GABAB Receptors Augment TRPC3-Mediated Slow Excitatory Postsynaptic Current to Regulate Cerebellar Purkinje Neuron Response to Type-1 Metabotropic Glutamate Receptor Activation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Brain Slice Preparation
2.2. Drug Delivery
2.3. Drugs
2.4. Electrophysiology
2.5. Data Acquisition and Analysis
3. Results
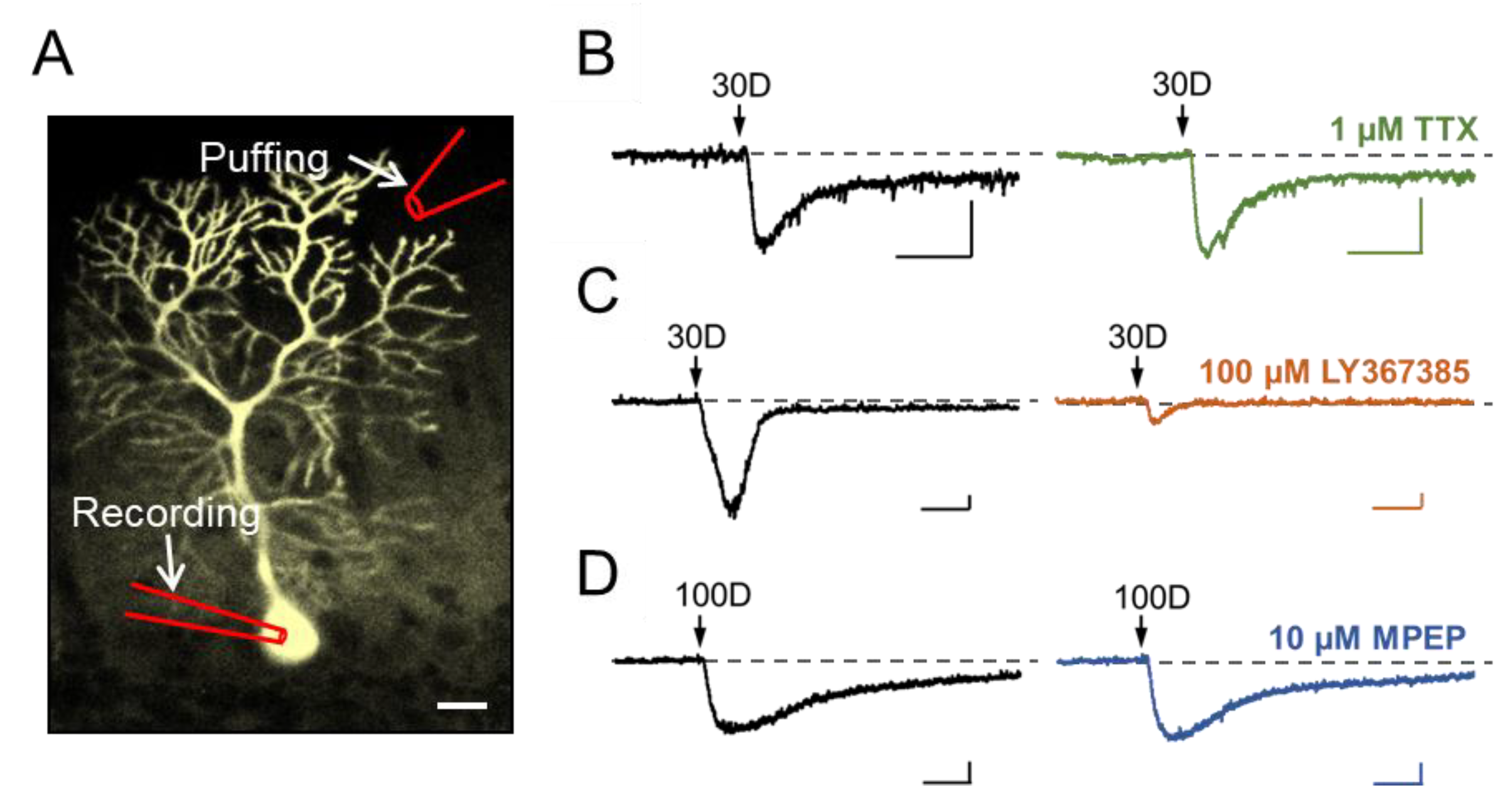
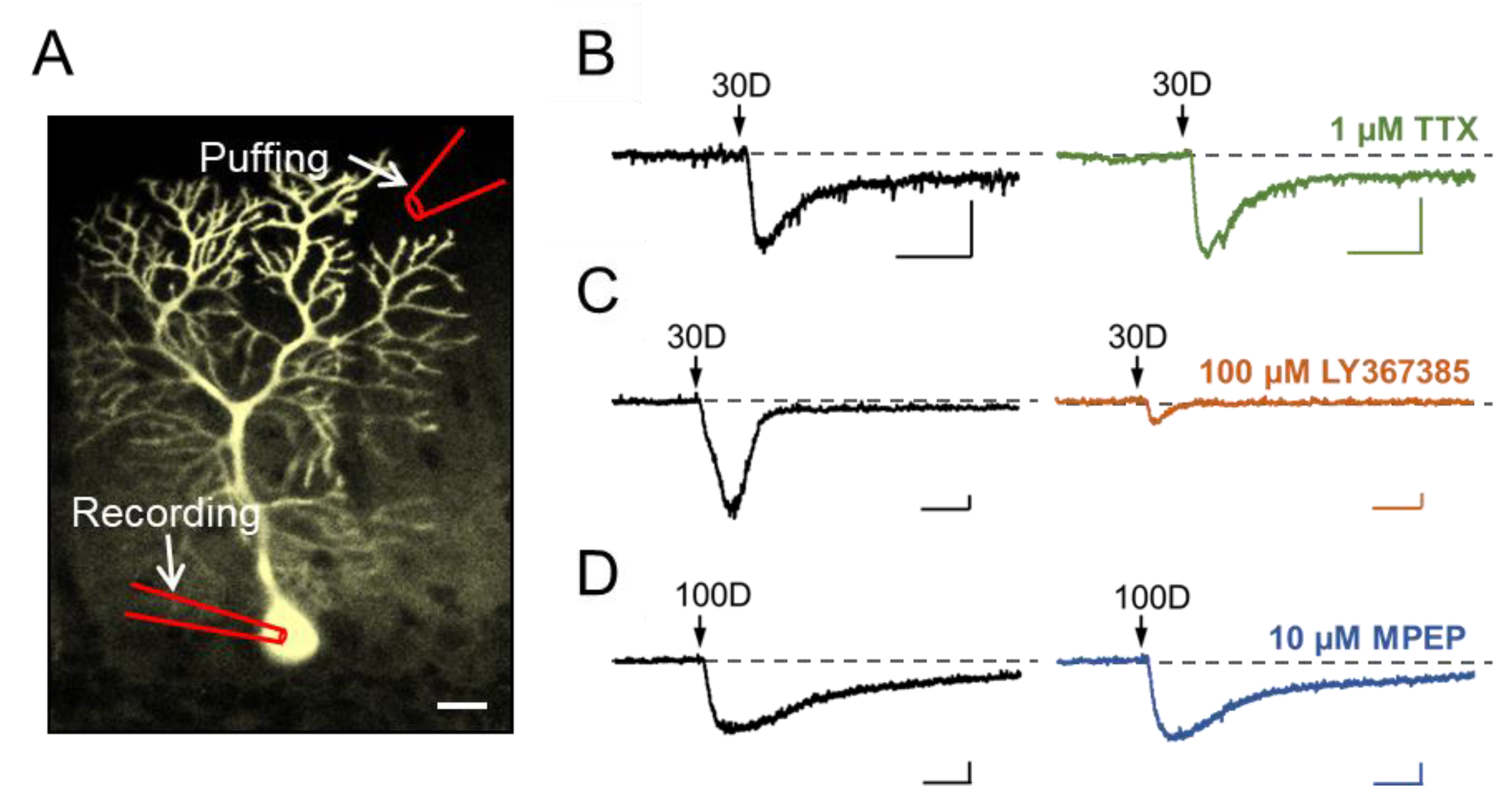
3.1. Stimulation of Dendritic mGluR1 Evokes an sEPSC-Like Inward Current in Cerebellar Purkinje Neurons
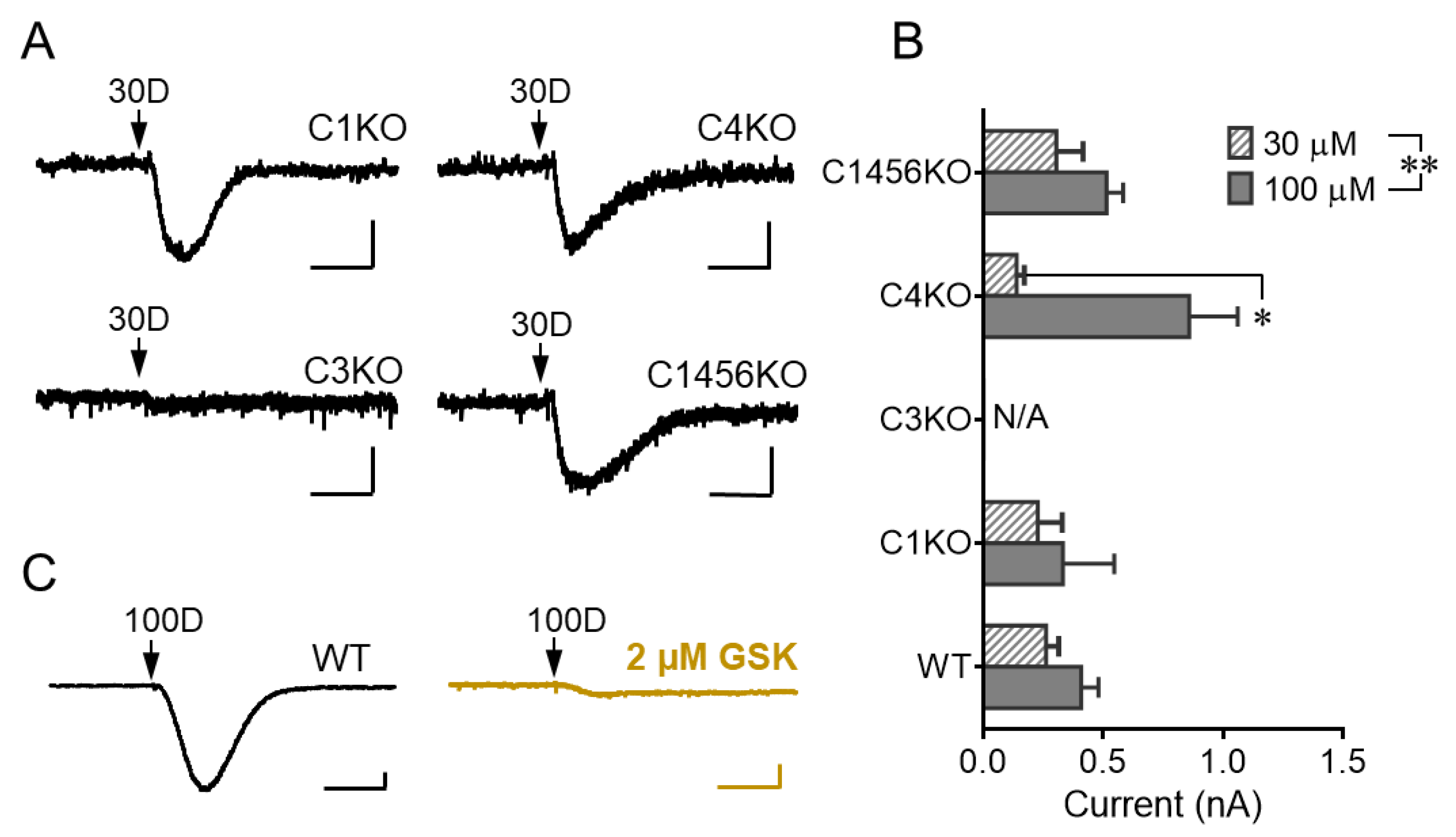
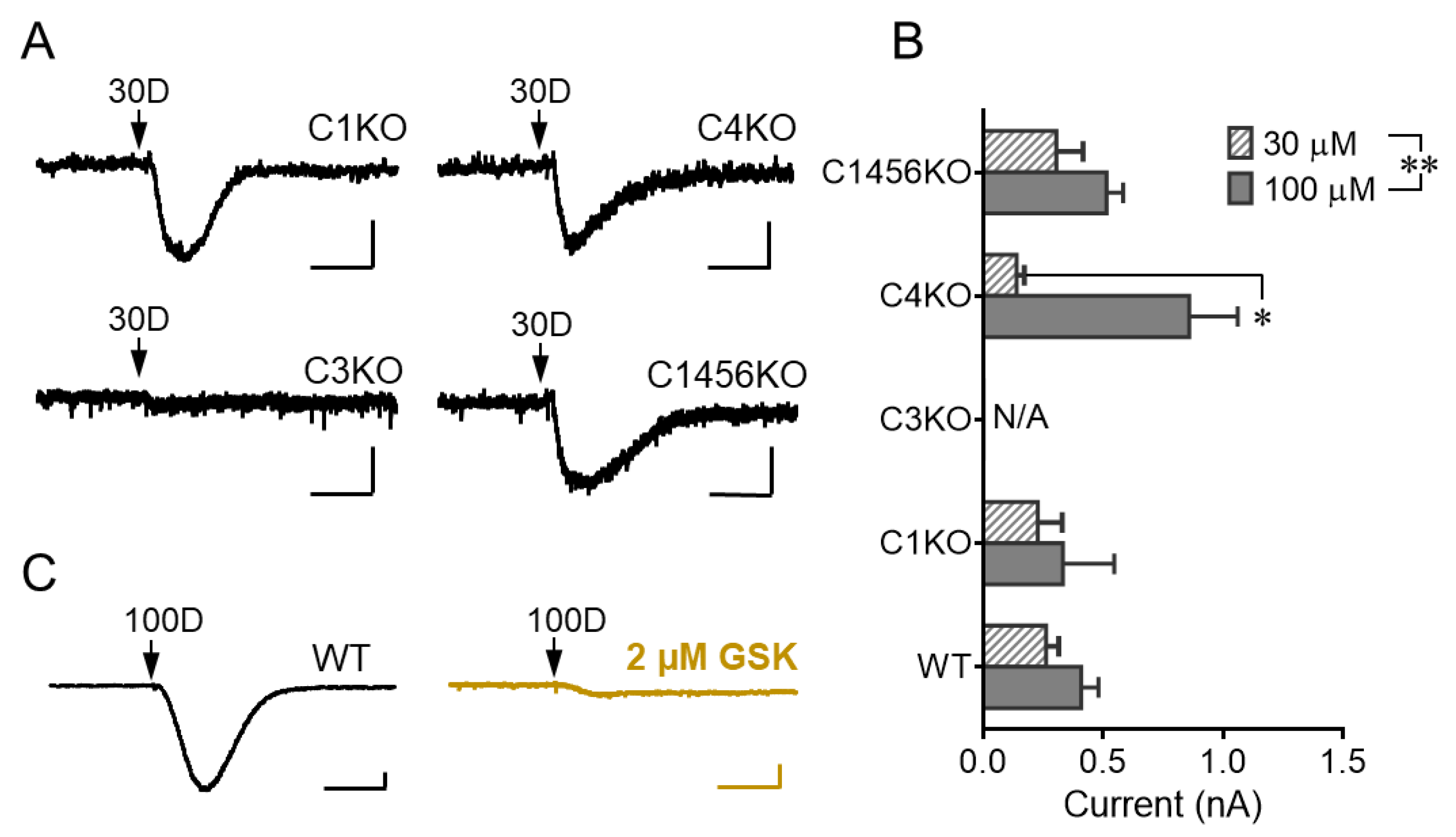
3.2. TRPC3 Mediates the mGluR1 Activation-Evoked sEPSC in Cerebellar Purkinje Neurons
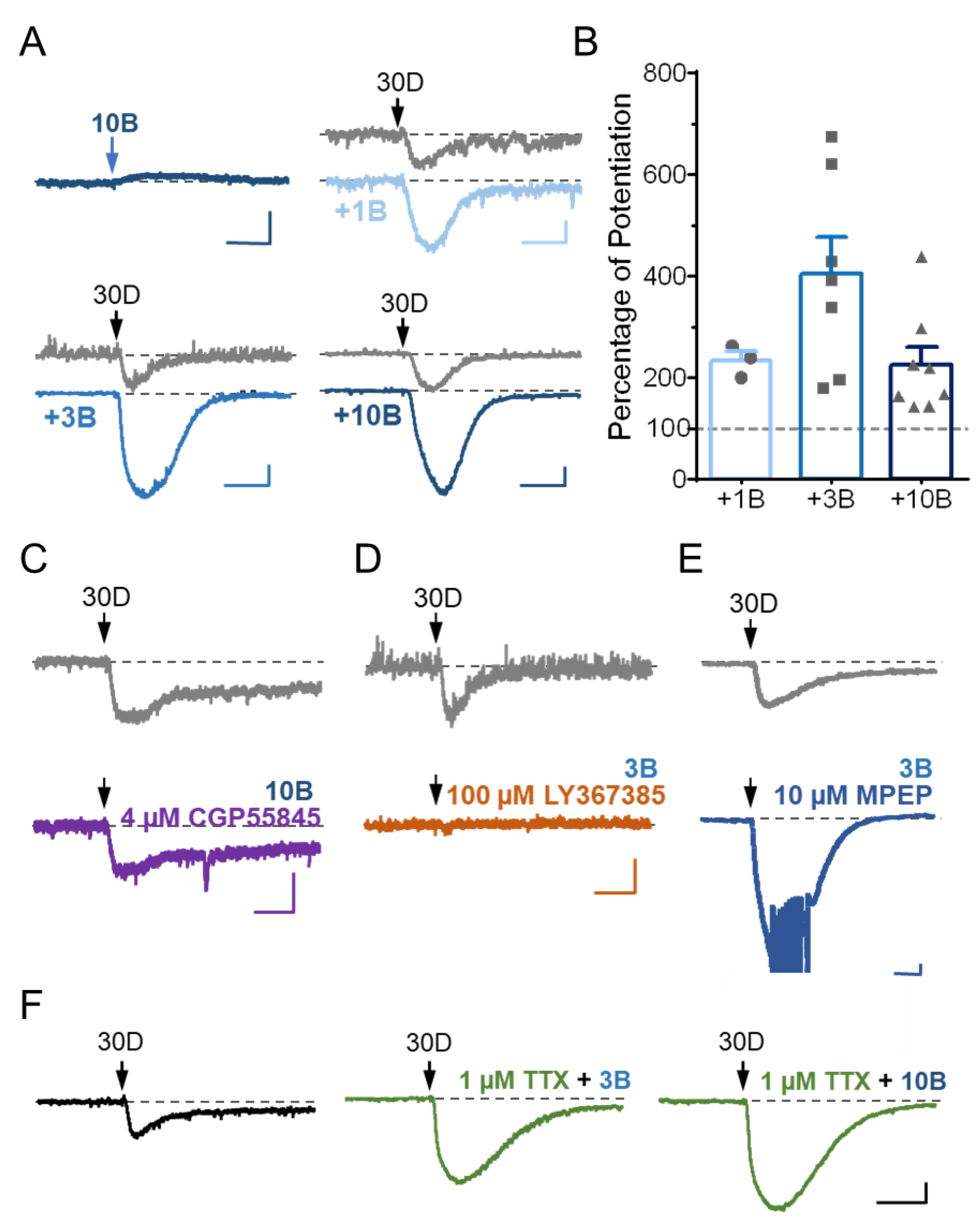
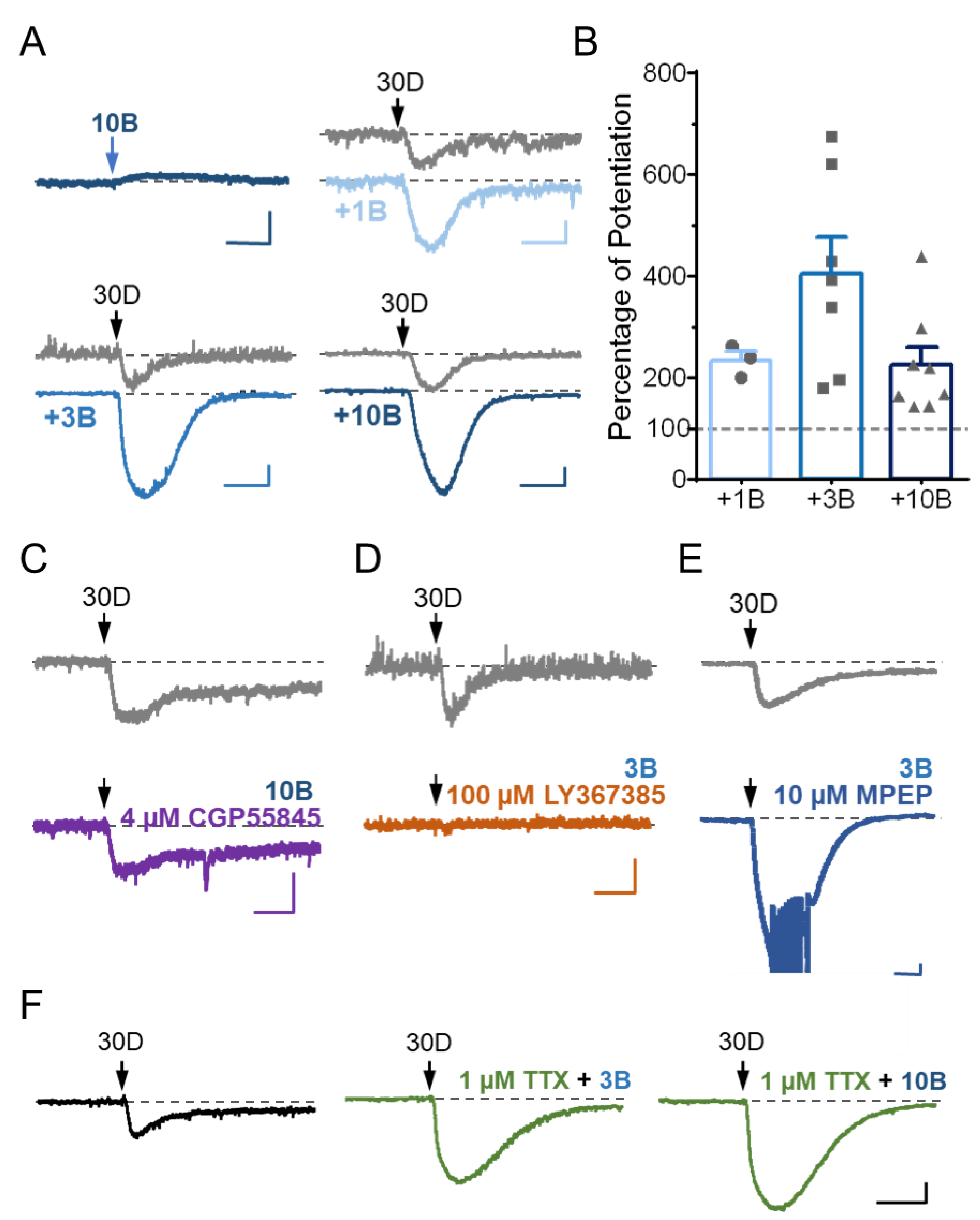
3.3. Stimulation of GABABR Potentiates mGluR1 Agonist-Evoked sEPSC in Cerebellar Purkinje Neurons via a Postsynaptic Mechanism
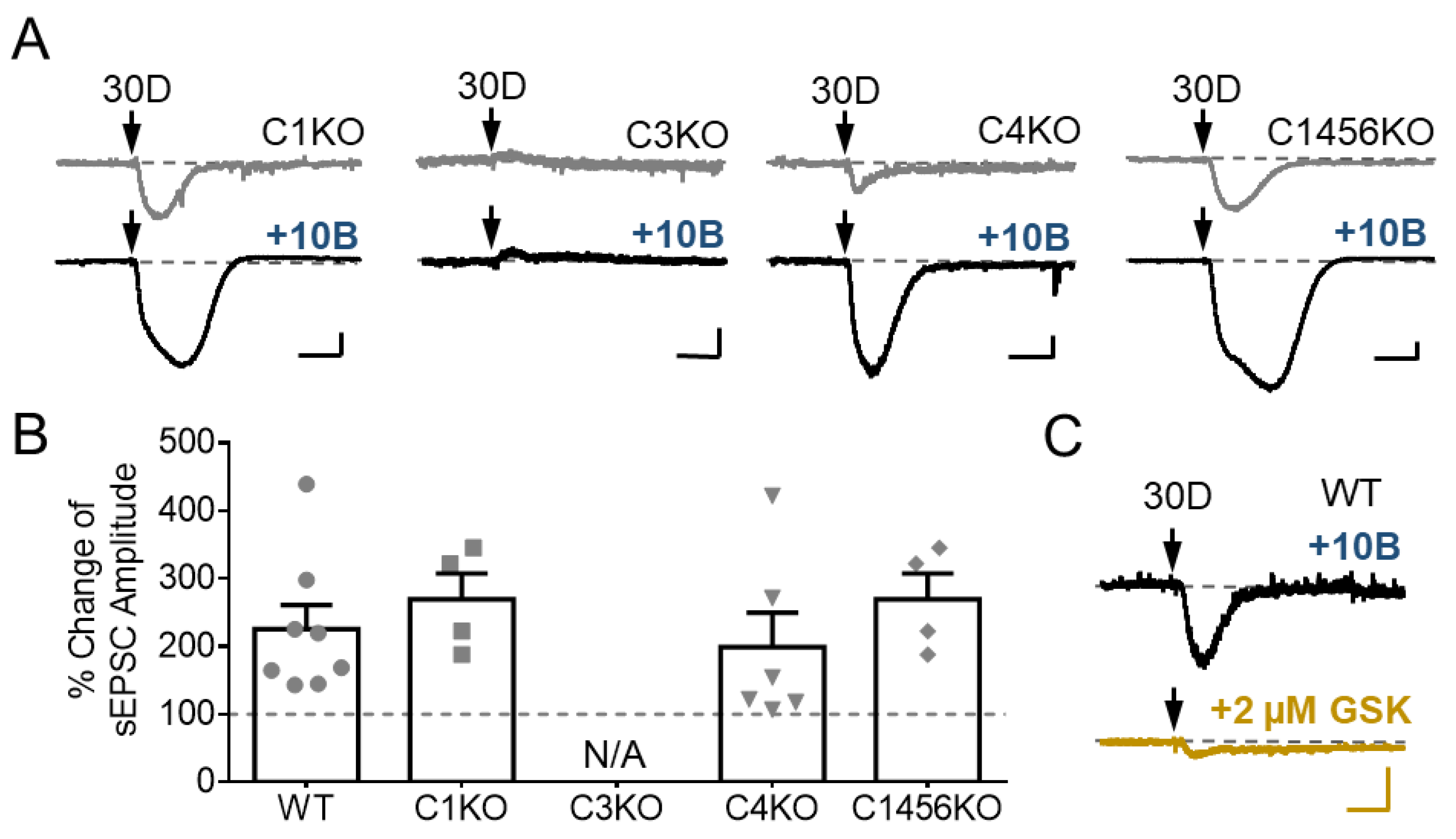
3.4. TRPC3 Underlies the Potentiation of sEPSC by GABABR Stimulation
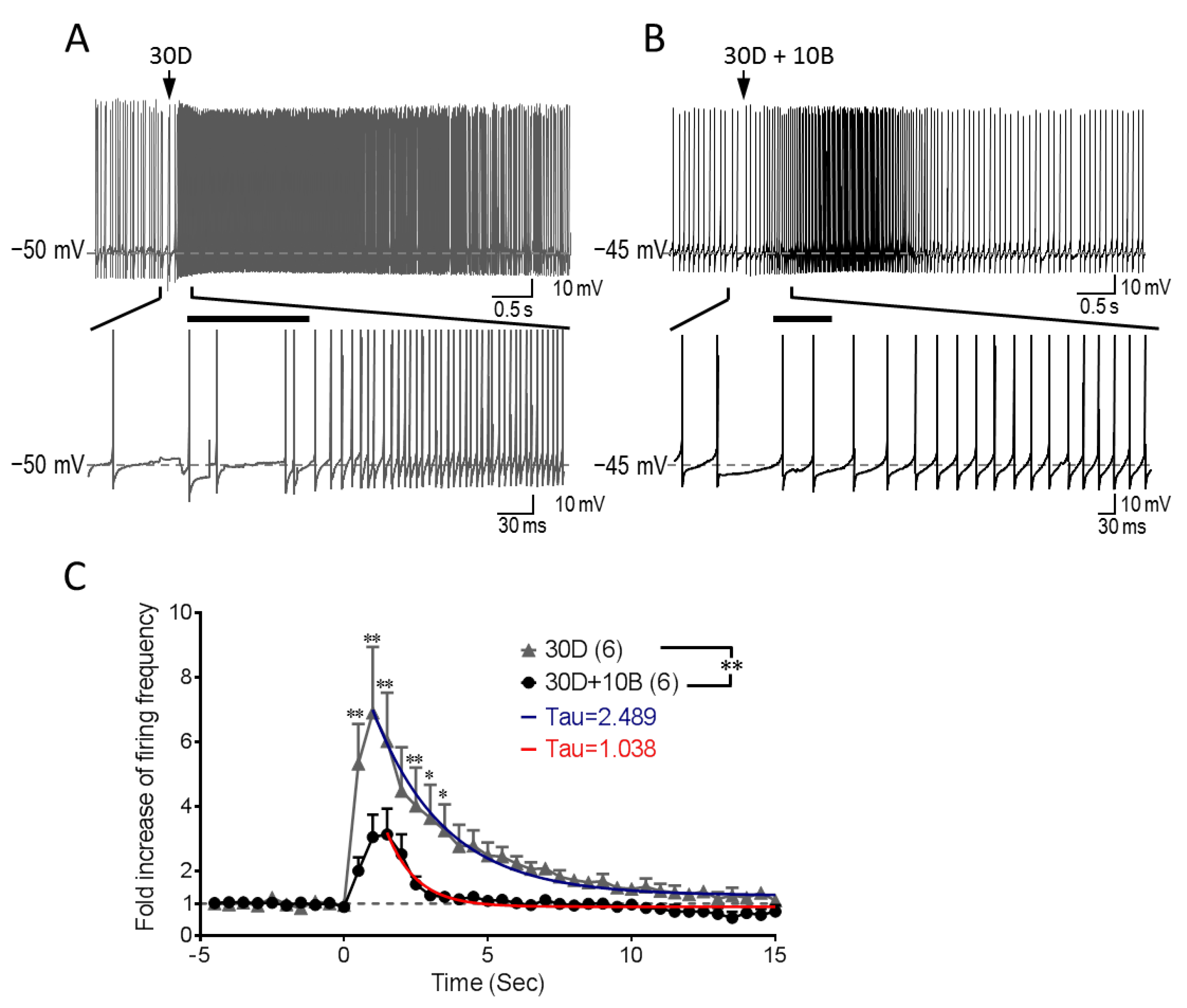
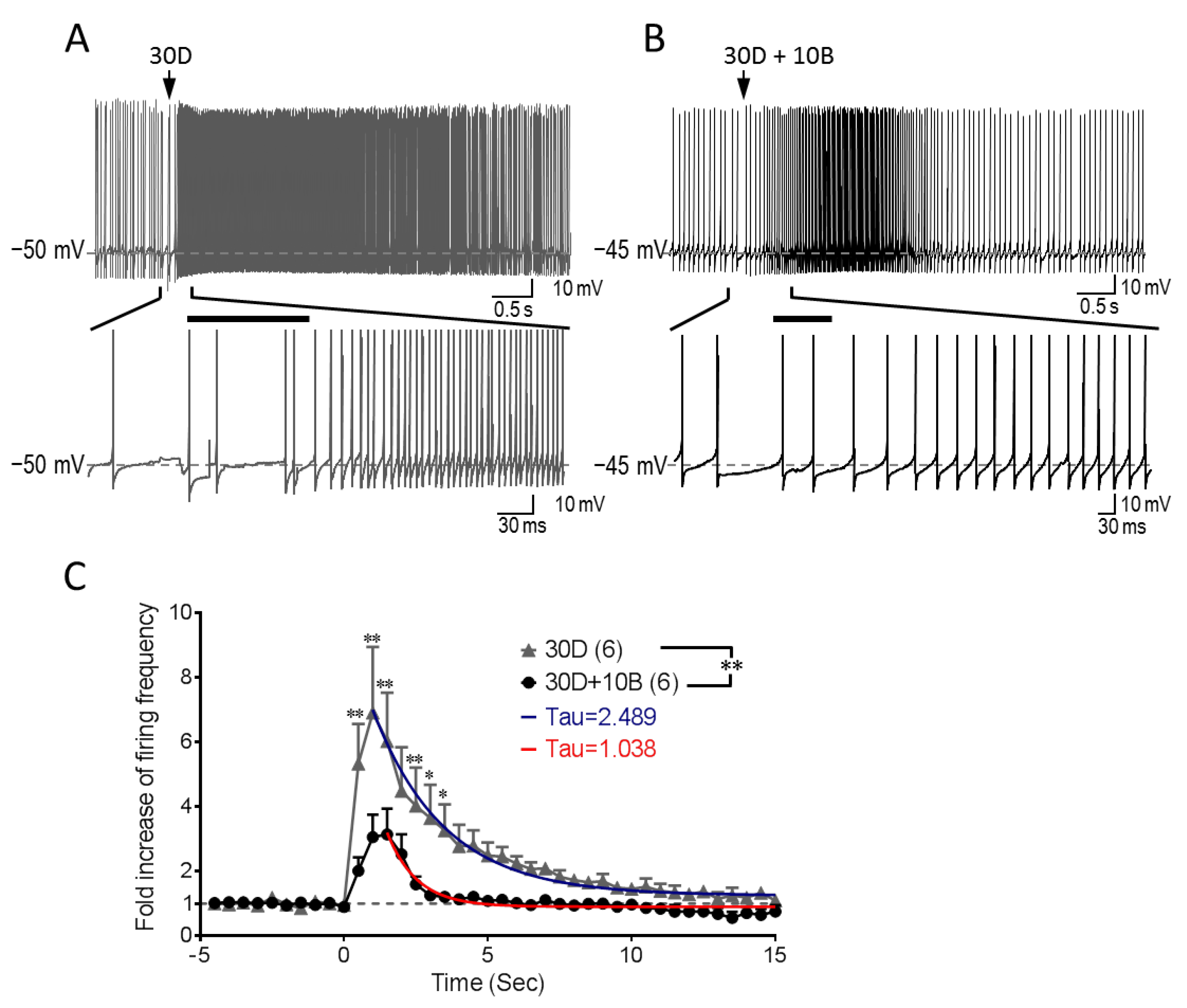
3.5. GABABR Co-Stimulation Reshapes mGluR1-mediated Increase of Purkinje Cell Firing
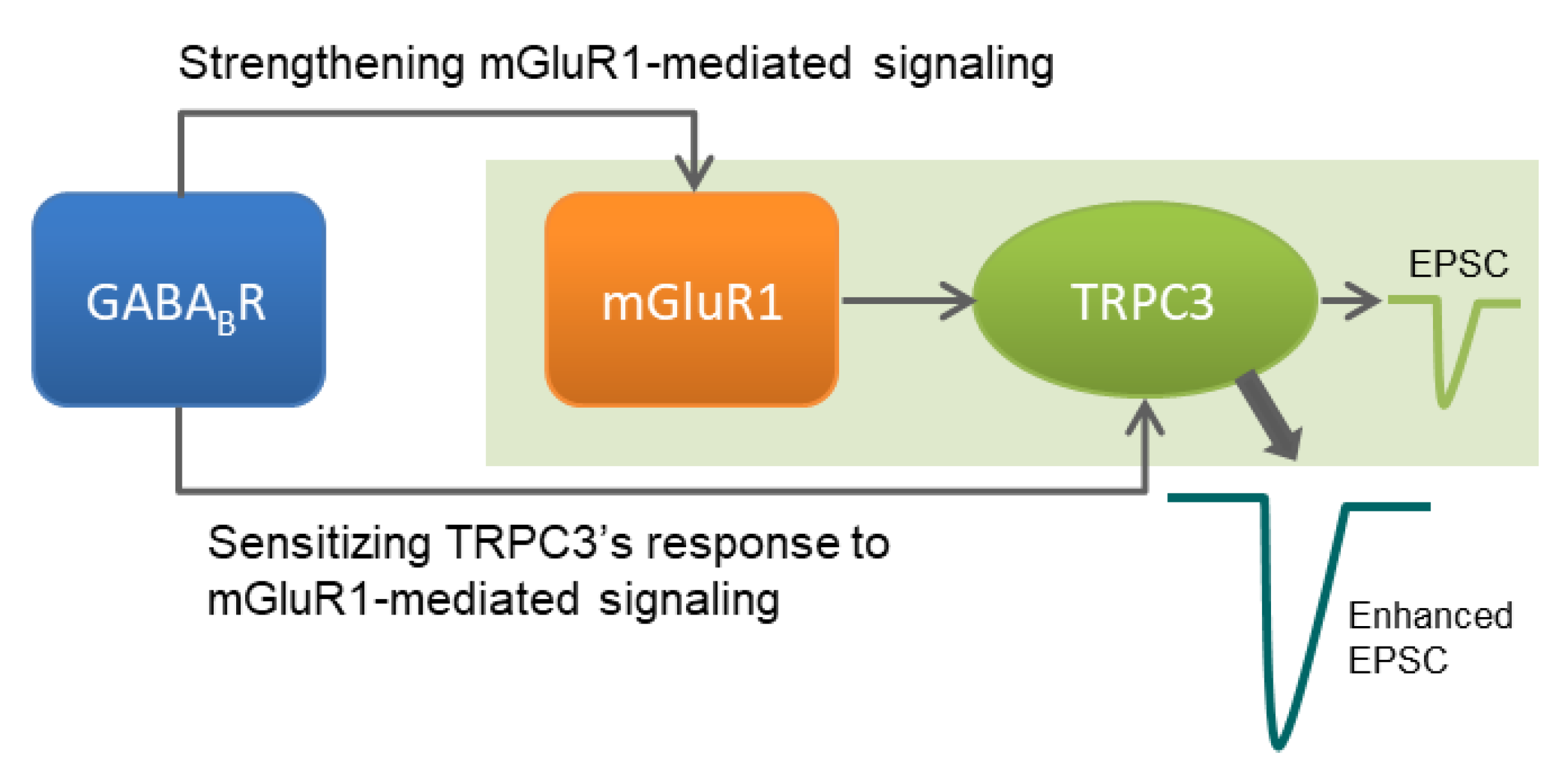
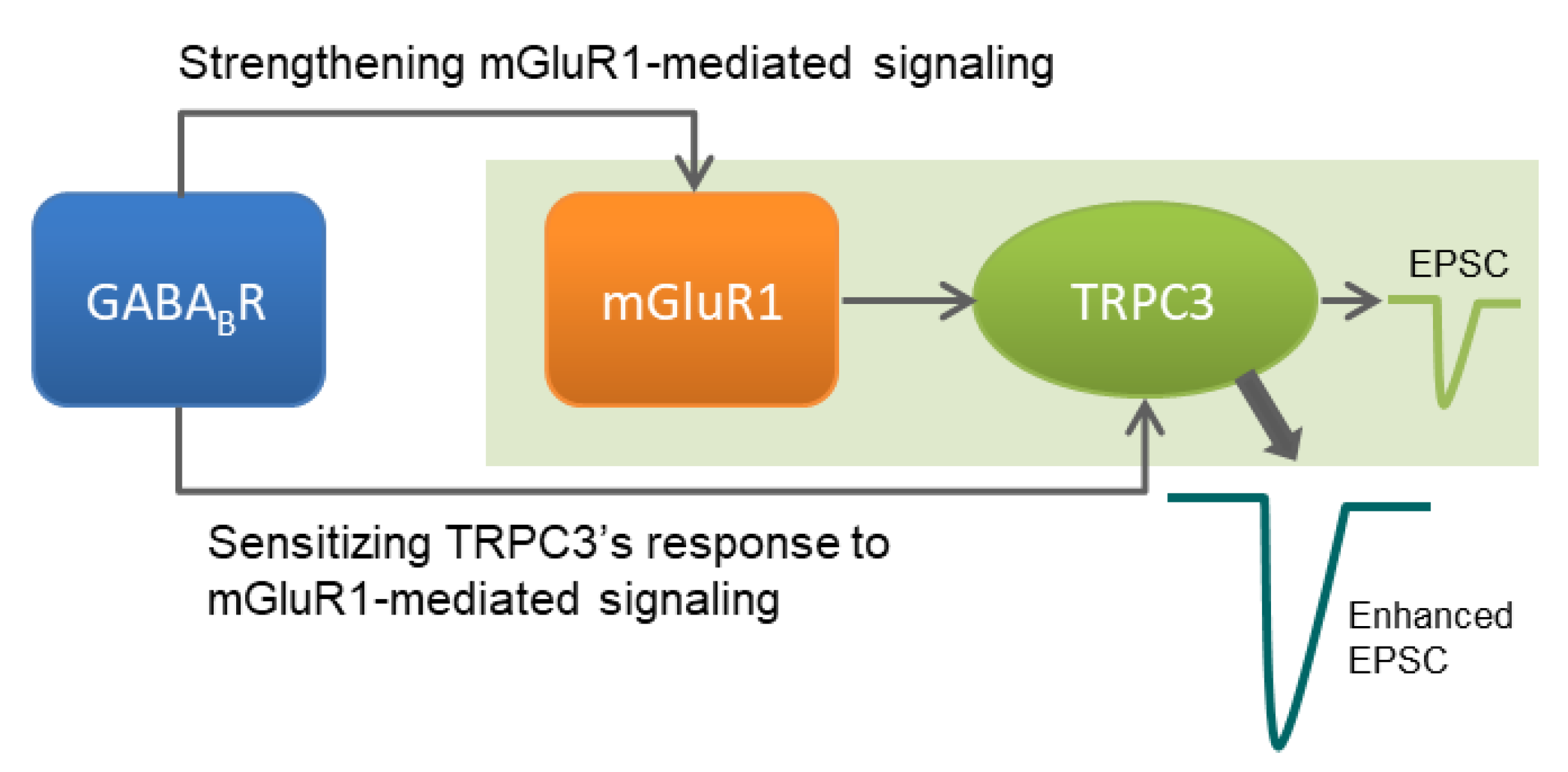
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Apps, R.; Garwicz, M. Anatomical and physiological foundations of cerebellar information processing. Nat. Rev. Neurosci. 2005, 6, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Voogd, J.; Glickstein, M. The anatomy of the cerebellum. Trends Neurosci. 1998, 21, 370–375. [Google Scholar] [CrossRef]
- Hartmann, J.; Dragicevic, E.; Adelsberger, H.; Henning, H.; Sumser, M.; Abramowitz, J.; Blum, R.; Dietrich, A.; Freichel, M.; Flockerzi, V.; et al. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 2008, 59, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Konnerth, A. TRPC3-dependent synaptic transmission in central mammalian neurons. J. Mol. Med. (Berl.) 2015, 93, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Hirono, M.; Yoshioka, T.; Konishi, S. GABAB receptor activation enhances mGluR-mediated responses at cerebellar excitatory synapses. Nat. Neurosci. 2001, 4, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Henning, H.; Konnerth, A. mGluR1/TRPC3-mediated synaptic transmission and calcium signaling in mammalian central neurons. Cold Spring Harb. Perspect. Biol. 2011, 3, a006726. [Google Scholar] [CrossRef] [PubMed]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Shigemoto, R.; Nakanishi, S.; Mizuno, N. Distribution of the mRNA for a metabotropic glutamate receptor (mGluR1) in the central nervous system: An in situ hybridization study in adult and developing rat. J. Comp. Neurol. 1992, 322, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Yamaguchi, K.; Nagao, S.; Yamazaki, T. Long-term depression as a model of cerebellar plasticity. Prog. Brain Res. 2014, 210, 1–30. [Google Scholar] [PubMed]
- Abramowitz, J.; Birnbaumer, L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 2009, 23, 297–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.B.; Thakur, D.; Lu, Y.; Zhu, M. TRPC Channels. In Handbook of Ion Channels; Zheng, J., Trudeau, M.C., Eds.; CRC Press: Boca Raton, FL, USA, 2015; pp. 411–426. [Google Scholar]
- Chae, H.G.; Ahn, S.J.; Hong, Y.H.; Chang, W.S.; Kim, J.; Kim, S.J. Transient receptor potential canonical channels regulate the induction of cerebellar long-term depression. J. Neurosci. 2012, 32, 12909–12914. [Google Scholar] [CrossRef] [PubMed]
- Kaupmann, K.; Malitschek, B.; Schuler, V.; Heid, J.; Froestl, W.; Beck, P.; Mosbacher, J.; Bischoff, S.; Kulik, A.; Shigemoto, R.; et al. GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature 1998, 396, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Fritschy, J.M.; Meskenaite, V.; Weinmann, O.; Honer, M.; Benke, D.; Mohler, H. GABAB-receptor splice variants GB1a and GB1b in rat brain: Developmental regulation, cellular distribution and extrasynaptic localization. Eur. J. Neurosci. 1999, 11, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Lujan, R.; Shigemoto, R. Localization of metabotropic GABA receptor subunits GABAB1 and GABAB2 relative to synaptic sites in the rat developing cerebellum. Eur. J. Neurosci. 2006, 23, 1479–1490. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, Y.S.; Yuan, J.P.; Petralia, R.S.; Worley, P.F.; Linden, D.J. Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature 2003, 426, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Lozinskaya, I.; Costell, M.; Lin, Z.A.; Ball, J.; Bernard, R.; Behm, D.P.; Marino, J.; Schnackenberg, C. Characterization of small molecule TRPC3 and TRPC6 agonist and antagonists. Biophys. J. 2013, 104, 454. [Google Scholar] [CrossRef]
- Padgett, C.L.; Slesinger, P.A. GABAB receptor coupling to G-proteins and ion channels. Adv. Pharmacol. 2010, 58, 123–147. [Google Scholar] [PubMed]
- Hirano, T. GABA and synaptic transmission in the cerebellum. In Handbook of the Cerebellum and Cerebellar Disorders; Manto, M., Schmahmann, J., Rossi, F., Gruol, D., Koibuchi, N., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 881–893. [Google Scholar]
- Billinton, A.; Upton, N.; Bowery, N.G. GABA(B) receptor isoforms GBR1a and GBR1b, appear to be associated with pre- and post-synaptic elements respectively in rat and human cerebellum. Br. J. Pharmacol. 1999, 126, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lin, Z.; Voges, K.; Ju, C.; Gao, Z.; Bosman, L.W.; Ruigrok, T.J.; Hoebeek, F.E.; De Zeeuw, C.I.; Schonewille, M. Cerebellar modules operate at different frequencies. ELife 2014, 3, e02536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, J.P.; Hong, C.; Park, E.J.; Jeon, J.H.; Cho, N.H.; Kim, I.G.; Choe, H.; Muallem, S.; Kim, H.J.; So, I. Selective Gαi subunits as novel direct activators of transient receptor potential canonical (TRPC)4 and TRPC5 channels. J. Biol. Chem. 2012, 287, 17029–17039. [Google Scholar] [CrossRef] [PubMed]
- Otsuguro, K.-I.; Tang, J.; Tang, Y.; Xiao, R.; Freichel, M.; Tsvilovskyy, V.; Ito, S.; Flockerzi, V.; Zhu, M.; Zholos, A. Isoform-specific inhibition of TRPC4 channel by phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 2008, 283, 10026–10036. [Google Scholar] [CrossRef] [PubMed]
- Thakur, D.P.; Tian, J.B.; Jeon, J.; Xiong, J.; Huang, Y.; Flockerzi, V.; Zhu, M.X. Critical roles of Gi/o proteins and phospholipase C-δ1 in the activation of receptor-operated TRPC4 channels. Proc. Natl. Acad. Sci. USA 2016, 113, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Tabata, T.; Araishi, K.; Hashimoto, K.; Hashimotodani, Y.; van der Putten, H.; Bettler, B.; Kano, M. Ca2+ activity at GABAB receptors constitutively promotes metabotropic glutamate signaling in the absence of GABA. Proc. Natl. Acad. Sci. USA 2004, 101, 16952–16957. [Google Scholar] [CrossRef] [PubMed]
- Pin, J.P.; Bettler, B. Organization and functions of mGlu and GABAB receptor complexes. Nature 2016, 540, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Gerber, U.; Gee, C.; Benquet, P. Metabotropic glutamate receptors: Intracellular signaling pathways. Curr. Opin. Pharmacol. 2007, 7, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Gong, Z.; Liang, Z.L.; Liu, Z.X.; Yang, F.; Sun, Y.J.; Ma, M.L.; Wang, Y.J.; Ji, C.R.; Wang, Y.H.; et al. Arrestin-biased AT1R agonism induces acute catecholamine secretion through TRPC3 coupling. Nat. Commun. 2017, 8, 14335. [Google Scholar] [CrossRef] [PubMed]
- Ordaz, B.; Tang, J.; Xiao, R.; Salgado, A.; Sampieri, A.; Zhu, M.; Vaca, L. Calmodulin and calcium interplay in the modulation of TRPC5 channel activity. Identification of a novel C-terminal domain for calcium/calmodulin-mediated facilitation. J. Biol. Chem. 2005, 280, 30788–30796. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tang, J.; Tikunova, S.; Johnson, J.; Chen, Z.; Qin, N.; Dietrich, A.; Stefani, E.; Birnbaumer, L.; Zhu, M. Activation of TRP3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc. Natl. Acad. Sci. USA 2001, 98, 3168–3173. [Google Scholar] [CrossRef] [PubMed]
- Aiba, A.; Kano, M.; Chen, C.; Stanton, M.E.; Fox, G.D.; Herrup, K.; Zwingman, T.A.; Tonegawa, S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell 1994, 79, 377–388. [Google Scholar] [PubMed]
- Coesmans, M.; Smitt, P.A.; Linden, D.J.; Shigemoto, R.; Hirano, T.; Yamakawa, Y.; van Alphen, A.M.; Luo, C.; van der Geest, J.N.; Kros, J.M.; et al. Mechanisms underlying cerebellar motor deficits due to mGluR1-autoantibodies. Ann. Neurol. 2003, 53, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Fogel, B.L.; Hanson, S.M.; Becker, E.B. Do mutations in the murine ataxia gene trpc3 cause cerebellar ataxia in humans? Mov. Disord. 2015, 30, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Lichtenegger, M.; Groschner, K. TRPC3: A multifunctional signaling molecule. Handb. Exp. Pharmacol. 2014, 222, 67–84. [Google Scholar] [PubMed]
- Frisullo, G.; Della Marca, G.; Mirabella, M.; Caggiula, M.; Broccolini, A.; Rubino, M.; Mennuni, G.; Tonali, P.A.; Batocchi, A.P. A human anti-neuronal autoantibody against GABAB receptor induces experimental autoimmune agrypnia. Exp. Neurol. 2007, 204, 808–818. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, J.; Zhu, M.X. GABAB Receptors Augment TRPC3-Mediated Slow Excitatory Postsynaptic Current to Regulate Cerebellar Purkinje Neuron Response to Type-1 Metabotropic Glutamate Receptor Activation. Cells 2018, 7, 90. https://doi.org/10.3390/cells7080090
Tian J, Zhu MX. GABAB Receptors Augment TRPC3-Mediated Slow Excitatory Postsynaptic Current to Regulate Cerebellar Purkinje Neuron Response to Type-1 Metabotropic Glutamate Receptor Activation. Cells. 2018; 7(8):90. https://doi.org/10.3390/cells7080090
Chicago/Turabian StyleTian, Jinbin, and Michael X. Zhu. 2018. "GABAB Receptors Augment TRPC3-Mediated Slow Excitatory Postsynaptic Current to Regulate Cerebellar Purkinje Neuron Response to Type-1 Metabotropic Glutamate Receptor Activation" Cells 7, no. 8: 90. https://doi.org/10.3390/cells7080090