The Dual Immunoregulatory function of Nlrp12 in T Cell-Mediated Immune Response: Lessons from Experimental Autoimmune Encephalomyelitis

 ,
,
Abstract
:1. Introduction
2. Materials and methods
2.1. Mice
2.2. Induction of EAE and Tissue Collection
2.3. Intracellular Staining and Flow Cytometry
2.4. Quantitative RT-PCR
2.5. Cytokine Measurement
2.6. Differentiation of 2D2 T Cells Toward Th1 or Th17 In Vitro
2.7. T Cell Activation and Signaling Pathways
2.8. Phosphoflow Cytometry
2.9. SpEAE and Histological Analysis
2.10. Analysis of CNS-Infiltrating Mononuclear Cells by Flow Cytometry
2.11. Statistical Analysis
3. Results
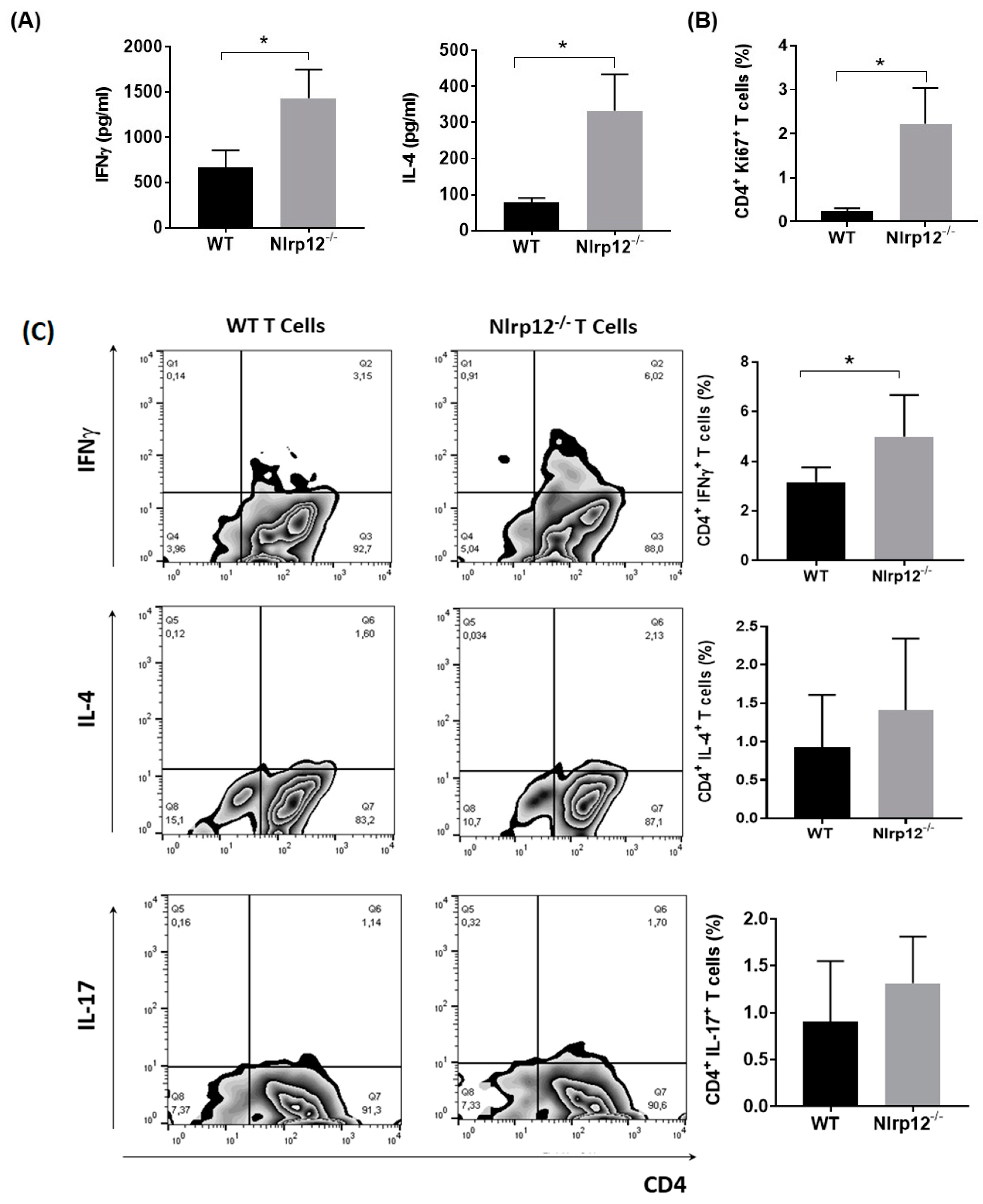
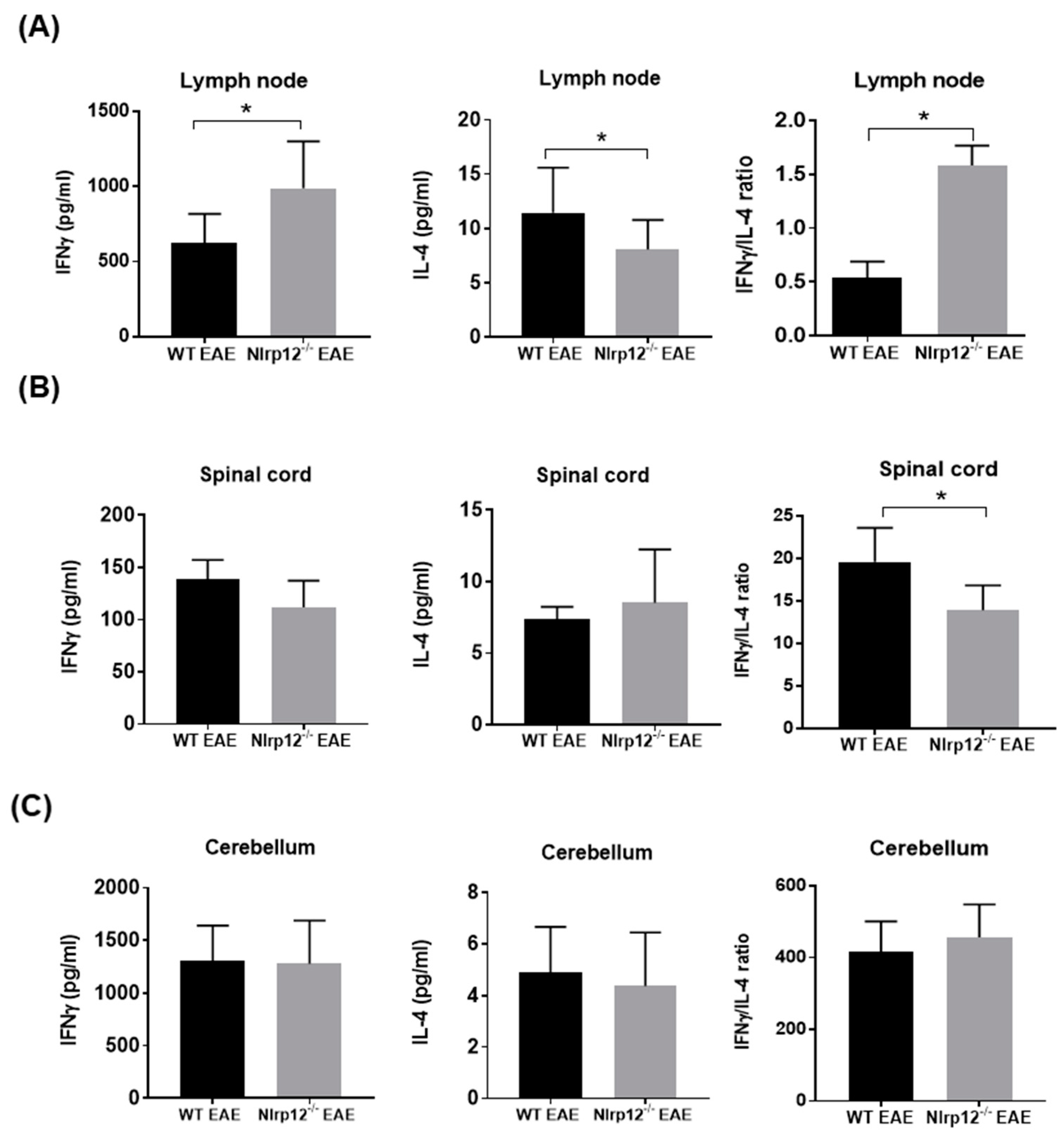
3.1. Nlrp12 Modulates Th1/Th2 Balance in EAE Mice
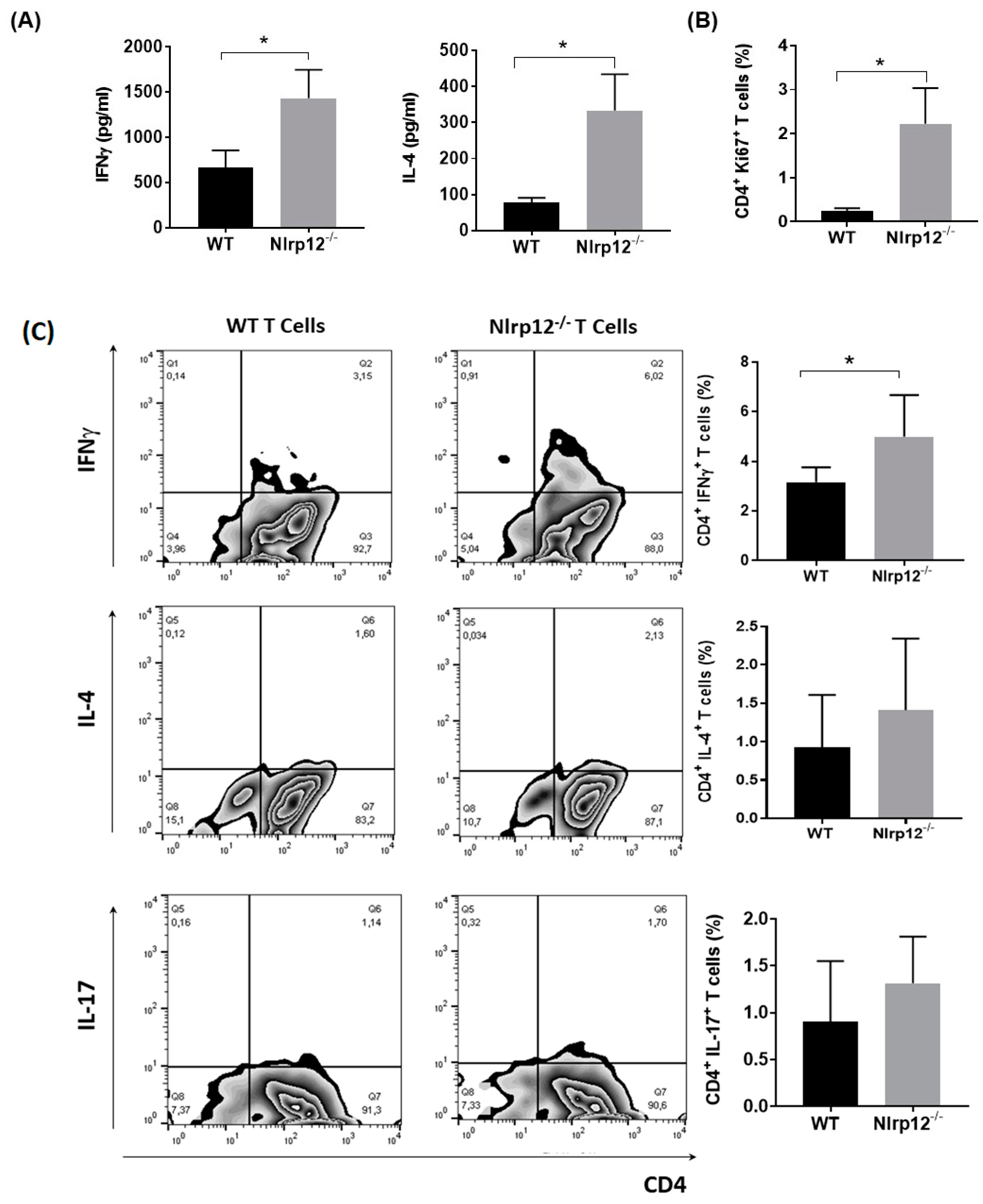
3.2. Nlrp12 Inhibits the Production of IFNγ by CD4+ T Cells In Vitro
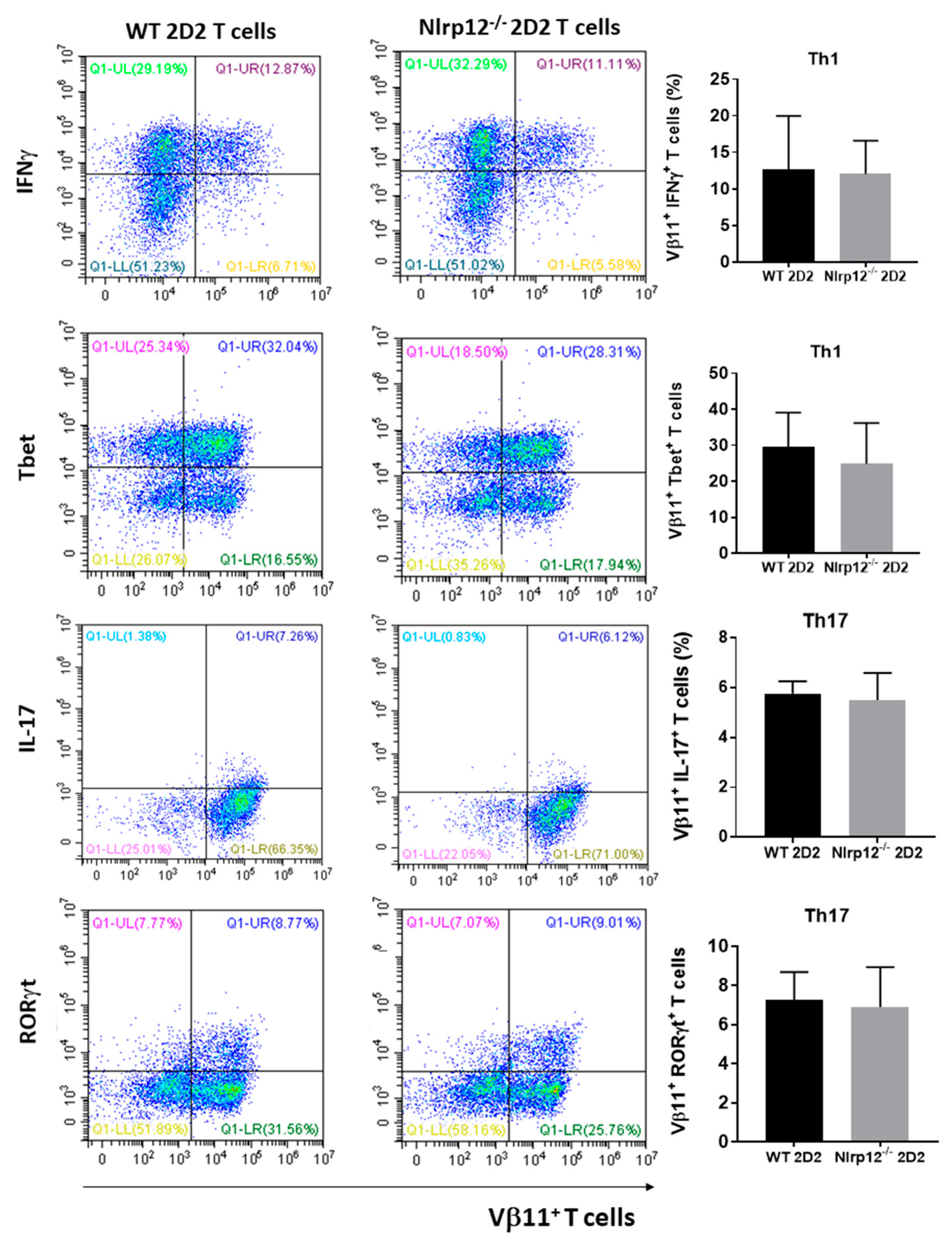
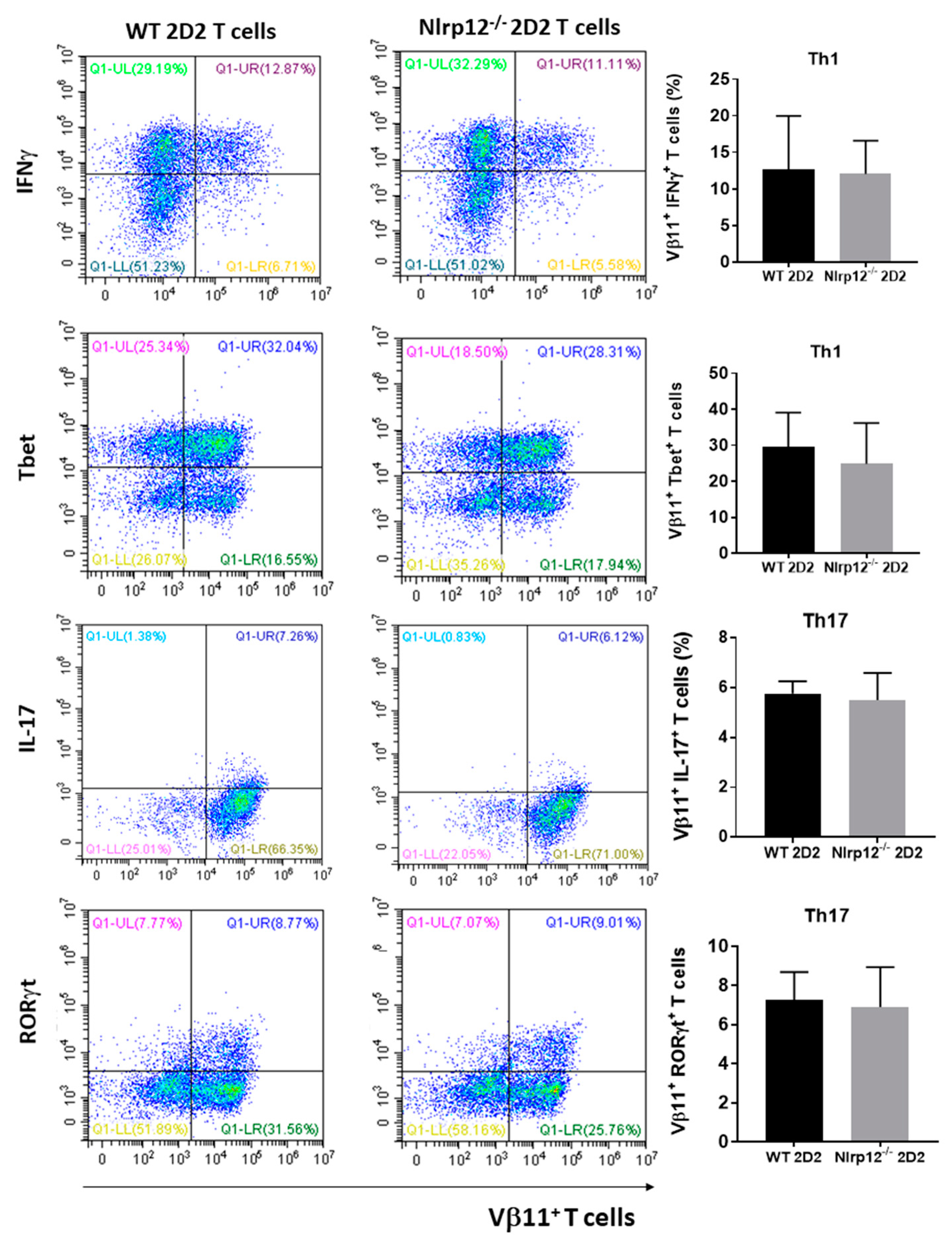
3.3. Nlrp12 Has No Effect on T Cell Differentiation toward Th1 and Th17
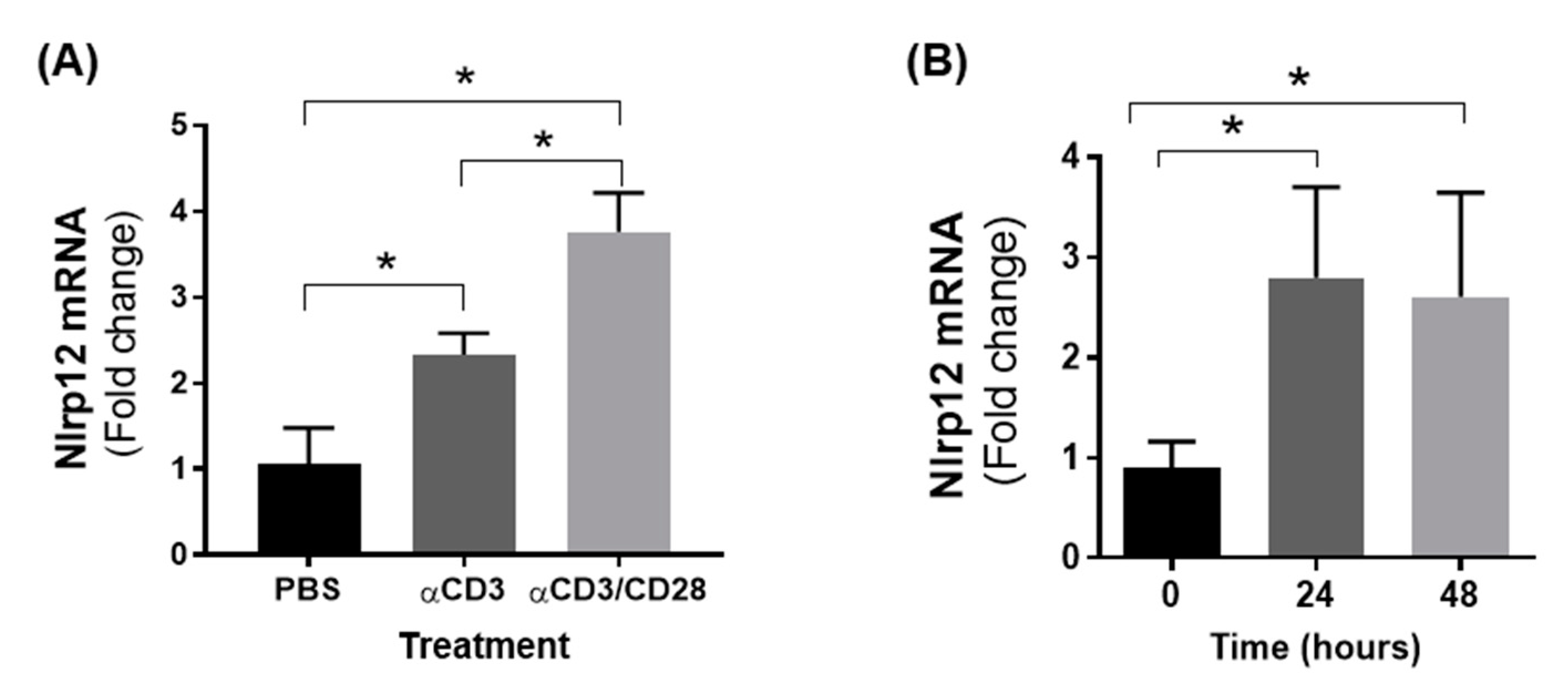
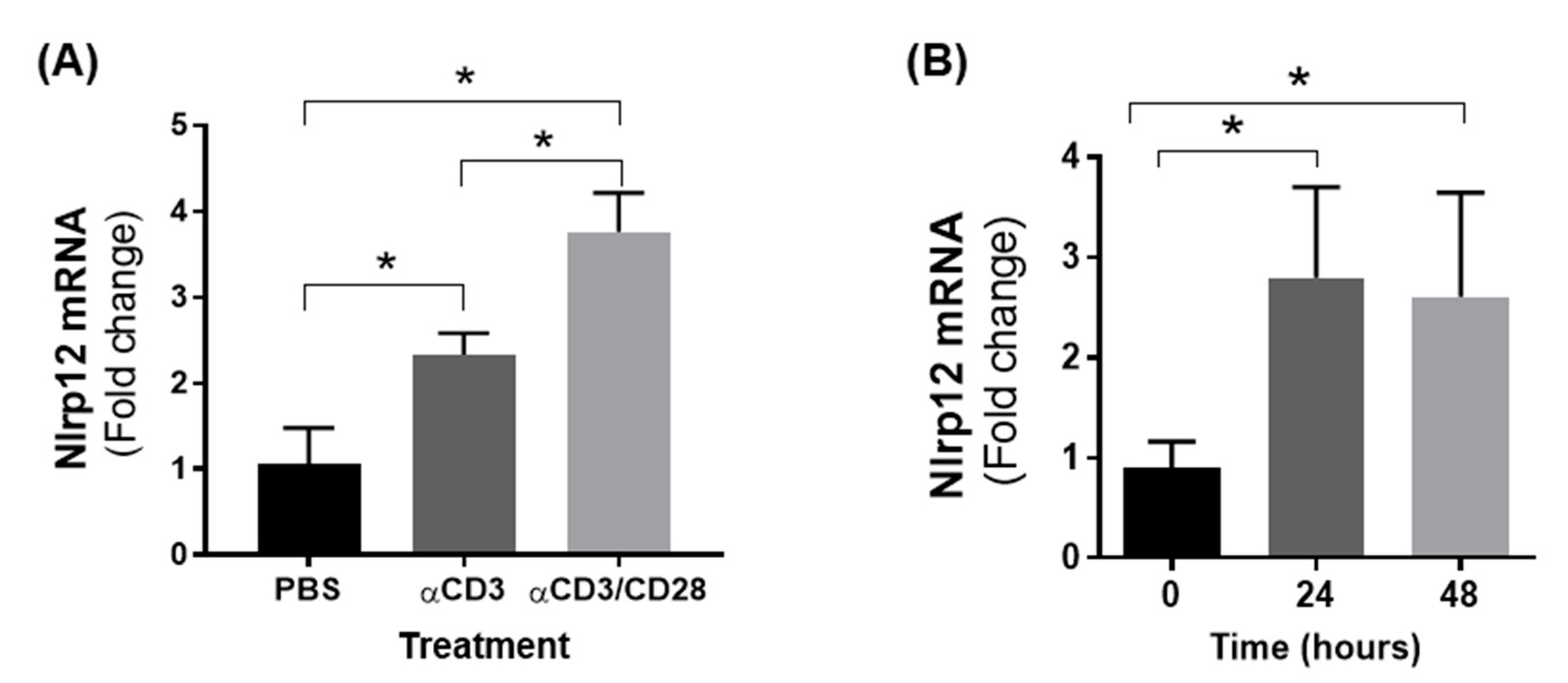
3.4. Nlrp12 Expression Is Increased in Activated T Cells
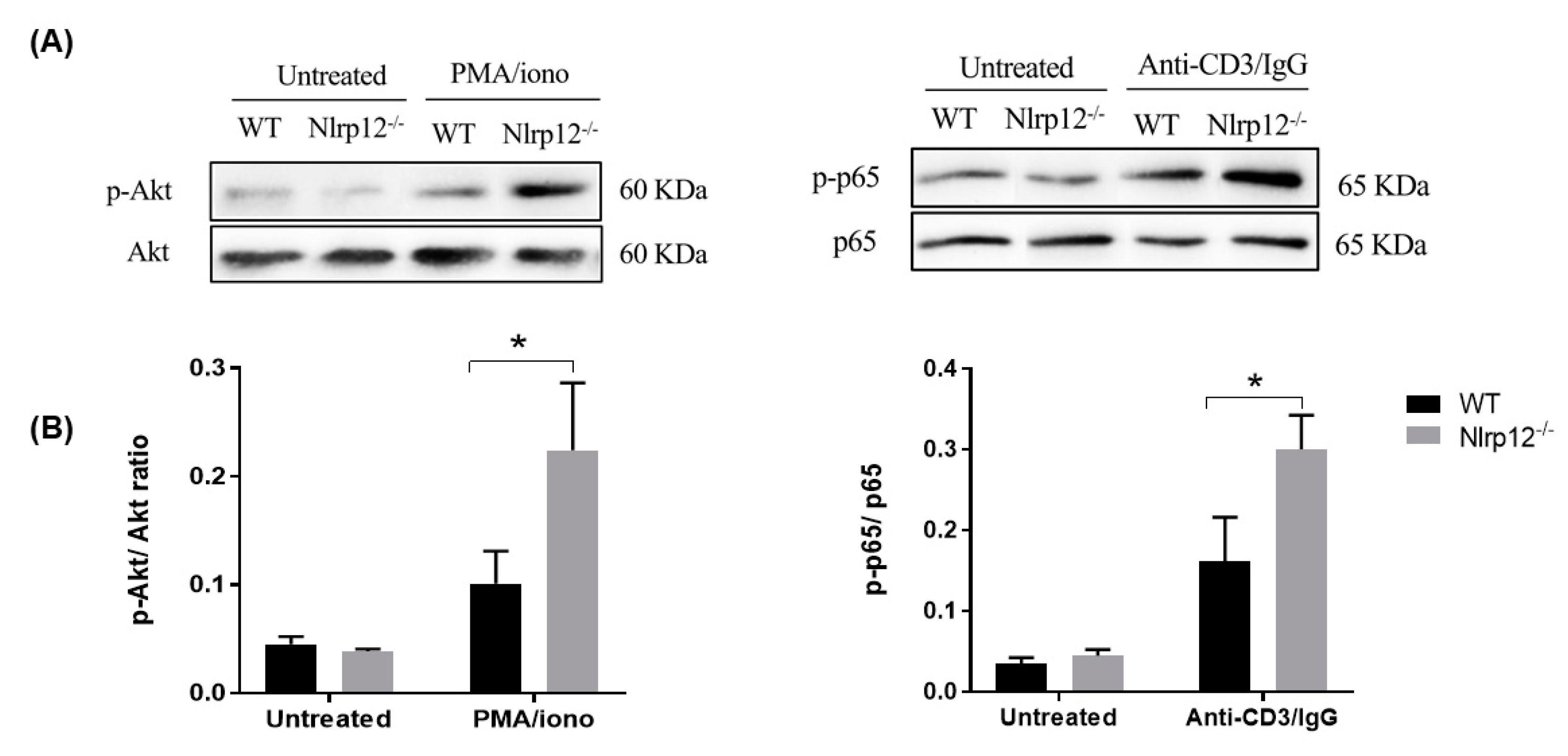
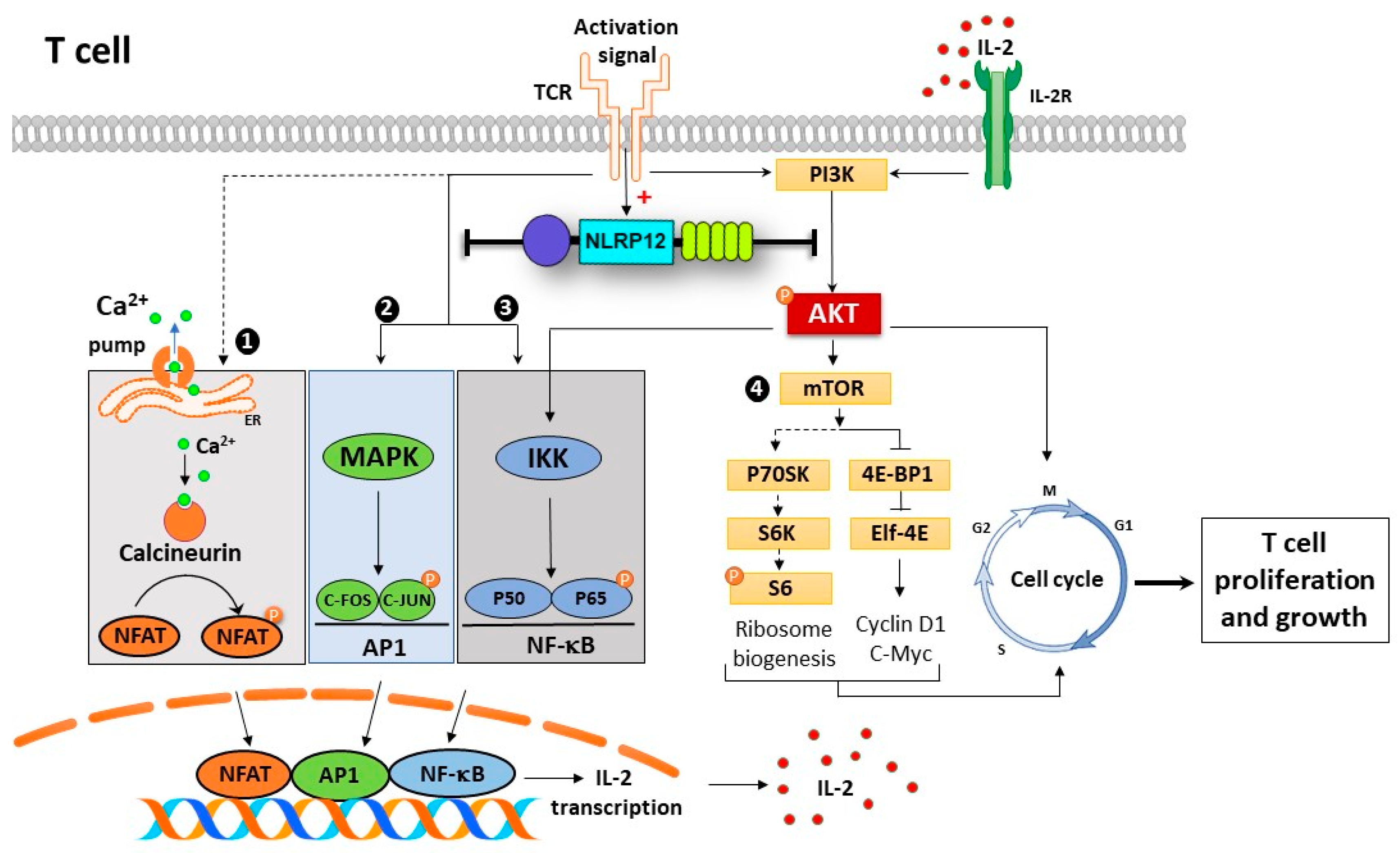
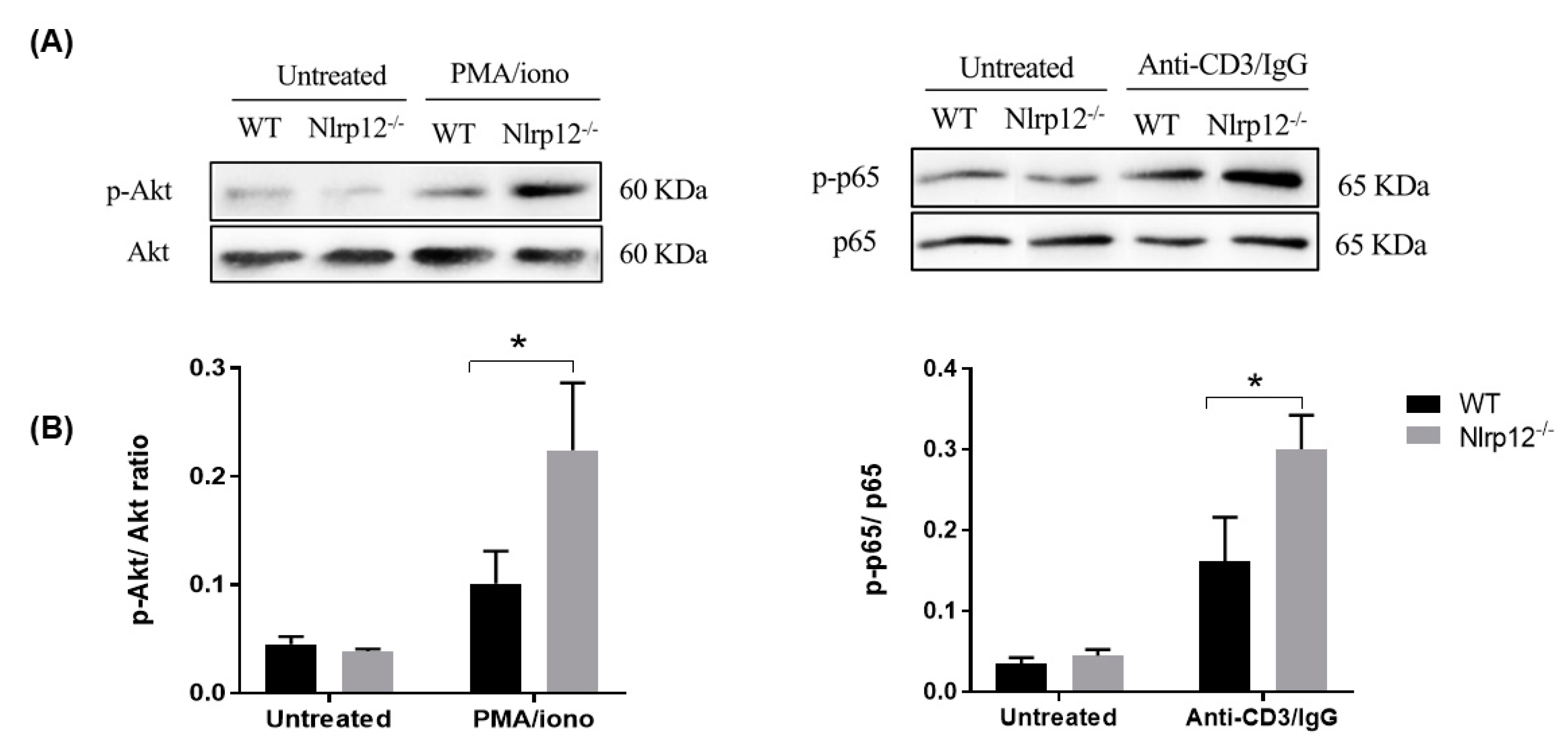
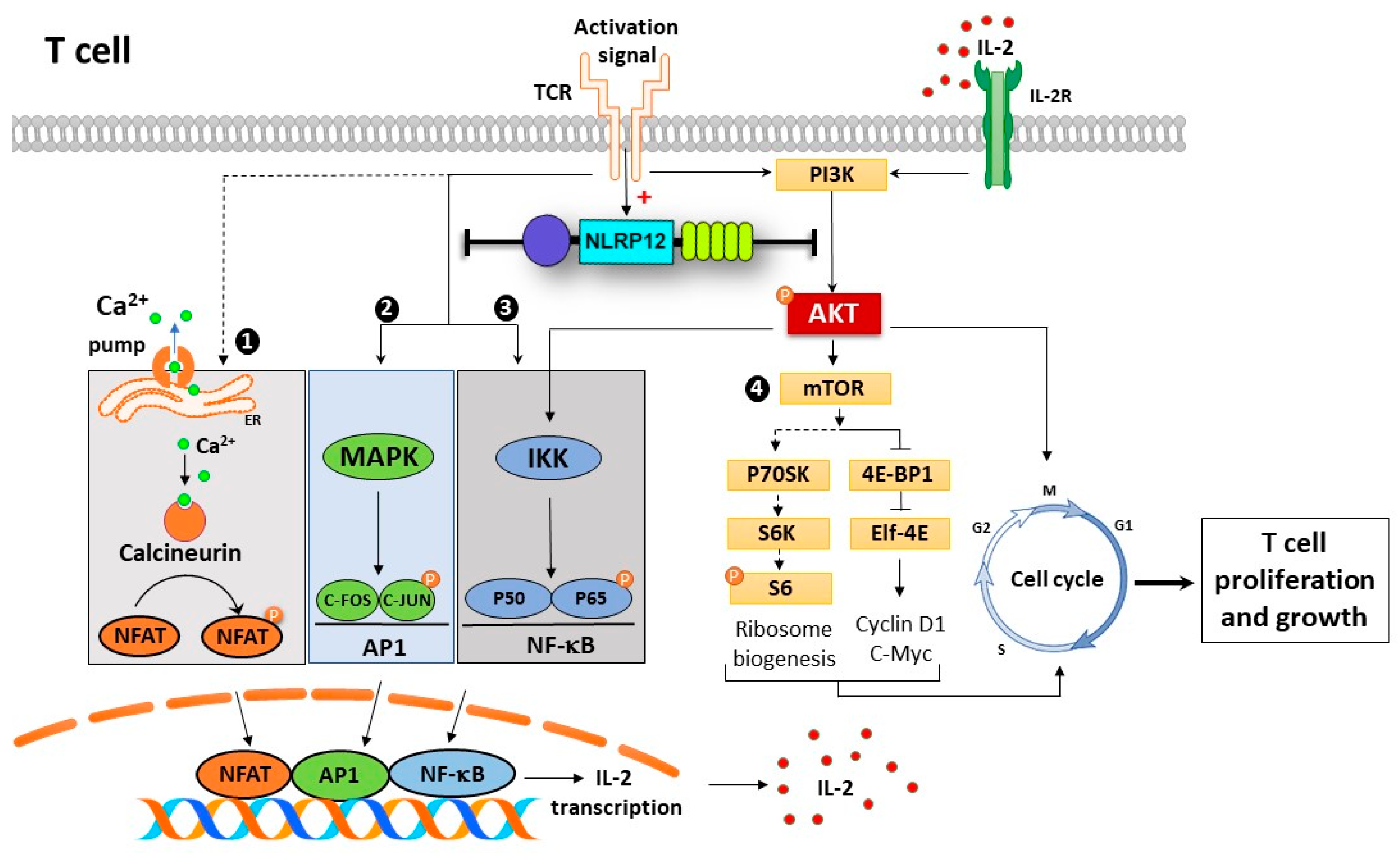
3.5. Nlrp12 Inhibits Phosphorylation of Akt and NF-κB Signaling in Activated T Cells
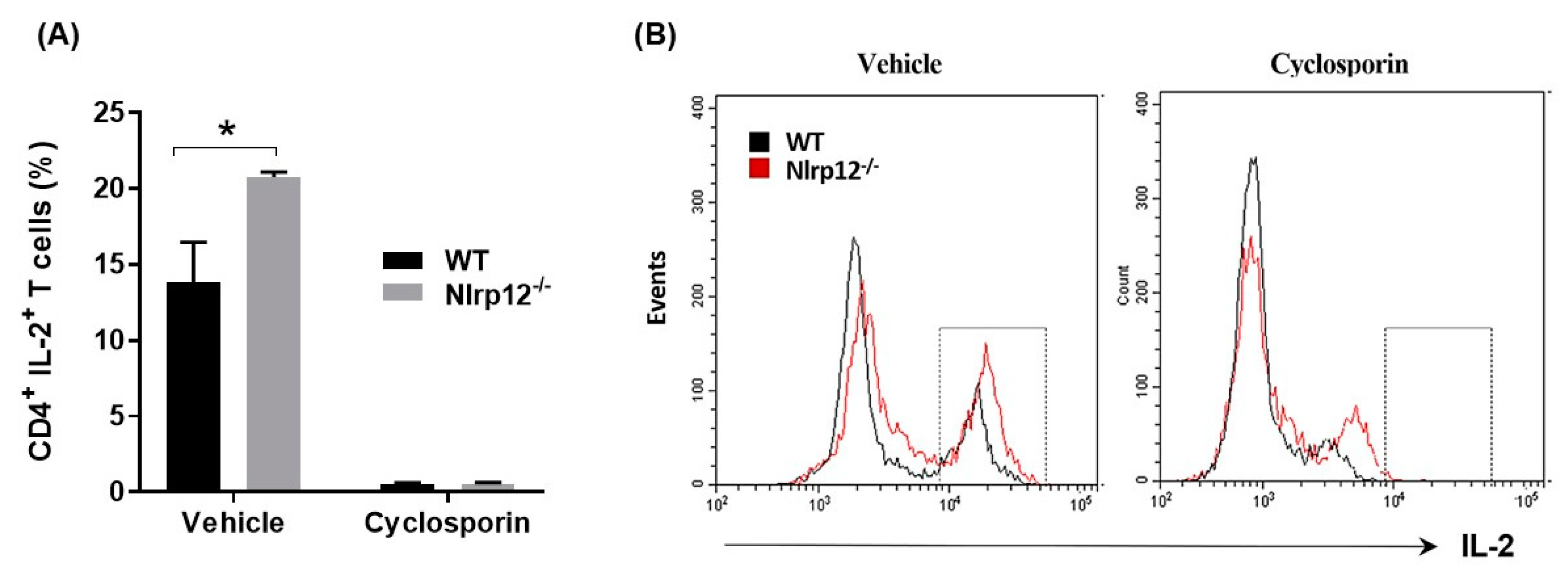
3.6. Nlrp12 Inhibits IL-2 Synthesis but Does Not Modify Ca2+/Calmodulin-Dependent T Cell Activation
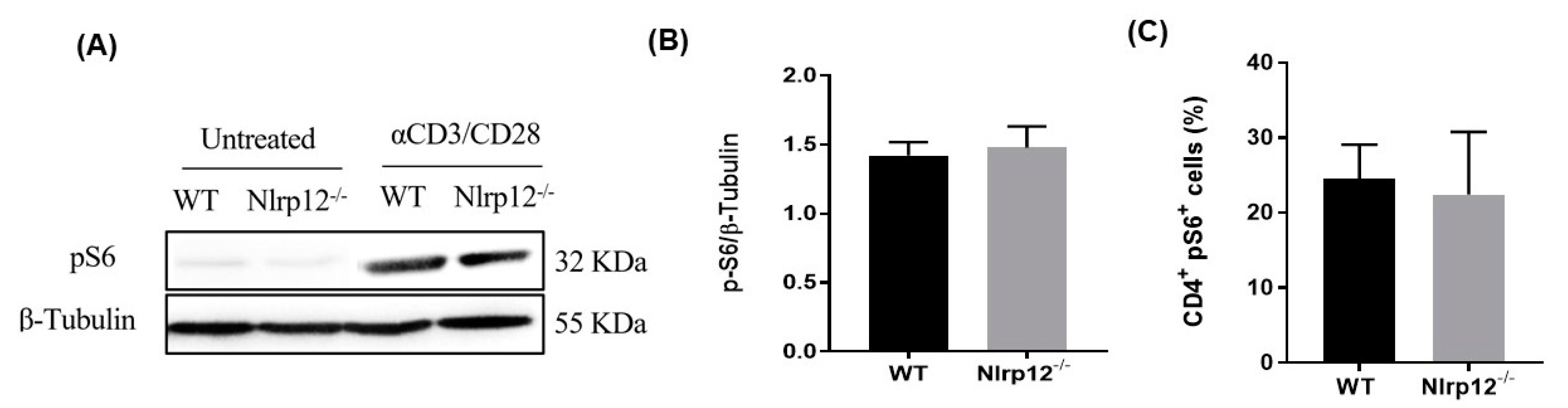
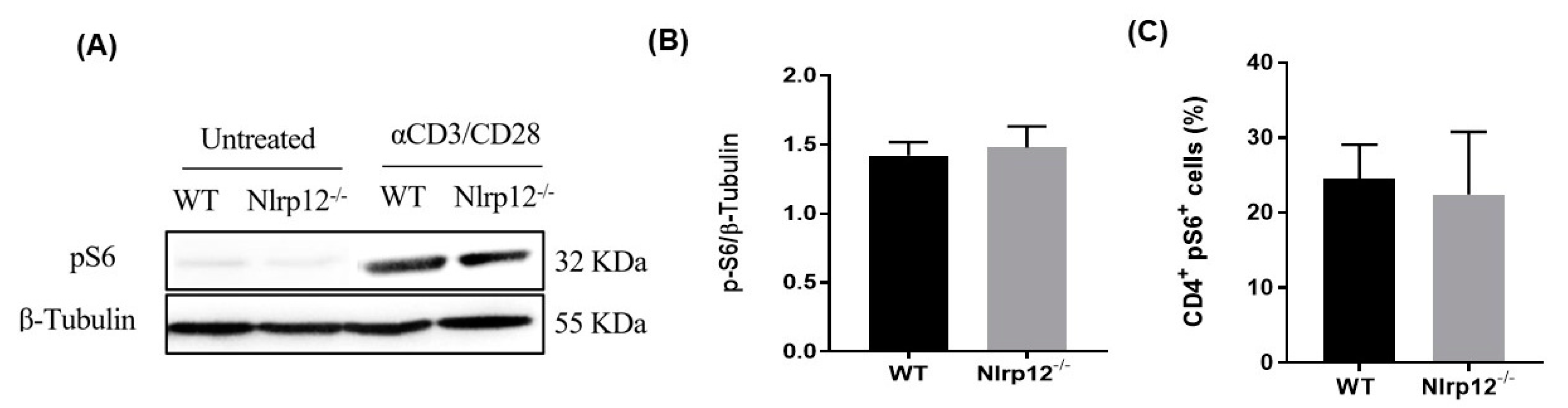
3.7. Nlrp12 Does Not Affect the Phosphorylation of S6 Ribosomal Protein in mTOR Pathway
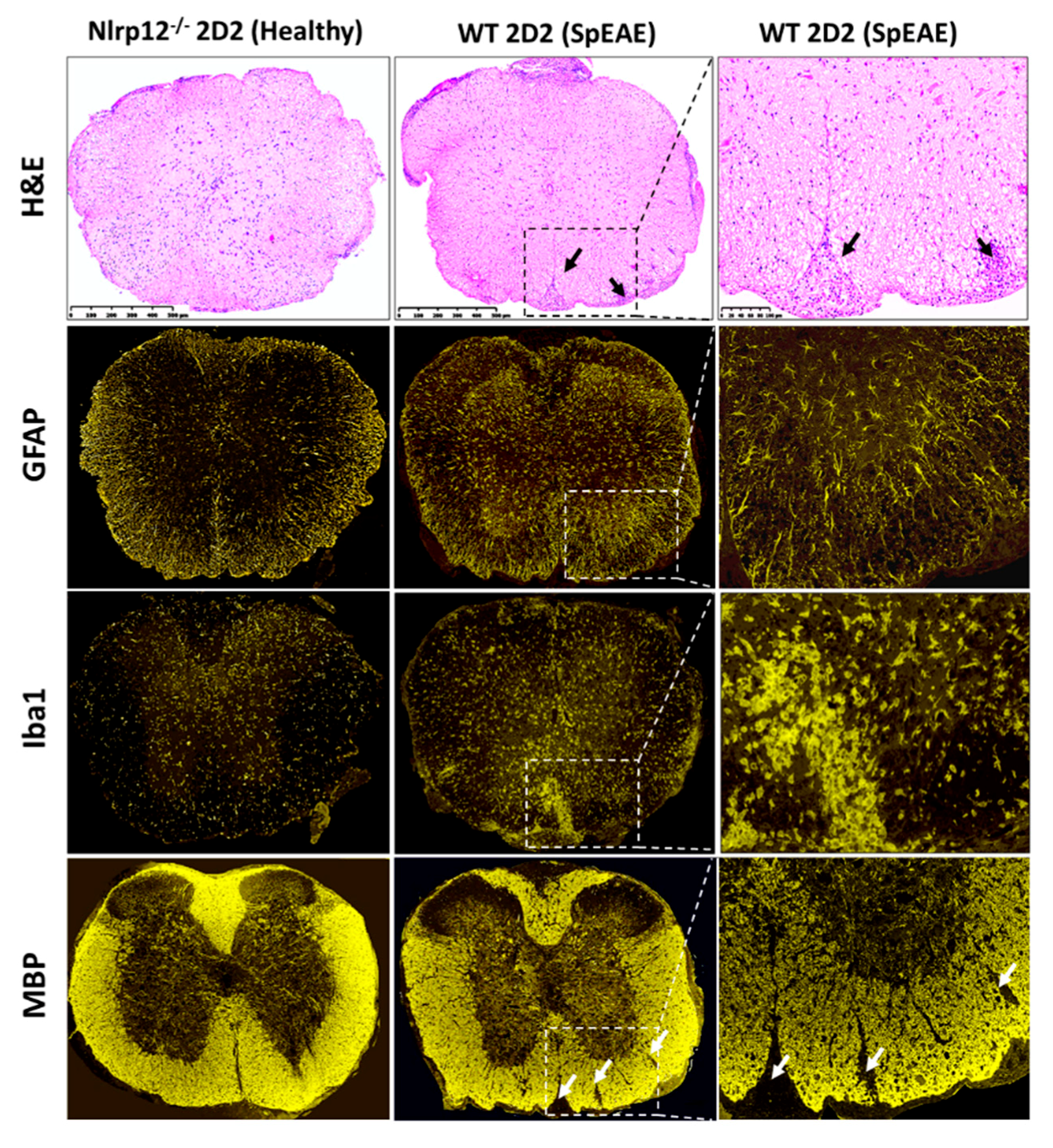
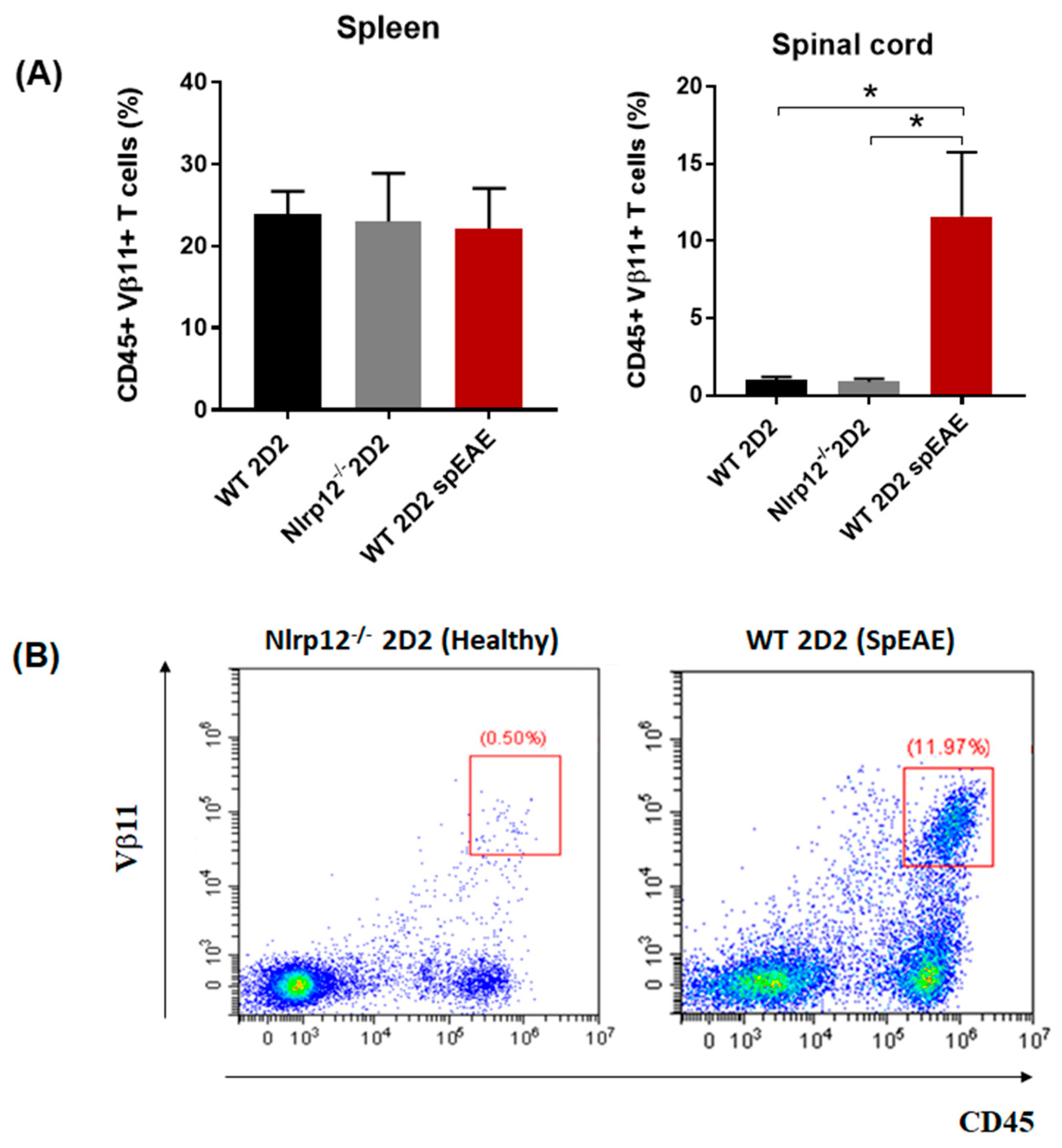
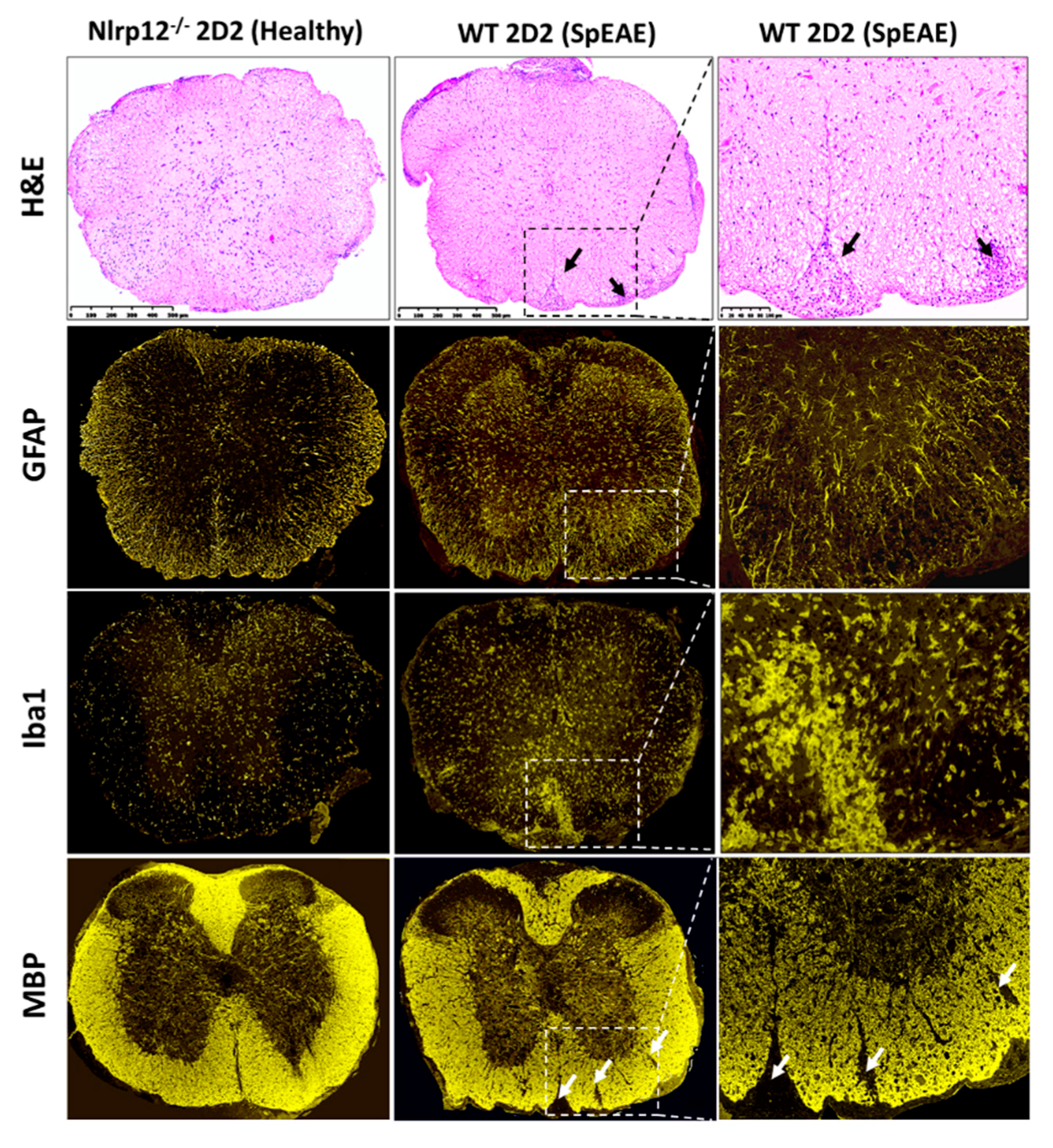
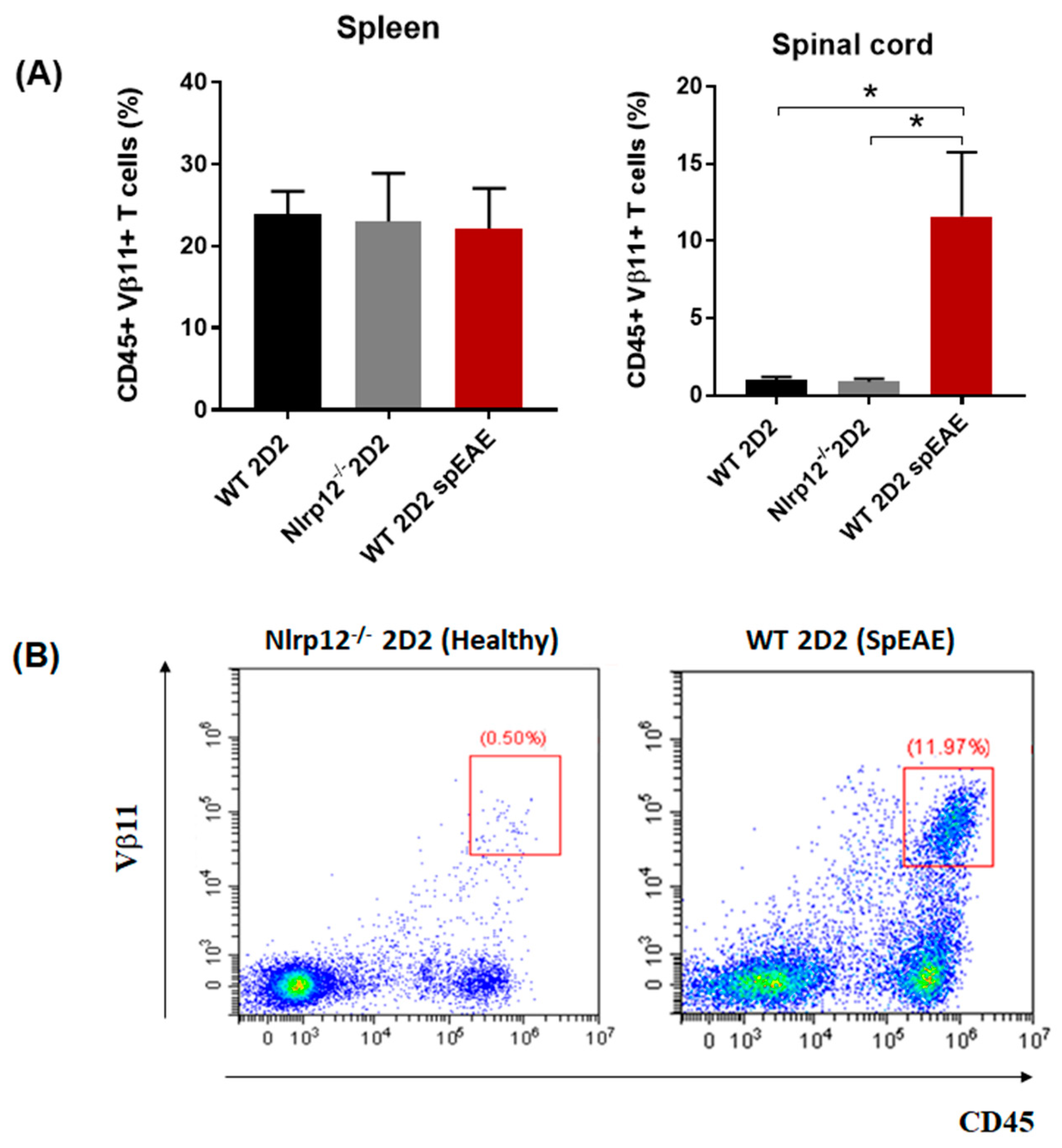
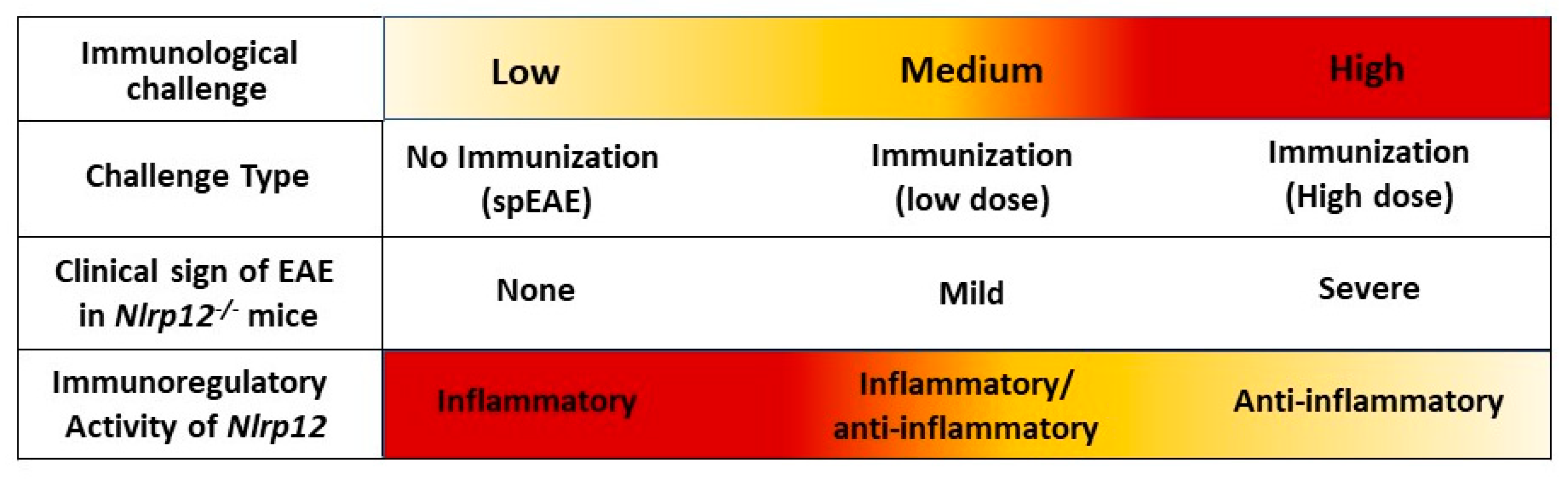
3.8. Nlrp12−/− 2D2 Mice Are Resistant to the Development of spEAE
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Weissert, R. The immune pathogenesis of multiple sclerosis. J. Neuroimmune Pharmacol. 2013, 8, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Kaskow, B.J.; Baecher-Allan, C. Effector T cells in multiple sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a029025. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Broux, B.; Stinissen, P.; Hellings, N. Which immune cells matter? The immunopathogenesis of multiple sclerosis. Crit. Rev. Immunol. 2013, 33, 283–306. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, T. The role of CD4 T cells in the pathogenesis of multiple sclerosis. Int. Rev. Neurobiol. 2007, 79, 43–72. [Google Scholar] [PubMed]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. Cd4+ T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, Y.; Nakae, S.; Saijo, S.; Ishigame, H. The roles of il-17a in inflammatory immune responses and host defense against pathogens. Immunol. Rev. 2008, 226, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Palm, N.W.; Medzhitov, R. Pattern recognition receptors and control of adaptive immunity. Immunol. Rev. 2009, 227, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Gharagozloo, M.; Gris, K.V.; Mahvelati, T.; Amrani, A.; Lukens, J.R.; Gris, D. Nlr-dependent regulation of inflammation in multiple sclerosis- dependent regulation of inflammation in multiple sclerosis. Front. Immunol. 2017, 8, 2012. [Google Scholar] [CrossRef] [PubMed]
- Reith, W.; LeibundGut-Landmann, S.; Waldburger, J.M. Regulation of MHC class II gene expression by the class II transactivator. Nat. Rev. Immunol. 2005, 5, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Neerincx, A.; Jakobshagen, K.; Utermohlen, O.; Buning, H.; Steimle, V.; Kufer, T.A. The N-terminal domain of Nlrc5 confers transcriptional activity for MHC class I and II gene expression. J. Immunol. 2014, 193, 3090–3100. [Google Scholar] [CrossRef] [PubMed]
- Meissner, T.B.; Li, A.; Kobayashi, K.S. Nlrc5: A newly discovered MHC class I transactivator (CITA). Microbes Infect. 2012, 14, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Bruchard, M.; Rebé, C.; Derangère, V.; Togbé, D.; Ryffel, B.; Boidot, R.; Humblin, E.; Hamman, A.; Chalmin, F.; Berger, H.; et al. The receptor Nlrp3 is a transcriptional regulator of Th2 differentiation. Nat. Immunol. 2015, 16, 859. [Google Scholar] [CrossRef] [PubMed]
- Gris, D.; Ye, Z.; Iocca, H.A.; Wen, H.; Craven, R.R.; Gris, P.; Huang, M.; Schneider, M.; Miller, S.D.; Ting, J.P. Nlrp3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J. Immunol. 2010, 185, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Wilson, J.E.; Schneider, M.; Lich, J.D.; Roberts, R.A.; Arthur, J.C.; Woodford, R.-M.T.; Davis, B.K.; Uronis, J.M.; Herfarth, H.H.; et al. Nlrp12 suppresses colon inflammation and tumorigenesis through the negative regulation of non-canonical NF-κB signaling and map kinase activation. Immunity 2012, 36, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wilson, J.E.; Koenigsknecht, M.J.; Chou, W.C.; Montgomery, S.A.; Truax, A.D.; Brickey, W.J.; Packey, C.D.; Maharshak, N.; Matsushima, G.K.; et al. Nlrp12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat. Immunol. 2017, 18, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.H.; Vogel, P.; Malireddi, R.K.; Body-Malapel, M.; Anand, P.K.; Bertin, J.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. The nod-like receptor Nlrp12 attenuates colon inflammation and tumorigenesis. Cancer Cell 2011, 20, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Gharagozloo, M.; Mahvelati, T.M.; Imbeault, E.; Gris, P.; Zerif, E.; Bobbala, D.; Ilangumaran, S.; Amrani, A.; Gris, D. The nod-like receptor, Nlrp12, plays an anti-inflammatory role in experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2015, 12, 198. [Google Scholar] [CrossRef] [PubMed]
- Lukens, J.R.; Gurung, P.; Shaw, P.J.; Barr, M.J.; Zaki, M.H.; Brown, S.A.; Vogel, P.; Chi, H.; Kanneganti, T.D. The Nlrp12 sensor negatively regulates autoinflammatory disease by modulating interleukin-4 production in T cells. Immunity 2015, 42, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Ann. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.; Govender, L.; Hughes, J.; Mavakla, W.; De Kock, M.; Barnard, C.; Pienaar, B.; Janse van Rensburg, E.; Jacobs, G.; Khomba, G.; et al. Novel application of ki67 to quantify antigen-specific in vitro lymphoproliferation. J. Immunol. Methods 2010, 362, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Chatila, T.; Silverman, L.; Miller, R.; Geha, R. Mechanisms of T cell activation by the calcium ionophore ionomycin. J. Immunol. 1989, 143, 1283–1289. [Google Scholar] [PubMed]
- Brennan, P.; Babbage, J.W.; Burgering, B.M.T.; Groner, B.; Reif, K.; Cantrell, D.A. Phosphatidylinositol 3-kinase couples the interleukin-2 receptor to the cell cycle regulator E2F. Immunity 1997, 7, 679–689. [Google Scholar] [CrossRef]
- Arthur, J.C.; Lich, J.D.; Wilson, J.E.; Ye, Z.; Allen, I.C.; Gris, D.; Schneider, M.; Roney, K.E.; O’Connor, B.P.; Moore, C.B.; et al. Nlrp12 controls dendritic and myeloid cell migration to affect contact hypersensitivity. J. Immunol. 2010, 185, 4515–4519. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.T.; De Castro, S.B.; Alves, C.C.; Mesquita, F.P.; De Figueiredo, N.S.; Evangelista, M.G.; Castanon, M.C.; Juliano, M.A.; Ferreira, A.P. Different MOG35–55 concentrations induce distinguishable inflammation through early regulatory response by IL-10 and TGF-β in mice CNS despite unchanged clinical course. Cell. Immunol. 2015, 293, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Palle, P.; Monaghan, K.L.; Milne, S.M.; Wan, E.C.K. Cytokine signaling in multiple sclerosis and its therapeutic applications. Med. Sci. 2017, 5, E0023. [Google Scholar] [CrossRef] [PubMed]
- Kroenke, M.A.; Chensue, S.W.; Segal, B.M. EAE mediated by a non-IFN-γ/non-IL-17 pathway. Eur. J. Immunol. 2010, 40, 2340–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulland, T.K.; Jain, N.; Hornick, E.E.; Elliott, E.I.; Clay, G.M.; Sadler, J.J.; Mills, K.A.; Janowski, A.M.; Volk, A.P.; Wang, K.; et al. Nlrp12 mutation causes C57BL/6J strain-specific defect in neutrophil recruitment. Nat. Commun. 2016, 7, 13180. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Batra, S.; Del Piero, F.; Jeyaseelan, S. Nlrp12 modulates host defense through IL-17A-CXCL1 axis. Mucosal Immunol. 2016, 9, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Lich, J.D.; Williams, K.L.; Moore, C.B.; Arthur, J.C.; Davis, B.K.; Taxman, D.J.; Ting, J.P.-Y. Cutting edge: Monarch-1 suppresses non-canonical NF-κB activation and p52-dependent chemokine expression in monocytes. J. Immunol. 2007, 178, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.L.; Lich, J.D.; Duncan, J.A.; Reed, W.; Rallabhandi, P.; Moore, C.; Kurtz, S.; Coffield, V.M.; Accavitti-Loper, M.A.; Su, L.; et al. The CATERPILLER protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor α- and mycobacterium tuberculosis-induced pro-inflammatory signals. J. Biol. Chem. 2005, 280, 39914–39924. [Google Scholar] [CrossRef] [PubMed]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Abukhdeir, A.M.; Park, B.H. P21 and p27: Roles in carcinogenesis and drug resistance. Expert Rev. Mol. Med. 2008, 10, e19. [Google Scholar] [CrossRef] [PubMed]
- Silveira, T.N.; Gomes, M.T.R.; Oliveira, L.S.; Campos, P.C.; Machado, G.G.; Oliveira, S.C. Nlrp12 negatively regulates proinflammatory cytokine production and host defense against brucella abortus. Eur. J. Immunol. 2017, 47, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Enderby, C.; Keller, C.A. An overview of immunosuppression in solid organ transplantation. Am. J. Manag. Care 2015, 21, s12–s23. [Google Scholar] [PubMed]
- Halloran, P.F. Immunosuppressive drugs for kidney transplantation. N. Engl. J. Med. 2004, 351, 2715–2729. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Pagany, M.; Weiner, H.L.; Linington, C.; Sobel, R.A.; Kuchroo, V.K. Myelin oligodendrocyte glycoprotein–specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 2003, 197, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Lafaille, J.J.; Nagashima, K.; Katsuki, M.; Tonegawa, S. High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice. Cell 1994, 78, 399–408. [Google Scholar] [CrossRef]
- Allen, I.C.; McElvania-TeKippe, E.; Wilson, J.E.; Lich, J.D.; Arthur, J.C.; Sullivan, J.T.; Braunstein, M.; Ting, J.P. Characterization of Nlrp12 during the in vivo host immune response to klebsiella pneumoniae and mycobacterium tuberculosis. PLoS ONE 2013, 8, e60842. [Google Scholar] [CrossRef]
- Vladimer, G.I.; Weng, D.; Paquette, S.W.; Vanaja, S.K.; Rathinam, V.A.; Aune, M.H.; Conlon, J.E.; Burbage, J.J.; Proulx, M.K.; Liu, Q.; et al. The Nlrp12 inflammasome recognizes yersinia pestis. Immunity 2012, 37, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Manji, G.A.; Grenier, J.M.; Al-Garawi, A.; Merriam, S.; Lora, J.M.; Geddes, B.J.; Briskin, M.; DiStefano, P.S.; Bertin, J. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-κB and caspase-1-dependent cytokine processing. J. Biol. Chem. 2002, 277, 29874–29880. [Google Scholar] [CrossRef] [PubMed]
- Ataide, M.A.; Andrade, W.A.; Zamboni, D.S.; Wang, D.; Do Carmo Souza, M.; Franklin, B.S.; Elian, S.; Martins, F.S.; Pereira, D.; Reed, G. Malaria-induced Nlrp12/Nlrp3-dependent caspase-1 activation mediates inflammation and hypersensitivity to bacterial superinfection. PLoS Pathog. 2014, 10, e1003885. [Google Scholar] [CrossRef] [PubMed]
- Gris, K.V.; Yamamoto, K.; Gharagozloo, M.; Mahmoud, S.; Simard, C.; Gris, P.; Gris, D. Exhaustive behavioral profile assay to detect genotype differences between wild-type, inflammasome-deficient and Nlrp12 knock-out mice. AIMS Med. Sci. 2018, 5, 238–251. [Google Scholar] [CrossRef]
- Conti, B.J.; Davis, B.K.; Zhang, J.; O’Connor, W., Jr.; Williams, K.L.; Ting, J.P. CATERPILLER 16.2 (CLR16.2), a novel NBD/LRR family member that negatively regulates T cell function. J. Biol. Chem. 2005, 280, 18375–18385. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Total (n) | SpEAE (%) | Age of Onset (Weeks) | EAE Score |
|---|---|---|---|---|
| WT 2D2 | 101 | 6 | 10.1 ± 4.7 | 3.6 ± 0.5 |
| Nlrp12−/− 2D2 | 30 | 0 | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gharagozloo, M.; Mahmoud, S.; Simard, C.; Mahvelati, T.M.; Amrani, A.; Gris, D. The Dual Immunoregulatory function of Nlrp12 in T Cell-Mediated Immune Response: Lessons from Experimental Autoimmune Encephalomyelitis. Cells 2018, 7, 119. https://doi.org/10.3390/cells7090119
Gharagozloo M, Mahmoud S, Simard C, Mahvelati TM, Amrani A, Gris D. The Dual Immunoregulatory function of Nlrp12 in T Cell-Mediated Immune Response: Lessons from Experimental Autoimmune Encephalomyelitis. Cells. 2018; 7(9):119. https://doi.org/10.3390/cells7090119
Chicago/Turabian StyleGharagozloo, Marjan, Shaimaa Mahmoud, Camille Simard, Tara M. Mahvelati, Abdelaziz Amrani, and Denis Gris. 2018. "The Dual Immunoregulatory function of Nlrp12 in T Cell-Mediated Immune Response: Lessons from Experimental Autoimmune Encephalomyelitis" Cells 7, no. 9: 119. https://doi.org/10.3390/cells7090119
APA StyleGharagozloo, M., Mahmoud, S., Simard, C., Mahvelati, T. M., Amrani, A., & Gris, D. (2018). The Dual Immunoregulatory function of Nlrp12 in T Cell-Mediated Immune Response: Lessons from Experimental Autoimmune Encephalomyelitis. Cells, 7(9), 119. https://doi.org/10.3390/cells7090119