Estrogen-Receptor Expression and Function in Female Reproductive Disease




{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Estrogen Receptors
2.1. ERα and ERβ
2.2. G Protein-Coupled Estrogen Receptor
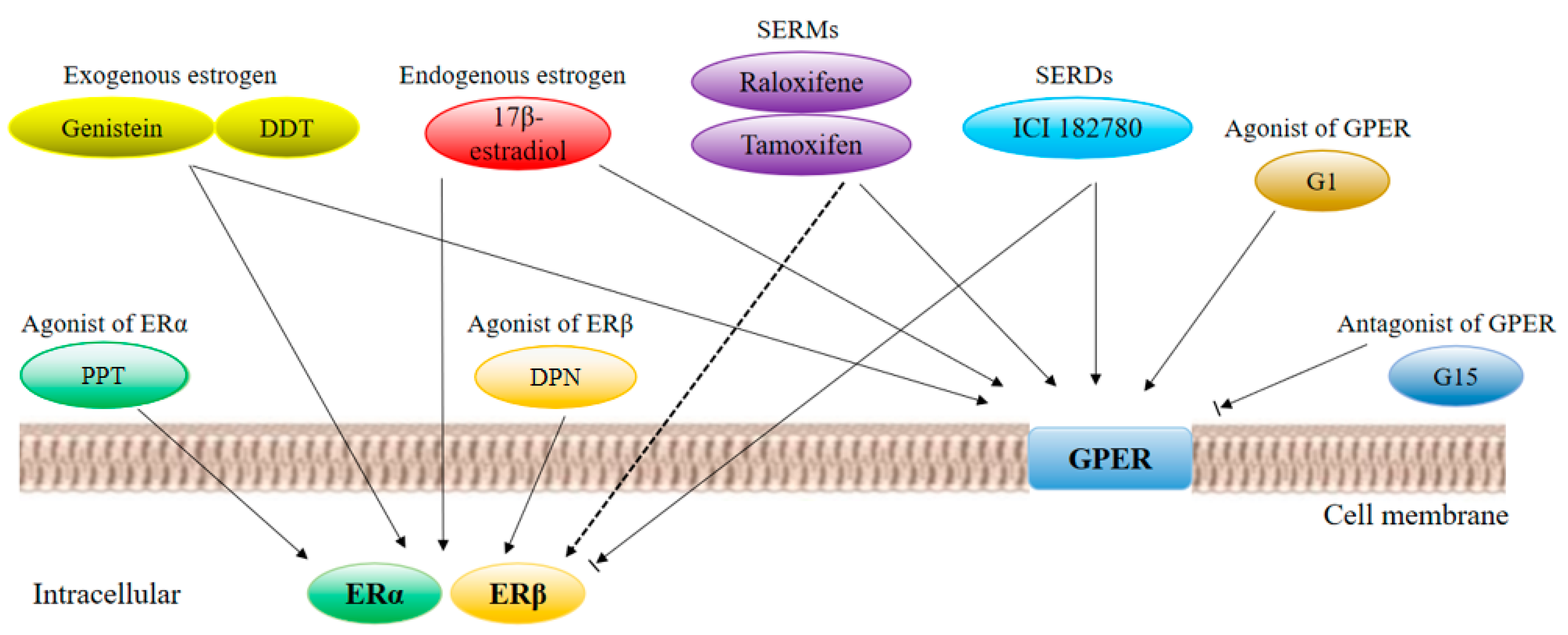
2.3. Estrogen Receptor Ligands
2.4. Cell Mechanisms
3. ERs and Female Reproductive Diseases
3.1. ERs and Ovarian Cancer
3.2. ERs and Endometriosis
3.3. ERs and Polycystic Ovary Syndrome
4. Therapeutic Drugs
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Callard, G.V.; Tarrant, A.M.; Novillo, A.; Yacci, P.; Ciaccia, L.; Vajda, S.; Chuang, G.; Kozakov, D.; Greytak, S.R.; Sawyer, S.; et al. Evolutionary origins of the estrogen signaling system: Insights from amphioxus. J. Steroid Biochem. Mol. Biol. 2011, 127, 176–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, E.V.; Jacobson, H.I. Basic guides to the mechanism of. Estrogen action. Recent Progr. Hormone Res. 1962, 18. [Google Scholar]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. P. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Burris, T.P.; Solt, L.A.; Wang, Y.; Crumbley, C.; Banerjee, S.; Griffett, K.; Lundasen, T.; Hughes, T.; Kojetin, D.J. Nuclear receptors and their selective pharmacologic modulators. Pharmacol. Rev. 2013, 65, 710–778. [Google Scholar] [CrossRef]
- Barton, M.; Prossnitz, E.R. Emerging roles of GPER in diabetes and atherosclerosis. Trends Endocrinol. Metab. 2015, 26, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lappano, R.; Pisano, A.; Maggiolini, M. GPER Function in Breast Cancer: An Overview. Front Endocrinol. 2014, 5, 66. [Google Scholar] [CrossRef] [Green Version]
- Prossnitz, E.R.; Barton, M. Estrogen biology: New insights into GPER function and clinical opportunities. Mol. Cell Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Casals-Casas, C.; Desvergne, B. Endocrine disruptors: From endocrine to metabolic disruption. Annu. Rev. Physiol. 2011, 73, 135–162. [Google Scholar] [CrossRef] [PubMed]
- Ciruelos, E.; Pascual, T.; Arroyo, V.M.; Blanco, M.; Manso, L.; Parrilla, L.; Munoz, C.; Vega, E.; Calderon, M.J.; Sancho, B.; et al. The therapeutic role of fulvestrant in the management of patients with hormone receptor-positive breast cancer. Breast 2014, 23, 201–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riggs, B.L.; Hartmann, L.C. Selective estrogen-receptor modulators—mechanisms of action and application toclinical practice. N. Engl. J. Med. 2003, 348, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Haematol. 2015, 29, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, K.J.; Hewitt, S.C.; Arao, Y.; Korach, K.S. Estrogen Hormone Biology. Curr. Top Dev. Biol. 2017, 125, 109–146. [Google Scholar] [PubMed]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef] [Green Version]
- Eyster, K.M. The Estrogen Receptors: An Overview from Different Perspectives. Methods Mol. Biol. 2016, 1366, 1–10. [Google Scholar]
- Germain, P.; Staels, B.; Dacquet, C.; Spedding, M.; Laudet, V. Overview of nomenclature of nuclear receptors. Pharmacol. Rev. 2006, 58, 685–704. [Google Scholar] [CrossRef]
- Bourguet, W.; Germain, P.; Gronemeyer, H. Nuclear receptor ligand-binding domains: Three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol. Sci. 2000, 21, 381–388. [Google Scholar] [CrossRef]
- Hager, G.L.; Lim, C.S.; Elbi, C.; Baumann, C.T. Trafficking of nuclear receptors in living cells. J. Steroid Biochem. Mol. Biol. 2000, 74, 249–254. [Google Scholar] [CrossRef]
- Evinger, A.J.R.; Levin, E.R. Requirements for estrogen receptor alpha membrane localization and function. Steroids 2005, 70, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.R.; Wardell, S.E.; McDonnell, D.P. The molecular mechanisms underlying the pharmacological actions of estrogens, SERMs and oxysterols: Implications for the treatment and prevention of osteoporosis. Bone 2013, 53, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; O’Malley, B.W. Coregulator function: A key to understanding tissue specificity of selectivereceptor modulators. Endocr. Rev. 2004, 25, 45–71. [Google Scholar] [CrossRef] [PubMed]
- Dahlman-Wright, K.; Cavailles, V.; Fuqua, S.A.; Jordan, V.C.; Katzenellenbogen, J.A.; Korach, K.S.; Maggi, A.; Muramatsu, M.; Parker, M.G.; Gustafsson, J. International Union of Pharmacology. LXIV. Estrogen receptors. Pharmacol. Rev. 2006, 58, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Charn, T.H.; Liu, E.T.; Chang, E.C.; Lee, Y.K.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Genome-wide dynamics of chromatin binding of estrogen receptors alpha and beta:mutual restriction and competitive site selection. Mol. Endocrinol. 2010, 24, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Arterburn, J.B.; Sklar, L.A. GPR30: A G protein-coupled receptor for estrogen. Mol. Cell Endocrinol. 2007, 265–266, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.H.; Benson, H.E.; Faccenda, E.; Pawson, A.J.; Sharman, J.L.; McGrath, J.C.; Catterall, W.A.; Spedding, M.; Peters, J.A.; Harmar, A.J.; et al. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Brit. J. Pharmacol. 2013, 170, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.J.; Liao, J.K. Nonnuclear actions of estrogen. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1952–1961. [Google Scholar] [CrossRef]
- Gurevich, E.V.; Tesmer, J.J.G.; Mushegian, A.; Gurevich, V.V. G protein-coupled receptor kinases: More than just kinases and not only forGPCRs. Pharmacol. Therapeut. 2012, 133, 40–69. [Google Scholar] [CrossRef]
- Terasawa, E.; Noel, S.D.; Keen, K.L. Rapid action of oestrogen in luteinising hormone-releasing hormone neurones: The role of GPR30. J. Neuroendocrinol. 2009, 21, 316–321. [Google Scholar] [CrossRef]
- Liverman, C.S.; Brown, J.W.; Sandhir, R.; McCarson, K.E.; Berman, N.E.J. Role of the oestrogen receptors GPR30 and ERalpha in peripheral sensitization:relevance to trigeminal pain disorders in women. Cephalalgia 2009, 29, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Sakamoto, H.; Mori, H.; Hosokawa, K.; Kawamura, A.; Itose, M.; Nishi, M.; Prossnitz, E.R.; Kawata, M. Expression and intracellular distribution of the G protein-coupled receptor 30 inrat hippocampal formation. Neurosci. Lett. 2008, 441, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Pupo, M.; Vivacqua, A.; Perrotta, I.; Pisano, A.; Aquila, S.; Abonante, S.; Gasperi-Campani, A.; Pezzi, V.; Maggiolini, M. The nuclear localization signal is required for nuclear GPER translocation andfunction in breast Cancer-Associated Fibroblasts (CAFs). Mol. Cell Endocrinol. 2013, 376, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Madeo, A.; Maggiolini, M. Nuclear alternate estrogen receptor GPR30 mediates 17beta-estradiol-induced gene expression and migration in breast cancer-associated fibroblasts. Cancer Res. 2010, 70, 6036–6046. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R.J. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupledreceptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.J.; Levin, E.R. Rapid actions of plasma membrane estrogen receptors. Trends Endocrinol. Metab. 2001, 12, 152–156. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R.J.; Bland, K.I. Estrogen action via the G protein-coupled receptor, GPR30: stimulation ofadenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factorreceptor-to-MAPK signaling axis. Mol. Endocrinol. (Baltimore, Md.) 2002, 16, 70–84. [Google Scholar] [CrossRef]
- Lindsey, S.H.; Liu, L.; Chappell, M.C. Vasodilation by GPER in mesenteric arteries involves both endothelial nitricoxide and smooth muscle cAMP signaling. Steroids 2014, 81, 99–102. [Google Scholar] [CrossRef]
- Kanda, N.; Watanabe, S. 17beta-estradiol stimulates the growth of human keratinocytes by inducing cyclin D2 expression. J. Investig. Dermatol. 2004, 123, 319–328. [Google Scholar] [CrossRef]
- Kanda, N.; Watanabe, S. 17Beta-estradiol enhances the production of nerve growth factor in THP-1-derived macrophages or peripheral blood monocyte-derived macrophages. J. Investig. Dermatol. 2003, 121, 771–780. [Google Scholar] [CrossRef]
- Albanito, L.; Lappano, R.; Madeo, A.; Chimento, A.; Prossnitz, E.R.; Cappello, A.R.; Dolce, V.; Abonante, S.; Pezzi, V.; Maggiolini, M. Effects of atrazine on estrogen receptor alpha- and G protein-coupled receptor30-mediated signaling and proliferation in cancer cells and cancer-associatedfibroblasts. Environ. Health Persp. 2015, 123, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Vivacqua, A.; Bonofiglio, D.; Albanito, L.; Madeo, A.; Rago, V.; Carpino, A.; Musti, A.M.; Picard, D.; Ando, S.; Maggiolini, M. 17beta-estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation ofthyroid cancer cells through the g protein-coupled receptor GPR30. Mol. Pharmacol. 2006, 70, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Albanito, L.; Madeo, A.; Lappano, R.; Vivacqua, A.; Rago, V.; Carpino, A.; Oprea, T.I.; Prossnitz, E.R.; Musti, A.M.; Ando, S.; et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growthresponse to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancercells. Cancer Res. 2007, 67, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Santolla, M.F.; Lappano, R.; De Marco, P.; Pupo, M.; Vivacqua, A.; Sisci, D.; Abonante, S.; Iacopetta, D.; Cappello, A.R.; Dolce, V.; et al. G protein-coupled estrogen receptor mediates the up-regulation of fatty acidsynthase induced by 17beta-estradiol in cancer cells and cancer-associatedfibroblasts. J. Biol. Chem. 2012, 287, 43234–43245. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Pellegrino, M.; Santolla, M.F.; Lappano, R.; Ricchio, E.; Abonante, S.; Maggiolini, M. GPER mediates activation of HIF1alpha/VEGF signaling by estrogens. Cancer Res. 2014, 74, 4053–4064. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.C.; Winuthayanon, W.; Korach, K.S. What’s new in estrogen receptor action in the female reproductive tract. J. Mol. Endocrinol. 2016, 56, R55–R71. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.; Rainville, J.; Zhao, X.; Katzenellenbogen, B.S.; Pfaff, D.; Vasudevan, N. Estrogen receptor-mediated transcription involves the activation of multiple kinase pathways in neuroblastoma cells. J. Steroid Biochem. Mol. Biol. 2014, 139, 45–53. [Google Scholar] [CrossRef]
- Kalyanaraman, H.; Schwappacher, R.; Joshua, J.; Zhuang, S.; Scott, B.T.; Klos, M.; Casteel, D.E.; Frangos, J.A.; Dillmann, W.; Boss, G.R.; et al. Nongenomic thyroid hormone signaling occurs through a plasma membrane-localizedreceptor. Sci. Signal 2014, 7, a48. [Google Scholar] [CrossRef]
- Razandi, M.; Oh, P.; Pedram, A.; Schnitzer, J.; Levin, E.R. ERs associate with and regulate the production of caveolin: Implications forsignaling and cellular actions. Mol. Endocrinol. 2002, 16, 100–115. [Google Scholar] [CrossRef]
- Razandi, M.; Pedram, A.; Greene, G.L.; Levin, E.R. Cell membrane and nuclear estrogen receptors (ERs) originate from a singletranscript: Studies of ERalpha and ERbeta expressed in Chinese hamster ovarycells. Mol. Endocrinol. 1999, 13, 307–319. [Google Scholar]
- Kumar, P.; Wu, Q.; Chambliss, K.L.; Yuhanna, I.S.; Mumby, S.M.; Mineo, C.; Tall, G.G.; Shaul, P.W. Direct interactions with G alpha i and G betagamma mediate nongenomic signalingby estrogen receptor alpha. Mol. Endocrinol. 2007, 21, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.V.; DeSombre, E.R. Estrogen-receptor interaction. Science 1973, 182, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Green, K.A.; Carroll, J.S. Oestrogen-receptor-mediated transcription and the influence of co-factors and chromatin state. Nat. Rev. Cancer 2007, 7, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-wide mapping of estrogen receptor binding reveals long-rangeregulation requiring the forkhead protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.C.; Li, L.; Grimm, S.A.; Chen, Y.; Liu, L.; Li, Y.; Bushel, P.R.; Fargo, D.; Korach, K.S. Research resource: Whole-genome estrogen receptor alpha binding in mouse uterine tissue revealed by ChIP-seq. Mol. Endocrinol. 2012, 26, 887–898. [Google Scholar] [CrossRef]
- Carroll, J.S.; Brown, M. Estrogen receptor target gene: An evolving concept. Mol. Endocrinol. 2006, 20, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Jakacka, M.; Ito, M.; Weiss, J.; Chien, P.Y.; Gehm, B.D.; Jameson, J.L. Estrogen receptor binding to DNA is not required for its activity through thenonclassical AP1 pathway. J. Biol. Chem. 2001, 276, 13615–13621. [Google Scholar] [CrossRef]
- Smith, C.L. Cross-talk between peptide growth factor and estrogen receptor signalingpathways. Biol. Reprod. 1998, 58, 627–632. [Google Scholar] [CrossRef]
- Maselli, A.; Pierdominici, M.; Vitale, C.; Ortona, E. Membrane lipid rafts and estrogenic signalling: A functional role in themodulation of cell homeostasis. Apoptosis 2015, 20, 671–678. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Wen, Z.; Kong, F.; Guan, X.; Liu, W. microRNA-206 overexpression inhibits cellular proliferation and invasion ofestrogen receptor alpha-positive ovarian cancer cells. Mol. Med. Rep. 2014, 9, 1703–1708. [Google Scholar] [CrossRef]
- Adams, B. Elucidating microRNA-Mediated Regulation of Estrogen Signaling and Response in Human Breast Cancer Cells. Ph.D.Thesis, University of Connecticut, Mansfield, CT, USA, 1 January 2009. [Google Scholar]
- Qiu, J.; Ye, L.; Ding, J.; Feng, W.; Jin, H.; Zhang, Y.; Li, Q.; Hua, K. Expression and clinical significance of estrogen-regulated long non-coding RNAsin estrogen receptor alpha-positive ovarian cancer progression. Oncol. Rep. 2014, 31, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Bossard, C.; Busson, M.; Vindrieux, D.; Gaudin, F.; Machelon, V.; Brigitte, M.; Jacquard, C.; Pillon, A.; Balaguer, P.; Balabanian, K.; et al. Potential role of estrogen receptor beta as a tumor suppressor of epithelialovarian cancer. PLoS ONE 2012, 7, e44787. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, I.; Zannoni, G.F.; Prisco, M.G.; Fagotti, A.; Tortorella, L.; Vizzielli, G.; Mencaglia, L.; Scambia, G.; Gallo, D. Cytoplasmic expression of estrogen receptor beta (ERbeta) predicts poor clinical outcome in advanced serous ovarian cancer. Gynecol. Oncol. 2011, 122, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Ciucci, A.; Zannoni, G.F.; Travaglia, D.; Petrillo, M.; Scambia, G.; Gallo, D. Prognostic significance of the estrogen receptor beta (ERbeta) isoforms ERbeta1, ERbeta2, and ERbeta5 in advanced serous ovarian cancer. Gynecol. Oncol. 2014, 132, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Viswanadhapalli, S.; Garcia, L.; Zhou, M.; Nair, B.C.; Kost, E.; Rao Tekmal, R.; Li, R.; Rao, M.K.; Curiel, T.; et al. Therapeutic utility of natural estrogen receptor beta agonists on ovarian cancer. Oncotarget 2017, 8, 50002–50014. [Google Scholar] [CrossRef] [PubMed]
- Giudice, L.C. Clinical practice. Endometriosis. N. Engl. J. Med. 2010, 362, 2389–2398. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, S.; Murk, W.; Arici, A. Endometriosis and infertility: Epidemiology and evidence-based treatments. Ann. Ny. Acad. Sci. 2008, 1127, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, L.; Yu, Q.; Zhang, Y.; Yan, L.; Chen, Z. The estrogen-regulated lncRNA H19/miR-216a-5p axis alters stromal cell invasionand migration via ACTA2 in endometriosis. Mol. Hum. Reprod. 2019. [Google Scholar] [CrossRef]
- Delvoux, B.; Groothuis, P.; D’Hooghe, T.; Kyama, C.; Dunselman, G.; Romano, A. Increased production of 17beta-estradiol in endometriosis lesions is the resultof impaired metabolism. J. Clin. Endocrinol. Metab. 2009, 94, 876–883. [Google Scholar] [CrossRef]
- Hewitt, S.C.; Korach, K.S. Oestrogen receptor knockout mice: Roles for oestrogen receptors alpha and beta inreproductive tissues. Reproduction 2003, 125, 143–149. [Google Scholar] [CrossRef]
- Zhao, Y.; Gong, P.; Chen, Y.; Nwachukwu, J.C.; Srinivasan, S.; Ko, C.; Bagchi, M.K.; Taylor, R.N.; Korach, K.S.; Nettles, K.W.; et al. Dual suppression of estrogenic and inflammatory activities for targeting of endometriosis. Sci. Transl. Med. 2015, 7, 271r–279r. [Google Scholar] [CrossRef] [PubMed]
- Burney, R.O. The genetics and biochemistry of endometriosis. Curr. Opin. Obstet. Gyn. 2013, 25, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.A.; Rodriguez, K.F.; Hewitt, S.C.; Janardhan, K.S.; Young, S.L.; Korach, K.S. Role of estrogen receptor signaling required for endometriosis-like lesion establishment in a mouse model. Endocrinology 2012, 153, 3960–3971. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; O’Malley, B.W. The dynamics of nuclear receptors and nuclear receptor coregulators in the pathogenesis of endometriosis. Hum. Reprod. Update 2014, 20, 467–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.J.; Jung, S.Y.; Wu, S.; Hawkins, S.M.; Park, M.J.; Kyo, S.; Qin, J.; Lydon, J.P.; Tsai, S.Y.; Tsai, M.; et al. Estrogen Receptor beta Modulates Apoptosis Complexes and the Inflammasome toDrive the Pathogenesis of Endometriosis. Cell 2015, 163, 960–974. [Google Scholar] [CrossRef]
- Monsivais, D.; Dyson, M.T.; Yin, P.; Navarro, A.; Coon, J.S.T.; Pavone, M.E.; Bulun, S.E. Estrogen receptor beta regulates endometriotic cell survival through serum andglucocorticoid-regulated kinase activation. Fertil. Steril. 2016, 105, 1266–1273. [Google Scholar] [CrossRef]
- Monsivais, D.; Dyson, M.T.; Yin, P.; Coon, J.S.; Navarro, A.; Feng, G.; Malpani, S.S.; Ono, M.; Ercan, C.M.; Wei, J.J.; et al. ERbeta- and prostaglandin E2-regulated pathways integrate cell proliferation via Ras-like and estrogen-regulated growth inhibitor in endometriosis. Mol. Endocrinol. 2014, 28, 1304–1315. [Google Scholar] [CrossRef]
- Bulun, S.E.; Yilmaz, B.D.; Sison, C.; Miyazaki, K.; Bernardi, L.; Liu, S.; Kohlmeier, A.; Yin, P.; Milad, M.; Wei, J. Endometriosis. Endocr. Rev. 2019, 40, 1048–1079. [Google Scholar] [CrossRef]
- Heublein, S.; Lenhard, M.; Vrekoussis, T.; Schoepfer, J.; Kuhn, C.; Friese, K.; Makrigiannakis, A.; Mayr, D.; Jeschke, U. The G-protein-coupled estrogen receptor (GPER) is expressed in normal humanovaries and is upregulated in ovarian endometriosis and pelvic inflammatorydisease involving the ovary. Reprod. Sci. 2012, 19, 1197–1204. [Google Scholar] [CrossRef]
- Plante, B.J.; Lessey, B.A.; Taylor, R.N.; Wang, W.; Bagchi, M.K.; Yuan, L.; Scotchie, J.; Fritz, M.A.; Young, S.L. G protein-coupled estrogen receptor (GPER) expression in normal and abnormalendometrium. Reprod. Sci. 2012, 19, 684–693. [Google Scholar] [CrossRef]
- Koppitz, M.; Brauer, N.; Ter Laak, A.; Irlbacher, H.; Rotgeri, A.; Coelho, A.; Walter, D.; Steinmeyer, A.; Zollner, T.M.; Peters, M.; et al. Discovery and optimization of pyridyl-cycloalkyl-carboxylic acids as inhibitorsof microsomal prostaglandin E synthase-1 for the treatment of endometriosis. Bioorg. Med. Chem. Lett. 2019. [Google Scholar] [CrossRef]
- Farrell, K.; Antoni, M.H. Insulin resistance, obesity, inflammation, and depression in polycystic ovary syndrome: Biobehavioral mechanisms and interventions. Fertil. Steril. 2010, 94, 1565–1574. [Google Scholar] [CrossRef]
- Azziz, R.; Carmina, E.; Dewailly, D.; Diamanti-Kandarakis, E.; Escobar-Morreale, H.F.; Futterweit, W.; Janssen, O.E.; Legro, R.S.; Norman, R.J.; Taylor, A.E.; et al. Positions statement: Criteria for defining polycystic ovary syndrome as apredominantly hyperandrogenic syndrome: An Androgen Excess Society guideline. J. Clin. Endocrinol. Metab. 2006, 91, 4237–4245. [Google Scholar] [CrossRef]
- Hiam, D.; Simar, D.; Laker, R.; Altintas, A.; Gibson-Helm, M.; Fletcher, E.; Moreno-Asso, A.; Trewin, A.J.; Barres, R.; Stepto, N.K. Epigenetic reprogramming of immune cells in women with PCOS impact genescontrolling reproductive function. J. Clin. Endocrinol. Metab. 2019. [Google Scholar] [CrossRef]
- Cobin, R.H. Cardiovascular and metabolic risks associated with PCOS. Intern. Emerg. Med. 2013, 8, 61–64. [Google Scholar] [CrossRef]
- Hart, R.; Doherty, D.A. The potential implications of a PCOS diagnosis on a woman’s long-term healthusing data linkage. J. Clin. Endocrinol. Metab. 2015, 100, 911–919. [Google Scholar] [CrossRef]
- Barry, J.A.; Azizia, M.M.; Hardiman, P.J. Risk of endometrial, ovarian and breast cancer in women with polycystic ovary syndrome: A systematic review and meta-analysis. Hum. Reprod. Update 2014, 20, 748–758. [Google Scholar] [CrossRef]
- Garcia, E.; Bouchard, P.; De Brux, J.; Berdah, J.; Frydman, R.; Schaison, G.; Milgrom, E.; Perrot-Applanat, M. Use of immunocytochemistry of progesterone and estrogen receptors for endometrialdating. J. Clin. Endocrinol. Metab. 1998, 67, 80–87. [Google Scholar] [CrossRef]
- Lecce, G.; Meduri, G.; Ancelin, M.; Bergeron, C.; Perrot-Applanat, M. Presence of estrogen receptor beta in the human endometrium through the cycle: Expression in glandular, stromal, and vascular cells. J. Clin. Endocrinol. Metab. 2001, 86, 1379–1386. [Google Scholar]
- Mehasseb, M.K.; Panchal, R.; Taylor, A.H.; Brown, L.; Bell, S.C.; Habiba, M. Estrogen and progesterone receptor isoform distribution through the menstrualcycle in uteri with and without adenomyosis. Fertil. Steril. 2011, 95, 2228–2235. [Google Scholar] [CrossRef]
- Gregory, C.W.; Wilson, E.M.; Apparao, K.B.C.; Lininger, R.A.; Meyer, W.R.; Kowalik, A.; Fritz, M.A.; Lessey, B.A. Steroid receptor coactivator expression throughout the menstrual cycle in normal and abnormal endometrium. J. Clin. Endocrinol. Metab. 2006, 87, 2960–2966. [Google Scholar] [CrossRef]
- Quezada, S.; Avellaira, C.; Johnson, M.C.; Gabler, F.; Fuentes, A.; Vega, M. Evaluation of steroid receptors, coregulators, and molecules associated withuterine receptivity in secretory endometria from untreated women with polycystic ovary syndrome. Fertil. Steril. 2006, 85, 1017–1026. [Google Scholar] [CrossRef]
- Kim, J.Y.; Song, H.; Kim, H.; Kang, H.J.; Jun, J.H.; Hong, S.R.; Koong, M.K.; Kim, I.S. Transcriptional profiling with a pathway-oriented analysis identifiesdysregulated molecular phenotypes in the endometrium of patients with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2009, 94, 1416–1426. [Google Scholar] [CrossRef]
- Piltonen, T.T.; Chen, J.; Erikson, D.W.; Spitzer, T.L.B.; Barragan, F.; Rabban, J.T.; Huddleston, H.; Irwin, J.C.; Giudice, L.C. Mesenchymal stem/progenitors and other endometrial cell types from women withpolycystic ovary syndrome (PCOS) display inflammatory and oncogenic potential. J. Clin. Endocrinol. Metab. 2013, 98, 3765–3775. [Google Scholar] [CrossRef]
- Rothenberg, S.S.; Beverley, R.; Barnard, E.; Baradaran-Shoraka, M.; Sanfilippo, J.S. Polycystic ovary syndrome in adolescents. Best Pract. Res. Clin. Haematol. 2018, 48, 103–114. [Google Scholar] [CrossRef]
- Witchel, S.F.; Oberfield, S.E.; Pena, A.S. Polycystic Ovary Syndrome: Pathophysiology, Presentation, and Treatment WithEmphasis on Adolescent Girls. J. Endocrine Soc. 2019, 3, 1545–1573. [Google Scholar] [CrossRef]
- Hosseini, E.; Shahhoseini, M.; Afsharian, P.; Karimian, L.; Ashrafi, M.; Mehraein, F.; Afatoonian, R. Role of epigenetic modifications in the aberrant CYP19A1 gene expression inpolycystic ovary syndrome. Arch. Med. Sci. 2019, 15, 887–895. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Carlsson, B.; Grandien, K.; Enmark, E.; Haggblad, J.; Nilsson, S.; Gustafsson, J.A. Comparison of the ligand binding specificity and transcript tissue distributionof estrogen receptors alpha and beta. Endocrinology 1997, 138, 863–870. [Google Scholar] [CrossRef]
- Ariazi, E.A.; Ariazi, J.L.; Cordera, F.; Jordan, V.C. Estrogen receptors as therapeutic targets in breast cancer. Curr. Top Med. Chem. 2006, 6, 181–202. [Google Scholar] [CrossRef]
- Petrie, W.K.; Dennis, M.K.; Hu, C.; Dai, D.; Arterburn, J.B.; Smith, H.O.; Hathaway, H.J.; Prossnitz, E.R. G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth. Obst. Gynecol. Int. 2013, 2013, 472720. [Google Scholar] [CrossRef]
- Blair, R.M.; Fang, H.; Branham, W.S.; Hass, B.S.; Dial, S.L.; Moland, C.L.; Tong, W.; Shi, L.; Perkins, R.; Sheehan, D.M. The estrogen receptor relative binding affinities of 188 natural andxenochemicals: Structural diversity of ligands. Toxicol. Sci. 2000, 54, 138–153. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Z.-R.; Zhang, R.; Lian, Z.-X.; Deng, S.-L.; Yu, K. Estrogen-Receptor Expression and Function in Female Reproductive Disease. Cells 2019, 8, 1123. https://doi.org/10.3390/cells8101123
Tang Z-R, Zhang R, Lian Z-X, Deng S-L, Yu K. Estrogen-Receptor Expression and Function in Female Reproductive Disease. Cells. 2019; 8(10):1123. https://doi.org/10.3390/cells8101123
Chicago/Turabian StyleTang, Zi-Run, Rui Zhang, Zheng-Xing Lian, Shou-Long Deng, and Kun Yu. 2019. "Estrogen-Receptor Expression and Function in Female Reproductive Disease" Cells 8, no. 10: 1123. https://doi.org/10.3390/cells8101123