Deubiquitinating Enzymes Related to Autophagy: New Therapeutic Opportunities?


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Autophagy and Ubiquitin System: A Brief Overview
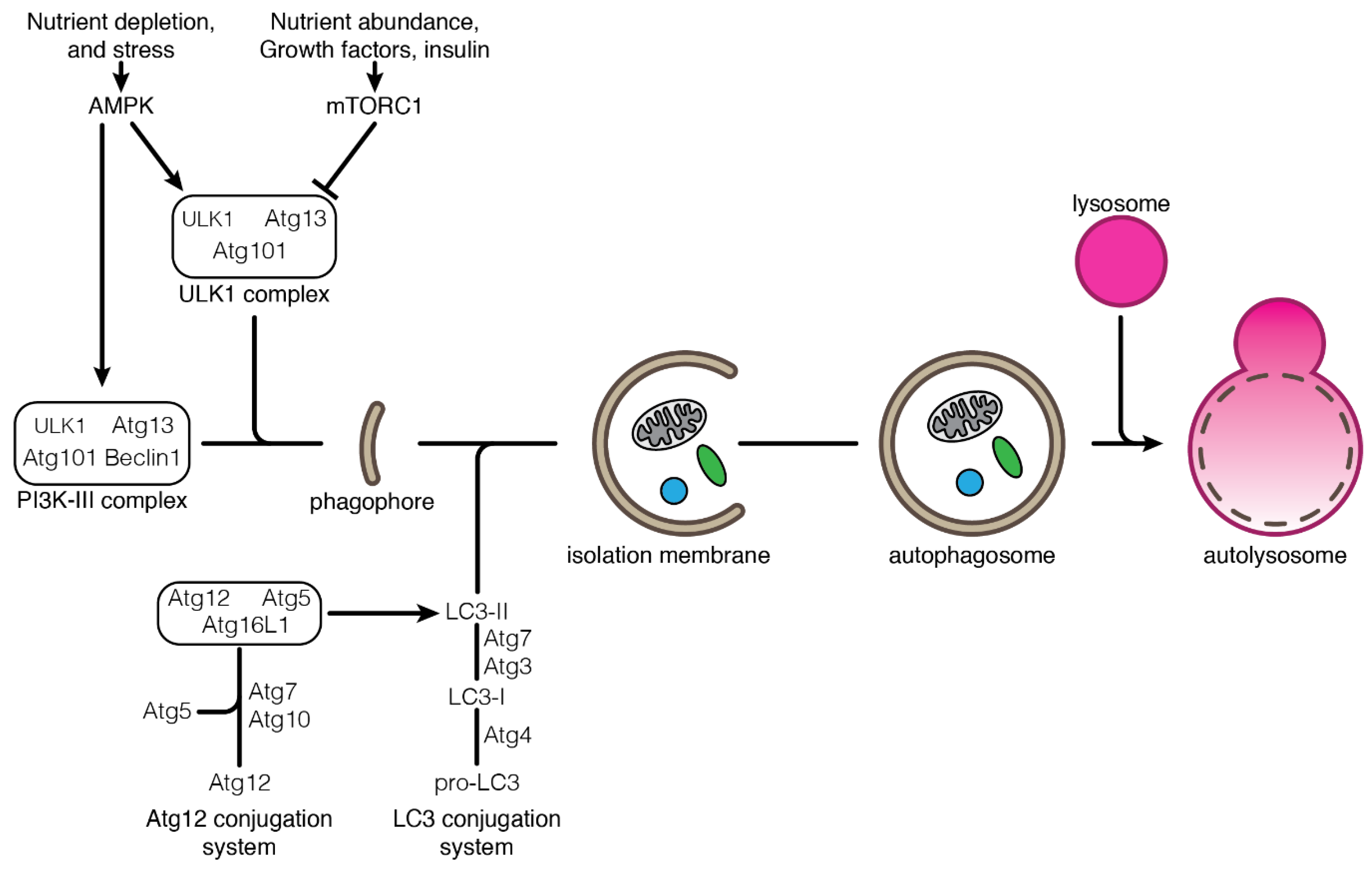
2.1. Autophagy
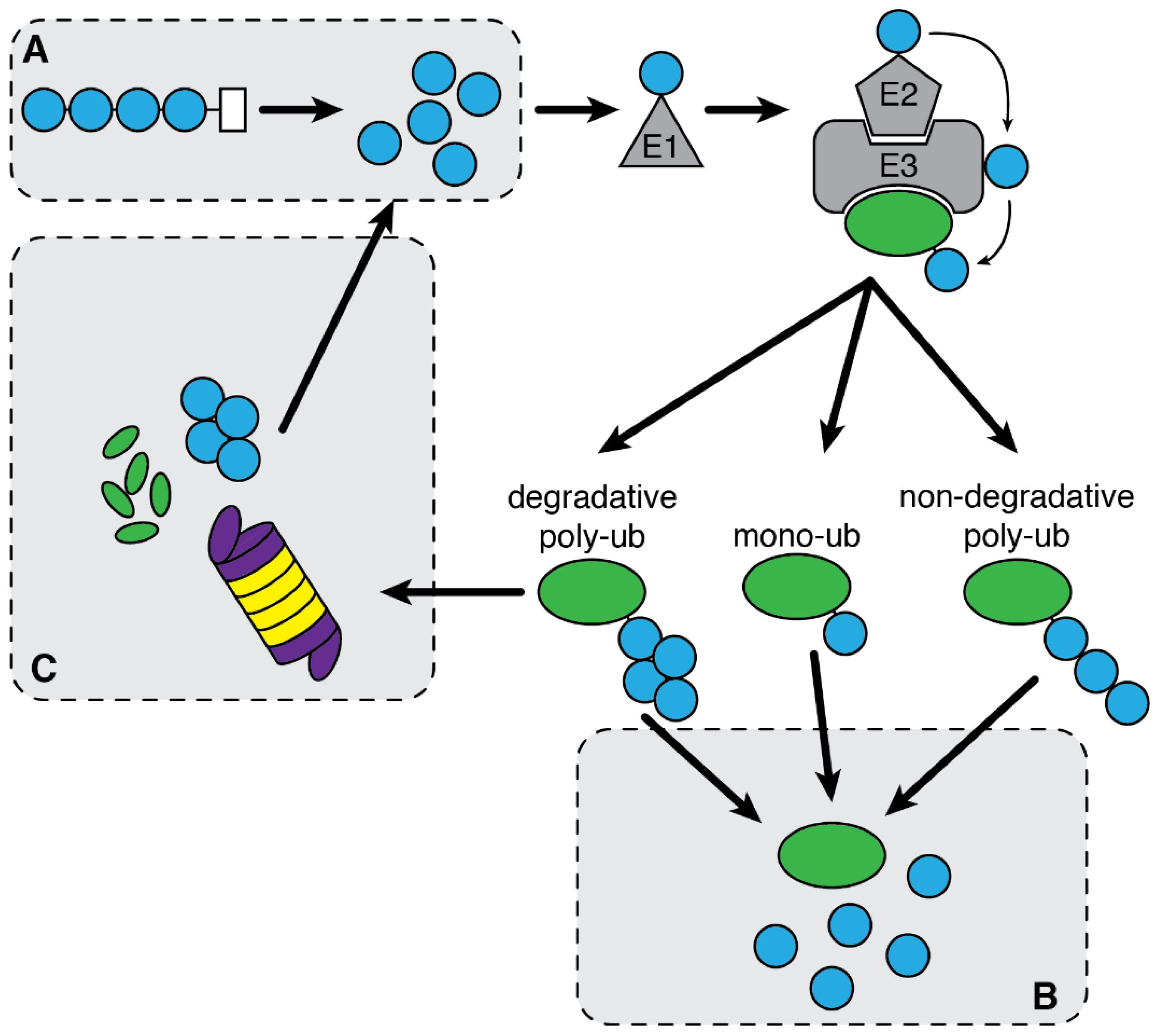
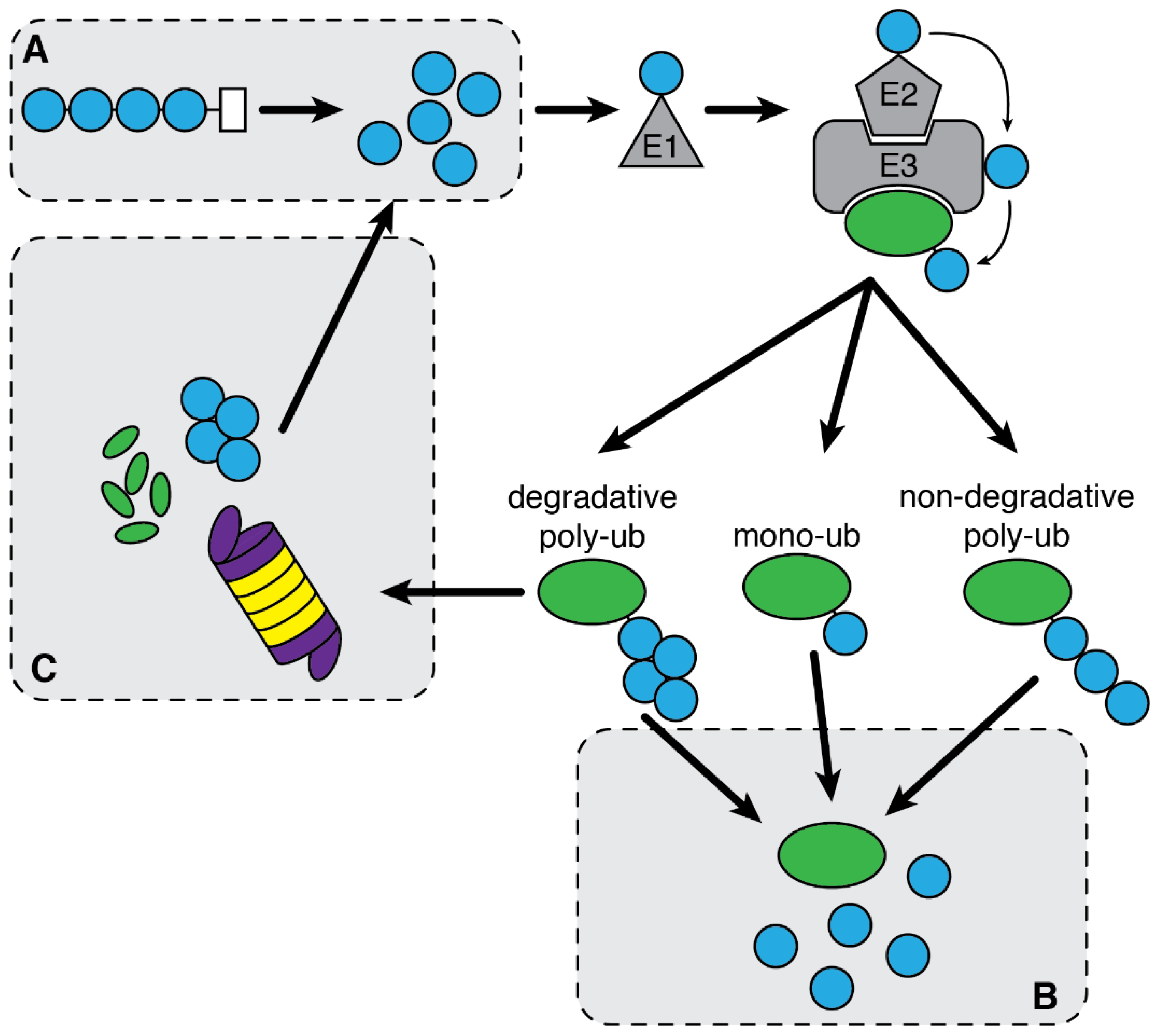
2.2. Ubiquitin System
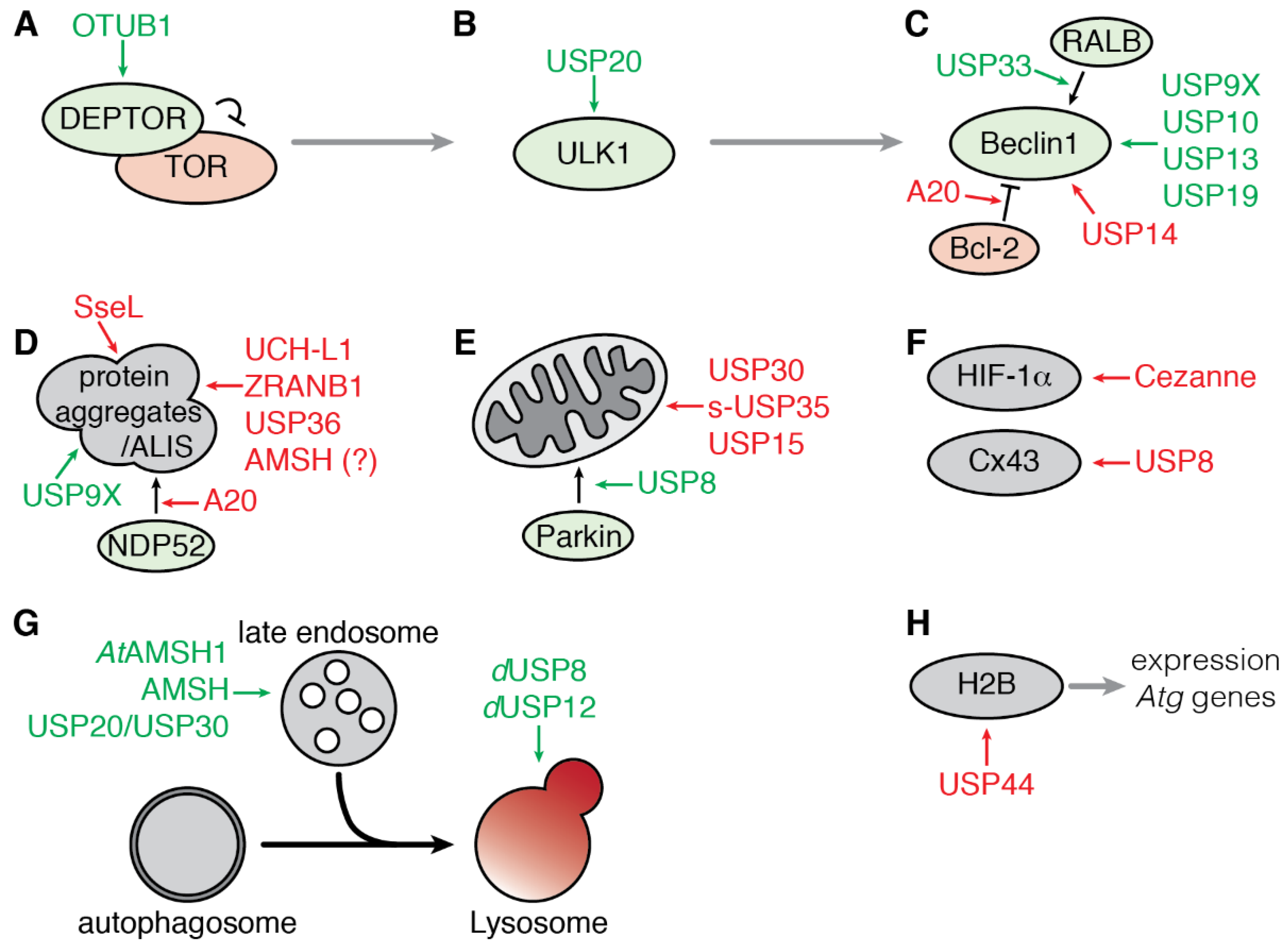
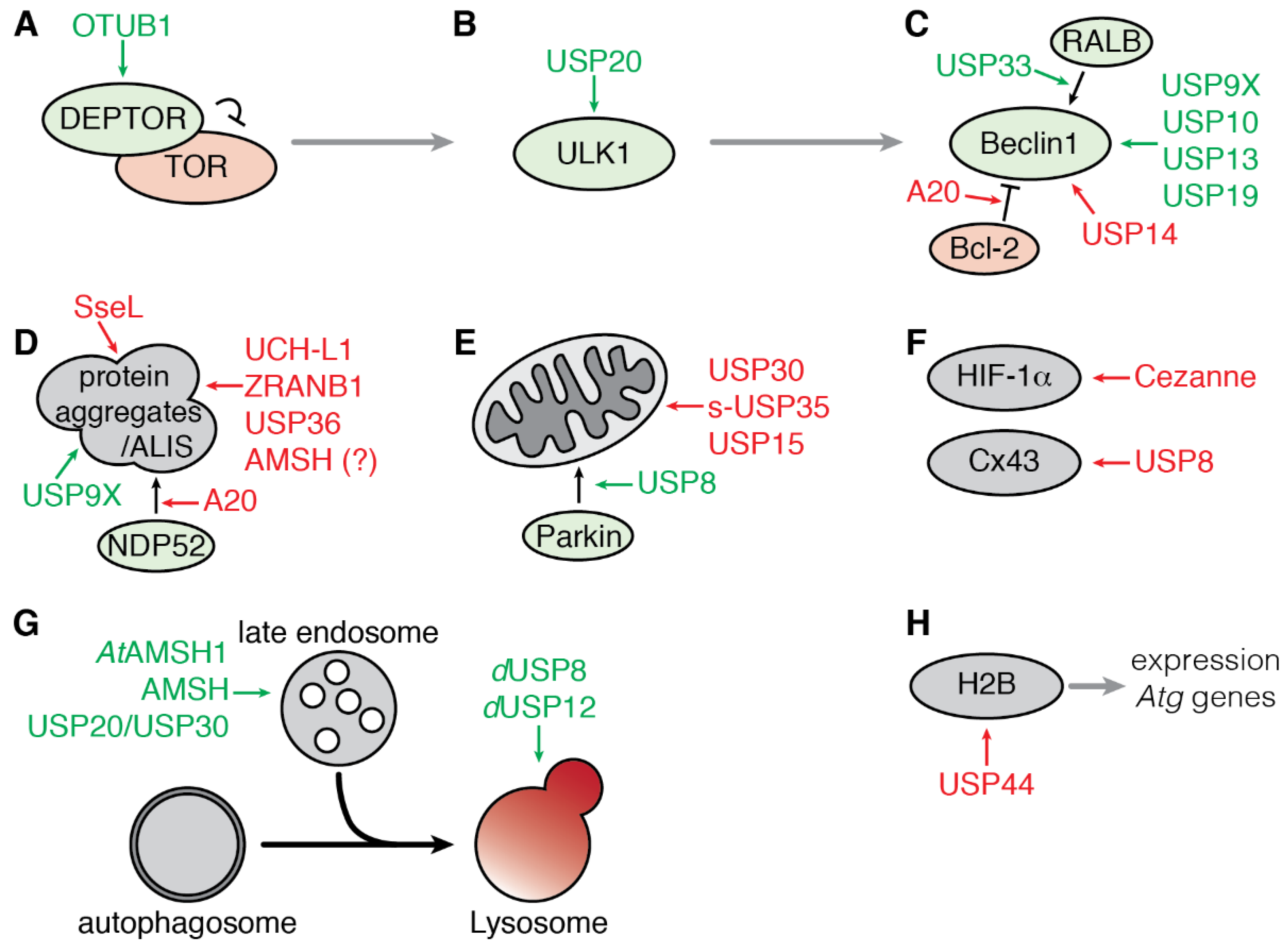
3. Deubiquitinating Enzymes Involved in Autophagy
3.1. Regulation of Early Steps of Autophagy
3.1.1. Regulation of the mTOR Complex 1 by OTUB1
3.1.2. Regulation of ULK1 by USP20
3.1.3. Regulation of the Beclin1 Complex
3.2. Regulation of Selective Autophagy
3.2.1. Aggrephagy
3.2.2. Mitophagy
3.2.3. Targeted Degradation of Cargoes
3.3. Regulation of the Fusion of Endosome to Autophagosome
3.4. Transcriptional Regulation of Autophagy by USP44
3.5. Regulation of Autophagy by Bacterial and Viral DUB-Like Enzymes
4. Targeting Deubiquitinating Enzymes Acting in Autophagy for Therapeutic Purpose
4.1. The Challenge of Developing Drugs Targeting DUBs
4.2. Characterised Inhibitors of DUBs Acting in Autophagy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Johansen, T. Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Wani, W.Y.; Boyer-Guittaut, M.; Dodson, M.; Chatham, J.; Darley-Usmar, V.; Zhang, J. Regulation of autophagy by protein post-translational modification. Lab. Investig. 2015, 95, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kang, R.; Sun, X.; Zhong, M.; Huang, J.; Klionsky, D.J.; Tang, D. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy 2015, 11, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-T.; Chen, G.-C. The role of ubiquitin system in autophagy. In Autophagy in Current Trends in Cellular Physiology and Pathology; Gurbunov, N.V., Schneider, M., Eds.; InTech: Rijeka, Croatia, 2016. [Google Scholar]
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Heride, C.; Urbe, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Komander, D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009, 37, 937–953. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.T.; Komander, D. Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Courtois, G.; Fauvarque, M.O. The many roles of ubiquitin in NF-κB signaling. Biomedicines 2018, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.A. Ubiquitination: Friend and foe in cancer. Int. J. Biochem. Cell Biol. 2018, 101, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–78. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Agarraberes, F.A.; Terlecky, S.R.; Dice, J.F. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J. Cell Biol. 1997, 137, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Patel, B.; Koga, H.; Cuervo, A.M.; Jenny, A. Selective endosomal microautophagy is starvation-inducible in drosophila. Autophagy 2016, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Uytterhoeven, V.; Lauwers, E.; Maes, I.; Miskiewicz, K.; Melo, M.N.; Swerts, J.; Kuenen, S.; Wittocx, R.; Corthout, N.; Marrink, S.J.; et al. Hsc70-4 deforms membranes to promote synaptic protein turnover by endosomal microautophagy. Neuron 2015, 88, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Tekirdag, K.A.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mtor network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase y sorting in saccharomyces cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutouja, F.; Brinkmeier, R.; Mastalski, T.; El Magraoui, F.; Platta, H.W. Regulation of the tumor-suppressor BECLIN 1 by distinct ubiquitination cascades. Int. J. Mol. Sci. 2017, 18, 2541. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Vijay-Kumar, S.; Bugg, C.E.; Cook, W.J. Structure of ubiquitin refined at 1.8 Å resolution. J. Mol. Biol. 1987, 194, 531–544. [Google Scholar] [CrossRef]
- Kiel, C.; Serrano, L. The ubiquitin domain superfold: Structure-based sequence alignments and characterization of binding epitopes. J. Mol. Biol. 2006, 355, 821–844. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Passmore, L.A.; Barford, D. Getting into position: The catalytic mechanisms of protein ubiquitylation. Biochem. J. 2004, 379, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Shabek, N. Ubiquitin ligases: Structure, function, and regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Herr, R.A.; Hansen, T.H. Ubiquitination of substrates by esterification. Traffic 2012, 13, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Finley, D.; Varshavsky, A. Ubiquitin dependence of selective protein degradation demonstrated in the mammalian cell cycle mutant ts85. Cell 1984, 37, 57–66. [Google Scholar] [CrossRef]
- Haglund, K.; Di Fiore, P.P.; Dikic, I. Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem. Sci. 2003, 28, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Kirisako, T.; Kamei, K.; Murata, S.; Kato, M.; Fukumoto, H.; Kanie, M.; Sano, S.; Tokunaga, F.; Tanaka, K.; Iwai, K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006, 25, 4877–4887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rittinger, K.; Ikeda, F. Linear ubiquitin chains: Enzymes, mechanisms and biology. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Eddins, M.J.; Varadan, R.; Fushman, D.; Pickart, C.M.; Wolberger, C. Crystal structure and solution NMR studies of Lys48-linked tetraubiquitin at neutral pH. J. Mol. Biol. 2007, 367, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Varadan, R.; Assfalg, M.; Haririnia, A.; Raasi, S.; Pickart, C.; Fushman, D. Solution conformation of Lys63-linked di-ubiquitin chain provides clues to functional diversity of polyubiquitin signaling. J. Biol. Chem. 2004, 279, 7055–7063. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. Deubiquitylation and regulation of the immune response. Nat. Rev. Immunol. 2008, 8, 501–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Barsukov, I.; Coulson, J.M.; Liu, H.; Rigden, D.J.; Urbe, S. Deubiquitylases from genes to organism. Physiol. Rev. 2013, 93, 1289–1315. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates, J.R., III; Koonin, E.V.; Deshaies, R.J. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002, 298, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Engel, E.; Viargues, P.; Mortier, M.; Taillebourg, E.; Coute, Y.; Thevenon, D.; Fauvarque, M.O. Identifying USPs regulating immune signals in drosophila: USP2 deubiquitinates Imd and promotes its degradation by interacting with the proteasome. Cell Commun. Signal. 2014, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Kuang, E.; Qi, J.; Ronai, Z. Emerging roles of E3 ubiquitin ligases in autophagy. Trends Biochem. Sci. 2013, 38, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Wang, X.; Yu, Y.; Deng, L.; Chen, L.; Peng, X.; Jiao, C.; Gao, G.; Tan, X.; Pan, W.; et al. OTUB1 protein suppresses mTOR complex 1 (mTORC1) activity by deubiquitinating the mTORC1 inhibitor DEPTOR. J. Biol. Chem. 2018, 293, 4883–4892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, H.; Su, G.Q.; Dong, W.; Zhang, L.; Xie, W.; Yao, L.M.; Chen, P.; Wang, Z.X.; Liou, Y.C.; You, H. Chaperone-like protein p32 regulates ULK1 stability and autophagy. Cell Death Differ. 2015, 22, 1812–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazio, F.; Carinci, M.; Valacca, C.; Bielli, P.; Strappazzon, F.; Antonioli, M.; Ciccosanti, F.; Rodolfo, C.; Campello, S.; Fimia, G.M.; et al. Fine-tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J. Cell Biol. 2016, 215, 841–856. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Seo, D.; Kim, S.J.; Choi, D.W.; Park, J.S.; Ha, J.; Choi, J.; Lee, J.H.; Jung, S.M.; Seo, K.W.; et al. The deubiquitinating enzyme USP20 stabilizes ULK1 and promotes autophagy initiation. EMBO Rep. 2018, 19, e44378. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Glover, K.; Su, M.; Sinha, S.C. Conformational flexibility of BECN1: Essential to its key role in autophagy and beyond. Protein Sci. 2016, 25, 1767–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.H.; Lee, M.J.; Park, S.; Oh, D.C.; Elsasser, S.; Chen, P.C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, J.; Hathaway, N.A.; Tone, Y.; Crosas, B.; Elsasser, S.; Kirkpatrick, D.S.; Leggett, D.S.; Gygi, S.P.; King, R.W.; Finley, D. Deubiquitinating enzyme UBP6 functions noncatalytically to delay proteasomal degradation. Cell 2006, 127, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, B.G.; Han, W.H.; Lee, J.H.; Cho, J.Y.; Park, W.S.; Maurice, M.M.; Han, J.K.; Lee, M.J.; Finley, D.; et al. Deubiquitination of dishevelled by USP14 is required for wnt signaling. Oncogenesis 2013, 2, e64. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Shan, B.; Lee, B.H.; Zhu, K.; Zhang, T.; Sun, H.; Liu, M.; Shi, L.; Liang, W.; Qian, L.; et al. Phosphorylation and activation of ubiquitin-specific protease-14 by Akt regulates the ubiquitin-proteasome system. eLife 2015, 4, e10510. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Shan, B.; Sun, H.; Xiao, J.; Zhu, K.; Xie, X.; Li, X.; Liang, W.; Lu, X.; Qian, L.; et al. Usp14 regulates autophagy by suppressing K63 ubiquitination of Beclin 1. Genes Dev. 2016, 30, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kehrl, J.H. TRAF6 and A20 regulate Lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal 2010, 3, ra42. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Lee, S.; Kim, M.J.; Chun, E.; Lee, K.Y. Ubiquitin-specific protease 14 negatively regulates toll-like receptor 4-mediated signaling and autophagy induction by inhibiting ubiquitination of TAK1-binding protein 2 and Beclin 1. Front Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Elgendy, M.; Ciro, M.; Abdel-Aziz, A.K.; Belmonte, G.; Dal Zuffo, R.; Mercurio, C.; Miracco, C.; Lanfrancone, L.; Foiani, M.; Minucci, S. Beclin 1 restrains tumorigenesis through Mcl-1 destabilization in an autophagy-independent reciprocal manner. Nat. Commun. 2014, 5, 5637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwickart, M.; Huang, X.; Lill, J.R.; Liu, J.; Ferrando, R.; French, D.M.; Maecker, H.; O’Rourke, K.; Bazan, F.; Eastham-Anderson, J.; et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010, 463, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Perez-Mancera, P.A.; Rust, A.G.; van der Weyden, L.; Kristiansen, G.; Li, A.; Sarver, A.L.; Silverstein, K.A.; Grutzmann, R.; Aust, D.; Rummele, P.; et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 2012, 486, 266–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platta, H.W.; Abrahamsen, H.; Thoresen, S.B.; Stenmark, H. Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin 1. Biochem. J. 2012, 441, 399–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Tian, S.; Chen, Y.; Zhang, C.; Xie, W.; Xia, X.; Cui, J.; Wang, R.F. Usp19 modulates autophagy and antiviral immune responses by deubiquitinating beclin-1. EMBO J. 2016, 35, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Simicek, M.; Lievens, S.; Laga, M.; Guzenko, D.; Aushev, V.N.; Kalev, P.; Baietti, M.F.; Strelkov, S.V.; Gevaert, K.; Tavernier, J.; et al. The deubiquitylase USP33 discriminates between RALB functions in autophagy and innate immune response. Nat. Cell Biol. 2013, 15, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Wooten, M.W.; Geetha, T.; Babu, J.R.; Seibenhener, M.L.; Peng, J.; Cox, N.; Diaz-Meco, M.T.; Moscat, J. Essential role of sequestosome 1/p62 in regulating accumulation of lys63-ubiquitinated proteins. J. Biol. Chem. 2008, 283, 6783–6789. [Google Scholar] [CrossRef] [PubMed]
- Zatloukal, K.; Stumptner, C.; Fuchsbichler, A.; Heid, H.; Schnoelzer, M.; Kenner, L.; Kleinert, R.; Prinz, M.; Aguzzi, A.; Denk, H. P62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am. J. Pathol. 2002, 160, 255–263. [Google Scholar] [CrossRef]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjorkoy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The lewy body in parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Engelender, S. Ubiquitination of α-synuclein and autophagy in parkinson’s disease. Autophagy 2014, 4, 372–374. [Google Scholar] [CrossRef]
- Rott, R.; Szargel, R.; Haskin, J.; Bandopadhyay, R.; Lees, A.J.; Shani, V.; Engelender, S. α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 18666–18671. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Hsu, S.D. Familial mutations and post-translational modifications of UCH-L1 in parkinson’s disease and neurodegenerative disorders. Curr. Protein Pept. Sci. 2017, 18, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Pukass, K.; Richter-Landsberg, C. Inhibition of UCH-L1 in oligodendroglial cells results in microtubule stabilization and prevents α-synuclein aggregate formation by activating the autophagic pathway: Implications for multiple system atrophy. Front Cell Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Setsuie, R.; Wada, K. The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem. Int. 2007, 51, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Meray, R.K.; Grammatopoulos, T.N.; Fredenburg, R.A.; Cookson, M.R.; Liu, Y.; Logan, T.; Lansbury, P.T., Jr. Membrane-associated farnesylated UCH-L1 promotes α-synuclein neurotoxicity and is a therapeutic target for parkinson’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 4635–4640. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, Z.; Lang, J.; Perrett, R.M.; Elschami, M.; Hurry, M.E.; Kim, H.T.; Mazaraki, D.; Szabo, A.; Kessler, B.M.; Goldberg, A.L.; et al. Deubiquitinase USP8 regulates α-synuclein clearance and modifies its toxicity in lewy body disease. Proc. Natl. Acad. Sci. USA 2016, 113, E4688–E4697. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.M.M.; Wong, E.S.P.; Dawson, V.L.; Dawson, T.; Lim, K.-L. Lysine 63-linked polyubiquitin potentially partners with p62 to promote the clearance of protein inclusions by autophagy. Autophagy 2014, 4, 251–253. [Google Scholar] [CrossRef]
- Nibe, Y.; Oshima, S.; Kobayashi, M.; Maeyashiki, C.; Matsuzawa, Y.; Otsubo, K.; Matsuda, H.; Aonuma, E.; Nemoto, Y.; Nagaishi, T.; et al. Novel polyubiquitin imaging system, PolyUb-FC, reveals that K33-linked polyubiquitin is recruited by SQSTM1/p62. Autophagy 2018, 14, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Licchesi, J.D.; Mieszczanek, J.; Mevissen, T.E.; Rutherford, T.J.; Akutsu, M.; Virdee, S.; El Oualid, F.; Chin, J.W.; Ovaa, H.; Bienz, M.; et al. An ankyrin-repeat ubiquitin-binding domain determines trabid’s specificity for atypical ubiquitin chains. Nat. Struct. Mol. Biol. 2011, 19, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Nezis, I.P.; Simonsen, A.; Sagona, A.P.; Finley, K.; Gaumer, S.; Contamine, D.; Rusten, T.E.; Stenmark, H.; Brech, A. Ref(2)p, the drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J. Cell Biol. 2008, 180, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, B.J.; Isakson, P.; Lewerenz, J.; Sanchez, H.; Kotzebue, R.W.; Cumming, R.C.; Harris, G.L.; Nezis, I.P.; Schubert, D.R.; Simonsen, A.; et al. P62, Ref(2)p and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy 2014, 7, 572–583. [Google Scholar] [CrossRef]
- Taillebourg, E.; Gregoire, I.; Viargues, P.; Jacomin, A.C.; Thevenon, D.; Faure, M.; Fauvarque, M.O. The deubiquitinating enzyme USP36 controls selective autophagy activation by ubiquitinated proteins. Autophagy 2012, 8, 767–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Muhlinen, N.; Akutsu, M.; Ravenhill, B.J.; Foeglein, A.; Bloor, S.; Rutherford, T.J.; Freund, S.M.; Komander, D.; Randow, F. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol. Cell 2012, 48, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Thurston, T.L.; Ryzhakov, G.; Bloor, S.; von Muhlinen, N.; Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009, 10, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Inomata, M.; Niida, S.; Shibata, K.; Into, T. Regulation of toll-like receptor signaling by NDP52-mediated selective autophagy is normally inactivated by A20. Cell Mol. Life Sci. 2012, 69, 963–979. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014, 460, 127–139. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, T.G.; Muqit, M.M. PINK1 and parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Fon, E.A. The three ‘p’s of mitophagy: Parkin, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Hirose, S. Regulation of mitochondrial morphology by USP30, a deubiquitinating enzyme present in the mitochondrial outer membrane. Mol. Biol. Cell 2008, 19, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.R.; Martinez, A.; Lane, J.D.; Mayor, U.; Clague, M.J.; Urbe, S. Usp30 deubiquitylates mitochondrial parkin substrates and restricts apoptotic cell death. EMBO Rep. 2015, 16, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.N.; Baughman, J.M.; Phu, L.; Tea, J.S.; Yu, C.; Coons, M.; Kirkpatrick, D.S.; Bingol, B.; Corn, J.E. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol. 2015, 17, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Serricchio, M.; Jauregui, M.; Shanbhag, R.; Stoltz, T.; Di Paolo, C.T.; Kim, P.K.; McQuibban, G.A. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 2015, 11, 595–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, M.A.; Swatek, K.N.; Hospenthal, M.K.; Komander, D. Ubiquitin linkage-specific affimers reveal insights into K6-linked ubiquitin signaling. Mol. Cell 2017, 68, 233–246.e5. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G. Organelle turnover: A USP30 safety catch restrains the trigger for mitophagy and pexophagy. Curr. Biol. 2018, 28, R842–R845. [Google Scholar] [CrossRef] [PubMed]
- Marcassa, E.; Kallinos, A.; Jardine, J.; Rusilowicz-Jones, E.V.; Martinez, A.; Kuehl, S.; Islinger, M.; Clague, M.J.; Urbe, S. Dual role of USP30 in controlling basal pexophagy and mitophagy. EMBO Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, C.M.; Lodhi, I.J. Peroxisomal dysfunction in age-related diseases. Trends Endocrinol. Metab. 2017, 28, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; Mattie, S.; Prudent, J.; McBride, H.M. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature 2017, 542, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, T.; Haddad, D.; Wauters, F.; Van Humbeeck, C.; Mandemakers, W.; Koentjoro, B.; Sue, C.; Gevaert, K.; De Strooper, B.; Verstreken, P.; et al. The deubiquitinase USP15 antagonizes parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 2014, 23, 5227–5242. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Tang, M.Y.; Perusse, J.R.; Dashti, E.A.; Aguileta, M.A.; McLelland, G.L.; Gros, P.; Shaler, T.A.; Faubert, D.; Coulombe, B.; et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 2014, 33, 2473–2491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbi, M.E.; Hu, H.; Kshitiz; Ahmed, I.; Levchenko, A.; Semenza, G.L. Chaperone-mediated autophagy targets hypoxia-inducible factor-1α (HIF-1α) for lysosomal degradation. J. Biol. Chem. 2013, 288, 10703–10714. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.V.; Fofo, H.; Bejarano, E.; Bento, C.F.; Ramalho, J.S.; Girao, H.; Pereira, P. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 2013, 9, 1349–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremm, A.; Moniz, S.; Mader, J.; Rocha, S.; Komander, D. Cezanne (OTUD7B) regulates HIF-1α homeostasis in a proteasome-independent manner. EMBO Rep. 2014, 15, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Troilo, A.; Alexander, I.; Muehl, S.; Jaramillo, D.; Knobeloch, K.P.; Krek, W. HIF1α deubiquitination by USP8 is essential for ciliogenesis in normoxia. EMBO Rep. 2014, 15, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Leithe, E. Regulation of connexins by the ubiquitin system: Implications for intercellular communication and cancer. Biochim. Biophys. Acta 2016, 1865, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Leithe, E.; Rivedal, E. Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J. Cell Sci. 2004, 117, 1211–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.M.; Baker, S.M.; Gumpert, A.M.; Segretain, D.; Buckheit, R.W., 3rd. Gap junction turnover is achieved by the internalization of small endocytic double-membrane vesicles. Mol. Biol. Cell 2009, 20, 3342–3352. [Google Scholar] [CrossRef] [PubMed]
- Fong, J.T.; Kells, R.M.; Gumpert, A.M.; Marzillier, J.Y.; Davidson, M.W.; Falk, M.M. Internalized gap junctions are degraded by autophagy. Autophagy 2012, 8, 794–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Hu, Q.; Peng, H.; Peng, C.; Zhou, L.; Lu, J.; Huang, C. The ubiquitin-specific protease USP8 deubiquitinates and stabilizes Cx43. J. Biol. Chem. 2018, 293, 8275–8284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlin, I.; Schwartz, H.; Nash, P.D. Regulation of epidermal growth factor receptor ubiquitination and trafficking by the USP8.STAM complex. J. Biol. Chem. 2010, 285, 34909–34921. [Google Scholar] [CrossRef] [PubMed]
- Niendorf, S.; Oksche, A.; Kisser, A.; Lohler, J.; Prinz, M.; Schorle, H.; Feller, S.; Lewitzky, M.; Horak, I.; Knobeloch, K.P. Essential role of ubiquitin-specific protease 8 for receptor tyrosine kinase stability and endocytic trafficking in vivo. Mol. Cell Biol. 2007, 27, 5029–5039. [Google Scholar] [CrossRef] [PubMed]
- Row, P.E.; Liu, H.; Hayes, S.; Welchman, R.; Charalabous, P.; Hofmann, K.; Clague, M.J.; Sanderson, C.M.; Urbé, S. The mit domain of UBPY constitutes a CHMP binding and endosomal localization signal required for efficient epidermal growth factor receptor degradation. J. Biol. Chem. 2007, 282, 30929–30937. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Sanchez, D.; Furlan, M.; Colombo, M.I. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in K562 cells. Traffic 2008, 9, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Kyuuma, M.; Kikuchi, K.; Kojima, K.; Sugawara, Y.; Sato, M.; Mano, N.; Goto, J.; Takeshita, T.; Yamamoto, A.; Sugamura, K.; et al. AMSH, an ESCRT-Ш associated enzyme, deubiquitinates cargo on MVB/late endosomes. Cell Struct. Funct. 2007, 31, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.M.; Boucrot, E.; Villen, J.; Affar El, B.; Gygi, S.P.; Gottlinger, H.G.; Kirchhausen, T. Targeting of AMSH to endosomes is required for epidermal growth factor receptor degradation. J. Biol. Chem. 2007, 282, 9805–9812. [Google Scholar] [CrossRef] [PubMed]
- McCullough, J.; Row, P.E.; Lorenzo, O.; Doherty, M.; Beynon, R.; Clague, M.J.; Urbe, S. Activation of the endosome-associated ubiquitin isopeptidase AMSH by STAM, a component of the multivesicular body-sorting machinery. Curr. Biol. 2006, 16, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Sierra, M.I.; Wright, M.H.; Nash, P.D. AMSH interacts with ESCRT-0 to regulate the stability and trafficking of CXCR4. J. Biol. Chem. 2010, 285, 13990–14004. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbe, S. Endocytosis: The dub version. Trends Cell Biol. 2006, 16, 551–559. [Google Scholar] [CrossRef] [PubMed]
- McCullough, J.; Clague, M.J.; Urbe, S. AMSH is an endosome-associated ubiquitin isopeptidase. J. Cell Biol. 2004, 166, 487–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, N.; Owada, Y.; Yamada, M.; Miura, S.; Murata, K.; Asao, H.; Kondo, H.; Sugamura, K. Loss of neurons in the hippocampus and cerebral cortex of AMSH-deficient mice. Mol. Cell Biol. 2001, 21, 8626–8637. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Tamai, K.; Watanabe, M.; Kyuuma, M.; Ono, M.; Sugamura, K.; Tanaka, N. AMSH is required to degrade ubiquitinated proteins in the central nervous system. Biochem. Biophys. Res. Commun. 2011, 408, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Katsiarimpa, A.; Kalinowska, K.; Anzenberger, F.; Weis, C.; Ostertag, M.; Tsutsumi, C.; Schwechheimer, C.; Brunner, F.; Huckelhoven, R.; Isono, E. The deubiquitinating enzyme AMSH1 and the ESCRT-Ш subunit VSP2.1 are required for autophagic degradation in arabidopsis. Plant Cell 2013, 25, 2236–2252. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Yanez, X.; Aguilar-Gurrieri, C.; Jacomin, A.C.; Journet, A.; Mortier, M.; Taillebourg, E.; Soleilhac, E.; Weissenhorn, W.; Fauvarque, M.O. CHMP1B is a target of USP8/UBPY regulated by ubiquitin during endocytosis. PLoS Genet. 2018, 14, e1007456. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.H.; Berlin, I.; Nash, P.D. Regulation of endocytic sorting by ESCRT-DUB-mediated deubiquitination. Cell Biochem. Biophys. 2011, 60, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Du, J.; Lei, C.; Liu, M.; Zhu, A.J. UBPY controls the stability of the ESCRT-0 subunit Hrs in development. Development 2014, 141, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Row, P.E.; Prior, I.A.; McCullough, J.; Clague, M.J.; Urbe, S. The ubiquitin isopeptidase UBPY regulates endosomal ubiquitin dynamics and is essential for receptor down-regulation. J. Biol. Chem. 2006, 281, 12618–12624. [Google Scholar] [CrossRef] [PubMed]
- Jacomin, A.C.; Bescond, A.; Soleilhac, E.; Gallet, B.; Schoehn, G.; Fauvarque, M.O.; Taillebourg, E. The deubiquitinating enzyme UBPY is required for lysosomal biogenesis and productive autophagy in drosophila. PLoS ONE 2015, 10, e0143078. [Google Scholar] [CrossRef] [PubMed]
- Moretti, J.; Chastagner, P.; Liang, C.C.; Cohn, M.A.; Israel, A.; Brou, C. The ubiquitin-specific protease 12 (USP12) is a negative regulator of notch signaling acting on notch receptor trafficking toward degradation. J. Biol. Chem. 2012, 287, 29429–29441. [Google Scholar] [CrossRef] [PubMed]
- Jacomin, A.C.; Fauvarque, M.O.; Taillebourg, E. A functional endosomal pathway is necessary for lysosome biogenesis in Drosophila. BMC Cell Biol. 2016, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Xiang, Y.K. Trafficking of β-adrenergic receptors: Implications in intracellular receptor signaling. In Progress in Molecular Biology and Translational Science; Wu, G., Ed.; Academic Press: New York, NY, USA, 2015; Volume 132, pp. 151–188. [Google Scholar]
- Kommaddi, R.P.; Jean-Charles, P.Y.; Shenoy, S.K. Phosphorylation of the deubiquitinase USP20 by Protein Kinase A regulates post-endocytic trafficking of β2 adrenergic receptors to autophagosomes during physiological stress. J. Biol. Chem. 2015, 290, 8888–8903. [Google Scholar] [CrossRef] [PubMed]
- Berthouze, M.; Venkataramanan, V.; Li, Y.; Shenoy, S.K. The deubiquitinases USP33 and USP20 coordinate β2 adrenergic receptor recycling and resensitization. EMBO J. 2009, 28, 1684–1696. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; He, H.; Tang, Z.; Hattori, T.; Liu, Y.; Young, M.M.; Serfass, J.M.; Chen, L.; Gebru, M.; Chen, C.; et al. An autophagy assay reveals the ESCRT-Ш component CHMP2A as a regulator of phagophore closure. Nat. Commun. 2018, 9, 2855. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Jing, Y.; Kang, X.; Yang, L.; Wang, D.L.; Zhang, W.; Zhang, L.; Chen, P.; Chang, J.F.; Yang, X.M.; et al. Histone H2B monoubiquitination is a critical epigenetic switch for the regulation of autophagy. Nucleic. Acids Res. 2017, 45, 1144–1158. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, F.S.; Thomas, M.; Sachse, M.; Santos, A.J.; Figueira, R.; Holden, D.W. The salmonella deubiquitinase Ssel inhibits selective autophagy of cytosolic aggregates. PLoS Pathog. 2012, 8, e1002743. [Google Scholar] [CrossRef] [PubMed]
- Rytkonen, A.; Poh, J.; Garmendia, J.; Boyle, C.; Thompson, A.; Liu, M.; Freemont, P.; Hinton, J.C.; Holden, D.W. Ssel, a salmonella deubiquitinase required for macrophage killing and virulence. Proc. Natl. Acad. Sci. USA 2007, 104, 3502–3507. [Google Scholar] [CrossRef] [PubMed]
- Choy, A.; Dancourt, J.; Mugo, B.; O’Connor, T.J.; Isberg, R.R.; Melia, T.J.; Roy, C.R. The legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 2012, 338, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Pantoom, S.; Wu, Y.W. Elucidation of the anti-autophagy mechanism of the legionella effector RavZ using semisynthetic LC3 proteins. eLife 2017, 6, e23905. [Google Scholar] [CrossRef] [PubMed]
- Kubori, T.; Bui, X.T.; Hubber, A.; Nagai, H. Legionella RavZ plays a role in preventing ubiquitin recruitment to bacteria-containing vacuoles. Front Cell Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Horenkamp, F.A.; Kauffman, K.J.; Kohler, L.J.; Sherwood, R.K.; Krueger, K.P.; Shteyn, V.; Roy, C.R.; Melia, T.J.; Reinisch, K.M. The legionella anti-autophagy effector RavZ targets the autophagosome via PI3P- and curvature-sensing motifs. Dev. Cell 2015, 34, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479–480, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.J.; Britton, P. Involvement of autophagy in coronavirus replication. Viruses 2012, 4, 3440–3451. [Google Scholar] [CrossRef] [PubMed]
- Clementz, M.A.; Chen, Z.; Banach, B.S.; Wang, Y.; Sun, L.; Ratia, K.; Baez-Santos, Y.M.; Wang, J.; Takayama, J.; Ghosh, A.K.; et al. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 2010, 84, 4619–4629. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, K.; Xing, Y.; Tu, J.; Yang, X.; Zhao, Q.; Li, K.; Chen, Z. Coronavirus membrane-associated papain-like proteases induce autophagy through interacting with beclin1 to negatively regulate antiviral innate immunity. Protein Cell 2014, 5, 912–927. [Google Scholar] [CrossRef] [PubMed]
- Munz, C. Beclin-1 targeting for viral immune escape. Viruses 2011, 3, 1166–1178. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Xing, Y.; Chen, X.; Zheng, Y.; Yang, Y.; Nichols, D.B.; Clementz, M.A.; Banach, B.S.; Li, K.; Baker, S.C.; et al. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of sting-mediated signaling. PLoS ONE 2012, 7, e30802. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, P.; Li, M.; Li, W.; Yao, T.; Wu, J.W.; Gu, W.; Cohen, R.E.; Shi, Y. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell 2002, 111, 1041–1054. [Google Scholar] [CrossRef]
- Hu, M.; Li, P.; Song, L.; Jeffrey, P.D.; Chenova, T.A.; Wilkinson, K.D.; Cohen, R.E.; Shi, Y. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005, 24, 3747–3756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avvakumov, G.V.; Walker, J.R.; Xue, S.; Finerty, P.J., Jr.; Mackenzie, F.; Newman, E.M.; Dhe-Paganon, S. Amino-terminal dimerization, NRDP1-rhodanese interaction, and inhibited catalytic domain conformation of the ubiquitin-specific protease 8 (USP8). J. Biol. Chem. 2006, 281, 38061–38070. [Google Scholar] [CrossRef] [PubMed]
- Das, C.; Hoang, Q.Q.; Kreinbring, C.A.; Luchansky, S.J.; Meray, R.K.; Ray, S.S.; Lansbury, P.T.; Ringe, D.; Petsko, G.A. Structural basis for conformational plasticity of the parkinson’s disease-associated ubiquitin hydrolase UCH-L1. Proc. Natl. Acad. Sci. USA 2006, 103, 4675–4680. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, M.J.; Iphofer, A.; Akutsu, M.; Altun, M.; di Gleria, K.; Kramer, H.B.; Fiebiger, E.; Dhe-Paganon, S.; Kessler, B.M. Structural basis and specificity of human otubain 1-mediated deubiquitination. Biochem. J. 2009, 418, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, G.; Habeck, M.; Masino, L.; Svergun, D.I.; Pastore, A. Structure validation of the Josephin domain of ataxin-3: Conclusive evidence for an open conformation. J. Biomol. NMR 2006, 36, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Faesen, A.C.; Dirac, A.M.; Shanmugham, A.; Ovaa, H.; Perrakis, A.; Sixma, T.K. Mechanism of USP7/HAUSP activation by its C-terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol. Cell 2011, 44, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Pozhidaeva, A.; Valles, G.; Wang, F.; Wu, J.; Sterner, D.E.; Nguyen, P.; Weinstock, J.; Kumar, K.G.S.; Kanyo, J.; Wright, D.; et al. USP7-specific inhibitors target and modify the enzyme’s active site via distinct chemical mechanisms. Cell Chem. Biol. 2017, 24, 1501–1512.e5. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, L.; Wu, J.; Sokirniy, I.; Nguyen, P.; Bregnard, T.; Weinstock, J.; Mattern, M.; Bezsonova, I.; Hancock, W.W.; et al. Active site-targeted covalent irreversible inhibitors of USP7 impair the functions of Foxp3+ T-regulatory cells by promoting ubiquitination of Tip60. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Kim, W.; Gygi, S.; Ye, Y. Characterization of the deubiquitinating activity of USP19 and its role in endoplasmic reticulum-associated degradation. J. Biol. Chem. 2014, 289, 3510–3517. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.M.; Bhattacharyya, B.; Rachel, R.A.; Coppola, V.; Tessarollo, L.; Householder, D.B.; Fletcher, C.F.; Miller, R.J.; Copeland, N.G.; Jenkins, N.A. Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat. Genet. 2002, 32, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Boselli, M.; Lee, B.H.; Robert, J.; Prado, M.A.; Min, S.W.; Cheng, C.; Silva, M.C.; Seong, C.; Elsasser, S.; Hatle, K.M.; et al. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J. Biol. Chem. 2017, 292, 19209–19225. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Yukiue, H.; Moriyama, S.; Kobayashi, Y.; Nakashima, Y.; Kaji, M.; Fukai, I.; Kiriyama, M.; Yamakawa, Y.; Fujii, Y. Expression of the protein gene product 9.5, PGP9.5, is correlated with T-status in non-small cell lung cancer. Jpn. J. Clin. Oncol. 2001, 31, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lashuel, H.A.; Choi, S.; Xing, X.; Case, A.; Ni, J.; Yeh, L.A.; Cuny, G.D.; Stein, R.L.; Lansbury, P.T., Jr. Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell line. Chem. Biol. 2003, 10, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Huo, H.; Yang, C.; Zhang, T.; Chu, Y.; Liu, Y. Ubiquitin C-terminal hydrolase L1 regulates autophagy by inhibiting autophagosome formation through its deubiquitinating enzyme activity. Biochem. Biophys. Res. Commun. 2018, 497, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.; Leach, C.A.; Goldenberg, S.J.; Francis, D.M.; Kodrasov, M.P.; Tian, X.; Shanks, J.; Sterner, D.E.; Bernal, A.; Mattern, M.R.; et al. Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein Sci. 2008, 17, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driessen, S.; Berleth, N.; Friesen, O.; Loffler, A.S.; Bohler, P.; Hieke, N.; Stuhldreier, F.; Peter, C.; Schink, K.O.; Schultz, S.W.; et al. Deubiquitinase inhibition by WP1130 leads to ULK1 aggregation and blockade of autophagy. Autophagy 2015, 11, 1458–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapuria, V.; Peterson, L.F.; Fang, D.; Bornmann, W.G.; Talpaz, M.; Donato, N.J. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 2010, 70, 9265–9276. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Chen, Z.; Liu, H.; Yan, C.; Chen, M.; Feng, D.; Yan, C.; Wu, H.; Du, L.; Wang, Y.; et al. A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res. 2014, 24, 482–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluge, A.F.; Lagu, B.R.; Maiti, P.; Jaleel, M.; Webb, M.; Malhotra, J.; Mallat, A.; Srinivas, P.A.; Thompson, J.E. Novel highly selective inhibitors of ubiquitin specific protease 30 (USP30) accelerate mitophagy. Bioorg. Med. Chem. Lett. 2018, 28, 2655–2659. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacomin, A.-C.; Taillebourg, E.; Fauvarque, M.-O. Deubiquitinating Enzymes Related to Autophagy: New Therapeutic Opportunities? Cells 2018, 7, 112. https://doi.org/10.3390/cells7080112
Jacomin A-C, Taillebourg E, Fauvarque M-O. Deubiquitinating Enzymes Related to Autophagy: New Therapeutic Opportunities? Cells. 2018; 7(8):112. https://doi.org/10.3390/cells7080112
Chicago/Turabian StyleJacomin, Anne-Claire, Emmanuel Taillebourg, and Marie-Odile Fauvarque. 2018. "Deubiquitinating Enzymes Related to Autophagy: New Therapeutic Opportunities?" Cells 7, no. 8: 112. https://doi.org/10.3390/cells7080112