transcription factor Arid3a. J. Exp. Med. 2015, 212, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Oldham, A.L.; Miner, C.A.; Wang, H.C.; Webb, C.F. The transcription factor Bright plays a role in marginal zone B lymphocyte development and autoantibody production. Mol. Immunol. 2011, 49, 367–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, M.; Nixon, J.C.; Maier, S.; Workman, J.; Farris, A.D.; Webb, C.F. Anti-nuclear antibody production and autoimmunity in transgenic mice that overexpress the transcription factor Bright. J. Immunol. 2007, 178, 2996–3006. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Li, J.; Wu, Q.; Yang, P.; Pawar, R.D.; Xie, S.; Timares, L.; Raman, C.; Chaplin, D.D.; Lu, L.; et al. Marginal zone precursor B cells as cellular agents for type I IFN-promoted antigen transport in autoimmunity. J. Immunol. 2010, 184, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Y.; Georg, I.; Diaz-Barreiro, A.; Varela, N.; Lauwerys, B.; Kumar, R.; Bagavant, H.; Castillo-Martin, M.; El Salem, F.; Maranon, C.; et al. Concordance of increased B1 cell subset and lupus phenotypes in mice and humans is dependent on BLK expression levels. J. Immunol. 2015, 194, 5692–5702. [Google Scholar] [CrossRef] [PubMed]
- Bendelac, A.; Bonneville, M.; Kearney, J.F. Autoreactivity by design: Innate B and T lymphocytes. Nat. Rev. Immunol. 2001, 1, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Molano-Gonzalez, N.; Rojas, M.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Rodriguez, Y.; Rodriguez-Jimenez, M.; Ramirez-Santana, C.; Anaya, J.M. Cluster analysis of autoimmune rheumatic diseases based on autoantibodies. New insights for polyautoimmunity. J. Autoimmun. 2019, 98, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Dieudonne, Y.; Gies, V.; Guffroy, A.; Keime, C.; Bird, A.K.; Liesveld, J.; Barnas, J.L.; Poindron, V.; Douiri, N.; Soulas-Sprauel, P.; et al. Transitional B cells in quiescent SLE: An early checkpoint imprinted by IFN. J. Autoimmun. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, H.M.; Thouvenel, C.D.; Leach, S.; Arkatkar, T.; Metzler, G.; Scharping, N.E.; Kolhatkar, N.S.; Rawlings, D.J.; Jackson, S.W. Cutting Edge: BAFF Promotes Autoantibody Production via TACI-Dependent Activation of Transitional B Cells. J. Immunol. 2016, 196, 3525–3531. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Kearney, J.F. Marginal-zone B cells. Nat. Rev. Immunol. 2002, 2, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.M.; Rose, K.; Montgomery, C.; Adrianto, I.; James, J.A.; Merrill, J.T.; Webb, C.F. Disease activity in systemic lupus erythematosus correlates with expression of the transcription factor AT-rich-interactive domain 3A. Arthritis Rheumatol. 2014, 66, 3404–3412. [Google Scholar] [CrossRef]
- Ward, J.M.; James, J.A.; Zhao, Y.D.; Webb, C.F. Antibody Reactivity of B Cells in Lupus Patients with Increased Disease Activity and ARID3a Expression. Antibodies 2015, 4, 354–368. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.M.; Ratliff, M.L.; Dozmorov, M.G.; Wiley, G.; Guthridge, J.M.; Gaffney, P.M.; James, J.A.; Webb, C.F. Human effector B lymphocytes express ARID3a and secrete interferon alpha. J. Autoimmun. 2016, 75, 130–140. [Google Scholar] [CrossRef] [Green Version]
- James, J.A.; Robertson, J.M. Lupus and Epstein-Barr. Curr. Opin. Rheumatol. 2012, 24, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Sundar, K.; Jacques, S.; Gottlieb, P.; Villars, R.; Benito, M.-E.; Taylor, D.K.; Spatz, L.A. Expression of the Epstein-Barr virus nuclear antigen-1 (EBNA-1) in the mouse can elicit the production of anti-dsDNA and anti-Sm antibodies. J. Autoimmun. 2004, 23, 127–140. [Google Scholar] [CrossRef]
- Borestrom, C.; Forsman, A.; Ruetschi, U.; Rymo, L. E2F1, ARID3A/Bright and Oct-2 factors bind to the Epstein-Barr virus C promoter, EBNA1 and oriP, participating in long-distance promoter-enhancer interactions. J. Gen. Virol. 2012, 93, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.M.; Ratliff, M.L.; Dozmorov, M.G.; Wiley, G.; Guthridge, J.M.; Gaffney, P.M.; James, J.A.; Webb, C.F. Expression and methylation data from SLE patient and healthy control blood samples subdivided with respect to ARID3a levels. Data Brief. 2016, 9, 213–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calame, K.; Atchison, M. YY1 helps to bring loose ends together. Genes Dev. 2007, 21, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Scharer, C.D.; Blalock, E.L.; Barwick, B.G.; Haines, R.R.; Wei, C.; Sanz, I.; Boss, J.M. ATAC-seq on biobanked specimens defines a unique chromatin accessibility structure in naive SLE B cells. Sci. Rep. 2016, 6, 27030. [Google Scholar] [CrossRef] [PubMed]
- Scharer, C.D.; Blalock, E.L.; Mi, T.; Barwick, B.G.; Jenks, S.A.; Deguchi, T.; Cashman, K.S.; Neary, B.E.; Patterson, D.G.; Hicks, S.L.; et al. Epigenetic programming underpins B cell dysfunction in human SLE. Nat. Immunol. 2019, 20, 1071–1082. [Google Scholar] [CrossRef]
- Nascimbeni, M.; Perie, L.; Chorro, L.; Diocou, S.; Kreitmann, L.; Louis, S.; Garderet, L.; Fabiani, B.; Berger, A.; Schmitz, J.; et al. Plasmacytoid dendritic cells accumulate in spleens from chronically HIV-infected patients but barely participate in interferon-alpha expression. Blood 2009, 113, 6112–6119. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef]
- Liu, Y.J. IPC: Professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [CrossRef]
- Ratliff, M.L.; Garton, J.; Garman, L.; Barron, M.D.; Georgescu, C.; White, K.A.; Chakravarty, E.; Wren, J.D.; Montgomery, C.G.; James, J.A.; et al. ARID3a gene profiles are strongly associated with human interferon alpha production. J. Autoimmun. 2019, 96, 158–167. [Google Scholar] [CrossRef]
- Denny, M.F.; Yalavarthi, S.; Zhao, W.; Thacker, S.G.; Anderson, M.; Sandy, A.R.; McCune, W.J.; Kaplan, M.J. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J. Immunol. 2010, 184, 3284–3297. [Google Scholar] [CrossRef]
- Lindau, D.; Mussard, J.; Rabsteyn, A.; Ribon, M.; Kotter, I.; Igney, A.; Adema, G.J.; Boissier, M.C.; Rammensee, H.G.; Decker, P. TLR9 independent interferon alpha production by neutrophils on NETosis in response to circulating chromatin, a key lupus autoantigen. Ann. Rheum. Dis. 2014, 73, 2199–2207. [Google Scholar] [CrossRef] [PubMed]
- Landolt-Marticorena, C.; Bonventi, G.; Lubovich, A.; Ferguson, C.; Unnithan, T.; Su, J.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Wither, J. Lack of association between the interferon-alpha signature and longitudinal changes in disease activity in systemic lupus erythematosus. Ann. Rheum. Dis. 2009, 68, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rivera, C.; Kaplan, M.J. Low-density granulocytes: A distinct class of neutrophils in systemic autoimmunity. Semin. Immunopathol. 2013, 35, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.H.; Homey, B.; Cao, W.; Wang, Y.H.; Su, B.; Nestle, F.O.; et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 2007, 449, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Gestermann, N.; Di Domizio, J.; Lande, R.; Demaria, O.; Frasca, L.; Feldmeyer, L.; Di Lucca, J.; Gilliet, M. Netting Neutrophils Activate Autoreactive B Cells in Lupus. J. Immunol. 2018, 200, 3364–3371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puga, I.; Cols, M.; Barra, C.M.; He, B.; Cassis, L.; Gentile, M.; Comerma, L.; Chorny, A.; Shan, M.; Xu, W.; et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat. Immunol. 2011, 13, 170–180. [Google Scholar] [CrossRef]
- Coquery, C.M.; Wade, N.S.; Loo, W.M.; Kinchen, J.M.; Cox, K.M.; Jiang, C.; Tung, K.S.; Erickson, L.D. Neutrophils contribute to excess serum BAFF levels and promote CD4+ T cell and B cell responses in lupus-prone mice. PLoS ONE 2014, 9, e102284. [Google Scholar] [CrossRef]
- Scapini, P.; Bazzoni, F.; Cassatella, M.A. Regulation of B-cell-activating factor (BAFF)/B lymphocyte stimulator (BLyS) expression in human neutrophils. Immunol. Lett. 2008, 116, 1–6. [Google Scholar] [CrossRef]
- Scapini, P.; Nardelli, B.; Nadali, G.; Calzetti, F.; Pizzolo, G.; Montecucco, C.; Cassatella, M.A. G-CSF-stimulated neutrophils are a prominent source of functional BLyS. J. Exp. Med. 2003, 197, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Parsa, R.; Lund, H.; Georgoudaki, A.M.; Zhang, X.M.; Ortlieb Guerreiro-Cacais, A.; Grommisch, D.; Warnecke, A.; Croxford, A.L.; Jagodic, M.; Becher, B.; et al. BAFF-secreting neutrophils drive plasma cell responses during emergency granulopoiesis. J. Exp. Med. 2016, 213, 1537–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marini, O.; Costa, S.; Bevilacqua, D.; Calzetti, F.; Tamassia, N.; Spina, C.; De Sabata, D.; Tinazzi, E.; Lunardi, C.; Scupoli, M.T.; et al. Mature CD10(+) and immature CD10(-) neutrophils present in G-CSF-treated donors display opposite effects on T cells. Blood 2017, 129, 1343–1356. [Google Scholar] [CrossRef] [PubMed]
- Palanichamy, A.; Bauer, J.W.; Yalavarthi, S.; Meednu, N.; Barnard, J.; Owen, T.; Cistrone, C.; Bird, A.; Rabinovich, A.; Nevarez, S.; et al. Neutrophil-mediated IFN activation in the bone marrow alters B cell development in human and murine systemic lupus erythematosus. J. Immunol. 2014, 192, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Hirano, K.; Ogino, H.; Ochi, H. Arid3a regulates nephric tubule regeneration via evolutionarily conserved regeneration signal-response enhancers. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Rhee, C.; Lee, B.K.; Beck, S.; Anjum, A.; Cook, K.R.; Popowski, M.; Tucker, H.O.; Kim, J. Arid3a is essential to execution of the first cell fate decision via direct embryonic and extraembryonic transcriptional regulation. Genes Dev. 2014, 28, 2219–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, C.; Edwards, M.; Dang, C.; Harris, J.; Brown, M.; Kim, J.; Tucker, H.O. ARID3A is required for mammalian placenta development. Dev. Biol. 2017, 422, 83–91. [Google Scholar] [CrossRef] [PubMed]
- An, G.; Miner, C.A.; Nixon, J.C.; Kincade, P.W.; Bryant, J.; Tucker, P.W.; Webb, C.F. Loss of Bright/ARID3a function promotes developmental plasticity. Stem Cells 2010, 28, 1560–1567. [Google Scholar] [CrossRef]
- Fairhurst, A.M.; Xie, C.; Fu, Y.; Wang, A.; Boudreaux, C.; Zhou, X.J.; Cibotti, R.; Coyle, A.; Connolly, J.E.; Wakeland, E.K.; et al. Type I interferons produced by resident renal cells may promote end-organ disease in autoantibody-mediated glomerulonephritis. J. Immunol. 2009, 183, 6831–6838. [Google Scholar] [CrossRef]
- Ratliff, M.L.; Mishra, M.; Frank, M.B.; Guthridge, J.M.; Webb, C.F. The Transcription Factor ARID3a Is Important for In Vitro Differentiation of Human Hematopoietic Progenitors. J. Immunol. 2016, 196, 614–623. [Google Scholar] [CrossRef]
- Ratliff, M.L.; Ward, J.M.; Merrill, J.T.; James, J.A.; Webb, C.F. Differential expression of the transcription factor ARID3a in lupus patient hematopoietic progenitor cells. J. Immunol. 2015, 194, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.F.; Bryant, J.; Popowski, M.; Allred, L.; Kim, D.; Harriss, J.; Schmidt, C.; Miner, C.A.; Rose, K.; Cheng, H.L.; et al. The ARID Family Transcription Factor Bright Is Required for both Hematopoietic Stem Cell and B Lineage Development. Mol. Cell. Biol. 2011, 31, 1041–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moonen, J.R.; de Leeuw, K.; van Seijen, X.J.; Kallenberg, C.G.; van Luyn, M.J.; Bijl, M.; Harmsen, M.C. Reduced number and impaired function of circulating progenitor cells in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2007, 9, R84. [Google Scholar] [CrossRef] [PubMed]
- Westerweel, P.E.; Luijten, R.K.; Hoefer, I.E.; Koomans, H.A.; Derksen, R.H.; Verhaar, M.C. Haematopoietic and endothelial progenitor cells are deficient in quiescent systemic lupus erythematosus. Ann. Rheum. Dis. 2007, 66, 865–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Chen, W.; Ren, G.; Zhao, L.; Guo, J.; Gong, D.; Zeng, C.; Hu, W.; Liu, Z. Autologous Hematopoietic Stem Cell Transplantation for Refractory Lupus Nephritis. Clin. J. Am. Soc. Nephrol. 2019, 14, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Marmont du Haut Champ, A.M. Hematopoietic stem cell transplantation for systemic lupus erythematosus. Clin. Dev. Immunol. 2012, 2012, 380391. [Google Scholar] [CrossRef] [PubMed]
- Essers, M.A.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908. [Google Scholar] [CrossRef]
- Sato, T.; Onai, N.; Yoshihara, H.; Arai, F.; Suda, T.; Ohteki, T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat. Med. 2009, 15, 696–700. [Google Scholar] [CrossRef]
- Pietras, E.M.; Lakshminarasimhan, R.; Techner, J.M.; Fong, S.; Flach, J.; Binnewies, M.; Passegue, E. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J. Exp. Med. 2014, 211, 245–262. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Herrada, A.A.; Escobedo, N.; Iruretagoyena, M.; Valenzuela, R.A.; Burgos, P.I.; Cuitino, L.; Llanos, C. Innate Immune Cells’ Contribution to Systemic Lupus Erythematosus. Front. Immunol. 2019, 10, 772. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Tsai, L.M.; Leong, Y.A.; Hu, X.; Ma, C.S.; Chevalier, N.; Sun, X.; Vandenberg, K.; Rockman, S.; Ding, Y.; et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity 2013, 39, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Klarquist, J.; Zhou, Z.; Shen, N.; Janssen, E.M. Dendritic Cells in Systemic Lupus Erythematosus: From Pathogenic Players to Therapeutic Tools. Mediat. Inflamm. 2016, 2016, 5045248. [Google Scholar] [CrossRef] [PubMed]
- Orme, J.; Mohan, C. Macrophage subpopulations in systemic lupus erythematosus. Discov. Med. 2012, 13, 151–158. [Google Scholar] [PubMed]
- Spada, R.; Rojas, J.M.; Barber, D.F. Recent findings on the role of natural killer cells in the pathogenesis of systemic lupus erythematosus. J. Leukoc. Biol. 2015, 98, 479–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez-Fueyo, A.; Bradley, S.J.; Tsokos, G.C. T cells in Systemic Lupus Erythematosus. Curr. Opin. Immunol. 2016, 43, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Fike, A.J.; Elcheva, I.; Rahman, Z.S.M. The Post-GWAS Era: How to Validate the Contribution of Gene Variants in Lupus. Curr. Rheumatol. Rep. 2019, 21, 3. [Google Scholar] [CrossRef]
- Callery, E.M.; Smith, J.C.; Thomsen, G.H. The ARID domain protein dril1 is necessary for TGFbeta signaling in Xenopus embryos. Dev. Biol. 2005, 278, 542–559. [Google Scholar] [CrossRef]
- Shandala, T.; Kortschak, R.D.; Gregory, S.; Saint, R. The Drosophila dead ringer gene is required for early embryonic patterning through regulation of argos and buttonhead expression. Development 1999, 126, 4341–4349. [Google Scholar]
- Ren, J.; Panther, E.; Liao, X.; Grammer, A.C.; Lipsky, P.E.; Reilly, C.M. The Impact of Protein Acetylation/Deacetylation on Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2018, 19, 4007. [Google Scholar] [CrossRef]
- White, C.A.; Pone, E.J.; Lam, T.; Tat, C.; Hayama, K.L.; Li, G.; Zan, H.; Casali, P. Histone deacetylase inhibitors upregulate B cell microRNAs that silence AID and Blimp-1 expression for epigenetic modulation of antibody and autoantibody responses. J. Immunol. 2014, 193, 5933–5950. [Google Scholar] [CrossRef] [PubMed]
- Chamilos, G.; Gregorio, J.; Meller, S.; Lande, R.; Kontoyiannis, D.P.; Modlin, R.L.; Gilliet, M. Cytosolic sensing of extracellular self-DNA transported into monocytes by the antimicrobial peptide LL37. Blood 2012, 120, 3699–3707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puissegur, M.P.; Eichner, R.; Quelen, C.; Coyaud, E.; Mari, B.; Lebrigand, K.; Broccardo, C.; Nguyen-Khac, F.; Bousquet, M.; Brousset, P. B-cell regulator of immunoglobulin heavy-chain transcription (Bright)/ARID3a is a direct target of the oncomir microRNA-125b in progenitor B-cells. Leukemia 2012, 26, 2224–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honarpisheh, M.; Kohler, P.; von Rauchhaupt, E.; Lech, M. The Involvement of MicroRNAs in Modulation of Innate and Adaptive Immunity in Systemic Lupus Erythematosus and Lupus Nephritis. J. Immunol. Res. 2018, 2018, 4126106. [Google Scholar] [CrossRef] [PubMed]
- Yoon, G.; Park, J.Y.; Kim, H.J.; Choi, G.S.; Kim, J.G.; Kang, B.W.; Kang, M.K.; Seo, A.N. ARID3A Positivity Correlated With Favorable Prognosis in Patients With Residual Rectal Cancer After Neoadjuvant Chemoradiotherapy. Anticancer Res. 2019, 39, 2845–2853. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Chirshev, E.; Oberg, K.C.; Ioffe, Y.J.; Unternaehrer, J.J. Let-7 as biomarker, prognostic indicator, and therapy for precision medicine in cancer. Clin. Transl. Med. 2019, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Samotij, D.; Reich, A. Biologics in the Treatment of Lupus Erythematosus: A Critical Literature Review. BioMed Res. Int. 2019, 2019, 8142368. [Google Scholar] [CrossRef]
- Khamashta, M.; Merrill, J.T.; Werth, V.P.; Furie, R.; Kalunian, K.; Illei, G.G.; Drappa, J.; Wang, L.; Greth, W.; CD1067 Study Investigators. Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: A randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 1909–1916. [Google Scholar] [CrossRef]
- Lauwerys, B.R.; Ducreux, J.; Houssiau, F.A. Type I interferon blockade in systemic lupus erythematosus: Where do we stand? Rheumatology 2014, 53, 1369–1376. [Google Scholar] [CrossRef]
- Sanz, I. New Perspectives in Rheumatology: May You Live in Interesting Times: Challenges and Opportunities in Lupus Research. Arthritis Rheumatol. 2017, 69, 1552–1559. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garton, J.; Barron, M.D.; Ratliff, M.L.; Webb, C.F. New Frontiers: ARID3a in SLE. Cells 2019, 8, 1136. https://doi.org/10.3390/cells8101136
Garton J, Barron MD, Ratliff ML, Webb CF. New Frontiers: ARID3a in SLE. Cells. 2019; 8(10):1136. https://doi.org/10.3390/cells8101136
Chicago/Turabian StyleGarton, Joshua, M. David Barron, Michelle L. Ratliff, and Carol F. Webb. 2019. "New Frontiers: ARID3a in SLE" Cells 8, no. 10: 1136. https://doi.org/10.3390/cells8101136
APA StyleGarton, J., Barron, M. D., Ratliff, M. L., & Webb, C. F. (2019). New Frontiers: ARID3a in SLE. Cells, 8(10), 1136. https://doi.org/10.3390/cells8101136