Leptin Promotes Expression of EMT-Related Transcription Factors and Invasion in a Src and FAK-Dependent Pathway in MCF10A Mammary Epithelial Cells

 , ,
, ,  , , and
, , and 
Abstract
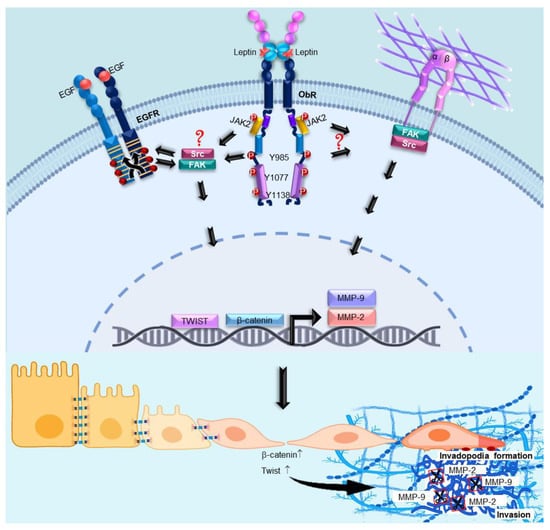
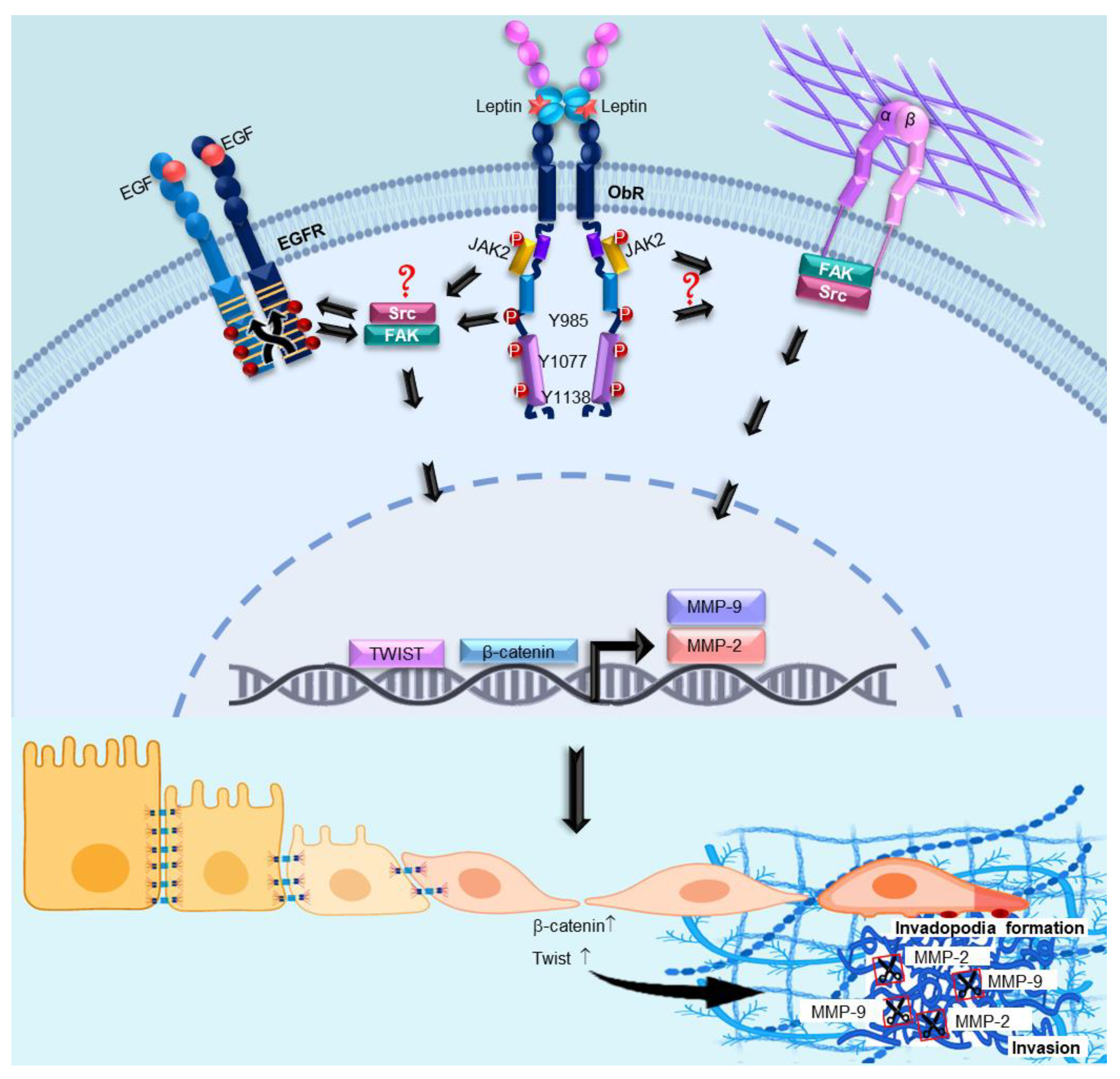
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cell Stimulation
2.4. Western Blot
2.5. Immunofluorescence and F-Actin Staining
2.6. Gelatin Zymograms
2.7. Invadopodia Formation Assays
2.8. Cell Invasion Assays
2.9. Statistical Analysis
3. Results
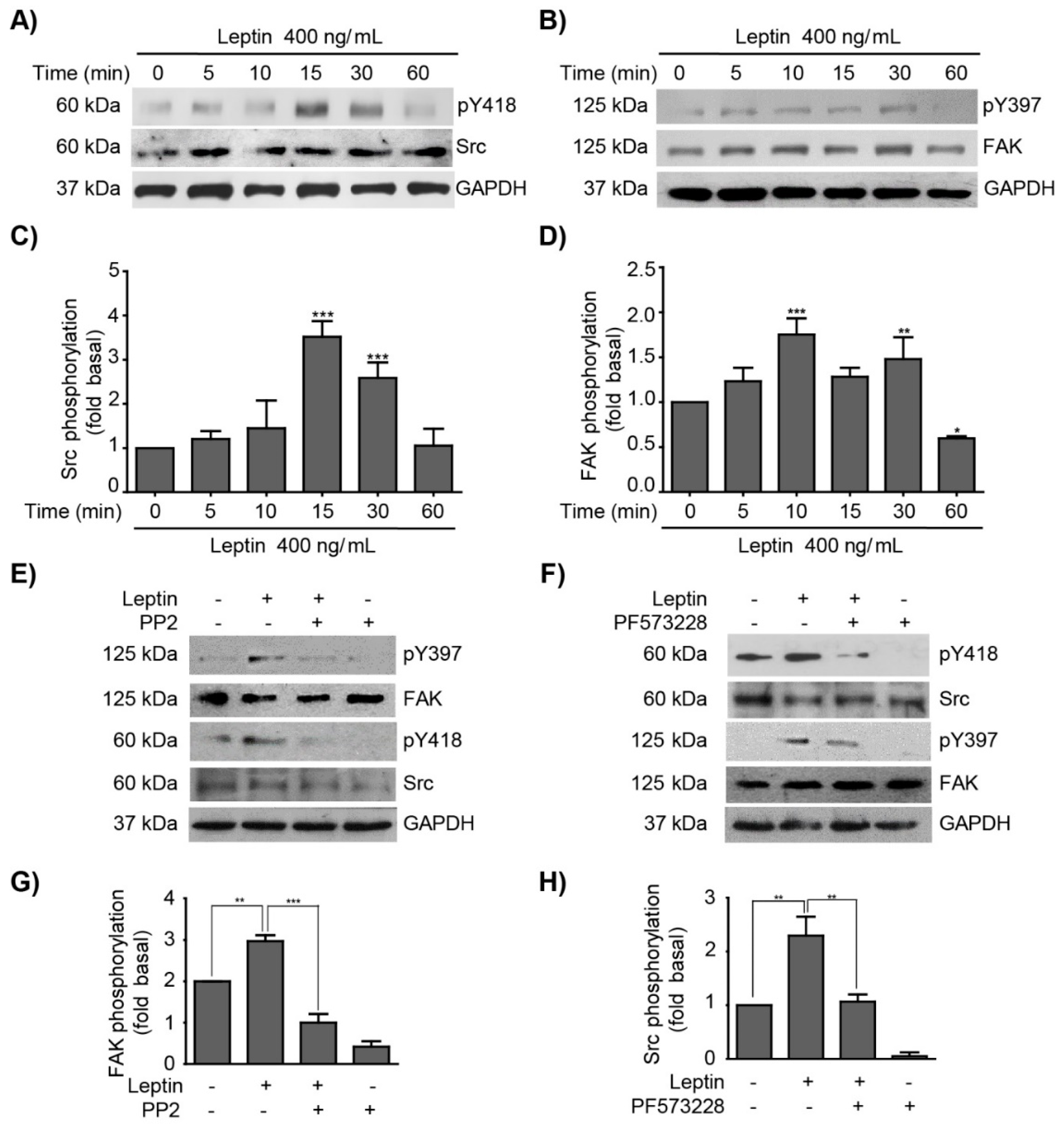
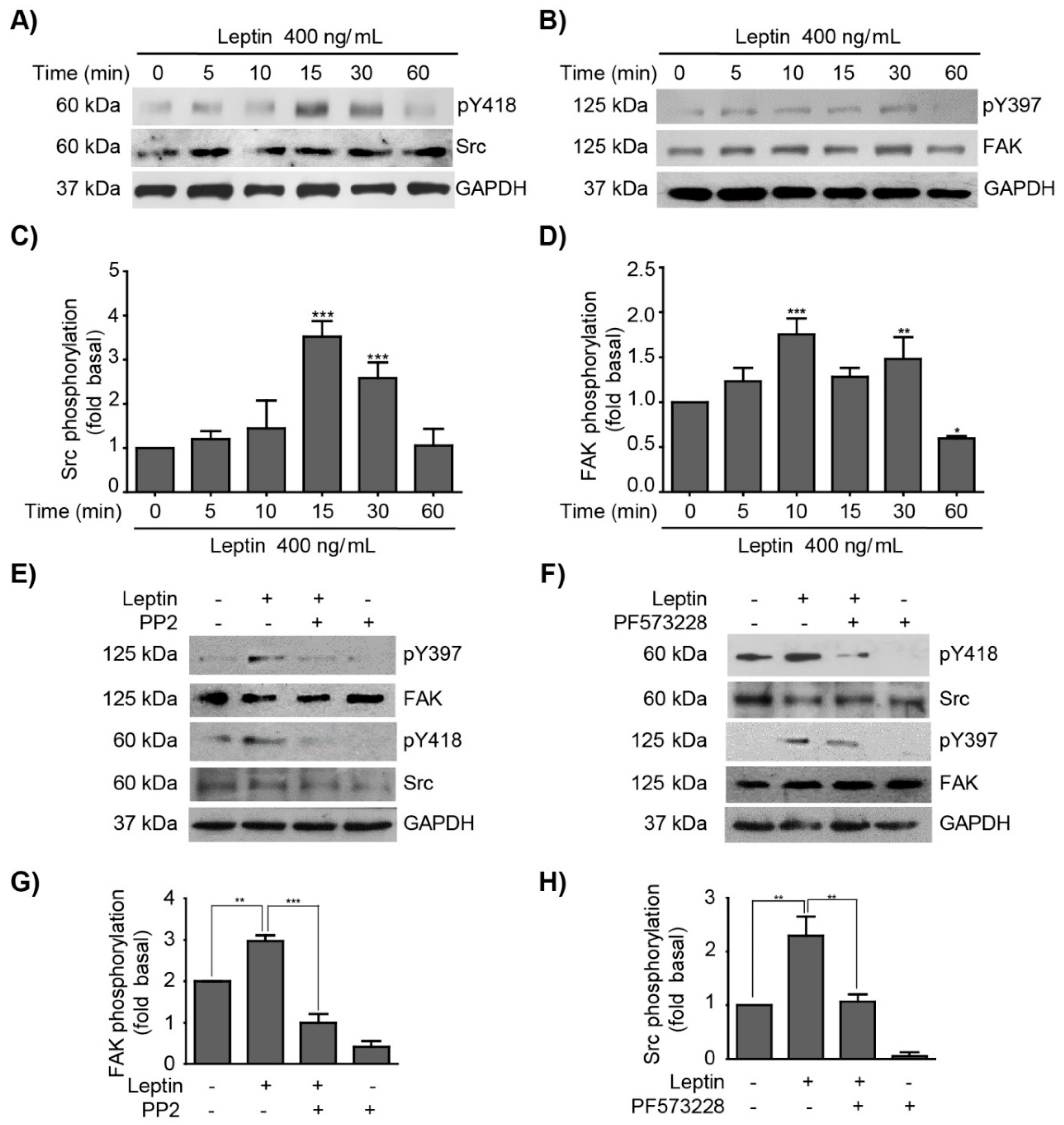
3.1. Leptin Induces FAK and Src Activation in MCF10A Non-Tumorigenic Epithelial Breast Cells
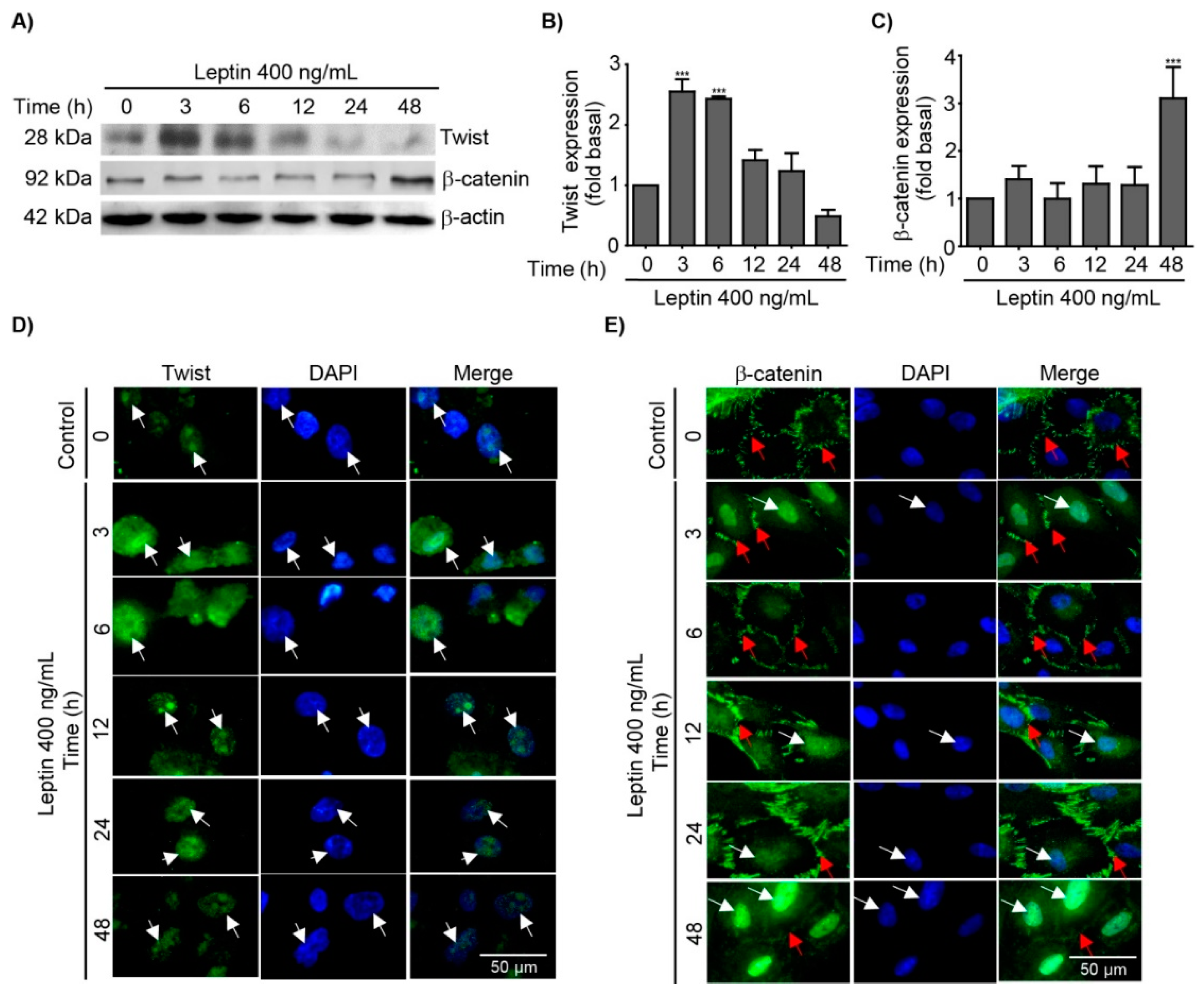
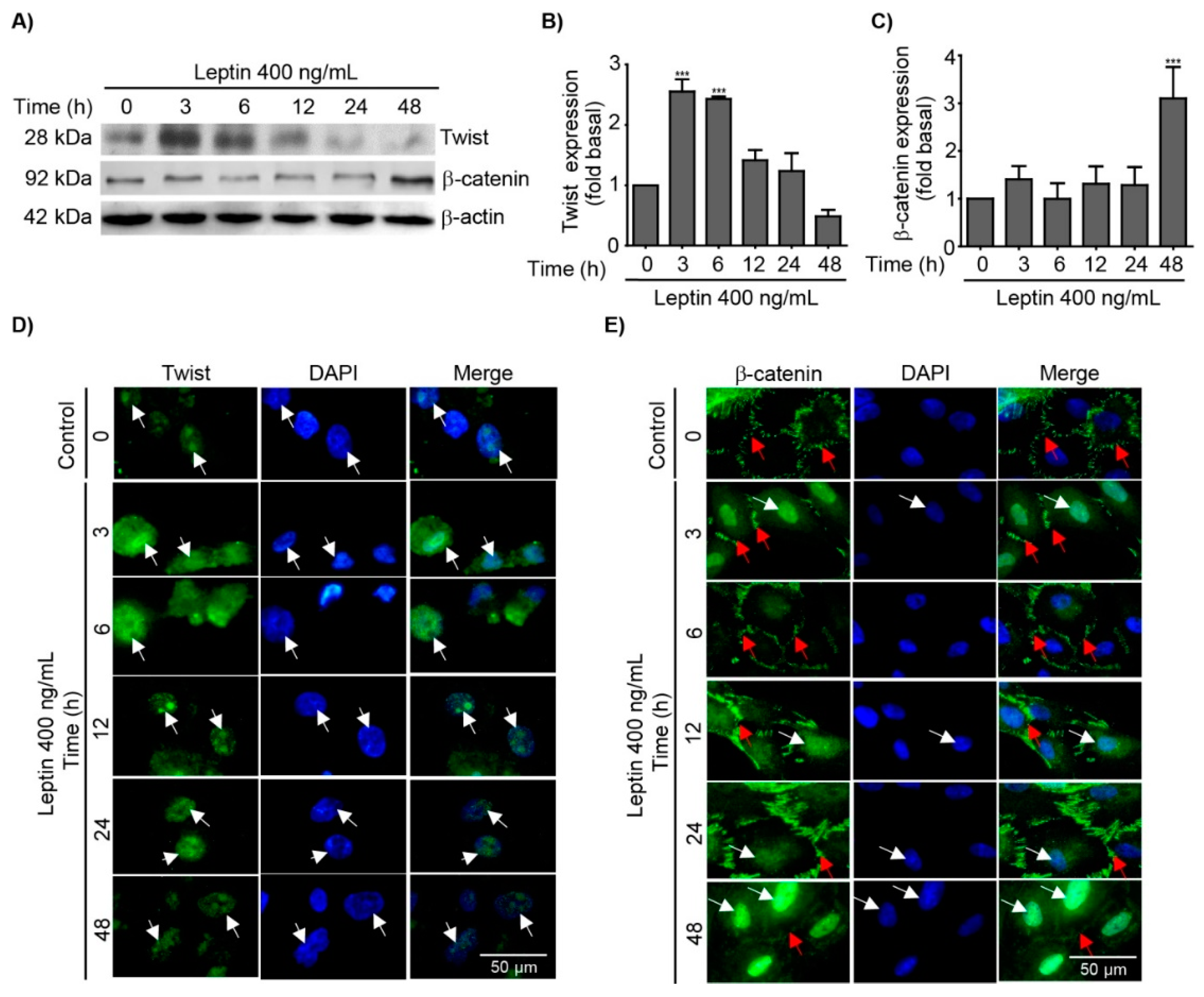
3.2. Leptin Regulates the Expression and Subcellular Localization of Twist and β-Catenin in MCF10A Epithelial Breast Cells
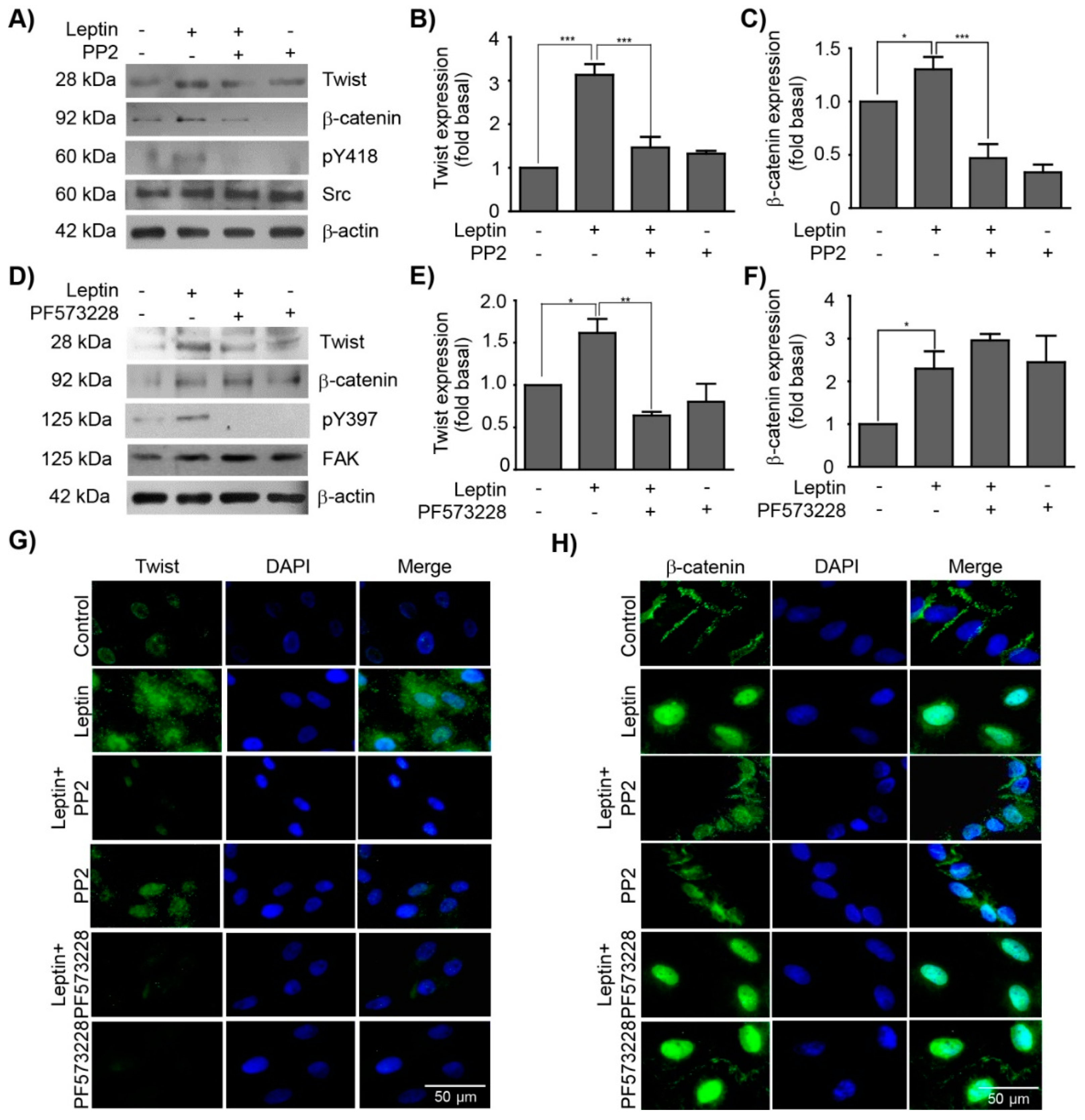
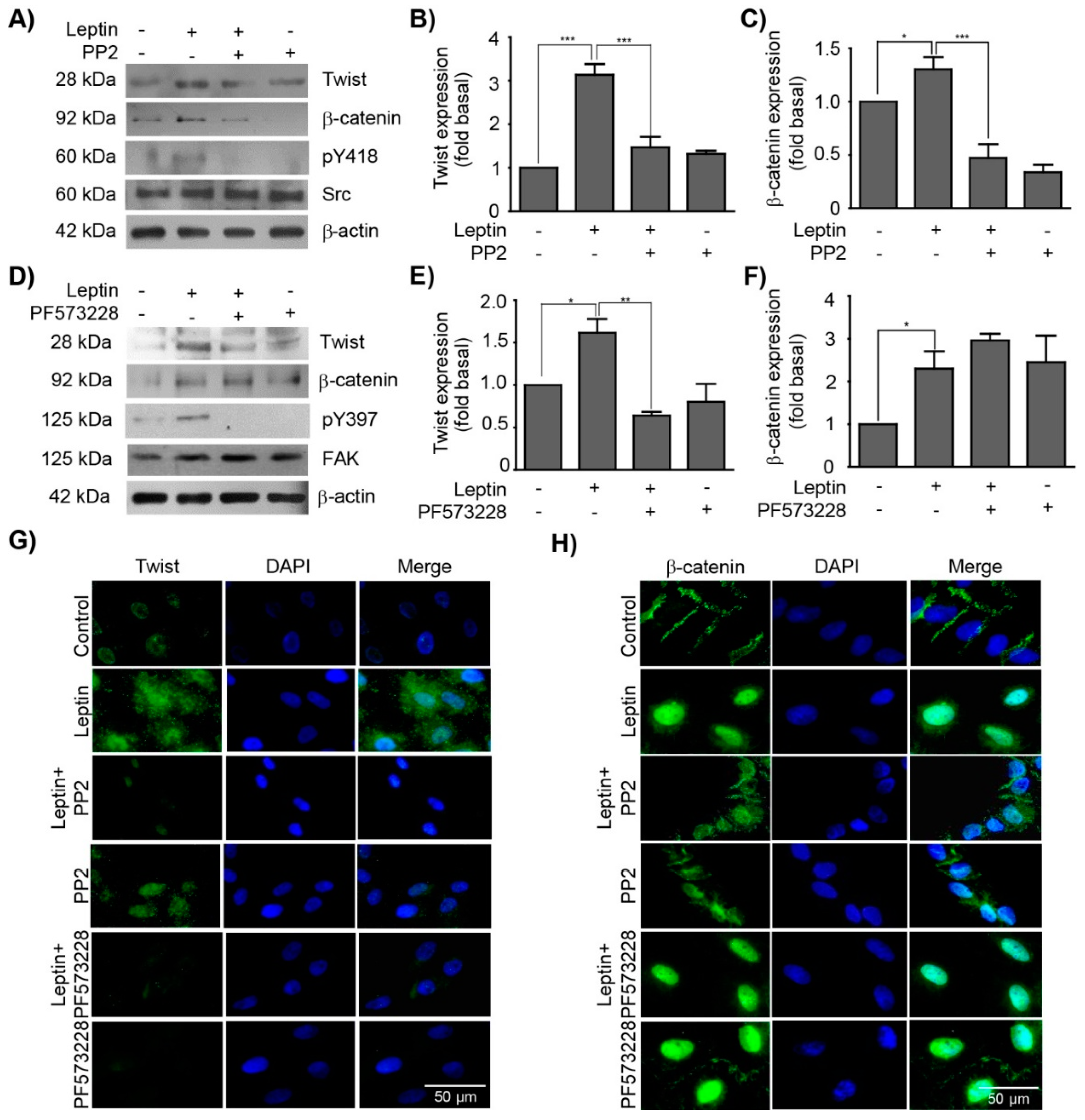
3.3. Leptin Regulates the Expression and Subcellular Localization of Twist and β-Catenin in a Src and FAK-Dependent Manner in MCF10A Cells
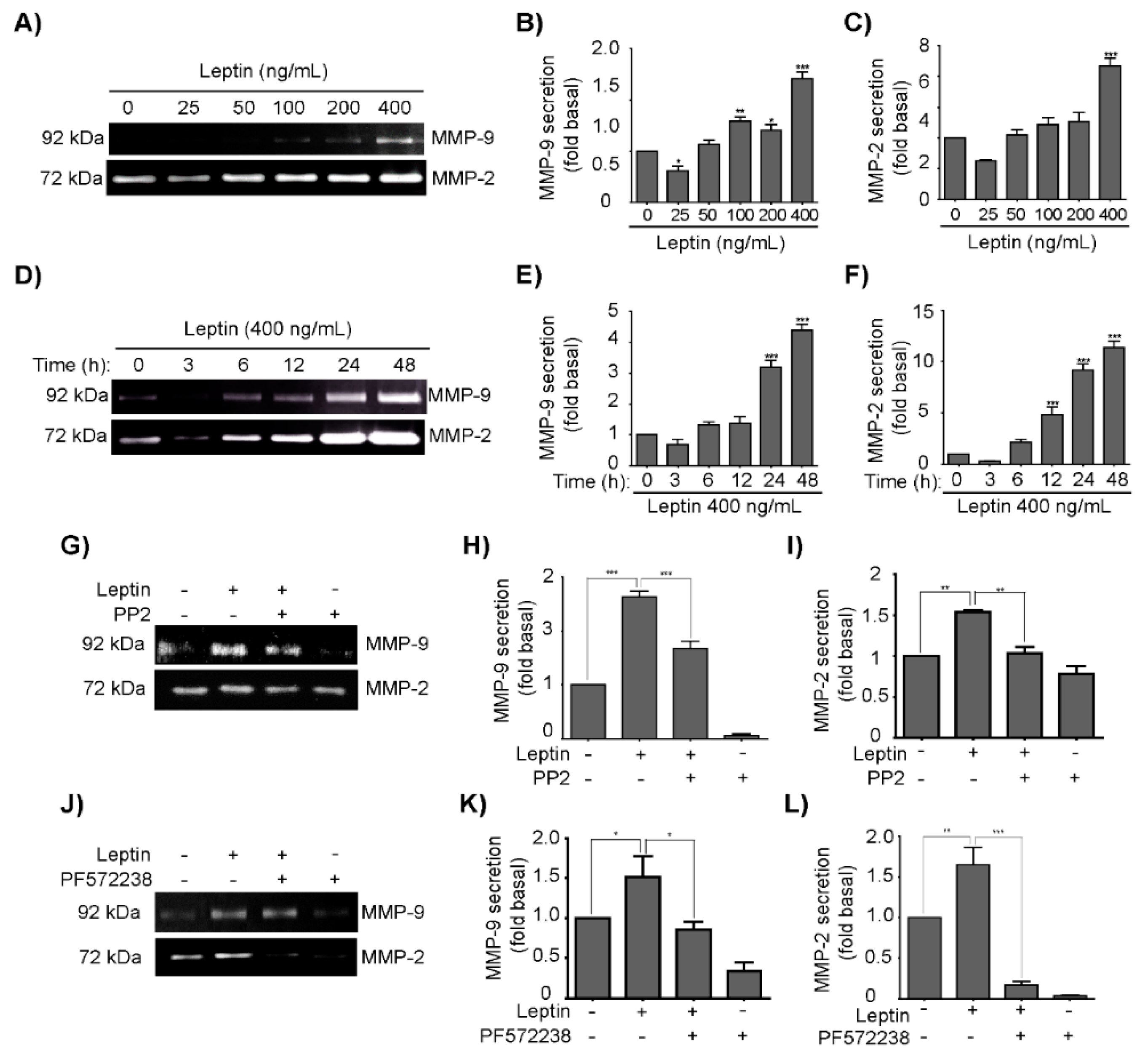
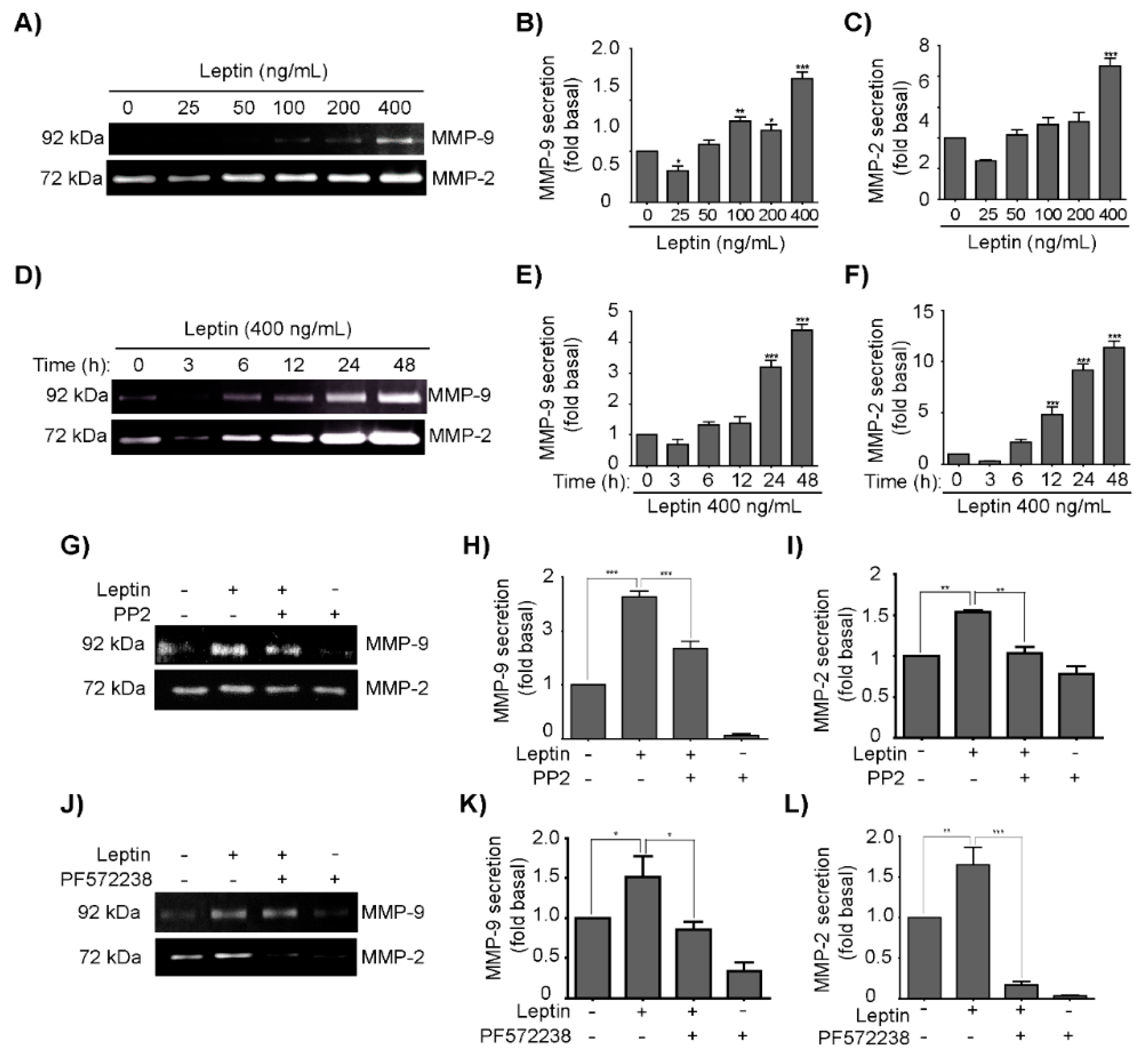
3.4. Leptin Induces MMP-2 and MMP-9 Secretion in a Src- and FAK-Dependent Fashion in MCF10A Cells
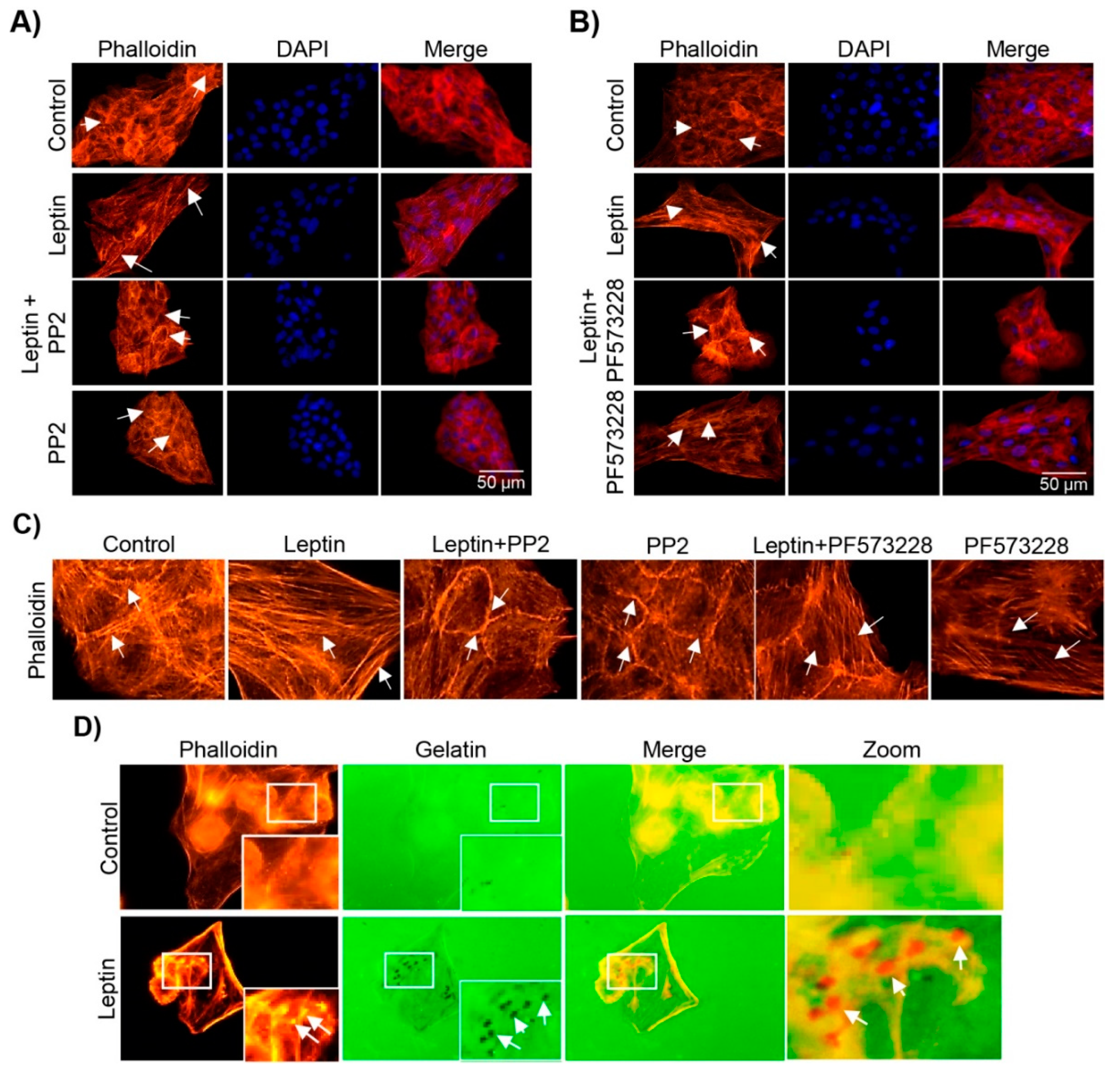
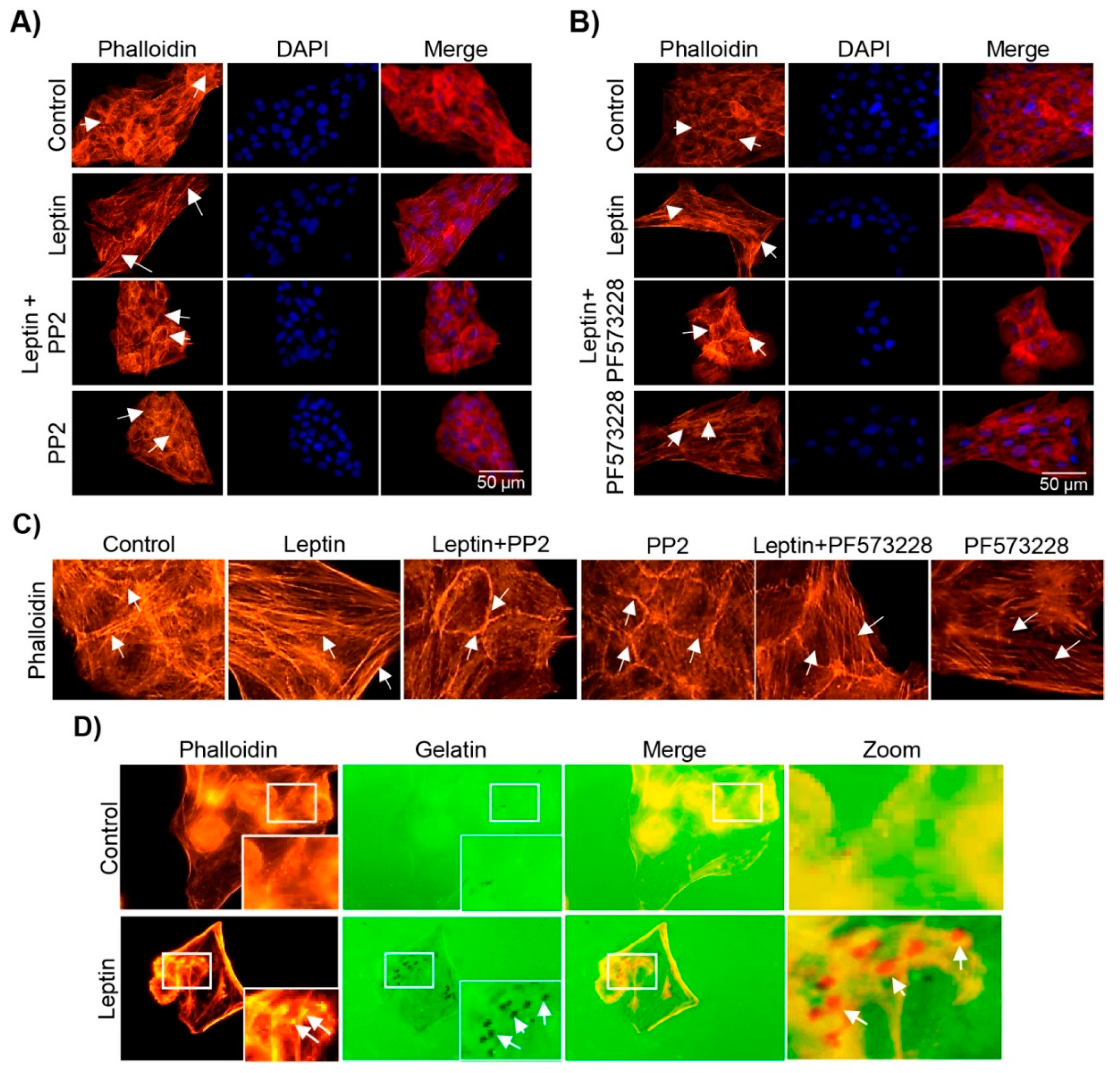
3.5. Leptin Induces the Formation of Stress Fibers in a Src- and FAK-Dependent Manner in MCF10A Cells
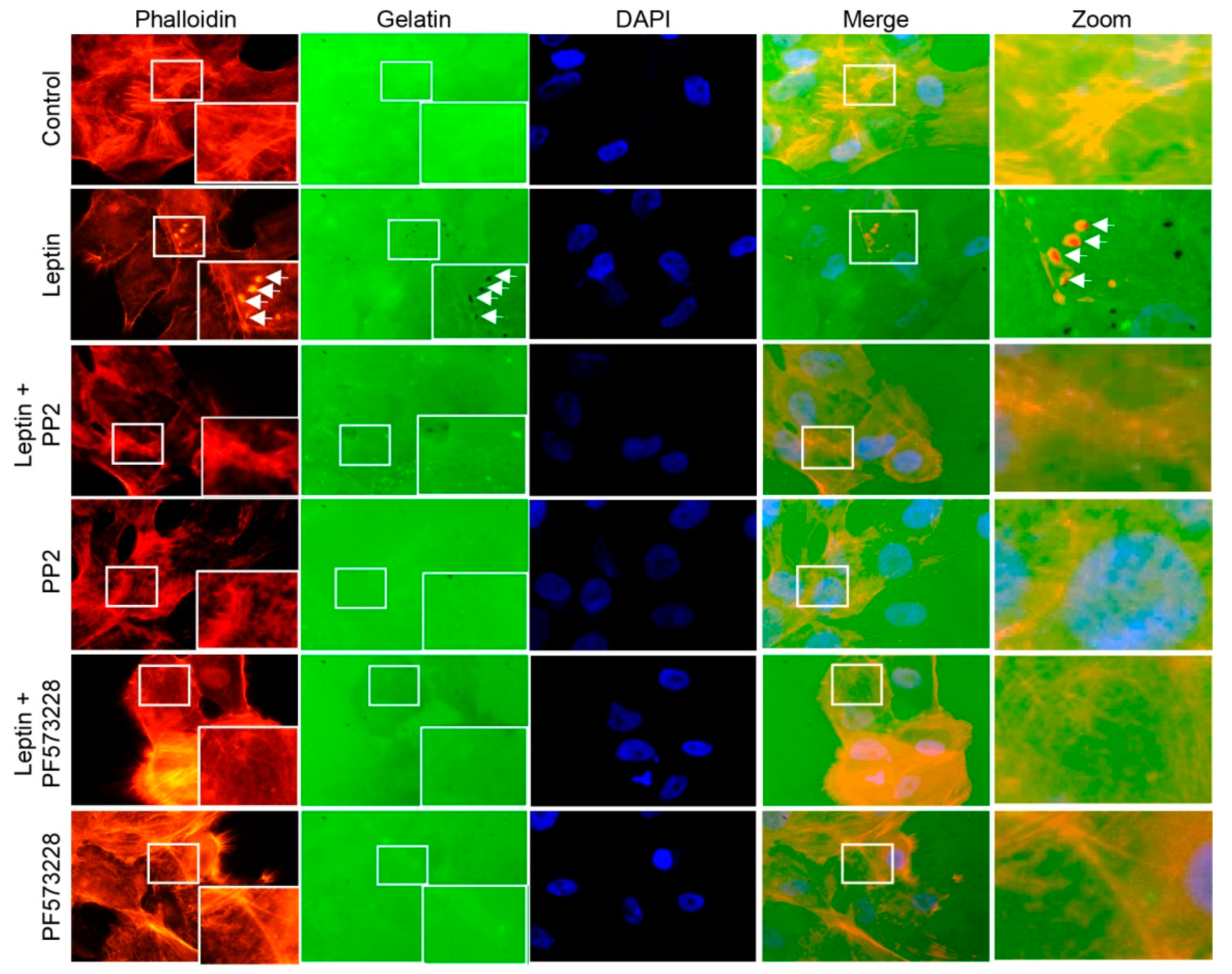
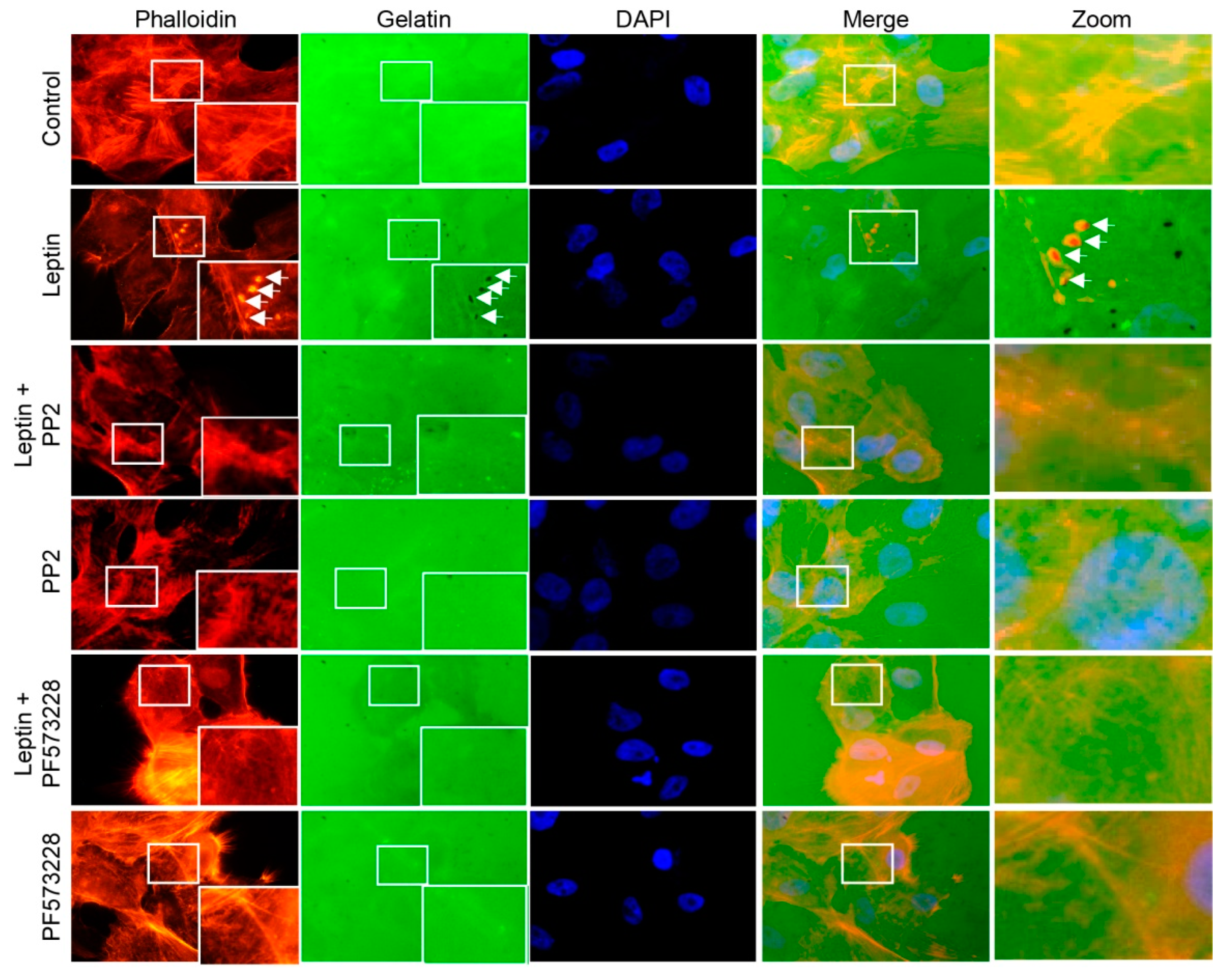
3.6. Leptin Induces Invadopodia Formation in a FAK and Src-Dependent Pathway in MCF10A Cells
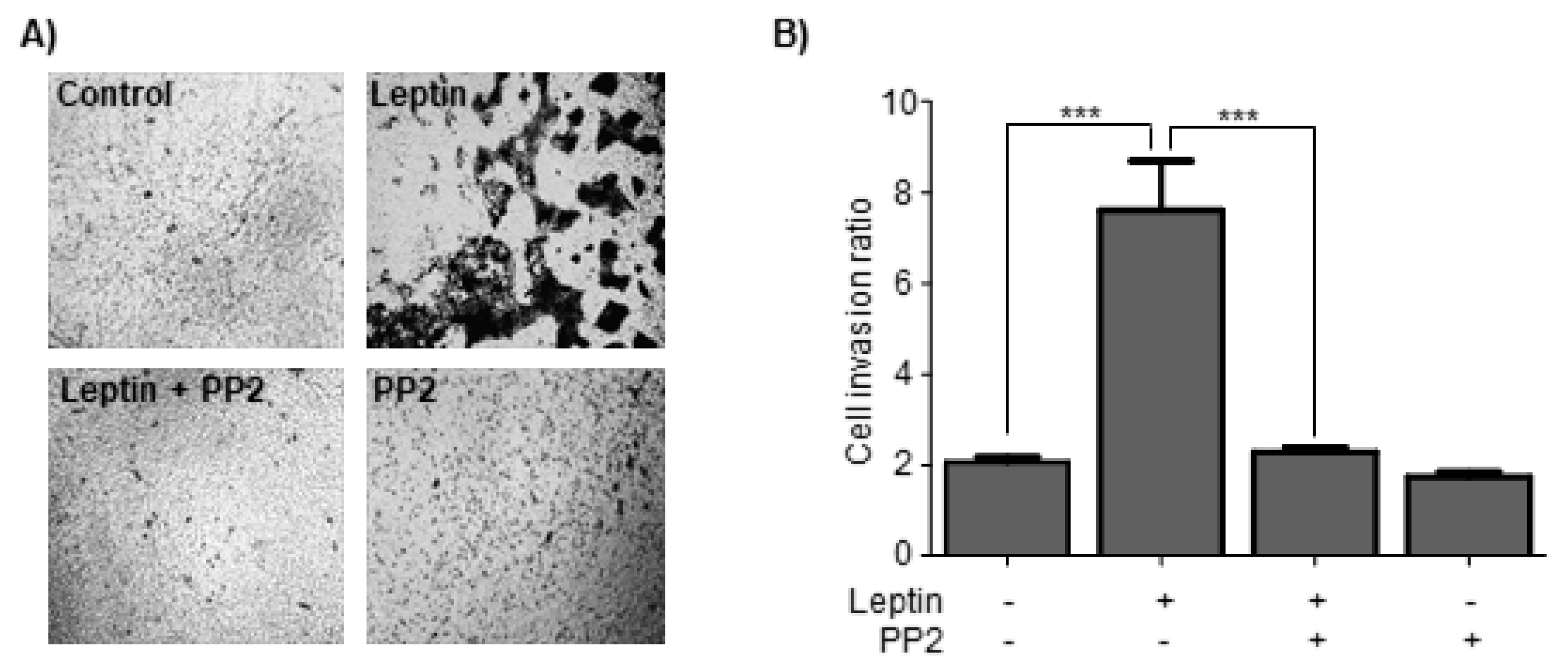
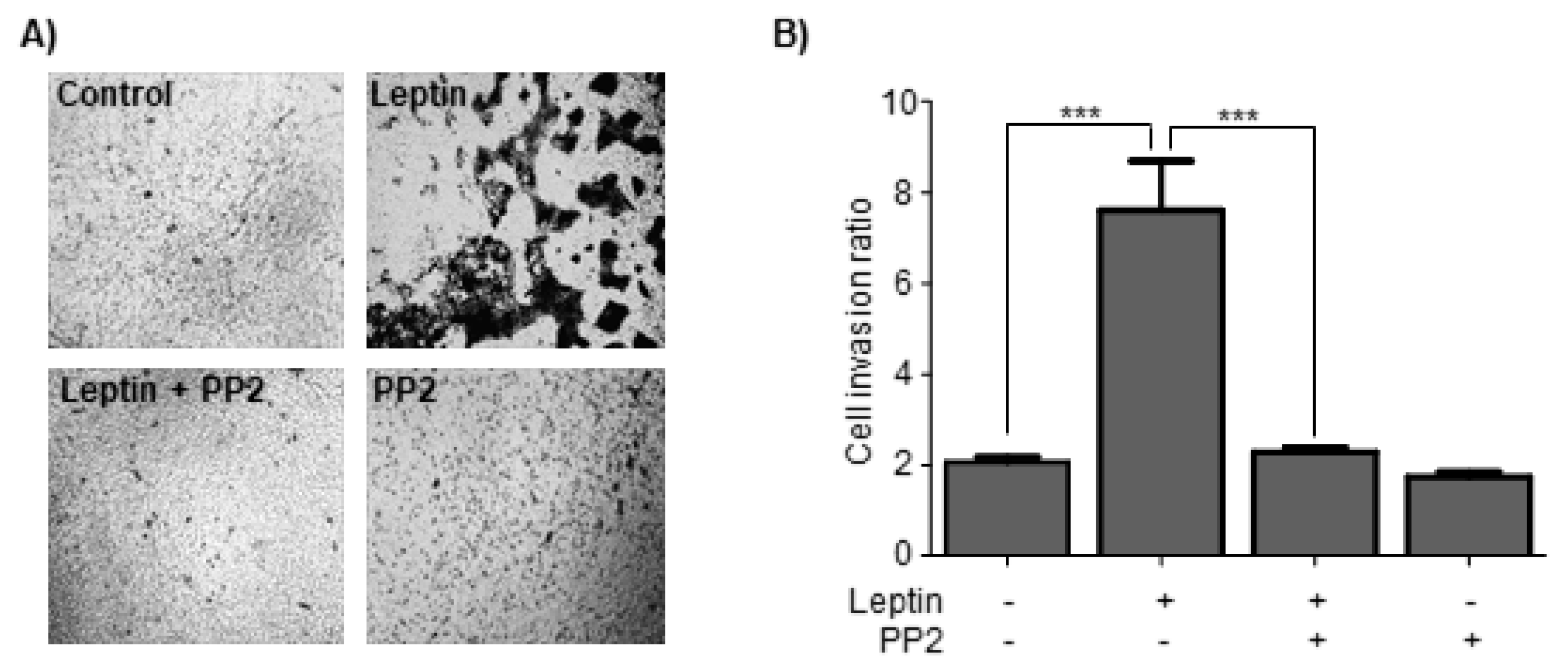
3.7. Leptin Induces Cell Invasion in a Src-Dependent Pathway in MCF10A Cells
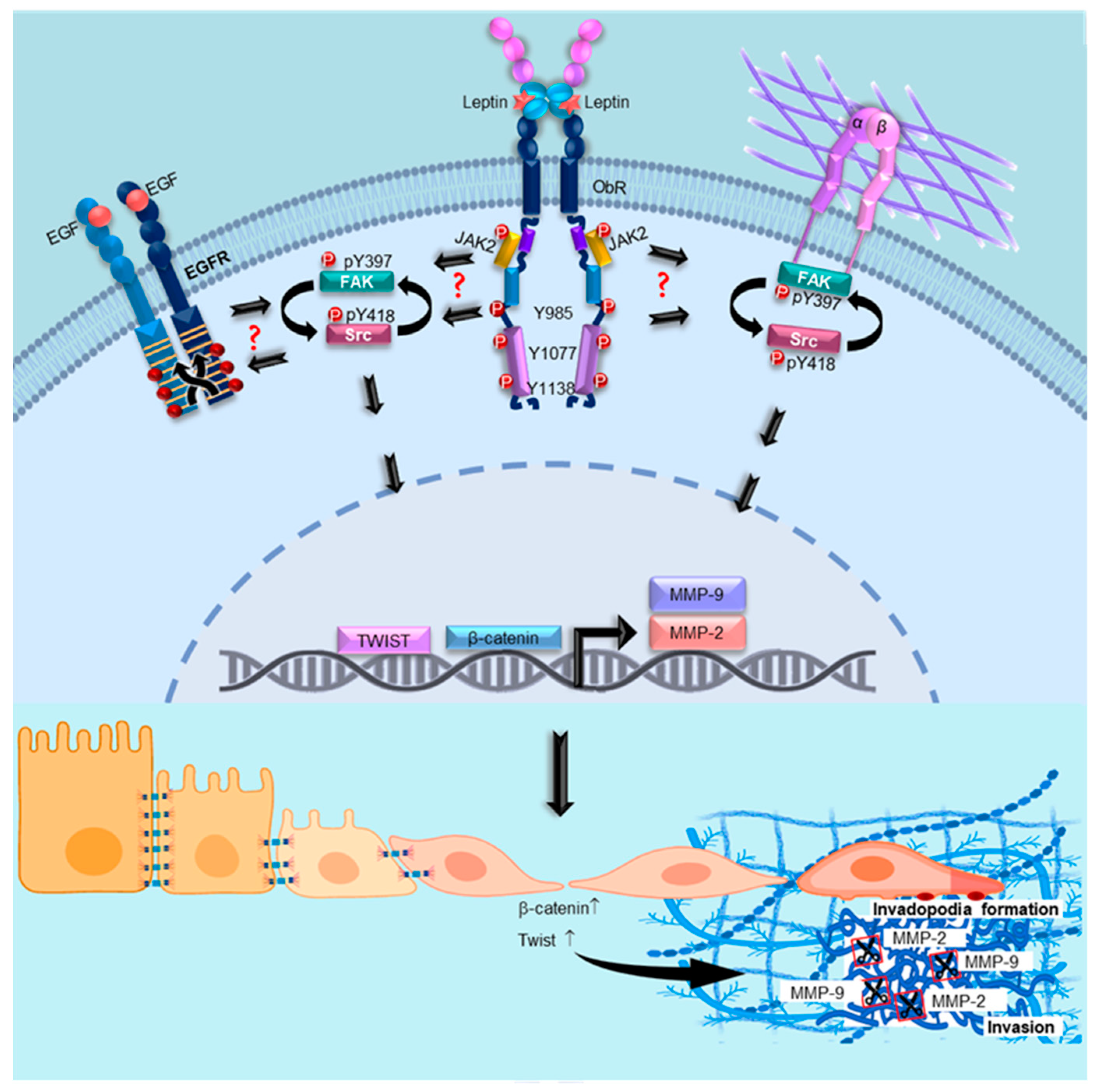
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cirillo, D.; Rachiglio, A.M.; La Montagna, R.; Giordano, A.; Normanno, N. Leptin signaling in breast cancer: An overview. J. Cell. Biochem 2008, 105, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Ando, S.; Barone, I.; Giordano, C.; Bonofiglio, D.; Catalano, S. The Multifaceted Mechanism of Leptin Signaling within Tumor Microenvironment in Driving Breast Cancer Growth and Progression. Front. Oncol. 2014, 4, 1–6. [Google Scholar] [CrossRef]
- Olea-Flores, M.; Juárez-Cruz, J.C.; Mendoza-Catalán, M.A.; Padilla-Benavides, T.; Navarro-Tito, N. Signaling Pathways Induced by Leptin during Epithelial-Mesenchymal Transition in Breast Cancer. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Wang, C.-D.; Cao, C.; Cai, L.-R.; Li, D.-H.; Zheng, Y.-Z. Association of serum leptin with breast cancer. Medicine 2019, 98, e14094. [Google Scholar] [CrossRef] [PubMed]
- Saxena, N.K.; Sharma, D. Multifaceted Leptin network: The molecular connection between obesity and breast cancer. J. Mammary Gland Biol. Neoplasia 2016, 25, 289–313. [Google Scholar] [CrossRef]
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2018, 69, 11–20. [Google Scholar] [CrossRef]
- Mullen, M.; Gonzalez-Perez, R. Leptin-Induced JAK/STAT Signaling and Cancer Growth. Vaccines 2016, 4, 26. [Google Scholar] [CrossRef]
- Candelaria, P.V.; Rampoldi, A.; Harbuzariu, A.; Gonzalez-Perez, R.R. Leptin signaling and cancer chemoresistance: Perspectives. World J. Clin. Oncol. 2017, 8, 106. [Google Scholar] [CrossRef]
- Yan, D.; Avtanski, D.; Saxena, N.K.; Sharma, D. Leptin-induced epithelial-mesenchymal transition in breast cancer cells requires β-catenin activation via Akt/GSK3- and MTA1/Wnt1 protein-dependent pathways. J. Biol. Chem. 2012, 287, 8598–8612. [Google Scholar] [CrossRef]
- Wei, L.; Li, K.; Pang, X.; Guo, B.; Su, M.; Huang, Y.; Wang, N.; Ji, F.; Zhong, C.; Yang, J.; et al. Leptin promotes epithelial-mesenchymal transition of breast cancer via the upregulation of pyruvate kinase M2. J. Exp. Clin. Cancer Res. 2016, 35, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Tang, C.; Cao, H.; Li, K.; Pang, X.; Zhong, L.; Dang, W.; Tang, H.; Huang, Y.; Wei, L.; et al. Activation of IL-8 via PI3K/Akt-dependent pathway is involved in leptin-mediated epithelial-mesenchymal transition in human breast cancer cells. Cancer Biol. Ther. 2015, 16, 1220–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A. The Ins and Outs of the Epithelial to Mesenchymal Transition in Health and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.Y.J.; Jackson, R.A.A.; Thiery, J.P.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Isaías-Tizapa, R.; Acosta, E.; Tacuba-Saavedra, A.; Mendoza-Catalán, M.; Navarro-Tito, N. Leptina promueve la expresión de Hic-5 y la formación de puntos de actina de manera dependiente de las cinasas FAK y Src en células epiteliales mamarias MCF10A. Biomédica 2019, 39. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Goossens, S.; Vandamme, N.; Van Vlierberghe, P.; Berx, G. EMT transcription factors in cancer development re-evaluated: Beyond EMT and MET. Rev. Cancer 2017, 1868, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Review Emerging Biological Principles of Metastasis. Cell 2016, 168, 670–691. [Google Scholar] [CrossRef]
- Skrypek, N.; Goossens, S.; De Smedt, E.; Vandamme, N.; Berx, G. Epithelial-to-Mesenchymal Transition: Epigenetic Reprogramming Driving Cellular Plasticity. Trends Genet. 2017, 33, 943–959. [Google Scholar] [CrossRef] [PubMed]
- Kimelman, D.; Xu, W. β-Catenin destruction complex: Insights and questions from a structural perspective. Oncogene 2006, 25, 7482–7491. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, K.; Clevers, H. Wnt/β-catenin signaling in adult mammalian epithelial stem cells. Dev. Biol. 2017, 428, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, E.H.; Ward, C.; Muller, H.K.; Mahmood, M.Q.; Weston, S.; Shukla, S.D.; Sohal, S.S. β-catenin, Twist and Snail: Transcriptional regulation of EMT in smokers and COPD, and relation to airflow obstruction. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Zhao, Z.; Rahman, M.A.; Chen, Z.G.; Dong, M. Multiple biological functions of Twist1 in various cancers. Oncotarget 2017, 8, 20380–20393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Massi, D.; Hemmings, B.A.; Mandalà, M.; Wicki, A.; Xue, G. AKT-ions with a TWIST between EMT and MET. Oncotarget 2016, 7, 62767–62777. [Google Scholar] [CrossRef] [PubMed]
- Eckert, M.A.; Lwin, T.M.; Chang, A.T.; Kim, J.; Danis, E.; Ohno-Machado, L.; Yang, J. Twist1-Induced Invadopodia Formation Promotes Tumor Metastasis. Cancer Cell 2011, 19, 372–386. [Google Scholar] [CrossRef]
- Guan, X. Cancer metastases: Challenges and opportunities. Acta Pharm. Sin. B. 2015, 5, 402–418. [Google Scholar] [CrossRef]
- Yousef, E.M.; Tahir, M.R.; St-Pierre, Y.; Gaboury, L.A. MMP-9 expression varies according to molecular subtypes of breast cancer. BMC Cancer 2014. [Google Scholar] [CrossRef]
- Farina, A.R.; Mackay, A.R. Gelatinase B/MMP-9 in Tumour Pathogenesis and Progression. Cancers 2014, 6, 240–296. [Google Scholar] [CrossRef]
- Artym, V.V.; Zhang, Y.; Yamada, K.M.; Mueller, S.C. Dynamic Interactions of Cortactin and Membrane Type 1 Matrix Metalloproteinase at Invadopodia: Defining the Stages of Invadopodia Formation and Function. Cancer Res. 2006, 66, 3034–3044. [Google Scholar] [CrossRef]
- Stetler-Stevenson, W.G. Type IV collagenases in tumor invasion and metastasis. Cancer Metastasis Rev. 1990, 9, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Maguire, T.M.; Hill, A.; McDermott, E.; O’Higgins, N. Metalloproteinases: Role in breast carcinogenesis, invasion and metastasis. Breast Cancer Res. 2000, 2, 252–257. [Google Scholar] [CrossRef] [PubMed]
- La Rocca, G.; Marrazzo, A.; Taormina, P.; Minafra, S.; Maddalena, L.; Lorenzo, V.S.; Scienze, V.; Specialita, A. Zymographic detection and clinical correlations of MMP-2 and MMP-9 in breast cancer sera. Br. J. Cancer 2004, 90, 1414–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Yaqoob, U.; Mehta, D.; Shah, V.H.; Motility, F.E.C.; Fak, T. FXR Promotes Endothelial Cell Motility Through Coordinated Regulation of FAK and MMP-9. Cell Biol. 2009, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Jena, M.K.; Janjanam, J. Role of extracellular matrix in breast cancer development: A brief update. F1000Research 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Sabbineni, H.; Clarke, A.; Somanath, P.R. Novel roles of Src in cancer cell epithelial-to-mesenchymal transition, vascular permeability, microinvasion and metastasis. Life Sci. 2016, 157, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Yu, D. Targeting Src family kinases in anti-cancer therapies: Turning promise into triumph. Trends Pharmacol. Sci. 2012, 33, 122–128. [Google Scholar] [CrossRef]
- Cortes-Reynosa, P.; Robledo, T.; Macias-Silva, M.; Wu, S.V.; Salazar, E.P. Src kinase regulates metalloproteinase-9 secretion induced by type IV collagen in MCF-7 human breast cancer cells. Matrix Biol. 2008, 27, 220–231. [Google Scholar] [CrossRef]
- Wu, X.; Yang, L.; Zheng, Z.; Li, Z.; Shi, J.; Li, Y.; Han, S.; Gao, J.; Tang, C.; Su, L.; et al. Src promotes cutaneous wound healing by regulating MMP-2 through the ERK pathway. Int. J. Mol. Med. 2016, 37, 639–648. [Google Scholar] [CrossRef]
- Tai, Y.L.; Chen, L.C.; Shen, T.L. Emerging Roles of Focal Adhesion Kinase in Cancer. Biomed. Res. Int. 2015, 2015, 1–13. [Google Scholar] [CrossRef]
- Frame, M.C.; Patel, H.; Serrels, B.; Lietha, D.; Eck, M.J. The FERM domain: Organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 2010, 11, 802–814. [Google Scholar] [CrossRef] [PubMed]
- Elsberger, B. Translational evidence on the role of Src kinase and activated Src kinase in invasive breast cancer. Crit. Rev. Oncol. Hematol. 2014, 89, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Villanueva Duque, A.; Zuniga Eulogio, M.D.; Dena Beltran, J.; Saucedo Castaneda, E.; Calixto Galvez, M.; Mendoza Catalán, M.A.; Ortuno Pineda, C.; Navarro Tito, N. Leptin induces partial epithelial-mesenchymal transition in a FAK-ERK dependent pathway in MCF10A mammary non-tumorigenic cells. Int. J. Clin. Exp. Pathol. 2017, 10, 10334–10342. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Kruger, N.J. The Bradford Method for Protein Quantitation. In The Protein Protocols Handbook; Humana Press: Totowa, NJ, USA, 2009; pp. 17–24. [Google Scholar]
- Toth, R.; Sohail, M.; Fridman, A. Assessment of Gelatinases (MMP-2 and MMP-9) by Gelatin Zymography. Metastasis Res. Protoc. 2012, 878, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Pignatelli, J.; Tumbarello, D.A.; Schmidt, R.P.; Turner, C.E. Hic-5 promotes Invadopodia formation and invasion during TGF-β–induced epithelial–mesenchymal transition. J. Cell Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Kramer, N.; Walzl, A.; Unger, C.; Rosner, M.; Krupitza, G.; Hengstschläger, M.; Dolznig, H. In vitro cell migration and invasion assays. Mutat. Res. Rev. Mutat. Res. 2013, 752, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Tito, N.; Robledo, T.; Salazar, E.P. Arachidonic acid promotes FAK activation and migration in MDA-MB-231 breast cancer cells. Exp. Cell Res. 2008, 314, 3340–3355. [Google Scholar] [CrossRef]
- Lai, I.R.; Chu, P.Y.; Lin, H.S.; Liou, J.Y.; Jan, Y.J.; Lee, J.C.; Shen, T.L. Phosphorylation of focal adhesion kinase at Tyr397 in gastric carcinomas and its clinical significance. Am. J. Pathol. 2010, 177, 1629–1637. [Google Scholar] [CrossRef]
- Juárez-Cruz, J.C.; Zuñiga-Eulogio, M.D.; Olea-Flores, M.; Castañeda-Saucedo, E.; Mendoza-catalán, M.Á.; Ortuño-Pineda, C.; Moreno-Godínez, M.E.; Villegas-Comonfort, S.; Padilla Benavides, T.; Navarro-Tito, N. Leptin induces cell migration and invasion in a FAK-Src- dependent manner in breast cancer cells. BioRxiv 2019. [Google Scholar] [CrossRef]
- Choma, D.P.; Milano, V.; Pumiglia, K.M.; Dipersio, C.M. Integrin α3β1-Dependent Activation of FAK/Src Regulates Rac1-Mediated Keratinocyte Polarization on Laminin-5. J. Investig. Dermatol. 2007, 127, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Cicchini, C.; Laudadio, I.; Citarella, F.; Corazzari, M.; Steindler, C.; Conigliaro, A.; Fantoni, A.; Amicone, L.; Tripodi, M. TGF β -induced EMT requires focal adhesion kinase. Exp. Cell Res. 2007, 14. [Google Scholar] [CrossRef]
- Taliaferro-smith, L.; Oberlick, E.; Liu, T.; Mcglothen, T.; Alcaide, T.; Tobin, R.; Donnelly, S.; Commander, R.; Kline, E.; Nagaraju, G.P.; et al. FAK activation is required for IGF1R-mediated regulation of EMT, migration, and invasion in mesenchymal triple negative breast cancer cells. Oncotarget 2015, 6, 4757. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Nicholes, K.; Bustos, D.; Lin, E.; Song, Q.; Stephan, J.; Kirkpatrick, D.S.; Settleman, J. Overcoming EMT-associated resistance to anti-cancer drugs via Src/FAK pathway inhibition. Oncotarget 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Wong, M.; Tan, T.; Kuay, K.; Ng, A.; Chung, V.; Chu, Y.; Matsumura, N.; Lai, H.; Lee, Y.; et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013. [Google Scholar] [CrossRef]
- Yang, J.; Hou, Y.; Zhou, M.; Wen, S.; Zhou, J.; Xu, L.; Tang, X.; Du, Y.E.; Hu, P.; Liu, M. Twist induces epithelial-mesenchymal transition and cell motility in breast cancer via ITGB1-FAK/ILK signaling axis and its associated downstream network. Int. J. Biochem. Cell Biol. 2016, 7, 62–71. [Google Scholar] [CrossRef]
- Kim, M.S.; Lee, W.S.; Jeong, J.; Kim, S.; Jin, W. Induction of metastatic potential by TrkB via activation of IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget 2015, 6, 40158–40171. [Google Scholar] [CrossRef] [Green Version]
- Vesuna, F.; Lisok, A.; Kimble, B.; Raman, V. Twist Modulates Breast Cancer Stem Cells by Transcriptional Regulation of CD24 Expression. Neoplasia 2009, 11, 1318–1328. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.; Yang, J. Previews Unraveling the TWIST between EMT and Cancer Stemness. Stem Cell 2015, 16, 1–2. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Shay, G.; Lynch, C.C.; Fingleton, B. Moving targets: Emerging roles for MMPs in cancer progression and metastasis. Matrix Biol. 2015, 44, 200–206. [Google Scholar] [CrossRef]
- Gagliano, N. Tumor—Stroma Cross-Talk in Human Pancreatic Ductal Adenocarcinoma: A Focus on the Effect of the Extracellular Matrix on Tumor Cell Phenotype and Invasive Potential. Cells 2018. [Google Scholar] [CrossRef]
- Walker, C.; Mojares, E. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef]
- Lee, Y.; Albig, A.R.; Regner, M.; Schiemann, B.J.; Schiemann, W.P. Fibulin-5 initiates epithelial–mesenchymal transition (EMT) and enhances EMT induced by TGF- b in mammary epithelial cells via a MMP-dependent mechanism. Carcinogenesis 2008, 29, 2243–2251. [Google Scholar] [CrossRef]
- Hotulainen, P.; Lappalainen, P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J. Cell Biol. 2006, 173, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Haynes, J.; Srivastava, J.; Madson, N.; Wittmann, T.; Barber, D.L. Dynamic actin remodeling during epithelial—Mesenchymal transition depends on increased moesin expression. Mol. Biol. Cell. 2011. [Google Scholar] [CrossRef]
- Kato, S.; Abarzua-catalan, L.; Trigo, C.; Delpiano, A.; García, K.; Ibañez, C.; Hormazábal, K.; Diaz, D.; Brañes, J.; Castellón, E.; et al. Leptin stimulates migration and invasion and maintains cancer stem-like properties in ovarian cancer cells: An explanation for poor outcomes in obese women. Oncotarget 2015, 6, 211000. [Google Scholar] [CrossRef]
- Jaffe, T.; Schwartz, B. Leptin promotes motility and invasiveness in human colon cancer cells by activating multiple signal-transduction pathways. Int. J. Cancer 2008, 2556, 2543–2556. [Google Scholar] [CrossRef]
- He, J.; Wei, X.; Li, S.; Liu, Y.; Hu, H.; Li, Z.; Kuang, X.; Wang, L. Adipocyte-derived IL-6 and leptin promote breast Cancer metastasis via upregulation of Lysyl Hydroxylase-2 expression. Cell Commun. Signal. 2018, 16, 100. [Google Scholar] [CrossRef]
- Liang, J.; Feng, J.; Ka, W.; Wu, K.; Xiao, J.; Wu, Z.; Han, D.; Zhu, Y.; Qiu, G. Leptin-Mediated Cytoskeletal Remodeling in Chondrocytes Occurs Via the RhoA/ROCK Pathway. J. Orthop. Res. 2011, 29, 369–374. [Google Scholar] [CrossRef]
- Albiges-Rizo, C.; Destaing, O.; Fourcade, B.; Planus, E.; Block, M.R. Actin machinery and mechanosensitivity in invadopodia, podosomes and focal adhesions. J. Cell Sci. 2009, 122, 3037–3049. [Google Scholar] [CrossRef] [Green Version]
- Desai, B.; Chellaiah, M.A. Invadopodia and Matrix Degradation, a New Property of Prostate Cancer Cells during Migration and Invasion. J. Biol. Chem. 2008, 283, 13856–13866. [Google Scholar] [CrossRef]
- Weaver, A.M. Invadopodia: Specialized cell structures for cancer invasion. Clin. Exp. Metas. 2006, 23, 97–105. [Google Scholar] [CrossRef]
- Jacob, A.; Prekeris, R. The regulation of MMP targeting to Invadopodia during cancer metastasis. Front. Cell Dev. Biol. 2015, 3, 1–9. [Google Scholar] [CrossRef]
- Detection, I.; Assay, G.D. Invadopodia Detection and Gelatin Degradation Assay. HHS Pub. Access 2018, 3, 1–9. [Google Scholar] [CrossRef]
- Beghein, E.; Devriese, D.; Van Hoey, E.; Gettemans, J. Cortactin and fascin-1 regulate extracellular vesicle release by controlling endosomal trafficking or Invadopodia formation and function. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Bailey, K.M.; Airik, M.; Krook, M.A.; Pedersen, E.A.; Lawlor, E.R. Micro-Environmental Stress Induces Src-Dependent Activation of Invadopodia and Cell Migration in Ewing Sarcoma. NEO 2016, 18, 480–488. [Google Scholar] [CrossRef]
- Parekh, A.; Weaver, A.M. Regulation of Invadopodia by mechanical signaling. HHS Pub. Access 2017, 343, 89–95. [Google Scholar] [CrossRef]
- Argolo, D.F.; Hudis, C.A.; Iyengar, N.M. The Impact of Obesity on Breast Cancer. Curr. Oncol. Rep. 2018, 20. [Google Scholar] [CrossRef]
- MITRA, S.K.; Schlaepfer, D.D. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef]
- Heida, N.; Leifheit-Nestler, M.; Schroeter, M.R.; Mu, J.; Cheng, I.; Henkel, S.; Limbourg, A.; Limbourg, F.P.; Alves, F.; Quigley, J.P.; et al. Leptin Enhances the Potency of Circulating Angiogenic Cells Via Src Kinase and Integrin αvβ5 Implications for Angiogenesis in Human Obesity. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 200–206. [Google Scholar] [CrossRef]
- Ratke, J.; Entschladen, F.; Niggemann, B.; Zänker, K.S.; Lang, K. Leptin stimulates the migration of colon carcinoma cells by multiple signaling pathways. Endocr. Relat. Cancer. 2010, 17, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Alshaker, H.; Krell, J.; Frampton, A.E.; Waxman, J.; Blyuss, O.; Zaikin, A.; Winkler, M.; Stebbing, J.; Yagï, E.; Pchejetski, D. Leptin induces upregulation of sphingosine kinase 1 in oestrogen receptor-negative breast cancer via Src family kinase-mediated, janus kinase 2-independent pathway. Breast Cancer Res. 2014, 16, 1–15. [Google Scholar] [CrossRef]
- Chen, W.; Gao, Q.; Han, S.; Pan, F.; Fan, W. The CCL2/CCR2 axis enhances IL-6-induced epithelial-mesenchymal transition by cooperatively activating STAT3-Twist signaling. Tumor Biol. 2015, 973–981. [Google Scholar] [CrossRef]
- Zhang, C.; Guo, F.; Xu, G.; Ma, J.; Shao, F. STAT3 cooperates with Twist to mediate epithelial-mesenchymal transition in human hepatocellular carcinoma cells. Oncol. Rep. 2015, 33, 1872–1882. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-C.; Tai, H.-C.; Lin, T.-H.; Wang, S.-W.; Lin, C.-Y.; Chao, C.-C.; Yu, H.-J.; Tsai, Y.-C.; Lai, Y.-W.; Lin, C.-W.; et al. CCN3 promotes epithelial-mesenchymal transition in prostate cancer via FAK/Akt/HIF-1α-induced Twist expression. Oncotarget 2017, 8, 74506–74518. [Google Scholar] [CrossRef]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.H.; Zheng, G. E-Cadherin/β-catenin complex and the epithelial barrier. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef]
- Mishra, A.K.; Parish, C.R.; Wong, M.L.; Licinio, J.; Blackburn, A.C. Leptin signals via TGFB1 to promote metastatic potential and stemness in breast cancer. PLoS ONE 2017, 12, e0178454. [Google Scholar] [CrossRef]
- Schmalhofer, O.; Brabletz, S.; Brabletz, T. E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009, 28, 151–166. [Google Scholar] [CrossRef]
- Nadanaka, S.; Kinouchi, H.; Kitagawa, H. Chondroitin sulfate–mediated N-cadherin/B-catenin signaling is associated with basal-like breast cancer cell invasion. J. Biol. Chem. 2018, 293, 444–465. [Google Scholar] [CrossRef]
- Wang, D.; Rai, B.; Qi, F.; Liu, T.; Wang, J. Influence of the Twist gene on the invasion and metastasis of colon cancer. Oncol. Rep. 2018, 39, 31–44. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, X.; Liang, Y.; Chen, W.; Zhang, F. Over-Expressed Twist Associates with Markers of Epithelial Mesenchymal Transition and Predicts Poor Prognosis in Breast Cancers via ERK and Akt Activation. PLoS ONE 2015, 10, e0135851. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metas. Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [Green Version]
- Mori, M.; Nakagami, H.; Koibuchi, N.; Miura, K.; Takami, Y.; Koriyama, H.; Hayashi, H.; Sabe, H.; Mochizuki, N.; Morishita, R.; et al. Zyxin Mediates Actin Fiber Reorganization in Epithelial-Mesenchymal Transition and Contributes to Endocardial Morphogenesis. Cardiovascular 2009, 17, S353. [Google Scholar] [CrossRef]
- Xiong, N.; Li, S.; Tang, K.; Bai, H.; Peng, Y.; Yang, H.; Wu, C.; Liu, Y. Involvement of caveolin-1 in low shear stress-induced breast cancer cell motility and adhesion: Roles of FAK/Src and ROCK/p-MLC pathways. Biochim. Biophys. Acta BBA Mol. Cell Res. 2017, 1864, 12–22. [Google Scholar] [CrossRef]
- Kim, S.; Choi, J.H.; Lim, H.I.; Lee, S.K.; Kim, W.W.; Cho, S.; Kim, J.S.; Kim, J.H.; Choe, J.H.; Nam, S.J.; et al. EGF-induced MMP-9 expression is mediated by the JAK3/ERK pathway, but not by the JAK3/STAT-3 pathway in a SKBR3 breast cancer cell line. Cell Signal. 2009, 21, 892–898. [Google Scholar] [CrossRef]
- Cavallo, F.; De Giovanni, C.; Nanni, P.; Forni, G.; Lollini, P.-L. 2011: The immune hallmarks of cancer. Cell 2011, 60, 319–326. [Google Scholar] [CrossRef]
- Jeong, Y.; Cho, H.; Chung, F.; Wang, X. Isothiocyanates suppress the invasion and metastasis of tumors by targeting FAK/MMP-9 activity. Oncotarget 2017, 8, 63949–63962. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, N.; Pang, B.; Tong, D.; Sun, D.; Sun, H. TRIB1 promotes colorectal cancer cell migration and invasion through activation MMP-2 via FAK/Src and ERK pathways. Oncotarget 2017, 8, 47931–47942. [Google Scholar] [CrossRef]
- Jung, O.; Lee, J.; Jin, Y.; Yun, J.; Son, Y.; Youl, J.; Ryou, C.; Yeol, S. Timosaponin AIII inhibits migration and invasion of A549 human non-small-cell lung cancer cells via attenuations of MMP-2 and MMP-9 by inhibitions of ERK1/2, Src/FAK and β-catenin signaling pathways. Bioorg. Med. Chem. Lett. 2016, 26, 7–11. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olea-Flores, M.; Zuñiga-Eulogio, M.; Tacuba-Saavedra, A.; Bueno-Salgado, M.; Sánchez-Carvajal, A.; Vargas-Santiago, Y.; Mendoza-Catalán, M.A.; Pérez Salazar, E.; García-Hernández, A.; Padilla-Benavides, T.; et al. Leptin Promotes Expression of EMT-Related Transcription Factors and Invasion in a Src and FAK-Dependent Pathway in MCF10A Mammary Epithelial Cells. Cells 2019, 8, 1133. https://doi.org/10.3390/cells8101133
Olea-Flores M, Zuñiga-Eulogio M, Tacuba-Saavedra A, Bueno-Salgado M, Sánchez-Carvajal A, Vargas-Santiago Y, Mendoza-Catalán MA, Pérez Salazar E, García-Hernández A, Padilla-Benavides T, et al. Leptin Promotes Expression of EMT-Related Transcription Factors and Invasion in a Src and FAK-Dependent Pathway in MCF10A Mammary Epithelial Cells. Cells. 2019; 8(10):1133. https://doi.org/10.3390/cells8101133
Chicago/Turabian StyleOlea-Flores, Monserrat, Miriam Zuñiga-Eulogio, Arvey Tacuba-Saavedra, Magdalena Bueno-Salgado, Andrea Sánchez-Carvajal, Yovani Vargas-Santiago, Miguel A. Mendoza-Catalán, Eduardo Pérez Salazar, Alejandra García-Hernández, Teresita Padilla-Benavides, and et al. 2019. "Leptin Promotes Expression of EMT-Related Transcription Factors and Invasion in a Src and FAK-Dependent Pathway in MCF10A Mammary Epithelial Cells" Cells 8, no. 10: 1133. https://doi.org/10.3390/cells8101133