The Core Proteome of Biofilm-Grown Clinical Pseudomonas aeruginosa Isolates

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Sample Preparation and Proteome Analysis
2.3. Data Processing
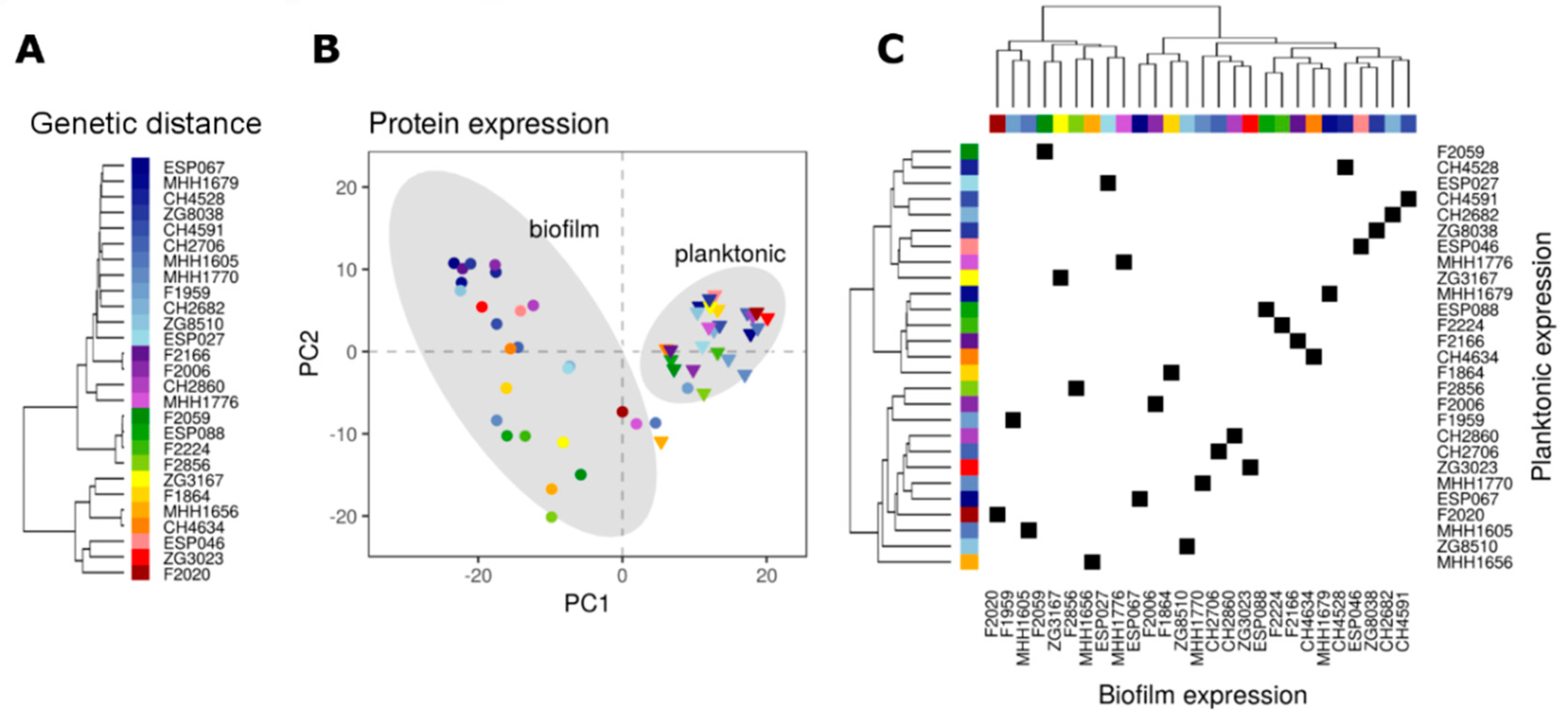
3. Results
3.1. SWATH-MS Characterizes Protein Expression Profiles in Multiple P. aeruginosaIsolates
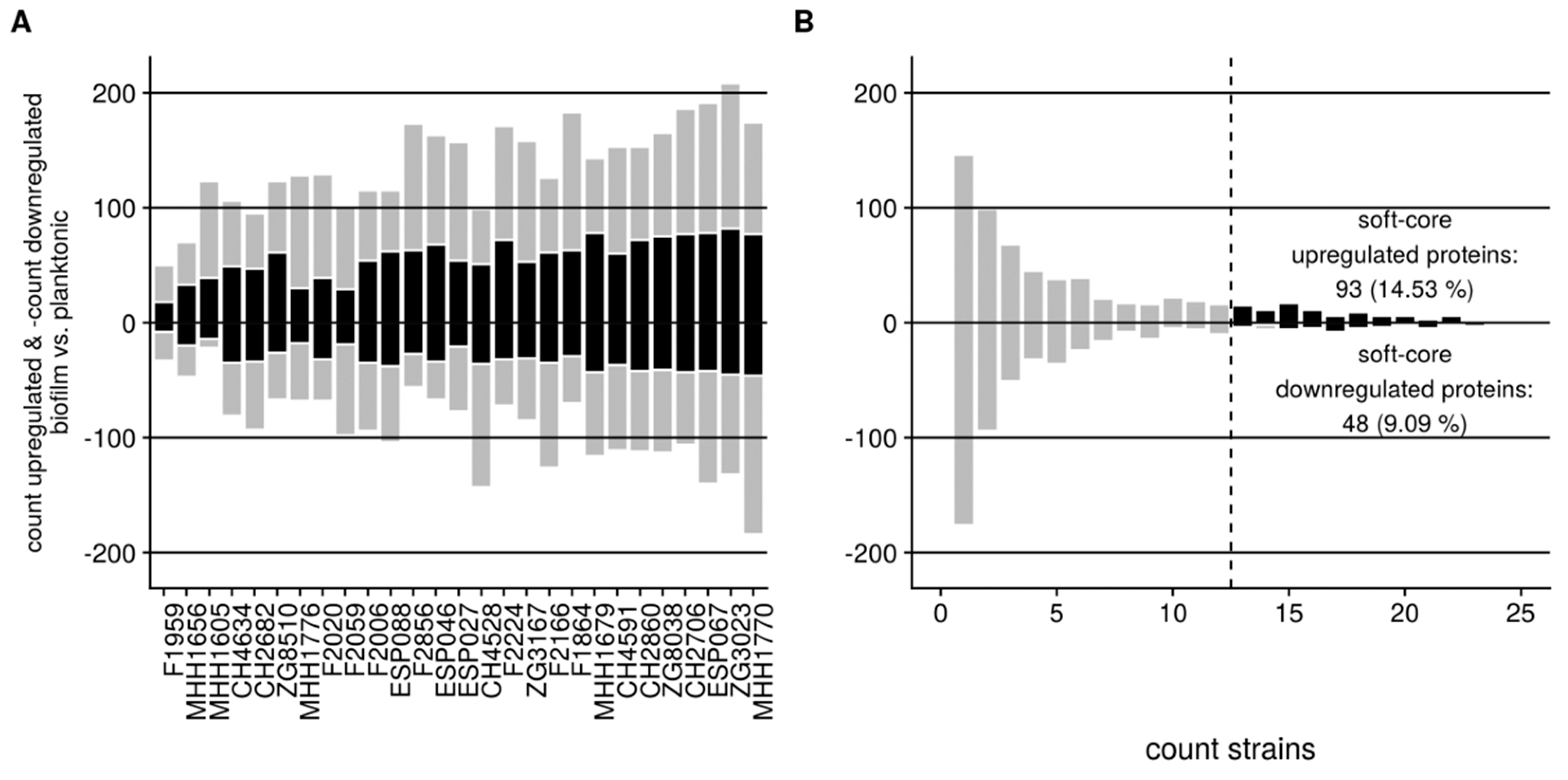
3.2. Proteome Expression Patterns Differ Between Physiological States
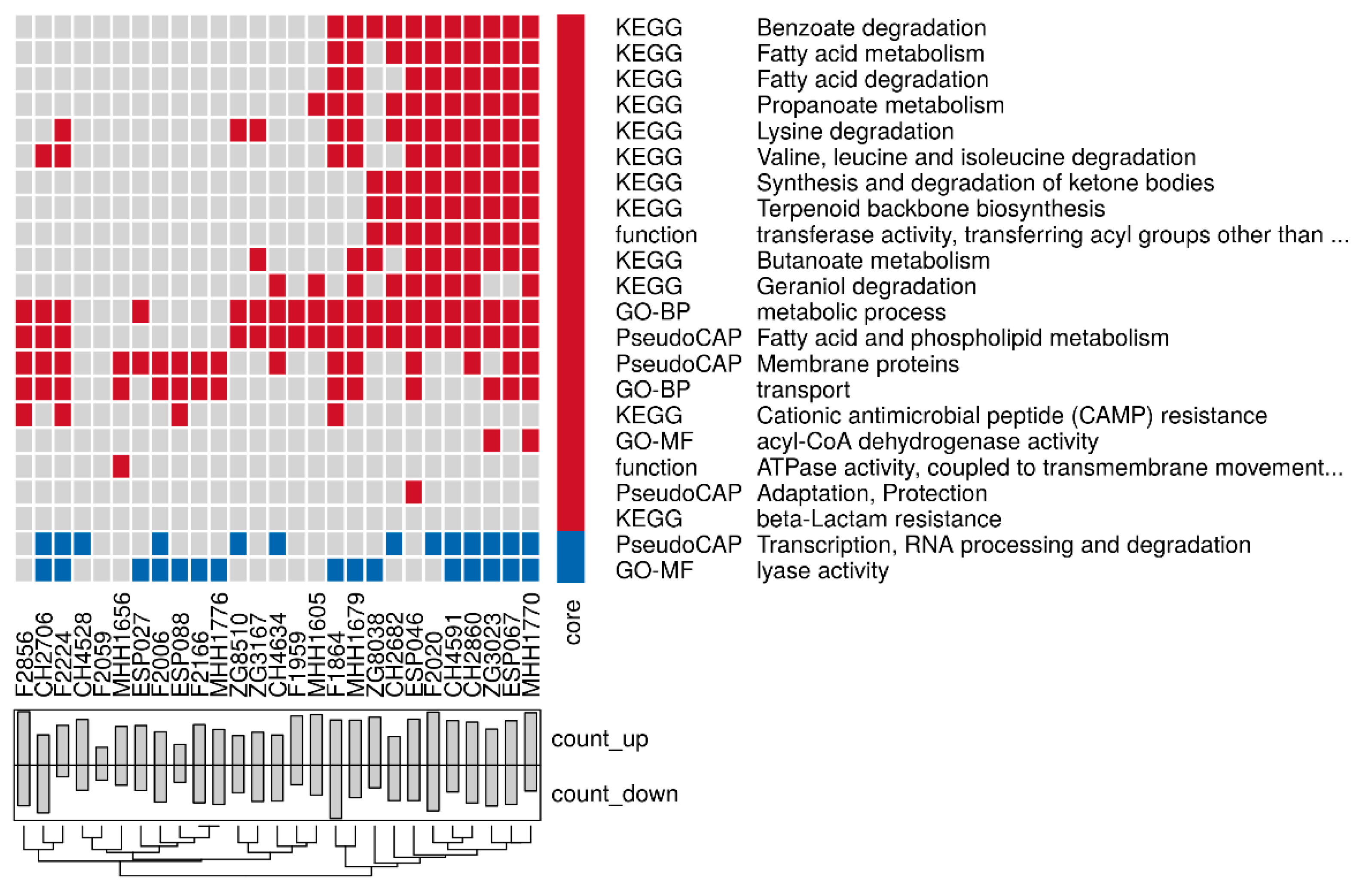
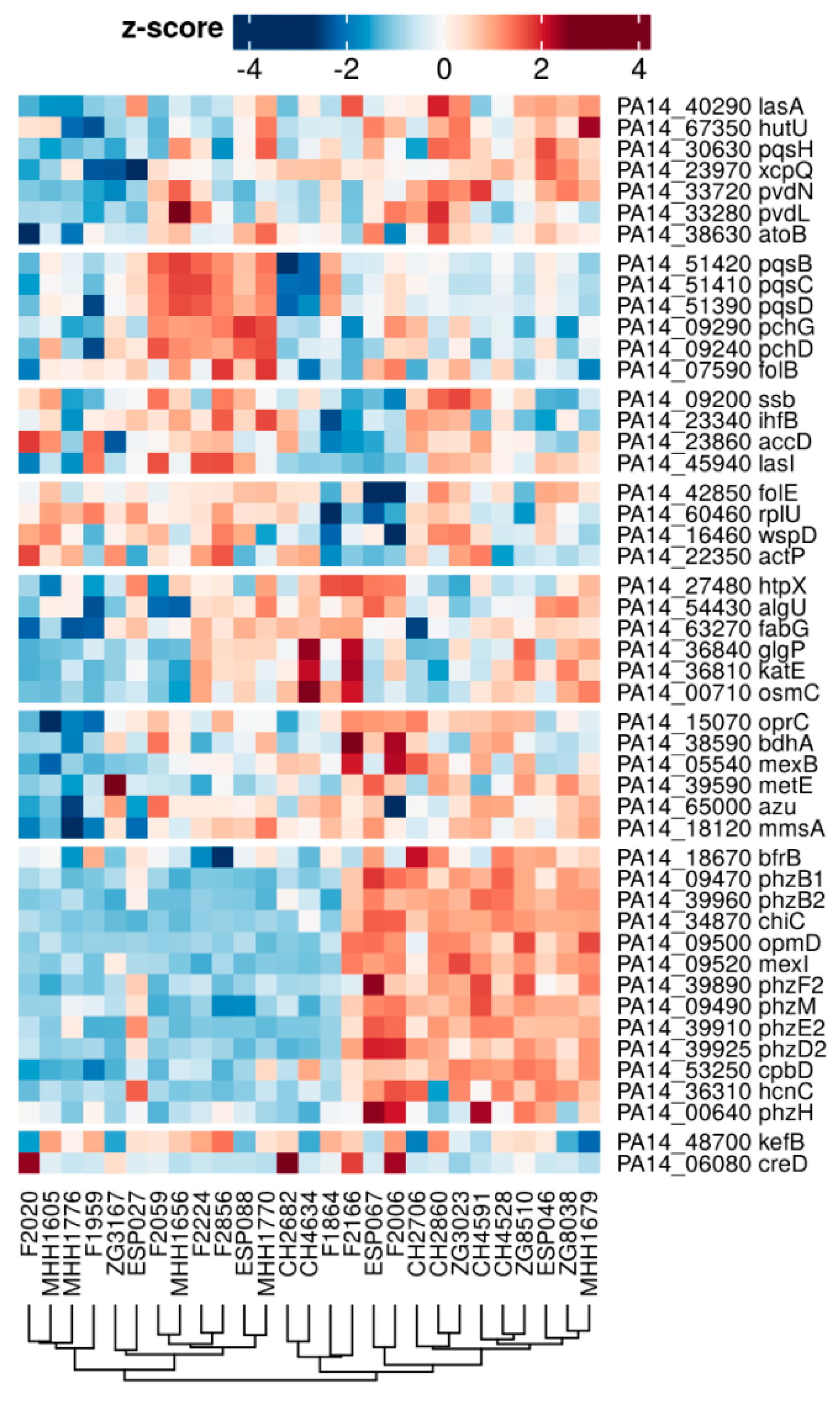
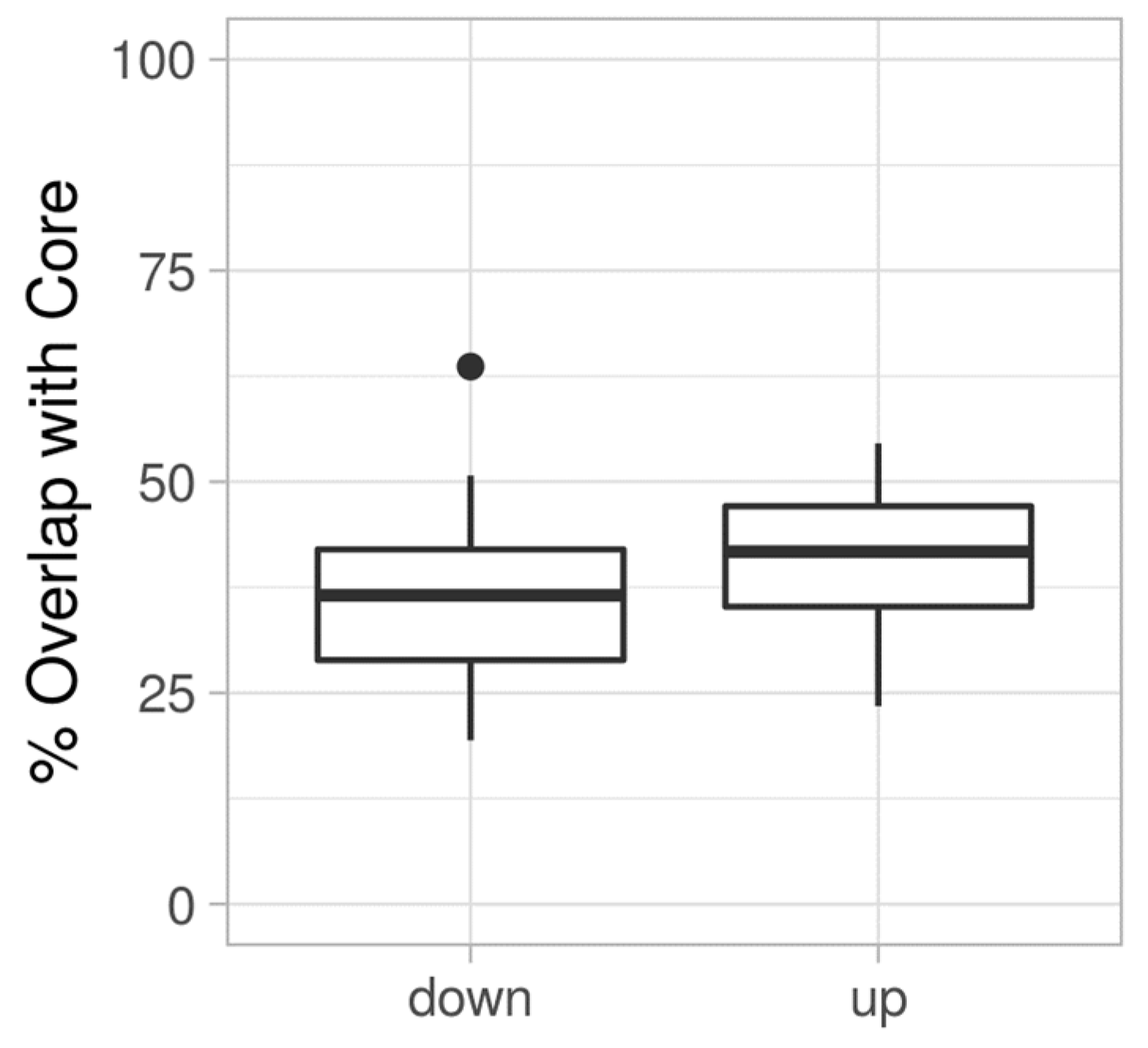
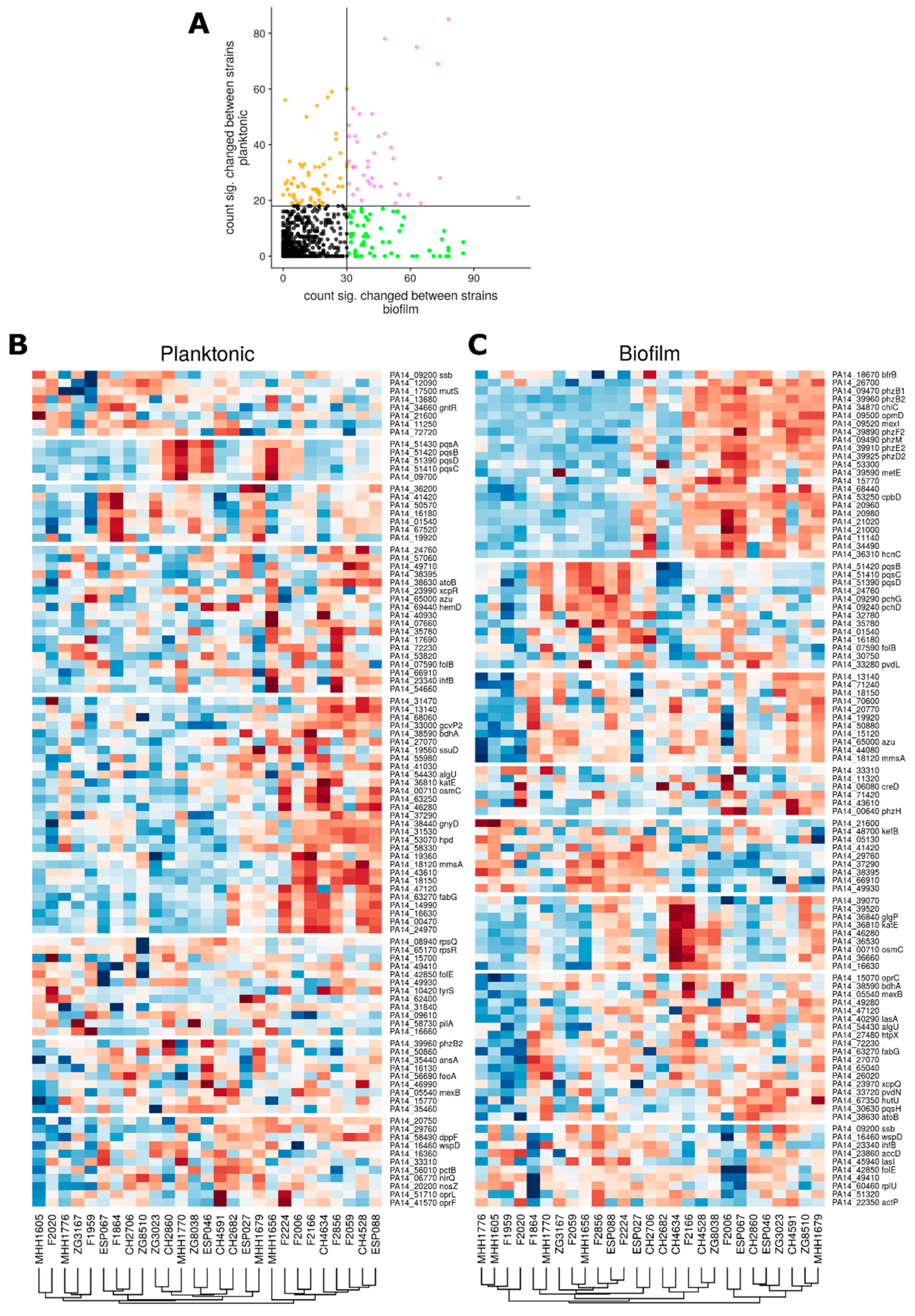
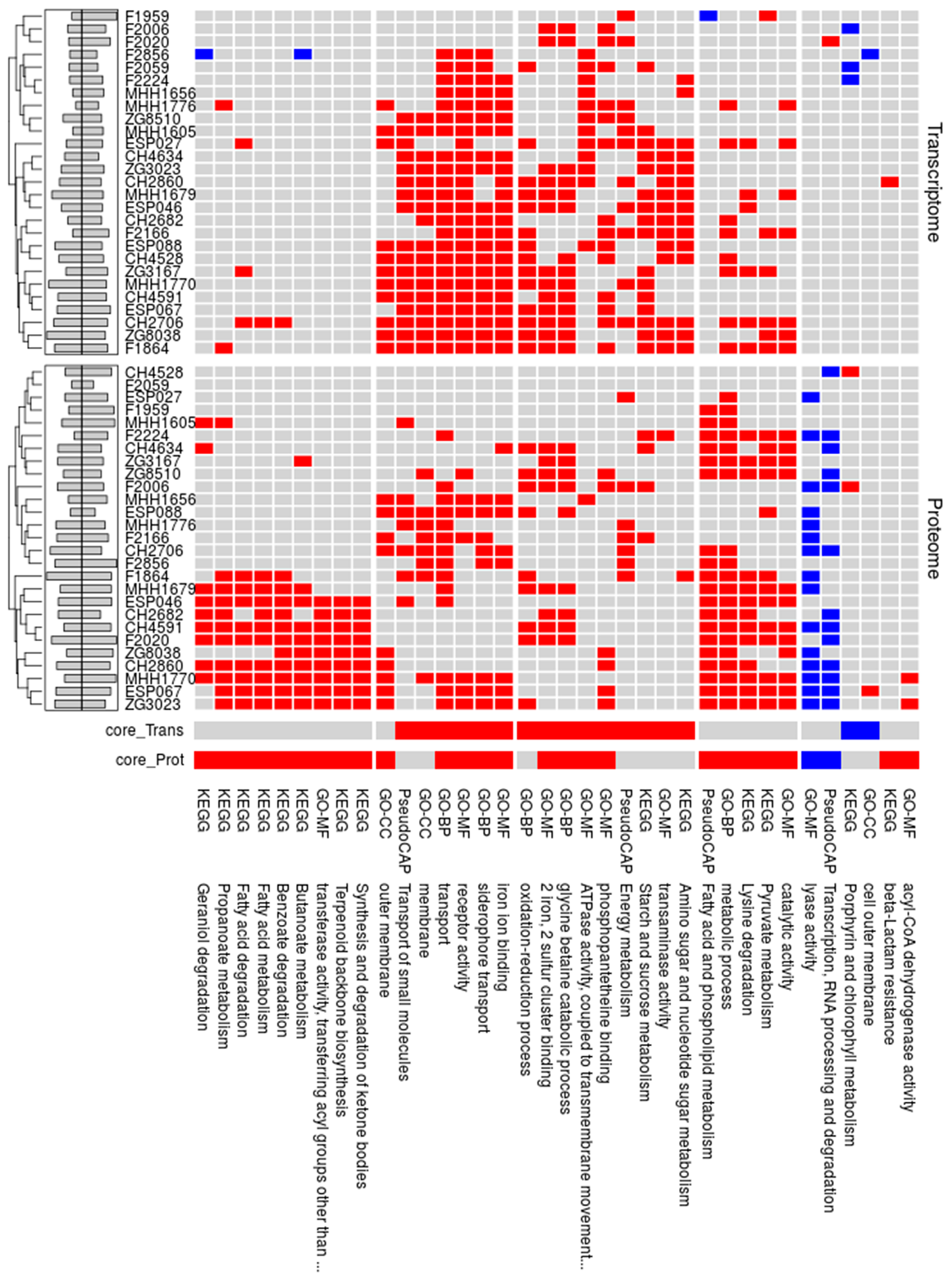
3.3. Proteome Expression Patterns Differ Between Groups of Clinical Isolates
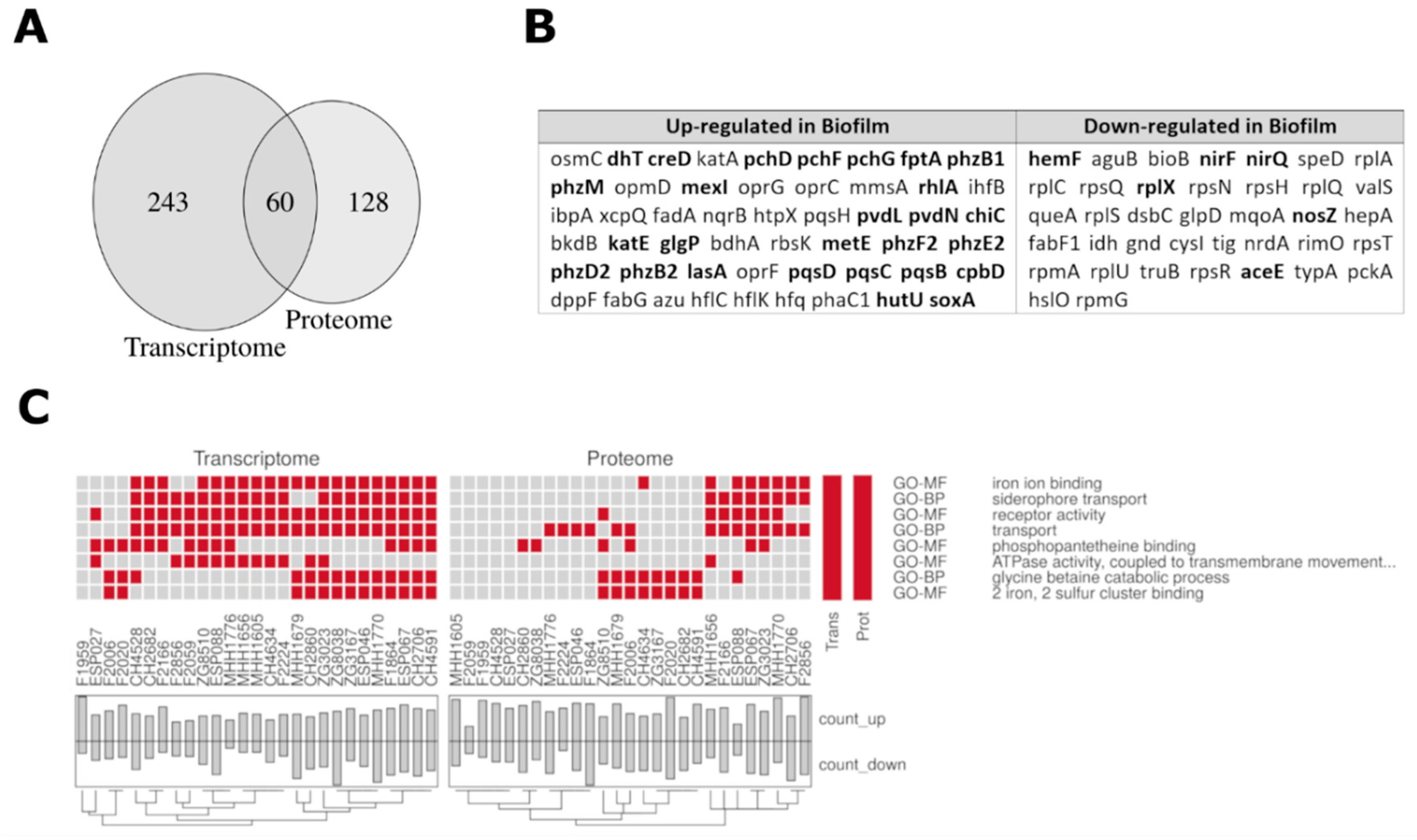
3.4. Comparison of the Proteome and Transcriptome Biofilm Profiles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein/mRNA (median) | Condition | PA14_ID | Gene.Name | Product.Name |
|---|---|---|---|---|
| 3.06 | pl | PA14_21010 | FAD-dependent monooxygenase | |
| 2.67 | pl | PA14_17930 | glpD | glycerol-3-phosphate dehydrogenase |
| 2.64 | pl | PA14_34840 | non-ribosomal peptide synthetase | |
| 2.63 | pl | PA14_21020 | non-ribosomal peptide synthetase | |
| 2.60 | bf | PA14_35790 | homospermidine synthase | |
| 2.41 | pl | PA14_33280 | pvdL | peptide synthase |
| 2.38 | pl | PA14_09280 | pchF | pyochelinsynthetase |
| 2.36 | pl | PA14_00640 | phzH | potential phenazine-modifying enzyme |
| 2.29 | pl | PA14_27370 | ATP-dependent RNA helicase | |
| 2.26 | bf | PA14_15580 | Type II restriction enzyme, methylase subunit | |
| −2.96 | bf | PA14_15980 | rimM | 16S rRNA-processing protein RimM |
| −2.99 | pl | PA14_15980 | rimM | 16S rRNA-processing protein RimM |
| −3.08 | bf | PA14_18670 | bfrB | bacterioferritin |
| −3.12 | pl | PA14_64520 | bacterioferritin | |
| −3.15 | pl | PA14_24770 | hypothetical protein | |
| −3.26 | pl | PA14_68260 | c4-dicarboxylate-binding protein | |
| −3.53 | pl | PA14_54430 | algU | RNA polymerase sigma factor AlgU |
| −3.64 | pl | PA14_18670 | bfrB | bacterioferritin |
| −3.65 | pl | PA14_23340 | ihfB | integration host factor subunit beta |
| −4.22 | pl | PA14_65000 | azu | azurin |



References
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the Natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Watnick, P.; Kolter, R. Biofilm, city of microbes. J. Bacteriol. 2000, 182, 2675–2679. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.-C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Teschler, J.K.; Zamorano-Sánchez, D.; Utada, A.S.; Warner, C.J.A.; Wong, G.C.L.; Linington, R.G.; Yildiz, F.H. Living in the matrix: Assembly and control of Vibrio cholerae biofilms. Nat. Rev. Microbiol. 2015, 13, 255. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S.; Franklin, M.J.; Williamson, K.S.; Folsom, J.P.; Boegli, L.; James, G.A. Contribution of Stress Responses to Antibiotic Tolerance in Pseudomonas aeruginosa Biofilms. Antimicrob. Agents Chemother. 2015, 59, 3838–3847. [Google Scholar] [CrossRef] [PubMed]
- Townsley, L.; Sison Mangus, M.P.; Mehic, S.; Yildiz, F.H. Response of Vibrio cholerae to Low-Temperature Shifts: CspV Regulation of Type VI Secretion, Biofilm Formation, and Association with Zooplankton. Appl. Environ. Microbiol. 2016, 82, 4441–4452. [Google Scholar] [CrossRef] [PubMed]
- Hughes, G.; Webber, M.A. Novel approaches to the treatment of bacterial biofilm infections. Br. J. Pharmacol. 2017, 174, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections; American Association for the Advancement of Science: Washington, DC, USA, 1999; Volume 284, pp. 1318–1322. [Google Scholar]
- Reisner, A.; Høiby, N.; Tolker-Nielsen, T.; Molin, S. Microbial Pathogenesis and Biofilm Development. In Concepts in Bacterial Virulence; KARGER: Basel, Switzerland, 2004; Volume 12, pp. 114–131. [Google Scholar]
- Costerton, J.W. Cystic fibrosis pathogenesis and the role of biofilms in persistent infection. Trends Microbiol. 2001, 9, 50–52. [Google Scholar] [CrossRef]
- Khatoon, Z.; McTiernan, C.D.; Suuronen, E.J.; Mah, T.-F.; Alarcon, E.I. Bacterial biofilm formation on implantable devices and approaches to its treatment and prevention. Heliyon 2018, 4, e01067. [Google Scholar] [CrossRef]
- Smith, W.D.; Bardin, E.; Cameron, L.; Edmondson, C.L.; Farrant, K.V.; Martin, I.; Murphy, R.A.; Soren, O.; Turnbull, A.R.; Wierre-Gore, N.; et al. Current and future therapies for Pseudomonas aeruginosa infection in patients with cystic fibrosis. FEMS Microbiol. Lett. 2017, 364. [Google Scholar] [CrossRef]
- McCarthy, R.R.; Mazon-Moya, M.J.; Moscoso, J.A.; Hao, Y.; Lam, J.S.; Bordi, C.; Mostowy, S.; Filloux, A. Cyclic-di-GMP regulates lipopolysaccharide modification and contributes to Pseudomonas aeruginosa immune evasion. Nat. Microbiol. 2017, 2, 17027. [Google Scholar] [CrossRef] [PubMed]
- Valentini, M.; Gonzalez, D.; Mavridou, D.A.; Filloux, A. Lifestyle transitions and adaptive pathogenesis of Pseudomonas aeruginosa. Curr. Opin. Microbiol. 2018, 41, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Park, A.J.; Murphy, K.; Krieger, J.R.; Brewer, D.; Taylor, P.; Habash, M.; Khursigara, C.M. A temporal examination of the planktonic and biofilm proteome of whole cell Pseudomonas aeruginosa PAO1 using quantitative mass spectrometry. Mol. Cell. Proteomics 2014, 13, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Herbst, F.A.; Søndergaard, M.T.; Kjeldal, H.; Stensballe, A.; Nielsen, P.H.; Dueholm, M.S. Major proteomic changes associated with amyloid-induced biofilm formation in Pseudomonas aeruginosa PAO1. J. Proteome Res. 2015, 14, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Guilbaud, M.; Bruzaud, J.; Bouffartigues, E.; Orange, N.; Guillot, A.; Aubert-Frambourg, A.; Monnet, V.; Herry, J.-M.; Chevalier, S.; Bellon-Fontaine, M.-N. Proteomic Response of Pseudomonas aeruginosa PAO1 Adhering to Solid Surfaces. Front. Microbiol. 2017, 8, 1465. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, C.; Filloux, A.; Ghigo, J.-M.; Visick, K.L. Biofilms 2018: A diversity of microbes and mechanisms. J. Bacteriol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Cromie, G.A.; Tan, Z.; Hays, M.; Sirr, A.; Jeffery, E.W.; Dudley, A.M. Transcriptional Profiling of Biofilm Regulators Identified by an Overexpression Screen in Saccharomyces cerevisiae. G3 (Bethesda). 2017, 7, 2845–2854. [Google Scholar] [CrossRef]
- Suriyanarayanan, T.; Qingsong, L.; Kwang, L.T.; Mun, L.Y.; Truong, T.; Seneviratne, C.J. Quantitative Proteomics of Strong and Weak Biofilm Formers of Enterococcus faecalis Reveals Novel Regulators of Biofilm Formation. Mol. Cell. Proteomics 2018, 17, 643–654. [Google Scholar] [CrossRef]
- Wu, X.; Siehnel, R.J.; Garudathri, J.; Staudinger, B.J.; Hisert, K.B.; Ozer, E.A.; Hauser, A.R.; Eng, J.K.; Manoil, C.; Singh, P.K.; et al. In vivo proteome of Pseudomonas aeruginosa in airways of cystic fibrosis patients. J. Proteome Res. 2019, 18, 2601–2612. [Google Scholar] [CrossRef]
- Ormsby, M.J.; Grahame, E.; Burchmore, R.; Davies, R.L. Comparative bioinformatic and proteomic approaches to evaluate the outer membrane proteome of the fish pathogen Yersinia ruckeri. J. Proteomics 2019, 199, 135–147. [Google Scholar] [CrossRef]
- Ayllón, N.; Jiménez-Marín, Á.; Argüello, H.; Zaldívar-López, S.; Villar, M.; Aguilar, C.; Moreno, A.; De La Fuente, J.; Garrido, J.J. Comparative Proteomics Reveals Differences in Host-Pathogen Interaction between Infectious and Commensal Relationship with Campylobacter jejuni. Front. Cell. Infect. Microbiol. 2017, 7, 145. [Google Scholar] [CrossRef]
- Muthusamy, S.; Lundin, D.; Mamede Branca, R.M.; Baltar, F.; González, J.M.; Lehtiö, J.; Pinhassi, J. Comparative proteomics reveals signature metabolisms of exponentially growing and stationary phase marine bacteria. Environ. Microbiol. 2017, 19, 2301–2319. [Google Scholar] [CrossRef]
- Frohnmeyer, E.; Deptula, P.; Nyman, T.A.; Laine, P.K.S.; Vihinen, H.; Paulin, L.; Auvinen, P.; Jokitalo, E.; Piironen, V.; Varmanen, P.; et al. Secretome profiling of Propionibacterium freudenreichii reveals highly variable responses even among the closely related strains. Microb. Biotechnol. 2018, 11, 510–526. [Google Scholar] [CrossRef]
- Koo, H.; Allan, R.N.; Howlin, R.P.; Stoodley, P.; Hall-Stoodley, L. Targeting microbial biofilms: current and prospective therapeutic strategies. Nat. Rev. Microbiol. 2017, 15, 740–755. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteomics 2012, 11, O111.016717. [Google Scholar] [CrossRef]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Miladinović, S.M.; Cheng, L.-Y.; Messner, S.; Ehrenberger, T.; Zanotelli, V.; Butscheid, Y.; Escher, C.; et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics 2015, 14, 1400–1410. [Google Scholar] [CrossRef]
- Collins, B.C.; Hunter, C.L.; Liu, Y.; Schilling, B.; Rosenberger, G.; Bader, S.L.; Chan, D.W.; Gibson, B.W.; Gingras, A.-C.C.; Held, J.M.; et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 2017, 8, 291. [Google Scholar] [CrossRef]
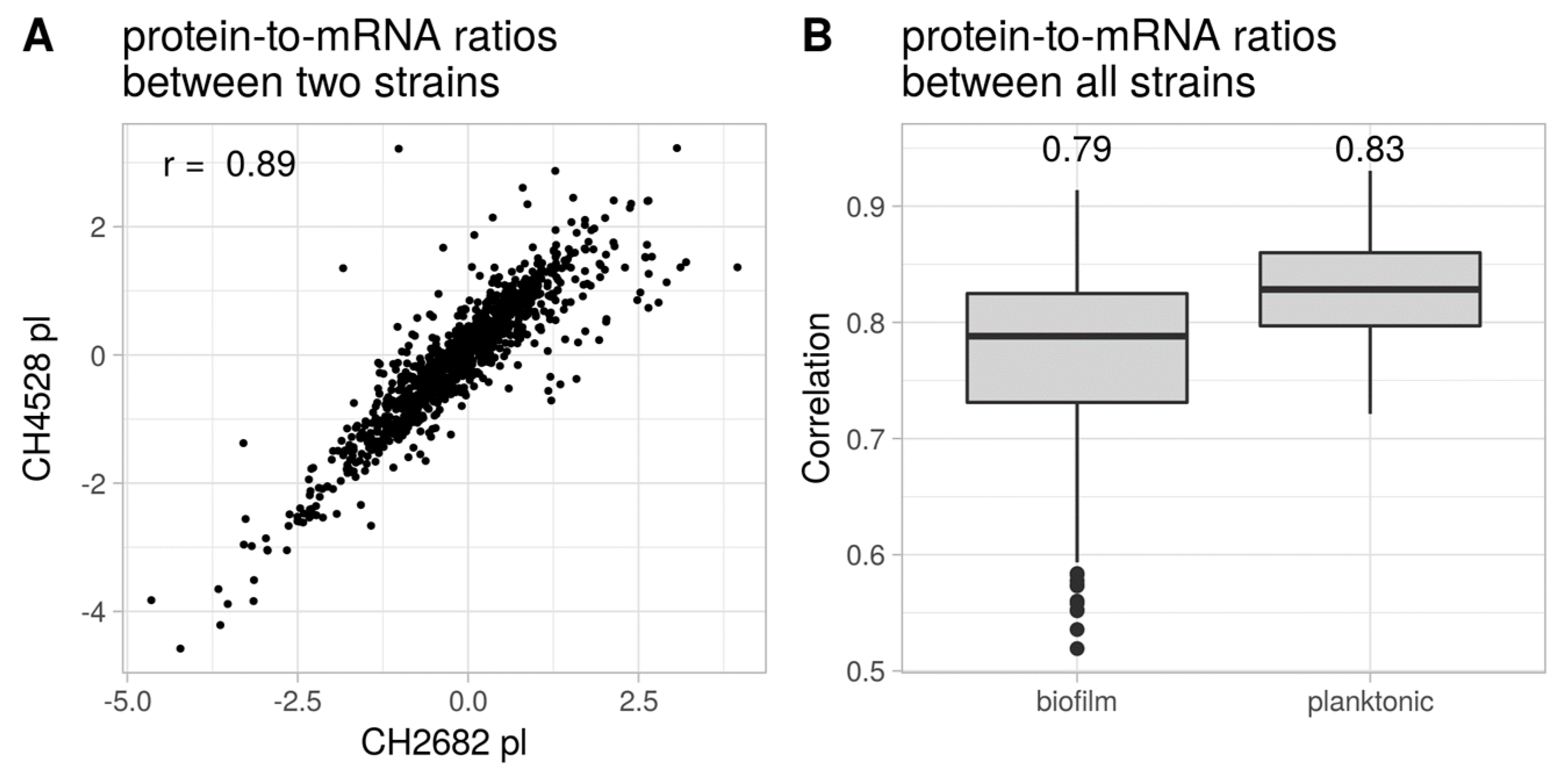
- Kwon, T.; Huse, H.K.; Vogel, C.; Whiteley, M.; Marcotte, E.M. Protein-to-mRNA ratios are conserved between Pseudomonas aeruginosa strains. J. Proteome Res. 2014, 13, 2370–2380. [Google Scholar] [CrossRef]
- Erdmann, J.; Preusse, M.; Khaledi, A.; Pich, A.; Häussler, S. Environment-driven changes of mRNA and protein levels in Pseudomonas aeruginosa. Environ. Microbiol. 2018, 20, 3952–3963. [Google Scholar] [CrossRef]
- Krueger, J.; Pohl, S.; Preusse, M.; Kordes, A.; Rugen, N.; Schniederjans, M.; Pich, A.; Häussler, S. Unravelling post-transcriptional PrmC-dependent regulatory mechanisms in Pseudomonas aeruginosa. Environ. Microbiol. 2016, 18, 3583–3592. [Google Scholar] [CrossRef]
- Vowinckel, J.; Capuano, F.; Campbell, K.; Deery, M.J.; Lilley, K.S.; Ralser, M. The beauty of being (label)-free: sample preparation methods for SWATH-MS and next-generation targeted proteomics. F1000Research 2014, 2, 272. [Google Scholar] [CrossRef]
- Atanassov, I.; Urlaub, H. Increased proteome coverage by combining PAGE and peptide isoelectric focusing: comparative study of gel-based separation approaches. Proteomics 2013, 13, 2947–2955. [Google Scholar] [CrossRef]
- Zhang, Y.; Bilbao, A.; Bruderer, T.; Luban, J.; Strambio-De-Castillia, C.; Lisacek, F.; Hopfgartner, G.; Varesio, E. The Use of Variable Q1 Isolation Windows Improves Selectivity in LC–SWATH–MS Acquisition. J. Proteome Res. 2015, 14, 4359–4371. [Google Scholar] [CrossRef]
- Winsor, G.L.; Lo, R.; Ho Sui, S.J.; Ung, K.S.E.; Huang, S.; Cheng, D.; Ching, W.K.H.; Hancock, R.E.W.; Brinkman, F.S.L. Pseudomonas aeruginosa Genome Database and PseudoCAP: Facilitating community-based, continually updated, genome annotation. Nucleic Acids Res. 2005, 33, D338–D343. [Google Scholar] [CrossRef][Green Version]
- Lambert, J.-P.; Ivosev, G.; Couzens, A.L.; Larsen, B.; Taipale, M.; Lin, Z.-Y.; Zhong, Q.; Lindquist, S.; Vidal, M.; Aebersold, R.; et al. Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat. Methods 2013, 10, 1239–1245. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Khaledi, A.; Weimann, A.; Schniederjans, M.; Asgari, E.; Kuo, T.-H.; Oliver, A.; Cabot, G.; Kola, A.; Gastmeier, P.; Hogardt, M.; et al. Fighting antimicrobial resistance in Pseudomonas aeruginosa with machine learning-enabled molecular diagnostics. bioRxiv 2019, 643676. [Google Scholar]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
- Kordes, A.; Grahl, N.; Koska, M.; Preusse, M.; Arce-Rodriguez, A.; Abraham, W.R.; Kaever, V.; Häussler, S. Establishment of an induced memory response in Pseudomonas aeruginosa during infection of a eukaryotic host. ISME J. 2019, 1. [Google Scholar] [CrossRef]
- Lunter, G.; Goodson, M. Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011, 21, 936–939. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- R Development Core Team R: A Language and Environment for Statistical Computing. 2016. Available online: https://www.R-project.org/ (accessed on 1 April 2019).
- Dötsch, A.; Schniederjans, M.; Khaledi, A.; Hornischer, K.; Schulz, S.; Bielecka, A.; Eckweiler, D.; Pohl, S.; Häussler, S. The Pseudomonas aeruginosa Transcriptional Landscape Is Shaped by Environmental Heterogeneity and Genetic Variation. MBio 2015, 6, e00749. [Google Scholar] [CrossRef]
- de Matos Simoes, R.; Emmert-Streib, F. Bagging Statistical Network Inference from Large-Scale Gene Expression Data. PLoS ONE 2012, 7, e33624. [Google Scholar] [CrossRef]
- Corona, F.; Reales-Calderón, J.A.; Gil, C.; Martínez, J.L. The development of a new parameter for tracking post-transcriptional regulation allows the detailed map of the Pseudomonas aeruginosa Crc regulon. Sci. Rep. 2018, 8, 16793. [Google Scholar] [CrossRef]
- Losensky, G.; Jung, K.; Urlaub, H.; Pfeifer, F.; Fröls, S.; Lenz, C. Shedding light on biofilm formation of Halobacterium salinarum R1 by SWATH-LC/MS/MS analysis of planktonic and sessile cells. Proteomics 2017, 17, 1600111. [Google Scholar] [CrossRef]
- Hornischer, K.; Khaledi, A.; Pohl, S.; Schniederjans, M.; Pezoldt, L.; Casilag, F.; Muthukumarasamy, U.; Bruchmann, S.; Thöming, J.; Kordes, A.; et al. BACTOME - a reference database to explore the sequence- and gene expression-variation landscape of Pseudomonas aeruginosa clinical isolates. Nucleic Acids Res. 2019, 47, D716–D720. [Google Scholar] [CrossRef]
- Yates, J.R., III. Recent technical advances in proteomics. F1000Research 2019, 8, 351. [Google Scholar] [CrossRef]
- Malmström, L.; Bakochi, A.; Svensson, G.; Kilsgård, O.; Lantz, H.; Petersson, A.C.; Hauri, S.; Karlsson, C.; Malmström, J. Quantitative proteogenomics of human pathogens using DIA-MS. J. Proteomics 2015, 129, 98–107. [Google Scholar] [CrossRef]
- Kamath, K.S.; Krisp, C.; Chick, J.; Pascovici, D.; Gygi, S.P.; Molloy, M.P. Pseudomonas aeruginosa Proteome under Hypoxic Stress Conditions Mimicking the Cystic Fibrosis Lung. J. Proteome Res. 2017, 16, 3917–3928. [Google Scholar] [CrossRef]
- Penesyan, A.; Kumar, S.S.; Kamath, K.; Shathili, A.M.; Venkatakrishnan, V.; Krisp, C.; Packer, N.H.; Molloy, M.P.; Paulsen, I.T. Genetically and Phenotypically Distinct Pseudomonas aeruginosa Cystic Fibrosis Isolates Share a Core Proteomic Signature. PLoS ONE 2015, 10, e0138527. [Google Scholar] [CrossRef]
- Patell, S.; Gu, M.; Davenport, P.; Givskov, M.; Waite, R.D.; Welch, M. Comparative microarray analysis reveals that the core biofilm-associated transcriptome of Pseudomonas aeruginosa comprises relatively few genes. Environ. Microbiol. Rep. 2010, 2, 440–448. [Google Scholar] [CrossRef]
- Tolker-Nielsen, T. Biofilm Development. Microbiol. Spectr. 2015, 3, MB-0001-2014. [Google Scholar] [CrossRef]
- Malone, K.M.; Rue-Albrecht, K.; Magee, D.A.; Conlon, K.; Schubert, O.T.; Nalpas, N.C.; Browne, J.A.; Smyth, A.; Gormley, E.; Aebersold, R.; et al. Comparative ’omics analyses differentiate Mycobacterium tuberculosis and Mycobacterium bovis and reveal distinct macrophage responses to infection with the human and bovine tubercle bacilli. Microb. Genomics 2018, 4, e000163. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erdmann, J.; Thöming, J.G.; Pohl, S.; Pich, A.; Lenz, C.; Häussler, S. The Core Proteome of Biofilm-Grown Clinical Pseudomonas aeruginosa Isolates. Cells 2019, 8, 1129. https://doi.org/10.3390/cells8101129
Erdmann J, Thöming JG, Pohl S, Pich A, Lenz C, Häussler S. The Core Proteome of Biofilm-Grown Clinical Pseudomonas aeruginosa Isolates. Cells. 2019; 8(10):1129. https://doi.org/10.3390/cells8101129
Chicago/Turabian StyleErdmann, Jelena, Janne G. Thöming, Sarah Pohl, Andreas Pich, Christof Lenz, and Susanne Häussler. 2019. "The Core Proteome of Biofilm-Grown Clinical Pseudomonas aeruginosa Isolates" Cells 8, no. 10: 1129. https://doi.org/10.3390/cells8101129
APA StyleErdmann, J., Thöming, J. G., Pohl, S., Pich, A., Lenz, C., & Häussler, S. (2019). The Core Proteome of Biofilm-Grown Clinical Pseudomonas aeruginosa Isolates. Cells, 8(10), 1129. https://doi.org/10.3390/cells8101129